

Abstract
X‐linked adrenoleukodystrophy (X‐ALD) is the most common peroxisomal disorder. The disease is characterized by the accumulation of very long‐chain fatty acids (VLCFA; >C22) in plasma and tissues. X‐ALD is caused by mutations in the ABCD1 gene encoding ALDP, an adenosine triphosphate (ATP)‐binding‐cassette (ABC) transporter located in the peroxisomal membrane. In this paper, we describe the current knowledge on the function of ALDP, its role in peroxisomal VLCFA beta‐oxidation and the consequences of a defect in ALDP on VLCFA metabolism. Furthermore, we pay special attention to the role of the VLCFA elongation system in VLCFA homeostasis, with elongation of very long‐chain fatty acids like‐1 (ELOVL1) as key player, and its relevance to X‐ALD.
Keywords: ALDP, ELOVL, peroxisome, VLCFA
INTRODUCTION
X‐linked adrenoleukodystrophy (X‐ALD) is a progressive neurodegenerative disease that is caused by mutations in the ABCD1 gene (52). The disease affects the cerebral white matter, peripheral nerves, adrenal cortex and testis (51). It is the most common peroxisomal disorder with a hemizygote frequency of 1:21.000 (6). X‐ALD is characterized clinically by a striking and unpredictable variation in phenotypic expression, ranging from the rapidly progressive childhood cerebral form (CCALD) to the more slowly progressive adult form adrenomyeloneuropathy (AMN) and variants without neurological involvement (“Addison‐only” phenotype). Males in the same family may express different phenotypes 4, 34. X‐ALD is characterized biochemically by elevated plasma and tissue levels of saturated straight chain very long‐chain fatty acids (VLCFA: C24:0 and C26:0) (49) and monounsaturated VLCFA (C26:1) (76). The elevation of VLCFA is associated with reduced beta‐oxidation of VLCFA in peroxisomes.
VLCFA: A BIOMARKER FOR X‐ALD
In the early 1970s, Powers and Schaumburg reported the presence of intracytoplasmic lamellae and lamellar lipid inclusions in adrenal cells, testicular cells, Schwann cells and brain macrophages from patients with X‐ALD (61). Biochemical analysis of these inclusion bodies revealed that they contained cholesterol, phospholipids and gangliosides esterified with saturated VLCFA (29). The subsequent discovery by Moser and colleagues that these elevated VLCFA, in particular C26:0, were also detectable in fibroblasts, blood cells and especially plasma of X‐ALD patients has been of crucial importance as this finding allowed unequivocal identification of male patients (50). Plasma VLCFA analysis is still the best initial biomarker of X‐ALD.
DISCOVERY OF THE ABCD1 GENE AND RESOLUTION OF THE FUNCTION OF ALDP
Genetic linkage with glucose‐6‐phosphate dehydrogenase (G6PD) pointed the X‐ALD locus to the end of the long arm of the X‐chromosome, Xq28 (45). In 1993, the X‐ALD gene (ABCD1) was identified using positional cloning strategies (52). The ABCD1 gene is 19.9 kb long, contains 10 exons and encodes a protein, ALDP, of 745 amino acids.
The capacity to degrade VLCFA with 22 or more carbons is impaired in X‐ALD both in vitro 44, 67, 82 and in vivo (22). Free fatty acids, including VLCFA, are, in general, metabolically inactive. The activation of VLCFA by thioesterification to coenzyme A (VLCFA‐CoA) is essential for degrading VLCFA via peroxisomal beta‐oxidation. In 1986, Hashmi and coworkers had demonstrated that the activation of VLCFA to their respective CoA esters in peroxisomes is deficient in fibroblasts from X‐ALD patients (25). Based on these findings, it was anticipated that ALDP would have VLCFacyl‐CoA synthetase activity. It therefore came as a surprise that when the ABCD1 gene was identified, its product was not a VLCFacyl‐CoA synthetase (52). Recently, it was demonstrated that the fatty acid transport protein 4 is the fatty acyl‐CoA synthetase that preferentially activates VLCFA to their CoA derivatives (31).
Based on sequence homology, ALDP belongs to the ATP‐binding cassette (ABC) superfamily of transmembrane transporter proteins and it shares no homology with VLCFacyl‐CoA synthetases. The proteins of the ABC superfamily are membrane‐bound proteins. Their structure has been highly conserved from eubacteria to mammals. They catalyze the ATP‐dependent transmembrane transport of a wide variety of substrates across extra‐ and intracellular membranes (16). Most ABC transporters are expressed as full‐transporter proteins like the cystic fibrosis transmembrane regulator (ABCC7) and the multidrug resistance protein (MDR1, ABCB1). ALDP is a half‐size ABC transporter. The amino‐terminal half of ALDP contains the transmembrane domain which consists of six putative transmembrane segments. The carboxyl‐terminal half contains a single hydrophilic nucleotide‐binding domain. A functional ABC transporter is composed of two transmembrane domains and two nucleotide‐binding domains. Therefore, dimerization is a prerequisite for ABC half‐transporters (reviewed in (35)). Besides ALDP, the ABCD subfamily contains three additional ABC half‐transporters with significant homology to ALDP: the ALD‐related protein (ALDRP, 66% identity) is encoded by the ABCD2 gene, the more distantly related 70‐kDa peroxisomal membrane protein (PMP70) is encoded by the ABCD3 gene and the PMP70‐related protein (P70R) is encoded by the ABCD4 gene. The proteins belonging to the ABCD subfamily of ABC transporters are also known as peroxisomal ABC proteins. Peroxisomal localization has been demonstrated for ALDP, ALDRP and PMP70. Based on its homology, it was anticipated that P70R would be localized to the peroxisome as well. Recently, however, Kashiwayama and colleagues demonstrated that endogenous P70R localizes to the endoplasmic reticulum and not to the peroxisome (33). ALDP and other peroxisomal membrane proteins are synthesized on cytosolic ribosomes and posttranslationally targeted to peroxisomes. The targeting of peroxisomal membrane proteins to peroxisomes is mediated by PEX19 (24). The PEX19‐binding site is located in the amino‐terminal hydrophilic region preceding the first transmembrane segment. While this region is present in ALDP, ALDRP and PMP70, it is absent in P70R (33).
