

Recently sustained virologic response can be achieved by direct-acting antiviral (DAA) therapy in most hepatitis C patients. Unfortunately, DAA therapy does not completely eliminate a risk of hepatocellular carcinoma (HCC).
KEYWORDS: HCV, HOX gene, PRC1, H2A, monoubiquitination
ABSTRACT
Hepatitis C virus (HCV) infection causes liver pathologies, including hepatocellular carcinoma (HCC). Homeobox (HOX) gene products regulate embryonic development and are associated with tumorigenesis, although the regulation of HOX genes by HCV infection has not been clarified in detail. We examined the effect of HCV infection on HOX gene expression. In this study, HCV infection induced more than half of the HOX genes and reduced the level of histone H2A monoubiquitination on lysine 119 (K119) (H2Aub), which represses HOX gene promoter activity. HCV infection also promoted proteasome-dependent degradation of RNF2, which is an E3 ligase mediating H2A monoubiquitination as a component of polycomb repressive complex 1. Since full-genomic replicon cells but not subgenomic replicon cells exhibited reduced RNF2 and H2Aub levels and induction of HOX genes, we focused on the core protein. Expression of the core protein reduced the amounts of RNF2 and H2Aub and induced HOX genes. Treatment with LY-411575, which can reduce HCV core protein expression via signal peptide peptidase (SPP) inhibition without affecting other viral proteins, dose-dependently restored the amounts of RNF2 and H2Aub in HCV-infected cells and impaired the induction of HOX genes and production of viral particles but not viral replication. The chromatin immunoprecipitation assay results also indicated infection- and proteasome-dependent reductions in H2Aub located in HOX gene promoters. These results suggest that HCV infection or core protein induces HOX genes by impairing histone H2A monoubiquitination via a reduction in the RNF2 level.
IMPORTANCE Recently sustained virologic response can be achieved by direct-acting antiviral (DAA) therapy in most hepatitis C patients. Unfortunately, DAA therapy does not completely eliminate a risk of hepatocellular carcinoma (HCC). Several epigenetic factors, including histone modifications, are well known to contribute to hepatitis C virus (HCV)-associated HCC. However, the regulation of histone modifications by HCV infection has not been clarified in detail. In this study, our data suggest that HCV infection or HCV core protein expression impairs monoubiquitination of histone H2A K119 in the homeobox (HOX) gene promoter via destabilization of RNF2 and then induces HOX genes. Several lines of evidence suggest that the expression of several HOX genes is dysregulated in certain types of tumors. These findings reveal a novel mechanism of HCV-related histone modification and may provide information about new targets for diagnosis and prevention of HCC occurrence.
INTRODUCTION
Hepatitis C virus (HCV) is a causative factor of hepatitis C, which affects approximately 71 million individuals worldwide and is a leading cause of liver pathologies, including chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma (HCC) (1). HCV is an enveloped RNA virus belonging to the genus Hepacivirus of the Flaviviridae family. The positive-strand RNA genome of HCV, which is 9.6 kb in length, encodes a single polyprotein that is cleaved by viral and host proteases to produce 10 viral proteins. The nonstructural proteins—p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B—play important roles in viral genome replication, assembly, and budding, while the structural proteins—the core protein, E1, and E2—form the viral particles enclosing the viral genome. The core protein, a component of the nucleocapsid, has been implicated in cellular transformation and immortalization of human primary hepatocytes (2, 3). Furthermore, mice with hepatic expression of the core protein (core Tg mice) develop insulin resistance (4), steatosis (5) and HCC (6), suggesting that the core protein plays crucial roles in HCV pathogenesis.
Several reports suggest that alterations in gene expression patterns in HCV-infected cells or liver tissue are involved in the mechanism underlying HCV pathogenesis (7–9). Several epigenetic factors, including genomic DNA methylation, histone modifications, and microRNA (miRNA) regulation, contribute to HCV-associated HCC via the following mechanisms: the core protein can promote the expression of DNA methyltransferase 1 and 3B, both of which lead to epigenetic alterations in hepatocytes in hepatitis C patients (10, 11). Expression of viral proteins or infection with cell culture-derived HCV (HCVcc) suppresses histone H4 acetylation and H2AX phosphorylation, followed by alterations in the gene expression pattern associated with HCC development (12). However, the regulation of other histone modifications by HCV infection has not been clarified in detail.
Chromatin comprises DNA, histones, and nonhistone proteins (13). A DNA strand with a length of 146 bp is wrapped around an octamer of core histones, which consists of one H3-H3-H4-H4 tetramer and two H2A-H2B dimers, to form nucleosomal core particles (13). Amino acid residues in histone tails are acetylated, methylated, phosphorylated and/or ubiquitinated to affect chromatin structure and gene function (13). H2A ubiquitination is one of the general modifications, occurring on 10% of all H2A proteins (14). Although lysine 129 (K129) and K15 of H2A are ubiquitinated, monoubiquitination occurs predominantly on K119 of H2A (15). H2A monoubiquitination on K119 (H2Aub) affects chromatin structure and nucleosome composition, leading to transcriptional repression of target genes (15). H2Aub slightly facilitates binding of the linker histone H1 to the nucleosome (16), while deubiquitination of H2Aub coordinates histone acetylation and H1 dissociation to activate the related genes (17). H2Aub interferes with recruitment of the histone chaperone complex “facilitates chromatin transcription” (FACT), resulting in blockade of transcriptional elongation (18). In addition, H2Aub blocks subsequent activation of genes via the histone modifications H3K4 di- and trimethylation (19). Polycomb repressive complex (PRC1) catalyzes H2A K119 monoubiquitination to silence several developmental genes and then plays essential roles in cell lineage commitment, stem cell identity, tumorigenesis, and genomic imprinting (15, 20). HOX genes are classical targets of PRC1 (20).
HOX genes were originally identified in Drosophila melanogaster, and mutations in these genes were found to result in radical alterations in body parts (21). The HOX gene family is a well-known family of genes that provide cell position information during embryogenesis. The HOX gene family contains a common element of 183 bp, the “homeobox,” which encodes a highly conserved protein domain containing 61 amino acid residues, the “homeodomain” (22). The homeodomain is responsible for recognition and binding of a specific DNA motif. Mammalian genomes contain 39 HOX genes, which are classified into four clusters termed HOX A, B, C, and D that are located on separate chromosomes (23). Each HOX gene exhibits organ-specific expression in adult individuals, suggesting that HOX genes regulate organ and tissue differentiation and maturation in the postnatal body as well as the embryo (23). Furthermore, many accumulating lines of evidence suggest that the expression of several HOX genes is dysregulated in certain types of tumors (24, 25).
Mukherjee et al. reported that HCV infection promoted the expression of HOXA1 via miR-181c downregulation (26). However, the effect of HCV infection on other HOX genes is unknown. In this study, we examined the effect of HCV infection on the induction of HOX gene expression. The expression of more than half of the HOX genes was induced in HCV-infected cells compared to mock-infected cells. Furthermore, we examined the effect of HCV infection on H2A monoubiquitination and RNF2 stability to clarify the mechanism by which HCV infection regulates HOX gene induction.
RESULTS
Impact of HCV infection on the expression of HOX genes.
We evaluated the effect of HCV infection on HOX gene expression. Huh7OK1 cells were infected with HCV and were then harvested at 9 days postinfection (dpi). We then prepared total RNA from the harvested cells and evaluated the transcription levels of 39 HOX genes in HCV-infected and mock-infected cells by reverse transcription-semiquantitative PCR (RT-sqPCR) (Fig. 1). Figure 1A shows that PCR products for 39 HOX genes were amplified by RT-sqPCR and then subjected to an agarose electrophoresis. The columns are arranged in corresponding clusters as HOXA (A), HOXB (B), HOXC (C), and HOXD (D). The rows are arranged in gene numbers, HOX A1 to HOXA13 (1 to 13), HOXB1 to HOXB13 (1 to 13), HOXC4 to HOXC13 (4 to 13), and HOXD1 to HOXD13 (1 to 13). The expression levels of more than half of the HOX genes were increased by HCV infection, although those of some of the genes were reduced or not affected (Fig. 1A); moreover, HCV RNA was detected in HCV-infected cells but not in mock-infected cells (Fig. 1B). The value of HOX gene induction was calculated as the ratio of each HOX gene-PCR product in an HCV lane to the same region in a mock lane (Fig. 1A) and then represented as a heat map (Fig. 1C). White color indicates no change, red color indicates an increase, and blue color indicates a decrease in HOX transcription level in HCV-infected cells. The mRNA levels of HOX A4, A6, A9, B1, B2, B3, B8, B9, B13, C4, C12, C13, D4, D8, D10, D12, and D13 were markedly increased by at least 2-fold in HCV-infected cells compared with mock-infected cells (Fig. 1C). Furthermore, the transcription of 13 HOX genes tended to increase by 1.2- to 2-fold in HCV-infected cells compared with mock-infected cells (Fig. 1C). Notably, HOXB9 and C13 gene expression was potently induced in HCV-infected cells but was not detected in mock-infected cells (Fig. 1C). Thus, HCV infection-dependent induction of HOXB9 and C13 was prominently observed, suggesting that both genes may be silenced under uninfected conditions. HOXB9, C13, and D13 were induced in Huh7.5.1. cells by HCV infection in a similar way to the data using Huh7OK1 (Fig. 1D). HOXB9 mRNA and HCV RNA were detected in human primary hepatocytes (PHHs) infected with HCV at 1 dpi but not at 2 dpi or later (Fig. 1E), while other HOX genes, HOXB1, C13, and D13, were also induced at 1 dpi in the infected PHHs (Fig. 1E). These data suggest that HCV infection activates the expression of silenced HOX gene alleles.
