

Abstract
Inflammation in the periventricular white matter (PWM) of hypoxic neonatal brain causes myelination disturbances. In this connection, macrophage colony‐stimulating factor (M‐CSF) has been reported to regulate release of proinflammatory cytokines that may be linked to PWM damage. We sought to determine if M‐CSF derived from amoeboid microglial cells (AMC) would promote proinflammatory cytokine production by astrocytes in the PWM following hypoxic exposure, and, if so, whether it is associated with axon degeneration and myelination disturbances. In 1‐day hypoxic rats, expression of M‐CSF was upregulated in AMC. This was coupled with increased expression of CSF‐1 receptor, tumor necrosis factor‐α (TNF‐α) and interleukin‐1β (IL‐1β) in astrocytes, and TNF‐receptor 1 and IL‐receptor 1 on the axons. Neurofilament‐200 immunopositive axons and myelin basic protein immunopositive processes appeared to undergo disruption in 14‐days hypoxic rats. By electron microscopy, some axons showed degenerative changes affecting the microtubules and myelin sheath. Primary cultured microglial cells subjected to hypoxia showed enhanced release of M‐CSF. Remarkably, primary cultured astrocytes treated with conditioned‐medium derived from hypoxic microglia or M‐CSF exhibited increased production of TNF‐α and IL‐1β. Our results suggest that AMC‐derived M‐CSF promotes astrocytes to generate proinflammatory cytokines, which may be involved in axonal damage following a hypoxic insult.
Keywords: amoeboid microglial cells, astrocytes, IL‐1β, M‐CSF, PWMD, TNF‐α
INTRODUCTION
Periventricular white matter damage (PWMD) is one of the most common forms of brain injury in the premature infants and is characterized by periventricular white matter (PWM) focal necrosis and hypomyelination in association with diffuse reactive astrogliosis, microglial activation 10, 27, 31, 38 and axon damage (13). It is well known that injury to premyelinating oligodendrocytes (preOLs) and subsequent reduction in mature myelinating oligodendrocytes result in hypomyelination in long‐term survivors of PWMD (12). There are several possible perinatal risk factors such as hypoxia/ischemia, infection, oxidative stress and inflammation that have been reported to be associated with PWMD 12, 16, 17, 37, 40, 42. In this regard, inflammation induced by hypoxia‐ischemia plays a pivotal role 12, 37. PreOLs and axons are likely to be key targets of excessive inflammatory mediators, which result in myelin deficits.
It has been reported that microglial cells and astrocytes are instrumental in the modulation of inflammatory response in the central nervous system (CNS) 9, 39. Microglial cells are resident immune cells in the CNS and are present in large numbers in the human brain during the window of vulnerability to PWM 2, 22. They play an important role in the development of an inflammatory response by releasing TNF‐α, IL‐1β and MCP‐1 in the developing brain (43). Besides microglia, another cellular source of proinflammatory cytokine production in the PWM would be the astrocytes (35). TNF‐α is reported to be produced by astrocytes besides microglial cells in brain pathology (26). Since both microglial cells and astrocytes are involved in cytokine production, Sawada et al (34) investigated the differences in roles of these cells and concluded that microglia might contribute to the early phase of cytokine production, whereas astrocytes to a late phase in brain pathologies. Our previous study has shown that protein expression of TNF‐α and IL‐1β in the PWM was increased up to 14 days after hypoxic exposure, suggesting a strong and persistent inflammatory response in the PWM of neonatal rats after hypoxic insult (7). By immunofluorescence microscopy, enhanced expression of TNF‐α and IL‐1β was detected in amoeboid microglial cells (AMC) up to 7 days after hypoxic exposure (7). Therefore, AMC were considered to be the main source of the proinflammatory cytokines TNF‐α and IL‐1β at early phase after hypoxic exposure in neonatal rats (7). It was surmised that activated astrocytes may be the main cellular source of TNF‐α and IL‐1β in the protracted period of hypoxia that is, beyond 7 days. However, the mechanism by which astrocytes are activated and release proinflammatory cytokines in the later phase of inflammatory response in the PWM in the hypoxic neonatal rats has not been fully elucidated. It also needs to be further ascertained whether these proinflammatory cytokines are implicated in disruption of developing oligodendroglial processes and axons through their respective receptors.
The macrophage colony‐stimulating factor (M‐CSF), also referred to as CSF‐1, is a hematopoietic cytokine that induces proliferation, migration and activation of microglia, and is released mainly by macrophages, T cells, B cells and microglia 21, 28, 37. M‐CSF has also been identified as an important mediator of the inflammatory response, and can also regulate the release of proinflammatory cytokines from macrophages (11). M‐CSF exerts its pleiotropic effects by binding to its high‐affinity receptor CSF‐1R, which is encoded by the c‐fms proto‐oncogene (32). Arising from the above, we hypothesized that M‐CSF produced by AMC in the early phase following hypoxic injury may activate astrocytes and regulate the release of proinflammatory cytokines from them. This may then sustain and exacerbate the inflammatory response in hypoxic neonatal rats in the late phase. Overproduction of proinflammatory cytokines may cause axonal injury and hypomyelination. To test this hypothesis, this study sought to determine if AMC cross‐talk with astrocytes through M‐CSF in the PWM under hypoxic conditions in the neonatal brain. The expression of M‐CSF in AMC and CSF‐1R in astrocytes was first examined. We then ascertained whether conditioned medium derived from hypoxic microglia or M‐CSF protein can induce the release of proinflammatory cytokines from primary cultured astrocytes in vitro. Furthermore, we also investigated whether the developing axons in the PWM in hypoxic neonatal rats would be a potential target of persistent inflammatory response by expressing TNF receptor 1 (TNF‐R1) and IL‐1 receptor 1 (IL‐1R1), and if so, whether they would undergo damage and degeneration.
MATERIALS AND METHODS
Animals
One‐day old Wistar rats were used in this study because it has been described that the developmental stage of white matter in rats at birth is equivalent to mid‐gestation in humans (36). Seventy‐one 1‐day old Wistar rats were subjected to hypoxia by placing them in a chamber filled with a gas mixture of 5% oxygen and the remainder 95% nitrogen (Model: MCO 18M; Sanyo® Biomedical Electrical Co., Ltd, Tokyo, Japan.) for 2 h. The rats were then allowed to recover under normoxic conditions for 3 and 24 h, 3, 7 or 14 days before being killed. Another group of 71 rats kept outside the chamber was used as age‐matched controls. Animal handling and experiments were approved by Institutional Animal Care and Use Committee, National University of Singapore.
Mixed glial cell culture
Mixed glial cells separated from the cerebral cortex of 1‐day old postnatal Wistar rats were plated on a 75 cm2 flask at a density of 1.2 × 106 cells/mL of Dulbecco's Modified Eagle's Medium (DMEM, Sigma, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS, Hyclone®, Logan, UT, USA). The culture medium was changed after 24 h and then twice a week.
Microglia purification and treatment
Two weeks later, the mixed glial cells were purified with mild trypsinization (0.05%–0.12%) in the presence of 0.2–0.5 mM EDTA and 0.5–0.8 mM Ca2+ for 10 minutes as described before by Saura et al (33). For immunocytochemistry and ELISA, purified microglial cells in the 75 cm2 flask were isolated by trypsinization, and the detached cells were plated at 2.5 × 105 per well on a 24 multi‐well culture dish and incubated at 37°C in a humidified atmosphere of 95% air and 5% CO2 for 24 h. For mRNA extraction, purified cells were plated at 1 × 106 cells per T75 flask. On the following day, the cells were subjected to different treatments. The purity of microglia was assessed by cytochemical detection of microglia using tomato (Lycopersicon esculentum) lectin (1:100: Sigma), a widely used marker for rat microglia, and DAPI (20 µg/mL; Sigma), a nuclear marker of all cells. The microglial cultures with above 96% purity were used in this study.
To study the effects of hypoxia on expression of M‐CSF in microglial cells, the cells were exposed to hypoxia in a chamber (Model 18M; multi‐gas incubator, Sanyo® Company Pte Ltd., Tokyo, Japan) for 1, 2, 4 and 6 h at 3% oxygen, 5% CO2 and 92% nitrogen at 37°C. In all the experiments, microglial cells in matching controls were incubated at 37°C incubator with 95% air and 5% CO2.
Astrocyte purification and treatment
Two weeks later, nonastroglial cells were removed by shaking 20, 25, 32. For immunocytochemistry and ELISA, purified astrocytes in the 75 cm2 flask were isolated by trypsinization, and the detached cells were plated at 2.5 × 105 per well on a 24 multi‐well culture dish and incubated at 37°C in a humidified atmosphere of 95% air and 5% CO2 for 24 h. For Western blotting and mRNA extraction, purified cells were plated at 1 × 106 cells per flask. On the following day, the cells were subjected to different treatments. The purity of astrocyte culture was determined by double labeling of cultured cells with antiglial fibrillary acidic protein (GFAP) (1:1000: Chemicon® International Temecula, CA, USA), a widely used marker for rat astrocytes, and DAPI (20 µg/mL; Sigma), a nuclear marker of all cells. This immunocytochemical analysis indicated that more than 99% of the confluent cells were GFAP‐positive astrocytes.
