

Abstract
Oxidative stress is a key feature during progression of chronic neurodegenerative conditions such as Alzheimer's disease. In aging humans and animals, voluntary exercise lowers oxidative stress reactions. Additionally, recent work in our lab demonstrated that cognitive and physical stimulation (termed environmental enrichment) counteracts amyloid beta pathology, neurovascular dysfunction and behavioral symptoms in mice with Alzheimer‐like disease. Based on these facts, we hypothesized that cognitive and physical activity can also protect against oxidative stress in Alzheimer‐diseased brain. We, therefore, kept female TgCRND8 mice under standard and enriched housing from day 30 until 5 months of age. Environmental stimulation attenuated pro‐oxidative processes and triggered anti‐oxidative defense mechanisms as indicated by diminished biomarkers for reactive oxygen and nitrogen species, downregulation of pro‐inflammatory and pro‐oxidative mediators, decreased expression of pro‐apoptotic caspases, and upregulation of SOD1 and SOD2. This study identifies a thus far undescribed antagonizing effect of environmental stimulation on Alzheimer's disease‐related oxidative damage.
Keywords: Alzheimer's disease, apoptosis, environmental enrichment, inflammation, oxidative stress, TgCRND8 mice
INTRODUCTION
Alzheimer's disease (AD), an age‐related neurodegenerative disorder, is pathologically characterized by accumulated cerebral amyloid beta (Aβ) and abnormal hyperphosphorylated tau protein in the form of senile plaques and neurofibrillary tangles, respectively. Clinically, AD patients exhibit a progressive decline in cognitive functions due to vast neuronal and synaptic loss (37).
Oxidative stress resulting from an imbalance of increased production of reactive oxygen and nitrogen species (ROS/RNS) and their impaired detoxification plays a prominent role in the pathogenesis of AD (27), and possibly precedes Aβ and tau aggregation (8). Furthermore, oxidative stress is closely linked to inflammatory reactions and promotes cellular degeneration via apoptosis, which in turn emerge during AD progression (8).
Voluntary physical exercise has been shown to lower peripheral oxidative stress in both humans and animals (19) and cerebral oxidative damage in aging animals 13, 32, thereby revealing neuro‐/cardioprotective abilities 9, 33. Until now, data on the effects of physical (and/or cognitive) stimulation on oxidative stress under neurodegenerative conditions, in particular in AD brain, are lacking.
In transgenic mice with Alzheimer‐like pathology, physical and cognitive stimulation in the form of environmental enrichment improves cognitive performances and reduces anxiety‐related behavior 2, 12, 18. Furthermore, we and others have shown that enrichment also interferes with other aspects of AD pathology as it reduces Aβ plaque burden and the extent of cerebral amyloid angiopathy (CAA) 2, 5, 21. This effect is obviously independent from amyloid precursor protein (APP) expression or processing and is rather associated with reduced aggregation and enhanced clearance of Aβ. The mechanism appears to be mediated by multiple pathways, in particular enhanced enzymatic activity of Aβ degrading endopeptidase, neprilysin (21), reduced inflammatory response, enhanced microglial phagocytosis and proteasomal degradation (5), as well as increased angiogenesis and differential regulation of Aβ receptor/transporter molecules promoting Aβ efflux across the blood brain barrier (20).
In continuation of our own previous findings on the multifaceted effects of cognitive and physical stimulation on various molecular aspects of AD and in view of the prominent role of oxidative stress in the pathogenesis of the disease, we asked whether enriched housing (EH) of transgenic CRND8 mice also has an impact on the cerebral oxidative stress status, its associated pro‐inflammatory mediators, signal transducers, and resulting apoptosis, and whether it influences the endogenous anti‐oxidative defense mechanisms.
MATERIALS AND METHODS
Animals and housing conditions
We examined 18 female TgCRND8 mice carrying the human APP 695 including the “Swedish” and “Indiana” mutations, controlled by the Syrian hamster prion promoter, on a hybrid C57BL/6‐C3H/HeJ background. TgCRND8 mice can be distinguished from other murine models of AD by early exhibition (3 months) of Aβ pathology, accompanied by astrogliosis/microgliosis, and cognitive deficits (11). At 30 days of age, animals were conveyed to the experimental housing conditions. Nine mice were hold in groups of three in standard housing (SH), while nine were housed in equally composed groups in EH. SH consisted of transparent polycarbonate cages (38 × 22 × 15 cm) including sawdust as bedding material. A wooden scaffolding, a plastic inset and further nesting material supplemented the so‐called enriched “home cages.” During the dark phase, EH animals additionally gained access to a second “stimulus cage.” The “stimulus cage” contained different cognitive and physical stimulating objects divided into five categories. (i) Permanently, a gnawing wood and sisal rope were available. Accessorily, objects of the categories; (ii) tunnels, (iii) balls; (iv) soft materials; and (v) varied locomotive substrates including wooden ramps and ladders, plastic stairs as well as running wheels, were included. Every day, one stimulus object of a daily switching category was replaced to expose EH mice to novel environmental stimulation as previously described (20). A photoperiod of 12 h light/dark cycle was sustained. All experimental procedures were conducted in accordance with international standards on animal welfare and accepted by the district government of Muenster/Germany.
Tissue preparation
At 150 days of age, mice were decapitated. One brain hemisphere was immediately snap‐frozen in liquid nitrogen. Total RNA and subsequently protein was extracted from the same homogenized tissue of the whole cerebral hemisphere (without cerebellum and brain stem) using TRIzol Reagent (Invitrogen, Karlsruhe, Germany) following manufacturer's instructions. RNA was DNase treated, cleaned and its quality was assessed by Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). Proteins were dissolved in 1% sodium dodecyl sulfate, stored at −80°C until use, and diluted before using in a lysis buffer containing 1 mM of the metal chelators ethylene diamine tetraacetic acid (EDTA) and ethylene glycol tetraacetic acid (EGTA), each to avoid ex vivo protein degradation or oxidation. For RNA and protein analyses, the number of EH mice was nine and the number of SH mice (due to loss of one pellet) eight. Caused by material limitation, enzyme‐linked immunosorbent assay (ELISA) analyses were performed in six SH and six EH mice. The other brain hemisphere was fixed in 4% buffered formaldehyde followed by paraffin embedding for morphometry analyses.
