

Abstract
Glioblastomas, like other cancers, harbor small cell populations with the capability of sustaining tumor formation. These cells are referred to as cancer stem cells. We isolated cells expressing the surface marker A2B5 from three human glioblastomas (GBM) and showed that after grafting into nude mice, they generated dense and highly infiltrative tumors. Then, we extensively studied A2B5+ cells isolated from 11 human GBM. These cells display neurosphere‐like, self‐renewal, asymmetrical cell division properties and have multipotency capability. Stereotactic xenografts of dissociated A2B5+‐derived secondary spheres revealed that as few as 1000 cells produced a tumor. Moreover, flow cytometry characterization of A2B5+‐derived spheres revealed three distinct populations of cells: A2B5+/CD133+, A2B5+/CD133‐ and A2B5‐/CD133‐, with striking proportion differences among GBM. Both A2B5+/CD133+ and A2B5+/CD133‐ cell fractions displayed a high proliferative index, the potential to generate spheres and produced tumors in nude mice. Finally, we generated two green fluorescent protein‐cell lines that display—after serum induction—distinct proliferative and migratory properties, and differ in their CD133 level of expression. Taken together, our results suggest that transformed A2B5+ cells are crucial for the initiation and maintenance of GBM, although CD133 expression is more involved in determining the tumor's behavior.
Keywords: A2B5, CD133, tumor spheres, glioblastomas, stem cells
INTRODUCTION
Although glioblastomas (GBM) show similar histological phenotypes, differences in biology and clinical prognosis suggest that these tumors might harbor different entities arising from cells of different origin and/or at different stages of differentiation. Multipotent neural stem cells (NSC) and progenitors derived from developing or adult central nervous system (CNS) can be isolated and propagated in culture in the presence of mitogens. This involves the generation of free‐floating spherical clusters, which are termed “neurospheres”(15). Neurospheres consist of both multipotent stem cells and restricted progenitors at different stages of differentiation. In addition to the heterogeneity that occurs within neurospheres, there is evidence that neurospheres issued from different regions of the developing or adult brain display unique characteristics with regard to growth, differentiation and gene expression (13).
Previous studies have shown that primary human brain tumors, especially GBM, contain cells with NSC features, suggesting that GBM arise after malignant transformation of NSC or progenitors 5, 6, 7, 16. It was first admitted that prominin‐1, also called CD133, was a stem cell marker as only CD133+ cells from GBM were able to initiate brain cancer in a mouse model (17). However, this contention is now questioned (2) as GBM contain heterogeneous stem cell and precursor populations, all being putative brain tumor‐initiating cells. As a consequence, the exact role of CD133+ cells vs. other cell populations in GBM initiation and progression needs to be further explored. In a previous study, we analyzed the cell composition of human gliomas and demonstrated the presence of glial precursor cells expressing GT3 gangliosides recognized by the A2B5 monoclonal antibody (4). This antigen is known to characterize a small fraction of human subcortical white matter cells that have NSC properties (11). We showed that in GBM, A2B5+ cells display the potential to migrate, divide, differentiate into oligodendroglial and type‐1 and type‐2 astroglial cells, and are genetically transformed as they showed recurrent epidermal growth factor receptor (EGFR) gene amplification (4). Therefore, it was tempting to speculate that A2B5+ cells isolated from GBM might represent brain‐tumor initiating cells as also suggested by a recent study (12).
Here, we first confirmed that A2B5+ cells, but not the A2B5‐ fraction isolated from GBM, are able to form tumors when injected into nude mice. In order to better understand the role of A2B5, the in vitro characteristics of these cells isolated from GBM were defined and compared with those of cells isolated from the human fetal subventricular zone (SVZ) known to be enriched in NSC. We demonstrate that the A2B5+ cells, but not the A2B5‐ cells isolated from GBM, have neural stem‐like cell properties. Moreover, the CD133 antigen distinguishes two populations among the A2B5+ pool: the A2B5+/CD133+ and the A2B5+/CD133‐ cells, which are both able to generate tumors when grafted into nude mice. Finally, we derived cell lines from two different parental GBM that originate—one from SVZ and the other from cortex. These cell lines differ in their CD133 cell content and have different behaviors in vitro.
MATERIALS AND METHODS
Human samples
Seventeen glioblastoma (GBM) samples (World Health Organization, grade IV) were obtained after informed consent from patients who did not receive chemotherapy or radiotherapy before surgery (Timone hospital, Marseille, France). For culture or xenograft experiments, smears were done to exclude samples containing normal brain or necrosis. For the first series of experiments, three GBM cases were used for A2B5 magnetic cell sorting, followed by injection in nude mice (Table 1, see footnote *). Eleven additional primary fresh GBM samples were selected and placed under sterile conditions in Dulbecco's Modified Eagles Medium with fetal calf serum 0.1% medium (DMEM‐FCS 0.1%) until dissociation, cell selection and culture. The main clinical pathological features are reported in Table 2. In particular, we analyzed whether GBM samples contact the lateral ventricle (LV) or not. Fluorescencein situ hybridization (FISH) analysis was performed as previously described (4). Finally, three fresh GBM samples were used exclusively for flow cytometry cell fractions analysis.
Table 1.
Main data regarding xenograft experiments (only tumor masses were recorded).
| Type of cells injected | Number of cells injected | Site of injection | Number of animals injected | Time between injection and sacrifice (weeks) | Tumor mass formation after xenograft | |
|---|---|---|---|---|---|---|
| A2B5+ and A2B5‐ cells from FACS‐sorted fresh GBM sample* | A2B5+ cells | 5 × 105 | Corpus callosum | 8 | 5–10 | 7/8 |
| A2B5– cells | 5 × 105 | Corpus callosum | 7 | 5–14 | 0/7§ | |
| Dissociated secondary spheres from | A2B5+ cells† | 1 × 105 | Corpus callosum | 4 | 12 | 3/4 |
| 1 × 103 | Corpus callosum | 4 | 10–20 | 2/4 | ||
| A2B5+/CD133+, A2B5+/CD133‐ cells from FACS‐sorted spheres obtained from A2B5+ cells‡ | A2B5+/CD133+ cells | 1 × 103 | Corpus callosum | 2 | 35 | 2/2 |
| 1 × 103 | Striatum | 4 | 19 | 1/2 | ||
| 35 | 2/2 | |||||
| A2B5+/CD133‐ cells | 1 × 103 | Corpus callosum | 4 | 35 | 4/4 | |
| 1 × 103 | Striatum | 2 | 19 | 2/2 | ||
Cells were isolated from three fresh GBM samples (these cases were not included in the in vitro study).
