

Abstract
Diffuse astrocytoma of World Health Organization (WHO) grade II has an inherent tendency to spontaneously progress to anaplastic astrocytoma WHO grade III or secondary glioblastoma WHO grade IV. We explored the role of microRNAs (miRNAs) in glioma progression by investigating the expression profiles of 157 miRNAs in four patients with primary WHO grade II gliomas that spontaneously progressed to WHO grade IV secondary glioblastomas. Thereby, we identified 12 miRNAs (miR‐9, miR‐15a, miR‐16, miR‐17, miR‐19a, miR‐20a, miR‐21, miR‐25, miR‐28, miR‐130b, miR‐140 and miR‐210) showing increased expression, and two miRNAs (miR‐184 and miR‐328) showing reduced expression upon progression. Validation experiments on independent series of primary low‐grade and secondary high‐grade astrocytomas confirmed miR‐17 and miR‐184 as promising candidates, which were selected for functional analyses. These studies revealed miRNA‐specific influences on the viability, proliferation, apoptosis and invasive growth properties of A172 and T98G glioma cells in vitro. Using mRNA and protein expression profiling, we identified distinct sets of transcripts and proteins that were differentially expressed after inhibition of miR‐17 or overexpression of miR‐184 in glioma cells. Taken together, our results support an important role of altered miRNA expression in gliomas, and suggest miR‐17 and miR‐184 as interesting candidates contributing to glioma progression.
Keywords: astrocytoma, glioma progression, microRNA, miR‐17‐92 cluster, miR‐184
INTRODUCTION
Diffuse astrocytoma of World Health Organization (WHO) grade II inherently tends to locally recur and spontaneously progress to anaplastic astrocytoma WHO grade III and eventually secondary glioblastoma WHO grade IV (35). The molecular basis of astrocytoma progression has been investigated in previous studies by analyzing chromosomal and genetic aberrations, as well as changes in mRNA expression levels [for review, see (48)]. Collectively, these studies revealed that up to 60% of diffuse astrocytomas carry TP53 mutations, usually accompanied by loss of heterozygosity (LOH) on 17p, thus resulting in complete abrogation of wild‐type p53 function in the tumor cells (48). In addition, IDH1 or IDH2 gene mutations and gains on chromosome 7 are commonly found in these tumors 2, 48, 63. Anaplastic astrocytomas often carry additional chromosomal alterations, such as deletions on chromosomes 6, 9p, 11p, 19q and 22q. Relevant target genes on 9p are the CDKN2A, CDKN2B and p14ARF tumor suppressor genes. Moreover, CDK4 or CDK6 amplification or inactivating alterations of RB1 are detectable in a subset of anaplastic astrocytomas (25). Glioblastomas show various chromosomal and genetic alterations that lead to the inactivation of different tumor suppressor genes, as well as the aberrant activation of proto‐oncogenes (48). Interestingly, the pattern of genetic aberrations in primary glioblastomas, which present de novo with a short clinical history, is distinct from that of secondary glioblastomas, which develop by progression from pre‐existing lower grade gliomas (43). For example, primary glioblastomas show frequent EGFR amplification and PTEN mutation, while secondary glioblastomas, similar to diffuse and anaplastic astrocytomas, are characterized by frequent mutations in the TP53 and IDH1 genes 2, 43, 63. Notably, most of the molecular alterations at the gene and transcript levels that have been detected in glioblastomas can be assigned to certain functional pathways, in particular the phosphoinositol 3 kinase/Akt, mitogen‐activated kinase, p53 and pRb signaling cascades 7, 48.
MicroRNAs (miRNAs) are approximately 22‐nucleotide‐long, single‐stranded, noncoding RNA molecules that posttranscriptionally regulate gene expression by translational inhibition or destabilization of mRNA transcripts (13). A specific miRNA may simultaneously regulate multiple targets, thereby enabling complex changes in protein expression profiles. Furthermore, a single target can be regulated by multiple miRNAs, and upstream regulation of a given miRNA can involve multiple regulators at different steps of miRNA biogenesis. Thus, miRNAs take part in complex regulatory networks that may influence almost every cellular process (13). We explored the role of miRNAs in the malignant progression of human gliomas by comparing miRNA expression profiles in primary low‐grade gliomas and secondary high‐grade gliomas from individual patients. Two glioma progression‐associated miRNAs (miR‐17 and miR‐184) were selected for further functional and molecular characterization using targeted inhibition of miR‐17 or overexpression of miR‐184 in cultured glioma cells.
MATERIALS AND METHODS
Patient samples and extraction of nucleic acids
All tumors were selected from the collection of frozen brain tumor tissue specimens at the Department of Neuropathology, Heinrich‐Heine‐University, Düsseldorf, Germany, and investigated in accordance with protocols approved by the institutional review board (study number 2767). The tumors were classified according to the WHO classification of tumors of the central nervous system (35). Parts of each tumor were snap‐frozen immediately after operation, and stored at −80°C. Only specimens with a histologically estimated tumor cell content of 80% or more were used for the molecular analyses. MiRNA expression profiles were determined in primary low‐grade and recurrent high‐grade tumor pairs derived from four independent patients. These patients had been originally operated on WHO grade II gliomas (three diffuse astrocytomas and one astrocytoma‐predominant oligoastrocytoma), and then developed spontaneous recurrences that were histologically classified as glioblastoma (WHO grade IV). None of the patients had been treated by radio‐ or chemotherapy in between both operations. For validation purposes, we investigated tumors from three additional patients with primary WHO grade II gliomas (two diffuse astrocytomas and one astrocytoma‐predominant oligoastrocytoma) and recurrent high‐grade gliomas (two anaplastic astrocytomas and one astrocytoma‐predominant anaplastic oligoastrocytoma, all corresponding to WHO grade III). All seven patients were included in our previous studies addressing genetic alterations and differential mRNA expression associated with astrocytoma progression 56, 57. An additional validation step involved the analysis of 14 diffuse astrocytomas and 13 secondary glioblastomas from 27 independent patients. DNA and RNA were extracted from unfixed frozen tumor samples by ultracentrifugation over cesium chloride as described (57). In the initial screening experiments, we used two commercially available RNA samples obtained from adult human brain tissue (Clontech, Mountain View, CA; Stratagene, Cedar Creek, TX) as non‐neoplastic reference samples. For the second validation step, these control samples were supplemented by an additional set of nine non‐neoplastic brain RNAs obtained from different commercial sources (Ambion, Austin, TX; Biochain, Hayward, CA). Commercially available universal human RNA (Stratagene) was used to calibrate the real‐time polymerase chain reaction (PCR) experiments.
MiRNA sequences and miRNA target prediction
MiRNA sequences and potential target genes were retrieved from the Sanger Institute mirBase registry and mirBase target database release 10.1 (http://microrna.sanger.ac.uk/index.shtml).
Real‐time reverse transcription PCR (RT–PCR) analyses
Reverse transcription of mature miRNAs was carried out with stem‐loop primers specific for each investigated miRNA. Real‐time PCR was performed on an ABI PRISM® 5700 system using the Applied Biosystems Early Access miRNA Kit with dye‐labeled TaqMan® probes to monitor amplification according to the manufacturer's protocol (Applied Biosystems, Foster City, CA). Fluorescent data were converted into cycle threshold measurements by the SDS system software, and exported to Microsoft Excel. Fold expression changes relative to universal human RNA were calculated with the 2−ΔΔCT method. As recommended by the manufacturer, the expression level of let‐7a was used as a reference, which showed robust expression in our samples, as well as in glioma samples of a previous study (52). The miRNA sequences that could be detected using the Early Access miRNA Kit are listed in Supporting Information Table S1. Sequences of all primers used for miRNA amplification are listed at http://www.appliedbiosystems.com. The miRNAs were regarded as showing progression‐associated increased expression if a more than two‐fold higher expression level was detected in the secondary glioblastoma as compared to the respective primary low‐grade glioma with a raised expression level in the secondary glioblastoma sample as compared to a non‐neoplastic brain tissue. Progression‐associated decreased expression was assumed if the recurrent tumor demonstrated a more than two‐fold lower expression level as compared to the respective primary tumor with a lowered expression level in the secondary glioblastoma sample as compared to a non‐neoplastic brain tissue. In the validation experiments performed for selected miRNAs (miR‐16, miR‐17, miR‐19a, miR‐20a, miR‐140, miR‐184), amplification of let‐7a cDNA was measured by incorporation of SYBR green fluorescent dye (Applied Biosystems) into the double‐stranded DNA, while the individual target miRNAs were detected by TaqMan technology. The independent validation series of 14 diffuse astrocytomas, 13 secondary glioblastomas and nine non‐neoplastic brain tissue samples was investigated for expression of the six selected candidate miRNAs using TaqMan‐based assays. For expression analysis of potential miRNA target transcripts, 1 µg of total RNA of each tumor was reverse transcribed into cDNA using SuperScript® Reverse Transcriptase (Invitrogen, Carlsbad, CA) and random primers. The mRNA expression levels were detected by real‐time PCR on the StepOnePlus™ system (Applied Biosystems) using incorporation of SYBR green fluorescent dye into the double‐stranded PCR products. The expression level of each transcript was normalized to the expression level of ARF1 transcripts (ADP‐ribosylation factor 1, NCBI GenBank accession no. M36340; for primer sequences, see Supporting Information Table S2).