Astrocyte cultures were divided into three groups: Group I: To study the effects of M‐CSF treatment on expression of TNF‐α, IL‐1β and CSF‐1R in the astrocytes. The cells were treated with M‐CSF (Antigenix® America Inc., Huntington Station, NY, USA; Cat. No. RC333140; 25 ng/mL) for 3, 6, 12 and 24 h. Group II: To study the effects of M‐CSF treatment on MAP kinase pathway. In this, the astrocytes were treated with M‐CSF (25 ng/mL) for 15, 30 minutes, 1 and 2 h. Group III: To examine the effects of MAP kinase inhibitors on M‐CSF treatment‐induced TNF‐α and IL‐1β expression in the astrocytes. Astrocyte cultures were treated with either SP600125 (1 µM; USA® Scientific Inc., Ocala, FL, USA) or SB203580 (1 µM; Merck™, Darmstadt, Germany), inhibitors of JNK and p38, respectively, 30 minutes before M‐CSF (25 ng/mL) administration. In all the experiments, astrocytes were incubated in an incubator filled with 95% air and 5% CO2 at 37°C.
Neutralization test
The cultured microglial cells were treated with 3% oxygen for 4 h and medium was collected for neutralization test. To study the effects of conditioned medium derived from hypoxic microglia on release of TNF‐α and IL‐1β from the astrocytes, the cells were exposed to conditioned medium for 3 h in the absence or presence of M‐CSF antibody at different concentration (5, 10, 15 µg/mL), and the medium was collected for TNF‐α and IL‐1β assay.
Real‐time RT‐PCR
The PWM (supraventricular part of the corpus callosum) was dissected from the hypoxic rats (n = 5 at each time point), and their age‐matched controls (n = 5 at each time point) under a Leica stereomicroscope (MZ 6; Leica™ Pte Ltd, Singapore). Total RNA was extracted from the PWM, cultured microglial cells or cultured astrocytes using RNeasy mini kit (Qiagen™, Valencia, CA, USA) according to the manufacturer's protocol. Standard real time RT‐PCR procedures were applied as described earlier (7). Table 1 summarizes the sequences of specific gene primers used in this study. The expression of target genes was measured in triplicate, and was normalized to β‐actin as an internal control. Gene expression was quantified using a modification of the 2−ΔΔct method as previously described (23).
Table 1.
Sequence of specific primers.
| Primer | Forward | Reverse | Amplicon size |
|---|---|---|---|
| M‐CSF | agagctcctgcctaccaagac | tcctaaaggaaagggtcctga | 195 bp |
| CSF‐1R | ggagatcttctcgcttggtct | ggaaggtgggtcttctggtag | 173 bp |
| TNF‐α | ccaacaaggaggagaagttcc | ctctgcttggtggtttgctac | 134 bp |
| IL‐1β | ggaacccgtgtcttcctaaag | ctgacttggcagaggacaaag | 123 bp |
| β‐actin | tcatgaagtgtgacgttgacatccgt | cctagaagcatttgcggtgcaggatg | 285 bp |
Western blotting
The PWM (supraventricular part of the corpus callosum) was dissected from control and hypoxic rats (n = 5 at each time point) under the Leica stereo microscope (MZ 6; Leica™ Pte Ltd.). Proteins were extracted from the PWM, microglial cells or astrocytes from various groups of cells using protein extraction kit according to the manufacturer's protocol. Protein concentrations were determined by the Bradford's method (3) using bovine serum albumin (Sigma‐Aldrich®, Saint Louis, MO, USA) as a standard. Standard Western blotting procedures were carried out as described earlier (7). The primary antibodies used (Table 2) were as follows: phospho‐JNK, JNK, phospho‐p38, p38, phospho‐ERK1/2, ERK1/2, TNF‐α, IL‐1β, M‐CSF, CSF‐1R, MBP and β‐actin. The HRP‐conjugated secondary antibodies were used in this study (cell signaling technology; Cat. no. 7074). The signal intensity levels of MAP kinase, TNF‐α, IL‐1β, M‐CSF, CSF‐1R and MBP levels compared with the control were measured with Quantity One® software, version 4.4.1 (Bio‐Rad Laboratories Inc., Hercules, CA, USA).
Table 2.
Antibodies and lectin used in experiments.
| Antibody | Host | Company | Cat. No. | Concentration |
|---|---|---|---|---|
| TNF‐α | Rabbit | Chemicon, Temecula, CA, USA | AB1837P | 1:1000 |
| IL‐1β | Rabbit | Chemicon, Temecula, CA, USA | AB1832P | 1:5000 |
| TNF‐R1 | Rabbit | Santa Cruz, Santacruz, CA, USA | sc‐7895 | 1:500 |
| IL‐1R1 | Rabbit | Santa Cruz, Santacruz, CA, USA | sc‐689 | 1:500 |
| M‐CSF | Rabbit | Santa Cruz, Santacruz, CA, USA | sc‐13103 | 1:200 |
| CSF‐1R | Rabbit | Santa Cruz, Santacruz, CA, USA | sc‐692 | 1:500 |
| GFAP | Mouse | Chemicon, Temecula, CA, USA | MAB360 | 1:1000 |
| phospho‐JNK | Rabbit | Cell Signaling Technology, Beverly, MA, USA | 9251s | 1:500 |
| JNK | Rabbit | Cell Signaling Technology, Beverly, MA, USA | 9252 | 1:500 |
| phospho‐p38 | Rabbit | Cell Signaling Technology, Beverly, MA, USA | 9211s | 1:500 |
| p38 | Rabbit | Cell Signaling Technology, Beverly, MA, USA | 9212 | 1:500 |
| phospho‐ERK1/2 | Rabbit | Cell Signaling Technology, Beverly, MA, USA | 9101s | 1:500 |
| ERK1/2 | Rabbit | Cell Signaling Technology, Beverly, MA, USA | 9102 | 1:500 |
| MBP | Goat | Santa Cruz, Santacruz, CA, USA | sc‐13912 | 1:1000 |
| NF‐200 | Mouse | Sigma‐Aldrich, Saint Louis, MO, USA | N0142 | 1:1000 |
| Lectin | Biotinylated | Sigma‐Aldrich, Saint Louis, MO, USA | L0401 | 1:100 |
| β‐actin | Mouse | Sigma‐Aldrich, Saint Louis, MO, USA | A1978 | 1:10 000 |
Double immunofluorescence
All the sections from control and hypoxic rats (n = 3 at each time point) were divided into four groups. The sections in group I were then incubated with antibodies directed against M‐CSF and lectin (Lycopersicon esculentum). The sections in group II were incubated with a mix of anti‐CSF‐1R, anti‐TNF‐α, anti‐IL‐1β and anti‐GFAP. The sections in group III were incubated with a mix of anti‐TNF‐R1, anti‐IL‐1R1 and anti‐neurofilament 200 (NF‐200) antibodies. NF‐200 antibody specifically stains the axons. The sections in the group IV were incubated with a mix of anti‐MBP and anti‐NF‐200 antibodies and processed according to the method described earlier (7). Finally, these sections were mounted with a fluorescent mounting medium (DAKO Cytomation, Glostrup, Denmark). Cellular colocalization was examined by confocal microscopy (LSM FV1000; Olympus® Company Pte Ltd., Tokyo, Japan).
The cultured microglial cells were treated with 3% oxygen for 4 h. Subsequently, they were processed for immunocytochemical analysis as described earlier (7). These cells were incubated with antibodies directed against M‐CSF and lectin. Finally, the cells were counterstained with DAPI and examined under a confocal microscope.
The cultured astrocytes were treated with M‐CSF (25 ng/mL) for 12 h, and immunocytochemical analysis was carried out as mentioned above. These cells were incubated with antibodies directed against CSF‐1R, TNF‐α, IL‐1β and GFAP. Finally, the cells were counterstained with DAPI and examined under a confocal microscope.
The antibodies and lectin used in double immunofluorescence are summarized in Table 2.
ELISA
For microglial cultures, cells were subjected to hypoxia for 1, 2, 4 and 6 h, and the culture medium from each group was collected. M‐CSF concentration released from microglial cells in the culture medium was measured using the rat M‐CSF ELISA kit (Uscn Life™ Science Inc., Wuhan, China; Cat. No. E0090r) according to the manufacturer's protocol. For astrocyte cultures, cells were treated with conditioned medium derived from hypoxic microglia or conditioned medium + M‐CSF antibody (5, 10 or 15 µg/mL) for 3 h, and the culture medium from each group was collected. TNF‐α and IL‐1β concentration in the culture medium was measured using the rat TNF‐α (IBL, Hamburg, Germany; Cat. No.BE 45471) and IL‐1β (IBL; Cat. No.JP27193) ELISA kit according to the manufacturer's protocols. Finally, the reaction plates were read within 30 minutes in an ELISA plate reader (Molecular Devices®, Eugene, OR, USA) at 450 nm.
Electron microscopy
The hypoxic (n = 3 at 7 days and 14 days) and their corresponding controls (n = 3 at each time point) were perfused with a mixed aldehyde fixative composed of 2% paraformaldehyde and 3% glutaraldehyde in 0.1 M phosphate buffer, pH 7.2. After perfusion, the brain was removed and coronal slices (approximately 1 mm thickness) were cut. Blocks of PWM were trimmed from these slices. Vibratome sections (Model 3000™, The Vibratome™ Company, St Louis, MO, USA) of 80–100 µm thickness were prepared from these blocks and rinsed overnight in 0.1 M phosphate buffer. They were then postfixed for 2 h in 1% osmium tetroxide, dehydrated and embedded in Araldite mixture. Ultrathin sections were cut and viewed in a Philips CM 120 electron microscope (FEI™ Company, Hillsboro, OR, USA).