Cells were obtained from GBM 6 and GBM 7.
A2B5+ cells were obtained from GBM 6.
In 2/7 animals, rare, isolated cells were recorded within the corpus callosum.
Table 2.
Main clinical characteristics of the 11 GBM patients and summary of the spheres obtained from the A2B5+ fractions isolated from these samples. Abbreviations: EGFR: epidermal growth factor receptor; NA: not analyzed; SS: secondary spheres; SVZ: subventricular zone; TP: temporo parietal; F: frontal; S: sphenoidal.
| Number | Date of birth | Topography | Contact with SVZ | Date of surgery | Date and state at the last follow up | Time (days) between cell culture and primary sphere formation | Maximum passages of SS | Genetic alterations |
|---|---|---|---|---|---|---|---|---|
| 1 | 08/06/1926 | Right frontal | – | 06/02/2006 | 16/10/2007 (dead) | 3 | 2 | monosomy 10, EGFR amplification |
| 2 | 17/08/1947 | Left parieto‐occipital | – | 27/03/2006 | 2/06/2008 (dead) | 7 | 2 | monosomy 10, EGFR amplification |
| 3 | 20/04/1943 | Right occipital inferior | + TP | 11/04/2006 | 21/11/2006 (dead) | 12 | 3 | monosomy 10 |
| 4 | 19/05/1959 | Right frontal | – | 17/05/2006 | 29/04/2008 (alive) | 3 | 8 | monosomy 10, EGFR amplification |
| 5 | 12/12/1945 | Left temporal | + TP | 19/06/2006 | 14/03/2007 (dead) | 7 | 7 | monosomy 10, EGFR amplification |
| 6 | 04/11/1949 | Fronto rolandic | + F | 03/07/2006 | 10/02/2007 (dead) | 2 | 36 | monosomy 10, EGFR amplification |
| 7 | 22/02/1953 | Right temporal | + S | 13/09/2006 | 29/05/2007 (dead) | 10 | 18 | monosomy 10 |
| 8 | 01/01/1935 | Left frontal | – | 29/12/2006 | 26/04/2007 (dead) | 4 | 2 | monosomy 10, EGFR amplification |
| 9 | 15/09/1947 | Left frontal | – | 22/01/2007 | 3/07/2008 (dead) | 6 | 34 | monosomy 10, EGFR amplification |
| 10 | 15/05/1938 | Left temporal | – | 07/02/2007 | 22/05/2007 (dead) | 9 | 10 | monosomy 10, EGFR amplification |
| 11 | 03/03/1933 | Right temporal | + S | 09/08/2007 | 06/06/2008 (alive) | 10 | 7 | monosomy 10, trisomy 7 |
Human fetal brain tissues were obtained from the Pathological Department. Consents were obtained from their parents. The forebrain SVZ was dissected from four fetuses (18–24 weeks old) and placed in DMEM‐FCS 0.1% medium until dissociation.
Cell dissociation and magnetic separation of A2B5+ cells
Within 4 h after obtention, tumors were washed, dissected, automatically sectioned using a McIlwain tissue chopper and enzymatically dissociated with both 5 mg/mL of Trypsin (Sigma‐Aldrich, Paris, France) and 200 U/mL of DNAse (Sigma‐Aldrich) for 10 minutes at 37°C. Trypan blue staining confirmed more than 80% cell viability. Cells were resuspended in DMEM‐FCS 10% and incubated with anti‐A2B5 mouse IgM antibody (cell supernatant 1/2 diluted, ATCC, Manassas, VA, USA) for 30 minutes at 4°C, washed and incubated with magnetic microbead‐tagged mouse‐specific IgM rat antibody for 30 minutes at 4°C. Positive magnetic cell separation (MACS Miltenyi Biotec, Paris, France) was performed using MACS column with an average purity of 93% (ranging from 85% to 98%) as assessed by A2B5 control‐immunostaining.
Culture of A2B5+ and A2B5‐ cells obtained from human GBM and SVZ
A2B5+ and A2B5‐ cells were plated at the density of 20 000 cells/25 cm3 flask in 3.5 mL of DMEM/F12 medium supplemented with hormones (5 µg/mL of Insulin, 0.1 mM of Putrescin, 100 µg/mL of Transferrin, 2.10−8 M of Progesterone; all purchased from Sigma‐Aldrich), 50 mg/mL of Penicillin‐Streptomycin (Invitrogen Life Technologies, Cergy Pontoise, France) and growth factors including 10 ng/mL of basic fibroblast growth factor (bFGF, Sigma‐Aldrich), 20 ng/mL of epidermal growth factor (EGF, R&D systems, Lille, France) and B27 (Invitrogen Life Technologies) as a stem cell‐permissive medium and maintained in 5% CO2/95% O2 atmosphere. The number and size of spheres were assessed twice a week using a 10× magnification and a calibrated micrometer reticule.
Clonal analyses of A2B5+ and A2B5‐ cells, freshly isolated from two GBM cases, were performed in 96‐well plates. Experiments were done in triplicate both for A2B5+ and A2B5‐ for each case.
Subsphere‐forming assay (secondary spheres)
After primary spheres formed and reached a size of 150 ± 50 cells each, spheres were harvested, dissociated into single cells and plated at the density of 20 000 cells/25 cm3 in the presence of 3.5 mL of stem cell‐permissive medium. Cultures were fed by changing half of the medium every 3 days. Subsphere‐forming assay (also called passage) was repeated every 2 weeks for GBM and each week for SVZ. After each passage, the number and size of spheres were assessed at days 7 and 14.
In order to assess clonal frequency of secondary spheres obtained from GBM (n = 3) single cells were plated in limiting dilution conditions (≤1 cell/well) in 96 well plates. Experiments were done in triplicate for each case.