Duplex PCR analyses
The miR‐184 locus was analyzed for homozygous deletion, and the miR‐17 locus for gene amplification by duplex PCR assays (for primer sequences, see Supporting Information Table S2). The PCR products were separated by electrophoresis in 3% agarose gels, and the ethidium bromide‐stained bands were recorded with the Gel Doc™ 1000 system (Bio‐Rad, Hercules, CA). Quantitative analysis of the signals obtained for each miRNA locus and a reference gene [adenine phosphoribosyltransferase (APRT), NCBI GenBank accession no. AC092384] was performed with the Molecular Analyst software (Bio‐Rad). Increases in the target gene/reference gene ratio of more than three‐fold of the ratio obtained for constitutional DNA were considered as evidence for gene amplification. Reduction in the target gene/reference gene ratio below 0.3‐fold was considered as evidence for homozygous deletion.
Transfection of cultured glioma cells
The glioma cell lines A172 and T98G were obtained from American Type Culture Collection (Manassas, VA), and grown as monolayer cultures in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat‐inactivated fetal bovine serum and penicillin/streptomycin (Invitrogen/GIBCO, Carlsbad, CA) in 5% CO2 humidified incubator at 37°C. Then, miR‐184 was overexpressed in glioma cells by transient transfection of a 25 nM miR‐184 pre‐miR miRNA precursor (Ambion) with siPORT™ NeoFX™ transfection reagent (Ambion) according to the manufacturer's protocol. MiR‐17 was silenced by transfection of 50 nM anti‐miR miRNA inhibitors (Ambion). To normalize for side‐effects not caused by specific miRNA overexpression or inhibition, cells were transfected with commercially available negative controls (pre‐miRNA negative control, pre‐NC, anti‐miRNA negative control, anti‐NC; Ambion).
In vitro assays for functional analyses
To determine the influence of miRNA modulation on cell viability, a standard colorimetric assay was used (Sigma, Seelze, Germany). Seventy‐two hours after transfection, 10 µL of 5 mg/mL 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide was added to each well of a 96‐well cell culture plate. After incubation at 37°C for 4 h, the reaction was stopped by solubilization with 150 µL dimethylsulfoxide. Absorbance at a wavelength of 570 nm was measured using an enzyme‐linked immunosorbent assay plate reader (Tecan, Crailsheim, Germany) to determine the amount of formazan that was produced. Absorbance at a wavelength of 630 nm was subtracted to normalize for different amounts of cell debris.
The proliferation rate of cells transfected with pre‐miRNA or anti‐miRNA was compared to cells transfected with the respective negative controls using a commercially available BrdU incorporation assay (Roche, Mannheim, Germany). BrdU labeling solution was added 48 h after transfection, and cells were fixed after 24 h of incubation at 37°C.
Apoptotic activity in transfected and control cells was determined with a fluorometric caspase‐3/7 assay (Promega, Mannheim, Germany). Caspase substrate was added 72 h after transfection, and fluorometric measurements were performed after an incubation time of 2 h at room temperature.
Invasive growth properties of transfected and control cells were quantified with a transwell assay (BD Biosciences, San Jose, CA). BD BioCoat™ Matrigel™ invasion chambers 8.0 µm, and BD BioCoat 8.0 µm control inserts were rehydrated in a 24‐well companion plate with serum‐free DMEM at 37°C for 2 h. Cells were mechanically detached from 10 cm dishes 48 h after transfection, and suspended in serum‐free DMEM at a concentration of 70 000 cells/mL. Then, 750 µL of medium containing 10% fetal bovine serum as chemoattractant was pipetted into each well of an additional 24‐well plate. Inserts were transferred to this plate, and 500 µL of cell suspension was dispensed in each well followed by incubation at 37°C for 24 h. Afterwards, cells that had not migrated through the membrane (and the Matrigel coating) were removed with cotton swabs. The inserts were put into methanol for 1 minute to fix the migrated cells at the other side of the membrane. Membranes were stained with hematoxylin and eosin, sliced out of the inserts and fixed on a glass slide. A representative sector was counted under the microscope at 100‐fold magnification. To normalize for different proliferation and unspecific migration through the pores of the membrane, the number of cells that migrated through each Matrigel‐coated membrane was divided by the average number of cells that migrated through control membranes without Matrigel coating.
Each experimental group in the individual functional tests consisted of six replicates. Two‐sided Student's t‐tests were applied to compare results between transfected and control cells. P values of less than 0.05 were considered to indicate significant differences. The results of each in vitro experiment were converted into arbitrary units (a.u.) for better comparability of different experiments, setting the average measurement of the control cells at 100 a.u.
Microarray‐based expression profiling
A172 and T98G glioma cells were washed twice with phosphate‐buffered saline (PBS) 72 h after transfection, and then lysed with Trizol reagent (Invitrogen) to extract the RNA according to the manufacturer's protocol. Expression profiles of specifically transfected vs. control‐transfected cells were determined by hybridization to Affymetrix Human Genome U133 Plus 2.0 Array (Affymetrix, Santa Clara, CA). We used a global scaling strategy that sets the average signal intensity of the array to a default target signal of 500, and performed a comparative analysis of the specifically transfected samples vs. the control samples. Changes with P values <0.05 were considered statistically significant. Genes with a signal log ratio (transfected/negative control) >1 or <−1 were regarded as being differentially regulated.
Proteomic analyses using two‐dimensional difference gel electrophoresis (2D‐DIGE) and mass spectrometry (MS)
A172 and T98G glioma cells were put on ice 72 h after transfection, washed with cold PBS twice and then suspended mechanically in 1 mL PBS, followed by centrifugation at 1000 × g for 5 minutes. After removal of the supernatant, the cell pellet was snap‐frozen in liquid nitrogen and stored at −80°C. For cell lysis, 100 µg pellet was mixed with 148 µL of lysis buffer (Tris–HCl 30 mmol/L, thiourea 2 mol/L, urea 7 mol/L, CHAPS 4%, pH 8.5). Cell lysis was completed by subsequent sonification (6 × 10 s pulses on ice). Centrifugation at 12 000 × g for 15 minutes was performed to remove cell debris. The protein samples with miRNA overexpression or inhibition of miRNA function were labeled with Cy3 minimal CyDye™ (GE Healthcare, Munich, Germany), and the control‐transfected samples were labeled with Cy5 minimal CyDye (GE Healthcare) according to the manufacturer's instructions. An internal standard composed of equal amounts of all samples of one cell line was then labeled with Cy2 minimal CyDye (GE Healthcare). Two‐dimensional electrophoresis analysis was performed using carrier ampholyte‐based isoelectric focusing as described elsewhere (29). After gel scanning, the protein patterns were differentially analyzed using DeCyder™ (GE Healthcare) as described elsewhere (53). For protein identification by matrix‐assisted laser desorption/ionization (MALDI)–MS, silver post‐staining was performed using MS‐compatible protocol (40). In‐gel digestion of proteins was performed with trypsin, and obtained peptides were subjected to MALDI–MS analysis and identified using the Mascot algorithm for searching the IPI protein database (http://www.ebi.ac.uk).