Aβ immunohistochemistry
For Aβ staining, three pairs of 2 µm sagittal brain sections of each transgenic animal were pre‐treated with formic acid and automatically stained in a TechMate instrument (DAKO, Hamburg, Germany) with 6F/3D anti‐Aβ monoclonal antibody (1:100, DAKO) followed by the DAKO StreptABC complex‐horseradish peroxidase conjugated Duet anti‐mouse/rabbit antibody kit and development with 3,3′‐diaminobenzidine. Counterstaining was performed with hematoxylin. The pairs of sections (10 µm distance) were located between 100 µm and 300 µm lateral from the mid‐sagittal fissure. For further details of Aβ plaque burden and CAA quantification, see (5).
mRNA analyses
Cerebral gene expression of Caspase 3 (Casp3), Casp6, Casp8 and Casp9 was determined by TaqMan assay. cDNA samples of each mouse were synthesized separately (iScriptTMcDNA Synthesis Kit, Bio‐Rad, Munich, Germany). Primer Express software (version 2.0, Applied Biosystems, Foster City, CA, USA) was applied for designing polymerase chain reaction primers and TaqMan probes. A BLAST search was conducted to guarantee amplicon specificity. Glyceraldehyde‐3‐phosphate dehydrogenase (Gapdh) was utilized for normalization. Assays were run in triplicate. Primers and cycling conditions are available as supplementary material (Table S1).
Protein analyses
Protein concentration was determined by the detergent compatible (DC) Protein Assay (Bio‐Rad).
Cerebral protein carbonyl amounts were determined individually using ELISA (STA‐310, Cell Biolabs, San Diego, CA, USA). Samples were incubated with polyethyleneimine (P3143, Sigma—Aldrich, Munich, Germany) at a final concentration of 0.5% overnight at 4°C followed by centrifugation at 13 000 rpm for 10 minutes at 4°C for DNA precipitation prior to ELISA. Supernatant was collected and protein amounts were determined spectrometrically. Subsequently, carbonyl groups where derivatized by 2,4‐dinitrophenol hydrazine, resulting in the corresponding hydrazone product (DNP) which was detected by specific anti‐DNP antibodies. The most common products of protein oxidation in biological samples are derivatives of proline, arginine, lysine and threonine (31). ELISA assays were run in duplicate following manufacturer's instructions.
Nitrotyrosine levels (levels of nitrated tyrosine residues in tissue proteins), prostaglandin E receptor 2 (PTGER2, alias EP2), prostaglandin‐endoperoxide synthase 2 (PTGS2, alias COX2), the dually phosphorylated (threonine 183 and tyrosine 185) stress‐activated protein kinase/Jun‐amino‐terminal kinases 1 and 2/3 (p∼SAPK/JNK1 and JNK2/3), superoxide dismutase 1 (SOD1), superoxide dismutase 2 (SOD2), and glutathione peroxidase 1 (GPX1) were quantified individually in each SH vs. EH mice by Western Blot analysis. A total of 20 µg protein of each animal was loaded on a 10% or 15% sodium dodecyl sulfate—polyacrylamide gel for nitrotyrosine, PTGER2, PTGS2, p∼SAPK/JNK, GPX1, SOD2 and SOD1, respectively. After electrophoresis and wet blotting, membranes were blocked with 5% non‐fat milk in tris sodium tween (TST) buffer (10 mM Tris‐HCl pH 7.6, 150 mM NaCl, 0.05% Tween 20) for nitrotyrosine, SOD1 and GPX1, with 0.5% bovine serum albumin (BSA) in TST for PTGER2 and PTGS2, with 2% BSA in TST for SOD2, or with 5% BSA in TST for p∼SAPK/JNK for 1 h at room temperature (RT). Subsequent incubation with nitrotyrosine antibody (AB5411, 1:500, Millipore, Billerica, MA, USA), PTGER2 antibody (ABIN154102, 1:500, Alpha Diagnostic, San Antonio, TX, USA), PTGS2 antibody (#610204, 1:1000, BD Biosciences, San Jose, CA, USA), p∼SAPK/JNK antibody (#9251, 1:1000, Cell Signaling Technology, Danvers, MA, USA), SOD1 antibody (#574597, 1:10 000, Calbiochem, Darmstadt, Germany), SOD2 antibody (#574596, 1:5000, Calbiochem), or GPX1 antibody (LF‐PA0019, 1:4000, Lab Frontier, Seoul, Korea) were performed overnight at 4°C followed by incubation with secondary antibodies (A2074, 1:5000, Sigma‐Aldrich, for nitrotyrosine; A2074, 1:3000, for PTGER2; A3682, 1:2000, Sigma—Aldrich, for PTGS2; #111‐035‐003, 1:2000, Jackson ImmunoResearch, West Grove, PA, USA, for p∼SAPK/JNK; A9452, 1:15 000, Sigma‐Aldrich, for SOD1; A9452, 1:7500, Sigma‐Aldrich, for SOD2, and #111‐035‐003, 1:10 000, for GPX1). Detection was done by the use of peroxidase‐catalyzed enhanced chemiluminescence (Amersham enhanced chemiluminescence (ECL) PlusTM Western Blotting Detection Reagents, GE Healthcare, Chicago, Il, USA). Evaluation of protein expression levels was achieved by densitometry software Gel‐Pro Analyzer (Media Cybernetics, Bethesda, MD, USA). β—actin control immunoblotting was conducted for normalization (primary and secondary antibody 1:10 000 each, Sigma‐Aldrich). Specificity of all utilized antibodies was tested by the omission of primary antibodies. Furthermore, nitrotyrosine antibody specificity was evaluated by additional pre‐incubating with 3‐nitro‐L‐tyrosine (N7389, 10 mM, Sigma‐Aldrich) for 30 minutes at RT. All samples were analyzed individually and in triplicate.
Statistical analyses
Data are presented as means ± SEM. One‐sample Kolmogorov‐Smirnov test and Q‐Q‐plots validated normal distribution of all datasets. As all data were normally distributed, SH and EH groups were compared using an unpaired Student's t‐test. Tests were applied two‐tailed for the majority of experiments. As these experiments revealed an inhibitory role of EH on oxidative stress, tests for Western blot analyses of SOD1/2 and SAPK were applied one‐tailed. Pearson correlation was used to test the strength of association between Aβ plaque load and CAA (data published in (5)) obtained from the paraffin‐embedded brain hemispheres, with oxidative damage and anti‐oxidant status measured in the other brain hemisphere of the same animals. All tests were performed using the software package SPSS (version 12.0.1, SPSS Inc., Chicago, IL, USA). Differences were considered significant at P < 0.05.