ALDP is an integral peroxisomal membrane protein with the nucleotide‐binding domain located toward the cytoplasmic surface of the peroxisomal membrane (14). ATP‐binding and ATPase activity has been demonstrated for ALDP (71), and Roerig et al demonstrated that two disease causing missense mutations located in the ATP‐binding domain resulted in either decreased ATP‐binding capacity (p.Ser606Leu) or reduced ATPase activity (p.Gly512Ser) (64).
Evidence that ABCD1 is indeed the gene responsible for X‐ALD came initially from identification of mutations in the ABCD1 gene (52). Complementation studies demonstrated that expression of wild‐type ABCD1 cDNA in fibroblasts derived from X‐ALD patients restored VLCFA beta‐oxidation 8, 65. Furthermore, stable expression of ABCD1 cDNA in X‐ALD fibroblasts reduced VLCFA to normal levels (11). It has been well established that ALDP, ALDRP and PMP70 are at least partially functionally redundant and have overlapping substrate specificities (reviewed in (35)). Indeed, overexpression of PMP70 in X‐ALD fibroblasts partially corrected VLCFA beta‐oxidation, whereas overexpression of ALDRP resulted in complete correction of VLCFA degradation (36).
Recent publications have brought insight in: (i) the physiological function of ALDP; (ii) the primary metabolite transported by ALDP; and (iii) ALDP's role in VLCFA metabolism. In yeast Saccharomyces cerevisiae, peroxisomes are the exclusive site of fatty acid beta‐oxidation. The uptake of fatty acids into peroxisomes occurs via two routes, either as free fatty acids or as long‐chain acyl‐CoA esters (26). The acyl‐CoA route involves the two peroxisomal ABC half‐transporters Pxa1p and Pxa2p that form a heterodimeric complex in the peroxisomal membrane. Yeast mutants lacking both Pxa1 and Pxa2 are unable to grow on oleate‐containing medium, are deficient in the beta‐oxidation of oleic acid and deficient in the peroxisomal import of acyl‐CoA esters (78). Expression of ALDP in these Pxa1/Pxa2 double mutants partially restored the mutant phenotype (78). More recently, we demonstrated using fibroblasts from X‐ALD patients and controls that a deficiency of ALDP impairs peroxisomal β‐oxidation of VLCFA but also raises cytosolic levels of VLCFacyl‐CoA (54). Taken together, these data show that ALDP is active as a homodimer, is involved in the transport of VLCFacyl‐CoA esters across the peroxisomal membrane and that a defect in ALDP results in increased levels of VLCFacyl‐CoA esters in the cytosol.
PEROXISOMAL VLCFA BETA‐OXIDATION
The peroxisomal beta‐oxidation system has the same basic structure as that in mitochondria and is made up of four subsequent steps of dehydrogenation, hydration, dehydrogenation again and thiolytic cleavage (reviewed in (81))). The beta‐oxidation of VLCFA involves the concerted action of ACOX1, DBP and either PTH1 or PTH2 (SCPx). The ultimate result is that the first two carbon atoms of the fatty acyl‐CoA ester are released as acetyl‐CoA leaving a shortened acyl‐CoA ester which can undergo subsequent rounds of beta‐oxidation. As peroxisomes are not able to degrade fatty acyl‐CoAs to completion, the chain‐shortened acyl‐CoAs are shuttled to mitochondria for full beta‐oxidation, after which, the acetyl‐CoA units are converted into CO2 and H2O in the citric acid (Krebs) cycle. For VLCFA like C26:0, it has not been established definitively how many cycles of beta‐oxidation take place in the peroxisome.
THE ROLE OF VLCFA IN THE PATHOGENESIS OF X‐ALD
The role of VLCFA in the pathogenesis of X‐ALD is still largely unknown. VLCFA levels in plasma do not correlate with the patient's phenotype. However, as VLCFA accumulation is the only biochemical abnormality identified in X‐ALD, it seems plausible that this accumulation is somehow related to the development of symptoms as postulated from human studies (59). Further support comes from the demonstration of the disruptive effects of C26:0 on cell membrane structure, stability and function 27, 39 and the direct toxic effect of C26:0 on adrenocortical cells resulting in a decreased response to adrenocorticotropic hormone (ACTH) stimulation (84). In addition, C26:0 accumulation results in oxidative stress (21) and both the adrenal cortex and brain of X‐ALD patients show evidence of oxidative stress and oxidative damage particularly from lipid peroxidation (60).
Some insight into how mutations in the ABCD1 gene eventually result in the demyelinating process comes from the study of the expression pattern of ALDP (20). In human and mouse brain, ALDP is expressed in astrocytes, microglial cells and oligodendrocytes. However, only oligodendrocytes located in the corpus callosum, internal capsules and the anterior commissure express ALDP. Interestingly, these are the first regions affected in patients with cerebral ALD. Peroxisomes are abundant in cultured oligodendrocyte processes where myelin lipids are incorporated into the myelin membrane during the ensheathment of axons (20). Normal myelin contains mostly long‐chain fatty acids (C16 to C20). In contrast, myelin in X‐ALD brain contains large amounts of VLCFA. What process causes the inflammatory reaction leading to loss of myelin remains to be resolved. Several scenarios have been proposed which include: unfolding of the myelin sheet due to the increased incorporation of VLCFA in the multilamellar myelin membrane (27); VLCFA induced apoptosis of oligodendrocytes resulting in activation of microglia and secretion of cytokines; or altered functions of central nervous system (CNS)‐cell membrane receptors due to structural changes in the membrane bilayer caused by high amounts of VLCFA (reviewed in (66)). Unfortunately, these scenarios can not be investigated using the available mouse model for X‐ALD (42), as X‐ALD mice do not develop cerebral demyelination. Instead, they have a late onset and progressive neurodegenerative phenotype that resembles AMN in patients (62).