SDS–polyacrylamide gel electrophoresis (PAGE) and Western blot analysis
For protein extraction from cell cultures, cells were put on ice and washed with cold PBS twice. Then, 400 µL lysis buffer [50 mmol/L Tris–Cl (pH 8.0), 150 mmol/L NaCl, 0.5% Triton X‐100, 0.5% deoxycholate] was added to each 10 cm dish. Cell lysates were centrifuged to remove cell debris. Supernatants were separated on 10% Tris–glycin SDS polyacrylamide gels. For Western blot analysis of tumor tissues, we used the protein fraction obtained by ultracentrifugation over cesium chloride. Excess of guanidine hydrochloride salt load was removed by replacement of solvent with sample buffer using ultrafiltration applying Microcon™ (Millipore, Schwalbach, Germany; 3 kDa cutoff). The proteins were then denatured and separated by gel electrophoresis on precasted NuPAGE™ Novex 4%–12% Bis‐Tris Midi gels (Invitrogen, Karlsruhe, Germany). Separated proteins were transferred to Hybond‐P™ PVDF membranes (Amersham Biosciences, Piscataway, NJ) using a Trans Blot® Cell Blot module (Bio‐Rad). The membranes were blocked with 5% bovine serum albumine (BSA) in PBS‐T (0.05% Tween 20 in PBS), and probed with antibodies against Akt2 (1:100, Cell Signaling, Danvers, MA: #2962) and α‐tubulin (1:100, Sigma, Taufkirchen, Germany: T9026) or β‐actin (1:10 000, Sigma‐Aldrich, St. Louis, MO: A5541). Primary antibody binding was detected by anti‐mouse or anti‐rabbit antibodies linked to horseradish peroxidase followed by incubation with Immobilon® Western HRP Substrate luminol reagent and peroxide solution (Millipore, Billerica, MA). Chemiluminescence was recorded with the LAS‐3000 mini system (Fujifilm Life Science, Stanford, CT).
RESULTS
Identification of miRNAs that are differentially expressed between diffuse astrocytoma and secondary glioblastoma
We determined the expression levels of 157 miRNAs in four primary gliomas of WHO grade II and their corresponding recurrent secondary glioblastomas of WHO grade IV by stem‐loop real‐time RT–PCR (Supporting Information Table S1 provides a list of the 157 miRNAs that were analyzed). Two miRNAs (miR‐184 and miR‐328) exhibited a progression‐associated down‐regulation, that is, a more than two‐fold lower expression in the secondary glioblastoma as compared to the corresponding primary diffuse astrocytoma, in the majority of patients, that is, two of three or three of four investigated patients, respectively (Table 1). Twelve miRNAs (miR‐9, miR‐15a, miR‐16, miR‐17, miR‐19a, miR‐20a, miR‐21, miR‐25, miR‐28, miR‐130b, miR‐140 and miR‐210) showed a progression‐associated up‐regulation in the majority of investigated patients (Table 1).
Table 1.
MicroRNAs (miRNAs) with progression‐associated differential expression in four patients with primary low‐grade glioma that recurred as secondary glioblastoma. Expression levels of 157 miRNAs were determined. MiRNAs were regarded as showing progression‐associated up‐regulation if they showed a fold change (recurrent/primary tumor) >2 with a raised expression level in the secondary glioblastoma sample. Progression‐associated down‐regulation was defined in an analogous manner as a fold change (recurrent/primary tumor) <0.5 with a lowered expression level in the secondary glioblastoma sample. Only miRNAs with progression‐associated expression in the majority of patients are listed. Patient numbers 1–4 encode the individual patient; case numbers encode primary and recurrent gliomas.
| Fold change (recurrent/primary tumor) | |||||
|---|---|---|---|---|---|
| Patient no. | 1 | 2 | 3 | 4 | |
| Case no. (primary tumor) | A72D | A201D | A128D | OA23D | |
| Case no. (recurrent tumor) | GB239D | GB240D | GB119D | GB175D | |
| Progression‐associated up‐regulation | miR‐9 | 3.16 | 5.03 | 3.75 | 3.19 |
| miR‐15a | 2.50 | 4.50 | 1.36 | 7.81 | |
| miR‐16 | 3.72 | 4.82 | 2.69 | 3.24 | |
| miR‐17 | 3.89 | 12.64 | 7.52 | n.d. | |
| miR‐19a | 1.46 | 2.85 | 3.88 | 16.28 | |
| miR‐20a | 1.27 | 7.06 | 5.21 | 5.43 | |
| miR‐21 | 0.75 | 5.98 | 3.64 | n.d. | |
| miR‐25 | 0.72 | 6.13 | 4.44 | 6.66 | |
| miR‐28 | 0.75 | 4.47 | 7.46 | 4.58 | |
| miR‐130b | 2.94 | 5.39 | 7.04 | 11.88 | |
| mir‐140 | 3.56 | 1.54 | 5.72 | 4.21 | |
| miR‐210 | n.d. | 1.49 | 17.45 | 6.89 | |
| Progression‐associated down‐regulation | miR‐184 | 0.28 | 0.17 | 0.56 | 0.02 |
| miR‐328 | 0.13 | 8.06 | 0.41 | 0.30 | |
Validation of differentially expressed miRNAs
From the set of 14 miRNAs demonstrating progression‐associated differential expression, we selected miR‐16, miR‐17, miR‐19a, miR‐20a, miR‐140 and miR‐184 for validation experiments. We did not further analyze miRNAs that had been investigated in gliomas before. In addition, we restricted the number of candidate miRNAs for validation experiments taking into account the fraction of investigated patients demonstrating progression‐associated expression, reported differential expression in other tumor entities, genomic organization and predicted targets of each miRNA. By targeted real‐time stem‐loop RT–PCR, we determined expression levels in the four original patients' tumor samples and in tumor samples from three additional patients whose gliomas progressed from WHO grade II to WHO grade III on recurrence. These analyses confirmed progression‐associated expression of all selected miRNAs in the majority of the four original patients. Furthermore, miR‐17, miR‐20a and miR‐184 exhibited progression‐associated differential expression in at least two of the three additional patients (Table 2).
Table 2.
Results of validation experiments performed for six selected microRNAs (miRNAs) in the original four patients, and three additional patients with primary low‐grade and recurrent anaplastic astrocytoma. Expression of each miRNA was determined by real‐time reverse transcription polymerase chain reaction using a modified experimental setup as compared to the screening experiments (see Materials and Methods).
| Original patients (n = 4) recurrent/primary tumor | Additional patients (n = 3) recurrent/primary tumor | ||
|---|---|---|---|
| Screening | Validation | ||
| miR‐16 | 4/4 Up | 3/4 Up | 1/3 Up |
| miR‐17 | 3/3 Up | 3/4 Up | 3/3 Up |
| miR‐19a | 3/4 Up | 2/3 Up | 0/2 Up |
| miR‐20a | 3/4 Up | 3/3 Up | 2/3 Up |
| miR‐140 | 3/4 Up | 3/4 Up | 1/3 Up |
| miR‐184 | 3/4 Down | 3/4 Down | 3/3 Down |
A second validation step based on the analysis of an independent series of 14 diffuse astrocytomas and 13 secondary glioblastomas (Figure 1) demonstrated a significantly higher expression of miR‐17 in secondary glioblastomas as compared to diffuse astrocytomas. Median expression levels of miR‐16, miR‐19a, miR‐20a and miR‐140 were also increased in secondary glioblastomas as compared to low‐grade gliomas; however, for miR‐16 and miR‐19a, the differences were not statistically significant (Figure 1). Expression levels of miR‐16, miR‐19a, miR‐20a and miR‐140 were significantly lower in the tumors as compared to the non‐neoplastic brain tissues. The median expression level of miR‐184 was significantly decreased in secondary glioblastomas relative to diffuse astrocytomas and non‐neoplastic brain tissue, respectively (Figure 1). Based on these results, we decided to select miR‐17 and miR‐184 as candidates for further analyses.
Figure 1.
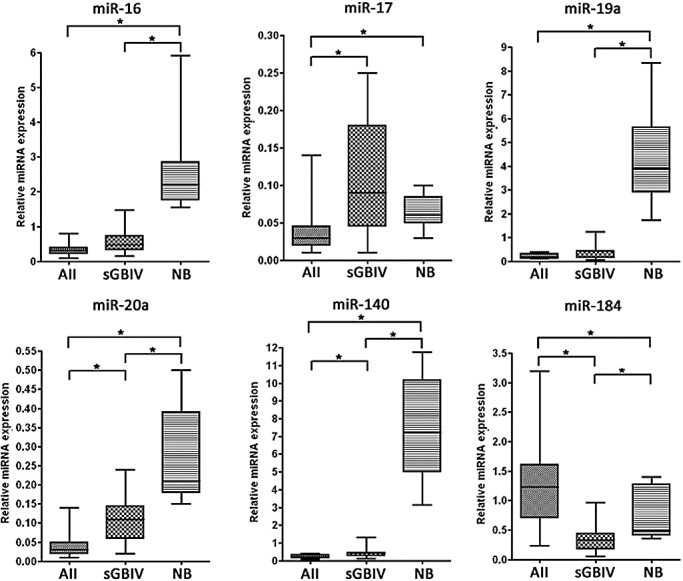
Results of validation experiments performed for the six selected microRNA (miRNA) candidates in an independent series of 14 diffuse astrocytomas (AII), 13 secondary glioblastomas (sGBIV) and nine non‐neoplastic brain tissue samples (NB). Expression levels of the individual miRNAs were determined by using TaqMan‐based polymerase chain reaction assays. Box plots are depicted indicating median, lower and upper quartile, as well as sample maximum and sample minimum of normalized expression values. Asterisks indicate significant expression differences (P < 0.05).