Statistical analysis
The data were presented as mean ± SEM. For in vivo data, statistical significance was evaluated by the Mann–Whitney test (GraphPad Prism 5® (GraphPad Software Inc., La Jolla, CA, USA)). For in vitro data, statistical significance was evaluated by two‐way analysis of variance (GraphPad Prism® 5 software). Results were considered as significant at P < 0.05.
RESULTS
M‐CSF, CSF‐1R mRNA and protein expression in the PWM
M‐CSF mRNA expression was significantly increased in the PWM at 3 and 24 h, 3 and 7 days after the hypoxic exposure in comparison with the corresponding controls (Figure 1A). There was no significant difference between the mRNA expression of M‐CSF at 14 days when compared with the corresponding control rats (Figure 1A). CSF‐1R mRNA expression remained relatively unchanged at 3 h (Figure 1B), but was markedly increased at 24 h and was sustained till 14 days in hypoxic rats when compared with the controls (Figure 1B). The immunoreactive band of M‐CSF protein levels that appeared at approximately 18.5 kDa (Figure 1C), increased significantly in optical density at 3, 24 h, 3, 7 and 14 days after hypoxic exposure as compared with the controls (Figure 1C,D). The double immunoreactive bands of CSF‐1R protein level appeared at approximately 170 kDa (Figure 1C). It was augmented significantly at 24 h, 3, 7 and 14 days after hypoxic exposure when compared with the controls (Figure 1C,E). There was no significant difference between the protein expression of CSF‐1R at 3 h after the hypoxic exposure when compared with the corresponding control rats.
Figure 1.
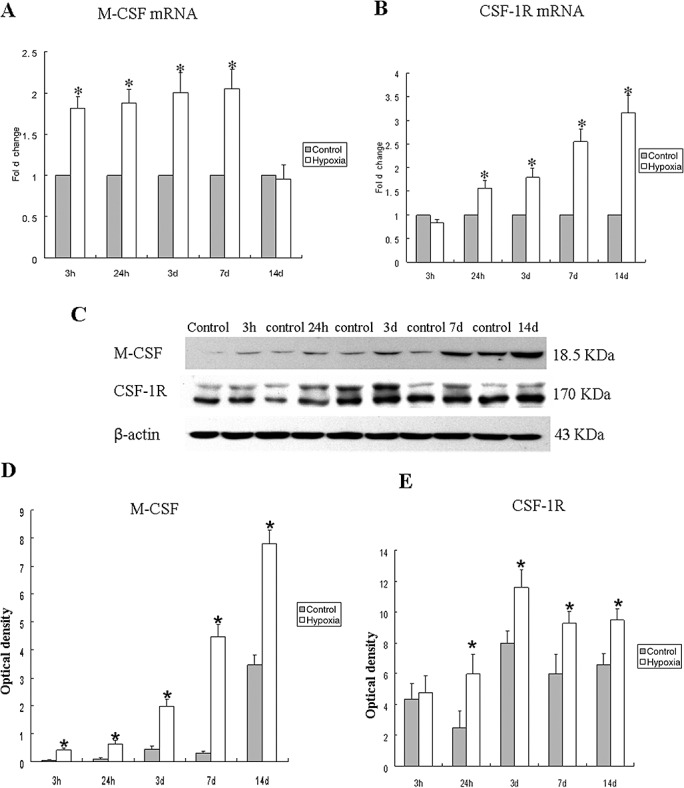
M‐CSF, CSF‐1R mRNA and protein expression in the PWM at 3, 24 h, 3, 7 and 14 days after hypoxic exposure and corresponding control rats. Panels A and B show the graphical representation of the fold changes in M‐CSF and CSF‐1R mRNA, respectively, as quantified by normalization to the β‐actin as an internal control. Panel C shows M‐CSF (18.5 kDa), CSF‐1R (170 kDa) and β‐actin (42 kDa) immunoreactive bands, respectively. Panels D–E show bar graphs depicting significant changes in the optical density of M‐CSF and CSF‐1R, respectively, following hypoxic exposure when compared with their corresponding controls. Significant difference in mRNA and protein levels in the PWM after the hypoxic exposure is evident when compared with controls. *P < 0.05. Abbreviations: M‐CSF = macrophage colony‐stimulating factor; PWM = periventricular white matter.
Cellular localization of M‐CSF and CSF‐1R protein expression in the PWM by double labeling
In the PWM of the control rats, M‐CSF expression was specifically detected in cells, confirmed to be the AMC by double labeling with lectin staining (Figure 2A–C,G–I). At 3 and 7 days following hypoxic exposure, M‐CSF immunoreactivity was enhanced in AMC (Figure 2D–F,J–L) when compared with the controls (Figure 2A–C,G–I). CSF‐1R expression was barely detected in the astrocytes in the control rats (Figure 3A–C,G–I), as confirmed by its co‐localization with GFAP. At 7 and 14 days following hypoxic exposure, CSF‐1R immunoreactivity was markedly enhanced in the astrocytes (Figure 3D–F,J–L). Furthermore, the incidence of CSF‐1R immunoreactive cells was noticeably increased and the cells appeared hypertrophic (Figure 3E,K).
Figure 2.
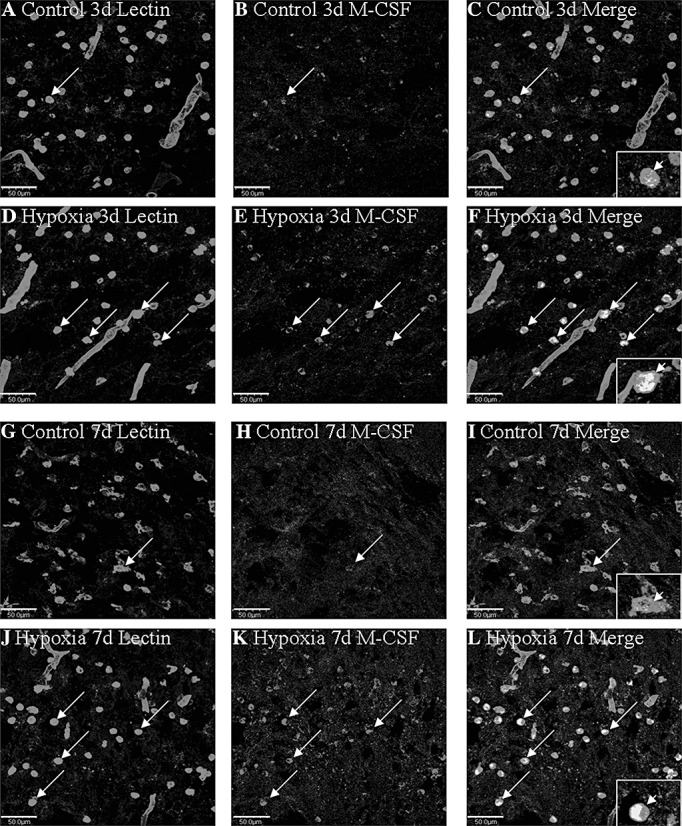
Confocal images showing the distribution of lectin labeled (A,D,G,J, green), and M‐CSF (B,E,H,K red) immunoreactive amoeboid microglial cells (AMC; arrows) in the PWM at 3 and 7 days after the hypoxic exposure and in the corresponding control rat. The co‐localized expression of lectin and M‐CSF in AMC can be seen in C, F, I and L. Note M‐CSF expression in AMC (arrows) is markedly enhanced after the hypoxic exposure. Scale bars: A–L, 50 µm. Abbreviations: M‐CSF = macrophage colony‐stimulating factor; AMC = amoeboid microglial cells.
Figure 3.
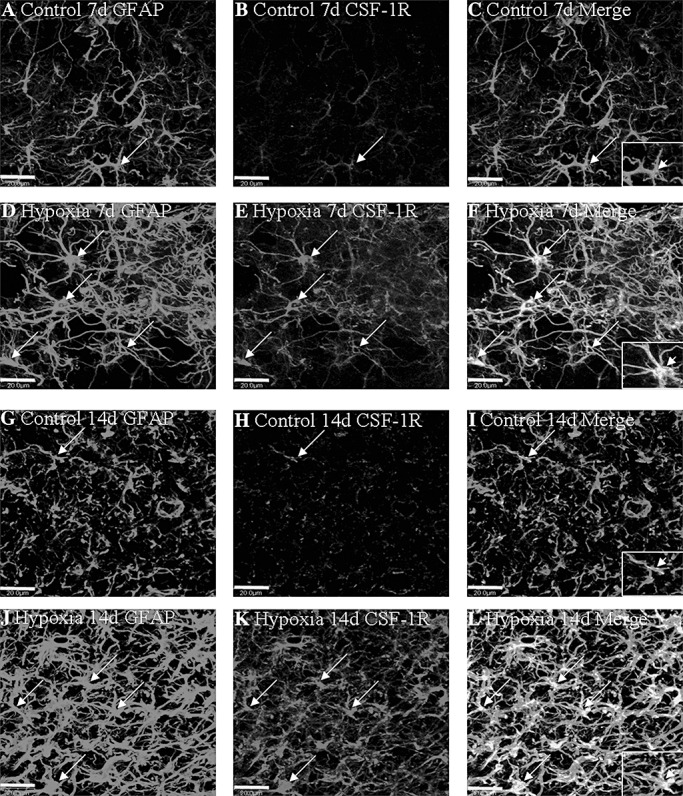
Confocal images showing the distribution of GFAP‐labeled (A,D,G,J, green), and CSF‐1R (B,E,H,K red) immunoreactive astrocytes (arrows) in the PWM at 7 and 14 days after the hypoxic exposure and the corresponding control rats. The colocalized expression of GFAP and CSF‐1R astrocytes can be seen in C, F, I and L. Note CSF‐1R expression in astrocytes (arrows) is markedly enhanced after the hypoxic exposure. Scale bars: A–L, 20 µm. Abbreviations: GFAP = glial fibrillary acidic protein; CSF = colony‐stimulating factor; PWM = periventricular white matter.