VLCFA are present in many different lipid species in X‐ALD patients. A mild to moderate excess of VLCFA is present in all brain lipid species. The greatest excess, however, occurs in the ganglioside, phosphatidylcholine and cholesterol ester fractions. Although in active demyelinating lesions the cholesterol fraction contains the greatest excess of VLCFA, this appears to be a consequence rather than a cause of demyelination because the fatty acid composition of this fraction was normal in regions of X‐ALD brain in which myelin was still intact (72). The gangliosides in X‐ALD brain contain 28%–50% of fatty acids with chain lengths exceeding 21 carbons (23). VLCFA‐containing gangliosides are virtually absent in normal brain. Interestingly, gangliosides have been implicated in a variety of neuroimmunological disorders (57). Furthermore, the immunologic properties of gangliosides vary with their fatty acid composition (32). Phosphatidylcholine is another potential trigger. Theda et al correlated the fatty acid composition of the various lipid fractions in X‐ALD brain with histopathological findings reasoning that abnormalities in trigger molecules should precede histopathological alterations (72). They found that in regions in which myelin was intact, the phosphatidylcholine fraction showed the highest VLCFA excess (16‐fold higher) which led these authors to conclude that the phosphatidylcholine abnormalities in X‐ALD may well be a critical factor in the pathogenesis of the disease. A more recent study by Asheuer and colleagues points to a direct correlation between VLCFA accumulation and disease severity (2). While normal myelin contains mostly fatty acids with chain lengths up to C20, myelin in X‐ALD brain contains large amounts of VLCFA. More importantly, myelin from CCALD patients contains higher VLCFA levels than myelin from AMN patients (2). This suggests that the level of VLCFA accumulation in the white matter may play a crucial role in the initiation of both the inflammatory response and the demyelinating process.
ORIGIN OF VLCFA
The VLCFA which accumulate in X‐ALD are partly absorbed from the diet (38), but mostly result from endogenous synthesis through elongation of long‐chain fatty acids as was first demonstrated by Tsuji et al in X‐ALD fibroblasts using radioactively labeled C18:0 (74). In line with this, Rizzo et al found no differences in C26:0 levels of X‐ALD fibroblasts cultured in lipid‐free media or under standard tissue culture conditions (63). Furthermore, dietary restriction of VLCFA did not lower plasma C26:0 levels in patients with X‐ALD (9). We recently demonstrated that VLCFA elongation is elevated in X‐ALD (37).
Over 90% of all fatty acids in humans are long‐chain fatty acids with a chain length of 16–18 carbon atoms. Long‐chain fatty acids (mainly C16:0) are the end product of the cytosolic fatty acid synthase complex which utilizes acetyl‐CoA, malonyl‐CoA and NADPH to elongate fatty acids in two‐carbon increments 13, 70, 80. The synthesis of saturated VLCFA, monounsaturated VLCFA and polyunsaturated fatty acids (PUFA) takes place at the cytoplasmic side of the endoplasmic membrane 17, 56. Fatty acid elongation requires four sequential reaction steps: (i) condensation between the fatty acyl‐CoA and malonyl‐CoA to form 3‐ketoacyl‐CoA; (ii) reduction using NADPH to form 3‐hydroxyacyl‐CoA; (iii) dehydration to trans‐2‐enoyl‐CoA; and (iv) reduction to fully elongated fatty acyl‐CoA. The initial condensation reaction is catalyzed by the enzyme referred to as “elongation of very long‐chain fatty acids” (ELOVLs) and is considered to be rate limiting 12, 53. Seven elongases have been identified in mammals and are designated ELOVL17 30, 41. Interestingly, each subsequent reaction step is catalyzed by single enzymes only 5, 15, 47 which supports the theory that elongases determine the specificity of each elongation reaction. It was previously established that ELOVL1, ELOVL3, ELOVL4 and ELOVL6 are involved in the synthesis of saturated and monounsaturated fatty acids 1, 48, 54, 75, 79, 83. ELOVL2, ELOVL4 and ELOVL5 are essential for PUFA metabolism 40, 46. ELOVL7's substrate has remained unclear.
For the identification of the human VLCFA‐specific elongase(s), S. cerevisiae proved to be a helpful model system. In yeast, three elongases (Elo1p, Elo2p and Elo3p) have been identified and characterized 55, 73. Elo1p is involved in the elongation of C14:0 to C16:0 fatty acids (73). Elo2p is needed for the elongation of fatty acids up to C24:0 (55). Elo3p is the elongase primarily involved in the synthesis of C26:0 from C22:0 (55). Elo3p knockout yeast do not contain C26:0 (54). Using these knockouts, we have recently identified ELOVL1 as the single elongase catalyzing the synthesis of both saturated VLCFA (C26:0) and monounsaturated VLCFA (C26:1) in humans (54). This conclusion is in line with a previous study on the effect of mouse Elovl1 expression on C26:0 synthesis in Elo3p deficient yeast cells (75).
Human ELOVL3 and mouse Elovl3 are involved in the synthesis of C20:0 and C22:0 54, 83. Human ELOVL4 is involved in the synthesis of >C28 fatty acids and C28‐C38 VLC‐PUFAs 1, 79. Earlier studies demonstrated that ELOVL6 exhibits fatty acyl‐CoA elongase activity specific for saturated and monounsaturated long‐chain fatty acids with chain lengths ranging from C12 to C16 43, 48. We have recently measured fatty acid synthesis using D3‐C16:0 as substrate in Chinese hamster ovary cells expressing ELOVL6 and demonstrated that these cells produced significant amounts of both saturated and monounsaturated C18, C20 and C22 fatty acids. This indicates a broader substrate specificity range for ELOVL6 than previously appreciated. While both ELOVL1 and ELOVL6 show a ubiquitous expression pattern 48, 75, the expression pattern of ELOVL3 is restricted to brown adipose tissue, liver and skin (75), and the expression pattern of ELOVL4 is expressed predominantly in retina, brain and sperm 75, 79, 85. Therefore, we concluded that saturated and monounsaturated VLCFA are synthesized from long‐chain fatty acids via the concerted action of ELOVL6 (C18:0–C22:0) and ELOVL1 (C24:0–C26:0) (54) (Figure 1).
Figure 1.
