

Abstract
We report on three adult patients with primary glioblastomas showing prominent adipocytic (lipomatous) differentiation, hence referred to as “glioblastomas with adipocyte‐like tumor cell differentiation.” Histologically, the tumors demonstrated typical features of glioblastoma but additionally contained areas consisting of glial fibrillary acidic protein (GFAP)‐positive astrocytic tumor cells resembling adipocytes, that is, containing large intracellular lipid vacuoles. Comparative genomic hybridization (CGH) and focused molecular genetic analyses demonstrated gains of chromosomes 7, losses of chromosomes 9 and 10, as well as homozygous deletion of p14ARF in one of the tumors. The second tumor showed gains of chromosomes 3, 4, 8q and 12 as well as losses of chromosomes 10, 13, 15q, 19 and 22. In addition, this tumor carried homozygous deletions of CDKN2A and p14ARF as well as point mutations in the TP53 and PTEN genes. The third tumor also had a mutation in the PTEN gene. None of the tumors demonstrated EGFR, CDK4 or MDM2 amplification. Taken together, our results define a rare glioblastoma differentiation pattern and indicate that glioblastomas with adipocyte‐like tumor cell differentiation share common molecular genetic features with other primary glioblastomas.
Keywords: glioblastoma, immunohistochemistry, lipidization, lipomatous metaplasia, molecular genetics
INTRODUCTION
Lipidization is a well‐known but infrequent feature in primary neuroepithelial neoplasms of the central nervous system (11). Lipids may accumulate within the cytoplasm of tumor cells as multiple small droplets giving rise to a foamy or xanthomatous appearance as exemplified by pleomorphic xanthoastrocytoma. Alternatively, lipidization may result in an adipocyte‐like appearance of neuroepithelial tumor cells as typically seen in cerebellar liponeurocytoma 1, 9, as well as rare cases of central neurocytoma 12, 17, primitive neuroectodermal tumors (30) and medulloblastoma (32). Though gliomas are not generally noted for the presence of extensive fatty changes, individual cases of lipidized ependymoma 29, 31 and low‐grade astrocytoma 2, 11, 27, 36 have been encountered. Also, a single case of astroblastoma with an unusual signet‐ring/adipocyte‐like cell component has been reported (33).
Glioblastoma is the most common and most malignant primary brain tumor, accounting for approximately 12%–15% of all intracranial neoplasms and 50%–60% of all astrocytic tumors (20). Occasionally, glioblastomas may contain cells with foamy cytoplasm that very rarely dominate the histological appearance. These tumors have been referred to as “malignant gliomas with heavily lipidized tumor cells” or “lipid‐rich glioblastomas”10, 15, 28, 34. However, to the best of our knowledge, glioblastomas with adipocytic differentiation have not been reported to date. Molecular genetic analyses of astrocytic tumors with adipocytic differentiation are restricted to a single case of low‐grade lipoastrocytoma (11). Here, we report on three patients with glioblastomas showing prominent lipomatous differentiation with astrocytic tumor cells resembling mature adipocytes. We designate these cases as “glioblastomas with adipocyte‐like tumor cell differentiation” and delineate their histological characteristics and molecular genetic profiles.
MATERIALS AND METHODS
Tissue specimens, histology, immunohistochemistry and electron microscopy
Tumor tissue specimens were fixed in 4% buffered formalin and were routinely embedded in paraffin. From each case, paraffin sections were stained with hematoxylin and eosin (HE) as well as with reticulin stains. In addition, immunohistochemistry was performed by using an avidin–biotin–peroxidase complex (ABC) technique with 3′3‐diaminobenzidine as the chromogen. As primary antibodies, we used rabbit polyclonal antibodies against glial fibrillary acidic protein (GFAP; Dako, Hamburg, Germany), as well as mouse monoclonal antibodies against the proliferation marker Ki‐67 (clone MIB‐1) and the tumor suppressor protein p53 (clone DO‐7) (both from Dako). The tumors were classified according to the World Health Organization (WHO) classification of tumors of the central nervous system (20) as glioblastoma (WHO grade IV) with adipocytic (lipomatous) metaplasia, and the term “glioblastomas with adipocyte‐like tumor cell differentiation” was suggested for these tumors. From each tumor, we extracted DNA from formalin‐fixed and paraffin‐embedded tumor specimens as described before (24). Formalin‐fixed tumor tissue from patient 3 was postfixed in glutaraldehyde and was routinely processed for electron microscopy.
Comparative genomic hybridization (CGH) analysis
The tumors from patients 1 and 2 were subjected to CGH analyses. With minor modifications, CGH analysis was performed as described (25). Briefly, tumor DNA was labeled with biotin‐16‐dUTP (Boehringer, Mannheim, Germany) and reference DNA from a healthy male donor with digoxigenin‐11‐dUTP (Boehringer) in a standard nick translation reaction. Biotinylated and digoxigenated sequences were detected simultaneously using anti‐avidin‐FITC (fluorescein isothiocyanate, dilution 1:200, Boehringer) and anti‐digoxigenin–rhodamine (dilution 1:40, Boehringer). The slides were counterstained with 4′,6‐Diamidino‐2‐phenylindole (DAPI) and were mounted with an antifade solution (Vectashield, Vector Laboratories, Burlingame, CA, USA). Separate gray‐level images of rhodamine, FITC and DAPI fluorescence were taken by a CCD camera. Image processing was carried out and the average red/green ratios were calculated for each chromosome in at least 10 metaphases. Chromosomal regions with CGH ratio profiles surpassing the upper threshold of 1.25 and the lower threshold of 0.75 were defined as copy number gains or losses (Figure 1).
