

Abstract
Activating β‐catenin (CTNNB1) mutations can be identified in the majority of adamantinomatous craniopharyngiomas (adaCP), suggesting an aberrant Wnt signaling pathway in this histopathologically peculiar tumor entity. However, there is no proven evidence that nuclear translocation of β‐catenin is associated with CTNNB1 mutations and target gene activation. We performed a laser‐microdissection‐based study comparing β‐catenin accumulating vs. non‐accumulating tumor cells. Mutational analysis and gene expression profiling using real‐time polymerase chain reaction were conducted in adamantinomatous and papillary tumor specimens. Target gene activation, that is, over‐expression of Axin2 could be detected in adaCP, especially in tumor cells with nuclear β‐catenin accumulation. In addition, increased expression of BMP4 was identified in the accumulating cell population, which supports the hypothesis of an oral ectodermal origin. Interestingly, accumulating and non‐accumulating tumor cell populations carried CTNNB1 mutations within exon 3. We extended the analysis, therefore, towards genetic regions encoding for membrane linkage and active/passive nuclear transport mechanisms (exon 4 and exon 8–13), but could not detect any alteration. This is the first report demonstrating an association between nuclear β‐catenin accumulation and target gene activation in adaCP. The results confirm the Wnt signaling pathway as molecular basis of the distinct and challenging clinical and morphological phenotype of adaCP.
Keywords: Axin2, BMP4, Conductin, CTNNB1, β‐catenin
INTRODUCTION
Craniopharyngiomas (CP) are rare, benign epithelial tumors of the sellar region, to be distinguished by their clinical occurrence, outcome and histopathology into an adamantinomatous (adaCP) and a rare papillary (papCP) variant. AdaCP constitute about 10% of childhood intracranial neoplasms, whereas papCP occur predominantly in adults. Moreover, papCP are characterized by well‐differentiated, solid squamous epithelium‐lacking cystic areas, palisaded cells and regressive changes, that is, keratin nodules (wet keratin), cholesterol clefts as well as calcifications. The latter represent histological hallmarks of the adamantinomatous subtype 9, 12. AdaCP seem to arise from remnants of Rathke's pouch or misplaced enamel organ as indicated by striking histological similarities to odontogenic tumors and cysts 27, 32. Recently, nuclear β‐catenin accumulation was shown as a highly significant finding in adaCP (10). As a key player of the canonical Wnt signaling pathway, β‐catenin plays an important role during development, cellular proliferation, differentiation and migration (23). It is located either at the cell membrane establishing adherens junctions, or in the cytoplasm tagged for proteosomal degradation when Wnt signaling is inactive (16). The cytoplasmatic amount is strictly regulated by a multiprotein‐complex containing the tumor suppressor adenomatous polyposis coli (APC), the scaffold proteins Axin and Axin2, and the phosphokinases GSK3β and CK1. Following binding to this multiprotein‐complex, β‐catenin becomes phosphorylated and tagged for its β‐transducing repeat‐containing protein driven ubiquitinylation and subsequent proteosomal clearance 1, 2, 29, 30. Noteworthy, N‐terminal phosphorylation sites are essential for β‐catenin functional regulation and encoded by exon 3 of the β‐catenin gene (CTNNB1). Activation of Wnt signaling leads to inhibition of GSK3β, which in turn, effects cytoplasmatic stabilization and subsequent nuclear translocation of β‐catenin (22). Within the nucleus, β‐catenin initiates gene expression via interaction with transcription factors of the Lef1/TCF family (3). Aberrant β‐catenin stabilization can be the result of an impaired degradation process and causes carcinogenesis. In many malignancies, genes encoding for the multiprotein complex components, that is, APC, Axin and Axin2, exhibit genetic loss of function alterations, whereas medulloblastomas, hepatoblastomas, hepatocellular carcinomas, pilomatricomas and ovarian carcinomas reveal activating mutations in CTNNB1 4, 8, 14, 28, 37, 39. These mutations are thought to entail cytoplasmatic and corresponding nuclear accumulation of the protein by its stabilization. Such activating mutations within the region encoding the phosphorylation sites in exon 3 of the CTNNB1 gene have also been described for adaCP 7, 15, 25, 33, 34. However, the relation between activating mutations in exon 3, nuclear β‐catenin accumulation and resultant target gene activation remains to be shown, and may involve active and passive nuclear transport mechanisms. A divergent interaction between members of the canonical Wnt signaling pathway is challenged by the histopathological heterogeneity. For that, neoplastic cells with nuclear accumulation of β‐catenin are the clear minority and appear not equally distributed throughout adaCP (7). In addition, until now there is no proven evidence that nuclear β‐catenin accumulation activates target genes of the Wnt signaling cascade in affected tumor cells of adaCP.
