

Abstract
Several recent reports proposed that astrocyte death might precede neuronal demise after focal ischemia, contrary to the conventional view that astrocytes are more resistant to injury than neurons. Interestingly, there are findings supporting each of these opposing views. To clarify these controversies, we assessed astrocyte viability after 2‐h middle cerebral artery occlusion in mice. In contrast to neighboring neurons, astrocytes were alive and contained glycogen across the ischemic area 6 h after reperfusion, and at the expanding outer border of the infarct at later time points. These glycogen‐positive astrocytes had intact plasma membranes. Astrocytes lost plasmalemma integrity much later than neurons: 19 ± 22 (mean ± standard deviation), 58 ± 14 and 69 ± 3% of astrocytes in the perifocal region became permeable to propidium iodide (PI) at 6, 24, 72 h after ischemia, respectively, in contrast to 81 ± 2, 96 ± 3, 97 ± 2% of neurons. Although more astrocytes in the cortical and subcortical core regions were PI‐positive, their numbers were considerably less than those of neurons. Lysosomal rupture (monitored by deoxyribonuclease II immunoreactivity) followed a similar time course. Cytochrome‐c immunohistochemistry showed that astrocytes maintained mitochondrial integrity longer than neurons. EM confirmed that astrocyte ultrastructure including mitochondria and lysosomes disintegrated much later than that of neurons. We also found that astrocytes died by a delayed necrosis without significantly activating apoptotic mechanisms although they rapidly swelled at the onset of ischemia.
Keywords: apoptosis, astrocyte, focal cerebral ischemia, glycogen, necrosis
INTRODUCTION
When ischemia is severe, all types of brain cells including astrocytes die; hence, the term pan‐necrosis is coined to describe brain infarction. In contrast, only neurons die and astrocytes remain viable after focal or global ischemia of short duration. This latter pathology is therefore termed selective neuronal necrosis. Unlike neurons, astrocytes in primary cultures do not die until almost all energy sources are exhausted 15, 33. Accordingly, astrocytes were traditionally considered to be more resistant to energy failure than neurons based on these important observations. However, this view has recently been challenged. Several groups have reported that ischemia can induce loss of astrocyte‐specific proteins before histological evidence of neuronal death is present 13, 26, 27). Moreover, Lukaszevicz et al noted that protoplasmic astrocytes were particularly vulnerable to ischemia (27). Several groups showed that cultured astrocytes were more sensitive than neurons to acidosis [e.g. 3, 14]. Therefore, it was suggested that the early swelling combined with acidity might impair astrocyte function and compromise vital neuron–astrocyte interactions 6, 30.
Astrocytes remove extracellular glutamate, lactate, hydrogen and potassium ions (8). Astrocytes store glycogen and may provide lactate as an alternative energy substrate to neurons (32). They are a rich source of reduced glutathione, which is an important antioxidant (28). Thus, disturbances in these astroglial functions can exacerbate neuronal injury. Glycogen‐derived glucose and lactate in astrocytes could supply the energy demand in ischemic astrocytes and perhaps also in surviving neurons. Anaerobic glycolysis in the absence of oxygen and oxidation of the energy substrates after reperfusion as well as in the penumbra during ischemia might generate sufficient adenosine‐5′‐triphosphate (ATP) to maintain ionic gradients and important astrocytic functions. However, lactic acid overproduction might offset any benefit gained and promote further damage (35). Astrocytic syncytium may also contribute to the expansion of infarct by propagating cell death mediators via gap junctions (24).
Astrocytic swelling is a prominent, early response to ischemia 12, 31, 33. It has been proposed that astrocytes die via osmotic swelling and necrosis at the onset of ischemia 26, 27. This idea was further strengthened by the observation that, after middle cerebral artery occlusion (MCAo), apoptotic mechanisms were activated in only a minority of ischemic astrocytes, although the involvement of apoptosis in several forms of astrocytic death, including in vitro or neonatal ischemia, has been demonstrated 11, 13, 29, 38. Considering the fact that neither the loss of synthesis of selective proteins nor swelling (astrocytes considerably change their volume during physiological activities) necessarily indicates cell death, we investigated astrocyte viability after transient MCAo and compared it with the pace of neuronal demise. We used glycogen and CellTracker® (Molecular Probes, Eugene, OR, USA) to monitor the metabolic activity of astrocytes. Cell death was assessed by monitoring plasmalemma integrity with propidium iodide (PI) and the lysosomal membrane integrity with deoxyribonuclease II (DNase II) leakage (an enzyme that is normally confined to lysosomes) to the cytoplasm. We separately evaluated perifocal and core ischemic regions because differing conditions in these areas might impact astrocyte and neuron viability differentially. For the above purposes, we adapted CellTracker® (Molecular Probes) and PI staining to in vivo conditions and modified periodic acid‐Schiff's base (PAS) method to track glycogen metabolism. Electron microscopy (EM) was also used to evaluate cellular and mitochondrial integrity, and the latter was additionally assessed by cytochrome‐c release.
MATERIALS AND METHODS
MCAo
Swiss albino mice weighing 32–44 g (Hacettepe University, Laboratory Animal Breeding and Research Facility) were housed under diurnal lighting conditions and fasted overnight, but were allowed free access to water before experiments. Animal housing, care and application of experimental procedures were all done in accordance with Ethics Committee of Hacettepe University (2002/44‐2) and all efforts were made to minimize animal suffering and to use only the number of animals necessary to produce reliable scientific data.
Mice were intraperitoneally anesthetized with ketamine (100 mg/kg) and xylazine HCl (10 mg/kg). Body temperature was monitored by a rectal probe and maintained at 37.0 ± 0.1°C via a homoeothermic blanket. Only animals with arterial blood pressure within the physiological range were included in the study. Proximal occlusion of the right MCA was performed with a filament as described before (16). Briefly, a nylon filament (8‐0) was inserted into the common carotid through a small incision proximal to the bifurcation and advanced into the internal carotid artery up to the origin of MCA (10 mm from the bifurcation). The distal 3 mm of the 8‐0 filament was coated with silicon. A flexible probe (PF‐318 of PeriFlux PF 2B; Perimed, Stockholm, Sweden) was placed over the skull (2 mm posterior, 6 mm lateral to bregma), away from large pial vessels, to monitor the regional cerebral blood flow (rCBF) by laser‐Doppler flowmetry. After obtaining a stable 10 minutes epoch of preischemic rCBF, the MCA was occluded and rCBF was monitored for the duration of ischemia (typically 10%–20% of baseline) and for the first 5–10 minutes of reperfusion until the rCBF was recovered. Reperfusion was accomplished by pulling the filament back. Mice were subjected to 2 h of proximal MCAo and 6 (n = 23), 24 (n = 18) and 72 h (n = 18) of reperfusion.
Glycogen histochemistry
Glycogen was detected histochemically in tissue sections using the PAS method after treatment with dimedone 6 (n = 4 mice), 24 (n = 6) and 72 h (n = 4) after reperfusion following 2 h of ischemia. Dimedone is required to block aldehyde groups on non‐glycogen substances such as glycolipids, glycoproteins and connective tissue mucopolysaccharides, because Schiff 's reagent also reacts with non‐glycogen aldehyde groups and may yield false‐positive staining in the brain, where the glycogen concentration is relatively low 5, 22. Brain sections were oxidized with 0.5% periodic acid for 10 minutes at room temperature followed by treatment with a saturated solution of dimedone in distilled water for 20 minutes at 60°C. After rinsing in distilled water, sections were reacted with Schiff's reagent. The specificity of the dimedone–PAS reaction was confirmed by digestion with diastase (5). Serial sections were also stained with hematoxylin. The PAS‐negative area and the infarct area detected by hematoxylin were measured by image analysis software (NIH Image 1.59; NIMH, Bethesda, MD, USA) on the posterior surface of each section, and the volumes were calculated by summing the areas of sequential 2‐mm thick sections.