Immunofluorescence
The expression of NSC markers and neural lineage markers was analyzed by immunofluorescence in three GBM cases for two different conditions: (i) at day 7 after secondary spheres were plated on 3 µg/mL poly‐L‐lysine coated glass coverslips supplemented with DMEM‐FCS 2%, conditions assumed to induce cell differentiation; and (ii) at day 14 on secondary spheres cultured in stem cell‐permissive medium. Immunofluorescence was performed as previously described (4), with the following primary antibodies for A2B5 (mouse IgM, supernatant 1/2 diluted, ATCC), CD133/2‐biotin (mouse IgG, 0.5 µg/mL, Miltenyi Biotec), Ki67 (mouse IgG, 5 µg/mL, Dako, Trappes, France), nestin (mouse IgG, 5 µg/mL, Abcam, Paris, France), polysialylated neural cell adhesion molecule (PSA‐NCAM, mouse IgM, supernatant 1/2 diluted, kindly provided by G. Rougon—IBDML, Marseille—France), glial fibrillary acid protein (GFAP, rabbit IgG, 10 µg/mL, Dako), βIII‐tubulin (mouse IgG, 2 µg/mL, Covance, Denver, PA, USA), GalC (mouse IgG, supernatant 1/2 diluted, ATCC), OLIG2 (goat IgG, 1 µg/mL, R&D systems). Fluorochrome conjugated‐secondary antibodies, Texas‐red anti‐mouse IgM, fluorescein isothiocyanate (FITC) anti‐rabbit IgG, Texas‐red anti‐mouse IgG, FITC anti‐mouse IgG, FITC anti‐goat IgG were purchased from Jackson Immunoresearch (Newmarket, Suffolk, UK) and anti‐biotin‐FITC from Miltenyi Biotec were diluted 1/100. Double stainings, A2B5/nestin, A2B5/Ki67 were performed by sequential incubation of primary antibodies.
Intracranial cell transplantation into nude mice
Cell populations of interest were resuspended in 1.5 µL of phosphate buffered saline (PBS). These aliquots were injected stereotactically in the corpus callosum or the striatum of 6‐ to 8‐week‐old athymic nude mice (Harlan France, Gannat, France). The injection coordinates were 1 mm anterior to bregma, −1 mm lateral and 2 mm in deep from the cortex surface for the corpus callosum and 1 mm anterior to bregma, −2 mm lateral and 2.5 mm in deep from surface for the striatum. Animals were anesthesized before all procedures and were observed until they fully recovered. The type of injected cells, their number, the site of injection and the time between injection and sacrifice are summarized in Table 1.
Mice were sacrificed as soon as they started to develop clinical symptoms (ataxia and loss of weight) and their brains were immediately removed and frozen in isopentan or fixed in 10% formalin for 24 h before being embedded in paraffin. Experimental protocols including animals were reviewed and approved by the Institutional Animal Care Committee of the School of Medicine.
Processing of mice brains
A total of about 200 brain sections were performed in all cases at 5‐µm thickness. Every 10 sections, a hematoxylin and eosin stain was performed for standard histopathological analysis. In addition, immunostaining with the anti‐EGFR25 (mouse IgG, Novocastra, Newcastle, UK) and anti‐Ki67 (mouse IgG, Dako) antibodies was performed using the Benchmark XT automate (Ventana Medical Systems SA, Illkirch, France) according to manufacturer's instructions.
Cell dissociation, fluorescence‐activated cell sorting and separation of A2B5+/CD133+ and A2B5+/CD133‐ cells
Fluorescence‐activated cell sorting (FACS) was performed on fresh GBM (n = 3) and on secondary spheres obtained from A2B5+ cells isolated from three different GBM (GBM 6 at passages 15 and 34, GBM 7 at passage 13, and GBM 9 at passages 15 and 32) to quantify the number of cells expressing A2B5 epitope and CD133 antigen, and to quantify their proliferation rate. Cells were dissociated, resuspended in PBS containing bovine serum albumin 0.5% and ethylenediaminetetraacetic acid 2 mM and stained with A2B5 (followed by FITC‐anti‐mouse IgM, Jackson Immunoresearch), CD133/2‐phycoerythrin (PE) (Miltenyi Biotec) or Ki67 (followed by allophycocyanin (APC)‐anti‐mouse IgG, Jackson Immunoresearch). The cells were processed on a FACSCalibur (Becton Dickinson, Le Pont‐De‐Claix, France). Data were analyzed using FlowJo software (Becton Dickinson). For each experiment, one million cells were analyzed. Cancer cells exhibiting large size were easily distinguished from dead cells and debris.
Generation of tumor cell lines from GBM 6 and GBM 9
We chose to expand enhanced green fluorescent protein (EGFP) primary cell lines from GBM 6 and GBM 9 because the GBM from which they derived, originated from a distinctive part of the brain (GBM 6 originates from the wall of the LV and GBM 9 from cortex) and contained a distinctive cell population with regard to the expression of A2B5 and CD133 markers recorded in secondary spheres. Therefore, these differences might reflect distinct GBM subgroups. Moreover, because both GBM 6 and GBM 9 primary cell lines demonstrated high self‐renewal capacities in vitro, the likelihood to obtain stable EGFP‐cell lines was higher than other primary cell lines. Cells were electroporated with pCX‐EGFP‐N1 (CMV‐IE enhancer and chicken β‐actin/rabbit β‐globin hybrid promoter). Adherent culture conditions were set up by coating the flasks with 10 µg/mL poly‐DL‐ornithin (Sigma‐Aldrich). Under these conditions, cells were able to grow attached to the bottom of the flasks without differentiating. Selection was achieved using 500 µg/mL neomycin in stem cell‐permissive medium and maintained in 5% CO2/95% O2 atmosphere. After a 2‐month selection, stable EGFP cell lines were obtained.