Figure 1.
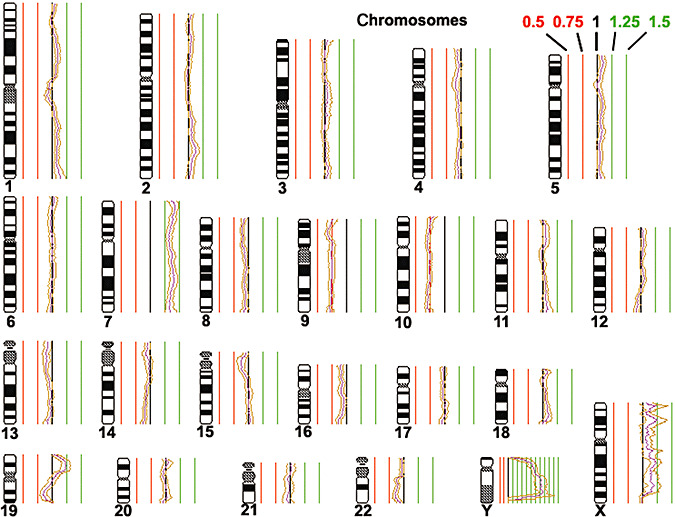
Comparative genomic hybridization (CGH) profile obtained for the tumor of patient 1. Thresholds for genomic gains are indicated in green, while thresholds for genomic losses are indicated in red. Note that the tumor shows a gain of chromosome 7 and losses of chromosomes 9 and 10.
Molecular genetic analyses
The TP53 (exons 4–10) and PTEN (exons 1–9) tumor suppressor genes were screened for mutations by single‐strand conformation polymorphism (SSCP) analysis as described 4, 24. In brief, PCR products were separated by electrophoresis on non‐denaturing polyacrylamide gels. Each PCR product was analyzed under two different conditions with variations in temperature and/or acrylamid/bisacrylamid ratios. The SSCP band patterns were visualized by silver staining. Aberrant SSCP bands were excised from the gel, re‐amplified using the same set of primers and sequenced in both directions using the BigDye cycle sequencing kit (Applied Biosystems, Foster City, CA, USA) and an ABI PRISM 377 DNA sequencer (Applied Biosystems). Analyses for amplification of the proto‐oncogenes MDM2 (12q14‐q15), CDK4 (12q13‐q14) and EGFR (7p12), as well as deletion of the CDKN2A and p14ARF genes (both on 9p21), were performed by duplex PCR assays as reported elsewhere 26, 37. DNA samples extracted from non‐neoplastic brain tissue and peripheral blood leukocytes were used as constitutional controls. As positive controls, we employed the human epithelial carcinoma cell line A431 with known EGFR amplification, the glioblastoma cell line TP365MG with CDK4 and MDM2 amplification, and the glioblastoma cell line U118MG with homozygous CDKN2A and p14ARF deletion. The PCR products were separated on 2% agarose gels, and ethidium bromide‐stained bands were recorded by the Gel‐Doc 1000 system (Bio‐Rad, Hercules, CA, USA). Quantitative analysis of the signal intensities obtained for each target gene and the corresponding reference gene was performed with the Molecular Analyst® software (version 2.1, Bio‐Rad). Only increases in the target/reference gene ratio of more than three times the ratio obtained for constitutional DNA were considered as gene amplification. A target/reference gene ratio of <0.3 relative to the control tissue was considered as homozygous deletion, whereas a ratio between 0.3 and 0.7 was judged as evidence for a hemizygous deletion.
RESULTS
Clinical findings
Patient 1
At hospital admission, a 63‐year‐old male patient presented with paresthesia in the face and, later on, in the whole body. Magnetic resonance imaging (MRI) showed a garland‐like, contrast‐enhancing pathologic structure (3 cm in the maximum diameter) in the right temporal lobe. A right pterional approach was chosen to remove the strongly vascularized tumor from the edematous brain and its attachments to the temporal dura mater. The immediate postoperative course was free from complications and neurological deficits. Adjuvant radiation therapy was administered. The patient died from his tumor 3 years after diagnosis.
Patient 2
A 63‐year‐old woman presented with a 2‐month history of frontal headaches and nausea as well as slowly developing cognitive deficits and aphasia. The relatives had additionally noticed a change of personality with an increasingly depressed mood. Upon hospital admission, the patient was only partially oriented, in a reduced general state and showed an unbalanced slow gait as well as bilateral bradydiadochokinesis. She appeared intellectually and motorically impaired and showed incomplete aphasia. Cranial computer tomography (CCT) and MRI scans revealed a left‐sided fronto‐temporal cystic lesion (5 × 5 cm in diameter) with marginal contrast enhancement and accompanying perifocal edema as well as midline lateralization. After surgical intervention, the patient did not recover significantly, remained confined to bed and had to be fed artificially. Because of the patient's reduced general state, adjuvant radiotherapy was not administered. Four months after surgery, the patient deceased from her disease in a hospice.