RESULTS
Reduction of oxidative stress biomarkers
To determine the cerebral oxidative stress status of SH and EH mice, the amount of nitrotyrosine and carbonylated protein was quantified. Following EH, both markers displayed a significant decrease. Nitrotyrosine levels decreased by 66% (Figure 1A,B) (SH: 2.99 ± 1.78, EH: 1 ± 0.43, P = 0.003) and protein carbonylation was attenuated by 37% (Figure 1C) (SH: 5.18 ± 0.61 nmol/mg protein, EH: 3.26 ± 0.38, P = 0.017). Additionally, pre‐incubating the nitrotyrosine antibody with nitrotyrosine or omission of primary antibody totally blocked ECL signal, verifying the antibody specificity (data not shown).
Figure 1.
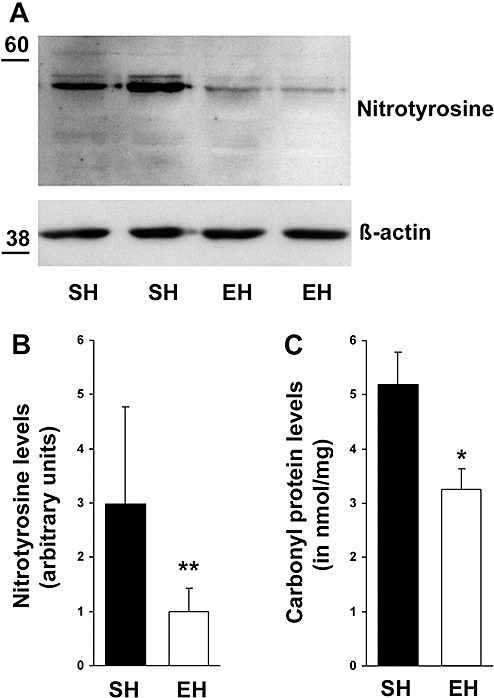
Reduction of oxidative stress markers following environmental enrichment. (A) Western Blot analysis displayed a decrease in nitrotyrosine levels due to enrichment. (B) Densitometry analysis of immunoblots identified a significant downregulation of cerebral nitrotyrosine by 66% (P = 0.003). (C) Additionally, enzyme‐linked immunosorbent assay revealed attenuated protein carbonylation by 37% (P = 0.017). Data are given as mean ± SEM, t‐test. EH = enriched housing; SH = standard housing. *P < 0.05, **P < 0.01.
Downregulation of pro‐inflammatory and pro‐oxidative mediators
To uncover whether prominent pro‐inflammatory and pro‐oxidative molecules exhibited differential regulation after EH, we performed quantitative Western blot analyses against PTGER2 (alias EP2), PTGS2 (alias COX2) and further quantified the levels of activated form of SAPK/JNK1 and JNK2/3. Following EH, the pro‐inflammatory and pro‐oxidative PTGER2 (22) revealed a significant downregulation by 53% (Figure 2A,B) (SH: 2.12 ± 0.33, EH: 1 ± 0.13, P = 0.0006), whereas PTGS2, a molecule known to activate PTGER2 expression (1), was not differentially regulated (Figure 2C,D) (SH: 1 ± 0.29, EH: 1.08 ± 0.16, P = 0.361). Furthermore, the pro‐inflammatory and pro‐apoptotic p∼JNK2/3 (25) (Figure 2E,F), but not p∼JNK1 (Figure 2E,F), was significantly diminished after EH (p∼JNK2/3: −49%, SH: 1.97 ± 0.96, EH: 1 ± 0.49, P = 0.047; p∼JNK1: SH: 1 ± 0.21, EH: 1.17 ± 0.3; P = 0.245). Omitting primary antibodies resulted in ECL signal absence, verifying antibody specificity (data not shown).
Figure 2.
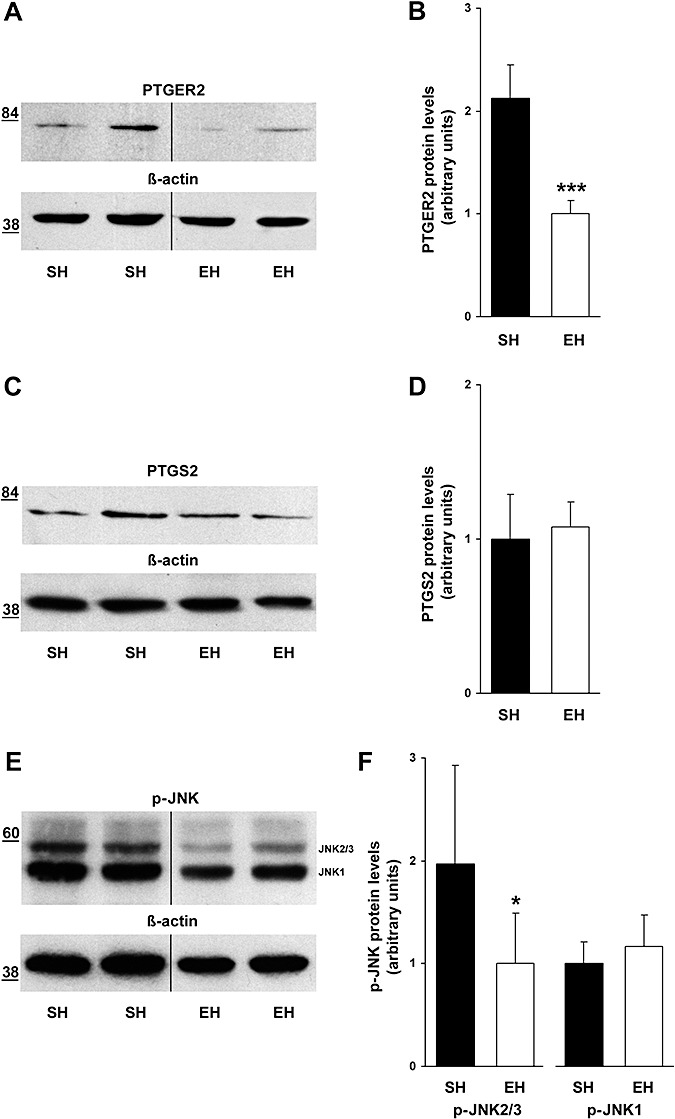
Downregulation of pro‐inflammatory and pro‐oxidative mediators due to enriched housing. (A) Western Blot analysis showed a decrease in cerebral prostaglandin E receptor 2 (PTGER2) amounts following enrichment (cropped images). (B) Densitometry analysis revealed a significant downregulation of PTGER2 by 53% (P = 0.0006). (C,D) In contrast, prostaglandin‐endoperoxide synthase 2 (PTGS2) Western Blot analysis identified unaltered cerebral protein amounts in both housing conditions (P = 0.361). (E,F) However, Western Blot analysis revealed diminished levels of activated Jun‐amino‐terminal kinases 2/3 (JNK2/3) (−49%, P = 0.047), whereas phosphorylated (p)‐JNK1 was not altered following enrichment (P = 0.245) (cropped images). Data are given as mean ± SEM, t‐test. EH = enriched housing; SH = standard housing. *P < 0.05, ***P < 0.001.