Proliferation and migration rate analysis
For proliferation and migration rate analysis, dissociated EGFP‐cells were seeded at a density of 2000 cells per well in 96‐well black plates (Greiner Bio‐One, Courtaboeuf, France) coated with 10 µg/mL poly‐DL‐ornithin. Cell content of each well was analyzed using an association of an automatic time‐lapse microscope (Flash‐cytometer, Trophos, Marseille, France) which automatically captures the fluorescence of the entire well plate together with the TINA software (Trophos). For proliferation analysis, EGFP‐cell lines were seeded on two distinct black plates. At day 0, for both cell lines, image acquisition was done for 12 wells. Images of the 12 remaining wells were taken 3 days later. Proliferation rate was calculated by dividing for each well the average cell content at day 3, by the average cell content at day 0. Experiments were done in triplicate. For migration analysis, a semi‐automated scratch (VP 408FH, VP‐Scientific, San Diego, CA, USA) was done in each well at day 0. Image acquisition was then performed. For each well, the scratch area acquired at day 0 was reported on the image of the same well acquired at day 3 and the number of cells recorded in the scratch area was evaluated. Because proliferation rate may influence migration, the migration rate was obtained by dividing the number of cells recorded in the scratch area at day 3 by the proliferation rate. Statistical analysis of results for both proliferation and migration was performed using the Mann–Whitney U test.
RESULTS
A2B5+ cells but not A2B5‐ cells are able to generate tumors when implanted into nude mice
When injected in the corpus callosum, A2B5+ cells, freshly isolated by magnetic cell sorting from three GBM samples, were able to generate dense tumors in 7/8 animals (n = 2 or 3 per case; Table 1, see footnote *). Injected cells infiltrated the whole hemisphere, reaching the pia matter and giving rise to a larger hemisphere (Figure 1A, a). Tumor cells were pleiomorphic and highly mitotic. Vessels showed hyperplasia of endothelial cells but neither microvascular proliferation nor necrosis was recorded (Figure 1A, b). Ki67 staining revealed about 15% of positive cells (Figure 1A, c and d). Moreover, tumor cells crossed the corpus callosum to colonize the contra‐lateral hemisphere (Figure 1A, c). In contrast, A2B5‐ cells were unable to generate huge tumor, although rare tumor cells were recorded within the corpus callosum in two out of seven animals. This might be caused by contamination with A2B5+ cells as MACS column allows an average purity of 93%.
Figure 1.
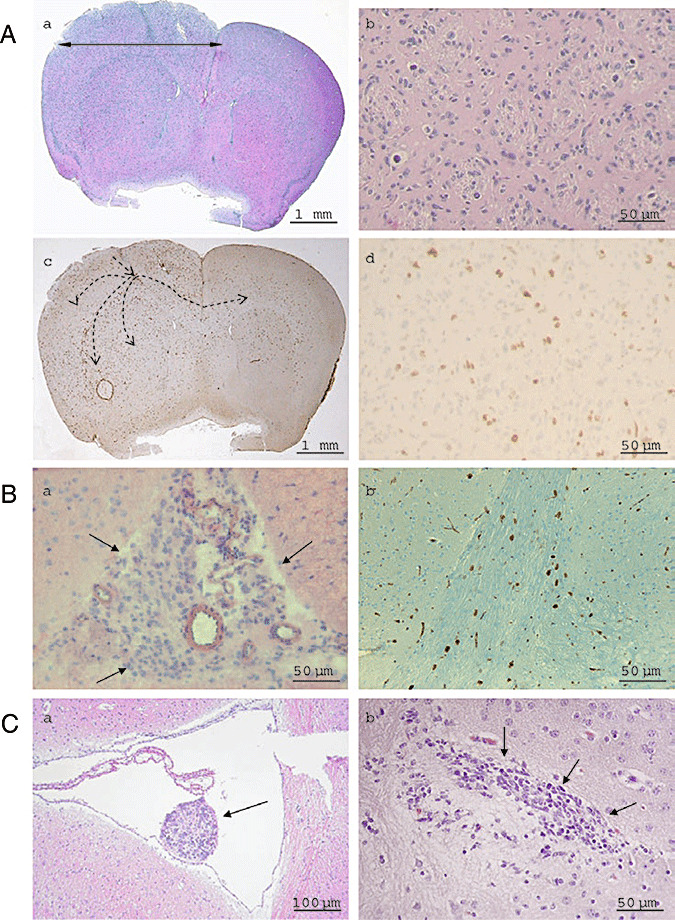
A. A2B5+ cells are tumorigenic after transplantation in nude mice brain: hematoxylin‐eosin histology (a, b) and anti‐Ki67 immunohistochemistry (c, d). Dotted arrows illustrate injected cells crossing the corpus callosum to colonize the contra‐lateral hemisphere (c). Dense tumors, made by polymorphic and highly mitotic cells (b) which infiltrate the whole hemisphere are observed, giving rise to a larger hemisphere (a). B. Secondary spheres obtained from cultured A2B5+ GBM cells are tumorigenic in vivo (GBM 6, 100 000 cells injected). Subarachnoidal spaces are filled with tumor (a). Tumor cells infiltrate the corpus callosum and express the proliferative marker Ki67 (b). C. Hematoxylin‐eosin histology of tumors obtained from injection of 1 000 cells A2B5+/CD133‐ (a) and 1000 cells A2B5+/CD133+ (b) isolated by flow cytometry from secondary spheres derived from GBM 6.
The GBM cells exhibit differential ability to form spheres according to A2B5 expression
After magnetic cell‐sorting, plating and culture in stem cell‐permissive medium, primary spheres were recorded in the A2B5+ fraction in 11/11 GBM cases within a week on average (from 2 days to 12 days). These spheres (Figure 2A, a) were abundant (40 to 455 spheres per flask), slightly irregular in shape and their volume increased continuously. It was worth noticing that in all cases when spheres were obtained within 9 days (Table 2), parental GBM demonstrated EGFR amplification. In contrast, there was no relationship between primary sphere formation time and the location of the GBM (lateral ventricular GBM vs. non‐lateral ventricular GBM). However, GBM 6 which originates from the frontal horn of the LV (SVZ area) generates spheres within 2 days (Table 2). As a comparison, in only one sample out of the 11 GBM (case 10, Table 2), a few spheres (<40) were observed at day 7 in the A2B5‐ fraction. These spheres are likely caused by the contamination of the A2B5‐ fraction by few A2B5+ cells as MACS column allows an average purity of 93%. Moreover, clonal analysis of both A2B5+ and A2B5‐ cells, freshly isolated from two GBM, confirmed that 2.5% ± 0.5% A2B5+ cells formed primary spheres whereas A2B5‐ did not. These results are in agreement with those obtained in vivo, when A2B5+ and A2B5‐ fractions from these two GBM were xenografted.