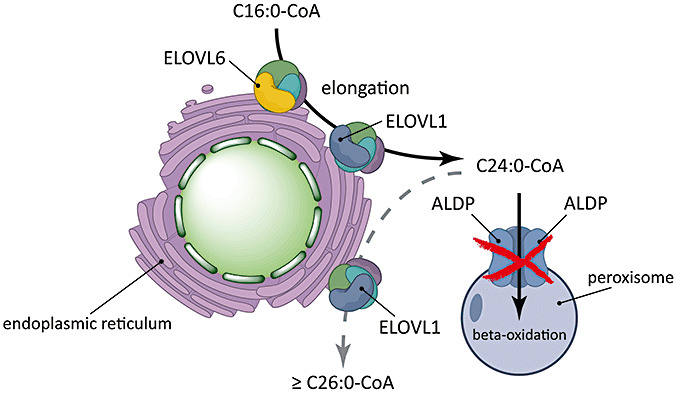
Very long‐chain fatty acids (VLCFA) are synthesized from elongation of long‐chain fatty acids via the concerted action of elongation of very long‐chain fatty acids (ELOVL)6 (C18:0–C22:0) and ELOVL1 (C24:0–C26:0). ALDP transports VLCFacyl‐coenzyme A (CoA) across the peroxisomal membrane. A deficiency in ALDP impairs peroxisomal β‐oxidation of VLCFA but also raises cytosolic levels of VLCFacyl‐CoA which are substrate for further elongation by ELOVL1.
DIAGNOSTIC TESTS FOR X‐ALD
To date, plasma VLCFA analysis is still the most frequently used diagnostic test for X‐ALD. The three parameters analyzed are: the concentration of C26:0, the ratio of C24:0/C22:0 and the ratio of C26:0/C22:0 (50). Plasma VLCFA analysis is recommended in all male patients with progressive spastic paraparesis of unknown cause or with Addison's disease. In males, unambiguous diagnosis of X‐ALD can be achieved by demonstration of elevated levels of VLCFA in plasma. Plasma VLCFA levels often are increased in women who are heterozygous for X‐ALD, but studies in obligate heterozygotes have shown false‐negative results in approximately 20% of obligate heterozygotes (51). Thus, a normal plasma VLCFA level does not exclude heterozygosity for X‐ALD. Mutation analysis is the most reliable method for the identification of heterozygotes, provided that the mutation in the family has been defined in an affected male or obligate heterozygote relative (7).
All X‐ALD patients investigated have mutations in the ABCD1 gene. As of February 2010, the X‐ALD mutation database ( http://www.x‐ald.nl) contains 1065 mutations, which have been identified by various groups (for all mutation references, see http://www.x‐ald.nl/references.htm). Of these 1065 mutations, 652 (61%) are missense mutations, 239 (22%) are frame shifts, 103 (10%) are nonsense, 39 (4%) are amino acid insertions or deletions and 32 (3%) are large deletions of one or more exons. The majority of X‐ALD kindreds have a unique mutation, 522 (49%) nonrecurrent mutations have been identified. The fact that the majority of X‐ALD kindreds have unique mutations makes DNA testing time‐consuming and laborious.
Sequence analysis of genomic DNA is made difficult because of the presence of ABCD1 paralogs on four autosomes 18, 69. At some moment in primate evolution (about 5–10 million years ago), a 9.7 kb DNA segment encompassing exons 7 through 10 of the ABCD1 gene was duplicated from the X‐chromosome to the chromosomes 2 (2p11), 10 (10p11), 16 (16p11) and 22 (22q11). Sequence comparison shows 92%–96% nucleotide identity among the paralogs (18) (http://www.x‐ald.nl/pseudogenes.htm). Because of this very high homology between the pseudogenes and the ABCD1 gene, great care should be taken when setting up mutation analysis using genomic DNA. In 1999, Boehm and colleagues developed and validated a robust genomic DNA‐based diagnostic test for ABCD1 mutation analysis without interference of the pseudogenes (7).
In order to facilitate mutation analysis, we keep track of the incidence of identified mutations in each of the 10 genomic amplicons (http://www.x‐ald.nl/stats.htm). Using this genomic sequence strategy and these mutation incidence statistics, approximately 70% of all mutations can be identified by analysis of only four amplicons which together cover 40% of the coding region. Approximately 40% of all mutations have been identified in the amplicons exon1b and exon1c, 15% in amplicon exon8/9 and 11% in amplicon exon6.
NEWBORN SCREENING
The Kennedy Krieger Institute has developed a validated liquid chromatography–tandem mass spectrometric (LC–MS/MS) method for newborn screening that is based on the measurement of C26:0 lysophosphatidylcholine (26:0‐lyso‐PC) in dried blood spots (28). The levels of 26:0‐lyso‐PC in X‐ALD are more than fivefold the levels in blood spots from normal subjects and there is no overlap between controls and X‐ALD patients. The test can be automated to achieve very high sample throughput which will be needed for the implementation and inclusion of X‐ALD in national screening programs. The availability of a diagnostic marker of X‐ALD in newborns offers tremendous potential for identification of X‐ALD at birth and long before the onset of clinical manifestations.
CONCLUDING REMARKS
We recently established that ALDP transports VLCFacyl‐CoA across the peroxisomal membrane 54, 78. A deficiency in ALDP has two major effects: on the one hand, it impairs peroxisomal β‐oxidation of VLCFA, and on the other hand, it raises cytosolic levels of VLCFacyl‐CoA. These VLCFacyl‐CoA esters are then further elongated by ELOVL1, the human C26 specific elongase (Figure 1). Hence, increased substrate availability caused by the primary deficiency in ALDP appears to underlie our previous finding of enhanced VLCFA elongation in X‐ALD (37) and the elevated C26 levels in X‐ALD (54).
Currently, treatment options are very limited and are mostly symptomatic. Lorenzo's oil reduces plasma C26:0 but does not halt progression of the disease 3, 77. Lovastatin also lowered plasma VLCFA (68), but a recent placebo‐controlled trial revealed that lovastatin has neither effect on C26:0 levels in peripheral blood lymphocytes and erythrocytes nor on the VLCFA content of the low‐density lipoprotein (LDL) fraction (19). Hematopoietic stem cell transplantation can halt or reverse clinical deterioration (58). However, it is only effective in patients with the earliest stage of CCALD. The recent breakthrough in gene therapy has currently only been applied to CCALD (10). The recent demonstration that ELOVL1 knockdown reduces the synthesis of C26:0 and lowers C26:0 levels in X‐ALD fibroblasts (54) offers exciting new opportunities for the development of therapeutic options for X‐ALD aimed at the inhibition of ELOVL1 activity.