Downregulation of pro‐apoptotic caspases
TaqMan assays were conducted to elucidate whether EH had an impact on the cerebral gene expression of apoptosis‐regulating caspases. EH resulted in significant downregulation of the apoptosis executioner caspases Casp3 and Casp6 (7) by 32% and 15%, respectively (Figure 3) (Casp3: SH: 1.47 ± 0.02, EH: 1 ± 0.07, P = 0.0001; Casp6: SH: 1.18 ± 0.06, EH: 1 ± 0.01, P = 0.023). Furthermore, the apoptosis initiating caspase Casp9 but not Casp8 (7) showed a trend toward reduced expression following EH (Figure 3) (Casp9: −16%, SH: 1.19 ± 0.06, EH: 1 ± 0.07, P = 0.051; Casp8: SH: 1.03 ± 0.06, EH: 1 ± 0.1, P = 0.389).
Figure 3.
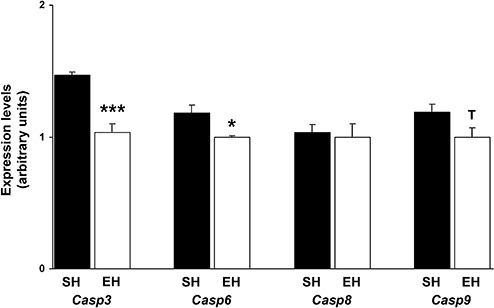
Downregulation of pro‐apoptotic caspases following enrichment. TaqMan quantitative real‐time PCR revealed decreased gene expression of the executioner caspases Casp3 and Casp6 by 32% (P = 0.0001) and 15% (P = 0.023), respectively. Furthermore, the apoptosis initiating caspase Casp9 but not Casp8 showed a trend toward reduced expression following enriched housing (Casp9: −16%, P = 0.051; Casp8: P = 0.389). Data are given as mean ± SEM, t‐test. Casp = caspase; EH = enriched housing; SH = standard housing. *P < 0.05, ***P < 0.001.
Induction of anti‐oxidative defense
We further questioned whether EH was able to induce endogenous oxidative defense mechanisms. Therefore, the detoxifying enzymes SOD1 (cytosolic), SOD2 (mitochondrial) and GPX1 (24) were quantified by Western blot. SOD1 and SOD2 protein expression revealed significant induction after EH (Figure 4A–D), whereas GPX1 was not differentially regulated (Figure 4E,F) (SOD1: +32%, SH: 1 ± 0.2, EH: 1.32 ± 0.21, P = 0.032; SOD2: +65%, SH: 1 ± 0.37, EH: 1.65 ± 0.42, P = 0.044; GPX1: SH: 1.08 ± 0.19, EH: 1 ± 0.21, P = 0.348). ECL signal disappeared by membrane incubation without primary antibody (data not shown).
Figure 4.
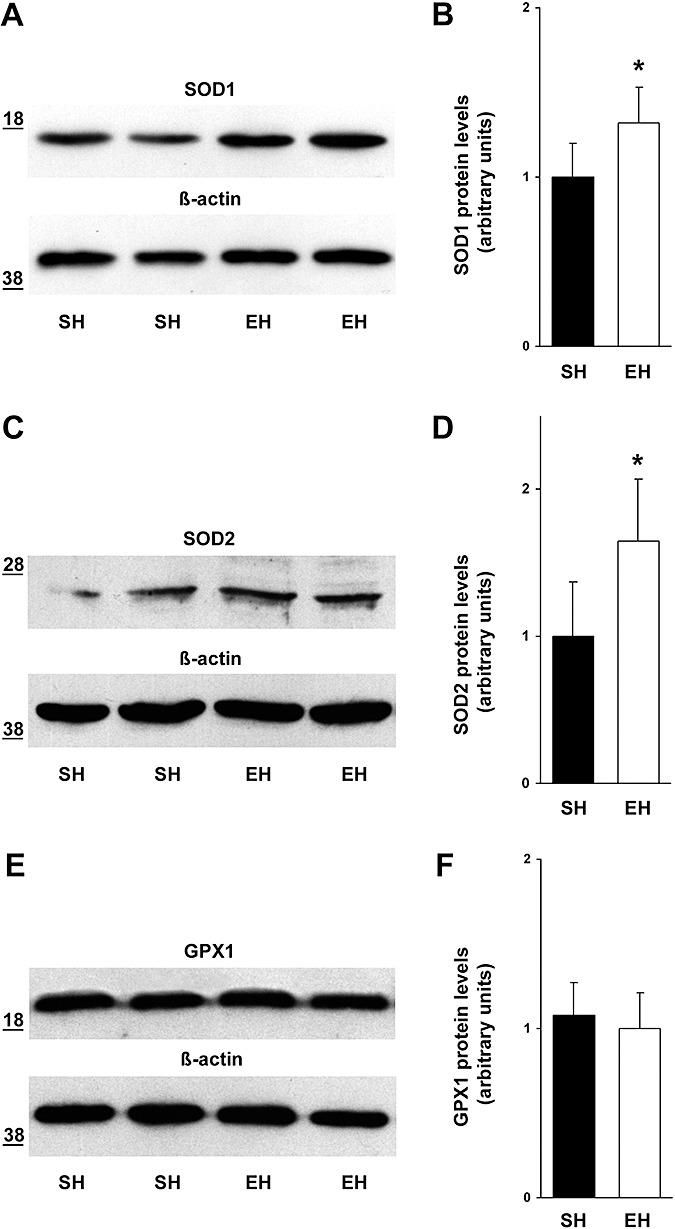
Induction of anti‐oxidative defense due to environmental enrichment. (A) Western Blot analysis identified an increase in cerebral SOD1 amounts following enrichment. (B) Densitometry analysis revealed a significant upregulation of SOD1 by 32% (P = 0.032). (C,D) Furthermore, SOD2 protein expression was enhanced by 65% due to EH (P = 0.044). (E,F) In contrast, GPX1 Western Blot analysis identified unaltered cerebral protein amounts in both housing conditions (P = 0.348). Data are given as mean ± SEM, t‐test. SOD1 = superoxide dismutase 1; SOD2 = superoxide dismutase 2; GPX1 = glutathione peroxidase 1; EH = enriched housing; SH = standard housing. *P < 0.05.