Figure 2.
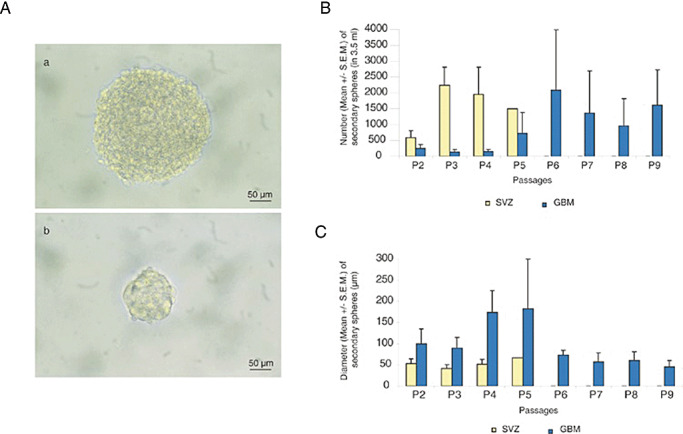
Persistence of self‐renewal and high proliferation capacity during passages for glioblastomas (GBM) secondary spheres derived from A2B5 + isolated cells compared with subventricular zone (SVZ). A. Phase contrast images of a sphere derived from GBM (a) and SVZ (b). B. Mean number of secondary spheres in 3.5 mL of stem‐cell permissive medium for 4 GBM (GBM 4, 5, 6 and 7) vs. 4 SVZ, showing a persistent self‐renewal capacity in GBM and a limited one in SVZ. C. Mean diameter of secondary spheres recorded in 4 GBM (GBM 4, 5, 6 and 7) compared with 4 SVZ. Mann–Whitney test showed that the diameter of GBM spheres is statistically higher than the diameter of SVZ spheres (P < 0.02). For B and C, results are expressed as mean ± standard error of the mean (SEM). Although bar error remains small for SVZ spheres, they are high for GBM spheres reflecting the heterogeneity of these tumors.
Both A2B5+ and A2B5‐ fractions of human embryo SVZ formed round small neurospheres [35 ± 18 µm, standard error of the mean (SEM)] as observed for 4/4 tested samples (Figure 2A, b). The number of spheres ranged from 50 to 100 per flask, suggesting that less than 1/200 dissociated cells were able to form neurospheres.
Self‐renewal potential of A2B5+ GBM cells
After the dissociation of primary spheres obtained from A2B5+ GBM cells, secondary spheres were observed in all cases (11/11). The secondary spheres started to form (two to four cells) as early as 1 day after dissociation. Clonal frequency was 71.25% ± 7.75% (SEM) of the total cells plated from dissociated secondary spheres (GBM 6). By comparison, secondary spheres obtained from either A2B5+ or A2B5‐ SVZ cells were recorded 2 ± 1 days after dissociation.
A smaller number of secondary spheres were obtained for GBM as compared with those obtained from SVZ (Figure 2B). However, the GBM secondary spheres exhibited a larger diameter than SVZ spheres (Figure 2C, P < 0.02). In subsphere‐forming assay, we observed that GBM secondary spheres demonstrated long‐term proliferation and self‐renewal. We may note that the total number of spheres as well as their diameter, measured 7 days after dissociation, varied from one passage to another and from one GBM to another (Figures 2B,C). In 7 out of 11 GBM cases, spheres can be passaged up to 7 times. This excludes the possibility that these spheres derive from normal residual NSC which exhibit limited self‐renewal properties (see below). Interestingly, three cases could be expanded for 36 (GBM 6), 18 (GBM 7) and 34 (GBM 9) passages (504, 252 and 476 days, respectively, in vitro), indicating a higher self‐renewal capacity in vitro. In contrast, for SVZ samples, the number of secondary spheres increased upon earlier passaging, then decreased gradually and propagation stopped after the fifth passage (Figure 2B). Their diameter however remained identical (Figure 2C).
Secondary spheres obtained from A2B5+ GBM cells and cultured in stem cell‐permissive medium contain CSC, proliferative A2B5+ cells and differentiated cells
In 3/11 GBM studied (GBM 6, 7 and 9), secondary spheres were positive for A2B5, nestin, CD133 and PSA‐NCAM as well as for the differentiation markers GFAP, βIII‐tubulin and GalC (Figure 3A). It is worth noticing that numerous A2B5+ cells expressed the Ki67 antigen (Figure 3A, b). Moreover, some A2B5+ cells co‐expressed nestin (Figure 3A, c). To quantify the number of A2B5+ cells in secondary spheres and further analyze their proliferation status, we performed flow cytometry analysis and showed that more than 70% of cells in GBM spheres were A2B5+ and also expressed the Ki67 antigen (data not shown), suggesting that they are in activated cell cycle status. Similar results were recorded in fresh GBM, suggesting that sphere culture conditions did not modify cell phenotype. It is worth noticing that the number of A2B5+ cells in secondary spheres remains stable through passages.
Figure 3.