Cellular localization of TNF‐α and IL‐1β protein expression in the PWM by double labeling
In the PWM of the control rats sacrificed at different time points, TNF‐α and IL‐1β immunoexpression was detected in cells, confirmed to be the astrocytes by double labeling with GFAP (Figure 4A–C,G–I,M–O; Figure S1A–C,G–I,M–O). There was no noticeable change in TNF‐α and IL‐1β expression at 24 h after the hypoxic exposure (Figure 4D–F; Figure S1D–F) when compared with the corresponding control rats (Figure 4A–C; Figure S1A–C). At 7 and 14 days following hypoxic exposure, very intense TNF‐α and IL‐1β immunoreactivity was detected in large numbers of astrocytes, which appeared hypertrophic (Figure 4J–L,P–R; Figure S1J–L,P–R) when compared with the controls (Figure 4G–I,M–O; Figure S1G–I,M–O).
Figure 4.

Confocal images showing the distribution of GFAP‐labeled (A,D,G,J,M,P, green), and TNF‐α (B,E,H,K,N,Q, red) immunoreactive astrocytes (arrows) in the PWM at 24 h, 7 and 14 days after the hypoxic exposure and the corresponding control rats. The co‐localized expression of GFAP and TNF‐α astrocytes can be seen in panels C, F, I, L, O and R. Note TNF‐α expression in astrocytes (arrows) is markedly enhanced at 7 and 14 days after the hypoxic exposure. Scale bars: A–R, 20 µm. Abbreviations: GFAP = glial fibrillary acidic protein; PWM = periventricular white matter.
Localization of TNF‐R1 and IL‐1R1 protein expression in the PWM by double labeling
In the control rats at 14 days, TNF‐R1 and IL‐1R1 immunofluorescence was localized in some small cells, as well as the axons as confirmed by double immunofluorescence showing colocalization of NF‐200 (Figure 5A–C,G–I). Following hypoxic exposure, TNF‐R1 and IL‐1R1 immunoreactivity was markedly enhanced being more conspicuous along the closely packed axons (Figure 5D–F,J–L).
Figure 5.
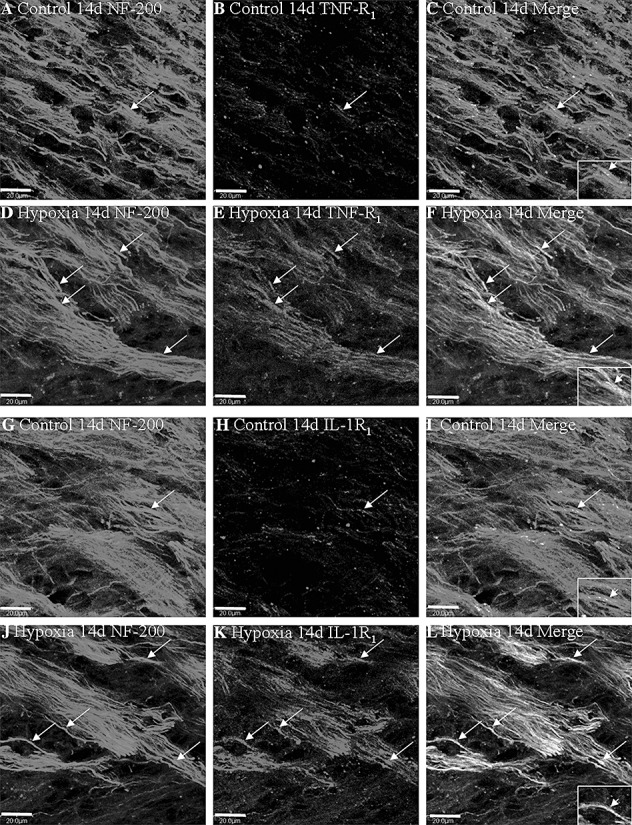
Confocal images showing the distribution of NF‐200 (A,D,G,J, green), TNF‐R1 (B,E red) and IL‐1R1 (H,K red) in axons (arrows) in the PWM at 14 days after the hypoxic exposure and the corresponding control. Co‐localized expression of NF‐200 with TNF‐R1 and IL‐1R1 is depicted in panels C and F, I and L. Note the expression of TNF‐R1 and IL‐1R1 is upregulated after the hypoxic exposure. Scale bars: A–L, 20 µm. Abbreviation: PWM = periventricular white matter.
MBP and NF‐200 protein expression in the PWM
MBP and NF‐200 protein expression was significantly decreased in the PWM at 14 days after the hypoxic exposure in comparison with the corresponding controls (Figure 6A–H). The immunoreactive bands of MBP protein levels (Figure 6A), decreased significantly (P < 0.05) in optical density at 14 days after hypoxic exposure as compared with the controls (Figure 6A,B). However, MBP protein expression was undetected at 3 and 24 h, 3 and 7 days both in the hypoxic and control rats. Confocal images showed that the MBP positive processes and NF‐200 positive axons were extensively disrupted following the hypoxic exposure (Figure 6F,G) as compared with the control (Figure 6C,D).
Figure 6.
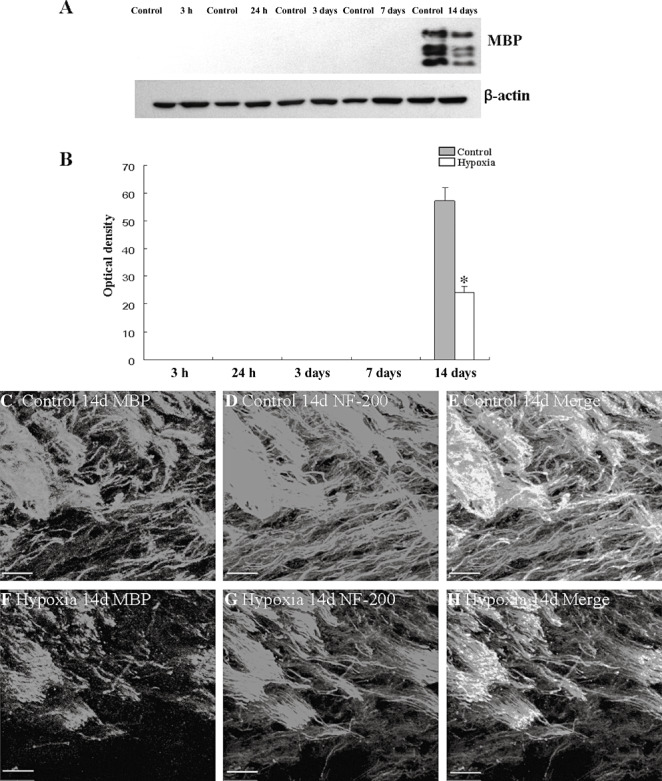
MBP and NF‐200 protein expression in the PWM at 14 days after the hypoxic exposure and the corresponding control rats. A shows MBP and β‐actin (42 KDa) immunoreactive bands, respectively. Bar graph in B shows significant decrease in the optical density of MBP following hypoxic exposure when compared with the corresponding controls (*P < 0.01). Confocal images showing the expression of MBP (C, green), NF‐200 (D, red), and co‐localized expression of MBP and NF‐200 (E) in the PWM at 14 days of control rats. F–H show the expression of MBP (F, green), NF‐200 (G, red), and co‐localized expression of MBP and NF‐200 (H) at 14 days after the hypoxic exposure. Note the MBP positive processes and NF‐200 positive axons are disrupted following the hypoxic exposure (F,G) as compared with the control (C,D). Scale bars: C–H, 50 µm. Abbreviation: PWM = periventricular white matter.
Ultrastructural observations
The most striking structural alteration in the PWM at 7 and 14 days after the hypoxic exposure was dilatation of a variable number of axons, which appeared club shaped in sections (Figure S2A,C). Many of the dilated axons were vesiculated (Figure S2A,C). Hypoxia also affected the myelinated axons, which appeared distorted (Figure S2B,D). In some sectional profiles, myelin‐like figures appeared to be internalized in the axoplasm (Figure S2B,D).
M‐CSF protein and mRNA expression in activated microglia under hypoxic conditions
M‐CSF protein release was augmented at 2, 4 and 6 h after the hypoxic exposure when compared with the controls (Figure 7A). M‐CSF mRNA (Figure 7B) expression was significantly upregulated at the above time intervals after the hypoxic exposure when compared with the controls. Double immunofluorescence labeling showed that the lectin labeling in microglial cells was completely co‐localized with M‐CSF expression (Figure 7C–H). At 4 h after hypoxia, M‐CSF immunofluorescence in microglial cells was markedly increased (Figure 7F–H) in comparison with the control cells (Figure 7C–E).