Copy number analyses of the miR‐17 and miR‐184 loci
Duplex PCR assays were performed to investigate aberrations in the gene copy number at the miR‐17 and miR‐184 loci. However, we neither detected homozygous deletion of the miR‐184 locus nor amplification of the miR‐17 locus in any of the seven primary or recurrent glioma samples from the individual patients with glioma progression (data not shown).
Functional effects of miR‐184 overexpression and miR‐17 inhibition in human glioma cells
To investigate the biological effects of progression‐associated miRNA expression, we overexpressed miR‐184 in A172 and T98G glioma cells by transfection of miR‐184 precursors. In addition, we inhibited miR‐17 in these glioma cell lines by transfection of complementary miRNA inhibitors. The glioma cell lines were efficiently transfectable with nearly 100% of cells showing an uptake of fluorescent‐labeled oligonucleotides. The effect of transfection on the respective miRNA expression levels was determined by real‐time stem‐loop RT–PCR. The miR‐184 expression level was markedly increased in cells transfected with miRNA precursors as compared to control‐transfected cells. The expression level of miR‐17 was effectively decreased in glioma cells transfected with the respective miRNA inhibitors as compared to glioma cells transfected with control oligonucleotides (data not shown).
Overexpression of miR‐184 in A172 and T98G glioma cells significantly (P < 0.05) decreased cell viability (Figure 2A) and proliferation (Figure 2C). In T98G cells, miR‐184 additionally reduced invasiveness in Matrigel assays (Figure 2B). Overexpression of miR‐184 increased apoptotic activity in A172 cells, but reduced apoptotic activity in T98G cells (Figure 2D). Inhibition of miR‐17 significantly reduced cell viability of A172 and T98G cells (Figure 2E), and increased apoptotic activity in T98G cells (Figure 2F). Invasiveness or proliferation was not significantly changed in cells with inhibited miR‐17 function.
Figure 2.
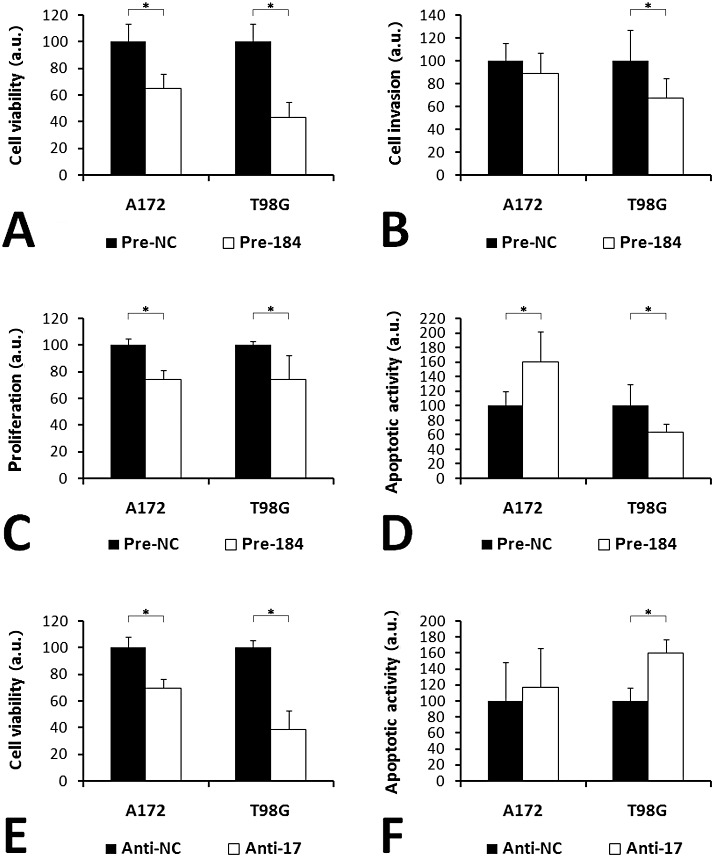
In vitro effects of miR‐184 overexpression (A–D) and miR‐17 inhibition (E,F) in glioma cells. (A,E) Cell viability was determined using an MTT assay. (B) Cell invasion was quantified using a transwell assay with Matrigel‐coated membranes. (C) Proliferation was measured using a BrdU assay. (D,F) For the comparison of apoptotic activity, a fluorometric caspase‐3/‐7 assay was used. The results of the in vitro assays were converted into arbitrary units (a.u.), setting the average result in the corresponding control cells as 100 a.u. Error bars show standard deviations; asterisks indicate significant differences (P < 0.05). Pre‐NC and Anti‐NC, cells transfected with negative controls; Pre‐184, cells transfected with miR‐184 precursors; Anti‐17, cells transfected with miR‐17 inhibitors.
Protein and mRNA expression profiling of glioma cells after miR‐184 overexpression or miR‐17 inhibition
We determined mRNA and protein expression profiles of glioma cells after miR‐184 overexpression and miR‐17 inhibition in order to identify transcripts and proteins that are regulated—either directly or indirectly—following specific miRNA inhibition or overexpression of these miRNAs. Proteomic analysis revealed 42 distinct protein spots that were regulated in A172 cells, and 74 distinct protein spots that were regulated in T98G cells following overexpression of miR‐184 (P < 0.05). MALDI–time‐of‐flight (TOF)/TOF MS identified 11 and 14 non‐redundant proteins being down‐regulated in A172 and T98G cells, respectively (Supporting Information Table S4, and Figures S1 and S2). In T98G cells, the predicted miR‐184 target nucleophosmin 1 (Npm1, OMIM 16040) was down‐regulated (average fold change = −1.6) (Figure 3A). The proteins Hnrpk and Hnrpm were up‐regulated in both cell lines. Affymetrix chip profiling revealed 1380 differentially regulated mRNAs in A172 cells, and 784 differentially regulated mRNAs in T98G cells transfected with miR‐184 precursors as compared to control cells (signal log ratio > 1 or < −1, P < 0.05). A total of 17 non‐redundant transcripts corresponding to predicted targets of miR‐184 showed decreased expression levels in both cell lines (Supporting Information Table S3). Real‐time RT–PCR analysis confirmed the differential mRNA expression of AKR1C3, AKT2, CDC25A, CTBP1, S100A16, MAZ, NRN1 and SH3GL1 when comparing miR‐184 precursor‐transfected vs. control‐transfected cells. Among these candidates, Akt2 was selected for additional Western blot analysis, which confirmed decreased expression of Akt2 in A172 and T98G after transfection of miR‐184 precursors (Figure 4). In addition, Western blot analysis of an independent series of five diffuse astrocytomas and five secondary glioblastomas revealed increased Akt2 protein expression in the secondary glioblastomas (Supporting Information Figure S5).
Figure 3.
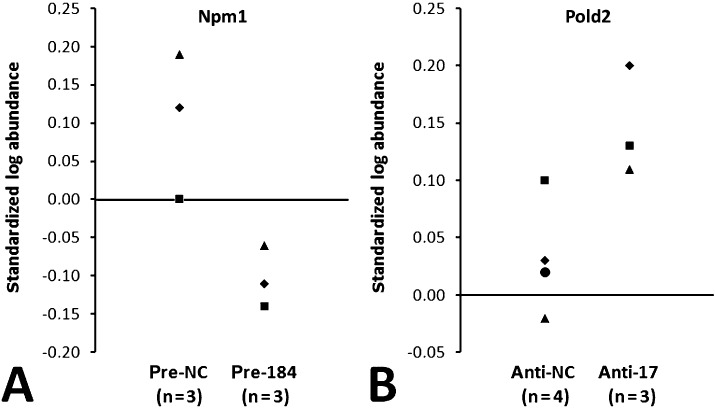
Proteome changes in T98G glioma cells following miR‐184 overexpression (A) or miR‐17 inhibition (B). (A) Expression of nucleophosmin 1 (Npm1) was found to be significantly (P < 0.05) down‐regulated in T98G cells transfected with miR‐184 precursors (Pre‐184) as compared to control‐transfected cells (Pre‐NC). (B) Protein expression of Pold2 was significantly (P < 0.05) up‐regulated in T98G cells transfected with miR‐17 inhibitors (Anti‐17) as compared to control‐transfected cells (Anti‐NC). Npm1 is a predicted target of miR‐184, while Pold2 is a predicted target of miR‐17 according to the Sanger Institute mirBase target database (http://microrna.sanger.ac.uk/index.shtml).