ACKNOWLEDGMENTS
This work was supported by grants from the European Leukodystrophy Association [ELA 2008‐05111A (RJW)], the Prinses Beatrix Fonds [WAR08‐20 (SK)] and the Netherlands Organization for Scientific Research [VIDI‐grant no. 016.086.328 (SK)].
REFERENCES
- 1. Agbaga MP, Brush RS, Mandal MN, Henry K, Elliott MH, Anderson RE (2008) Role of Stargardt‐3 macular dystrophy protein (ELOVL4) in the biosynthesis of very long chain fatty acids. Proc Natl Acad Sci U S A 105:12843–12848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Asheuer M, Bieche I, Laurendeau I, Moser A, Hainque B, Vidaud M, Aubourg P (2005) Decreased expression of ABCD4 and BG1 genes early in the pathogenesis of X‐linked adrenoleukodystrophy. Hum Mol Genet 14:1293–1303. [DOI] [PubMed] [Google Scholar]
- 3. Aubourg P, Adamsbaum C, Lavallard‐Rousseau MC, Rocchiccioli F, Cartier N, Jambaque I et al (1993) A two‐year trial of oleic and erucic acids (“Lorenzo's oil”) as treatment for adrenomyeloneuropathy. N Engl J Med 329:745–752. [DOI] [PubMed] [Google Scholar]
- 4. Berger J, Molzer B, Fae I, Bernheimer H (1994) X‐linked adrenoleukodystrophy (ALD): a novel mutation of the ALD gene in 6 members of a family presenting with 5 different phenotypes. Biochem Biophys Res Comm 205:1638–1643. [DOI] [PubMed] [Google Scholar]
- 5. Bernert JT, Jr , Sprecher H (1979) Solubilization and partial purification of an enzyme involved in rat liver microsomal fatty acid chain elongation: beta‐hydroxyacyl‐CoA dehydrase. J Biol Chem 254:11584–11590. [PubMed] [Google Scholar]
- 6. Bezman L, Moser AB, Raymond GV, Rinaldo P, Watkins PA, Smith KD et al (2001) Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann Neurol 49:512–517. [PubMed] [Google Scholar]
- 7. Boehm CD, Cutting GR, Lachtermacher MB, Moser HW, Chong SS (1999) Accurate DNA‐based diagnostic and carrier testing for X‐linked adrenoleukodystrophy. Mol Genet Metab 66:128–136. [DOI] [PubMed] [Google Scholar]
- 8. Braiterman LT, Zheng S, Watkins PA, Geraghty MT, Johnson G, McGuinness MC et al (1998) Suppression of peroxisomal membrane protein defects by peroxisomal ATP binding cassette (ABC) proteins. Hum Mol Genet 7:239–247. [DOI] [PubMed] [Google Scholar]
- 9. Brown FR, Van Duyn MA, Moser AB, Schulman JD, Rizzo WB, Snyder RD et al (1982) Adrenoleukodystrophy: effects of dietary restriction of very long chain fatty acids and of administration of carnitine and clofibrate on clinical status and plasma fatty acids. Johns Hopkins Med J 151:164–172. [PubMed] [Google Scholar]
- 10. Cartier N, Hacein‐Bey‐Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I et al (2009) Hematopoietic stem cell gene therapy with a lentiviral vector in X‐linked adrenoleukodystrophy. Science 326:818–823. [DOI] [PubMed] [Google Scholar]
- 11. Cartier N, Lopez J, Moullier P, Rocchiccioli F, Rolland MO, Jorge P et al (1995) Retroviral‐mediated gene transfer corrects very‐long‐chain fatty acid metabolism in adrenoleukodystrophy fibroblasts. Proc Natl Acad Sci U S A 92:1674–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cinti DL, Cook L, Nagi MN, Suneja SK (1992) The fatty acid chain elongation system of mammalian endoplasmic reticulum. Prog Lipid Res 31:1–51. [DOI] [PubMed] [Google Scholar]
- 13. Colli W, Hinkle PC, Pullman ME (1969) Characterization of the fatty acid elongation system in soluble extracts and membrane preparations of rat liver mitochondria. J Biol Chem 244:6432–6443. [PubMed] [Google Scholar]
- 14. Contreras M, Mosser J, Mandel JL, Aubourg P, Singh I (1994) The protein coded by the X‐adrenoleukodystrophy gene is a peroxisomal integral membrane protein. FEBS Lett 344:211–215. [DOI] [PubMed] [Google Scholar]
- 15. Cook L, Nagi MN, Suneja SK, Hand AR, Cinti DL (1992) Evidence that beta‐hydroxyacyl‐CoA dehydrase purified from rat liver microsomes is of peroxisomal origin. Biochem J 287:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dean M, Rzhetsky A, Allikmets R (2001) The human ATP‐binding cassette (ABC) transporter superfamily. Genome Res 11:1156–1166. [DOI] [PubMed] [Google Scholar]
- 17. Denic V, Weissman JS (2007) A molecular caliper mechanism for determining very long‐chain fatty acid length. Cell 130:663–677. [DOI] [PubMed] [Google Scholar]
- 18. Eichler EE, Budarf ML, Rocchi M, Deaven LL, Doggett NA, Baldini A et al (1997) Interchromosomal duplications of the adrenoleukodystrophy locus: a phenomenon of pericentromeric plasticity. Hum Mol Genet 6:991–1002. [DOI] [PubMed] [Google Scholar]
- 19. Engelen M, Ofman R, Dijkgraaf MGW, Hijzen M, Van Der Wardt LA, Van Geel BM et al (2010) Lovastatin in X‐linked adrenoleukodystrophy. N Engl J Med 362:276–277. [DOI] [PubMed] [Google Scholar]
- 20. Fouquet F, Zhou JM, Ralston E, Murray K, Troalen F, Magal E et al (1997) Expression of the adrenoleukodystrophy protein in the human and mouse central nervous system. Neurobiol Dis 3:271–285. [DOI] [PubMed] [Google Scholar]