To address this issue, we conducted reverse transcription polymerase chain reaction (RT‐PCR) analysis of Axin2 and bone morphogenetic protein 4 (BMP4) using total cDNA from snap frozen tissue samples of adaCP and papCP as well as cDNA from laser‐microdissected formalin fixed and paraffin embedded tumor cell fractions with and without nuclear β‐catenin accumulation. Whereas Axin2 is a well‐recognized inhibitor and target gene of β‐catenin (13), BMP4 is described to be enhanced in tumors with oncogenic β‐catenin elevation 17, 35. In addition, BMP4 plays a crucial role in tooth development, which seems to be imitated in adaCP 24, 27, 38. To proof the concept of genotypic heterogeneity within adaCP as potential cause of differential nuclear β‐catenin accumulation, we submitted laser‐microdissected cell clusters with or without accumulation also to a mutational analysis of exon 3 of CTNNB1. Besides exon 3, we extended our genetic analyses to exclude further alterations in regions of CTNNB1 encoding the ability to shuttle between the nucleus and the cytoplasm. Phosphorylation sites regulating dissociation from adherens junctions are encoded partly by exon 4 (20). In contrast, exon 8 to 13 encodes the armadillo‐repeats, which are not only crucial for the passive nuclear transport of β‐catenin, but represent also interaction sites to members of the destruction complex (eg, APC, Axin2), TCFs and cadherins 18, 40.
MATERIALS AND METHODS
Surgical specimens from 37 patients with histopathologically confirmed adaCP, and eight patients with papCP, were retrieved from the archives of the Departments of Neurosurgery and Neuropathology at the University of Erlangen‐Nuremberg, International Neuroscience Institute in Hannover, and the Evangelisches Krankenhaus in Bielefeld. There were 21 males, 16 females with adaCP, four males, and four females with papCP. The age ranged between two and 77 years in the group of adaCP (mean age 34, 4 years) and between 19 and 57 years in papCP (mean age 42, 4 years). Each specimen was classified according to World Health Organization guidelines using Haematoxylin and eosin as well as immunohistochemistry (IHC) (10).
IHC
Surgical samples were fixed overnight in 4% formaldehyde and routinely processed into liquid paraffin. Sections were cut at 4 µm with a microtome (Microm, Heidelberg, Germany) and mounted on positively charged slides (Menzel, Braunschweig, Germany). The slides were air dried in an incubator at 37°C overnight. Microwave pretreatment was performed in Citrate‐buffer at pH 6.0. β‐catenin immunoreactions were performed as described elsewhere (6). Immunohistochemical staining for Axin2 was performed at room temperature for 2 hours, using a polyclonal rabbit antibody (1:200; Abcam, Cambridge, UK). For detection of BMP4, we used the monoclonal mouse antibody clone 3H2 (1:50; Novocastra Laboratories Ltd, Newcastle, UK). BMP4 staining was operated at room temperature for 30 minutes. Both reactions were followed by avidin‐biotin labeling detected with daminobenzidine (Dako, Hamburg, Germany). Serial paraffin sections (1 µm) of tumor specimens were prepared and stained, either with antibodies against β‐catenin, Axin2 or BMP4, to examine intratumoral coexpression patterns.
DNA and cDNA preparation
For DNA extraction, snap frozen tissue samples were retrieved from our tissue bank (−80°C). Total cellular RNA was extracted from 10 patients (5 adaCP and 5 papCP). From all tissues, frozen sections were microscopically reviewed to confirm tumor tissue content. DNA was extracted using the DNeasy® tissue kit (Qiagen, Hilden, Germany). DNA from laser‐microdissected formalin fixed paraffin embedded material was applied using the QIAamp® DNA Micro kit (Qiagen) according to the manufacturer's instructions. The RNeasy® extraction kit (Qiagen) was used for RNA isolation. Subsequently, digestion with RNase‐free DNase I and purification via RNeasy® columns (Qiagen) was followed by reverse transcription using SuperScript™ First‐strand synthesis system for RT‐PCR (Invitrogen™, Carlsbad, California, USA) with oligo (dT) primers.
For RNA extraction of laser‐microdissected probes, cell fractions were collected in lyses buffer (36) and digested 16 h at 56°C with Proteinase K® (Qiagen). Using the TRIzol® reagent (Invitrogen™), RNA was isolated following the manufacturer's instructions. Digestion with RNase‐free DNase I and purification was followed by reverse transcription using SuperScript™ First‐strand synthesis system for RT‐PCR (Invitrogen™) with random hexamer primers.
Laser‐microdissection
Serial sections of 4 µm were prepared from eight different formalin fixed and paraffin embedded (FFPE) tissue tumor samples, harboring a substantial amount of β‐catenin accumulating cell clusters (Figure 1A indicated as +). Immunohistochemical staining procedure for β‐catenin was conducted as described above. Cell fractions of interest were separated using the laser‐microdissection microscope P.A.L.M. MicroBeam from Carl Zeiss (P.A.L.M. Microlaser Technologies GmbH, Bernried, Germany). DNA and RNA were isolated from up to four sections of each tumor sample (approximately 50.000 cells) as described above.