Figure 4.
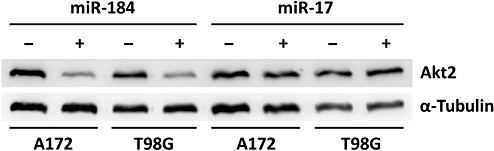
Down‐regulation of Akt2 in glioma cells overexpressing miR‐184. A172 and T98G cells were transfected with miR‐184 precursors (+) or corresponding controls (−). Western blots were probed with antibodies against Akt2 and α‐tubulin as a loading control. As a further control, glioma cells transfected with inhibitors of miR‐17 (−) or respective controls (+) were probed. Note markedly reduced Akt2 protein levels in A172 and T98G cells transfected with miR‐184 precursors, but not with miR‐17 inhibitors or controls.
Proteomic analysis revealed that inhibition of miR‐17 resulted in differential expression of 39 protein spots in A172 cells, and 83 protein spots in T98G cells (P < 0.05). MALDI–TOF/TOF MS identified 16 non‐redundant proteins up‐regulated in A172 cells, and 33 non‐redundant proteins up‐regulated in T98G cells (Supporting Information Table S5, and Figures S3 and S4). In T98G cells, the predicted miR‐17 target Pold2 showed elevated expression (average fold change = 1.3) (Figure 3B). Three non‐redundant proteins (Hspa1a/b, Eno1, Cct5) without predicted miR‐17 binding sites were up‐regulated in both cell lines. Expression profiling at the transcript level identified 475 regulated mRNAs in A172 cells, and 125 regulated mRNAs in T98G cells transfected with the anti‐17 miRNA inhibitors as compared to control‐transfected cells (signal log ratio > 1 or < −1, P < 0.05). However, none of these transcripts corresponded to a predicted target mRNA of miR‐17, and there was no overlap of regulated transcripts between the two cell lines.
DISCUSSION
Recent studies indicate that various miRNAs play important roles in human cancer [for review, see Lee and Dutta (33)]. In general, miRNAs may act as tumor suppressors by down‐regulating the expression of tumor‐promoting genes, or may have oncogenic functions by inhibiting the expression of tumor suppressor genes. In primary brain tumors, several miRNAs have been identified as being aberrantly expressed in gliomas, in particular glioblastomas. For example, miR‐221 and miR‐21 were found to be up‐regulated, while miR‐128, miR‐181a, miR‐181b and miR‐181c were down‐regulated in glioblastomas 8, 10. Furthermore, miR‐181a and miR‐181b showed lower expression in high‐grade as compared to low‐grade gliomas (51). Both miRNAs induced apoptosis, inhibited anchorage‐independent growth and reduced invasive growth of glioma cells (51). Overexpression of miR‐128 also reduced glioma cell proliferation (18). Additional miRNA species implicated in glioma pathogenesis include miR‐7 (28), miR‐124 and miR‐137 (52), miR‐221/222 (17), miR‐296 (61) and miR‐451 (16). Here, we report that miR‐184 and miR‐328 are down‐regulated, whereas miR‐9, miR‐15a, miR‐16, miR‐17, miR‐19a, miR‐20a, miR‐21, miR‐25, miR‐28, miR‐130b, miR‐140 and miR‐210 are up‐regulated during glioma progression in individual patients.
In line with our data, miR‐9 was reported as being up‐regulated in oligodendroglioma (39) and glioblastoma (10), thus suggesting a tumor‐promoting function in gliomas. However, studies on epithelial cancers revealed epigenetic silencing and/or down‐regulation of miR‐9‐1 in advanced breast and ovarian carcinomas 32, 34, which would fit to a tumor‐suppressive function. Similarly, the clustered miRNAs miR‐15a and miR‐16‐1 were reported to be deleted and/or down‐regulated in B‐CLL 4, 15. On the other hand, serous ovarian carcinomas showed increased miR‐16 expression relative to normal ovarian tissue (38), which is in accordance with our finding of miR‐15a and miR‐16 up‐regulation in high‐grade gliomas. We also detected higher expression of miR‐140 in secondary glioblastomas, while studies on ovarian cancer revealed reduced miR‐140 levels as compared to normal ovarian tissue (26). Taken together, these findings indicate tissue‐specific differences in the function of certain miRNAs, thus emphasizing the complexity of miRNA‐associated regulatory networks and the importance of validating miRNA function in the appropriate cell systems.
Our demonstration of a progression‐associated up‐regulation of miR‐21 is concordant with studies reporting on miR‐21 overexpression in glioblastomas 8, 10. Functional analyses of miR‐21 revealed oncogenic properties caused by inhibition of the p53, transforming growth factor‐β and mitochondrial apoptosis pathways (44). In addition, targeted inhibition of miR‐21 sensitized glioma cells to cytotoxic agents, pointing to a potential novel approach for targeted glioma therapy (11).
MiR‐25 is located on 7q22.1, a region frequently gained in diffuse astrocytomas (48). The observed up‐regulation in secondary glioblastomas is in line with a report on miR‐25 overexpression in glioblastomas (10). To analyze the functional significance of miR‐25 up‐regulation in glioma progression, however, a detailed analysis of the corresponding miRNA‐family, that is, miR‐25, miR‐92a and miR‐92b, would be necessary. Similarly, miR‐28 and miR‐130b, both demonstrating progression‐associated up‐regulation in our patients, each belong to distinct families of closely related miRNAs, that is, miR‐28 and miR‐151, as well as miR‐130a, miR‐130b, miR‐301a and miR‐301b, respectively. Hence, it remains to be investigated whether the up‐regulation of a single family member has significant biological effects or may be compensated for by loss of function of other family members. Nevertheless, in line with our findings, miR‐28 showed increased expression in renal cell carcinomas as compared to normal kidney tissue (19).
The up‐regulation of miR‐210 in malignant gliomas is interesting because miR‐210 was reported to be induced by hypoxia (6). In endothelial cells, inhibition of miR‐210 reduced cell growth, induced apoptosis and inhibited the formation of capillary‐like structures stimulated by hypoxia (12). Thus, elevated miR‐210 expression in glioblastomas may be triggered by the regional hypoxia in these tumors, a hypothesis that needs to be investigated in further studies.
MiR‐328 displayed a progression‐associated down‐regulation in our patients. It maps to 16q22.1, a region frequently lost in pediatric high‐grade astrocytomas (47). Functional studies implicated miR‐328 in Alzheimer's disease by regulating the β‐amyloid precursor protein‐converting enzyme (3). However, data on potential roles of this particular miRNA in cancer have not been reported to date. Own preliminary data suggest that miR‐328 expression levels decrease with increasing malignancy in astrocytic gliomas, but the median expression level in secondary glioblastomas is still higher than in non‐neoplastic brain tissue (data not shown).
MiR‐184 is a putative suppressor of glioma progression
The expression of miR‐184 (15q25.1) was down‐regulated during progression from low‐grade to anaplastic glioma and secondary glioblastoma in our patients. Furthermore, overexpression of miR‐184 significantly decreased cell viability and proliferation of glioma cells, suggesting a tumor‐suppressive role of this miRNA in gliomas. Interestingly, miR‐184 increased apoptotic activity in A172 cells, but reduced apoptotic activity and invasive growth in T98G cells. This divergent influence on apoptotic rates in T98G and A172 cells is probably related to the different genetic background of these cell lines, with the TP53 gene being mutant in T98G, but wild type in A172 cells (27). Cell type‐specific effects are also indicated by the fact that overexpression of miR‐184 in neuroblastoma cell lines decreased proliferation and induced apoptosis (9), while inhibition of miR‐184 in squamous cell carcinomas decreased proliferation (60). In neuroblastoma, inhibitory effects of N‐myc on miR‐184 transcription have been reported (9). More recently, epigenetic regulation of miR‐184 in the mouse brain has been analyzed and suggested a paternal‐specific expression of this particular miRNA (41). Therefore, the observed progression‐associated down‐regulation of miR‐184 may be caused by diverse molecular alterations.
With respect to miR‐184 targets, possible regulation of c‐Myc (60), Frizzeled 4 (50) and miR‐205 (65) has been reported. We identified Npm1 as being significantly down‐regulated in T98G cells after transfection with miR‐184 precursors. Npm1 is a ubiquitously expressed nucleolar phosphoprotein that has been implicated in the pathogenesis of epithelial and hematopoietic malignancies [for review, see Grisendi et al (20)]. Furthermore, increased NPM1 mRNA levels were found in glioblastomas (64), and siRNA‐mediated inhibition of Npm1 reduced the proliferation of neural stem cells (46). Thus, miR‐184‐mediated direct or indirect down‐regulation of Npm1 may contribute to its anti‐proliferative effects in glioma cells; however, this hypothesis warrants additional experimental validation.