Correlation among oxidative damage and anti‐oxidant status with the amount of Aβ deposits
Data (previously published in 5) on the extent of CAA and Aβ plaque load obtained from the formaldehyde fixed, paraffin‐embedded brain hemispheres were correlated with the protein levels of the opposite brain hemisphere of the same animals. Aβ plaque load was defined as Aβ plaque area and number in the neocortex and hippocampus. Absolute values of plaque burden were related to the investigated area. The severity of CAA was determined as percentage of Aβ positive vessels in relation to total counted vessel (for details, see (5)). Correlation analyses were performed among levels of carbonylated protein, nitrotyrosine (oxidative damage), SOD1 and SOD2 (detoxification) with the amount of Aβ deposits. All Aβ parameters were positively correlated with levels of protein carbonylation (Figure 5A–C) (Aβ plaque area vs. carbonyl∼protein: r = 0.543, P = 0.034; Aβ plaque number vs. carbonyl∼protein: r = 0.522, P = 0.041; CAA vs. carbonyl∼protein: r = 0.524, P = 0.040). Additionally, Aβ plaque area revealed a trend toward positive correlation with nitrotyrosine (Figure 5D) (r = 0.375, P = 0.084) and the extent of CAA was positively correlated with nitrotyrosine levels (Figure 5E) (r = 0.447, P = 0.047). On the other hand, the degree of CAA was inversely associated with levels of the anti‐oxidative SOD1 and SOD2 protein (Figure 5F,G) (CAA vs. SOD1: r = −0.476, P = 0.031; CAA vs. SOD2: r = −0.489, P = 0.063).
Figure 5.
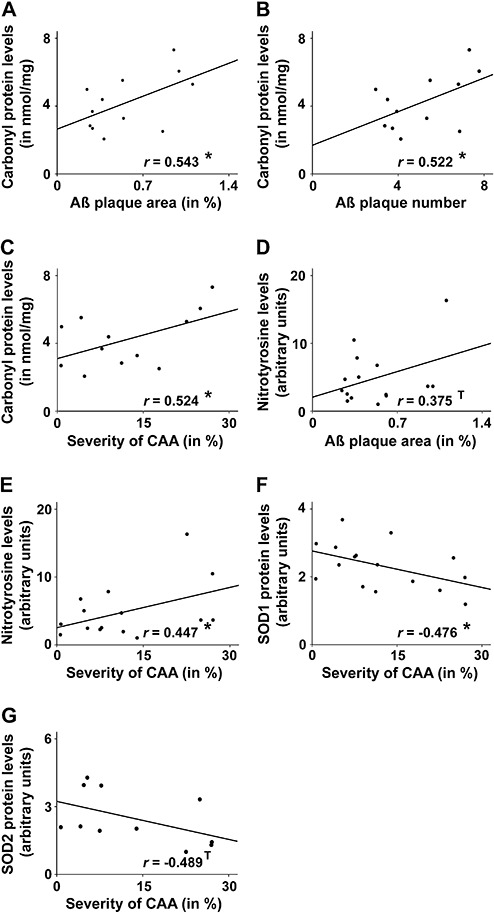
Correlation among oxidative damage and anti‐oxidant status with the amount of Aβ deposits. Pearson‐correlation analyses revealed a significant positive association between the amount of carbonyl∼protein and the Aβ parameters plaque area (A) (r = 0.543, P = 0.034), plaque number (B) (r = 0.522, P = 0.041) and cerebral amyloid angiopathy (CAA) (C) (r = 0.524, P = 0.040). Furthermore, Aβ plaque area showed a trend toward positive correlation with nitrotyrosine levels (D) (r = 0.375, P = 0.084) and the severity of CAA was positively correlated with the amount of nitrotyrosine (E) (r = 0.447, P = 0.047). On the other hand, the degree of CAA was inversely associated with levels of the anti‐oxidative SOD1 (F) (r = −0.476, P = 0.031) or SOD2 protein (G) (r = −0.489, P = 0.063). SOD1 = superoxide dismutase 1; SOD2 = superoxide dismutase 2. *P < 0.05.
DISCUSSION
Cerebral oxidative stress is one of the principle components of AD pathology (27), which is closely linked to inflammatory reactions and promotes cellular degeneration via apoptosis during AD progression (8).
In continuation of our previous findings on alleviation of anxiety, improvement of cognition (18), reduction of Aβ pathology (5) and associated neurovascular dysfunction (20), we demonstrate here that 4 months of continuous and diversified environmental stimulation (including enhanced physical activity in the form of voluntary wheel running) additionally reduces cerebral oxidative stress in TgCRND8 mice. In detail, we could detect a decrease in biomarkers for oxidative damage, namely nitrotyrosine and carbonylated protein, accompanied by (and probably due to) enhanced generation of the detoxifying enzymes SOD1 and SOD2, as well as downregulation of the pro‐inflammatory PTGER2 protein, decreased amount of p∼JNK2/3 and reduced expression of the pro‐apoptotic caspases Casp3, Casp6 and Casp9 (see also Figure 6). In contrast, the expression of PTGS2, GPX1 and Casp8 remained unchanged.
Figure 6.
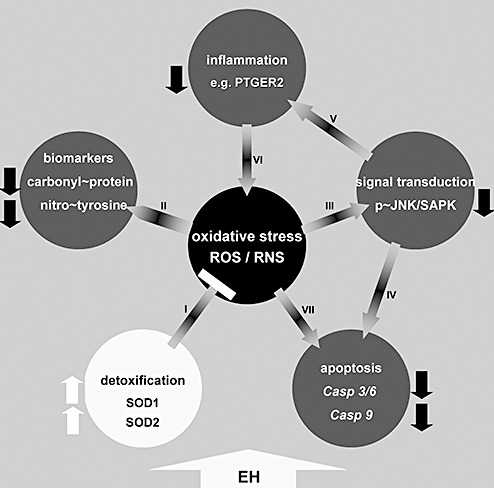
Schematic representation of EH induced effects on processes related to oxidative stress. EH results in upregulation of cerebral anti‐oxidative defense mechanisms (A), dampening oxidative stress as indicated by downregulation of associated biomarkers (B). Less oxidative stress leads to less Jun‐amino‐terminal kinase/stress‐activated protein kinase (JNK/SAPK) activation (C) which in turn attenuates pro‐apoptotic (D) and inflammatory (E) processes, again leading to diminished oxidative damage (F). Suppression of pro‐apoptotic caspases as a consequence of reduced oxidative stress can also be conducted by JNK/SAPK independent pathways (G).  = results in;
= results in;  = dampens;
= dampens;  = downregulation;
= downregulation;  = upregulation;
= upregulation;  = enriched housing induces. EH = enriched housing; SH = standard housing; ROS = reactive oxygen species; RNS = reactive nitrogen species; PTGER2 = prostaglandin E receptor 2.