Patient 3
After an initial single generalized seizure, a 75‐year‐old male patient was admitted to hospital in a postictal somnolent state. Imaging findings were those typical of glioblastoma: CCT and MRI scans revealed a centrally necrotic, marginally contrast‐enhancing tumor in the right occipital lobe. In the non‐contrast T1‐weighted MRI, the tumor appeared as a large, heterogeneous, hypointense white matter mass with an area of lower signal intensity in the center of the mass suggestive of necrosis (Figure 2J). A second non‐contrast‐enhancing focus was detected in the right temporal lobe. An initial surgical approach was undertaken to remove the right occipital focus after which the patient recovered well without focal neurological deficit. Surgical treatment was followed by radiotherapy and chemotherapy with temozolomide. With reappearance of seizures and focal neurological deficits about 5 months after the first operation, a second MRI scan revealed a marked progress of both the contrast‐enhancing, now partially cystic, as well as the non‐enhancing tumor parts with accompanying perifocal edema and infiltration of the corpus callosum. A second palliative surgery was performed in order to implant an Ommaya reservoir for drainage of the tumor‐associated cyst and for reduction of intracranial pressure. Also, brachytherapy with 209 MBq Yttrium 90 Kolloid was administered. Medication with steroids for reduction of edema and anti‐convulsives set the patient in a reasonable state for outpatient treatment in a retirement home. At the last follow‐up 8 months after diagnosis, edema and the size of the tumor cyst had decreased, and no deterioration of the general state and neurological symptoms were recorded.
Figure 2.
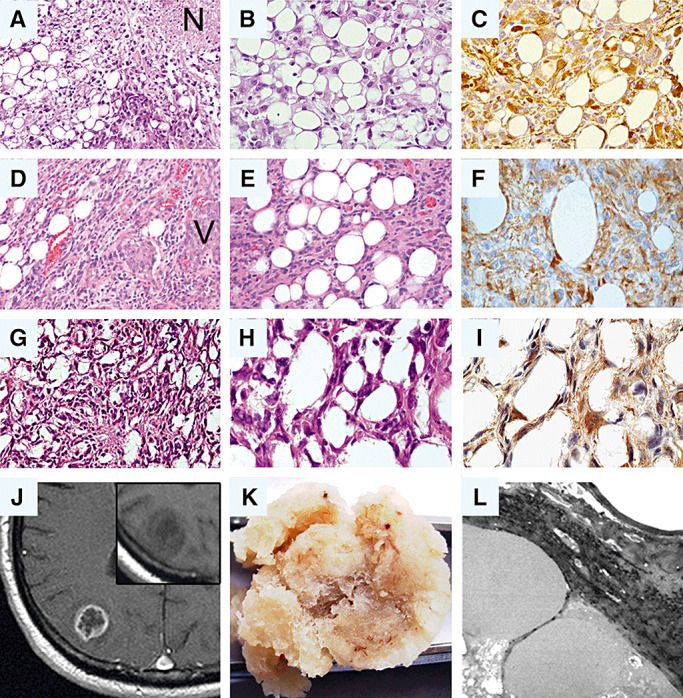
Synopsis of morphological features in glioblastomas with adipocyte‐like tumor cell differentiation. Note that the histological appearance of all three tumors (A–C: patient 1; D–F: patient 2; G–I: patient 3) is characterized by the presence of adipocyte‐like tumor cells intermingled with cells that resemble classic astrocytic tumor cells. (A,B,D,E,G,H: hematoxylin–eosin). Immunohistochemical staining for glial fibrillary acidic protein (GFAP) supports the glial origin of the adipocytic tumor cells in each case (C,F,I). (J–L) Radiological, macroscopic and ultrastructural features in case 3. (J) on contrast‐enhanced magnetic resonance imaging (MRI), the tumor shows ring enhancement as typical for glioblastoma. Insert: Though histologically dominated by adipocyte‐like cells, the tumor does not appear bright but is hypointense on non‐contrast T1‐weighted MRI. (K) The macroscopic appearance of the resected tumor specimen is similar to that of adipose tissue. (L) Electron microscopy reveals large fat vacuoles corroborating the adipocytic nature of the tumor cells. N = necrosis; V = pathological vessel.
Histopathological and immunohistochemical findings
Patient 1
Histological analysis of tumor 1 (Figure 2A,B) revealed a cellular, diffusely infiltrative glial tumor with marked regional heterogeneity. In addition to fibrillary and gemistocytic astrocytes, the tumor contained hypercellular areas with small anaplastic glioma cells as well as fascicular areas composed of spindle cells. Multiple typical and atypical mitoses, geographic necroses and microvascular proliferations were present (Figure 2A). In addition to these typical glioblastoma features, the tumor contained lipomatous areas consisting of adipocyte‐like cells with large intracytoplasmic vacuoles (Figure 2A,B). These lipomatous areas amounted to approximately 10% of the entire specimen. Immunohistochemically, the tumor cells expressed GFAP, with stronger reactivity in the gemistocytic than in the small anaplastic tumor cell component. Of note, the lipidized tumor component also showed strong GFAP immunoreactivity (Figure 2C). Approximately 30%–40% of the tumor cells displayed nuclear p53 staining, and the Ki‐67 (MIB‐1) proliferation index ranged around 20%–30%, with proliferative activity being most pronounced in the anaplastic small cell tumor component.