Figure 1.
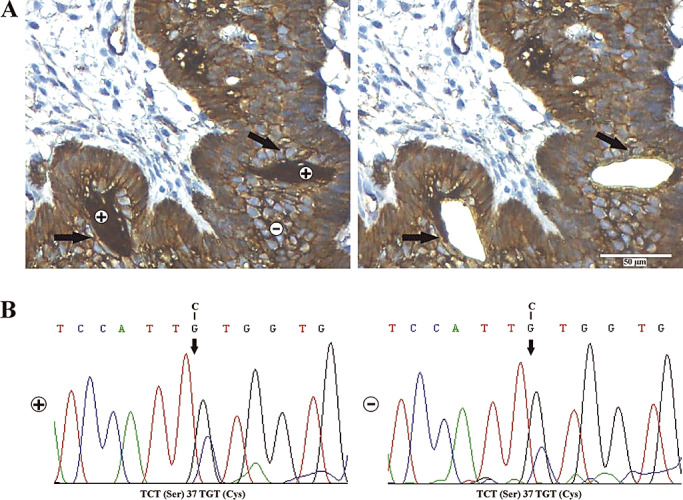
Molecular‐genetic analysis of laser‐microdissected tumor cells. For mutational analysis, we separated nuclear β‐catenin accumulating (+) vs. non‐accumulating (−) tumor cell fractions using laser microdissection. The arrows indicate accumulating cell clusters before and after dissection (A). Direct sequencing of exon 3 of CTNNB1 revealed a missense mutation at codon 37 encoding for a serine residue and phosphorylation site of GSK‐3β kinase in both fractions [accumulating (+) and non‐accumulating (−)] (B).
Mutational analysis
Exons 3, 4, 8, 9, 10, 11, 12 and 13 of the β‐catenin gene were amplified by PCR using genomic DNA extracted from fresh frozen tumor samples, for subsequent single‐strand conformation polymorphism (SSCP) analysis. PCR reactions were performed in a total volume of 10 µL with 10 ng genomic DNA in a PCR buffer containing, in a final concentration of 1.5 mM MgCl2 (Qiagen), 200 µM of each dNTP, 200 nM of forward and reverse primer (Table 1) and 0.25 U Taq polymerase (Qiagen).
Table 1.
Primer used for mutation analysis of CTNNB1.
| CTNNB1 | ||
|---|---|---|
| Exon | Forward primer 5′‐ 3′ | Reverse primer 5′‐ 3′ |
| 3 | gatttgatggagttggacatgg | tgttcttgagtgaaggactgag |
| 4‐a | aggtaaatgctgaactgtgg | gatctgcatgccctcatcta |
| 4‐b | tagatgagggcatgcagatc | gtcattgcttacctggtcct |
| 8 | gtggaatgcaagctttagga | tctgaacccagaagctgaac |
| 9‐a | aactggtgccatgggaatag | cacttggcagaccatcatct |
| 9‐b | gttcagcttctgggttcaga | gtcagatgacgaagagcaca |
| 9‐c | gacagggaagacatcactga | agccatccaacagctagaga |
| 10 | gttgagttgtatgccagttc | agatagccaggtatcactgt |
| 11 | tacagaggagaatgccctgt | cacttcacttaaaacatacctgc |
| 12 | gtgttttctccttagctgct | ctgctaaaggctttggttcc |
| 13 | ctccttgtctcttagcgaca | tgctcacatttccctacctc |
The amplified PCR products were denatured for 10 minutes at 95°C. SSCP electrophoresis of exons and exon fragments was conducted on polyacrylamide gels with different concentrations (10% and 14%) and varying bis‐acrylamide/acrylamide ratios (1:10, 1:29, 1:49, 1:99), with or without 5% glycerol in 0.5 × tris‐borate‐EDTA‐bufer (TBE; Merck, Darmstadt, Germany) at room temperature. The single and double strands of the PCR products were visualized by silver staining, as described previously (5). The shifted bands were excised from the wet gel, eluted and reamplified. DNA purification and sequencing was done by GATC Biotech AG (Konstanz, Germany) and the DNA sequencing facility of the Department of Human Genetics, University Hospital Cologne, Germany. Primers used for mutational screening are listed in Table 1.
For CTNNB1 gene exon 3 analysis of laser‐microdissected samples (β‐catenin accumulating vs. non‐accumulating cells), PCR amplicons were purified from agarose gels using the high pure PCR product purification kit (Roche, Mannheim, Germany) and cloned into the Topo TA cloning vector (Life Technologies) according to the manufacturer's instructions. Ten white colonies were picked, as blue white screening indicates plasmid integration. To confirm β‐catenin content, a colony PCR was conducted. Amplified plasmid‐DNA was extracted employing the plasmid midi kit® (Qiagen) and submitted to sequence analysis.