= enriched housing induces. EH = enriched housing; SH = standard housing; ROS = reactive oxygen species; RNS = reactive nitrogen species; PTGER2 = prostaglandin E receptor 2.
Biomarkers of oxidative stress reactions
Oxidative and nitrosative stresses are penetrative conditions of increasing amounts of ROS and RNS incompletely detoxified by the endogenous defense mechanisms and characteristic for various chronic diseases and the normal aging process. As ROS/RNS are generally too reactive to enable practicable direct measurement, stable metabolites resulting from reaction of ROS/RNS with biomolecules serve as quantifiable biomarkers.
Nitrotyrosine is a stable marker for nitric oxide‐derived oxidants, whereas carbonylated proteins are generated among others by the oxidation of different amino acid side chains and by oxidative stress‐induced peptide cleavage 14, 31. Both markers are early and prominent representatives for AD‐associated oxidative stress (14). Yet, the detected downregulation of both markers (Figure 1A–C) implies anti‐oxidative effects triggered by EH. In AD, the Aβ peptide plays a central role in the generation of free radicals and oxidative stress (27). Accordingly, we found a significant positive correlation between the extent of CAA and levels of both carbonylated proteins and nitrotyrosine and also a significant positive correlation between the amount of plaque load and levels of carbonylated proteins. There was also a trend toward positive correlation between the plaque load and levels of nitrotyrosine (Figure 5A–E). Besides this “traditional” view about the toxic effect of Aβ in producing oxidative damage, there are also some recent discussions on a rather adaptive role of this peptide and its aggregates in neuronal protection against oxidative stress (30). In light of this hypothesis, the oxidative stress decline found in this study would rather be the cause than the consequence of the reduction of Aβ deposits.
Oxidative stress‐related signal transduction
JNK/SAPK pathways are the central mediators that propagate oxidative stress signals, thereby inducing inflammation and apoptosis (25). It has been shown that cells containing the Swedish APP mutation reveal induction of the JNK pathway and activation of different caspases after exposure to ROS (23). Accordingly, JNK inhibition protects cells from Aβ‐induced cell death (42). With these issues in mind, the observed reduction of p∼JNK2/3 following EH (Figure 2E,F) is salutary, as it probably results in suppression of inflammation and apoptosis. Interestingly, the activation (ie, phosphorylation) of JNK2/3, but not JNK1, is associated with neurofibrillary tangles in AD cases (46). In our experiments, EH induced a significant reduction of p∼JNK2/3, but not p∼JNK1.
Pro‐inflammatory and pro‐oxidative mediators
Prolonged cognitive and physical stimulation in TgCRND8 mice resulted in significant downregulation of PTGER2 (alias EP2) protein (Figure 2A,B). Very well compatible with p∼JNK2/3 downregulation in this study, PTGER2 expression seems to be JNK2‐pathway dependent (1). Moreover, in line with our results, downregulation of PTGER2 in microglia and neuron co‐cultures (39) as well as in mice brains with (22) or without (28) Alzheimer pathology leads to reduction of oxidative damage, and in case of AD mice, decreases also Aβ burden (22).
In a previous study with an identical experimental design, we could demonstrate a downregulation of other pro‐inflammatory molecules, namely toll‐like receptor 2 (Tlr2) and macrophage scavenger receptor 1 (Msr1) as well as upregulation of scavenger receptor class B, member 1 (Scarb1), which has an anti‐inflammatory function (5). In detail, injection of Aβ in murine wild‐type hippocampi induces TLR2 expression (40), which in turn upregulates MSR1 production (26). Both TLR2 and MSR1 are known to elicit inflammation, induce apoptosis and stimulate the generation of ROS 4, 15. On the other hand, Scarb1 knockout mice suffer of increased oxidative stress (43). Taken together, environmental enrichment seems to suppress PTGER2 in concert with regulation of other pro‐/anti‐inflammatory molecules ending up in a less inflammatory micro‐environment.
PTGS2 (alias COX2), another prominent molecule involved in oxidative stress reactions (1), was not differentially regulated following EH (Figure 2C,D). The reason remains speculative, but what follows might explain this unexpected finding. First, PTGS2 seems to play a harmful role in AD pathogenesis mainly in very late stages of the disease (P450 old transgenic mice), and second, it is also involved in regulation of neuronal plasticity and spatial memory retention (for review, see (17)). Our mice were examined in an early stage of the Alzheimer‐like pathology (P150) and, maybe, even more cogent, probable ongoing plasticity processes widely accepted to be triggered by environmental enrichment (29) might have neutralized possible downregulation of PTGS2 linked to oxidative reactions.
Oxidative stress‐associated apoptosis
A pro‐oxidative environment induces apoptosis, a process known to be JNK dependent (38) or independent 6, 25. Particularly, in the context of AD and ROS, the executioner caspases 3 and 6 and the initiator caspases 8 and 9 seem to bear prominent JNK dependent pro‐apoptotic abilities 23, 45. Accordingly, following EH, Casp3 and Casp6 were significantly downregulated, and Casp9 revealed a trend toward suppression while Casp8 remained unchanged (Figure 3).
Cerebral endogenous ROS detoxification
SOD1, SOD2 and GPX1 represent important ROS‐detoxifying enzymes 3, 24. APP transgenic mice with compromised SOD1 activity display increased oxidative stress (36), whereas SOD1 over‐expression protects against Aβ‐related neurotoxicity in vitro (10). Furthermore, partial inactivation of SOD2 in transgenic mice with AD‐like pathology potentiates neuronal and vascular dysfunction and additionally impairs cognitive performance (16). We now show that EH upregulates SOD1 and SOD2 while GPX1 amounts remained unaltered (Figure 4A–F). The higher amount of SOD1 was associated with a lower extend of Aβ load in the form of CAA in a statistically significant way (Figure 5F). Moreover, SOD2 showed a trend toward negative correlation with the degree of CAA (Figure 5G).