Patient 2
Histologically (Figure 2D,E), the general architecture of the second tumor was that of a regionally heterogeneous, diffusely infiltrating astrocytic tumor with alternating highly cellular small cells areas as well as less cellular, partly microcystic tumor parts. Focally, the tumor also showed collagen‐rich desmoplastic areas. Apart from small tumor cells with eosinophilic fibrillary processes, larger and highly pleomorphic cells were encountered. Mitotic activity was prominent. Large geographic necroses, smaller necroses with perinecrotic pseudopalisading as well as prominent microvascular proliferations were present (Figure 2D). Dispersed throughout the biopsy specimen, areas containing tumor cells with adipocyte‐like cytoplasmic vacuolation caused by lipidization were found, which amounted for up to 5%–10% of the entire specimen (Figure 2E). Adipocyte‐like tumor cells were particularly common in the paucicellular, microcystic and less common in the highly cellular, small cell tumor parts. No adipocyte‐like cells were observed in the desmoplastic tumor areas. In many of the lipidized tumor cells, the cytoplasm was almost completely filled with fat, thus resulting in a morphological appearance similar to mature adipocytes. Both the astrocytic as well as the adipocyte‐like tumor cells showed variably strong and diffuse immunohistochemical reactivity for GFAP, which in the latter cells appeared as thin rims around the central vacuole (Figure 2F). The MIB‐1 index was variable throughout the tumor and focally reached up to 30%. While several proliferating adipocyte‐like cells were encountered, the MIB‐1 index taken for the adipocyte‐rich tumor cells alone was notably lower, ranging around 5%.
Patient 3
The resection specimen submitted for histopathological evaluation was exceptional as it macroscopically appeared like adipose tissue (Figure 2K). Microscopically (Figure 2G,H), the specimen was mainly composed of polygonal, vacuolated, adipocytic tumor cells amounting to about 80% of the entire specimen. Electron microscopy corroborated the adipocytic nature of the tumor cells by revealing large intracytoplasmatic vacuoles within tumor cells that corresponded to fat. Unfortunately, because of the preceding formalin fixation, the quality of the images was not sufficient to identify further ultrastructural features, such as, for example, intermediate filaments (Figure 2L). The lipid‐laden cells were arranged by small septa of fibrillary glial cells, which in parts of the tumor formed islets of solid glial tumor tissue with markedly increased cell density and the typical features of astrocytic differentiation. Frequent mitoses (up to two per high‐power field) and pseudopalisading necroses were encountered. Immunohistochemically, GFAP was highly expressed in the tumor parts with typical astrocytic differentiation as well as in the glial septa between the lipid‐laden cells (Figure 2I). In particular, single highly polymorphic, partly multinucleated tumor cells showed strong GFAP expression. More than 80% of tumor cell nuclei exhibited nuclear staining for the p53 tumor suppressor protein. The MIB‐1 proliferation index was about 5%, reaching focally up to 20% in the tumor parts with the highest cell density.
Molecular genetic findings
Patient 1
CGH analysis of the tumor of patient 1 revealed gains of chromosome 7 and losses of chromosomes 9 and 10 (Figure 1). In addition, molecular genetic analyses identified a homozygous deletion of the p14ARF gene on chromosome 9p (Figure 3). Neither mutations of the PTEN or TP53 gene, nor copy number gains or losses of the EGFR, CDK4, MDM2 or CDKN2A genes were detected.
Figure 3.
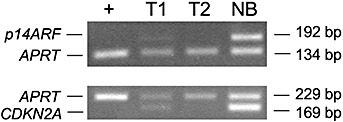
Duplex PCR analysis for homozygous deletion of CDKN2A and p14ARF. The lanes correspond to +, U118MG (control with homozygous deletion of CDKN2A and p14ARF); T1, tumor of patient 1; T2, tumor of patient 2; NB, non‐neoplastic brain tissue. Note homozygous deletion of both CDKN2A and p14ARF in the tumor of patient 2 and homozygous deletion of p14ARF in the tumor of patient 1. bp = length of the respective PCR products in base pairs.
Patient 2
The tumor of patient 2 showed gains of chromosomes 3, 4, 8q, 12p and 12q, as well as losses of chromosomes 10, 13, 15q, 19 and 22 by CGH analysis. Mutational screening detected a missense mutation within exon 7 of the TP53 gene (c.743G > A, W248Q; Figure 4) and a splice site mutation within intron 3 of the PTEN gene (IVS3 + 5G > A; Figure 4). Homozygous deletions were found in both the CDKN2A and the p14ARF gene (Figure 3), while the tumor lacked detectable amplification of the EGFR, CDK4 or MDM2 proto‐oncogenes.