RT‐PCR analysis
TaqMan® gene expression assays (Applied Biosystems, Darmstadt, Germany) were employed for relative quantification. Analyses were performed with the applied Biosystems 7500 fast RT‐PCR system. The RNA isolated from laser‐microdissected FFPE cells, was transcribed in cDNA and preamplified using the TaqMan® PreAmp Master Mix Kit (Applied Biosystems) as recommended. Uniformity of all assays used was checked previously as described in the manufacturer's instructions.
Relative quantification analysis of β‐catenin target genes Axin2/Conductin (Hs01063170_m1) and BMP4 (Hs00370078_m1) was performed using total tumor cDNA from adaCP (n = 5) and papCP (n = 5). To analyze whether nuclear β‐catenin accumulation in adaCP correlates with target gene activation, we analyzed the expression of Axin2/Conductin within accumulating and non‐accumulating cell fractions of three representative tumor samples. As endogenous control served ubiquitin (Hs00824723_m1) and β‐2‐microglobulin (Hs99999907_m1) using the letter for analysis. The experiment was replicated three times, whereas a non‐template control was carried along and always revealed a negative result.
RESULTS
AdaCP show target gene activation of the Wnt signaling pathway, particularly in cell clusters with nuclear β‐catenin accumulation
Total cDNAs obtained from snap frozen papCP (n = 5) as well as adaCP (n = 5) were analyzed employing TaqMan Axin2/Conductin and BMP4 gene expression assays. Samples were normalized to β‐2‐microglobulin, used as housekeeping control gene. Relative quantification of Axin2/Conductin and BMP4 expression in adaCP was performed using the average of papCP as calibrator. The expression of Axin2/Conductin was enhanced in adaCP between 28.8 fold (case 3239) and 288.4 fold (case 3598) compared with papCP. BMP4 expression was increased in a range of 1.3 fold (case 3239) to 4.3 fold (case 1551) in adaCP vs. papCP (Figure 2A).
Figure 2.

Elevated levels of Axin2/conductin and BMP4 in total tumor cDNA of adaCP vs. papCP as well as in accumulating vs. non‐accumulating tumor cell mRNA. A. Relative quantification TaqMan analysis of Axin2/Conductin and BMP4 cDNA obtained from snap frozen and histologically confirmed papCP (n = 5) and adaCP (n = 5), harboring a sufficient amount of β‐catenin accumulating tumor cell clusters. β‐2‐microglobulin served as endogenous control and papCP were used as calibrator. All analyses were performed in quadruplicates of each sample. B. mRNA expression of β‐catenin target genes Axin2/Conductin as well as BMP4 is increased in laser‐microdissected tumor cell clusters with nuclear β‐catenin accumulation (+) in relation to non‐accumulating cells (−) (TaqMan analysis). The experiments were conducted three times.
To examine the impact of nuclear β‐catenin accumulation on target gene expression, we performed the same TaqMan analysis using cDNA of laser‐microdissected fractions including β‐catenin accumulating vs. non‐accumulating epithelial tumor cells of three representative tumor samples (case numbers 2874, 3239 and 3598). β‐2‐microglobulin was used to equalize the samples. The β‐catenin accumulating cell fraction revealed an up‐regulation of Axin2/Conductin in a range of 3.9 fold (case 2874) to 24.6 (case 3239) compared with the non‐accumulating cells. The expression of BMP4 was enhanced between 1.2 fold (case 3239) and 20.9 (case 2874) in accumulating vs. non‐accumulating tumor cell mRNA (Figure 2B).
Correlation of Axin2 as well as BMP4 protein distribution and gene expression
Axin2, as well as BMP4 protein distribution, was immunohistochemically analyzed and compared with nuclear β‐catenin staining in adaCP using serial sections. As depicted in Figure 3, there was a distinct association between elevated Axin2 and BMP4 content and tumor cells with nuclear β‐catenin accumulation. In surrounding tumor cells without nuclear β‐catenin accumulation, both proteins were detectable, but were expressed obviously lower (data not shown) as indicated by RT‐PCR.
Figure 3.
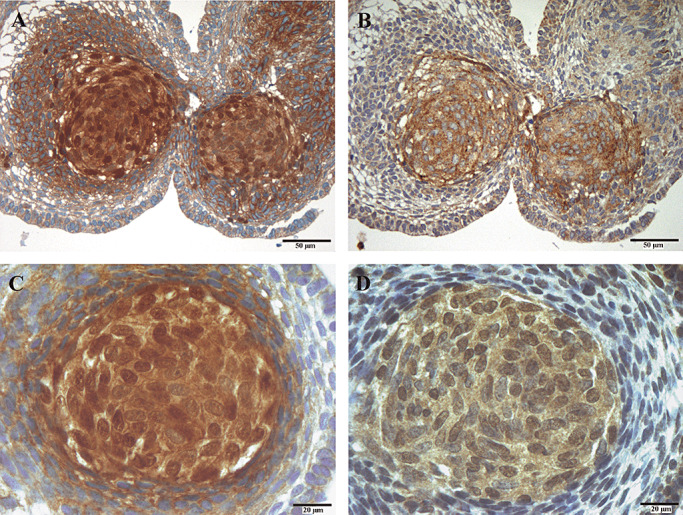
Immunohistochemical co‐localization of Axin2/BMP4 and β‐catenin. Using serial sections, whorl‐like tumor cell clusters showing nuclear β‐catenin accumulation (A,C), displayed elevated levels of Axin2 (B) and BMP4 (D). Magnification is 200× for A,B and 400× for C,D.