Exercise and oxidative stress
The exact mechanisms by which behavioral enrichment provides protection against oxidative damage in the brain with Alzheimer pathology remain speculative. The effects of behavioral enrichment including voluntary exercise on the brain with AD appear to be very complex. It reduces Aβ plaque burden and the extent of CAA 2, 5, 21, possibly mediated by the enhanced enzymatic activity of Aβ‐degrading neprilysin (21), reduced cholesterol levels and inflammatory response, enhanced microglial phagocytosis and proteasomal degradation (5), as well as increased angiogenesis and differential regulation of Aβ transporter molecules (20). Almost any of these alterations, in particular reduction of Aβ depositions, attenuated inflammatory response, and improved neurovascular function could accelerate the other and could lead to less oxidative damage 27, 41, 47. On the other hand, a broad array of investigations in man and animals has elucidated that voluntary and moderate exercise efficiently implements beneficial anti‐oxidative defense mechanisms independent from Alzheimer pathology 13, 19, 32. In healthy individuals, SOD1 and SOD2 are upregulated following moderate physical activity 34, 44, whereas JNK experiences deactivation (35). Thus, enrichment could also have primarily interfered with oxidative stress, which in turn would decelerate the cycle of oxidative damage—Aβ aggregation—inflammation—neurovascular dysfunction. In conclusion, EH seems to break this ongoing “vicious circle,” but which part of this loop is the initial target remains to be uncovered.
Supporting information
Table S1. Accession numbers, primer and probe sequences and cycling conditions for TaqMan assays.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
ACKNOWLEDGMENTS
This research was supported by a grant from Innovative Medical Research (IMF, KE 120517). The authors declare that they have no conflicts of interest. We thank B. Heuer and S. Peetz‐Dienhart for technical assistance and D. Westaway for providing the TgCRND8 mice.
REFERENCES
- 1. Abulencia JP, Gaspard R, Healy ZR, Gaarde WA, Quackenbush J, Konstantopoulos K (2003) Shear‐induced cyclooxygenase‐2 via a jnk2/c‐Jun‐dependent pathway regulates prostaglandin receptor expression in chondrocytic cells. J Biol Chem 278:28388–28394. [DOI] [PubMed] [Google Scholar]
- 2. Adlard PA, Perreau VM, Pop V, Cotman CW (2005) Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer's disease. J Neurosci 25:4217–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ali SS, Xiong C, Lucero J, Behrens MM, Dugan LL, Quick KL (2006) Gender differences in free radical homeostasis during aging: shorter‐lived female C57BL6 mice have increased oxidative stress. Aging Cell 5:565–574. [DOI] [PubMed] [Google Scholar]
- 4. Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD et al (1999) Cell activation and apoptosis by bacterial lipoproteins through toll‐like receptor‐2. Science 285:736–739. [DOI] [PubMed] [Google Scholar]
- 5. Ambrée O, Leimer U, Herring A, Goertz N, Sachser N, Heneka MT et al (2006) Reduction of amyloid angiopathy and Abeta plaque burden after enriched housing in TgCRND8 mice: involvement of multiple pathways. Am J Pathol 169:544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R et al (2006) TIGAR, a p53‐inducible regulator of glycolysis and apoptosis. Cell 126:107–120. [DOI] [PubMed] [Google Scholar]
- 7. Boatright KM, Salvesen GS (2003) Mechanisms of caspase activation. Curr Opin Cell Biol 15:725–731. [DOI] [PubMed] [Google Scholar]
- 8. Castellani RJ, Lee HG, Zhu X, Nunomura A, Perry G, Smith MA (2006) Neuropathology of Alzheimer disease: pathognomonic but not pathogenic. Acta Neuropathol 111:503–509. [DOI] [PubMed] [Google Scholar]
- 9. Cechetti F, Fochesatto C, Scopel D, Nardin P, Goncalves CA, Netto CA, Siqueira IR (2008) Effect of a neuroprotective exercise protocol on oxidative state and BDNF levels in the rat hippocampus. Brain Res 1188:182–188. [DOI] [PubMed] [Google Scholar]
- 10. Celsi F, Ferri A, Casciati A, D'Ambrosi N, Rotilio G, Costa A et al (2004) Overexpression of superoxide dismutase 1 protects against beta‐amyloid peptide toxicity: effect of estrogen and copper chelators. Neurochem Int 44:25–33. [DOI] [PubMed] [Google Scholar]
- 11. Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J et al (2001) Early‐onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 276:21562–21570. [DOI] [PubMed] [Google Scholar]
- 12. Costa DA, Cracchiolo JR, Bachstetter AD, Hughes TF, Bales KR, Paul SM et al (2007) Enrichment improves cognition in AD mice by amyloid‐related and unrelated mechanisms. Neurobiol Aging 28:831–844. [DOI] [PubMed] [Google Scholar]
- 13. Cui L, Hofer T, Rani A, Leeuwenburgh C, Foster TC (2007) Comparison of lifelong and late life exercise on oxidative stress in the cerebellum. Neurobiol Aging (in press). doi:10.1016/j.neurobiolaging.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dalle‐Donne I, Rossi R, Colombo R, Giustarini D, Milzani A (2006) Biomarkers of oxidative damage in human disease. Clin Chem 52:601–623. [DOI] [PubMed] [Google Scholar]
- 15. El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD (1996) Scavenger receptor‐mediated adhesion of microglia to beta‐amyloid fibrils. Nature 382:716–719. [DOI] [PubMed] [Google Scholar]
- 16. Esposito L, Raber J, Kekonius L, Yan F, Yu GQ, Bien‐Ly N et al (2006) Reduction in mitochondrial superoxide dismutase modulates Alzheimer's disease‐like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J Neurosci 26:5167–5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Firuzi O, Pratico D (2006) Coxibs and Alzheimer's disease: should they stay or should they go? Ann Neurol 59:219–228. [DOI] [PubMed] [Google Scholar]
- 18. Goertz N, Lewejohann L, Tomm M, Ambrée O, Keyvani K, Paulus W, Sachser N (2008) Effects of environmental enrichment on exploration, anxiety, and memory in female TgCRND8 Alzheimer mice. Behav Brain Res 191:43–48. [DOI] [PubMed] [Google Scholar]