Figure 7.
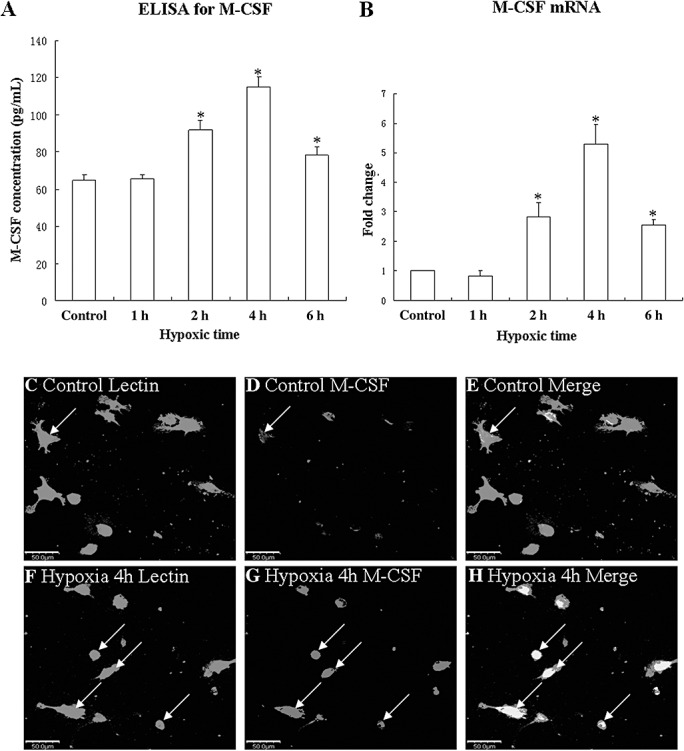
M‐CSF mRNA and protein expression in cultured microglia in the controls and at 1, 2, 4 and 6 h after hypoxic exposure. Bar graph in panel A shows that release of M‐CSF is significantly increased in the medium at 2, 4, and 6 h after hypoxic exposure. Bar graphs in panel B show changes in M‐CSF mRNA expression. Significant differences in mRNA and protein levels in hypoxic microglial cells are evident when compared with controls (*P < 0.05). Confocal images of cultured control microglia showing the expression of lectin (C, green), M‐CSF (D, red) and co‐localized expression of lectin and M‐CSF (E). F‐H show the expression of lectin (F, green), M‐CSF (G, red) and co‐localized expression of lectin and M‐CSF (H) after treatment with 3% oxygen for 4 h. Note the elevated expression of M‐CSF at 4 h after hypoxic exposure (G) as compared with the control cells (D). *P < 0.01. Scale bars: C–H, 50 µm. Abbreviation: M‐CSF = macrophage colony‐stimulating factor.
TNF‐α and IL‐1β release from astrocytes treated with microglial conditioned medium in the neutralization test
TNF‐α and IL‐1β release from astrocytes increased significantly after the treatment with conditioned medium derived from hypoxic microglia for 3 h as compared with the controls (Figure 8A,B). However, when the conditioned medium was neutralized with M‐CSF antibody in doses of 5, 10 or 15 µg/mL, TNF‐α and IL‐1β protein release from astrocytes decreased significantly (Figure 8A,B).
Figure 8.
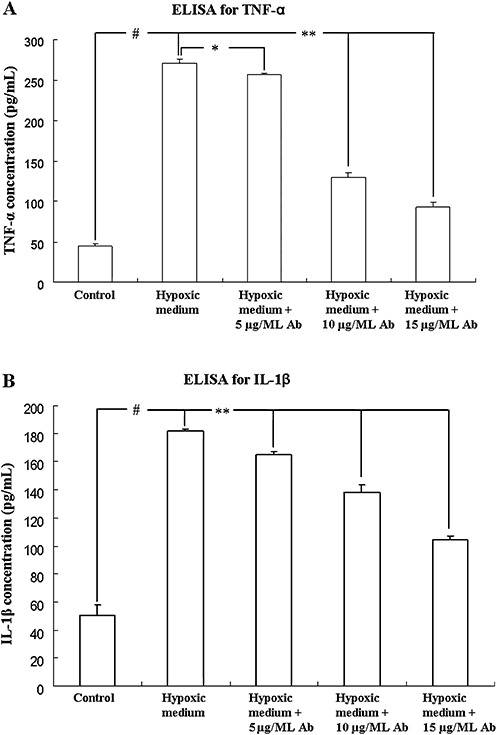
ELISA analysis shows release of TNF‐α and IL‐1β protein from astrocytes after treatment with microglia conditioned medium for 3 h in the neutralization test. Bar graph A shows that conditioned medium triggered release of TNF‐α is significantly increased and is neutralized with M‐CSF antibody in doses of 5, 10 and 15 µg/mL. Bar graph B shows that conditioned medium triggered release of IL‐1β is significantly increased and is neutralized with M‐CSF antibody in doses of 5, 10 and 15 µg/mL. # indicates significant differences between the control and astrocytes treated with conditioned medium. * or ** indicated significant differences between astrocytes treated with conditioned medium alone and neutralized with M‐CSF antibody. **P < 0.01, *P < 0.05, #P < 0.01. Abbreviation: M‐CSF = macrophage colony‐stimulating factor.
TNF‐α, IL‐1β and CSF‐1R mRNA and protein expression in activated astrocytes after M‐CSF treatment
An upregulated TNF‐α mRNA expression was observed at 3, 6 and 12 h after M‐CSF treatment peaking at 3 h when compared with the matching controls (Figure S3A). IL‐1β mRNA expression was concomitantly increased peaking at 3 h as compared with the controls (Figure S3B). There was no significant difference between the mRNA expression of TNF‐α and IL‐1β at 24 h when compared with the corresponding controls (Figure S3A,B). CSF‐1R mRNA expression was significantly increased at different time points following treatment, with M‐CSF peaking at 12 h when compared with the matching controls (Figure S3C). Upregulation of TNF‐α protein level peaking at 3 h after the hypoxic exposure was observed when compared with the controls (Figure S3D,E). IL‐1β protein level also increased significantly after the hypoxic exposure peaking at 3 h as compared with the controls (Figure S3D,F). CSF‐1R protein levels were increased significantly at 3, 6, 12 and 24 h with M‐CSF treatment when compared with the control (Figure S3D,G). Double immunofluorescence labeling showed that TNF‐α, IL‐1β and CSF‐1R expression in the astrocytes was completely co‐localized with GFAP labeling (Figure S4A–R). At 12 h, TNF‐α, IL‐1β and CSF‐1R immunofluorescence in the astrocytes was greatly intensified (Figure S4D–F,J–L,P–R) in comparison with the control cells (Figure S4A–C,G–I,M–O).
MAP Kinase signaling pathway was elicited in activated astrocytes after M‐CSF treatment
MAP kinase signaling transduction pathway plays a significant role in inducing the expression of inflammatory genes in many immune cell types. We therefore examined whether MAP kinases were activated in primary astrocytes cultures after M‐CSF treatment. Western blot analysis showed that the treatment with M‐CSF induced rapid and time‐dependent phosphorylation of JNK (Figure 9A,H) in the astrocytes. The phosphorylated JNK levels increased at 15 minutes and gradually declined to basal levels at 2 h. However, the time‐dependent phosphorylation of p38 was not observed in the astrocytes after M‐CSF treatment (Figure 9B,I). In addition, treatment with M‐CSF induced phosphorylation of ERK1/2 in the astrocytes (Figure 9C,J). The phosphorylated ERK level peaked at 15 minutes after M‐CSF treatment, but unlike JNK, ERK1/2 level was only moderately decreased at 1 and 2 h.
Figure 9.
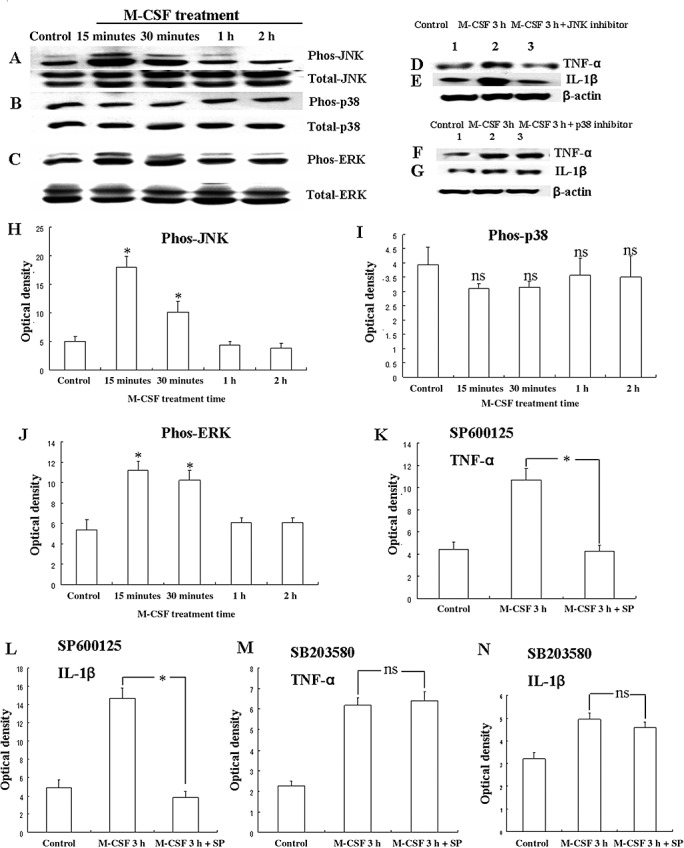
Western blot analysis showing M‐CSF induced TNF‐α and IL‐1β production via activation of MAPK pathway in astrocytes. Panels A, B and C show JNK, p38, ERK phosphorylation and total JNK, p38, ERK immunoreactive bands. Panels D and E show immunoreactive bands which indicate that SP600125 (a JNK inhibitor) suppress the expression of TNF‐α and IL‐1β, respectively, in the astrocytes after M‐CSF treatment for 3 h. Panels F and G show immunoreactive bands, which indicate that SB203580 (a p38 inhibitor) cannot suppress the expression of TNF‐α and IL‐1β, respectively, in the astrocytes after M‐CSF treatment for 3 h. Panels H and J are bar graphs showing significant changes in the optical density of JNK and ERK phosphorylation, respectively, following treatment with M‐CSF. Panels I is a bar graph showing no significant changes in the optical density of p38 phosphorylation following treatment with M‐CSF. Panels K–L are bar graphs showing significant suppression in expression of TNF‐α and IL‐1β by SP600125, respectively. Panels M–N are bar graphs showing no significant suppression in expression of TNF‐α and IL‐1β by SB203580, respectively. *P < 0.05, ns: P > 0.05. Abbreviation: M‐CSF = macrophage colony‐stimulating factor.