Mutations in exon 3 (CTNNB1) are not sufficient to induce nuclear β‐catenin accumulation in CP per se
Eight different FFPE tumor samples showing a representative number of cells with distinct nuclear β‐catenin accumulation (>50.000), as detected by IHC, were selected for mutational analysis. To have the possibility to analyze accumulating vs. non‐accumulating epithelial tumor cell clusters independently, both fractions were separated using laser‐microdissection (Figure 1A). Subsequent mutational analysis within the region encoding the phosphorylation sites (exon 3 of CTNNB1) revealed activating missense mutations in both fractions without discovering a significant pattern associated with nuclear accumulation (Figure 1B, Table 2). Experiments were repeated twice to control for paraffin‐induced aberrations.
Table 2.
Mutational analyses of exon 3 (CTNNB1) in accumulating vs. non‐accumulating tumor cells. DNA was extracted from laser‐microdissected, FFPE tissue pre‐stained immunohistochemically for β‐catenin (Figure 2). PCR product was cloned and subsequently sequenced. Abbreviations: F = female; M = male; FFPE = formalin fixed and paraffin embedded.
| Case | Age (years)/gender | Mutations in accumulating cells | Mutations in cells without accumulation |
|---|---|---|---|
| adaCP 1 | 48/F | TCT(Ser)37TGT(Cys) | GAC(Asp)32CAC(His) |
| adaCP 2 | 9/F | TCT(Ser)37TGT(Cys) | TCT(Ser)37TGT(Cys) |
| adaCP 3 | 9/F | ACC(Thr)41ATC(Ile) | ACC(Thr)41ATC(Ile) |
| GAC(Asp)32AAC(Asn) | |||
| adaCP 4 | 3/F | TCT(Ser)37TGT(Cys) | TCT(Ser)37TGT(Cys) |
| adaCP 5 | 25/F | GAC(Asp)32CAC(His) | GAC(Asp)32CAC(His) |
| ACC(Thr)41ATC(Ile) | TCT(Ser)37TGT(Cys) | ||
| adaCP 6 | 2/F | TCT(Ser)37TGT(Cys) | TCT(Ser)33TGT(Cys) |
| adaCP 7 | 14/F | GAC(Asp)32CAC(His) | TCT(Ser)37TGT(Cys) |
| adaCP 8 | 35/M | TCT(Ser)37TGT(Cys) | TCT(Ser)37TGT(Cys) |
Mutational analysis of exon 4 as well as exon 8–13 (CTNNB1) revealed no genetic alterations in CP
We used SSCP analysis in 32 adaCP and four papCP to detect mutations in exon 4 of the CTNNB1 gene encoding phosphorylation sites regulating dissociation of β‐catenin from adherens junctions. In addition, we examined exon 8–13 encoding the armadillo‐repeats which are essential for the passive nuclear im‐/ and export of β‐catenin. No mutations were identifiable in these gene regions (data not shown).
DISCUSSION
Constitutive Wnt signaling activation is recognized as an important pathomechanism in cancer origin and progression (4). Distinct nuclear β‐catenin accumulation represents a histopathological hallmark of adaCP, differentiating this variant from other cystic lesions in the sellar region (10). In addition, these tumor cells show specific morphologic features with arrangement in circumscribed whirl‐like clusters and simple epithelial differentiation (6). Activating mutations within exon 3 of the β‐catenin gene (CTNNB1) were likely to cause this nuclear β‐catenin translocation as a consequence of disordered degradation and intra‐cytoplasmatic accumulation 7, 33. However, molecular‐genetic studies demonstrated a broad range of missense mutations within CTNNB1 in adaCP, primarily affecting codons 32, 33, 37, 41, 43 and 45 7, 15, 33. To the best of our knowledge, confirmation of target gene activation in adaCP as well as the heterogeneous distribution of tumor cells with CTNNB1 mutations and nuclear β‐catenin accumulation remains to be shown. To address this intriguing issue, we conducted gene expression and mutational analyses. Our study design reliably identifies elevated levels of well‐accepted β‐catenin target genes, that is, Axin2 and BMP4 in adaCP compared with papCP. Furthermore, we could demonstrate that elevated gene expression levels were associated with nuclear β‐catenin accumulating tumor cell clusters and corresponding protein accumulation, as shown by IHC.