Immunohistochemistry
Neurons and astrocytes were labeled with antibodies against neuronal nuclei (NeuN), glial fibrillary acidic protein (GFAP), S100 or a cocktail of GFAP and S100 (all antibodies were obtained from Chemicon, Temecula, CA, USA and used at a 1:200 dilution) in frozen sections at 6 (n = 4), 24 (n = 5) and 72 h (n = 5) after reperfusion following 2 h of ischemia. Separate stainings of S100 or GFAP were used for cell type characterization, whereas a cocktail of GFAP and S100 immunostaining were used for astrocyte counts. Antigen retrieval for NeuN was achieved by microwaving for 10 minutes in 1 mmol/L ethylenediaminetetraacetic acid buffer (Ph 8). The loss of membrane integrity of lysosomes and mitochondria were monitored by detecting cytoplasmic spillage of the lysosomal enzyme DNase II [Rabbit Anti‐DNase II (C‐Terminal); Chemicon, 1:50] at 6 (n = 4), 24 (n = 5) and 72 h (n = 5); or the mitochondrial intermembrane protein cytochrome‐c (1:100; Abcam, Cambridge, UK) at 24 (n = 7) and 72 h (n = 5). In addition, caspase‐3 activation was assessed by immunolabeling the cleaved active form of caspase‐3 p20 (1:100; Cell Signaling, Danvers, MA, USA) at 72 h (n = 5).
Sections were incubated at 37°C for 90 minutes with primary and then with appropriate secondary antibodies (1:200) for 90 minutes. Alexa flour 488 or 586 goat anti‐mouse immunoglobulin G (IgG) (Molecular Probes), and Cy2 or Cy5 goat anti‐rabbit IgG (Jackson Immunoresearch, West Grove, PA, USA) were used as secondary antibodies as appropriate. The tyramide amplification method was used for caspase‐3 p20 immunofluorescence according to the manufacturer's recommendations (Molecular Probes). Primary antibody omission and incubations with either the blocking solution or phosphate‐buffered saline (PBS) were performed to test the specificity of immunoreactivity. Sections were mounted in glycerol/PBS medium containing 25 mg/mL sodium azide.
For identification of PAS‐positive cells, formalin‐fixed, paraffin‐embedded brain sections were deparaffinized overnight, dehydrated in graded xylene and alcohol solutions, and incubated with hydrogen peroxide in methanol for 10 minutes. The sections were rinsed with PBS and incubated in blocking solution (Zymed, South San Francisco, CA, USA) for 10 minutes, followed by incubation with primary neuron and astrocyte‐specific antibodies as indicated earlier. Sections were then labeled with biotinylated secondary antibody and streptavidin‐peroxidase (Zymed) for 10 minutes. 3,3′ Diaminobenzidine (DAB) was used as a chromogen and, finally, PAS‐labeling was used as a counterstain.
Bright‐field and fluorescent examinations were performed with a Nikon Eclipse E600 upright microscope (Nikon, Japan) using appropriate filter sets. Specimens were further examined using a Zeiss LSM‐510 confocal laser‐scanning microscope (Carl Zeiss, Jena, Germany). Single optical sections (2048 × 2048 pixels) were collected. Digitized images were pseudo‐colored according to their original fluorochromes.
Stereotaxic injection of PI and CellTracker®
To assess plasma membrane integrity, 0.5 µL of PI (P‐3566: Molecular Probes; 1 mg/mL in distilled water) was injected into the right lateral ventricle under ketamine/xylazine anesthesia 20 minutes before sacrifice, at the end of ischemia/reperfusion (6 h, n = 4; 24 h, n = 5; 72 h, n = 5). Mice were then perfused transcardially with 10% formaldehyde. Brains were removed and kept in formaldehyde solution for 24 h at 4°C. Later, the brains were bathed in 20% and then 30% sucrose solutions for 12 h at 4°C. Twenty‐µm thick coronal sections that passed through the anterior commissure were kept frozen at −200C until use. For identification of cells positively labeled with PI, sections were stained with antibodies against NeuN (Chemicon) and GFAP (Sigma, St. Louis, MO, USA) or S100 (Sigma) (6 h, n = 4; 24 h, n = 5; 72 h, n = 5) as described earlier (in the section on immunohistochemistry.
CellTracker® Green 5‐chloromethylfluorescein diacetate (CMFDA) was used as a vitality marker (Molecular Probes). Cell Tracker Green CMFDA freely passes through cell membranes and, once inside of a cell, it undergoes a series of metabolic reactions that transform it into a cell‐impermeant fluorescent dye. Under ketamine/xylazine anesthesia, CellTracker® was injected 1.5 mm deep into the cortex, corresponding to the perifocal ischemic region at 1.1 mm anterior and 1.2 mm lateral to bregma by means of a Hamilton syringe 1 h after 2 h of ischemia. Five hours later (i.e. 6 h after reperfusion; n = 6), mice were re‐anesthetized and transcardially perfused with 4% paraformaldehyde in PBS (pH 7.4). Brains were removed and postfixed in paraformaldehyde for 4 h, washed in PBS for 15 minutes and placed in a 30% sucrose solution in PBS for 24 h at 4°C. Fifteen‐µm thick coronal sections that pass through the anterior commissure were kept frozen at −20°C until PAS labeling.
PI‐ and CellTracker®‐labeled sections were also stained with PAS as described earlier. Because the fluorescence of PI and CellTracker® are lost after PAS staining, photographs of the PI‐ or CellTracker®‐labeled sections were captured before they were further processed for PAS staining. Subsequently, the bright field micrographs of PAS‐stained sections were taken at the same coordinates and superimposed with fluorescent images.
EM study
Brain tissues were perfused with 0.2 M phosphate buffer containing 2% (w/v) paraformaldehyde and 2.5% (v/v) glutaraldehyde at 6 h (n = 2) or 24 h (n = 2) after reperfusion or from naïve control mice (n = 2). Two areas corresponding to the ischemic penumbra and three areas from the core were removed and immersed in the same fixative for 3 h at 4°C. Samples were rinsed three times in 0.1 M phosphate buffer then postfixed with 1% osmium tetroxide in 0.1 M phosphate buffer for 1 h at room temperature. Following three washes in buffer, specimens were dehydrated in graded ethanol and finally embedded in Araldite® (Huntsman Advanced Materials, Basel, Switzerland). One‐micrometer semi‐thin sections were stained with1% toluidine blue for light microscopic localization and then ultra‐thin sections (60–80 nm) were cut with a Leica Ultracut R microtome (Leica, Vienna, Austria) and double‐stained with uranyl acetate and lead citrate. They were examined and photographed using a LEO 906 E electron microscope (80 kV; LEO Elektronenmikroskopie, Oberkohen, Germany).
Cell count and statistics
Labeled cells were counted manually under ×400 magnification in perifocal and core cortical regions as well as core subcortical regions. Three vertically adjacent, non‐overlapping circular microscopic grids were sampled from each region, and the mean number of NeuN/GFAP+S100/DNAse II/cytochrome‐c‐positive cells per mm2 of each section was calculated by averaging the counts obtained from these areas.
One section from the core (temporoparietal) and one from the penumbral (frontal) cortex were chosen randomly from each brain sample prepared for EM. Number of the normal‐looking mitochondria and of the mitochondria displaying minor (slightly swollen but inner and outer membranes are intact with minor damage to cristea) or severe damage (noticeably swollen, loose matrix, membrane and cristae integrity are lost) were counted in neurons and astrocytes.
Means were given with their standard deviations (SD) and compared with Kruskal–Wallis variance analysis followed by the Mann–Whitney U‐test. A value of P < 0.05 was considered to be significant.
RESULTS
Non‐ischemic brain areas were diffusely stained with PAS (Figure 1A). At higher magnifications, PAS‐positive cell bodies and processes were identified among several PAS‐negative larger cell bodies that looked like empty spherical spaces in cortical and subcortical areas (Figure 1F–H). Double‐staining for GFAP, S100 or NeuN demonstrated that PAS‐positive cells overlapped with astrocytes, whereas most PAS‐negative cell bodies corresponded to neurons (Figure 1E–H). Strong PAS staining that overlapped with GFAP was also observed around microvessels.