Figure 4.
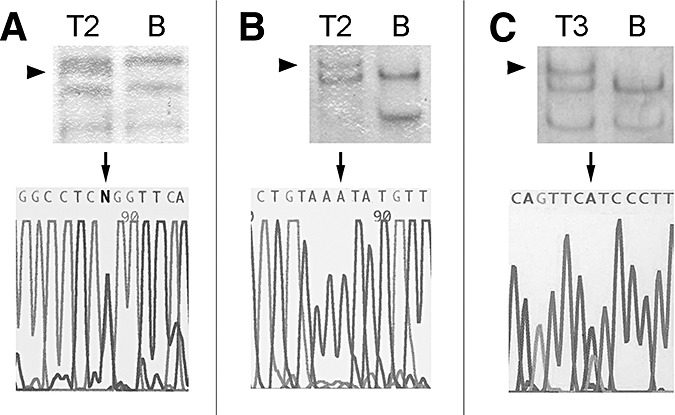
Results of the TP53 and PTEN mutation analyses. The tumor of patient 2 (T2) carried a missense mutation in exon 7 of the TP53 gene (c.743G > A, W248Q) (A) and a splice site mutation in intron 3 of the PTEN gene (IVS3 + 5G > A) (B). The tumor of patient 3 (T3) exhibited a nonsense mutation in exon 5 of the PTEN gene (c.388C > T, R130X) (C). Upper panel, single‐strand conformation polymorphism (SSCP) analysis; lower panel, DNA sequencing analysis. Aberrant SSCP bands are indicated by arrowheads. The depicted sequences are derived from the noncoding strand. The individual mutations are indicated by arrows. B = blood leukocyte DNA.
Patient 3
Mutational analysis of this patient's tumor exhibited a nonsense mutation in exon 5 of the PTEN gene (c.388C > T, R130X; Figure 4). Despite the widespread nuclear p53 immunopositivity, no TP53 mutation was identified by SSCP analysis of exons 4–10. Homozygous deletions of CDKN2A or p14ARF, and amplifications of EGFR, CDK4 or MDM2 were not detectable. CGH analysis was not performed.
DISCUSSION
Lipidization is a well‐known but infrequent feature in primary neuroepithelial neoplasms of the central nervous system (11). Occasionally, cells with foamy cytoplasms are observed in glioblastomas that in rare cases predominate the histological appearance and have in the past been referred to as “malignant gliomas with heavily lipidized tumor cells” or “lipid‐rich glioblastomas”10, 15, 28, 34. All of these glioblastomas were characterized by xanthomatous tumor cells which are a sign of fatty degeneration and can occur in any malignant neoplasm, in particular in cells immediately adjacent to necrotic areas. In contrast to these cases, the glioblastomas with adipocyte‐like tumor cell differentiation presented in this study do not show a xanthomatous but an adipocyte‐like differentiation with tumor cell cytoplasms being almost completely filled by fat, thus resulting in a morphological appearance similar to mature adipocytes.
The three reported tumors shared similar morphological features, with the general histological appearance being that of a diffusely infiltrating malignant astrocytic tumor composed of highly pleomorphic cells with obvious mitotic activity, microvascular proliferation and small filiform, as well geographic necroses. In addition, all three tumors contained areas of cells with cytoplasmic vacuolation caused by coalescing lipidization. These adipocyte‐like tumor cells were predominantly found in less cellular tumor areas. An association with necrosis was not observed. Of note, neither lesion presented with any xanthomatous tumor cells as reported in the previously published glioblastoma cases with extensive lipidization 10, 15, 28, 34. In each of the three tumors, adipocyte‐like tumor cells were immunohistochemically positive for GFAP, thus clearly pointing to their glial origin (Figure 2C,F,I).
The molecular mechanisms leading to lipidization or adipocyte‐like differentiation of glial tumor cells are still unclear. Non‐neoplastic astrocytes are supposed to go through a lipidized stage as part of their normal development in the course of myelinization of the white matter (18). Interestingly, in some cases of white matter injury, for example, periventricular leukomalacia, astrocytes may become markedly lipidized, which has been hypothesized to reflect metabolic changes (15). Furthermore, it has been demonstrated that gliomas can contain up to three times more lipids compared with normal brain tissue (21). Nevertheless, the reason why lipidization and adipocyte‐like differentiation may occur to such notable extent as in the cases of glioblastoma reported here remains elusive.