FIG 1.
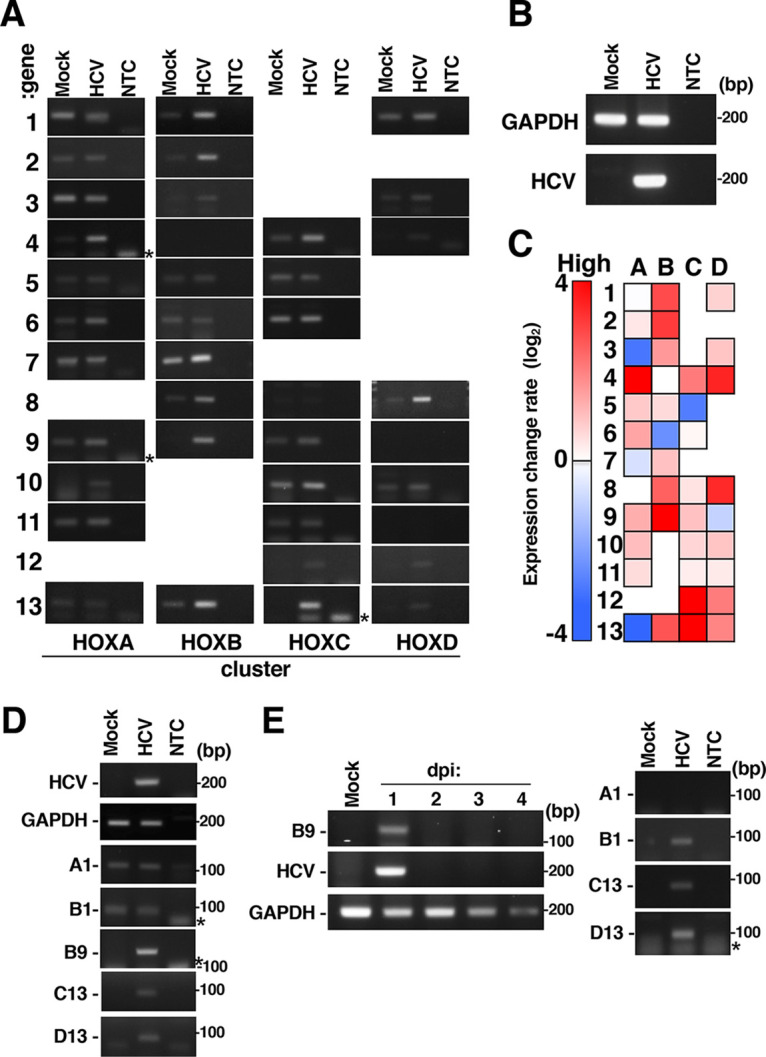
Effect of HCV infection on HOX gene expression. Huh7OK1 cells were infected with HCVcc at a multiplicity of infection (MOI) of 0.1 and were then harvested at 9 dpi. Total RNA was prepared from HCV-infected cells (HCV) or mock-infected cells (Mock). HCV RNA and HOX gene mRNA and GAPDH mRNA levels were evaluated by RT-sqPCR. (A) Expression of HOX genes. The gene numbers and the cluster names are indicated on the left and bottom, respectively, of the panels. NTC, no-template control. (B) Expression of GAPDH mRNA and HCV RNA. (C) Heatmap analysis of HOX gene expression in HCV-infected cells. The intensity of each band in panels A and B was quantified using ImageJ software. The data are presented as the ratio of each HOX gene mRNA level to the GAPDH mRNA level. The red and blue colors indicate higher and the lowest expression levels of each HOX gene, respectively. The data shown in this figure are representative of three independent experiments. (D) Huh7.5.1 cells were infected with HCVcc at an MOI of 0.1 and were then harvested at 9 dpi. HOX gene mRNA and GAPDH mRNA levels were evaluated by RT-sqPCR. A1, B1, B9, C13, and D13 indicate HOXA1, HOXB1, HOXB9, HOXC13, and HOXD13. (E) Primary human hepatocytes (PHHs) were infected with HCVcc at an MOI of 1 and were harvested at an indicated dpi (left half) or 1 dpi (right half). HOX gene mRNA and GAPDH mRNA levels were evaluated by RT-sqPCR. The asterisk indicates a product of primer dimer on the left side of the panel. A1, B1, B9, C13, and D13 indicate HOXA1, HOXB1, HOXB9, HOXC13, and HOXD13.
Effect of HCV infection on histone H2A K119 monoubiquitination.
H2Aub in the promoter region is a prevalent modification correlated with silencing of a HOX gene (15, 20). We examined the effect of HCV infection on the monoubiquitination of H2A K119. HCV-infected cells were fixed or harvested at 6 dpi. H2Aub was weakly stained in HCV core-positive cells compared to mock-infected cells or core-negative cells (Fig. 2A). The intensity of the H2Aub-staining signal was significantly lower in the core-positive cells than in the core-negative cells (Fig. 2A and B). The levels of H2Aub, H2A, and H3 in HCV-infected cells and mock-infected cells were evaluated by flow cytometry (Fig. 2C) and Western blotting (Fig. 2D). The level of H2Aub was decreased in HCV-infected cells compared to mock-infected cells, while infection did not affect the amounts of total H2A and H3 (Fig. 2C and D). These results suggest that HCV infection reduces the level of H2Aub.
FIG 2.

HCV infection reduced the level of H2A K119 monoubiquitination. (A) HCV-infected cells were fixed at 6 dpi and were then stained with DAPI. The core protein and H2Aub in HCV-infected cells (HCV) and mock-infected cells (Mock) were stained with a mouse anti-core protein antibody and a rabbit anti-H2Aub antibody and were then stained with an AF488-conjugated goat anti-mouse IgG antibody and an AF594-conjugated goat anti-rabbit IgG antibody. The stained samples were observed under a fluorescence microscope. Magnified microscopic images of the cells stained with anti-core and anti-H2ub antibodies are shown in the lower images in panel A. (B) The intensity of H2Aub in the core protein-positive or core protein-negative area in the images in panel A was estimated using software as described in Materials and Methods. The data are presented as the mean ± SD values, which were calculated from 15 fields of view. The significance of the differences is indicated for each bar pair (***, P < 0.005). (C and D) The levels of histone H2Aub, H2A, and H3 in HCV-infected and mock-infected cells were evaluated by flow cytometry (C) and Western blotting (D). The data shown in this figure are representative of three independent experiments.
Effect of HCV infection on H2A monoubiquitination in the HOXB9 gene promoter.
HOXB9 gene transcription was induced more potently than the transcription of other HOX genes in HCV-infected cells, but its transcription was not detected in mock-infected cells (Fig. 1). Thus, we examined the effect of HCV infection on the activation of the HOXB9 gene as a representative of HCV-induced HOX genes. HCV-infected cells and mock-infected cells were harvested at 3, 6, and 9 dpi for evaluation of HOXB9 mRNA expression. HOXB9 mRNA expression was increased in a time-dependent manner after infection, corresponding to HCV RNA replication (Fig. 3A). More than 80% of HCV-infected cells (9 dpi) exhibited positive staining for the core protein (Fig. 3B). Infected cells were subjected to flow cytometry. HOX gene expression was potentiated dependently on the level of NS5A (Fig. 3C), suggesting that the fluorescence intensity of HOXB9 protein is correlated with that of NS5A. Treatment with a direct antiviral agent, daclatasvir, reduced HCV replication as well as HOXB9 induction (Fig. 3D), suggesting that the induction of HOXB9 gene expression by HCV infection is associated with the HCV replication level and is reversible. The promoter region of the HOXB9 gene was identified and analyzed in detail by Yamagishi et al. (27). Then, the amount of H2Aub bound to the HOXB9 promoter region was evaluated using a chromatin immunoprecipitation (ChIP) assay. H2Aub accumulated in the HOXB9 promoter region under uninfected conditions but was significantly reduced by HCV infection (Fig. 3E). These data suggest that HCV infection reduces the level of H2Aub in the HOXB9 promoter region.