- 21. Fourcade S, Lopez‐Erauskin J, Galino J, Duval C, Naudi A, Jove M et al (2008) Early oxidative damage underlying neurodegeneration in X‐adrenoleukodystrophy. Hum Mol Genet 17:1762–1773. [DOI] [PubMed] [Google Scholar]
- 22. Fourcade S, Ruiz M, Camps C, Schluter A, Houten SM, Mooyer PAW et al (2009) A key role for the peroxisomal ABCD2 transporter in fatty acid homeostasis. Am J Physiol Endocrinol Metab 296:E211–E221. [DOI] [PubMed] [Google Scholar]
- 23. Garashi M, Belchis D, Suzuki K (1976) Brain gangliosides in adrenoleukodystrophy. J Neurochem 27:327–328. [DOI] [PubMed] [Google Scholar]
- 24. Halbach A, Lorenzen S, Landgraf C, Volkmer‐Engert R, Erdmann R, Rottensteiner H (2005) Function of the PEX19‐binding site of human adrenoleukodystrophy protein as targeting motif in man and yeast. PMP targeting is evolutionarily conserved. J Biol Chem 280:21176–21182. [DOI] [PubMed] [Google Scholar]
- 25. Hashmi M, Stanley W, Singh I (1986) Lignoceroyl‐CoASH ligase: enzyme defect in fatty acid beta‐oxidation system in X‐linked childhood adrenoleukodystrophy. FEBS Lett 196:247–250. [DOI] [PubMed] [Google Scholar]
- 26. Hettema EH, Van Roermund CW, Distel B, Van Den Berg M, Vilela C, Rodrigues‐Pousada C et al (1996) The ABC transporter proteins Pat1 and Pat2 are required for import of long‐chain fatty acids into peroxisomes of Saccharomyces cerevisiae . EMBO J 15:3813–3822. [PMC free article] [PubMed] [Google Scholar]
- 27. Ho JK, Moser H, Kishimoto Y, Hamilton JA (1995) Interactions of a very long chain fatty acid with model membranes and serum albumin. Implications for the pathogenesis of adrenoleukodystrophy. J Clin Invest 96:1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hubbard WC, Moser AB, Liu AC, Jones RO, Steinberg SJ, Lorey F et al (2009) Newborn screening for X‐linked adrenoleukodystrophy (X‐ALD): validation of a combined liquid chromatography‐tandem mass spectrometric (LC‐MS/MS) method. Mol Genet Metab 97:212–220. [DOI] [PubMed] [Google Scholar]
- 29. Igarashi M, Schaumburg HH, Powers J, Kishmoto Y, Kolodny E, Suzuki K (1976) Fatty acid abnormality in adrenoleukodystrophy. J Neurochem 26:851–860. [DOI] [PubMed] [Google Scholar]
- 30. Jakobsson A, Westerberg R, Jacobsson A (2006) Fatty acid elongases in mammals: their regulation and roles in metabolism. Prog Lipid Res 45:237–249. [DOI] [PubMed] [Google Scholar]
- 31. Jia Z, Moulson CL, Pei Z, Miner JH, Watkins PA (2007) Fatty acid transport protein 4 is the principal very long chain fatty acyl‐CoA synthetase in skin fibroblasts. J Biol Chem 282:20573–20583. [DOI] [PubMed] [Google Scholar]
- 32. Kannagi R, Nudelman E, Hakomori S (1982) Possible role of ceramide in defining structure and function of membrane glycolipids. Proc Natl Acad Sci U S A 79:3470–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kashiwayama Y, Seki M, Yasui A, Murasaki Y, Morita M, Yamashita Y et al (2009) 70‐kDa peroxisomal membrane protein related protein (P70R/ABCD4) localizes to endoplasmic reticulum not peroxisomes, and NH2‐terminal hydrophobic property determines the subcellular localization of ABC subfamily D proteins. Exp Cell Res 315:190–205. [DOI] [PubMed] [Google Scholar]
- 34. Kemp S, Ligtenberg MJL, VanGeel BM, Barth PG, Wolterman RA, Schoute F et al (1994) Identification of A 2 base‐pair deletion in 5 unrelated families with adrenoleukodystrophy—a possible hot‐spot for mutations. Biochem Biophys Res Commun 202:647–653. [DOI] [PubMed] [Google Scholar]
- 35. Kemp S, Wanders RJ (2007) X‐linked adrenoleukodystrophy: very long‐chain fatty acid metabolism, ABC half‐transporters and the complicated route to treatment. Mol Genet Metab 90:268–276. [DOI] [PubMed] [Google Scholar]
- 36. Kemp S, Wei HM, Lu JF, Braiterman LT, McGuinness MC, Moser AB et al (1998) Gene redundancy and pharmacological gene therapy: implications for X‐linked adrenoleukodystrophy. Nat Med 4:1261–1268. [DOI] [PubMed] [Google Scholar]
- 37. Kemp S, Valianpour F, Denis S, Ofman R, Sanders RJ, Mooyer P et al (2005) Elongation of very long‐chain fatty acids is enhanced in X‐linked adrenoleukodystrophy. Mol Genet Metab 84:144–151. [DOI] [PubMed] [Google Scholar]
- 38. Kishimoto Y, Moser HW, Kawamura N, Platt M, Pallante SL, Fenselau C (1980) Adrenoleukodystrophy: evidence that abnormal very long chain fatty acids of brain cholesterol esters are of exogenous origin. Biochem Biophys Res Commun 96:69–76. [DOI] [PubMed] [Google Scholar]
- 39. Knazek RA, Rizzo WB, Schulman JD, Dave JR (1983) Membrane microviscosity is increased in the erythrocytes of patients with adrenoleukodystrophy and adrenomyeloneuropathy. J Clin Invest 72:245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kobayashi T, Zadravec D, Jacobsson A (2007) ELOVL2 overexpression enhances triacylglycerol synthesis in 3T3‐L1 and F442A cells. FEBS Lett 581:3157–3163. [DOI] [PubMed] [Google Scholar]
- 41. Leonard AE, Pereira SL, Sprecher H, Huang YS (2004) Elongation of long‐chain fatty acids. Prog Lipid Res 43:36–54. [DOI] [PubMed] [Google Scholar]