Figure 1.
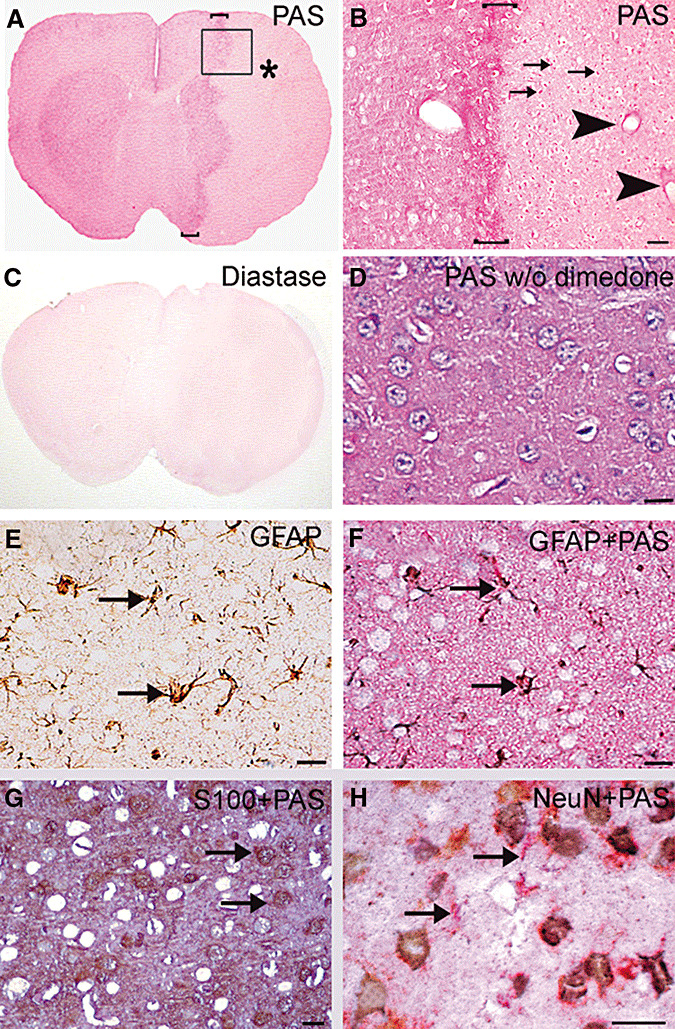
Glycogen is preserved in ischemic astrocytes after transient focal cerebral ischemia. Glycogen is identified with a magenta color on brain sections stained with periodic acid‐Schiff's base (PAS) after blocking non‐glycogen aldehyde groups in glycoproteins, glycolipids and glycosaminoglycans with dimedone. A. Glycogen staining was reduced in the ischemic middle cerebral artery territory [* in A, 24 h after a 2‐h ischemia). However, at higher magnifications, several PAS‐positive cells (three examples marked with arrows) and intense perivascular staining (arrowheads), reflecting lasting glycogen in some astrocytes and their endfeet, were clearly visible (B corresponds to the squared area in A). Also note the hyperintense staining surrounding the ischemic area (bracketed zones in A and B). Treatment of sections with the glycogen degrading enzyme diastase eliminated PAS positivity, confirming that PAS + dimedone‐staining labels only glycogen (C). In the absence of dimedone treatment, PAS reagent stained all cellular and extracellular structures (D). Note that the unstained spherical spaces (corresponding neurons) seen in dimedone‐treated sections have disappeared (F,G). To verify that PAS‐positive cells were astrocytes, PAS‐labeled sections were also stained with either glial fibrillary acidic protein (GFAP) (E,F) or S100 (G), or neuronal nuclei (NeuN) (H) immunohistochemistry (all micrographs were taken from the perifocal ischemic region in the frontoparietal cortex). E is adjacent to the F section and was only stained for GFAP (brown) to separately illustrate GFAP‐positive astrocytes that were co‐stained with PAS in F. Note the superimposed magenta‐ and brown‐colored, star‐shaped fibrillary astrocytes (arrows). Also note how well the brown and magenta backgrounds and unstained spherical spaces in E and F match. Brown‐colored cell bodies of S100‐positive protoplasmic astrocytes were also co‐stained with PAS (arrows in G). Confocal microscopy could not be used to identify PAS‐positive structures because PAS staining caused strong autofluorescence. H, NeuN‐positive cells (brown) were not stained with PAS (magenta, arrows), indicating that most of the PAS‐negative cell bodies (empty spherical spaces in E–G) are neurons. The neuropil in PAS‐stained sections has light magenta background staining similar to neuropilic GFAP staining in E, possibly caused by fine peripheral astrocyte processes, as treatment of these sections with diastase have eliminated PAS positivity (C). Scale bar: 50 µm in B, 20 µm in D–H.
PAS staining was reduced in the ischemic MCA territory compared with non‐ischemic brain areas, but it was strikingly increased in the peri‐ischemic area (Figure 1A,B). Interestingly, at higher magnifications, several PAS‐positive astrocytic bodies and endfeet were still noticeable in the ischemic area 6 h (n = 5) after reperfusion and occasionally at 24 h (n = 6) (Figure 1B). Therefore, we wanted to elucidate whether these PAS‐positive cells were alive and maintained at least part of its metabolism or if they were merely unable to use glycogen that led to its accumulation. For this purpose, we used the cell vitality indicator, CellTracker®, which is commonly employed to identify metabolically active cells in culture. In non‐ischemic areas, all parenchymal and vascular cells were positively stained with CellTracker®, whereas, in ischemic areas, most of the cells were not able to process CellTracker® to a fluorescent product, except in the case of PAS‐positive astrocytes 6 h after reperfusion (n = 6) (Figure 2A,B).
Figure 2.
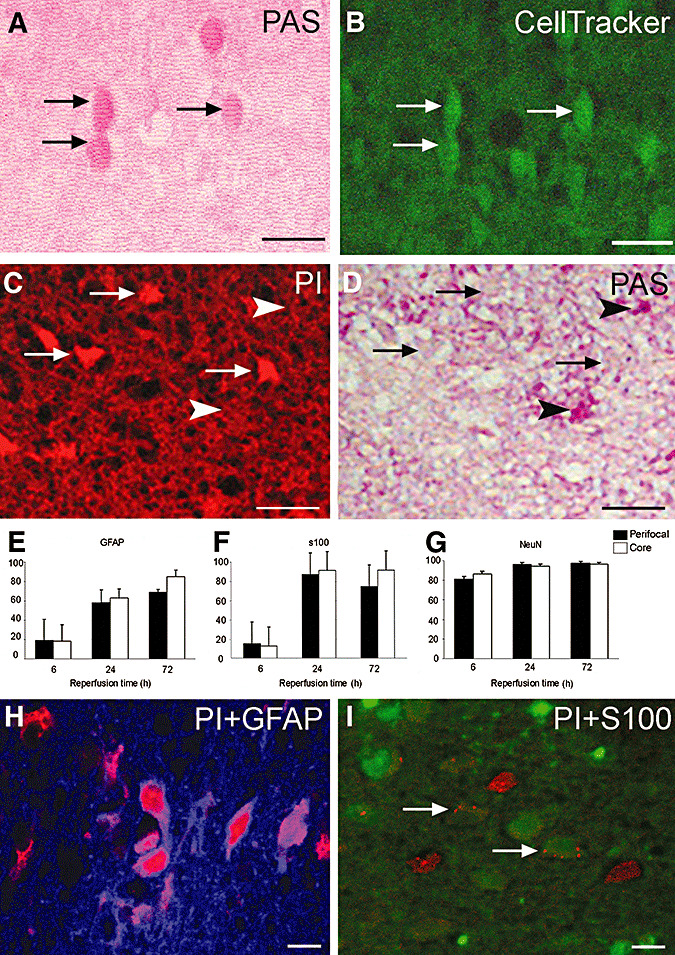
Glycogen‐bearing ischemic astrocytes are metabolically active and retain their plasma membrane integrity. Periodic acid‐Schiff's base (PAS)‐positive astrocytes (A) were able to take up and hydrolyze CellTracker® to a green fluorescent product (B, arrows denote corresponding astrocytes in A). Moreover, PAS‐positive astrocytes (arrowheads, D) were not labeled with the membrane impermeant fluorescent dye propidium iodide (PI) (red, C) in contrast to neighboring PAS‐negative cells (arrows, C,D). Astrocytes eventually became PI‐positive, but at a much later time than neurons. (E–G) Percentages of glial fibrillary acidic protein (GFAP)‐ or S100‐positive astrocytes or neuronal nuclei (NeuN)‐positive neurons that lost plasmalemma integrity in the perifocal (black columns) and core (white columns) areas at 6, 24 and 72 h after 2 h middle cerebral artery occlusion. The mean percentages of PI/GFAP‐positive astrocytes were significantly lower than corresponding PI/NeuN‐positive values at all time points, whereas only the mean percentage of PI/S100‐ positive astrocytes at 6 h was significantly lower than the corresponding NeuN value (see figure discussion in the text for n). H,I. Representative merged confocal images of PI (red) + GFAP (blue, H) and PI (red) + S100 (green, arrows in I)‐labeled astrocytes. Note limited PI leakage into astrocytes despite extensive labeling of neighboring cells (possibly neurons). All pictures were taken from the ischemic perifocal region in the frontal cortex at 6 (A,B) or 24 h (C,D,H,I) after reperfusion. Scale bar: 50 (A–D) or 20 µm (H,I).