FIG 3.

Effect of HCV infection on HOXB9 expression and H2A K119 monoubiquitination in the HOXB9 gene promoter. (A) HCV-infected cells were harvested at 3, 6, or 9 dpi. HOXB9 and GAPDH mRNAs were evaluated by RT-sqPCR in HCV-infected cells (+) and mock-infected cells (–). (B) HCV-infected (HCV) and mock-infected (Mock) cells were fixed at 9 dpi and were then stained with DAPI. The cells were stained with the mouse anti-core antibody and AF594-conjugated goat anti-mouse IgG antibodies. The stained samples were observed under a BZ-9000 fluorescence microscope. (C) HCV-infected and mock-infected cells were fixed, permeabilized, and stained with the mouse anti-NS5A IgG and rabbit anti-HOXB9 IgG and then with AF488-conjugated goat anti-mouse IgG antibody and AF647-conjugated goat anti-rabbit IgG antibody. The stained cells were subjected to flow cytometry. The data were analyzed with FlowJo software. Sorted cells were gated as NS5A-positive cells and then plotted based on fluorescence intensities of HOXB9 and NS5A. (D) HCV-infected and mock-infected cells were treated with 20 nM daclatasvir (DCV: +) or DMSO (–) from 9 to 15 dpi. The intracellular HCV RNA level and the HOXB9 and GAPDH mRNA levels were estimated by RT-qPCR. (E) The level of H2Aub in the HOXB9 gene promoter was evaluated by a ChIP assay using the antibody specific for H2Aub (H2Aub) or a nonspecific antibody (Control). Immunoprecipitates were analyzed by sqPCR (left panels) and qPCR (right graph) using the HOXB9 promoter-specific primer pair. The values of immunoprecipitates were normalized to that of input DNA and are presented as relative values with respect to mock-infected cells. The data shown in this figure are representative of three independent experiments and are presented as the mean ± SD values (n = 3). **, P < 0.01.
Effect of HCV infection on RNF2 expression and HOX gene induction.
PRC1, which is composed of BMI1, RNF1, RNF2, and other protein components, acts as an E3 ubiquitin ligase targeting K119 of H2A and silences its target genes by surrounding their promoters with H2Aub (15, 17, 18). Since RNF2 and BMI1 are essential components of PRC1 for H2A K119 monoubiquitination (15, 28), we examined the effect of HCV infection on the expression of RNF2 and BMI1. RNF2 protein expression was reduced in HCV-infected cells, while the expression of β-actin and BMI1 was not affected by HCV infection (Fig. 4A). However, RNF2 mRNA expression was significantly increased in HCV-infected cells (Fig. 4B), suggesting that the level of RNF2 is posttranscriptionally reduced by HCV infection. HOXB9 mRNA was decreased dependently of the RNF2 level (Fig. 4C). Then, we assessed the stability of endogenous and exogenous RNF2 with a cycloheximide chase assay (Fig. 5). The half-life of endogenous RNF2 was approximately 0.5 h in HCV-infected cells and 3.6 h in mock-infected cells (Fig. 5A and B), while the half-life of exogenous RNF2 was approximately 0.5 h in HCV-infected cells and longer than 6 h in mock-infected cells (Fig. 5C and D). These results suggest that HCV infection destabilizes the RNF2 protein.
FIG 4.
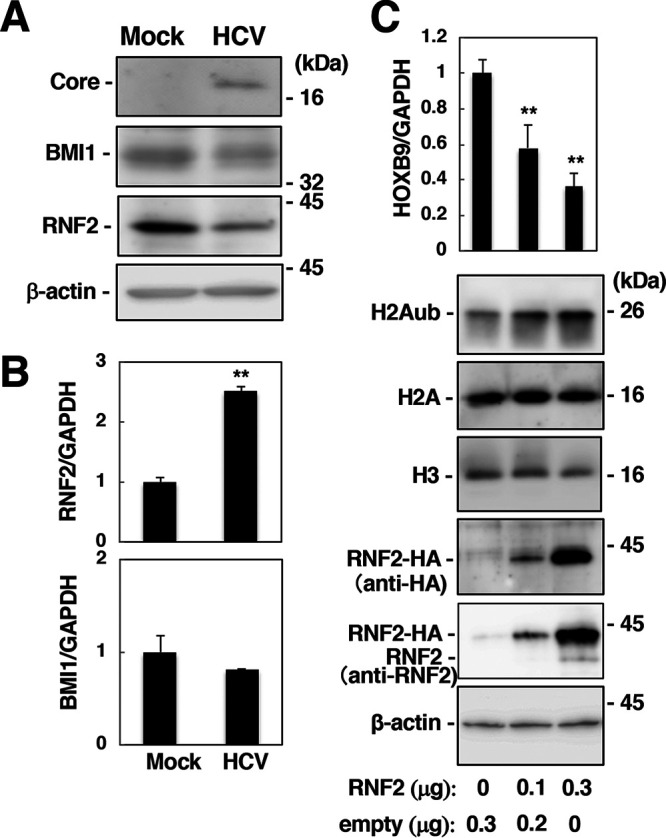
Effect of HCV infection on the expression of RNF2 in association with HOX gene induction. (A) Cell lysates were prepared at 9 dpi from mock-infected (Mock) and HCV-infected (HCV) cells and were then subjected to Western blotting using anti-HCV core, anti-BMI1, anti-RNF2, and anti-β-actin antibodies. (B) RNF2 and BMI1 mRNA levels were estimated with RT-qPCR and normalized to the GAPDH mRNA level. Each calculated value in the HCV-infected cells is presented as a relative value calculated based on the value in the mock-infected cells, which was set as 1.0. The data are representative of three independent experiments and are presented as the mean ± SD values (n = 3). (C) Huh7OK1 cells were transfected with the empty plasmid and/or the RNF2-HA-encoding plasmid. The HOXB9 mRNA level (top graph) was estimated with RT-qPCR. The HOXB9 mRNA level was normalized to the GAPDH mRNA level and is presented as a relative value compared to the empty plasmid control. The levels of H2Aub, H2A, H3, RNF2-HA, endogenous RNF2, and β-actin were evaluated by immunoblotting (lower panels). **, P < 0.01. The data shown in this figure are representative of three independent experiments and are presented as the mean ± SD values (n = 3).
FIG 5.

Cycloheximide chase assay to evaluate the stability of the RNF2 protein. (A) Mock-infected and HCV-infected cells were incubated in medium containing 10 μg/ml cycloheximide (CHX) and were then harvested at each indicated time. Cell lysates were prepared from the harvested cells and were then subjected to Western blotting. (B) The intensities of the RNF2 bands shown in panel A were quantified using ImageJ software and standardized with individual beta-actin intensity. The data shown are representative of three independent experiments and are presented as the mean ± SD values (n = 3). (C) A plasmid encoding RNF2-HA was transfected into mock-infected cells and HCV-infected cells. CHX was added to the medium at 36 h posttransfection. The resulting cells were harvested at the indicated times to prepare cell lysates, which were subjected to Western blotting. (D) The intensities of the RNF2-HA bands in each group shown in panel C were quantified using software and standardized with individual beta-actin intensity. The data shown are representative of three independent experiments and are presented as the mean ± SD values (n = 3).
We sought to determine whether HOXB9 expression is suppressed by RNF2 knockout. RNF2-knockout (RNF2KO) cell lines 1 and 2 were generated via the CRISPR/Cas9 system (Fig. 6A). The level of H2Aub but not total H2A was reduced significantly in RNF2KO cells (Fig. 6A). Expression of the HOXB1, B9, C13, and D13 genes was induced in RNF2KO cells, while that of HOXA1 and A3 was not affected by RNF2 knockout (Fig. 6B). Overexpression of RNF2 increased the level of H2Aub and impaired HOXB9 induction in HCV-infected cells in a dose-dependent manner (Fig. 4C), while RNF2 knockout impaired HCV propagation (Fig. 6C). RNF2 knockdown also showed a decrease in HCV-RNA levels in HCV-infected cells (Fig. 6D), although the suppression of HCV propagation by the RNF2 knockdown is weaker than that by knockout (Fig. 6C). The partial suppression by RNF2 breakdown in HCV-infected cells may contribute to the persistent infection in order to escape host immune system. Expression of RNF2 impaired the induction of HOXB1, B9, C13, and D13 gene expression in naive RNF2KO 2 cells and restored HCV propagation in HCV-infected RNF2KO 2 cells (Fig. 6E to G). Taken together, these results suggest that RNF2 plays essential roles in the regulation of both H2A K119 monoubiquitination and HOX gene induction in HCV-infected cells.