Noteworthy, this is the first report indicating BMP4 as a β‐catenin target gene in adaCP. Lef1 expression is activated by BMP4, and oncogenic β‐catenin is required for BMP4 expression in human cancers 17, 19. BMP4 is important in many developmental processes, particularly during early tooth development where its expression occurs downstream of Wnt signaling 21, 35, 38. In contrast to papCP, adaCP express various tooth specific genes and exhibit an odontogenic morphological appearance 27, 34. Elevated expression levels of BMP4 in adaCP, further confirm its probable oral ectodermal origin and that developmental processes mimicking tooth formation occur in adaCP. In particular, nuclear β‐catenin accumulating tumor cell clusters show an increased expression of the enamel knot marker p21 and are characterized by a specialized cytoarchitecture and differentiation resembling the early tooth anlage 6, 21.
Compared with Axin2 cDNA expression levels, the difference of BMP4 expression between the CP subtypes is significantly lower. A possible explanation for this observation could be that Axin2 was identified not only as a β‐catenin target gene but also being involved in cytoplasmatic β‐catenin destruction as a component of the multiprotein‐degradation‐complex 2, 13. In the latter role, increased levels of Axin2 could be an indicator for an elevated negative feedback loop to opposite the activated Wnt signaling pathway in adaCP.
After demonstrating target gene activation in nuclear β‐catenin expressing cell clusters, we were interested to characterize alterations involved in nuclear protein accumulation. Herein, we were able to demonstrate that both cell populations, β‐catenin accumulating as well as non‐accumulating cells, harbor activating mutations in exon 3 of the CTNNB1 gene, without identifying a cell specific mutation pattern. This implicates that alterations in β‐catenin gene regions encoding for phosphorylation sites crucial for protein degradation may not be sufficient to cause nuclear accumulation per se. This assumption is supported by a recent study of Sekine et al describing β‐catenin mutations within the mesenchymal component of adaCP, displaying no nuclear β‐catenin accumulation (33). In contrast, Kato et al were not able to verify mutations beyond the epithelial tumor cell component (15). Previous studies also identified multiple mutations in a single CP sample, which could suggest an increased genetic instability. However, chromosomal instability appears unlikely in CP 7, 41. Genetic heterogeneity within one tumor refers rather to a polyclonal origin of tumor cells and data from our, and previous, mutational analysis indicate that nearly all aberrations were heterozygous (31).
Cytoplasmatic β‐catenin levels and corresponding nuclear accumulation is not only determined by activating mutations. Many other mechanisms are likely to play a role, that is, E‐cadherin binding as well as active and passive nuclear shuttling processes. Nuclear localization and transcriptional activity of β‐catenin is prevented by binding to E‐cadherin (26). The membranous binding of β‐catenin to the intracellular part of cadherins and α‐catenins is regulated by phosphorylation of tyrosine residues Y142 encoded by exon 4, as well as Y489 and Y654 encoded by exons 9 and 13 of the CTNNB1 gene, the latter being partly identical to the armadillo‐repeat sequence (20). The armadillo repeat represents a core region of β‐catenin forming a super helix with a positively charged groove for protein binding, thus, regulating its activity (11). Identified binding partners include TCF‐family transcription factors, Axin2 and APC 11, 40. In addition, armadillo repeat units 10–12 are essential for the passive nuclear‐pore‐complex translocation of β‐catenin (18). Although compromised nuclear shuttling of β‐catenin may lead to nuclear accumulation, we were neither able to verify mutations within the CTNNB1 region encoding for the armadillo repeats (exon 8–13), thus indicating an intact passive nuclear transport machinery, nor in additional gene regions (exon 4) encoding interaction sites for membrane anchorage (20).
In conclusion, target genes of the Wnt signaling pathway are activated in adaCP, especially in cells with aberrant nuclear β‐catenin accumulation. The increased expression of inhibitors of the Wnt signaling pathway, that is, Axin2/Conductin indicate an activated negative feedback loop to opposite Wnt signaling in adaCP, whereas, elevated levels of BMP4 point to an oral ectodermal differentiation. Activating mutations in exon 3 of CTNNB1 are not restricted to nuclear β‐catenin accumulating tumor cells. Lack of additional alterations in the β‐catenin gene affecting important phosphorylation and interaction sites, points towards additional mechanisms being involved in the molecular trapping of β‐catenin within the nucleus. Further studies are mandatory to clarify the role of important components of the Wnt signaling pathway, that is, APC to be involved in this process.
ACKNOWLEDGMENTS
We thank V. Schmidt, S. Sterner, B. Rings, E. Hahnen, J. Hauke and S. Söllner for expert technical assistance. We thank Dr. K. Amman (University of Erlangen, Germany) for cooperation. This work was supported by the ELAN fund from the University of Erlangen Medical Faculty (to AH and RB).