- 19. Gomez‐Cabrera MC, Domenech E, Vina J (2008) Moderate exercise is an antioxidant: upregulation of antioxidant genes by training. Free Radic Biol Med 44:126–131. [DOI] [PubMed] [Google Scholar]
- 20. Herring A, Yasin H, Ambrée O, Sachser N, Paulus W, Keyvani K (2008) Environmental enrichment counteracts Alzheimer's neurovascular dysfunction in TgCRND8 mice. Brain Pathol 18:32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lazarov O, Robinson J, Tang YP, Hairston IS, Korade‐Mirnics Z, Lee VM et al (2005) Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell 120:701–713. [DOI] [PubMed] [Google Scholar]
- 22. Liang X, Wang Q, Hand T, Wu L, Breyer RM, Montine TJ, Andreasson K (2005) Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer's disease. J Neurosci 25:10180–10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marques CA, Keil U, Bonert A, Steiner B, Haass C, Muller WE, Eckert A (2003) Neurotoxic mechanisms caused by the Alzheimer's disease‐linked Swedish amyloid precursor protein mutation: oxidative stress, caspases, and the JNK pathway. J Biol Chem 278:28294– 28302. [DOI] [PubMed] [Google Scholar]
- 24. Mates JM, Sanchez‐Jimenez F (1999) Antioxidant enzymes and their implications in pathophysiologic processes. Front Biosci 4:D339–D345. [DOI] [PubMed] [Google Scholar]
- 25. Mielke K, Herdegen T (2000) JNK and p38 stresskinases— degenerative effectors of signal‐transduction‐cascades in the nervous system. Prog Neurobiol 61:45–60. [DOI] [PubMed] [Google Scholar]
- 26. Mietus‐Snyder M, Gowri MS, Pitas RE (2000) Class A scavenger receptor up‐regulation in smooth muscle cells by oxidized low density lipoprotein. Enhancement by calcium flux and concurrent cyclooxygenase‐2 up‐regulation. J Biol Chem 275:17661–17670. [DOI] [PubMed] [Google Scholar]
- 27. Miranda S, Opazo C, Larrondo LF, Munoz FJ, Ruiz F, Leighton F, Inestrosa NC (2000) The role of oxidative stress in the toxicity induced by amyloid beta‐peptide in Alzheimer's disease. Prog Neurobiol 62:633–648. [DOI] [PubMed] [Google Scholar]
- 28. Montine TJ, Milatovic D, Gupta RC, Valyi‐Nagy T, Morrow JD, Breyer RM (2002) Neuronal oxidative damage from activated innate immunity is EP2 receptor‐dependent. J Neurochem 83:463– 470. [DOI] [PubMed] [Google Scholar]
- 29. Nithianantharajah J, Hannan AJ (2006) Enriched environments, experience‐dependent plasticity and disorders of the nervous system. Nat Rev Neurosci 7:697–709. [DOI] [PubMed] [Google Scholar]
- 30. Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA (2006) Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp Neurol 65:631–641. [DOI] [PubMed] [Google Scholar]
- 31. Nystrom T (2005) Role of oxidative carbonylation in protein quality control and senescence. EMBO J 24:1311–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Opii WO, Joshi G, Head E, Milgram NW, Muggenburg BA, Klein JB et al (2008) Proteomic identification of brain proteins in the canine model of human aging following a long‐term treatment with antioxidants and a program of behavioral enrichment: relevance to Alzheimer's disease. Neurobiol Aging 29:51–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Powers SK, Quindry J, Hamilton K (2004) Aging, exercise, and cardioprotection. Ann N Y Acad Sci 1019:462–470. [DOI] [PubMed] [Google Scholar]
- 34. Rush JW, Turk JR, Laughlin MH (2003) Exercise training regulates SOD‐1 and oxidative stress in porcine aortic endothelium. Am J Physiol Heart Circ Physiol 284:H1378–H1387. [DOI] [PubMed] [Google Scholar]
- 35. Schenk S, Horowitz JF (2007) Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid‐induced insulin resistance. J Clin Invest 117:1690–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schuessel K, Schafer S, Bayer TA, Czech C, Pradier L, Muller‐Spahn F et al (2005) Impaired Cu/Zn‐SOD activity contributes to increased oxidative damage in APP transgenic mice. Neurobiol Dis 18:89–99. [DOI] [PubMed] [Google Scholar]
- 37. Selkoe DJ (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81:741–766. [DOI] [PubMed] [Google Scholar]
- 38. Shen HM, Liu ZG (2006) JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med 40:928–939. [DOI] [PubMed] [Google Scholar]
- 39. Shie FS, Montine KS, Breyer RM, Montine TJ (2005) Microglial EP2 is critical to neurotoxicity from activated cerebral innate immunity. Glia 52:70–77. [DOI] [PubMed] [Google Scholar]
- 40. Simard AR, Soulet D, Gowing G, Julien JP, Rivest S (2006) Bone marrow‐derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron 49:489–502. [DOI] [PubMed] [Google Scholar]
- 41. Standridge JB (2006) Vicious cycles within the neuropathophysiologic mechanisms of Alzheimer's disease. Curr Alzheimer Res 3:95–108. [DOI] [PubMed] [Google Scholar]
- 42. Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML, Greene LA (2001) beta‐Amyloid‐induced neuronal apoptosis requires c‐Jun N‐terminal kinase activation. J Neurochem 77:157–164. [DOI] [PubMed] [Google Scholar]
- 43. Van Eck M, Hoekstra M, Hildebrand RB, Yaong Y, Stengel D, Kruijt JK et al (2007) Increased oxidative stress in scavenger receptor BI knockout mice with dysfunctional HDL. Arterioscler Thromb Vasc Biol 27:2413–2419. [DOI] [PubMed] [Google Scholar]
- 44. Yamashita N, Hoshida S, Otsu K, Asahi M, Kuzuya T, Hori M (1999) Exercise provides direct biphasic cardioprotection via manganese superoxide dismutase activation. J Exp Med 189:1699–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Y, Goodyer C, LeBlanc A (2000) Selective and protracted apoptosis in human primary neurons microinjected with active caspase‐3, ‐6, ‐7, and ‐8. J Neurosci 20:8384–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhu X, Raina AK, Rottkamp CA, Aliev G, Perry G, Boux H, Smith MA (2001) Activation and redistribution of c‐jun N‐terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. J Neurochem 76:435–441. [DOI] [PubMed] [Google Scholar]
- 47. Zlokovic BV (2005) Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci 28:202–208. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Accession numbers, primer and probe sequences and cycling conditions for TaqMan assays.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item