Increased TNF‐α and IL‐β production in activated astrocytes after M‐CSF treatment was via activation of MAP kinase pathway
It was hypothesized that TNF‐α and IL‐1β production induced after M‐CSF treatment in activated astrocytes was mediated by the MAP kinase pathways. To ascertain this, TNF‐α and IL‐1β production was measured by Western blotting in M‐CSF‐treated astrocytes exposed to pharmacological agents, SP600125 and SB203580, which are selective inhibitors of JNK and p38, respectively. Incubation of astrocytes with SP600125, 30 minutes prior to treatment with M‐CSF for 3 h decreased the M‐CSF treatment‐induced TNF‐α and IL‐1β expression (Figure 9D,K,E,L). However, incubation of astrocytes with SB203580, 30 minutes prior to treatment with M‐CSF for 3 h did not alter the M‐CSF treatment‐induced TNF‐α and IL‐1β expression (Figure 9F,M,G,N). This indicates that TNF‐α and IL‐β production induced in activated astrocytes after M‐CSF treatment was via activation of JNK‐MAP kinase pathway.
DISCUSSION
The developing brain is extremely susceptible to hypoxic damage because of its high oxygen and energy requirement. Among different regions of the developing brain, the PWM is especially vulnerable in the perinatal period, which may be attributable to the existence of a widespread oligodendrocyte progenitors and lack of anastomoses of blood vessels in this area (10). The severity of PWM injury is dependent on the degree of severity and time of exposure to hypoxia. As long as the blood pressure and cerebral blood flow are maintained, mild hypoxia does not cause brain injury (30). Preconditioning with mild hypoxia can protect the brain against subsequent ischemic insults through a series of endogenous protective or preventative mechanisms resulting in the development of tolerance (15). Neonatal chronic hypoxia irreversibly reduces the extent of myelination in the PWM (18). Our previous studies have shown that the postnatal 1‐day pup subjected to hypoxia (5% oxygen and 95% nitrogen) demonstrated histological changes similar to PWMD in preterm infants, such as injuries of preOLs and axons, astrocytic and microglial reactivity 7, 8, 19.
Although the pathogenesis of PWMD is complex and is not fully elucidated, inflammatory response induced by various stimuli has been recognized as a common feature in the pathogenesis of PWMD (5). Production of inflammatory cytokines, chemokines and free radicals has been thought to be associated with PWMD 5, 10. Studies have shown that besides microglial cells, astrocytes have the ability to produce inflammatory mediators (such as TNF‐α) and free radicals (30). We reported recently a strong and persistent inflammatory response with enormous production of inflammatory cytokines namely, TNF‐α and IL‐1β up to 14 days in the PWM of neonatal rats after hypoxic insult (7). Additionally, we have shown that TNF‐α and IL‐1β immunoexpression was specifically detected in AMC from 24 h to 7 days after a hypoxic insult (7). The present study has extended this showing an upregulated TNF‐α and IL‐1β expression in the astrocytes in the same brain area at 7 and 14 days following a hypoxic exposure. This suggests that in the progression of inflammation in the white matter in the developing brain, while the AMC play major role at an early stage or acute phase in production of TNF‐α and IL‐1β, the astrocytes would be the main cellular source for proinflammatory cytokine production at a late stage i.e 7–14 days after a hypoxic insult.
M‐CSF, a multifunction cytokine, is expressed by activated microglia, cultured neurons and endothelia. It regulates cellular proliferation, migration, activation and proinflammatory response through its transmembrane receptor, CSF‐1R 4, 6. The present study has shown an upregulated expression of M‐CSF mRNA in the PWM of neonatal rats following a hypoxic exposure up to 7 days. Concurrently, CSF‐1R mRNA expression was upregulated and was progressively increased up to 14 days. Double immunofluorescence labeling has shown that M‐CSF expression was detected in AMC at 3 and 7 days after hypoxic exposure. However, CSF‐1R expression was localized in the astrocytes at 7 and 14 days. Western blotting results have further demonstrated that protein expression of M‐CSF and CSF‐1R in the PWM was increased throughout the period of study that is, up to 14 days in hypoxic rats. It is therefore, suggested that M‐CSF produced by AMC at early phase after hypoxia may promote the release of proinflammatory cytokines from astrocytes bearing CSF‐1R. Our in vitro study has shown that the mRNA expression and protein release of M‐CSF in cultured microglia was upregulated significantly after hypoxic exposure for 4 h. This suggests that hypoxia could activate isolated microglia to produce M‐CSF directly. Additionally, the present in vitro results have shown that release of TNF‐α and IL‐1β in cultured astrocytes was enhanced following the treatment with conditioned medium derived from hypoxic primary microglia. After neutralization with M‐CSF antibody, enhanced release of TNF‐α and IL‐1β in cultured astrocytes was dramatically reduced. These results indicated that conditioned medium derived from hypoxic primary microglia contained M‐CSF, which promoted release of TNF‐α and IL‐1β from astrocytes treated with conditioned medium. Furthermore, the present in vitro results have also shown that expression of TNF‐α, IL‐1β and CSF‐1R in cultured astrocytes was enhanced following the treatment with M‐CSF protein. The CSF‐1R consists of an extracellular ligand‐binding domain, a transmembrane domain and an intracellular tyrosine domain (29). M‐CSF binds to CSF‐1R and activates the receptor through autophosphorylation in transmembrane domain, thereby initiating a series of membrane‐proximal tyrosine phosphorylation cascades, leading to rapid stimulation of cytoskeletal remodeling, gene transcription and protein translation 29, 41. The CSF‐1R signaling pathways are complex and poorly understood at present. Recently, it has been reported that M‐CSF, by binding to its high‐affinity receptor CSF‐1R, induces the proliferation of monocyte/macrophages via activated ERK signaling pathway (32). The present results have demonstrated that the phosphorylated JNK and ERK1/2 levels were markedly increased at 15 minutes and gradually declined to basal levels at 2 h in the primary cultured astrocytes treated with M‐CSF protein. However, unlike JNK and ERK1/2, phosphorylated p38 level remained unchanged in the astrocytes after M‐CSF treatment at 15, 30 minutes, 1 and 2 h. Incubation of astrocytes with SP600125, a selective inhibitor of JNK, 30 minutes prior to M‐CSF protein treatment for 3 h decreased the M‐CSF‐induced TNF‐α and IL‐1β expression. However, incubation of astrocytes with SB203580, a selective inhibitors of p38, 30 minutes prior to M‐CSF protein treatment for 3 h did not affect the M‐CSF‐induced TNF‐α and IL‐1β expression. In the light of this, it is concluded that activated microglia generate M‐CSF, which would then induce astrocytes to produce proinflammatory cytokines via the CSF‐1R‐JNK kinase signaling pathway in hypoxic conditions.
A large number of cytokines and chemokines produced by activated microglial cells or astrocytes execute noxious effects on developing oligodendrocyte progenitors and axons through their respective receptors. Our previous results have shown that excessive expression of TNF‐α may induce oligodendrocyte apoptosis through binding to TNF‐R1, and IL‐1β can block oligodendrocyte proliferation at the late progenitor/pro‐oligodendrocyte stage via IL‐1R1 (7). The present study has further shown an upregulated TNF‐R1 and IL‐1R1 expression on the axons in the PWM in hypoxic rats. This was coupled with the disruption of MBP positive processes of oligodendrocytes and associated with NF‐200 positive axons in the PWM. Therefore, it is postulated that overproduction of local TNF‐α and IL‐1β may damage axons and delay the myelination of oligodendrocyte‐axonal unit in the same area via binding to their respective receptors. This notion lends its support from the ultrastructural observation.