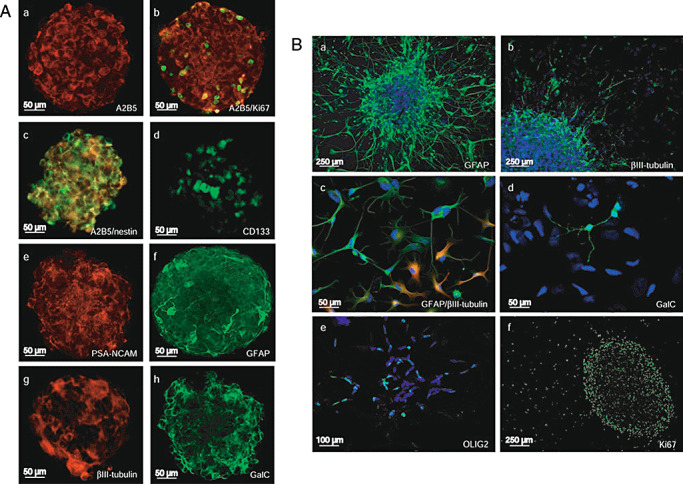
A. Neural stem cells (NSC) and differentiation markers in glioblastomas (GBM) secondary spheres cultured in stem cell‐permissive medium. (a) A2B5, (b) A2B5 (red)/Ki67 (green), (c) A2B5 (red)/nestin (green), (d) CD133, (e) polysialylated neural cell adhesion molecule (PSA‐NCAM), (f) glial fibrillary acid protein (GFAP), (g) βIII‐tubulin, (h) GalC. B. NSC and differentiation markers in GBM secondary spheres cultured in differentiation medium. (a) GFAP, (b) βIII‐tubulin, (c) GFAP (red)/βIII‐tubulin (green), (d) GalC, (e) OLIG2, (f) Ki67.
In differentiation assay in monolayer cultures, cells predominantly expressed the markers of the three cell lineage of CNS (Figure 3B): (a) GFAP, (b) βIII‐tubulin, (d) GalC (d) or (e) OLIG2. Few cells co‐expressed GFAP and βIII‐tubulin (Figure 3B, c). Ki67 expression was mainly restricted to the spheres (Figure 3B, f). Taken together, these results showed that A2B5+ cells derived from GBM are multipotent.
Secondary spheres obtained from A2B5+ GBM cells are tumorigenic in vivo
When A2B5+‐derived secondary spheres cultured for 6 (GBM 7) and 9 (GBM 6) passages in free‐floating sphere conditions were dissociated and then injected in nude mice, diffuse infiltrative tumors were observed in 6/10 mice (results are summarized in Table 1, see (2)). Tumor occurrence was not related to the number of injected cells nor to the parental GBM. As few as 1000 cells from secondary spheres obtained from A2B5+ cells derived from GBM 6 or GBM 7 were sufficient to generate tumors. Tumor cells were recorded in the subarachnoidal space and along the corpus callosum (Figure 1B, a). The migrating tumor cells expressed the Ki67 proliferation marker (Figure 1B, b) and the EGFR protein (data not shown). These injection experiments confirmed that after long term in vitro culture, A2B5+ cells are still able to generate tumors when grafted into nude mice.
A subset of A2B5+ GBM cells are CD133+ cells
In order to further define whether A2B5+ cells express the CD133 marker, the A2B5+ cells freshly isolated from three GBM samples were characterized by FACS. Sorted cell populations exhibited greater than 98% purity. In all cases, three distinct populations of cells were recorded: A2B5+/CD133+ cells, A2B5+/CD133‐ cells and A2B5‐/CD133‐ cells. We were unable to isolate any A2B5‐/CD133+ cells. In contrast, in the two cases of human fetal SVZ analyzed, 10% and 13% of A2B5‐/CD133+ cells were recorded, ruling out any technical difficulty in isolating A2B5‐/CD133+ cells from GBM tissue (data not shown). Although the number of A2B5+ cells recorded in all cases was always high (ranging from 69% to 98%), the number of CD133+ cells remained low, ranging from 1.3% to 9%.
Further characterization by flow cytometry of secondary spheres obtained from GBM 6, 7 and 9 showed major differences in their cell composition. The ratio of A2B5+/CD133+ cells was 5% (GBM 6, passage 15, Figure 4A), 13% (GBM 7, passage 13, Figure 4B) and 54% (GBM 9, passage 15, data not shown), whereas that of A2B5+/CD133‐ cells was 68% (GBM 6, Figure 4A), 84% (GBM 7, Figure 4B) and 89% (GBM 9, data not shown). In addition, we observed that all A2B5+/CD133+ cells proliferate (Figure 4). It is worth noticing that the percentage of A2B5 expressing cells remained stable through passages (GBM 6: passages 15 and 34; GBM 9: passages 15 and 32, data not shown).
Figure 4.
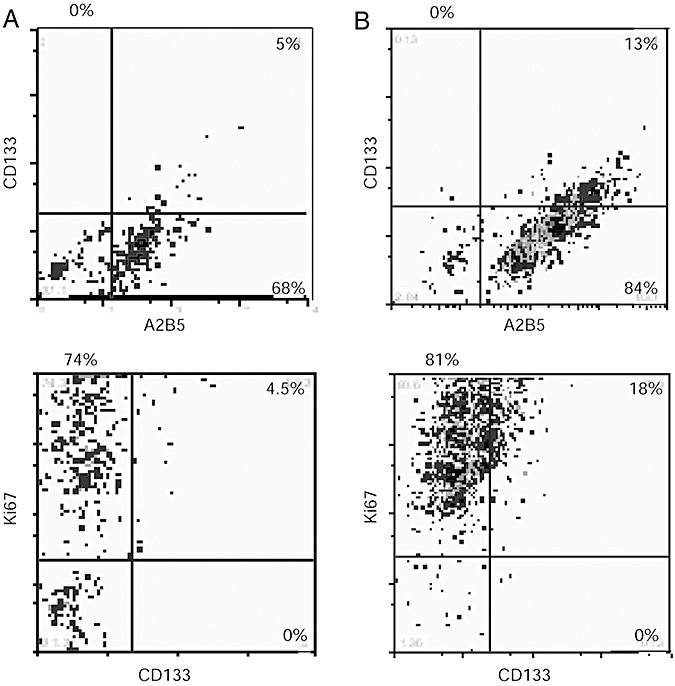
Flow cytometry analysis of secondary spheres derived from glioblastomas (GBM) A2B5+ cells A2B5, CD133 and Ki67 expression. A. GBM 6, passage 15: 5% of A2B5+/CD133+; 68% of A2B5+/CD133‐; 27% of A2B5‐/CD133‐. Similar results were obtained at passage 34. B. GBM 7, passage 13: 13% of A2B5+/CD133+; 84% of A2B5+/CD133‐; 3% of A2B5‐/CD133‐. All CD133+ cells express the Ki67 antigen but the major part of Ki67 expressing cells does not express CD133. Some CD133‐ cells do not proliferate (A,B).