After demonstrating that PAS‐positive cells in ischemic brain were live astrocytes with metabolic activity, we further elaborated this finding by showing that these astrocytes retained their plasma and lysosomal membrane integrity. Following the intracerebroventricular (i.c.v.) injection of a membrane impermeant fluorescent dye, PI, none of the cells with intact plasma membranes in the non‐ischemic brain were positively stained, whereas 81 ± 2%, 96 ± 2% and 97 ± 1% of neurons were PI‐positive in the perifocal ischemic region 6 (n = 4), 24 (n = 5) and 72 h (n = 5) after ischemia/reperfusion, respectively (Figure 2G). On the other hand, GFAP as well as S100‐positive astrocytes retained their membrane integrity longer than neurons: 19 ± 22%, 58 ± 14% and 69 ± 3% of GFAP(+) astrocytes were PI‐positive at corresponding sites and time points (Figure 2E–G). Similarly, 18 ± 16%, 63 ± 5% and 85 ± 3% of astrocytes in contrast to 86 ± 2%, 94 ± 3% and 96 ± 2 % of neurons were PI‐positive in core regions 6, 24 and 72 h after ischemia, respectively. Although ischemia‐induced loss of immunoreactivities of cellular markers used may partly underestimate the absolute values, the mean percentages of PI/GFAP‐positive astrocytes were significantly lower than corresponding PI/NeuN‐positive values at all time points, whereas only the mean percentage of PI/S100‐positive astrocytes at 6 h was significantly lower than the corresponding NeuN value. Co‐localization studies at 6 h revealed that PI‐positive and PAS‐positive cells were clearly separated (Figure 2C,D).
Loss of lysosomal membrane integrity in astrocytes followed a similar time course as PI labeling. Non‐ischemic neurons and astrocytes exhibited a fine granular DNase II immunoreactivity that was compatible with lysosomal location of this enzyme (Figure 3A inset). This pattern was replaced with a diffuse cytoplasmic immunostaining in ischemic cells following loss of lysosomal membrane integrity (Figure 3). The percentage of astrocytes showing DNase II leakage to the cytoplasm at the ischemic core increased from 6 (n = 4) to 24 h (n = 5) (35 ± 9%, 58 ± 29%, respectively) but decreased at 72 h (n = 5) (22 ± 10%), possibly because of the depletion of the protein or decreased immunoreactivity, whereas DNase leakage continued to increase in the perifocal ischemic area (18 ± 5%, 35 ± 17% and 49 ± 12%, respectively; the mean percentages at 24 h and 72 h were significantly higher than values at 6 h, Figure 3D).
Figure 3.
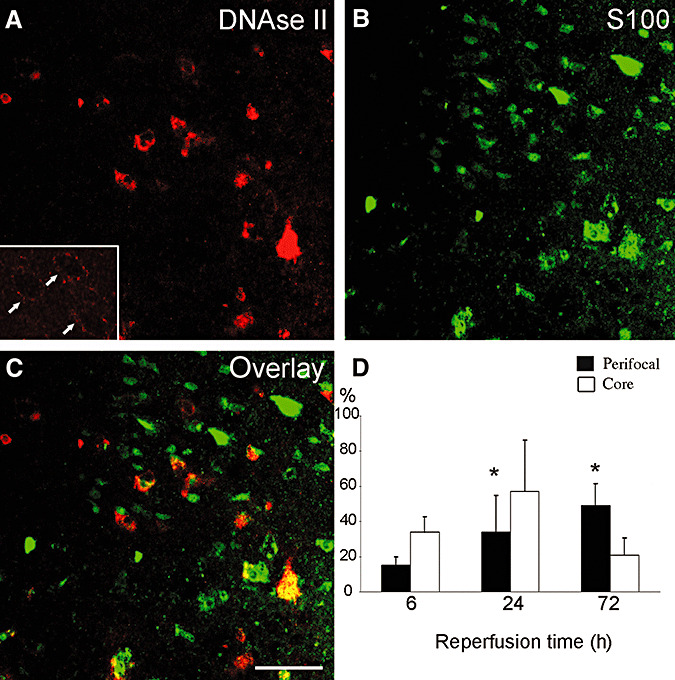
Rupture of the lysosomal membranes in astrocytes follows a similar time course as the loss of plasma membrane integrity. In ischemic astrocytes, diffuse cytoplasmic DNase II immunostaining (red) replaced fine granular immunoreactivity observed in non‐ischemic cells (arrows in inset), suggesting loss of lysosomal membrane integrity and spillage of DNase II, an enzyme normally confined to lysosomes, to the cytoplasm (A). C. The merged confocal image of A and B (S100‐positive astrocytes, green) illustrates that only a fraction of astrocytes exibited DNAse II spillage. Images were captured from the ischemic perifocal region of the frontal cortex at 6 h after 2 h middle cerebral artery (MCA) occlusion. Typically, astrocytes showing diffuse DNAse II immunostaining are less common than are they in this example, as the quantitative data in the graph indicates (D). The graph shows the percentages of astrocytes with diffuse DNase II immunoreactivity at 6, 24 and 72 h after ischemia in the core (white columns) and perifocal areas (black columns) of the MCA territory. The percentage of DNase II positive astrocytes seemed to decrease in the core at 72 h, whereas it continued to increase in the perifocal region, possibly because of reduced DNase II immunoreactivity in the core. The mean percentages of DNAse II/S100‐positive astrocytes in the penumbra at 24 h and 72 h were significantly higher than values at 6 h was (see figure discussion in the text for n). Scale bar: 50 µm.
Electron microscopic examination was in line with these findings (Figure 4). At 6 h (10 sections), plasma and nuclear membrane integrity of most astrocytes were preserved despite considerable swelling especially at endfeet. Lysosomes, infrequently encountered in these very thin sections, also had intact membranes at 6 h. At 24 h (10 sections), EM showed several astrocytes with ruptured plasma, nuclear and organelle membranes (Figure 4). Interestingly, several mitochondria displayed normal morphology in a watery astrocyte cytoplasm at 6 h and, although less frequently, at 24 h (see Table 1 for quantitative data and comparison of astrocytic and neuronal mitochondria). The latter finding conformed to the immunohistochemical data: only a few astrocytes (11 ± 2% of GFAP + S100‐labeled cells) displayed cytoplasmic cytochrome‐c immunostaining even 72 h (n = 4) after ischemia (Figure 5A,B,D arrowheads), whereas almost all neurons exhibited cytoplasmic cytochrome‐c labeling. Triple immunostaining with antibodies against cytochrome‐c, caspase‐3‐p20 and S100 indicated that only 5 ± 1% of astrocytes co‐localized cytochrome‐c and caspase‐3p20 in contrast to prevalent co‐localization of the two markers in neurons at 72 h (Figure 5). No immunoreactivity to cytochrome‐c and caspase‐3‐p20 was observed in non‐ischemic cells.