Molecular genetic analyses of lipidized astrocytic tumors are limited to just one case of low‐grade astrocytoma that neither displayed mutation of TP53 nor demonstrated deletion of CDKN2A or amplification of EGFR, CDK4 or MDM2 (11). The molecular findings in our three cases of glioblastomas with adipocyte‐like tumor cell differentiation are in line with previous data on primary glioblastomas. A review on CGH results of 184 patients revealed loss of genetic material of 10q as the most common chromosomal change in adult glioblastomas (25), an aberration also encountered in both glioblastomas with adipocyte‐like tumor cell differentiation that were subjected to CGH analysis. Deletion of 10q has previously been linked to the acquisition of the glioblastoma phenotype during astrocytoma progression (7). Moreover, multiple tumor suppressor gene loci on the long arm of chromosome 10 have been implicated in the development of astrocytic gliomas (13). Approximately one‐third of primary glioblastomas carries mutations in the PTEN tumor suppressor gene located at 10q23.3 8, 35, with PTEN mutations being rare in secondary glioblastomas and low‐grade gliomas (22). We detected PTEN mutations in two of the three glioblastomas with adipocyte‐like tumor cell differentiation, namely, an intronic splice site mutation within intron 3 in one tumor and a nonsense mutation within exon 5 in the other tumor. Both mutations had already been reported in sporadic cases of high‐grade astrocytomas 3, 5, 6, 22, 35, thus providing evidence for shared genetic aberrations between glioblastomas with adipocyte‐like tumor cell differentiation and other primary glioblastomas.
Mutations in the TP53 tumor suppressor gene are detectable in approximately 25%–30% of all glioblastomas, with a significantly higher percentage of mutant tumors among secondary glioblastomas as compared with primary glioblastomas (20). One glioblastoma with adipocyte‐like tumor cell differentiation of our series demonstrated a missense mutation within exon 7 of TP53. This particular mutation had been reported before in the glioblastoma cell line SF‐295 (14).
Gains of chromosome 7—containing the EGFR gene at 7p12, which is amplified in 30%–40% of primary glioblastomas—and losses of chromosome 9p—containing the tumor suppressor genes CDKN2A, p14ARF and CDKN2B at 9p21—are both frequent genomic alterations in glioblastomas 19, 23. Combined gain of chromosome 7 and loss of chromosome 9 was also detected in one of the glioblastomas with adipocyte‐like tumor cell differentiation (tumor 1). While all three tumors lacked EGFR amplification, homozygous deletions of CDKN2A and/or p14ARF were found in two glioblastomas with adipocyte‐like tumor cell differentiation (tumors 1 and 2). Amplification of CDK4 (12q14) and MDM2 (12q14.3‐15) was not encountered in our tumor panel. Nevertheless, case 2 presented with a gain of 12q in CGH, which is detectable in approximately 16% of glioblastomas (25).
The tumor of patient 2 had additional chromosomal losses affecting the chromosomal arms 13q, 19q and 22q, which are all well‐documented chromosomal changes in glioblastomas 19, 23. Further imbalances (gains on chromosomes 3, 4, 8q and 12p, losses on 15q) are restricted to less than 10% of glioblastomas (25). Taken together, our molecular findings clearly indicate that glioblastomas with adipocyte‐like tumor cell differentiation share similar genetic alterations with other primary glioblastomas. Potential molecular differences that may account for the peculiar lipomatous differentiation remain to be identified.
With respect to the clinical outcome of glioblastoma patients with adipocyte‐like tumor cell differentiation, one of our patients died rapidly within 4 months after surgery, while one patient showed a rather long survival of 3 years after diagnosis. The third patient was alive at the last follow‐up 8 months after surgery. Because of the small number of patients, we cannot draw any conclusions concerning a possible prognostic significance of adipocyte‐like differentiation in glioblastomas. However, it appears likely that survival is similarly poor as in patients with classic glioblastoma, in whom long‐term survival of 3 or more years is restricted to a small subset of patients (16).
Compared to the three glioblastomas presented in this study, the two previously reported low‐grade astrocytomas of pediatric age exhibited a nearly identical lipidization pattern with univacuolated, adipocytic GFAP‐positive tumor cells (11). In the classic astrocytic tumor component, the low‐grade lesions rather exhibited features of pilocytic than diffusely infiltrating astrocytoma, namely, eosinophilic granular bodies, Rosenthal fibers, cyst formation and well‐demarcated growth, though they lacked the biphasic appearance that characterizes most pilocytic neoplasms. Also, the single pediatric tumor that had undergone molecular genetic analyses lacked genetic changes typical for diffusely infiltrating astrocytoma, like TP53 mutations, loss of CDKN2A or amplifications of EGFR, CDK4 or MDM2. After surgery, both children exhibited a favorable clinical course (11). Thus, it was not yet clear if adipocytic changes could also coincide with the more aggressive histological phenotype of diffusely infiltrating astrocytic glioma, or if they were invariably associated with a more circumscript growth pattern and hence a more benign behavior. The three glioblastoma cases contributed in the present study clearly indicate that adipocytic tumor cell changes may also occur in diffusely infiltrating high‐grade astrocytic gliomas and are not restricted to low‐grade lesions. The histological, molecular genetic and clinical features of astrocytic gliomas with adipoctye‐like tumor cells thus seem to be defined by the astrocytic phenotype of these tumors and most likely do not differ from their conventional non‐adipocytic counterparts.
In summary, we report on three rare cases of glioblastomas with adipocyte‐like tumor cell differentiation that, in contrast to previously reported lipid‐rich glioblastomas, did not show a xanthomatous but an adipocyte‐like differentiation. The 2007 edition of the WHO classification of central nervous system tumors (20) clearly designates the criteria that define histological tumor variants (distinct histology with some relevance on clinical outcome) and histological patterns of differentiation (distinct histology but no distinct clinical or pathological significance). While histomorphologically different to classic glioblastomas, our results suggest that glioblastomas with adipocyte‐like tumor cell differentiation are associated with similar clinical and genetic features as conventional glioblastomas, and thus have to be regarded as a rare glioblastoma differentiation pattern.