The myelination process occurs via a complex interaction between axons and oligodendrocyte processes in association with a wide variety of signals (14). In vitro studies have shown that co‐culture with neurons significantly up‐regulates MBP gene expression of oligodendrocytes, suggesting that axons are indispensable for efficient myelination (24). In vivo experiments in the rat optic nerve demonstrated that oligodendrocyte proliferation and subsequent myelination depend on the electrical activity in axons (1). The present Western blotting results have shown that protein expression of MBP was decreased significantly in the PWM at 14 days after the hypoxic exposure. Therefore, it is postulated that injury to preOLs and axons in hypoxic rats results in hypomyelination in PWMD.
Taken together, the present results suggest that one of the causative factors of a strong and persistent inflammatory response in the PWM of neonatal rats after hypoxic insult may be attributed to M‐CSF produced by AMC. AMC‐released M‐CSF would then induce the neighboring astrocytes to express proinflammatory cytokines, such as TNF‐α and IL‐1β via JNK kinase signaling pathway. The prolonged release of TNF‐α and IL‐1β by AMC and astrocytes in temporal sequence over a protracted period may cause the PWM lesion.
CONCLUSION
This study has shown that the AMC in the PWM in the neonatal brain were the primary cellular source of M‐CSF at early phase following a hypoxic exposure. Concomitantly, the ambient astrocytes upregulated CSF‐1R, TNF‐α and IL‐1β expression. The possible interaction between AMC and astrocytes via the above‐mentioned M‐CSF and its receptor would lead to release of proinflammatory cytokines such as TNF‐α and IL‐1β from the latter cell type via the JNK kinase signaling pathway. It is suggested that the process would persist and augment the inflammatory response in the PWM of the hypoxic neonatal rats. The above‐mentioned proinflammatory cytokines interact with their respective receptors expressed on the axons and oligodendrocytes. This would lead to damage of the axons, as well as delay in their myelination. This was evidenced by the disruption of axons and reduction in the MBP expression following hypoxia. A better understanding of the above process will undoubtedly help to provide a platform for development of potential therapeutic strategies for mitigation of the hypoxia induced PWMD in neonatal brain.
Supporting information
Figure S1. Confocal images showing the distribution of GFAP labeled (A,D,G,J,M,P, green), and IL‐1β (B,E,H,K,N,Q, red) immunoreactive astrocytes (arrows) in the PWM at 24 h, 7 and 14 days after the hypoxic exposure and the corresponding control rats. The colocalized expression of GFAP and IL‐1β astrocytes can be seen in C, F, I, L, O and R. Note IL‐1β expression in astrocytes (arrows) is markedly enhanced at 7 and 14 days after the hypoxic exposure. Scale bars: A–R, 20 μm. Abbreviations: GFAP = glial fibrillary acidic protein; PWM = periventricular white matter.
Figure S2. Electron micrographs of non‐myelinated (A,C) and myelinated (B,D) axons (AX) in the PWM at 7 and 14 days after the hypoxic exposure. Note the swelling and vacuolation of nonmyelinated axons (A,C) and distortion of myelin sheaths (arrows, B,D). Scale bars A,C = 0.5 μm, B = 0.2 μm, D = 1 μm. Abbreviation: PWM = periventricular white matter.
Figure S3. mRNA and protein expression of TNF‐α, IL‐1β and CSF‐1R in cultured control astrocytes and at 3, 6, 12 and 24 h after treatment with M‐CSF in A–G. A (TNF‐α mRNA), B (IL‐1β mRNA) and C (CSF‐1R mRNA) show the graphical representation of the fold changes quantified by normalization to the β‐actin as an internal control. Panel D shows TNF‐α (30 kDa), IL‐1β (17 kDa) and CSF‐1R (170 kDa) immunoreactive protein bands. E, F and G are bar graphs showing changes in the optical density of TNF‐α, IL‐1β and CSF‐1R, respectively, following M‐CSF treatment. Significant differences in mRNA and protein levels in astrocytes with M‐CSF treatment are evident when compared with controls. *P < 0.05. Abbreviation: M‐CSF = macrophage colony‐stimulating factor.
Figure S4. Confocal images of cultured control astrocytes showing the expression of GFAP (A,G,M green), TNF‐α (B, red), IL‐1β (H, red), CSF‐1R (N, red) and co‐localized expression of GFAP and TNF‐α (C), GFAP and IL‐1β (I) as well as GFAP and CSF‐1R (O). D‐F show the expression of GFAP (D, green), TNF‐α (E, red) and co‐localized expression of GFAP and TNF‐α (F) after treatment with M‐CSF for 12 h. J‐L show the expression of GFAP (J, green), IL‐1β (K, red) and co‐localized expression of GFAP and IL‐1β (L) after treatment with M‐CSF for 12 h. P–R show the expression of GFAP (P, green), CSF‐1R (Q, red) and co‐localized expression of GFAP and CSF‐1R (R) after treatment with M‐CSF for 12 h. The expression of TNF‐α (E), IL‐1β (K) and CSF‐1R (Q) is greatly increased in the astrocytes after treatment with M‐CSF for 12 h as compared with the control cells (B,H,N).Scale bars: A–R, 50 μm. Abbreviations: GFAP = glial fibrillary acidic protein; M‐CSF = macrophage colony‐stimulating factor.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
ACKNOWLEDGMENTS
This study was supported by research grants (R‐181‐000‐098‐112) from the National University of Singapore and (R‐181‐000‐120‐213) from the National Medical research Council of Singapore. The technical assistance Ms Y.G. Chan and Dr V. Sivakumar is gratefully acknowledged.
REFERENCES
- 1. Barres BA, Raff MC (1993) Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature 361:258–260. [DOI] [PubMed] [Google Scholar]
- 2. Billiards SS, Haynes RL, Folkerth RD, Trachtenberg FL, Liu LG, Volpe JJ, Kinney HC (2006) Development of microglia in the cerebral white matter of the human fetus and infant. J Comp Neurol 497:199–208. [DOI] [PubMed] [Google Scholar]
- 3. Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem 72:248–254. [DOI] [PubMed] [Google Scholar]
- 4. Cecchini MG, Dominguez MG, Mocci S, Wetterwald A, Felix R, Fleisch H et al (1994) Role of colony stimulating factor‐1 in the establishment and regulation of tissue macrophages during postnatal development of the mouse. Development 120:1357–1372. [DOI] [PubMed] [Google Scholar]
- 5. Chew LJ, Takanohashi A, Bell M (2006) Microglia and inflammation: impact on developmental brain injuries. Ment Retard Dev Disabil Res Rev 12:105–112. [DOI] [PubMed] [Google Scholar]
- 6. Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S et al (2002) Targeted disruption of the mouse colony‐stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 99:111–120. [DOI] [PubMed] [Google Scholar]
- 7. Deng Y, Lu J, Sivakumar V, Ling EA, Kaur C (2008) Amoeboid microglia in the periventricular white matter induce oligodendrocyte damage through expression of proinflammatory cytokines via MAP kinase signaling pathway in hypoxic neonatal rats. Brain Pathol 18:387–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deng YY, Lu J, Ling EA, Kaur C (2009) Monocyte chemoattractant protein‐1 (MCP‐1) produced via NF‐kappaB signaling pathway mediates migration of amoeboid microglia in the periventricular white matter in hypoxic neonatal rats. Glia 57:604–621. [DOI] [PubMed] [Google Scholar]
- 9. Dheen ST, Jun Y, Yan Z, Tay SS, Ling EA (2005) Retinoic acid inhibits expression of TNF‐alpha and iNOS in activated rat microglia. Glia 50:21–31. [DOI] [PubMed] [Google Scholar]
- 10. Folkerth RD (2006) Periventricular leukomalacia: overview and recent findings. Pediatr Dev Pathol 9:3–13. [DOI] [PubMed] [Google Scholar]
- 11. Hao AJ, Dheen ST, Ling EA (2002) Expression of macrophage colony‐stimulating factor and its receptor in microglia activation is linked to teratogen‐induced neuronal damage. Neuroscience 112:889–900. [DOI] [PubMed] [Google Scholar]
- 12. Haynes RL, Folkerth RD, Keefe RJ, Sung I, Swzeda LI, Rosenberg PA et al (2003) Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol 62:441–450. [DOI] [PubMed] [Google Scholar]
- 13. Haynes RL, Billiards SS, Borenstein NS, Volpe JJ, Kinney HC (2008) Diffuse axonal injury in periventricular leukomalacia as determined by apoptotic marker fractin. Pediatr Res 63:656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jakovcevski I, Filipovic R, Mo Z, Rakic S, Zecevic N (2009) Oligodendrocyte development and the onset of myelination in the human fetal brain. Front Neuroanat 3:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jones NM, Lee EM, Brown TG, Jarrott B, Beart PM (2006) Hypoxic preconditioning produces differential expression of hypoxia‐inducible factor‐1alpha (HIF‐1alpha) and its regulatory enzyme HIF prolyl hydroxylase 2 in neonatal rat brain. Neurosci Lett 404:72–77. [DOI] [PubMed] [Google Scholar]