Figure 4.
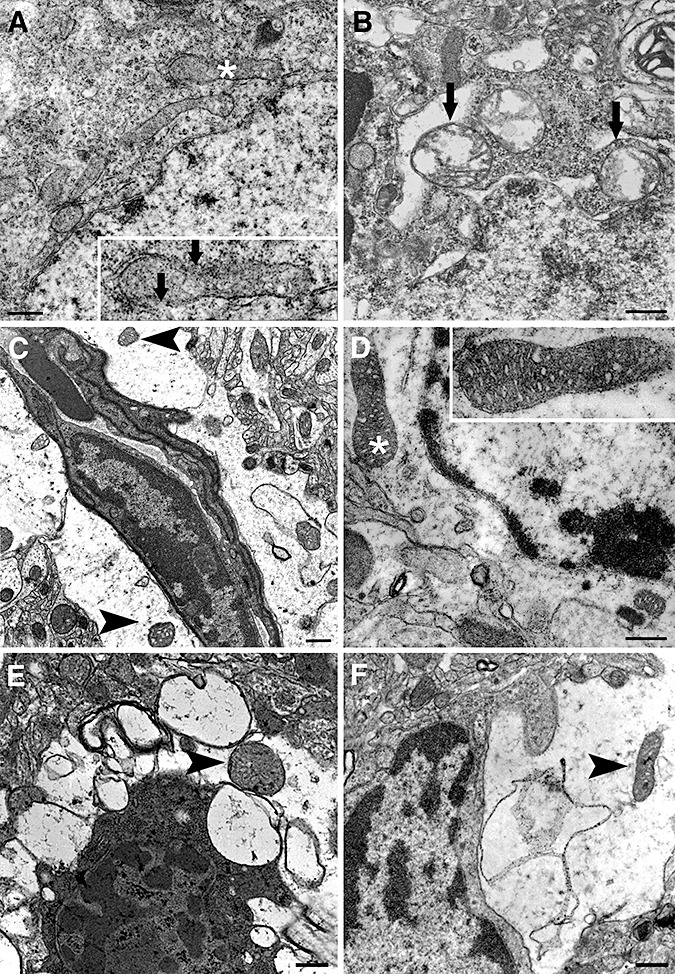
Neurons rapidly degenerate whereas astrocytes are better preserved after ischemia/reperfusion. Electron micrographs illustrate neurons (A,B,E) and astrocytes (C,D–F) from the ischemic frontoparietal core cortex at 6 (A–D,F) or 24 h (E) after 2 h middle cerebral artery occlusion. Neurons already displayed significant alterations in mitochondria at 6 h after reperfusion (A and B). The inset illustrates the asterisk‐labeled mitochondrion in A, exhibiting discontinuities in the outer and inner membrane (arrows) and loss of cristae. Swollen mitochondria with severely disorientated cristae are readily noticeable in B (arrows). In contrast, swollen astrocytic endfeet encircling a vessel contain intact round mitochondria with numerous cristae and are enclosed by a double membrane (arrowheads in C) despite watery astrocytic cytoplasm. D. Another astrocyte, distinguished by its relatively organelle‐free cytoplasm and euchromatic nucleus, exhibits well‐preserved mitochondria 6 h after reperfusion (arrowheads). The insets in C and D show the mitochondria labeled with asterisks at higher magnification. At 6 (F) and 24 h (E) after 2 h of ischemia, astrocytic processes harbor only mildly deteriorated mitochondria (arrowheads) despite considerable cytoplasmic swelling, whereas a neighboring neuron in E displays advanced degenerative features such as severely shrunken and condensed cytoplasm and heterochromatic nuclei. Scale bar: 5 µm.
Table 1.
Astrocytic mitochondria are better preserved than neuronal ones after ischemia/reperfusion.
| 2 + 6 h Normal (%) | Minor damage (%) | Swollen (%) | n | 2 + 24 h Normal (%) | Minor Damage (%) | Swollen (%) | n | |
|---|---|---|---|---|---|---|---|---|
| Astrocyte | ||||||||
| Penumbra | 91 ± 25 | 10 ± 25 | 0 ± 0 | (7, 25) | 28 ± 45 | 57 ± 43 | 15 ± 27 | (8, 23) |
| Core | 59 ± 43 | 35 ± 46 | 7 ± 18 | (7, 18) | 13 ± 30 | 80 ± 45 | 7 ± 15 | (5, 18) |
| Neuron | ||||||||
| Penumbra | 0 ± 0 | 33 ± 45 | 67 ± 35 | (10, 138) | 0 ± 0 | 26 ± 35 | 74 ± 35 | (9, 56) |
| Core | 0 ± 0 | 7 ± 3 | 98 ± 4 | (12, 85) | 0 ± 0 | 4 ± 8 | 96 ± 8 | (8, 98) |
Number in parentheses denote the number of cells and mitochondria screened, respectively. Data are expressed as mean ± standard deviation.
Figure 5.
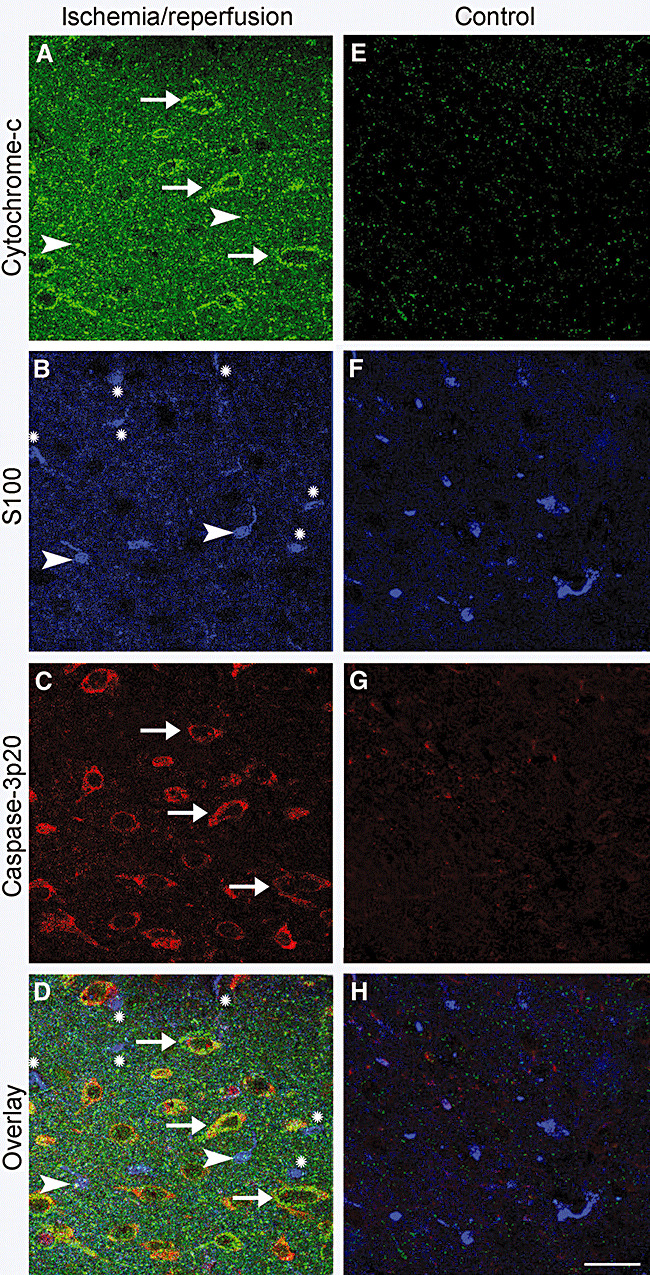
Release of cytochrome‐c from mitochondria and activation of caspase‐3 were much less prevalent in ischemic astrocytes compared with neurons. Brain sections were immunostained with antibodies against cytochrome‐c, caspase‐3‐p20 and S100. A–D. Obtained from the ischemic hemisphere after 2 h of ischemia and 72 h of reperfusion. E–H. Corresponding controls, which illustrate that there was not any cytochrome‐c or capase‐3p20 immunoreactivity in the non‐ischemic hemisphere. D,H. Overlay of images A–C and E–G, respectively. The mitochondrial intermembrane protein cytochrome‐c was released into the cytoplasm in neurons (A, green, arrows), whereas astrocytes identified with S100 immunoreactivity (B, blue) were seldom and faintly labeled with the anti‐cytochrome‐c antibody (note astrocytes marked with arrowheads in A,B,D). Astrocytes that were not labeled with the anti‐cytochrome‐c antibody, are marked with an (*) in B and D. In addition, astrocytes did not express the active cleaved form of caspase‐3, capase‐3p20, unlike neurons (C, red, arrows). Note that most of the caspase‐3p20 immunopositive neurons also showed cytoplasmic cytochrome‐c labeling (yellow, D, arrows). Scale bar: 50 µm.