REFERENCES
- 1. Aberle H, Bauer A, Stappert J, Kispert A, Kemler R (1997) Beta‐catenin is a target for the ubiquitin‐proteasome pathway. EMBO J 16:3797–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R et al (1998) Functional interaction of an axin homolog, conductin, with beta‐catenin, APC, and GSK3beta. Science 280:596–599. [DOI] [PubMed] [Google Scholar]
- 3. Behrens J, Von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W (1996) Functional interaction of beta‐catenin with the transcription factor LEF‐1. Nature 382:638–642. [DOI] [PubMed] [Google Scholar]
- 4. Behrens J, Lustig B (2004) The Wnt connection to tumorigenesis. Int J Dev Biol 48:477–487. [DOI] [PubMed] [Google Scholar]
- 5. Budowle B, Chakraborty R, Giusti AM, Eisenberg AJ, Allen RC (1991) Analysis of the VNTR locus D1S80 by the PCR followed by high‐resolution PAGE. Am J Hum Genet 48:137–144. [PMC free article] [PubMed] [Google Scholar]
- 6. Buslei R, Holsken A, Hofmann B, Kreutzer J, Siebzehnrubl F, Hans V et al (2007) Nuclear beta‐catenin accumulation associates with epithelial morphogenesis in craniopharyngiomas. Acta Neuropathol (Berl) 113:585–590. [DOI] [PubMed] [Google Scholar]
- 7. Buslei R, Nolde M, Hofmann B, Meissner S, Eyupoglu IY, Siebzehnrubl F et al (2005) Common mutations of beta‐catenin in adamantinomatous craniopharyngiomas but not in other tumours originating from the sellar region. Acta Neuropathol (Berl) 109:589–597. [DOI] [PubMed] [Google Scholar]
- 8. Chan EF (2000) Pilomatricomas contain activating mutations in beta‐catenin. J Am Acad Dermatol 43:701–702. [PubMed] [Google Scholar]
- 9. Giangaspero F, Burger PC, Osborne DR, Stein RB (1984) Suprasellar papillary squamous epithelioma (“papillary craniopharyngioma”). Am J Surg Pathol 8:57–64. [DOI] [PubMed] [Google Scholar]
- 10. Hofmann BM, Kreutzer J, Saeger W, Buchfelder M, Blumcke I, Fahlbusch R, Buslei R (2006) Nuclear beta‐catenin accumulation as reliable marker for the differentiation between cystic craniopharyngiomas and rathke cleft cysts: a clinico‐pathologic approach. Am J Surg Pathol 30:1595–1603. [DOI] [PubMed] [Google Scholar]
- 11. Huber AH, Nelson WJ, Weis WI (1997) Three‐dimensional structure of the armadillo repeat region of beta‐catenin. Cell 90:871–882. [DOI] [PubMed] [Google Scholar]
- 12. Jenkins JS (1972) The hypothalamus. Br Med J 2:99–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F (2002) Wnt/beta‐catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol 22:1172–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kajino Y, Yamaguchi A, Hashimoto N, Matsuura A, Sato N, Kikuchi K (2001) Beta‐Catenin gene mutation in human hair follicle‐related tumors. Pathol Int 51:543–548. [DOI] [PubMed] [Google Scholar]
- 15. Kato K, Nakatani Y, Kanno H, Inayama Y, Ijiri R, Nagahara N et al (2004) Possible linkage between specific histological structures and aberrant reactivation of the Wnt pathway in adamantinomatous craniopharyngioma. J Pathol 203:814–821. [DOI] [PubMed] [Google Scholar]
- 16. Kemler R (1993) From cadherins to catenins: cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet 9:317–321. [DOI] [PubMed] [Google Scholar]
- 17. Kim JS, Crooks H, Dracheva T, Nishanian TG, Singh B, Jen J, Waldman T (2002) Oncogenic beta‐catenin is required for bone morphogenetic protein 4 expression in human cancer cells. Cancer Res 62:2744–2748. [PubMed] [Google Scholar]
- 18. Koike M, Kose S, Furuta M, Taniguchi N, Yokoya F, Yoneda Y, Imamoto N (2004) Beta‐Catenin shows an overlapping sequence requirement but distinct molecular interactions for its bidirectional passage through nuclear pores. J Biol Chem 279:34038–34047. [DOI] [PubMed] [Google Scholar]
- 19. Kratochwil K, Dull M, Farinas I, Galceran J, Grosschedl R (1996) Lef1 expression is activated by BMP‐4 and regulates inductive tissue interactions in tooth and hair development. Genes Dev 10:1382–1394. [DOI] [PubMed] [Google Scholar]
- 20. Lilien J, Balsamo J (2005) The regulation of cadherin‐mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta‐catenin. Curr Opin Cell Biol 17:459–465. [DOI] [PubMed] [Google Scholar]