- 16. Kadhim H, Tabarki B, Verellen G, De Prez C, Rona AM, Sebire G (2001) Inflammatory cytokines in the pathogenesis of periventricular leukomalacia. Neurology 56:1278–1284. [DOI] [PubMed] [Google Scholar]
- 17. Kadhim H, Tabarki B, De Prez C, Rona AM, Sebire G (2002) Interleukin‐2 in the pathogenesis of perinatal white matter damage. Neurology 58:1125–1128. [DOI] [PubMed] [Google Scholar]
- 18. Kanaan A, Farahani R, Douglas RM, Lamanna JC, Haddad GG (2006) Effect of chronic continuous or intermittent hypoxia and reoxygenation on cerebral capillary density and myelination. Am J Physiol Regul Integr Comp Physiol 290:R1105–R1114. [DOI] [PubMed] [Google Scholar]
- 19. Kaur C, Sivakumar V, Ang LS, Sundaresan A (2006) Hypoxic damage to the periventricular white matter in neonatal brain: role of vascular endothelial growth factor, nitric oxide and excitotoxicity. J Neurochem 98:1200–1216. [DOI] [PubMed] [Google Scholar]
- 20. Kliot M, Smith GM, Siegal JD, Silver J (1990) Astrocyte‐polymer implants promote regeneration of dorsal root fibers into the adult mammalian spinal cord. Exp Neurol 109:57–69. [DOI] [PubMed] [Google Scholar]
- 21. Lee SC, Liu W, Roth P, Dickson DW, Berman JW, Brosnan CF (1993) Macrophage colony‐stimulating factor in human fetal astrocytes and microglia. Differential regulation by cytokines and lipopolysaccharide, and modulation of class II MHC on microglia. J Immunol 150:594–604. [PubMed] [Google Scholar]
- 22. Ling EA, Wong WC (1993) The origin and nature of ramified and amoeboid microglia: a historical review and current concepts. Glia 7:9–18. [DOI] [PubMed] [Google Scholar]
- 23. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C (T)). Method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- 24. Macklin WB, Weill CL, Deininger PL (1986) Expression of myelin proteolipid and basic protein mRNAs in cultured cells. J Neurosci Res 16:203–217. [DOI] [PubMed] [Google Scholar]
- 25. McCarthy KD, De Vellis J (1980) Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 85:890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Medana IM, Hunt NH, Chaudhri G (1997) Tumor necrosis factor‐alpha expression in the brain during fatal murine cerebral malaria: evidence for production by microglia and astrocytes. Am J Pathol 150:1473–1486. [PMC free article] [PubMed] [Google Scholar]
- 27. Mu D, Jiang X, Sheldon RA, Fox CK, Hamrick SE, Vexler ZS, Ferriero DM (2003) Regulation of hypoxia‐inducible factor 1alpha and induction of vascular endothelial growth factor in a rat neonatal stroke model. Neurobiol Dis 14:524–534. [DOI] [PubMed] [Google Scholar]
- 28. Nohava K, Malipiero U, Frei K, Fontana A (1992) Neurons and neuroblastoma as a source of macrophage colony‐stimulating factor. Eur J Immunol 22:2539–2545. [DOI] [PubMed] [Google Scholar]
- 29. Pixley FJ, Stanley ER (2004) CSF‐1 regulation of the wandering macrophage: complexity in action. Trends Cell Biol 14:628–638. [DOI] [PubMed] [Google Scholar]
- 30. Ran R, Xu H, Lu A, Bernaudin M, Sharp FR (2005) Hypoxia preconditioning in the brain. Dev Neurosci 27:87–92. [DOI] [PubMed] [Google Scholar]
- 31. Rezaie P, Dean A (2002) Periventricular leukomalacia, inflammation and white matter lesions within the developing nervous system. Neuropathology 22:106–132. [DOI] [PubMed] [Google Scholar]
- 32. Rovida E, Spinelli E, Sdelci S, Barbetti V, Morandi A, Giuntoli S, Dello SP (2008) ERK5/BMK1 is indispensable for optimal colony‐stimulating factor 1 (CSF‐1)‐induced proliferation in macrophages in a Src‐dependent fashion. J Immunol 180:4166–4172. [DOI] [PubMed] [Google Scholar]
- 33. Saura J, Tusell JM, Serratosa J (2003) High‐yield isolation of murine microglia by mild trypsinization. Glia 44:183–189. [DOI] [PubMed] [Google Scholar]
- 34. Sawada M, Suzumura A, Marunouchi T (1995a) Cytokine network in the central nervous system and its roles in growth and differentiation of glial and neuronal cells. Int J Dev Neurosci 13:253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sawada M, Suzumura A, Marunouchi T (1995b) Induction of functional interleukin‐2 receptor in mouse microglia. J Neurochem 64:1973–1979. [DOI] [PubMed] [Google Scholar]
- 36. Sheldon RA, Chuai J, Ferriero DM (1996) A rat model for hypoxic‐ischemic brain damage in very premature infants. Biol Neonate 69:327–341. [DOI] [PubMed] [Google Scholar]
- 37. Stanley ER, Berg KL, Einstein DB, Lee PS, Pixley FJ, Wang Y, Yeung YG (1997) Biology and action of colony—stimulating factor‐1. Mol Reprod Dev 46:4–10. [DOI] [PubMed] [Google Scholar]
- 38. Sugai K, Ito M, Tateishi I, Funabiki T, Nishikawa M (2006) Neonatal periventricular leukomalacia due to severe, poorly controlled asthma in the mother. Allergol Int 55:207–212. [DOI] [PubMed] [Google Scholar]
- 39. Vilhardt F (2005) Microglia: phagocyte and glia cell. Int J Biochem Cell Biol 37:17–21. [DOI] [PubMed] [Google Scholar]
- 40. Wu YW, Colford JM Jr (2000) Chorioamnionitis as a risk factor for cerebral palsy: a meta‐analysis. JAMA 284:1417–1424. [DOI] [PubMed] [Google Scholar]
- 41. Yeung YG, Stanley ER (2003) Proteomic approaches to the analysis of early events in colony‐stimulating factor‐1 signal transduction. Mol Cell Proteomics 2:1143–1155. [DOI] [PubMed] [Google Scholar]
- 42. Yoon BH, Kim CJ, Romero R, Jun JK, Park KH, Choi ST, Chi JG (1997) Experimentally induced intrauterine infection causes fetal brain white matter lesions in rabbits. Am J Obstet Gynecol 177:797–802. [DOI] [PubMed] [Google Scholar]
- 43. Zhou Y, Ling EA, Dheen ST (2007) Dexamethasone suppresses monocyte chemoattractant protein‐1 production via mitogen activated protein kinase phosphatase‐1 dependent inhibition of Jun N‐terminal kinase and p38 mitogen‐activated protein kinase in activated rat microglia. J Neurochem 102:667–678. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Confocal images showing the distribution of GFAP labeled (A,D,G,J,M,P, green), and IL‐1β (B,E,H,K,N,Q, red) immunoreactive astrocytes (arrows) in the PWM at 24 h, 7 and 14 days after the hypoxic exposure and the corresponding control rats. The colocalized expression of GFAP and IL‐1β astrocytes can be seen in C, F, I, L, O and R. Note IL‐1β expression in astrocytes (arrows) is markedly enhanced at 7 and 14 days after the hypoxic exposure. Scale bars: A–R, 20 μm. Abbreviations: GFAP = glial fibrillary acidic protein; PWM = periventricular white matter.
Figure S2. Electron micrographs of non‐myelinated (A,C) and myelinated (B,D) axons (AX) in the PWM at 7 and 14 days after the hypoxic exposure. Note the swelling and vacuolation of nonmyelinated axons (A,C) and distortion of myelin sheaths (arrows, B,D). Scale bars A,C = 0.5 μm, B = 0.2 μm, D = 1 μm. Abbreviation: PWM = periventricular white matter.
Figure S3. mRNA and protein expression of TNF‐α, IL‐1β and CSF‐1R in cultured control astrocytes and at 3, 6, 12 and 24 h after treatment with M‐CSF in A–G. A (TNF‐α mRNA), B (IL‐1β mRNA) and C (CSF‐1R mRNA) show the graphical representation of the fold changes quantified by normalization to the β‐actin as an internal control. Panel D shows TNF‐α (30 kDa), IL‐1β (17 kDa) and CSF‐1R (170 kDa) immunoreactive protein bands. E, F and G are bar graphs showing changes in the optical density of TNF‐α, IL‐1β and CSF‐1R, respectively, following M‐CSF treatment. Significant differences in mRNA and protein levels in astrocytes with M‐CSF treatment are evident when compared with controls. *P < 0.05. Abbreviation: M‐CSF = macrophage colony‐stimulating factor.
Figure S4. Confocal images of cultured control astrocytes showing the expression of GFAP (A,G,M green), TNF‐α (B, red), IL‐1β (H, red), CSF‐1R (N, red) and co‐localized expression of GFAP and TNF‐α (C), GFAP and IL‐1β (I) as well as GFAP and CSF‐1R (O). D‐F show the expression of GFAP (D, green), TNF‐α (E, red) and co‐localized expression of GFAP and TNF‐α (F) after treatment with M‐CSF for 12 h. J‐L show the expression of GFAP (J, green), IL‐1β (K, red) and co‐localized expression of GFAP and IL‐1β (L) after treatment with M‐CSF for 12 h. P–R show the expression of GFAP (P, green), CSF‐1R (Q, red) and co‐localized expression of GFAP and CSF‐1R (R) after treatment with M‐CSF for 12 h. The expression of TNF‐α (E), IL‐1β (K) and CSF‐1R (Q) is greatly increased in the astrocytes after treatment with M‐CSF for 12 h as compared with the control cells (B,H,N).Scale bars: A–R, 50 μm. Abbreviations: GFAP = glial fibrillary acidic protein; M‐CSF = macrophage colony‐stimulating factor.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item