FIG 6.

Involvement of RNF2 in HOX gene induction and HCV propagation. (A) Cell lysates were prepared from the parental cell line (wild type [WT]) and the RNF2KO#1 and RNF2KO#2 cell lines and were then subjected to Western blotting. (B) The mRNA levels of HOXA1, HOXA3, HOXB1, HOXB9, HOXC13, HOXD13, and GAPDH in these cells were evaluated by RT-sqPCR. A1, A3, B1, B9, C13, and D13 indicate HOXA1, HOXA3, HOXB1, HOXB9, HOXC13, and HOXD13. (C) Parental, RNF2KO#1, and RNF2KO#2 cells were infected with HCVcc at an MOI of 0.1. HCV-infected cells and mock-infected cells were harvested at 6 dpi. Intracellular HCV RNA and GAPDH mRNA levels were estimated by RT-qPCR. The data shown are representative of three independent experiments and are presented as the mean ± SD values (n = 3). (D) Two siRNAs targeting RNF2 (siRNF#1 and #2) were added to a culture supernatant of Huh7OK1 cells at 10 nM. Transfected cells were infected with HCVcc at 24 h posttransfection. HCV-infected cells were harvested at 2 dpi. HCV RNA and GAPDH mRNA levels were evaluated by RT-qPCR. (E and F) A plasmid encoding RNF2 was transfected into RNF2KO#2 cells. The transfected cells were harvested at 36 h for preparation of total RNA and cell lysates. HOX gene mRNA levels were estimated by RT-sqPCR. RNF2 and β-actin expression was evaluated by immunoblotting (E) HOX gene mRNA levels were analyzed by RT-sqPCR. (F) A1, A3, B1, B9, C13, and D13 indicate HOXA1, HOXA3, HOXB1, HOXB9, HOXC13, and HOXD13. (G) A plasmid encoding RNF2 (RNF2) or an empty plasmid (Control) was transfected into RNF2KO#2 cells. The transfected cells were infected with HCVcc at an MOI of 0.1 at 36 h posttransfection. HCV-infected cells and mock-infected cells were harvested at 6 dpi. (C, D, and G) The data shown are representative of three independent experiments and are presented as the mean ± SD values (n = 3). **, P < 0.01; *, P < 0.05; ND, not detected.
Activation of HOX genes by HCV core protein.
We next sought to determine which viral proteins are responsible for HOX gene induction and H2A K119 monoubiquitination. Full-genomic or subgenomic replicon RNA was transfected into Huh7OK1 cells. The cells harboring replicon RNA were selected in medium containing G418 for 10 days and prepared as crude cell populations. NS5A or each replicon RNA was detected in both full-genomic and subgenomic replicon cells, while the core protein was detected in full-genomic replicon cells but not in subgenomic replicon cells (Fig. 7A to C). The amounts of RNF2 and H2Aub were decreased in full-genomic replicon cells but not affected in subgenomic replicon cells (Fig. 7B), although the amounts of RNF1 and BMI1 in the replicon-containing cells were similar to those in mock-transfected cells (Fig. 7B). Furthermore, expression of the HOXB1, B8, B9, C13, and D13 genes was induced in full-genomic replicon cells but not in subgenomic replicon cells (Fig. 7A and F). Long-term treatment with daclatasvir eliminated replicon RNA from full-genomic or subgenomic replicon cells and suppressed the transcription of HOXB9 mRNA in full-genomic replicon cells (Fig. 7D and E). These data suggest that structural proteins are essential for HOX gene induction in HCV-infected cells.
FIG 7.

Activation of HOXB9 gene expression in cells harboring full-genomic replicon RNA but not subgenomic replicon RNA. (A) Huh7OK1 cells were transfected with full- or subgenomic replicon RNA. Cells harboring full-genomic replicon RNA (F) or subgenomic replicon RNA (S) were established in medium containing 400 μg/ml G418. Total RNA was prepared from mock-transfected cells and cells harboring each replicon. HOXB9 mRNA, intracellular HCV RNA and GAPDH mRNA levels were evaluated by RT-sqPCR. NTC, no-template control. (B) Cell lysates were prepared from mock-transfected cells or individual replicon cells and were then subjected to Western blotting. (C) NS5A and the core protein in the mock-transfected or full-genomic and subgenomic replicon cells were stained with a mouse anti-core antibody or anti-NS5A antibody and were then stained with an AF488-conjugated goat anti-mouse IgG antibody. The stained samples were observed under fluorescence microscopy. (D) Cells from which the full-genomic replicon RNA (FC) or subgenomic replicon RNA (SC) was removed by treatment with daclatasvir were also stained with the anti-NS5A antibody and were then stained with DAPI. (E) Total RNA was prepared from M, F, S, FC, and SC cells. HCV RNA, HOXB9 mRNA, and GAPDH mRNA levels were evaluated by RT-sqPCR. The data shown in this figure are representative of three independent experiments. (F) Total RNA was prepared from mock-transfected cells (Mock) and cells harboring full-genomic (F) or subgenomic (S) replicon RNA. The mRNA levels of HOXA1, HOXA3, HOXB1, HOXB8, HOXC13, HOXD13, and GAPDH were evaluated by RT-sqPCR. NTC, no-template control. A1, A3, B1, B8, B9, C13, and D13 indicate HOXA1, HOXA3, HOXB1, HOXB8, HOXB9, HOXC13, and HOXD13.
Several reports have suggested that the HCV core protein contributes to alterations in the expression of genes, including genes involved in lipid metabolism, cell proliferation and inflammatory responses (29–31). We next examined the effect of the HCV core protein on the induction of HOX genes. A plasmid encoding the core protein was transfected into the Huh7OK1 cells. The amount of core protein was increased in a plasmid-dose-dependent manner, whereas the amounts of RNF2 and H2Aub were reduced (Fig. 8A). Transcription of the HOXB9 gene was activated in cells expressing the core protein compared to untransfected cells. Furthermore, expression of the core protein activated the transcription of the HOXB1, B8, and C13 genes, but not that of HOXA1 or A3, similar to the data shown in Fig. 1 (Fig. 8B).
FIG 8.

Effect of the HCV core protein on HOX gene mRNA and histone levels. A plasmid encoding the core protein and/or the empty plasmid were introduced into Huh7OK1 cells. The transfected cells were harvested 36 h posttransfection. Total RNA or cell lysates were prepared from the transfected cells. (A) The core protein, β-actin, RNF2, H2A, H3, and H2Aub were evaluated by immunoblotting. The intensity of H2Aub or RNF2 is indicated on the bottom of each panel. (B) HOXB9, HOXA1, HOXA3, HOXB1, HOXB8, HOXC13, HOXD8, HOXD13, and GAPDH mRNA levels were evaluated by RT-sqPCR. A1, A3, B1, B8, B9, C13, and D13 indicate HOXA1, HOXA3, HOXB1, HOXB8, HOXB9, HOXC13, and HOXD13.
Signal peptide peptidase (SPP) is required for the maturation and stability of the core protein (32). Knockout of the SPP gene suppresses the maturation of the core protein and induces rapid degradation of the immature core protein by polyubiquitin-dependent proteasome activity but does not affect other viral proteins (32). Since treatment with LY-411575, which can inhibit SPP activity, reduced the amount of the core protein in the cultured cells (33), we evaluated the effect of LY-411575 treatment on the expression levels of RNF2 and H2Aub and transcription of HOX genes in HCV-infected cells. LY-411575 treatment decreased the amount of the core protein but not that of NS5A (Fig. 9A and B). The HCV RNA level in the supernatant was decreased significantly by LY-411575 treatment (Fig. 9C), although the intracellular HCV RNA level was not affected (Fig. 9C). The expression levels of RNF2 and H2Aub were restored in HCV-infected cells by LY-411575 treatment, corresponding to downregulation of the core protein (Fig. 9B), while HoxB9 transcription was reduced significantly in HCV-infected cells by LY-411575 treatment (Fig. 9C). These results suggest that the core protein is responsible for RNF2 downregulation, H2A K119 monoubiquitination, and HOX gene activation in HCV-infected cells.
FIG 9.