ACKNOWLEDGMENTS
The authors would like to thank Britta Friedensdorf and Beate Schröder for excellent technical assistance.
REFERENCES
- 1. Aker FV, Ozkara S, Eren P, Peker O, Armagan S, Hakan T (2005) Cerebellar liponeurocytoma/lipidized medulloblastoma. J Neurooncol 71:53–59. [DOI] [PubMed] [Google Scholar]
- 2. Aryan HE, Imbesi SG, Amjadi DK, Abshire BB (2003) Intramedullary spinal cord astrolipoma: case report. Neurosurgery 53:985–987; discussion 7–8. [DOI] [PubMed] [Google Scholar]
- 3. Bonneau D, Longy M (2000) Mutations of the human PTEN gene. Hum Mutat 16:109–122. [DOI] [PubMed] [Google Scholar]
- 4. Bostrom J, Cobbers JM, Wolter M, Tabatabai G, Weber RG, Lichter P et al (1998) Mutation of the PTEN (MMAC1) tumor suppressor gene in a subset of glioblastomas but not in meningiomas with loss of chromosome arm 10q. Cancer Res 58:29–33. [PubMed] [Google Scholar]
- 5. Davies MP, Gibbs FE, Halliwell N, Joyce KA, Roebuck MM, Rossi ML et al (1999) Mutation in the PTEN/MMAC1 gene in archival low grade and high grade gliomas. Br J Cancer 79:1542–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Duerr EM, Rollbrocker B, Hayashi Y, Peters N, Meyer‐Puttlitz B, Louis DN et al (1998) PTEN mutations in gliomas and glioneuronal tumors. Oncogene 16:2259–2264. [DOI] [PubMed] [Google Scholar]
- 7. Fujisawa H, Kurrer M, Reis RM, Yonekawa Y, Kleihues P, Ohgaki H (1999) Acquisition of the glioblastoma phenotype during astrocytoma progression is associated with loss of heterozygosity on 10q25‐qter. Am J Pathol 155:387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fujisawa H, Reis RM, Nakamura M, Colella S, Yonekawa Y, Kleihues P, Ohgaki H (2000) Loss of heterozygosity on chromosome 10 is more extensive in primary (de novo) than in secondary glioblastomas. Lab Invest 80:65–72. [DOI] [PubMed] [Google Scholar]
- 9. George DH, Scheithauer BW (2001) Central liponeurocytoma. Am J Surg Pathol 25:1551–1555. [DOI] [PubMed] [Google Scholar]
- 10. Gherardi R, Baudrimont M, Nguyen JP, Gaston A, Cesaro P, Degos JD et al (1986) Monstrocellular heavily lipidized malignant glioma. Acta Neuropathol 69:28–32. [DOI] [PubMed] [Google Scholar]
- 11. Giangaspero F, Kaulich K, Cenacchi G, Cerasoli S, Lerch KD, Breu H et al (2002) Lipoastrocytoma: a rare low‐grade astrocytoma variant of pediatric age. Acta Neuropathol 103:152–156. [DOI] [PubMed] [Google Scholar]
- 12. Horoupian DS, Shuster DL, Kaarsoo‐Herrick M, Shuer LM (1997) Central neurocytoma: one associated with a fourth ventricular PNET/medulloblastoma and the second mixed with adipose tissue. Hum Pathol 28:1111–1114. [DOI] [PubMed] [Google Scholar]
- 13. Ichimura K, Schmidt EE, Miyakawa A, Goike HM, Collins VP (1998) Distinct patterns of deletion on 10p and 10q suggest involvement of multiple tumor suppressor genes in the development of astrocytic gliomas of different malignancy grades. Genes Chromosomes Cancer 22:9–15. [DOI] [PubMed] [Google Scholar]
- 14. Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O'Meara S et al (2006) Mutation analysis of 24 known cancer genes in the NCI‐60 cell line set. Mol Cancer Ther 5:2606–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kepes JJ, Rubinstein LJ (1981) Malignant gliomas with heavily lipidized (foamy) tumor cells: a report of three cases with immunoperoxidase study. Cancer 47:2451–2459. [DOI] [PubMed] [Google Scholar]
- 16. Krex D, Klink B, Hartmann C, Von Deimling A, Pietsch T, Simon M et al (2007) Long‐term survival with glioblastoma multiforme. Brain 130:2596–2606. [DOI] [PubMed] [Google Scholar]
- 17. Kuchelmeister K, Nestler U, Siekmann R, Schachenmayr W (2006) Liponeurocytoma of the left lateral ventricle–case report and review of the literature. Clin Neuropathol 25:86–94. [PubMed] [Google Scholar]
- 18. Leech RW, Alvord EC Jr (1974) Glial fatty metamorphosis. An abnormal response of premyelin glia frequently accompanying periventricular leukomalacia. Am J Pathol 74:603–612. [PMC free article] [PubMed] [Google Scholar]
- 19. Louis DN (2006) Molecular pathology of malignant gliomas. Annu Rev Pathol 1:97–117. [DOI] [PubMed] [Google Scholar]