Affymetrix chip‐based expression profiling identified 17 predicted targets of miR‐184 whose mRNA levels were at least two‐fold reduced in A172 and T98G cells after transfection of miR‐184 precursors. We selected nine of these candidate targets (AKR1C3, CTBP1, S100A16, CENTG1, MAZ, NRN1, SH3GL1, CDC25A, AKT2) and validated their down‐regulation at the mRNA level by real‐time RT–PCR. In addition, we showed that Akt2 protein expression is decreased in miR‐184 overexpressing glioma cells, and increased in secondary glioblastomas relative to diffuse astrocytomas. Thus, down‐regulation of miR‐184 in high‐grade gliomas may directly or indirectly lead to increased Akt2 protein levels and thereby contribute to an aberrant activation of the phosphoinositol 3‐kinase/Akt pathway commonly seen in these tumors (30).
Increased expression of miRNAs encoded by the miR‐17‐92 cluster may promote astrocytoma progression
The miR‐17‐92 cluster at 13q31.3, comprising miR‐17, miR‐18a, miR‐19a, miR‐20a, miR‐19b‐1 and miR‐92a‐1, is transcribed as a polycistron. Screening for the expression of miR‐17, miR‐19a, miR‐20a and miR‐92a in our series revealed progression‐associated up‐regulation of miR‐17, miR‐19a and miR‐20a. MiR‐92a was up‐regulated in the recurrent glioblastomas of two of our patients, but down‐regulated in one patient. Such differences in the expression levels of individual members of the miR‐17‐92 cluster may be explained by independent posttranscriptional regulation or alterations of the paralogous miR‐106a‐92 cluster. In line with our findings, miRNAs of the miR‐17‐92 cluster were found to be overexpressed in advanced neuroblastomas with MYCN amplification (49), in hematopoietic malignancies 5, 22, 58, as well as in various types of carcinomas 19, 31, 59. In B‐cell lymphomas, overexpression of the miR‐17‐92 cluster was related to increased gene dosage (22). In contrast, none of our glioma patients showed amplification of the respective gene locus. However, other mechanisms leading to overexpression of the miR‐17‐92 cluster have been reported, including transcriptional transactivation by Myc proteins and E2F1‐3 (1), each of which may be overexpressed in malignant gliomas.
Our in vitro analyses of glioma cells after inhibition of miR‐17 demonstrated miRNA‐specific influences on cell viability and apoptotic activity. These results are in accordance with studies that have demonstrated oncogenic effects of the miR‐17‐92 cluster in different cell types. For example, overexpression of the miR‐17‐92 cluster in lung cancer cells significantly increased proliferation (21). In anaplastic thyroid cancer cell lines, inhibition of miR‐17, miR‐17* and miR‐19a reduced cell growth (55). Furthermore, inhibition of miR‐17 and miR‐20a induced apoptosis in lung cancer cells (37). In vitro or in vivo treatment with antagomir‐17 inhibited the growth of MYCN‐amplified neuroblastoma (14). On the other hand, the miR‐17‐92 locus is deleted in a fraction of breast cancers (66), and miR‐17 overexpression caused reduced proliferation in breast cancer cells (24). Taken together, these findings point to cell type‐specific roles of this particular miRNA cluster.
Several targets of miRNAs belonging to the miR‐17‐92 cluster have been experimentally validated, including members of the E2F family 42, 54, Rbl2 (36), Aib1 (24), p21 and Bim (14). In addition, the Pten tumor suppressor protein, which is frequently inactivated in glioblastoma, has been identified as a target of miR‐17 and miR‐19a in mouse embryonic fibroblasts (62). Using proteomic analyses, we identified Pold2 as a predicted target that was up‐regulated—directly or indirectly—by inhibition of miR‐17 in T98G cells. Pold2 is the 50 kdA regulatory subunit of the DNA polymerase delta complex and is involved in DNA repair mechanisms 23, 45. Thus, up‐regulation of miR‐17 may lead to alterations of DNA repair in gliomas; however, this hypothesis requires experimental validation.
In summary, we identified 14 miRNAs that were differentially expressed between primary low‐grade gliomas and secondary glioblastomas in an exploratory analysis of four individual patients. Following validation experiments in independent series of primary low‐grade and secondary high‐grade astrocytic gliomas, we concentrated on miR‐17 and miR‐184 as two promising candidates showing glioma progression‐associated up‐ or down‐regulation, respectively. In vitro analyses of glioma cell lines revealed further evidence for miR‐184 having tumor‐suppressive functions in gliomas, while miR‐17 likely promotes glioma progression. Using mRNA and protein expression profiling, we identified putative targets that were differentially expressed in glioma cells following overexpression of miR‐184 or inhibition of miR‐17, respectively, including Npm1 and Akt2 (miR‐184), as well as Pold2 (miR‐17). Additional studies are needed to characterize in detail the molecular mechanisms underlying the observed progression‐associated miRNA expression changes and to investigate whether the identified candidate targets are directly or indirectly regulated by the respective miRNAs. Nevertheless, our data support an important role of miRNA aberrations in glioma progression. The significance of miRNA expression profiles in the molecular diagnostics of gliomas and the role of miRNAs as novel therapeutic targets in these tumors warrant further exploration.
Supporting information
Figure S1. Representative proteome pattern of A172 cells transfected with miR‐184 precursors revealed by 2D‐DIGE. Image analysis and subsequent MS analyses led to the identification of 24 significantly regulated proteins.
Figure S2. Representative proteome pattern of T98G cells transfected with miR‐184 precursors revealed by 2D‐DIGE. Image analysis and subsequent MS analyses led to the identification of 54 significantly regulated proteins.
Figure S3. Representative proteome pattern of A172 cells transfected with miR‐17 inhibitors revealed by 2D‐DIGE. Image analysis and subsequent MS analyses led to the identification of 23 significantly regulated proteins.
Figure S4. Representative proteome pattern of T98G cells transfected with miR‐17 inhibitors revealed by 2D‐DIGE. Image analysis and subsequent MS analyses led to the identification of 48 significantly regulated proteins.
Figure S5. Expression of Akt2 protein in diffuse astrocytomas as compared to secondary glioblastomas. Western blots containing protein extracts from five diffuse astrocytomas (AII) and five secondary glioblastomas (sGBIV) were probed with antibodies against Akt2 and β‐actin as a loading control. Note increased Akt2 protein levels in secondary glioblastomas as compared to the low‐grade diffuse astrocytomas.
Table S1. List of investigated human microRNAs (miRNAs) that were detectable with the Applied Biosystems miRNA Early Access Kit.
Table S2. Primer sequences used for duplex polymerase chain reaction analysis of microRNA (miRNA) loci and expression analyses of putative miRNA targets.
Table S3. Potential target genes of miR‐184 identified by expression profiling using Affymetrix Human Genome U133 plus 2.0 arrays. The mRNA expression profiles of glioma cells with miR‐184 overexpression (pre‐184) and glioma cells transfected with negative controls (pre‐NC) were compared. The list contains targets of miR‐184 as predicted by the mirBase target database with signal log ratios (pre‐184/pre‐NC) < −1 and change P values <0.05 in both investigated glioma cell lines (A172 and T98G).
Table S4. Differential protein expression caused by miR‐184 overexpression. A172 and T98G cells were transfected with miR‐184 precursors (pre‐184) or negative controls (pre‐NC). 2D‐DIGE analyses were performed to identify differentially expressed proteins. Note that nucleophosmin 1 is a predicted target of miR‐184.
Table S5. Differential protein expression caused by inhibition of miR‐17. A172 and T98G cells were transfected with miR‐17 inhibitors (anti‐17) or negative controls (anti‐NC). 2D‐DIGE analyses were performed to identify differentially expressed proteins. Note that Pold2 is a predicted target of miR‐17.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
ACKNOWLEDGMENTS
This study was supported by grants from the German Ministry for Education and Research (BMBF) within the German National Genome Network (NGFNplus, grant no. 01GS0884 to M.W. and G.R.), and by the Research Commission of the Medical Faculty of Heinrich‐Heine‐University (grant no. 9772297 to M.W.). B.M. is supported by the German National Academic Foundation (Studienstiftung des Deutschen Volkes).