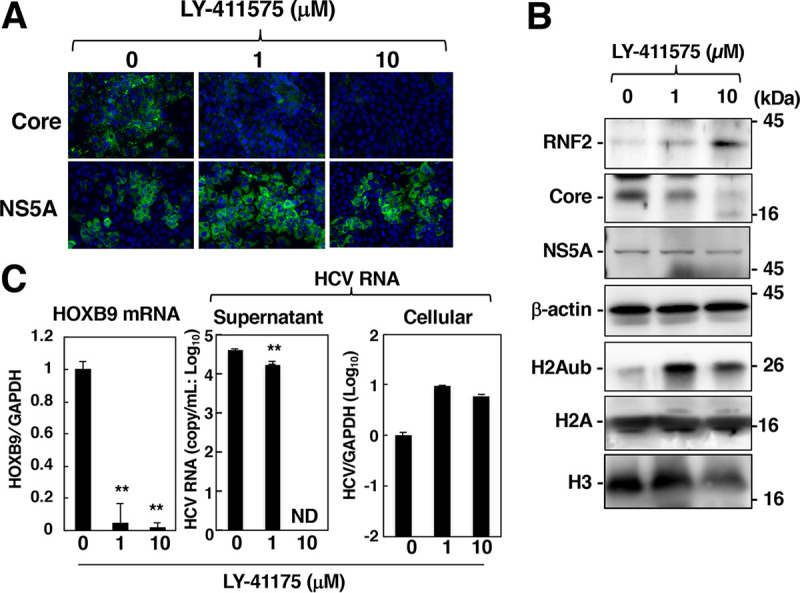
Effect of SPP inhibition on the RNF2 protein, H2Aub, and HOXB9 mRNA levels. HCV-infected cells were treated with 0, 1, or 10 μM LY-411575 and were then fixed or harvested at 72 h after treatment. (A) The core protein and NS5A were evaluated by immunostaining. (B) Cell lysates prepared from the harvested cells were subjected to Western blotting. (C) HOXB9 and GAPDH mRNA levels and supernatant and cellular HCV RNA levels were estimated by RT-qPCR. The data shown in this figure are representative of three independent experiments and are presented as the mean ± SD values (n = 3). **, P < 0.01; ND, not detected.
Involvement of proteasome activity in HCV-dependent regulation of HOX genes.
We next investigated the involvement of the ubiquitin-proteasome pathway in RNF2 protein degradation. HA-tagged ubiquitin was expressed in HCV-infected cells and mock-infected cells. The resulting cells were treated with MG132 or DMSO at 36 h posttransfection and were then harvested 40 h posttransfection. The cell lysates and the immunoprecipitates were subjected to Western blotting. The total ubiquitinated protein level was decreased slightly by infection (Fig. 10A, lanes 2, 3, 5, and 6), while polyubiquitination of RNF2 was also decreased slightly by infection (Fig. 10A, lanes 9 and 12). The infection-dependent reduction in polyubiquitinated RNF2 may correspond to the amount of total polyubiquitination (Fig. 10A, lanes 3, 6, 9, and 12). Knockout of E6AP, which has been reported to be an E3 ubiquitin ligase targeting RNF2, promoted accumulation of RNF2 in hepatocytes (34), suggesting that E6AP is responsible for polyubiquitination of RNF2. Thus, we investigated the effect of E6AP knockdown on HCV infection-dependent downregulation of RNF2. Transfection with E6AP-targeting small interfering RNA (siRNA) (siE6AP1 or siE6AP2) reduced the amount of E6PA but did not affect the amount of RNF2 in HCV-infected cells or mock-infected cells (Fig. 10B), suggesting that HCV infection destabilizes the RNF2 protein independently of polyubiquitination. Expression of the core protein reduced the level of RNF2, whereas MG132 treatment restored the level of RNF2 in cells expressing the core protein (Fig. 10C). These data suggest that HCV infection and core protein expression promote RNF2 degradation via a polyubiquitin-independent, proteasome-dependent pathway.
FIG 10.
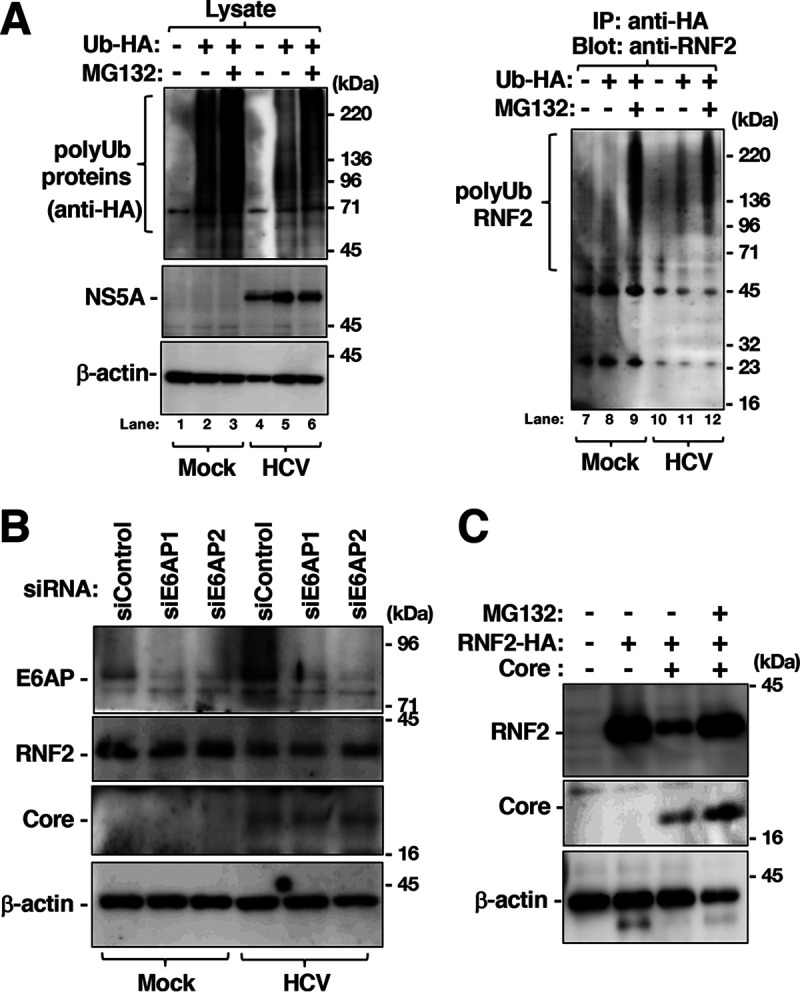
Involvement of protease activity in the degradation of RNF2 in HCV-infected cells or core-expressing cells. (A) HCV-infected and mock-infected Huh7OK1 cells were transfected with a plasmid encoding HA-tagged ubiquitin or the empty plasmid. The transfected cells were treated with 10 μM MG-132 or DMSO at 36 h posttransfection and harvested at 40 h posttransfection. RNF2, polyubiquitinated (polyUb) proteins, NS5A, and β-actin in cell lysates were evaluated by immunoblotting (left panels), while the proteins immunoprecipitated with an anti-HA antibody were evaluated by immunoblotting with an anti-RNF2 antibody (right panel). (B) HCV-infected cells and mock-infected cells were transfected with siRNA targeting E6AP mRNA, siE6AP1, or siE6AP2. The cells were harvested at 48 h posttransfection. The cell lysates were subjected to Western blotting. (C) Plasmids encoding the core protein and/or RNF2-HA were transfected into Huh7OK1 cells. The cells were treated with 10 μM MG-132 or DMSO at 36 h posttransfection and were then harvested at 40 h posttransfection. The cell lysates were subjected to Western blotting. The data shown in this figure are representative of three independent experiments.
Finally, we examined the impact of RNF2 degradation by the proteasomal pathway on the induction of HOX gene expression in HCV-infected cells. Treatment with MG132 impaired the enhancement of HOXB9 transcription in HCV-infected cells but did not affect HCV propagation (Fig. 11A and B). The ChIP assay results showed that the level of H2Aub in the HOXB9 promoter region was reduced by infection and was then restored by treatment with MG132 (Fig. 11C and D), although no significant change in the amount of H2Aub in the HOXA1 promoter region was observed regardless of the HCV infection status (Fig. 11D, right graph). These data suggest that HCV infection-dependent proteasome activity enhances HOX gene promoter activity via a reduction in H2Aub in its promoter region.
FIG 11.