The PAS‐negative MCA area was consistently smaller than the ischemic area detected by hematoxylin staining (i.e. the area showing sponginess, pallor, alterations in the shape and staining of nuclei and red or ghost neurons), suggesting that astrocytes at the expanding outer border of the infarct were still able to maintain glycogen levels unlike the core area (Figure 6). At 6 (n = 5) and 24 h (n = 6), more than 80% of these astrocytes had nuclei that appeared normal by hematoxylin and Hoechst‐33258 (Molecular Probes) staining in contrast to neighboring neurons with pyknotic nuclei (the mean percentages of astrocytes with pyknotic nuclei were significantly lower than corresponding neuronal values at 6 h and 24 h time points, see Figure 6G for quantitative data). On coronal sections passing through the anterior commissure, the PAS‐negative area expanded from 42 ± 7% of the hemisphere at 6 h (n = 5) to 47 ± 6% at 24 h (n = 6) and to 69 ± 6% at 72 h (n = 4), whereas the areas displaying overt ischemic changes with hematoxylin staining of the same sections were 54 ± 8%, 56 ± 8% and 74 ± 6% at 6, 24 and 72 h, respectively (Figure 6). In other words, the PAS‐positive ischemic area harboring live astrocytes shrunk from 12 ± 6% of the hemisphere at 6 h to 9 ± 9% at 24 h and to 4 ± 2% at 72 h. The same trend was observed when the tissue volumes were calculated based on area measurements from consecutive coronal slices (Figure 6). At the immediate periphery of this PAS‐positive ischemic area, there was an area where PAS staining intensity was greater than in the contralateral homologous regions (Figure 6). This PAS hyperintense area exhibited no ischemic changes and amounted to 28 ± 3%, 22 ± 2% and 24 ± 2% of the hemisphere at 6, 24 and 72 h, respectively. As the astrocytes in the core—where metabolism is severely compromised—lost PAS positivity, the intense staining in the peri‐infarct zone suggests an accelerated glycogen synthesis rather than passive accumulation of glycogen caused by inhibition of its use by astrocytes.
Figure 6.
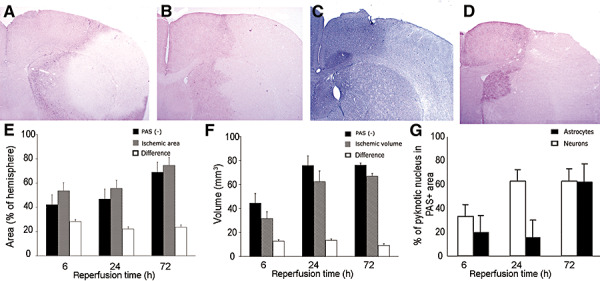
The astrocytes at the periphery of the expanding ischemic lesion were alive and metabolized glycogen in contrast to the neighboring degenerated neurons. The area showing reduced periodic acid‐Schiff's base (PAS) staining on brain sections obtained at 6 (A), 24 (B) and 72 (D) h after 2 h middle cerebral artery (MCA) occlusion was smaller than the MCA area displaying overt ischemic histological changes with hematoxylin staining (a typical example obtained at 24 h is given in C). E. Black, dashed and white columns represent the means (standard deviation) of PAS‐negative and ischemic areas, and of their differences, respectively. The corresponding tissue volumes calculated from five consecutive sections are shown in F. G. Percentages of neurons (white columns) and astrocytes (black columns) with pyknotic nuclei to the total number of neurons and astrocytes, respectively, counted in the PAS‐positive ischemic area at the periphery of the ischemic lesion. The mean percentages of astrocytes with pyknotic nuclei were significantly lower than corresponding neuronal values at 6 and 24 h time points (see discussion in the text for n).
DISCUSSION
We found that astrocytes at the periphery of the expanding ischemic core still harbored glycogen despite significant depletion of glycogen in the core. These glycogen‐positive astrocytes were located among shrunken ischemic neurons with pycnotic nuclei. They had normal nuclei and processed CellTracker®, suggesting that they were alive, unlike neighboring neurons. As expected, these astrocytes maintained their plasmalemma integrity. Disintegration of the plasma membrane developed much later than that of neurons. Rupture of lysosomal and mitochondrial membranes paralleled a similar course to that of plasmalemma disruption. Taken together, these findings strongly suggest that astrocytes tolerate ischemia better than neurons and that they die possibly with a delayed necrosis despite early swelling at the onset of ischemia.
The majority of glycogen is located in astrocytes in the adult brain. Glycogen phosphorylase can liberate energy metabolites from glycogen when needed (4). Astrocytic glycogen is thought to be utilized when glycolysis cannot match brain's energy demand during conditions such as intense glutamatergic synaptic activity, or when there is an insufficient supply of other energy substrates 8, 32, 37. It has been shown that glycogen turnover increased in hippocampal slices during hypoxia‐ischemia, and that glycogen was eventually depleted because of increased use whereas its synthesis was not inhibited (25). Although PAS/dimedone staining does not provide a quantitative assessment of in situ brain glycogen content, parallel findings were already obtained in vivo in a rodent 2‐h MCAo model, which is very similar to ours, by using biochemical detection methods (10). Astrocyte glycogen has also been reported to sustain neuronal activity during hypoglycemia (36). Similarly, in our study, the presence of live (CellTracker®‐positive) as well as PAS‐positive astrocytes in the ischemic region suggest an accelerated use of glycogen after focal ischemia/reperfusion. The increased energy demand in the reperfused brain may stimulate glycogen turnover. In addition to glycogen, astrocytes can oxidize alternative energy substrates including fatty acids, short‐chain monocarboxylic acids and amino acids (8). Astrocytes contain greater concentrations of glutathione and superoxide dismutase than do neurons, hence, they are more capable of scavenging reactive oxygen species 6, 28. Accordingly, astrocytes may survive longer than neurons and continue to support the ischemic tissue for some time after ischemia/reperfusion.