The two fractions, A2B5+/CD133+ and A2B5+/CD133‐, isolated by FACS from GBM 6 A2B5+ secondary spheres at passage 19 were both able to generate spheres.
The cell fractions A2B5+/CD133+ and A2B5+/CD133‐ isolated from GBM secondary spheres are tumorigenic in vivo
When 1000 cells were injected into the corpus callosum and the striatum of nude mice, both A2B5+/CD133+ and A2B5+/CD133‐ fractions were able to generate tumors (Table 1, see (3)). In all cases, A2B5+/CD133‐ tumors were highly infiltrative, often associated with subarachnoidal space and ventricles involvement with intraventricular nodules (Figure 1C, a). By contrast, A2B5+/CD133+ cells rather had a tendency to form well‐demarcated tumors at the injection site without subarachnoidal space or the ventricles' involvement (Figure 1C, b).
Stable EGFP‐GBM 6 and ‐GBM 9 cell lines displayed differential migratory and proliferative behaviors
We then generated EGFP‐transfected GBM 6 and GBM 9 cell lines to further characterize their in vitro behavior. GBM 6 originates from a parental GBM lining the SVZ area, whereas GBM 9 derives from a cortical GBM (Figure 5A). After transfection, GBM 6 and GBM 9 cell lines kept the ability to divide and differentiate.
Figure 5.

Correlation between in vitro behavior of glioblastomas (GBM) 6 and GBM 9 EGFP cell lines, and Magnetic Resonance Imaging (MRI) images of the primary tumor of the corresponding patient. A. MRI scans show that GBM 6 tumor contacts lateral ventricle (left) and GBM 9 tumor is located in the cortex (right). B. GBM 6 and GBM 9 cell lines migration and proliferation rates. Pictures were obtained after acquisition at day 0 and at day 3 of monolayer culture using the flash‐cytometer. C. GBM 6 cells show the higher migration rate (left) and GBM 9 cells the higher proliferation rate (right). *P < 0.005. Abbreviation: SEM = standard error of the mean.
When cultivated as adherent cultures, the two EGFP‐transfected cell lines strikingly differed in their behavior. Three days after scratch test experiment (Figure 5B), cells from GBM 6 had an 11.5‐fold higher migration rate than cells from GBM 9 (Figure 5C, left; P < 0.005), whereas cells from GBM 9 proliferated 4.3‐fold more than those from GBM 6 (Figure 5C, right), suggesting that these two cell lines displayed distinct intrinsic properties. These results are sustained by FACS screening of CD133 and Ki67 content of the GBM 6 and GBM 9‐derived spheres. The first exhibited 5% of CD133+ cells and 78.5% of Ki67+ cells (passage 15, Figure 4A), the last 52.5% of CD133+ cells and 95% of Ki67 cells (passage 15, data not shown).
DISCUSSION
In a previous study, we reported that all human gliomas harbor a pool of glial progenitor cells expressing GT3 gangliosides recognized by the monoclonal antibody A2B5. The A2B5+ cells isolated from human GBM demonstrate a high proliferation index and are genetically transformed (4). In the present study, we demonstrate that these cells fulfill the criteria of brain cancer stem cells (CSC) as defined by Vescovi et al (18). They share with normal NSC the ability to proliferate and generate multipotent clones of cells—neurospheres—when cultured with the appropriate growth factors including EGF and bFGF (15), but in contrast to normal NSC, they have an extensive self‐renewal capacity as demonstrated in vitro by subsphere‐forming assay. Moreover, they are tumorigenic as they are able to generate tumors when implanted in nude mice. In addition, these cells display genetic alterations (4) and demonstrate aberrant differentiation properties such as coexpression of GFAP and βIII‐tubulin. In contrast, A2B5‐ cells isolated from GBM are not able to generate spheres (present study) and when xenografted into animals, they are poorly tumorigenic.
Our screening of GBM cells for A2B5 and CD133 expression distinguishes three subpopulations in agreement with a previous study (12): the A2B5‐/CD133‐, the A2B5+/CD133‐ and the A2B5+/CD133+ cells. The number of A2B5+ cells in GBM is consistently high, whereas the number of cells co‐expressing A2B5 and CD133 varies remarkably among patients, reflecting tumor heterogeneity between GBM. Evidence for the tumorigenic potential of both A2B5+/CD133+ and A2B5+/CD133‐ contrasts with that from previous experiments, which suggested that proliferation in vitro and tumorigenicity in vivo were restricted to the CD133+ cell subpopulation 16, 17. The observation that both CD133+ and CD133‐ cells are able to generate tumors in rodents first reported by Beier et al (2) has been confirmed by further studies 8, 12, 19. Therefore, it is likely that there are at least two kinds of CSC (A2B5+/CD133+ and A2B5+/CD133‐) in GBM patients which demonstrate different biological and clinical characteristics, as evidenced by in vitro properties of the GBM 6 and GBM 9 cell lines we have generated. GBM 6 originates from a GBM located in the wall of the LV; it contains a low number of CD133+ cells, exhibit a high migrating capacity both in vitro and in vivo. In contrast, GBM 9 derives from a cortical GBM which recurs in situ, contains a high number of CD133+ cells and displays a much lower migrating capacity (both in vitro and in vivo). Our results are in total accordance with the recent study reported by Joo et al (8).
In vivo study shows that CD133‐ glioma cells give rise to CD133+ cells in xenografts (19) a feature that we also observed after in vitro passaging. Moreover, CD133 expression upregulation coincides with the onset of angiogenesis and a shorter survival (19). In addition, orthotopic GBM xenografts suggest that GBM CD133+ CSC secrete angiogenic factor that promotes the recruitment and formation of tumor blood vessels (1). More recently, it has been reported that endothelial cells interact closely with self‐renewing CD133+ brain tumor cells suggesting that tumor microvasculature generates specific microenvironment niche which promotes the formation and/or maintenance of brain CD133+ CSC (3). Targeting CSC will require a better understanding of the differences between normal and malignant stem cells. We have shown that in contrast to normal A2B5‐ cells isolated from SVZ, A2B5‐ isolated from GBM are unable to form spheres. Therefore, the A2B5 epitope is required for GBM sphere formation in vitro and tumor initiation in vivo. Besides, A2B5+/CD133‐ cells isolated from GBM differ from the A2B5+/CD133+ cells and it is likely that the acquisition of the CD133 expression by tumor cells creates a more permissive microenvironment for survival.