Proteasome inhibitor treatment increased H2Aub in the HOXB9 gene promoter and suppressed HOXB9 transcription. Mock-infected cells and HCV-infected cells were treated with 10 μM MG-132 or DMSO and were then harvested 4 h after treatment. (A) Total RNA was prepared from those cells. The levels of HCV RNA, HOX9 mRNA and GAPDH mRNA were evaluated by RT-sqPCR. (B) The values were quantified by RT-qPCR using total RNA samples described in panel A. These levels of HCV RNA and HOX9 mRNA were normalized to the level of GAPDH mRNA. (C) Nuclear fractions were subjected to a ChIP assay using normal IgG (NC) or anti-H2Aub IgG (H2Aub). The level of H2Aub bound to the HOXB9 gene promoter was estimated based on the level of immunoprecipitated fragments originating from the HOXB9 gene promoter. The levels of the HOXB9 gene promoter in the total fraction (left panel, input DNA) and in the immunoprecipitated fraction (right panel, ChIP) from each group were estimated by sqPCR. (D) The level of H2Aub bound to the HOXB9 or HOXA1 gene promoter in each group was estimated by a ChIP assay. The data shown in this figure are representative of three independent experiments and are presented as the mean ± SD values (n = 3). **, P < 0.01; ND, not detected.
DISCUSSION
Here, we showed that an HCV-induced reduction in H2Aub is responsible for one mechanism by which HCV infection induces alterations in host gene expression. H2Aub has been reported to occur on 10% of total cellular H2A (14), an event that has been linked primarily to PRC1-mediated gene silencing. H2Aub firmly compacts the structure and components of nucleosomes, leading to transcriptional repression of target genes (15–18). In this study, the expression of HOX genes, which are the classic targets of the PRC1-H2Aub pathway, was induced by HCV infection in the cell culture system (Fig. 1). The data shown in Fig. 1 indicate that HOXB9 and C13 are markedly activated by HCV infection. Furthermore, more than half of the HOX genes, including HOXA4, A6, A9, B1, B2, B3, B8, B13, C4, D4, D8, D10, D12, and D13, were expressed at a higher level in HCV-infected cells than in mock-infected cells. These results imply that HCV infection can activate silenced HOX genes via the PRC1-H2Aub pathway. However, some HOX genes were not affected by HCV infection (Fig. 1, 5, 6, and 8). The expression of HOX genes is regulated by not only PRC1 but also by several other factors, such as miRNA, in each tissue and at each developmental stage (35). HCV infection has been shown to upregulate HOXA1 expression via downregulation of miR-181c (26), although it was not affected by HCV infection in our system. Several HOX genes whose expression is not affected by HCV infection may be regulated by an unknown factor independent of the PRC1-H2Aub pathway.
We examined HCV-dependent induction of the HOXB9 gene as a representative of HCV infection-induced HOX genes because HOXB9 gene expression was clearly induced in HCV-infected cells compared to mock-infected cells in this study, and its promoter has been well characterized as an H2Aub target, as described in a previous report (27). Transcription of the HOXB9 gene was largely promoted by HCV infection in a time-dependent manner (Fig. 1 and 3), while this enhancement of transcription was reversibly abolished in HCV-infected cells by treatment with daclatasvir, which is associated with a decrease in HCV RNA (Fig. 3C). The chromatin immunoprecipitation (ChIP) assay data suggested that the level of H2Aub in the HOXB9 promoter region was reduced significantly by HCV infection (Fig. 3D). We next examined the impact of HCV infection on PRC1 activity, which is responsible for H2A K119 monoubiquitination in vitro and in vivo (15, 36). The RNF2 protein is a critical component for the activity of PRC1 as an E3 ubiquitin ligase targeting H2A K119 (15, 16, 37). The data shown in Fig. 4 indicate that HCV infection reduced the protein but not the mRNA level of RNF2 and that overexpression of RNF2 increased the level of H2Aub and impaired HOXB9 transcription. The protein level but not the mRNA level of RNF2 was decreased in HCV-infected cells (Fig. 4B), suggesting that HCV infection destabilizes the RNF2 protein. RNF2 was expected to be degraded via proteasome activity in HCV-infected cells or core-expressing cells because treatment with MG132 restored the amounts of RNF2 and H2Aub and impaired HOX gene activation in HCV-infected cells and core-expressing cells (Fig. 10 and 11). Gene expression of HOXB1, B8, B9, C13, and D13 was potently induced in full-genomic replicon cells but not in subgenomic replicon cells (Fig. 7). Furthermore, overexpression of the core protein reduced the H2Aub level but not the total H2A level and potentiated transcription of the HOXB1, B9, D8, D13, and C13 genes (Fig. 8), similar to the data shown in Fig. 1 and 7. Treatment with LY-411575, which inhibits the maturation of the core protein and promotes the proteasomal degradation of immature core protein but does not affect other viral proteins, restored the amount of RNF2 and suppressed the expression of the HOX gene, corresponding to a decrease in the amount of core protein (Fig. 9). Inductions of several HOX genes were observed at 1 dpi in PHHs by HCV infection (Fig. 1E), suggesting that HOX genes are induced as soon as the core protein is released into the cytosol after uncoating and that HCV infection could induce HOX genes in nontumor cells. Taken together, our data suggest that the HCV core protein disturbs the gene repression mechanism regulated by the PRC1-H2Aub pathway to induce HOX gene expression in HCV-infected cells.
Alterations in the chromatin structure affect the sensitivity of cells to chemical and irradiation-induced DNA damage (38). HCV infection or expression of the core protein has been reported to sensitize cultured cells or mice to DNA damage agent-induced cell death (39). The HCV-induced reduction in H2A monoubiquitination may be involved in some hepatitis C-related disorders. Genome-wide association studies (GWASs) can identify hundreds of common variants associated with cancer predisposition. According to systemic annotation of the GWAS catalog database, the majority (over 93%) of single nucleotide polymorphisms (SNPs) lie in noncoding regions (40). Several SNPs have been reported to influence the risk of carcinogenesis due to deregulation of HOX genes (41). Several HOX genes may contribute to carcinogenesis by regulation of cell growth, survival, migration, and invasion. HOX gene deregulation causes in cancer development is still obscure; however, HOX genes were reported to be deregulated tissue-specifically in tumors. The HOXD10 gene was silenced epigenetically in gastric cancer and was upregulated in colorectal cancer (42), while HOXA9 was silenced in lung cancers through epigenetic mechanisms but was upregulated in acute lymphocytic leukemia (43, 44). HOXB13 is upregulated in breast cancer and silenced in prostate cancer (45, 46). Induction of HOX genes by the core protein may be the first step of host regulation associated with hepatitis C-related liver disorders. Acetylation of histone H3K27 has been reported to be altered by HCV infection in patients’ liver, independent of indirect factors such as the inflammatory response and fibrosis, and these changes persist after a sustained virologic response (47). H3K27 methylation is catalyzed by methyl transferase EZH2, which is a main component of polycomb repressive complex 2 (PRC2). Kalb et al. suggest that H2Aub creates a docking site for PRC2 complex, leading to H2K27 trimethylation (48). HCV-dependent reduction in PRC1 activity during a chronic infection may contribute to impairment of H2K27 methylation and then acceleration of H3K27 acetylation, leading to induction of HOX genes.
The mechanism by which HCV infection or core protein reduces the RNF2 protein level remains unclear. Several reports suggest that HCV infection or viral proteins promote the activity of several major proteolysis pathways—autophagic, lysosomal, and proteasomal pathways (49–52). A report about proteasome activation by HCV core protein (53) is notable in connection with this study. In this study, the proteasome inhibitor MG132 restored the amount of RNF2 protein in core-expressing cells (Fig. 10). However, E6AP, which was reported to be the E3 ubiquitin ligase catalyzing polyubiquitination of RNF2 (34), did not affect HCV infection-induced downregulation of RNF2, suggesting that polyubiquitination-dependent proteasome activity is not predominantly associated with RNF2 downregulation in HCV-infected cells. Further study will be needed to clarify the mechanism by which HCV infection promotes RNF2 degradation.
This study shows a novel molecular mechanism by which HCV infection induces HOX gene expression via the PRC1-H2Aub pathway (The findings are summarized in Fig. 12.). H2A K119 monoubiquitination regulates silencing of not only HOX genes but also other genes (54, 55). Since PRC1 actively or repressively modulates the expression of metabolic and developmental genes in association with RNA polymerase II variants (55), HCV infection or core protein may regulate various genes via the PCR1-H2Aub pathway during long-term persistent infection. Thus, clarifying the observation of HCV-induced H2Aub suppression will be important for understanding the molecular mechanism of hepatitis C pathogenesis.
FIG 12.