Previous studies, which indicated that astrocytes are more vulnerable to ischemic injury than neurons, based this conclusion on rapid downregulation of astrocyte‐specific proteins before DNA fragmentation took place in neurons 26, 27. However, no evidence at the cellular level was provided to indicate that astrocytes with downregulated protein expression lost their vitality. In one study, protoplasmic astrocytes were reported to undergo rapid plasmalemma disintegration (27); however, no quantitative data were given and no comparison was made with the time course of neuronal plasmalemma rupture. Our data indicate that about 18% of astrocytes, contrary to 87% of neurons in the core, lose their membrane integrity 6 h after reperfusion following 2‐h ischemia. In the latter study, the authors may have encountered more frequent astrocyte rupture than we observed as they used a permanent MCAo model. However, it is likely that the neuronal membranes were also severely injured in their study, but this point was not investigated. Interestingly, in another study, degeneration of neurons was assessed with the neuronal marker TG2, which was lost with a slower time course than that of the astrocyte‐specific proteins (26). Had the authors used NeuN immunoreactivity, which rapidly disappears after ischemia (42) as a neuron‐specific marker, they might have concluded that neuronal vitality was lost soon after ischemia. In our study, we used two well‐established cell death and two vitality markers as well as EM to evaluate astrocytes' fate, and compared these data with that from neurons. PI uptake is an established method to evaluate plasmalemma integrity of astrocytes and other cells in vitro. All of the data obtained with these diverse approaches, including ultrastructural findings, were complementary and showed that astrocytes retained their vitality longer than neurons. Supporting these findings, it has recently been reported that astrocyte acetate metabolism was not significantly altered in perifocal tissue in rat cortex subjected to MCAo (40).
Ultrastructural studies show that astrocyte endfeet are the first cellular elements to swell during brain ischemia (6). Swelling can result from the uptake of extracellular K+, Cl‐, Na+ and glutamate and failure of the membrane Na+/K+ATPase to restore normal ion gradients (30). Importantly, astrocytic swelling does not necessarily indicate astrocyte death. Astrocytes swell during reversible pathological conditions (e.g. after seizures) (21). In fact, mild swelling promotes amino acid metabolism, glycogen synthesis and glutathione release in hepatocytes (17), all of which may support cell survival during ischemia. Similarly, swelling has been shown to enhance the glycogen content in cultured astrocytes (9). Certainly, further swelling developing over the course of ischemia will inevitably lead to astrocytic cell death because of the rupture of the plasmalemma and organelles, as shown at late time points in the present study.
Studies from our and several other laboratories consistently showed that, in vivo, only a small number of ischemic adult astrocytes exhibited markers of apoptosis that were widely expressed in ischemic neurons 11, 14, 23, 29, 38. These observations are rather surprising because astrocytes have all of the machinery necessary to activate the intrinsic and extrinsic apoptotic pathways as demonstrated in several pathologies including neonatal hypoxia/ischemia and in vitro ischemia 2, 39. Although mechanisms preventing apoptotic activation in ischemic adult astrocytes are currently not clear, it may be speculated that their high glutathione content may suppress activation of the death receptor‐mediated pathway (18). Lack of pancortin‐2 expression in adult astrocytes, which has recently been shown to play an important role in the formation of Bcl‐xL/Bax complexes and cytochrome‐c release after MCAo (7), or failure of activation of caspase‐9 and caspase‐3 because of the rapid inactivation of cytochrome‐c (34), may hinder initiation of the mitochondrial pathway. In fact, we detected diffuse cytochrome‐c immunoreactivity in only a small number of astrocytes even 72 h after ischemia. An interesting possibility is that low intensity cytochrome‐c immunoreactivity in astrocytes might simply be because of a lower density of mitochondria in astrocyte cell bodies. However, loss of cytochrome‐c from mitochondria along with the appearance of intense, diffuse cytoplasmic cytochrome‐c immunoreactivity in astrocytes have been reported by several groups after various types of insults in vitro[e.g. (34)] and in neonatal hypoxia‐ischemia in vivo (2). This uncertainty notwithstanding, EM clearly demonstrated that the loss of mitochondrial and cellular membrane integrity was significantly delayed in astrocytes as compared with neurons. Similarly, an in vitro study reported that metabolically perturbed astrocytes displayed low mitochondrial membrane potential and intact cellular membranes before they lost their mitochondrial membrane potential and plasmalemma integrity at later time points (19). It is likely that anaerobic energy sources, high anti‐oxidative capacity and higher calcium uptake capability of astrocytic mitochondria (1) may contribute to the preservation of the mitochondrial and plasmalemma integrity of astrocytes during ischemia and reperfusion injuries. Secondary mitochondrial failure after reperfusion (10) appears to develop in astrocytes less rapidly than in neurons. An acidic intracellular pH in astrocytes may delay the opening of permeablity transition pores and the progress of necrosis, whereas excess oxidative stress along with a higher pH may promote mitochondrial inner membrane permeabilization in neurons (20). Consequently, the ischemic death of astrocytes appears to be different than neuronal demise, in which apoptotic as well as necrotic pathways are concomitantly activated soon after the onset of ischemia (41). Further studies are warranted to clarify the mechanism of astrocytic death without activation of apoptotic mechanisms. This may extend our understanding of ischemic cell death in the brain beyond the initial simplifications that are based on the apoptosis–necrosis dichotomy.
In conclusion, this study demonstrates that astrocytes are more resistant to ischemia, as is conventionally accepted. After transient focal ischemia, astrocytes can maintain their metabolic and structural integrity longer than neurons and, consequently, they can support neuronal survival in the penumbra until they lose some of their important functions (31). Astrocytes in the infarct area eventually die possibly via delayed necrosis, despite their early swelling at the onset of ischemia.
ACKNOWLEDGMENTS
This paper is dedicated to the loving memory of Dr Günfer Gürer. This study is supported by Turkish Academy of Sciences (T. Dalkara and Y. Gursoy‐Ozdemir), TUBITAK/BAD (G. Gürer) and the Ankara University Institute of Biotechnology (A. Can). The authors are grateful to Dr Hülya Karatas for her help with the figures.
REFERENCES
- 1. Bambrick LL, Chandrasekaran K, Mehrabian Z, Wright C, Krueger BK, Fiskum G (2006) Cyclosporin a increases mitochondrial calcium uptake capacity in cortical astrocytes but not cerebellar granule neurons. J Bioenerg Biomembr 38:43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Benjelloun N, Joly LM, Palmier B, Plotkine M, Charriaut‐Marlangue C (2003) Apoptotic mitochondrial pathway in neurones and astrocytes after neonatal hypoxia‐ischaemia in the rat brain. Neuropathol Appl Neurobiol 29:350–360. [DOI] [PubMed] [Google Scholar]
- 3. Bondarenko A, Chesler M (2001) Rapid astrocyte death induced by transient hypoxia, acidosis, and extracellular ion shifts. Glia 34:134–142. [DOI] [PubMed] [Google Scholar]
- 4. Brown AM (2004) Brain glycogen re‐awakened. J Neurochem 89:537–552. [DOI] [PubMed] [Google Scholar]