REFERENCES
- 1. Aguda BD, Kim Y, Piper‐Hunter MG, Friedman A, Marsh CB (2008) MicroRNA regulation of a cancer network: consequences of the feedback loops involving miR‐17‐92, e2f, and myc. Proc Natl Acad Sci U S A 105:19678–19683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, Von Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116:597–602. [DOI] [PubMed] [Google Scholar]
- 3. Boissonneault V, Plante I, Rivest S, Provost P (2009) MicroRNA‐298 and microRNA‐328 regulate expression of mouse beta‐amyloid precursor protein converting enzyme 1. J Biol Chem 284:1971–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E et al (2002) Frequent deletions and down‐regulation of micro‐RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 99:15524–15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Calin GA, Ferracin M, Cimmino A, Leva GD, Shimizu M, Wojcik SE et al (2005) A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 353:1793–1801. [DOI] [PubMed] [Google Scholar]
- 6. Camps C, Buffa FM, Colella S, Moore J, Sotiriou C, Sheldon H et al (2008) Hsa‐miR‐210 is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res 14:1340–1348. [DOI] [PubMed] [Google Scholar]
- 7. Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chan JA, Krichevsky AM, Kosik KS (2005) MicroRNA‐21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 65:6029–6033. [DOI] [PubMed] [Google Scholar]
- 9. Chen Y, Stallings RL (2007) Differential patterns of microRNA expression in neuroblastoma are correlated with prognosis, differentiation, and apoptosis. Cancer Res 67:976–983. [DOI] [PubMed] [Google Scholar]
- 10. Ciafrè SA, Galardi S, Mangiola A, Ferracin M, Liu CG, Sabatino G et al (2005) Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Biophys Res Commun 334:1351–1358. [DOI] [PubMed] [Google Scholar]
- 11. Corsten MF, Miranda R, Kasmieh R, Krichevsky AM, Weissleder R, Shah K (2007) MicroRNA‐21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell delivered S‐TRAIL in human gliomas. Cancer Res 67:8994–9000. [DOI] [PubMed] [Google Scholar]
- 12. Fasanaro P, D'Alessandra Y, Stefano VD, Melchionna R, Romani S, Pompilio G et al (2008) MicroRNA‐210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine‐kinase ligand Ephrin‐A3. J Biol Chem 283:15878–15883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Filipowicz W, Bhattacharyya SN, Sonenberg N (2008) Mechanisms of post‐transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9:102–114. [DOI] [PubMed] [Google Scholar]
- 14. Fontana L, Fiori ME, Albini S, Cifaldi L, Giovinazzi S, Forloni M et al (2008) Antagomir‐17‐5p abolishes the growth of therapy‐resistant neuroblastoma through p21 and BIM. PLoS ONE 3:e2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fulci V, Chiaretti S, Goldoni M, Azzalin G, Carucci N, Tavolaro S et al (2007) Quantitative technologies establish a novel microRNA profile of chronic lymphocytic leukemia. Blood 109:4944–4951. [DOI] [PubMed] [Google Scholar]
- 16. Gal H, Pandi G, Kanner AA, Ram Z, Lithwick‐Yanai G, Amariglio N et al (2008) miR‐451 And imatinib mesylate inhibit tumor growth of glioblastoma stem cells. Biochem Biophys Res Commun 376:86–90. [DOI] [PubMed] [Google Scholar]
- 17. Gillies JK, Lorimer IAJ (2007) Regulation of p27Kip1 by miRNA 221/222 in glioblastoma. Cell Cycle 6:2005–2009. [DOI] [PubMed] [Google Scholar]
- 18. Godlewski J, Nowicki MO, Bronisz A, Williams S, Otsuki A, Nuovo G et al (2008) Targeting of the Bmi‐1 oncogene/stem cell renewal factor by microRNA‐128 inhibits glioma proliferation and self‐renewal. Cancer Res 68:9125–9130. [DOI] [PubMed] [Google Scholar]
- 19. Gottardo F, Liu CG, Ferracin M, Calin GA, Fassan M, Bassi P et al (2007) Micro‐RNA profiling in kidney and bladder cancers. Urol Oncol 25:387–392. [DOI] [PubMed] [Google Scholar]
- 20. Grisendi S, Mecucci C, Falini B, Pandolfi PP (2006) Nucleophosmin and cancer. Nat Rev Cancer 6:493–505. [DOI] [PubMed] [Google Scholar]
- 21. Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, Tomida S et al (2005) A polycistronic microRNA cluster, miR‐17‐92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 65:9628–9632. [DOI] [PubMed] [Google Scholar]
- 22. He L, Thomson JM, Hemann MT, Hernando‐Monge E, Mu D, Goodson S et al (2005) A microRNA polycistron as a potential human oncogene. Nature 435:828–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hindges R, Hübscher U (1997) DNA polymerase delta, an essential enzyme for DNA transactions. Biol Chem 378:345–362. [DOI] [PubMed] [Google Scholar]
- 24. Hossain A, Kuo MT, Saunders GF (2006) miR‐17‐5p Regulates breast cancer cell proliferation by inhibiting translation of AIB1 mRNA. Mol Cell Biol 26:8191–8201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ichimura K, Ohgaki H, Kleihues P, Collins VP (2004) Molecular pathogenesis of astrocytic tumours. J Neurooncol 70:137–160. [DOI] [PubMed] [Google Scholar]
- 26. Iorio MV, Visone R, Leva GD, Donati V, Petrocca F, Casalini P et al (2007) MicroRNA signatures in human ovarian cancer. Cancer Res 67:8699–8707. [DOI] [PubMed] [Google Scholar]
- 27. Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens AC, Meier EGV (1999) Frequent coalterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol 9:469–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kefas B, Godlewski J, Comeau L, Li Y, Abounader R, Hawkinson M et al (2008) MicroRNA‐7 inhibits the epidermal growth factor receptor and the Akt pathway and is down‐regulated in glioblastoma. Cancer Res 68:3566–3572. [DOI] [PubMed] [Google Scholar]
- 29. Klose J, Kobalz U (1995) Two‐dimensional electrophoresis of proteins: an updated protocol and implications for a functional analysis of the genome. Electrophoresis 16:1034–1059. [DOI] [PubMed] [Google Scholar]
- 30. Knobbe CB, Reifenberger G (2003) Genetic alterations and aberrant expression of genes related to the phosphatidyl‐inositol‐3′‐kinase/ protein kinase B (Akt) signal transduction pathway in glioblastomas. Brain Pathol 13:507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kutay H, Bai S, Datta J, Motiwala T, Pogribny I, Frankel W et al (2006) Downregulation of miR‐122 in the rodent and human hepatocellular carcinomas. J Cell Biochem 99:671–678. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32. Laios A, O'Toole S, Flavin R, Martin C, Kelly L, Ring M et al (2008) Potential role of miR‐9 and miR‐223 in recurrent ovarian cancer. Mol Cancer 7:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee YS, Dutta A (2009) MicroRNAs in cancer. Annu Rev Pathol 4:199–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lehmann U, Hasemeier B, Christgen M, Müller M, Römermann D, Länger F, Kreipe H (2008) Epigenetic inactivation of microRNA gene hsa‐mir‐9‐1 in human breast cancer. J Pathol 214:17–24. [DOI] [PubMed] [Google Scholar]
- 35. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) (2007) WHO Classification of Tumours of the Central Nervous System, 3rd edn. IARC Press: Lyon. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu Y, Thomson JM, Wong HYF, Hammond SM, Hogan BLM (2007) Transgenic over‐expression of the microRNA miR‐17‐92 cluster promotes proliferation and inhibits differentiation of lung epithelial progenitor cells. Dev Biol 310:442–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Matsubara H, Takeuchi T, Nishikawa E, Yanagisawa K, Hayashita Y, Ebi H et al (2007) Apoptosis induction by antisense oligonucleotides against miR‐17‐5p and miR‐20a in lung cancers overexpressing miR‐17‐92. Oncogene 26:6099–6105. [DOI] [PubMed] [Google Scholar]
- 38. Nam EJ, Yoon H, Kim SW, Kim H, Kim YT, Kim JH et al (2008) MicroRNA expression profiles in serous ovarian carcinoma. Clin Cancer Res 14:2690–2695. [DOI] [PubMed] [Google Scholar]
- 39. Nelson PT, Baldwin DA, Kloosterman WP, Kauppinen S, Plasterk RHA, Mourelatos Z (2006) RAKE and LNA‐ISH reveal microRNA expression and localization in archival human brain. RNA 12:187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nesterenko MV, Tilley M, Upton SJ (1994) A simple modification of Blum's silver stain method allows for 30 minute detection of proteins in polyacrylamide gels. J Biochem Biophys Methods 28:239–242. [DOI] [PubMed] [Google Scholar]