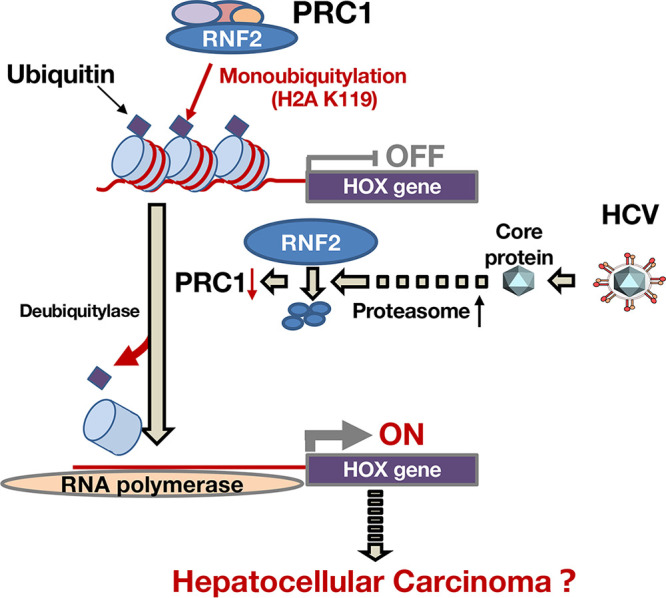
A schematic diagram of HOX gene induction in HCV-infected cells. The mechanism by which HCV infection induces expressions of HOX genes is summarized as a schematic diagram. PRC1 is known as a silencer of gene expression such as HOX gene expression. PRC1 monoubiquitinates histone H2A K119 as an E3 ubiquitin-ligase and then suppresses the transcription of the HOX gene in noninfected hepatocytes. HCV infects hepatocytes, and then the core protein is released into the cytosol. The core protein induces the activation of proteasome activity and the degradation of RNF2, which is a main component of PRC1. The degradation of RNF2 loses the balance between deubiquitylase and ubiquitin ligase with regard to H2A K119 monoubiquitination, leading to inductions of HOX genes. These findings may be a part of the mechanism associated with the hepatitis C-related disorders, including hepatocellular carcinoma.
MATERIALS AND METHODS
Cell lines, virus, and reagents.
The HCV strain JFH-1, which belongs to genotype 2a (56), was employed as cell culture-adapted HCV (HCVcc) in this study. The viral RNA derived from the plasmid pJFH1 was transcribed and introduced into Huh7OK1 cells according to the method reported by Wakita et al. (56). The infection procedure was reported previously (56) and is described in detail in each figure legend. Huh7OK1 and Huh7.5.1 cell lines, which are highly permissive for the JFH-1 strain, were cultured as reported previously (57, 58). The Huh7OK1 cell line was previously established by treatment of 9 to 13 replicon cells with interferon-alpha (57). Primary human hepatocytes (PHHs), PXB cells, were purchased from Phoenix Bio (Hiroshima, Japan) and cultured in the manufacturer’s medium for PXB cells (Phoenix Bio). Huh7OK1 cells harboring full-genomic or subgenomic replicon RNA derived from the genotype 1b strain Con 1 were prepared as reported previously (59). Each replicon RNA was transfected into Huh7OK1 cell lines. The replicon cell lines were established by limited dilution. The N-terminal HA-tagged RNF2 and FLAG-tagged BMI1 cDNAs were amplified by PCR using Pfu turbo DNA polymerase (Agilent, Santa Clara, CA) and cloned into pCAG-GS and pcDNA3.1, respectively. The plasmid encoding the HCV core protein was prepared based on pCAG-GS as described previously (60).
Transfection, immunoblotting, immunoprecipitation, and gene silencing.
Plasmid DNA was transfected into Huh7OK1 cells by using TransIT-LT1 (Mirus Bio, Madison, WI). The lysate preparation and the immunoprecipitation assay were carried out as described previously (60). Cell lysates were subjected to 12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and were electroblotted onto polyvinylidene difluoride membranes (Merck Millipore, Billerica, MA). Proteins on the membranes were reacted first with an appropriate antibody and then with a horseradish peroxidase-conjugated antibody specific for rabbit or mouse IgG, immersed in Super Signal West Femto (Thermo Fisher Scientific, Rockford, IL) and visualized using a LAS 4000 mini-imaging system (Cytiva, Marlborough, MA). The siRNAs targeting at RNF2 were purchased from Thermo Fisher Scientific, while the control siRNA (siControl nontargeting siRNA2, Darmacon) was from Horizon (Cambridge, UK). These siRNAs were introduced into the cell lines by using Lipofectamine RNAiMax (Thermo Fisher Scientific). siRNAs with the Ambion siRNA ID numbers s12068 and s12069 were designated siRNF#1 and siRNF#2, respectively. The cycloheximide chase assay was carried out by the method reported by Kao et al. (61).
Reverse transcription-quantitative PCR (RT-qPCR) and reverse transcription-semiquantitative PCR (RT-sqPCR).
Total RNA and first-strand cDNA were prepared by the method described previously (59). Host mRNAs and HCV RNA were evaluated by RT-qPCR as described previously (59, 60). The value of HCV RNA, or of each host mRNA, was normalized to that of GAPDH mRNA. HCV RNA and GAPDH mRNA were amplified using the primer pairs 5′-GAGTGTCGTGCAGCCTCCA-3′ and 5′-CACTCGCAAGCACCCTATCA-3′, and 5′-GAAGGTGAAGGTCGGAGTC-3′ and 5′- TGATGACAAGCTTCCCGTTCTC-3′, respectively, as described previously (62).
Each PCR product was detected as a single band of the correct size upon agarose gel electrophoresis. The amount of HCV in the culture supernatant was calculated as the copy number (60). HOX gene mRNAs were evaluated by RT-sqPCR (63) using the appropriate primer pairs listed in Table 1. The TaKaRa emerald ×2 mixture (TaKaRa Bio, Inc., Tokyo, Japan.) was used for semiquantitative PCR. The raw data were acquired at 25, 30, 35, and 40 cycles to confirm that the PCRs did not reach a plateau. HCV RNA and GAPDH mRNA were evaluated at 25 cycles, while HOX mRNAs were evaluated at 35 cycles. The reacted preparation with a volume of 5 μl (1/4 of the whole volume) was subjected to agarose electrophoresis. The remaining aliquot was subjected to further PCR running (5 cycles) to make sure that the amplification did not reach a plateau.
TABLE 1.