- 42. Lu JF, Lawler AM, Watkins PA, Powers JM, Moser AB, Moser HW, Smith KD (1997) A mouse model for X‐linked adrenoleukodystrophy. Proc Natl Acad Sci U S A 94:9366–9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Matsuzaka T, Shimano H, Yahagi N, Yoshikawa T, Memiya‐Kudo M, Hasty AH et al (2002) Cloning and characterization of a mammalian fatty acyl‐CoA elongase as a lipogenic enzyme regulated by SREBPs. J Lipid Res 43:911–920. [PubMed] [Google Scholar]
- 44. McGuinness MC, Zhang HP, Smith KD (2001) Evaluation of pharmacological induction of fatty acid beta‐oxidation in X‐linked adrenoleukodystrophy. Mol Genet Metab 74:256–263. [DOI] [PubMed] [Google Scholar]
- 45. Migeon BR, Moser HW, Moser AB, Axelman J, Sillence D, Norum RA (1981) Adrenoleukodystrophy: evidence for X linkage, inactivation, and selection favoring the mutant allele in heterozygous cells. Proc Natl Acad Sci U S A 78:5066–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Moon YA, Hammer RE, Horton JD (2009) Deletion of ELOVL5 leads to fatty liver through activation of SREBP‐1c in mice. J Lipid Res 50:412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moon YA, Horton JD (2003) Identification of two mammalian reductases involved in the two‐carbon fatty acyl elongation cascade. J Biol Chem 278:7335–7343. [DOI] [PubMed] [Google Scholar]
- 48. Moon YA, Shah NA, Mohapatra S, Warrington JA, Horton JD (2001) Identification of a mammalian long chain fatty acyl elongase regulated by sterol regulatory element‐binding proteins. J Biol Chem 276:45358–45366. [DOI] [PubMed] [Google Scholar]
- 49. Moser AB, Kreiter N, Bezman L, Lu S, Raymond GV, Naidu S, Moser HW (1999) Plasma very long chain fatty acids in 3000 peroxisome disease patients and 29 000 controls. Ann Neurol 45:100–110. [DOI] [PubMed] [Google Scholar]
- 50. Moser HW, Moser AB, Frayer KK, Chen W, Schulman JD, O'Neill BP, Kishimoto Y (1981) Adrenoleukodystrophy: increased plasma content of saturated very long chain fatty acids. Neurology 31:1241–1249. [DOI] [PubMed] [Google Scholar]
- 51. Moser HW, Smith KD, Watkins PA, Powers J, Moser AB (2001) X‐linked adrenoleukodystrophy. In: The Metabolic and Molecular Bases of Inherited Disease, Scriver CR, Beaudet AL, Sly WS, Valle D (eds), pp. 3257–3301. McGraw Hill: New York. [Google Scholar]
- 52. Mosser J, Douar AM, Sarde CO, Kioschis P, Feil R, Moser H et al (1993) Putative X‐linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 361:726–730. [DOI] [PubMed] [Google Scholar]
- 53. Nugteren DH (1965) The enzymic chain elongation of fatty acids by rat‐liver microsomes. Biochim Biophys Acta 106:280–290. [DOI] [PubMed] [Google Scholar]
- 54. Ofman R, Dijkstra IM, Van Roermund CW, Burger N, Turkenburg M, Van CA et al (2010) The role of ELOVL1 in very long‐chain fatty acid homeostasis and X‐linked adrenoleukodystrophy. EMBO Mol Med 2:90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Oh CS, Toke DA, Mandala S, Martin CE (1997) ELO2 and ELO3, homologues of the Saccharomyces cerevisiae ELO1 gene, function in fatty acid elongation and are required for sphingolipid formation. J Biol Chem 272:17376–17384. [DOI] [PubMed] [Google Scholar]
- 56. Osei P, Suneja SK, Laguna JC, Nagi MN, Cook L, Prasad MR, Cinti DL (1989) Topography of rat hepatic microsomal enzymatic components of the fatty acid chain elongation system. J Biol Chem 264:6844–6849. [PubMed] [Google Scholar]
- 57. Pestronk A (1991) Invited review: motor neuropathies, motor neuron disorders, and antiglycolipid antibodies. Muscle Nerve 14:927–936. [DOI] [PubMed] [Google Scholar]
- 58. Peters C, Charnas LR, Tan Y, Ziegler RS, Shapiro EG, DeFor T et al (2004) Cerebral X‐linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood 104:881–888. [DOI] [PubMed] [Google Scholar]
- 59. Powers JM, DeCiero DP, Ito M, Moser AB, Moser HW (2000) Adrenomyeloneuropathy: a neuropathologic review featuring its noninflammatory myelopathy. J Neuropathol Exp Neurol 59:89–102. [DOI] [PubMed] [Google Scholar]
- 60. Powers JM, Pei Z, Heinzer AK, Deering R, Moser AB, Moser HW et al (2005) Adreno‐leukodystrophy: oxidative stress of mice and men. J Neuropathol Exp Neurol 64:1067–1079. [DOI] [PubMed] [Google Scholar]
- 61. Powers JM, Schaumburg HH (1974) Adreno‐leukodystrophy (sex‐linked Schilder's disease). A pathogenetic hypothesis based on ultrastructural lesions in adrenal cortex, peripheral nerve and testis. Am J Pathol 76:481–491. [PMC free article] [PubMed] [Google Scholar]
- 62. Pujol A, Hindelang C, Callizot N, Bartsch U, Schachner M, Mandel JL (2002) Late onset neurological phenotype of the X‐ALD gene inactivation in mice: a mouse model for adrenomyeloneuropathy. Hum Mol Genet 11:499–505. [DOI] [PubMed] [Google Scholar]
- 63. Rizzo WB, Avigan J, Chemke J, Schulman JD (1984) Adrenoleukodystrophy: very long‐chain fatty acid metabolism in fibroblasts. Neurology 34:163–169. [DOI] [PubMed] [Google Scholar]
- 64. Roerig P, Mayerhofer P, Holzinger A, Gartner J (2001) Characterization and functional analysis of the nucleotide binding fold in human peroxisomal ATP binding cassette transporters. FEBS Lett 492:66–72. [DOI] [PubMed] [Google Scholar]