Pfenninger et al support the hypothesis that CSC arise from proliferative CD133‐ neurogenic astrocytes, but brain tumor development would involve the acquisition of CD133 antigen (14). The A2B5+ cells form a pool of multipotential neural progenitor cells from the subcortical white matter of the adult human brain (11). The A2B5+ expression is also recorded on radial glial cells (9) that give rise to adult NSC in the SVZ (10). The mitotically component A2B5+ expressing cells are probably the target of the oncogenic process in gliomagenesis.
In conclusion, we suggest that A2B5 expression is required for brain tumor initiation and maintenance, whereas CD133 plays a role in brain tumor progression by modulating the tumor's behavior. It is however likely that other cell surface markers might also influence the glioblastomas' behavior in keeping with their heterogeneity.
ACKNOWLEDGMENTS
We are grateful to the neurosurgeons Dr S. Fuentes, Professor H. Dufour and Prof. J.C. Peragut for providing tumor samples, to Drs C. Bouvier, C. Fernandez and A. Maues de Paula for their help in tumor and SVZ handling and pathological diagnoses, to Dr O. Chinot and C. Boucard for providing clinical information regarding the patients, to Dr B. Quilichini for FISH analysis and to C. Cognet, E. Cassotte, D. Intagliata, P. Morando, M. Auphan, L. Calvo and C. Prevot for technical assistance. Dr C. Berenguer is specially acknowledged for stereotactic injections and Dr P. Durbec for helpful discussions. Finally, we are deeply grateful to Dr G. Rougon for useful suggestions while reading the manuscript.
This work was supported by the Cancéropôle PACA Grants RS019, by the INCA platform 2007, by the Association pour la Recherche sur les Tumeurs Cérébrales (ARTC‐Sud) and by the Daher foundation.
REFERENCES
- 1. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB et al (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444:756–760. [DOI] [PubMed] [Google Scholar]
- 2. Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ et al (2007) CD133(+) and CD133(−) glioblastoma‐derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res 67:4010–4015. [DOI] [PubMed] [Google Scholar]
- 3. Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B et al (2007) A perivascular niche for brain tumor stem cells. Cancer Cell 11:69–82. [DOI] [PubMed] [Google Scholar]
- 4. Colin C, Baeza N, Tong S, Bouvier C, Quilichini B, Durbec P, Figarella‐Branger D (2006) In vitro identification and functional characterization of glial precursor cells in human gliomas. Neuropathol Appl Neurobiol 32:189–202. [DOI] [PubMed] [Google Scholar]
- 5. Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S et al (2004) Isolation and characterization of tumorigenic, stem‐like neural precursors from human glioblastomas. Cancer Res 64:7011–7021. [DOI] [PubMed] [Google Scholar]
- 6. Hemmati HD, Nakano I, Lazareff JA, Masterman‐Smith M, Geschwind DH, Bronner‐Fraser M, Kornblum HI (2003) Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA 100:15178–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA (2002) Human cortical glial tumors contain neural stem‐like cells expressing astroglial and neuronal markers in vitro . Glia 39:193–206. [DOI] [PubMed] [Google Scholar]
- 8. Joo KM, Kim SY, Jin X, Song SY, Kong DS, Lee JI et al (2008) Clinical and biological implications of CD133‐positive and CD133‐negative cells in glioblastomas. Lab Invest 88:808–815. [DOI] [PubMed] [Google Scholar]
- 9. Li H, Babiarz J, Woodbury J, Kane‐Goldsmith N, Grumet M (2004) Spatiotemporal heterogeneity of CNS radial glial cells and their transition to restricted precursors. Dev Biol 271:225–238. [DOI] [PubMed] [Google Scholar]
- 10. Merkle FT, Tramontin AD, Garcia‐Verdugo JM, Alvarez‐Buylla A (2004) Radial glia give rise to adult neural stem cells in the subventricular zone. Proc Natl Acad Sci USA 101:17528–17532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nunes MC, Roy NS, Keyoung HM, Goodman RR, McKhann G 2nd, Jiang L et al (2003) Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med 9:439–447. [DOI] [PubMed] [Google Scholar]
- 12. Ogden AT, Waziri AE, Lochhead RA, Fusco D, Lopez K, Ellis JA et al (2008) Identification of A2B5+CD133‐ tumor‐initiating cells in adult human gliomas. Neurosurgery 62:505–514. [DOI] [PubMed] [Google Scholar]
- 13. Ostenfeld T, Joly E, Tai YT, Peters A, Caldwell M, Jauniaux E, Svendsen CN (2002) Regional specification of rodent and human neurospheres. Brain Res Dev Brain Res 134:43–55. [DOI] [PubMed] [Google Scholar]
- 14. Pfenninger CV, Roschupkina T, Hertwig F, Kottwitz D, Englund E, Bengzon J et al (2007) CD133 is not present on neurogenic astrocytes in the adult subventricular zone, but on embryonic neural stem cells, ependymal cells, and glioblastoma cells. Cancer Res 67:5727–5736. [DOI] [PubMed] [Google Scholar]
- 15. Reynolds BA, Weiss S (1992) Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255:1707–1710. [DOI] [PubMed] [Google Scholar]
- 16. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB (2003) Identification of a cancer stem cell in human brain tumors. Cancer Res 63:5821–5828. [PubMed] [Google Scholar]
- 17. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T et al (2004) Identification of human brain tumour initiating cells. Nature 432:396–401. [DOI] [PubMed] [Google Scholar]
- 18. Vescovi AL, Galli R, Reynolds BA (2006) Brain tumour stem cells. Nat Rev Cancer 6:425–436. [DOI] [PubMed] [Google Scholar]
- 19. Wang J, Sakariassen P, Tsinkalovsky O, Immervoll H, Boe SO, Svendsen A et al (2008) CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int J Cancer 4:761–768. [DOI] [PubMed] [Google Scholar]