- 20. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) (2007) WHO Classification of Tumours of the Central Nervous System, 3rd edn. IARC Press: Lyon. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mangiardi JR, Yodice P (1990) Metabolism of the malignant astrocytoma. Neurosurgery 26:1–19. [DOI] [PubMed] [Google Scholar]
- 22. Rasheed BK, Stenzel TT, McLendon RE, Parsons R, Friedman AH, Friedman HS et al (1997) PTEN gene mutations are seen in high‐grade but not in low‐grade gliomas. Cancer Res 57:4187–4190. [PubMed] [Google Scholar]
- 23. Reifenberger G, Collins VP (2004) Pathology and molecular genetics of astrocytic gliomas. J Mol Med 82:656–670. [DOI] [PubMed] [Google Scholar]
- 24. Reifenberger J, Ring GU, Gies U, Cobbers L, Oberstrass J, An HX et al (1996) Analysis of p53 mutation and epidermal growth factor receptor amplification in recurrent gliomas with malignant progression. J Neuropathol Exp Neurol 55:822–831. [DOI] [PubMed] [Google Scholar]
- 25. Rickert CH, Strater R, Kaatsch P, Wassmann H, Jurgens H, Dockhorn‐Dworniczak B, Paulus W (2001) Pediatric high‐grade astrocytomas show chromosomal imbalances distinct from adult cases. Am J Pathol 158:1525–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Riemenschneider MJ, Buschges R, Wolter M, Reifenberger J, Bostrom J, Kraus JA et al (1999) Amplification and overexpression of the MDM4 (MDMX) gene from 1q32 in a subset of malignant gliomas without TP53 mutation or MDM2 amplification. Cancer Res 59:6091–6096. [PubMed] [Google Scholar]
- 27. Roda JM, Gutierrez‐Molina M (1995) Multiple intraspinal low‐grade astrocytomas mixed with lipoma (astrolipoma). Case report. J Neurosurg 82:891–894. [DOI] [PubMed] [Google Scholar]
- 28. Rosenblum MK, Erlandson RA, Budzilovich GN (1991) The lipid‐rich epithelioid glioblastoma. Am J Surg Pathol 15:925–934. [DOI] [PubMed] [Google Scholar]
- 29. Ruchoux MM, Kepes JJ, Dhellemmes P, Hamon M, Maurage CA, Lecomte M et al (1998) Lipomatous differentiation in ependymomas: a report of three cases and comparison with similar changes reported in other central nervous system neoplasms of neuroectodermal origin. Am J Surg Pathol 22:338–346. [DOI] [PubMed] [Google Scholar]
- 30. Selassie L, Rigotti R, Kepes JJ, Towfighi J (1994) Adipose tissue and smooth muscle in a primitive neuroectodermal tumor of cerebrum. Acta Neuropathol 87:217–222. [DOI] [PubMed] [Google Scholar]
- 31. Sharma MC, Arora R, Lakhtakia R, Mahapatra AK, Sarkar C (2000) Ependymoma with extensive lipidization mimicking adipose tissue: a report of five cases. Pathol Oncol Res 6:136–140. [DOI] [PubMed] [Google Scholar]
- 32. Sharma MC, Agarwal M, Suri A, Gaikwad S, Mukhopadhyay P, Sarkar C (2002) Lipomedulloblastoma in a child: a controversial entity. Hum Pathol 33:564–569. [DOI] [PubMed] [Google Scholar]
- 33. Sugita Y, Terasaki M, Shigemori M, Morimatsu M, Honda E, Oshima Y (2002) Astroblastoma with unusual signet‐ring‐like cell components: a case report and literature review. Neuropathology 22:200–205. [DOI] [PubMed] [Google Scholar]
- 34. Tanaka K, Waga S, Itho H, Shimizu DM, Namiki H (1989) Superficial location of malignant glioma with heavily lipidized (foamy) tumor cells: a case report. J Neurooncol 7:293–297. [DOI] [PubMed] [Google Scholar]
- 35. Tohma Y, Gratas C, Biernat W, Peraud A, Fukuda M, Yonekawa Y et al (1998) PTEN (MMAC1) mutations are frequent in primary glioblastomas (de novo) but not in secondary glioblastomas. J Neuropathol Exp Neurol 57:684–689. [DOI] [PubMed] [Google Scholar]
- 36. Walter A, Dingemans KP, Weinstein HC, Troost D (1994) Cerebellar astrocytoma with extensive lipidization mimicking adipose tissue. Acta Neuropathol 88:485–489. [DOI] [PubMed] [Google Scholar]
- 37. Wolter M, Reifenberger J, Blaschke B, Ichimura K, Schmidt EE, Collins VP, Reifenberger G (2001) Oligodendroglial tumors frequently demonstrate hypermethylation of the CDKN2A (MTS1, p16INK4a), p14ARF, and CDKN2B (MTS2, p15INK4b) tumor suppressor genes. J Neuropathol Exp Neurol 60:1170–1180. [DOI] [PubMed] [Google Scholar]