- 21. Liu F, Chu EY, Watt B, Zhang Y, Gallant NM, Andl T et al (2008) Wnt/beta‐catenin signaling directs multiple stages of tooth morphogenesis. Dev Biol 313:210–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu X, Rubin JS, Kimmel AR (2005) Rapid, Wnt‐induced changes in GSK3beta associations that regulate beta‐catenin stabilization are mediated by Galpha proteins. Curr Biol 15:1989–1997. [DOI] [PubMed] [Google Scholar]
- 23. Logan CY, Nusse R (2004) The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20:781–810. [DOI] [PubMed] [Google Scholar]
- 24. Ohazama A, Tucker A, Sharpe PT (2005) Organized tooth‐specific cellular differentiation stimulated by BMP4. J Dent Res 84:603–606. [DOI] [PubMed] [Google Scholar]
- 25. Oikonomou E, Barreto DC, Soares B, De Marco L, Buchfelder M, Adams EF (2005) Beta‐catenin mutations in craniopharyngiomas and pituitary adenomas. J Neurooncol 73:205–209. [DOI] [PubMed] [Google Scholar]
- 26. Orsulic S, Huber O, Aberle H, Arnold S, Kemler R (1999) E‐cadherin binding prevents beta‐catenin nuclear localization and beta‐catenin/LEF‐1‐mediated transactivation. J Cell Sci 112:1237–1245. [DOI] [PubMed] [Google Scholar]
- 27. Paulus W, Stockel C, Krauss J, Sorensen N, Roggendorf W (1997) Odontogenic classification of craniopharyngiomas: a clinicopathological study of 54 cases. Histopathology 30:172–176. [DOI] [PubMed] [Google Scholar]
- 28. Roh H, Green DW, Boswell CB, Pippin JA, Drebin JA (2001) Suppression of beta‐catenin inhibits the neoplastic growth of APC‐mutant colon cancer cells. Cancer Res 61:6563–6568. [PubMed] [Google Scholar]
- 29. Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P (1996) Binding of GSK3beta to the APC‐beta‐catenin complex and regulation of complex assembly. Science 272:1023–1026. [DOI] [PubMed] [Google Scholar]
- 30. Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain SH, Masiarz FR et al (1993) Association of the APC gene product with beta‐catenin. Science 262:1731–1734. [DOI] [PubMed] [Google Scholar]
- 31. Sarubi JC, Bei H, Adams EF, Boson WL, Friedman E, Brandao K et al (2001) Clonal composition of human adamantinomatous craniopharyngiomas and somatic mutation analyses of the patched (PTCH), Gsalpha and Gi2alpha genes. Neurosci Lett 310:5–8. [DOI] [PubMed] [Google Scholar]
- 32. Seemayer TA, Blundell JS, Wiglesworth FW (1972) Pituitary craniopharyngioma with tooth formation. Cancer 29:423–430. [DOI] [PubMed] [Google Scholar]
- 33. Sekine S, Shibata T, Kokubu A, Morishita Y, Noguchi M, Nakanishi Y et al (2002) Craniopharyngiomas of adamantinomatous type harbor beta‐catenin gene mutations. Am J Pathol 161:1997–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sekine S, Takata T, Shibata T, Mori M, Morishita Y, Noguchi M et al (2004) Expression of enamel proteins and LEF1 in adamantinomatous craniopharyngioma: evidence for its odontogenic epithelial differentiation. Histopathology 45:573–579. [DOI] [PubMed] [Google Scholar]
- 35. Shu W, Guttentag S, Wang Z, Andl T, Ballard P, Lu MM et al (2005) Wnt/beta‐catenin signaling acts upstream of N‐myc, BMP4, and FGF signaling to regulate proximal‐distal patterning in the lung. Dev Biol 283:226–239. [DOI] [PubMed] [Google Scholar]
- 36. Specht K, Richter T, Muller U, Walch A, Werner M, Hofler H (2001) Quantitative gene expression analysis in microdissected archival formalin‐fixed and paraffin‐embedded tumor tissue. Am J Pathol 158:419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Taniguchi K, Roberts LR, Aderca IN, Dong X, Qian C, Murphy LM et al (2002) Mutational spectrum of beta‐catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene 21:4863–4871. [DOI] [PubMed] [Google Scholar]
- 38. Thesleff I (2003) Epithelial‐mesenchymal signalling regulating tooth morphogenesis. J Cell Sci 116:1647–1648. [DOI] [PubMed] [Google Scholar]
- 39. Udatsu Y, Kusafuka T, Kuroda S, Miao J, Okada A (2001) High frequency of beta‐catenin mutations in hepatoblastoma. Pediatr Surg Int 17:508–512. [DOI] [PubMed] [Google Scholar]
- 40. Von Kries JP, Winbeck G, Asbrand C, Schwarz‐Romond T, Sochnikova N, Dell'Oro A et al (2000) Hot spots in beta‐catenin for interactions with LEF‐1, conductin and APC. Nat Struct Biol 7:800–807. [DOI] [PubMed] [Google Scholar]
- 41. Yoshimoto M, De Toledo SR, Da Silva NS, Bayani J, Bertozzi AP, Stavale JN et al (2004) Comparative genomic hybridization analysis of pediatric adamantinomatous craniopharyngiomas and a review of the literature. J Neurosurg 101(Suppl. 1):85–90. [DOI] [PubMed] [Google Scholar]