- 65. Shinnoh N, Yamada T, Yoshimura T, Furuya H, Yoshida Y, Suzuki Y et al (1995) Adrenoleukodystrophy: the restoration of peroxisomal beta‐oxidation by transfection of normal cDNA. Biochem Biophys Res Commun 210:830–836. [DOI] [PubMed] [Google Scholar]
- 66. Singh H, Brogan M, Johnson D, Poulos A (1992) Peroxisomal beta‐oxidation of branched chain fatty acids in human skin fibroblasts. J Lipid Res 33:1597–1605. [PubMed] [Google Scholar]
- 67. Singh I, Khan M, Key L, Pai S (1998) Lovastatin for X‐linked adrenoleukodystrophy. N Engl J Med 339:702–703. [DOI] [PubMed] [Google Scholar]
- 68. Singh I, Moser AE, Moser HW, Kishimoto Y (1984) Adrenoleukodystrophy: impaired oxidation of very long chain fatty acids in white blood cells, cultured skin fibroblasts, and amniocytes. Pediatr Res 18:286–290. [DOI] [PubMed] [Google Scholar]
- 69. Smith KD, Kemp S, Braiterman LT, Lu JF, Wei HM, Geraghty M et al (1999) X‐linked adrenoleukodystrophy: genes, mutations, and phenotypes. Neurochem Res 24:521–535. [DOI] [PubMed] [Google Scholar]
- 70. Smith S (1994) The animal fatty acid synthase: one gene, one polypeptide, seven enzymes. FASEB J 8:1248–1259. [PubMed] [Google Scholar]
- 71. Tanaka AR, Tanabe K, Morita M, Kurisu M, Kasiwayama Y, Matsuo M et al (2002) ATP binding/hydrolysis by and phosphorylation of peroxisomal ATP‐binding cassette proteins PMP70 (ABCD3) and adrenoleukodystrophy protein (ABCD1). J Biol Chem 277:40142–40147. [DOI] [PubMed] [Google Scholar]
- 72. Theda C, Moser AB, Powers JM, Moser HW (1992) Phospholipids in X‐linked adrenoleukodystrophy white matter: fatty acid abnormalities before the onset of demyelination. J Neurol Sci 110:195–204. [DOI] [PubMed] [Google Scholar]
- 73. Toke DA, Martin CE (1996) Isolation and characterization of a gene affecting fatty acid elongation in Saccharomyces cerevisiae . J Biol Chem 271:18413–18422. [DOI] [PubMed] [Google Scholar]
- 74. Tsuji S, Sano T, Ariga T, Miyatake T (1981) Increased synthesis of hexacosanoic acid (C26:0) by cultured skin fibroblasts from patients with adrenoleukodystrophy (ALD) and adrenomyeloneuropathy (AMN). J Biochem (Tokyo) 90:1233–1236. [DOI] [PubMed] [Google Scholar]
- 75. Tvrdik P, Westerberg R, Silve S, Asadi A, Jakobsson A, Cannon B et al (2000) Role of a new mammalian gene family in the biosynthesis of very long chain fatty acids and sphingolipids. J Cell Biol 149:707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Valianpour F, Selhorst JJ, Van Lint LE, Van Gennip AH, Wanders RJ, Kemp S (2003) Analysis of very long‐chain fatty acids using electrospray ionization mass spectrometry. Mol Genet Metab 79:189–196. [DOI] [PubMed] [Google Scholar]
- 77. Van Geel BM, Assies J, Haverkort EB, Koelman JH, Verbeeten B, Wanders RJ, Barth PG (1999) Progression of abnormalities in adrenomyeloneuropathy and neurologically asymptomatic X‐linked adrenoleukodystrophy despite treatment with “Lorenzo's oil.” J Neurol Neurosurg Psychiatry 67:290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Van Roermund CWT, Visser WF, IJlst L, Van Cruchten A, Boek M, Kulik W et al (2008) The human peroxisomal ABC half transporter ALDP functions as a homodimer and accepts acyl‐CoA esters. FASEB J 22:4201–4208. [DOI] [PubMed] [Google Scholar]
- 79. Vasireddy V, Uchida Y, Salem N Jr, Kim SY, Mandal MN, Reddy GB et al (2007) Loss of functional ELOVL4 depletes very long‐chain fatty acids (>or = C28) and the unique omega‐O‐acylceramides in skin leading to neonatal death. Hum Mol Genet 16:471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wakil SJ (1989) Fatty acid synthase, a proficient multifunctional enzyme. Biochemistry 28:4523–4530. [DOI] [PubMed] [Google Scholar]
- 81. Wanders RJ, Ferdinandusse S, Brites P, Kemp S (2010) Peroxisomes, lipid metabolism and lipotoxicity. Biochim Biophys Acta 1801:272–280. [DOI] [PubMed] [Google Scholar]
- 82. Wanders RJ, Van Roermund CW, Van Wijland MJ, Schutgens RB, Van Den BH, Schram AW, Tager JM (1988) Direct demonstration that the deficient oxidation of very long chain fatty acids in X‐linked adrenoleukodystrophy is due to an impaired ability of peroxisomes to activate very long chain fatty acids. Biochem Biophys Res Commun 153:618–624. [DOI] [PubMed] [Google Scholar]
- 83. Westerberg R, Tvrdik P, Unden AB, Mansson JE, Norlen L, Jakobsson A et al (2004) Role for ELOVL3 and fatty acid chain length in development of hair and skin function. J Biol Chem 279:5621–5629. [DOI] [PubMed] [Google Scholar]
- 84. Whitcomb RW, Linehan WM, Knazek RA (1988) Effects of long‐chain, saturated fatty acids on membrane microviscosity and adrenocorticotropin responsiveness of human adrenocortical cells in vitro . J Clin Invest 81:185–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhang K, Kniazeva M, Han M, Li W, Yu Z, Yang Z et al (2001) A 5‐bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet 27:89–93. [DOI] [PubMed] [Google Scholar]