- 5. Bulmer D (1959) Dimedone as an aldehyde blocking reagent to facilitate the histochemical demonstration of glycogen. Stain Technol 34:95–98. [DOI] [PubMed] [Google Scholar]
- 6. Chen Y, Swanson RA (2003) Astrocytes and brain injury. J Cereb Blood Flow Metab 23:137–149. [DOI] [PubMed] [Google Scholar]
- 7. Cheng A, Arumugam TV, Liu D, Khatri RG, Mustafa K, Kwak S et al (2007) Pancortin‐2 interacts with WAVE1 and Bcl‐xL in a mitochondria‐associated protein complex that mediates ischemic neuronal death. J Neurosci 27:1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dienel GA, Hertz L (2005) Astrocytic contributions to bioenergetics of cerebral ischemia. Glia 50:362–388. [DOI] [PubMed] [Google Scholar]
- 9. Dombro RS, Bender AS, Norenberg MD (2000) Association between cell swelling and glycogen content in cultured astrocytes. Int J Dev Neurosci 18:161–169. [DOI] [PubMed] [Google Scholar]
- 10. Folbergrova J, Zhao Q, Katsura K, Siesjo BK (1995) N‐tert‐butyl‐alpha‐phenylnitrone improves recovery of brain energy state in rats following transient focal ischemia. Proc Natl Acad Sci USA 92:5057–5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gabryel B, Adamczyk J, Huzarska M, Pudelko A, Trzeciak HI (2002) Aniracetam attenuates apoptosis of astrocytes subjected to simulated ischemia in vitro. Neurotoxicology 23:385–395. [DOI] [PubMed] [Google Scholar]
- 12. Garcia JH, Kalimo H, Kamijyo Y, Trump BF (1977) Cellular events during partial cerebral ischemia. I. Electron microscopy of feline cerebral cortex after middle‐cerebral‐artery occlusion. Virchows Arch B Cell Pathol 25:191–206. [DOI] [PubMed] [Google Scholar]
- 13. Giffard RG, Swanson RA (2005) Ischemia‐induced programmed cell death in astrocytes. Glia 50:299–306. [DOI] [PubMed] [Google Scholar]
- 14. Giffard RG, Monyer H, Choi DW (1990) Selective vulnerability of cultured cortical glia to injury by extracellular acidosis. Brain Res 530:138–141. [DOI] [PubMed] [Google Scholar]
- 15. Goldberg MP, Choi DW (1993) Combined oxygen and glucose deprivation in cortical cell culture: calcium‐dependent and calcium‐independent mechanisms of neuronal injury. J Neurosci 13:3510–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gursoy‐Ozdemir Y, Bolay H, Saribas O, Dalkara T (2000) Role of endothelial nitric oxide generation and peroxynitrite formation in reperfusion injury after focal cerebral ischemia. Stroke 31:1974–1980. discussion 81. [DOI] [PubMed] [Google Scholar]
- 17. Haussinger D, Lang F (1991) The mutual interaction between cell volume and cell function: a new principle of metabolic regulation. Biochem Cell Biol 69:1–4. [DOI] [PubMed] [Google Scholar]
- 18. Hentze H, Schmitz I, Latta M, Krueger A, Krammer PH, Wendel A (2002) Glutathione dependence of caspase‐8 activation at the death‐inducing signaling complex. J Biol Chem 277:5588–5595. [DOI] [PubMed] [Google Scholar]
- 19. Juurlink BH, Hertz L (1993) Ischemia‐induced death of astrocytes and neurons in primary culture: pitfalls in quantifying neuronal cell death. Brain Res Dev Brain Res 71:239–246. [DOI] [PubMed] [Google Scholar]
- 20. Kim JS, Ohshima S, Pediaditakis P, Lemasters JJ (2004) Nitric oxide: a signaling molecule against mitochondrial permeability transition‐ and pH‐dependent cell death after reperfusion. Free Radic Biol Med 37:1943–1950. [DOI] [PubMed] [Google Scholar]
- 21. Kimelberg H (2005) Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia 50:389–397. [DOI] [PubMed] [Google Scholar]
- 22. Kong J, Shepel PN, Holden CP, Mackiewicz M, Pack AI, Geiger JD (2002) Brain glycogen decreases with increased periods of wakefulness: implications for homeostatic drive to sleep. J Neurosci 22:5581–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li Y, Chopp M, Jiang N, Zhang ZG, Zaloga C (1995) Induction of DNA fragmentation after 10 to 120 minutes of focal cerebral ischemia in rats. Stroke 26:1252–1257. discussion 7–8. [DOI] [PubMed] [Google Scholar]
- 24. Lin JH, Weigel H, Cotrina ML, Liu S, Bueno E, Hansen AJ et al (1998) Gap‐junction‐mediated propagation and amplification of cell injury. Nat Neurosci 1:494–500. [DOI] [PubMed] [Google Scholar]
- 25. Lipton P (1989) Regulation of glycogen in the dentate gyrus of the in vitro guinea pig hippocampus; effect of combined deprivation of glucose and oxygen. J Neurosci Methods 28:147–154. [DOI] [PubMed] [Google Scholar]
- 26. Liu D, Smith CL, Barone FC, Ellison JA, Lysko PG, Li K, Simpson IA (1999) Astrocytic demise precedes delayed neuronal death in focal ischemic rat brain. Brain Res Mol Brain Res 68:29–41. [DOI] [PubMed] [Google Scholar]
- 27. Lukaszevicz AC, Sampaio N, Guegan C, Benchoua A, Couriaud C, Chevalier E et al (2002) High sensitivity of protoplasmic cortical astroglia to focal ischemia. J Cereb Blood Flow Metab 22: 289–298. [DOI] [PubMed] [Google Scholar]
- 28. Makar TK, Nedergaard M, Preuss A, Gelbard AS, Perumal AS, Cooper AJ (1994) Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: evidence that astrocytes play an important role in antioxidative processes in the brain. J Neurochem 62:45–53. [DOI] [PubMed] [Google Scholar]
- 29. Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ et al (1998) Activation and cleavage of caspase‐3 in apoptosis induced by experimental cerebral ischemia. J Neurosci 18:3659–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nedergaard M, Dirnagl U (2005) Role of glial cells in cerebral ischemia. Glia 50:281–286. [DOI] [PubMed] [Google Scholar]
- 31. Panickar KS, Norenberg MD (2005) Astrocytes in cerebral ischemic injury: morphological and general considerations. Glia 50:287–298. [DOI] [PubMed] [Google Scholar]
- 32. Pellerin L, Magistretti PJ (2004) Neuroscience. Let there be (NADH) light. Science 305:50–52. [DOI] [PubMed] [Google Scholar]
- 33. Petito CK, Babiak T (1982) Early proliferative changes in astrocytes in postischemic noninfarcted rat brain. Ann Neurol 11:510–518. [DOI] [PubMed] [Google Scholar]
- 34. Reichert SA, Kim‐Han JS, Dugan LL (2001) The mitochondrial permeability transition pore and nitric oxide synthase mediate early mitochondrial depolarization in astrocytes during oxygen‐glucose deprivation. J Neurosci 21:6608–6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rossi DJ, Bready JD, Mohr C (2007) Astrocyte metabolism and signaling during brain ischemia. Nature Neuroscience 10:1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Suh SW, Bergher JP, Anderson CM, Treadway JL, Fosgerau K, Swanson RA(2007) Astrocyte glycogen sustains neuronal activity during hypoglycemia: studies with the glycogen phosphorylase inhibitor CP‐316,819 ([R‐R*,S*]‐5‐chloro‐N‐[2‐hydroxy‐3‐(methoxymethylamino)‐3‐oxo‐1‐(phenylmethyl)propyl]‐1H‐indole‐2‐carboxamide). J Pharmacol Exp Ther 321:45–50. [DOI] [PubMed] [Google Scholar]
- 37. Swanson RA (1992) Physiologic coupling of glial glycogen metabolism to neuronal activity in brain. Can J Physiol Pharmacol 70(Suppl. 1):S138–S144. [DOI] [PubMed] [Google Scholar]
- 38. Takuma K, Lee E, Enomoto R, Mori K, Baba A, Matsuda T (2001) Ibudilast attenuates astrocyte apoptosis via cyclic GMP signalling pathway in an in vitro reperfusion model. Br J Pharmacol 133:841–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Takuma K, Baba A, Matsuda T (2004) Astrocyte apoptosis: implications for neuroprotection. Prog Neurobiol 72:111–127. [DOI] [PubMed] [Google Scholar]
- 40. Thoren AE, Helps SC, Nilsson M, Sims NR (2005) Astrocytic function assessed from 1‐14C‐acetate metabolism after temporary focal cerebral ischemia in rats. J Cereb Blood Flow Metab 25:440–450. [DOI] [PubMed] [Google Scholar]
- 41. Unal‐Cevik I, Kilinc M, Can A, Gursoy‐Ozdemir Y, Dalkara T (2004) Apoptotic and necrotic death mechanisms are concomitantly activated in the same cell after cerebral ischemia. Stroke 35:2189–2194. [DOI] [PubMed] [Google Scholar]
- 42. Unal‐Cevik I, Kilinc M, Gursoy‐Ozdemir Y, Gurer G, Dalkara T (2004) Loss of NeuN immunoreactivity after cerebral ischemia does not indicate neuronal cell loss: a cautionary note. Brain Res 1015:169–174. [DOI] [PubMed] [Google Scholar]