- 41. Nomura T, Kimura M, Horii T, Morita S, Soejima H, Kudo S, Hatada I (2008) MeCP2‐dependent repression of an imprinted miR‐184 released by depolarization. Hum Mol Genet 17:1192–1199. [DOI] [PubMed] [Google Scholar]
- 42. O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT (2005) c‐Myc‐regulated microRNAs modulate E2F1 expression. Nature 435:839–843. [DOI] [PubMed] [Google Scholar]
- 43. Ohgaki H, Kleihues P (2007) Genetic pathways to primary and secondary glioblastoma. Am J Pathol 170:1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Papagiannakopoulos T, Shapiro A, Kosik KS (2008) MicroRNA‐21 targets a network of key tumor‐suppressive pathways in glioblastoma cells. Cancer Res 68:8164–8172. [DOI] [PubMed] [Google Scholar]
- 45. Pavlov YI, Frahm C, McElhinny SAN, Niimi A, Suzuki M, Kunkel TA (2006) Evidence that errors made by DNA polymerase alpha are corrected by DNA polymerase delta. Curr Biol 16:202–207. [DOI] [PubMed] [Google Scholar]
- 46. Qing Y, Yingmao G, Lujun B, Shaoling L (2008) Role of Npm1 in proliferation, apoptosis and differentiation of neural stem cells. J Neurol Sci 266:131–137. [DOI] [PubMed] [Google Scholar]
- 47. Rickert CH, Sträter R, Kaatsch P, Wassmann H, Jürgens H, Dockhorn‐Dworniczak B, Paulus W (2001) Pediatric high‐grade astrocytomas show chromosomal imbalances distinct from adult cases. Am J Pathol 158:1525–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Riemenschneider MJ, Reifenberger G (2009) Astrocytic tumors. In: Gliomas. Recent Results in Cancer Research, Vol. 171. Von Deimling A (ed.), pp. 3–24. Springer: Berlin. [DOI] [PubMed] [Google Scholar]
- 49. Schulte JH, Horn S, Otto T, Samans B, Heukamp LC, Eilers UC et al (2008) MYCN regulates oncogenic microRNAs in neuroblastoma. Int J Cancer 122:699–704. [DOI] [PubMed] [Google Scholar]
- 50. Shen J, Yang X, Xie B, Chen Y, Swaim M, Hackett SF, Campochiaro PA (2008) MicroRNAs regulate ocular neovascularization. Mol Ther 16:1208–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shi L, Cheng Z, Zhang J, Li R, Zhao P, Fu Z, You Y (2008) Hsa‐mir‐181a and hsa‐mir‐181b function as tumor suppressors in human glioma cells. Brain Res 1236:185–193. [DOI] [PubMed] [Google Scholar]
- 52. Silber J, Lim DA, Petritsch C, Persson AI, Maunakea AK, Yu M et al (2008) miR‐124 And miR‐137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med 6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sitek B, Sipos B, Klöppel G, Schmiegel W, Hahn SA, Meyer HE, Stühler K (2008) Application of fluorescence dye saturation labeling for differential proteome analysis of 1,000 microdissected cells from pancreatic ductal adenocarcinoma precursor lesions. Methods Mol Biol 425:1–14. [DOI] [PubMed] [Google Scholar]
- 54. Sylvestre Y, Guire VD, Querido E, Mukhopadhyay UK, Bourdeau V, Major F et al (2007) An E2F/miR‐20a autoregulatory feedback loop. J Biol Chem 282:2135–2143. [DOI] [PubMed] [Google Scholar]
- 55. Takakura S, Mitsutake N, Nakashima M, Namba H, Saenko VA, Rogounovitch TI et al (2008) Oncogenic role of miR‐17‐92 cluster in anaplastic thyroid cancer cells. Cancer Sci 99:1147–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Van den Boom J, Wolter M, Blaschke B, Knobbe CB, Reifenberger G (2006) Identification of novel genes associated with astrocytoma progression using suppression subtractive hybridization and real‐time reverse transcription–polymerase chain reaction. Int J Cancer 119:2330–2338. [DOI] [PubMed] [Google Scholar]
- 57. Van den Boom J, Wolter M, Kuick R, Misek DE, Youkilis AS, Wechsler DS et al (2003) Characterization of gene expression profiles associated with glioma progression using oligonucleotide‐based microarray analysis and real‐time reverse transcription–polymerase chain reaction. Am J Pathol 163:1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Venturini L, Battmer K, Castoldi M, Schultheis B, Hochhaus A, Muckenthaler MU et al (2007) Expression of the miR‐17‐92 polycistron in chronic myeloid leukemia (CML) CD34+ cells. Blood 109:4399–4405. [DOI] [PubMed] [Google Scholar]
- 59. Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F et al (2006) A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 103:2257–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wong TS, Liu XB, Wong BYH, Ng RWM, Yuen APW, Wei WI (2008) Mature miR‐184 as potential oncogenic microRNA of squamous cell carcinoma of tongue. Clin Cancer Res 14:2588–2592. [DOI] [PubMed] [Google Scholar]
- 61. Würdinger T, Tannous BA, Saydam O, Skog J, Grau S, Soutschek J et al (2008) miR‐296 Regulates growth factor receptor overexpression in angiogenic endothelial cells. Cancer Cell 14:382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J et al (2008) Lymphoproliferative disease and autoimmunity in mice with increased miR‐17‐92 expression in lymphocytes. Nat Immunol 9:405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yokota T, Kouno J, Adachi K, Takahashi H, Teramoto A, Matsumoto K et al (2006) Identification of histological markers for malignant glioma by genome‐wide expression analysis: dynein, alpha‐PIX and sorcin. Acta Neuropathol 111:29–38. [DOI] [PubMed] [Google Scholar]
- 65. Yu J, Ryan DG, Getsios S, Oliveira‐Fernandes M, Fatima A, Lavker RM (2008) MicroRNA‐184 antagonizes microRNA‐205 to maintain SHIP2 levels in epithelia. Proc Natl Acad Sci U S A 105:19300–19305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang L, Huang J, Yang N, Greshock J, Megraw MS, Giannakakis A et al (2006) MicroRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci U S A 103:9136–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Representative proteome pattern of A172 cells transfected with miR‐184 precursors revealed by 2D‐DIGE. Image analysis and subsequent MS analyses led to the identification of 24 significantly regulated proteins.
Figure S2. Representative proteome pattern of T98G cells transfected with miR‐184 precursors revealed by 2D‐DIGE. Image analysis and subsequent MS analyses led to the identification of 54 significantly regulated proteins.
Figure S3. Representative proteome pattern of A172 cells transfected with miR‐17 inhibitors revealed by 2D‐DIGE. Image analysis and subsequent MS analyses led to the identification of 23 significantly regulated proteins.
Figure S4. Representative proteome pattern of T98G cells transfected with miR‐17 inhibitors revealed by 2D‐DIGE. Image analysis and subsequent MS analyses led to the identification of 48 significantly regulated proteins.
Figure S5. Expression of Akt2 protein in diffuse astrocytomas as compared to secondary glioblastomas. Western blots containing protein extracts from five diffuse astrocytomas (AII) and five secondary glioblastomas (sGBIV) were probed with antibodies against Akt2 and β‐actin as a loading control. Note increased Akt2 protein levels in secondary glioblastomas as compared to the low‐grade diffuse astrocytomas.
Table S1. List of investigated human microRNAs (miRNAs) that were detectable with the Applied Biosystems miRNA Early Access Kit.
Table S2. Primer sequences used for duplex polymerase chain reaction analysis of microRNA (miRNA) loci and expression analyses of putative miRNA targets.
Table S3. Potential target genes of miR‐184 identified by expression profiling using Affymetrix Human Genome U133 plus 2.0 arrays. The mRNA expression profiles of glioma cells with miR‐184 overexpression (pre‐184) and glioma cells transfected with negative controls (pre‐NC) were compared. The list contains targets of miR‐184 as predicted by the mirBase target database with signal log ratios (pre‐184/pre‐NC) < −1 and change P values <0.05 in both investigated glioma cell lines (A172 and T98G).
Table S4. Differential protein expression caused by miR‐184 overexpression. A172 and T98G cells were transfected with miR‐184 precursors (pre‐184) or negative controls (pre‐NC). 2D‐DIGE analyses were performed to identify differentially expressed proteins. Note that nucleophosmin 1 is a predicted target of miR‐184.
Table S5. Differential protein expression caused by inhibition of miR‐17. A172 and T98G cells were transfected with miR‐17 inhibitors (anti‐17) or negative controls (anti‐NC). 2D‐DIGE analyses were performed to identify differentially expressed proteins. Note that Pold2 is a predicted target of miR‐17.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item