List of primer pairs targeting HOX mRNAs
| Genes | Forward (5′ to 3′) | Reverse (5′ to 3′) | Size (bp) |
|---|---|---|---|
| HOXA1 | TCCTGGAATACCCCATACTTAGCA | GCCGCCGCAACTGTTG | 122 |
| HOXB1 | CTCCTCTCCGAGGACAAGGAA | CTGTCTTGGGTGGGTTTCTCTTAA | 103 |
| HOXD1 | CGACCCCCATCCCTATCTAGAC | TGGAACTCGGAAGCCAACTAAA | 115 |
| HOXA2 | ACAGCGAAGGGAAATGTAAAAGC | GGGCCCCAGAGACGCTAA | 102 |
| HOXB2 | TCCTTGGCCGTCTACTGGAA | AGTGGATTAAACGCTAATTCAGTAATACC | 105 |
| HOXA3 | TGCAAAAAGCGACCTACTACGA | CGTCGGCGCCCAAAG | 126 |
| HOXB3 | CCTGGCCTGAGAGGTTGCT | TCCCGGGCGTGGAATT | 145 |
| HOXD3 | CCATAAATCAGCCGCAAGGAT | ATGGGTCTCAGACTTACCTTTGG | 111 |
| HOXA4 | TCCCCATCTGGACCATAATAGG | GCAACCAGCACAGACTCTTAACC | 101 |
| HOXB4 | TTTTCAGCTTTGGCGAAGATG | ACCGAGGCCCGTCTTCTC | 102 |
| HOXC4 | GGCAGCTACCCCGGGTACT | TGTGAGTTATGTTTTATAACCTGGTAATGTC | 101 |
| HOXD4 | GGCGGGATTCTCTCTCTAAGTATATTATA | GAGCGGTGATTTTCATAAGTTTAATGA | 92 |
| HOXA5 | TCTCGTTGCCCTAATTCATCTTTT | CATTCAGGACAAAGAGATGAACAGAA | 101 |
| HOXB5 | AGCGCCAATTTCACCGAA | GGCTGCTTAGCTGGCTTGC | 76 |
| HOXC5 | AGGTGCAGGCATCCAGGTACT | GGGTTGGCAGCCATGTCTAC | 106 |
| HOXA6 | CCCTCTACCAGGCTGGCTATG | CAGGACCGAGTTGGACTGTTG | 119 |
| HOXB6 | AGCAGCCCCCGTTCCA | AAAGGAGGAACTGTTGCACGAAT | 143 |
| HOXC6 | TCAAACGTGGACCTGAAAGTCA | GGGAAAAGGGCCTGTAGACAA | 126 |
| HOXA7 | CAAAATGCCGAGCCGACTT | TAGCCGGACGCAAAGGG | 146 |
| HOXB7 | TCTTTAATGCTGTCTTTGTGGACTGT | GAACACGCGAGTGGTAGGTTTT | 131 |
| HOXB8 | AACTCACTGTTCTCCAAATACAAAACC | GACGGCCCGTGGTAGAACT | 167 |
| HOXC8 | CGCACCACGTTCAAGACTTCT | TAAGCGAGCACGGGTTCTG | 81 |
| HOXD8 | GATAACTTACAGAGACAGCCGATTTTTAC | CCAATATTACCACTGGACGATTTACA | 92 |
| HOXA9 | CCGAGAGGCAGGTCAAGATC | AAATAAGCCCAAATGGCATCA | 101 |
| HOXB9 | AGGCCGTGCTGTCTAATCAAA | CGAGCGTGCAGCCAGTT | 145 |
| HOXC9 | GGGCCCATCAGTAACTATTACGTG | CGGTGGCCGGAAACCT | 82 |
| HOXD9 | GGGACGCCTCAAAATGTCTTC | GTCGCCCTCATGGCCTATAA | 76 |
| HOXA10 | ACAAGAAATGTCAGCCAGAAAGG | GATGAGCGAGTCGACCAAAAA | 112 |
| HOXC10 | CCTCGCAATGTAACTCCGAACT | ACCCCGCAATTGAAGTCACT | 119 |
| HOXD10 | ATAAGCGCAACAAACTCATTTCG | ATATCGAGGGACGGGAACCT | 101 |
| HOXA11 | ACAGGCTTTCGACCAGTTTTTC | CCTTCTCGGCGCTCTTGTC | 92 |
| HOXC11 | GTGAAGGGAAGTGTCTGATGCA | AATCCGAGCAGCAAGACATTG | 108 |
| HOXD11 | CGGGCTGCGCCTACTATGT | AGGACGACGGTTGGGAAAG | 72 |
| HOXC12 | TAATCTCCTGAATCCCGGGTTT | TGGGTAGGACAGCGAAGGC | 124 |
| HOXD12 | TGTGTGAGCGCAGTCTCTACAGA | CGGCCTCAGGTTGGAGAAG | 92 |
| HOXA13 | AAATGTACTGCCCCAAAGAGCA | ATCCGAGGGATGGGAGACC | 81 |
| HOXB13 | CCACTGGCTGCTGGACTGTT | TATGACTGGGCCAGGTTCTTTG | 118 |
| HOXC13 | AAGGTGGTCAGCAAATCGAAAG | TGGTACAAAGCGGAGACATAAATAGA | 94 |
| HOXD13 | CTGGGCTACGGCTACCACTTC | GCGATGACTTGAGCGCATT | 91 |
Immunofluorescence microscopy.
Huh7OK1 cells were infected with HCVcc, passaged twice every 4 days, seeded at 0.5 × 104 cells per well on a glass cover slip, and incubated at 37°C for 24 h. The cells were washed twice with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde at room temperature for 20 min. The cells were washed twice with PBS after fixation, permeabilized by incubation for 15 min at room temperature in PBS containing 0.3% saponin, and then incubated in PBS containing 3% bovine serum albumin (PBS-BSA) to block nonspecific signals. These cells were stained with 50 μM DAPI and incubated at 4°C overnight in PBS-BSA containing rabbit anti-RNF2 (clone D22F2; Cell Signaling Technology, Denver, MA) and mouse anti-NS5A (clone 9E10) IgGs or containing rabbit anti-H2Aub (clone D27C4; Cell Signaling Technology) and mouse anti-HCV core protein IgGs (clone B2; Anogen, Mississauga, Canada). The cells were washed three times with PBS-BSA and incubated at room temperature for 2 h in PBS-BSA containing appropriate Alexa Fluor (AF)-conjugated secondary antibodies (Thermo Fisher Scientific). These cells were washed three times with PBS-BSA and observed using a confocal laser scanning microscope (FV1000; Olympus, Tokyo) or fluorescence microscope (BZ-9000; Keyence, Osaka, Japan). The core-positive and core-negative areas were determined and evaluated using fluorescence microscopy and software (Keyence, Osaka, Japan).
Flow cytometry analysis.
HCV-infected cells were fixed with 4% paraformaldehyde/PBS and were permeabilized with permeabilization buffer (eBioscience). For analyses of the expression of NS5A and HOXB9, HCV-infected cells were sequentially incubated with mouse anti-NS5A IgG (clone 9E10) and rabbit anti-HOXB9 IgG (Santa Cruz) at 4°C overnight and then with Alexa-488-conjugated goat anti-mouse IgG antibody and Alexa-488-conjugated mouse anti-rabbit IgG antibody on ice for 90 min. After washing, cells were suspended in PBS containing 2% fetal calf serum (FCS) and analyzed using a BD FACS Calibur instrument. The raw data captured by flow cytometry were analyzed using FlowJo software (FlowJo LLC, Ashland, OR).
Extraction of histones.
The cultured cells were harvested using a cell scraper, collected by centrifugation at 700 × g for 10 min, and washed once with PBS. The washed cells were lysed with ice-cold lysis buffer (20 mM Tris-HCl [pH 7.4], 135 mM sodium chloride, 1% Nonidet P-40, 10% glycerol). After 10 min incubation on ice, the resulting preparations were centrifuged at 1,000 × g for 10 min. The precipitates were washed three times with lysis buffer and once with TE buffer (10 mM Tris-HCl [pH 7.4], 13 mM EDTA). The resulting precipitates were suspended in 90 μl ice-cold H2O, and then mixed with 10 μl 4M H2SO4. After incubation at 4°C for 1 h, the suspensions were centrifuged for 5 min at 19,000 × g, and the supernatants were mixed with 1 ml of ice-cold acetone. After overnight incubation at −20°C, the coagulated material was collected by microcentrifugation and air-dried. The resulting preparations were dissolved in 0.05 mM Tris-HCl buffer (pH 6.8). The resulting mixtures were applied to Western blotting for histone detection.
ChIP assay.
ChIP assays were carried out according to the method reported by Sakurai et al. (64). Huh7OK1 cells were infected with HCVcc and passaged twice every 4 days. HCV-infected cells and mock-infected cells were harvested 8 dpi and incubated in fixation solution (1% formaldehyde, 4.5 mM HEPES [pH 8.0], 9 mM NaCl, 0.09 mM EDTA, and 0.04 mM EGTA) for 15 min at room temperature. Fixation was terminated by the addition of glycine at a final concentration of 150 mM. These fixed cells were washed with PBS containing 2% bovine serum and 0.05% NaN3 and were then sonicated in 0.25 ml of SDS lysis buffer (50 mM Tris-HCl [pH 8.0], 10 mM EDTA [pH 8.0], and 1% SDS) containing 0.01% protease inhibitor cocktail (Roche Molecular Biochemicals) until the cellular DNA was fragmented to a length shorter than 500 bp. The preparation was diluted twice with dilution buffer (50 mM Tris-HCl [pH 8.0], 167 mM NaCl, 1.1% Triton X-100, and 0.11% sodium deoxycholate). One-tenth of the preparation was kept at 4°C as an input fraction. The remaining preparation was divided into 9 aliquots. One aliquot (50 μl) of the preparation was incubated with 1 μg of the mouse anti-H2Aub antibody (clone D27C4; Cell Signaling Technology) or normal mouse IgG (Santa Cruz Biotechnology, Dallas, TX) for 12 h at 4°C. The protein-DNA-antibody complexes were mixed with 10 μl of protein G magnetic beads (Cell Signaling Technology) suspended at 50% (vol/vol) in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 8.0], 1 mM EDTA, 1% Triton X-100, 0.1% SDS, and 0.1% sodium deoxycholate) containing 150 mM NaCl, 100 μg/ml salmon sperm DNA (Sigma-Aldrich) and 1% BSA and were incubated at 4°C for 16 h. The resulting beads were separated magnetically and washed twice with RIPA buffer containing 150 mM NaCl, four times with RIPA buffer containing 500 mM NaCl, twice with LiCl buffer (10 mM Tris-HCl [pH 8.0], 0.25 M LiCl, 1 mM EDTA [pH 8.0], 0.5% NP-40, and 0.5% sodium deoxycholate), and finally, twice with TE buffer (10 mM Tris-HCl [pH 8.0] and 1 mM EDTA). The resulting bead complexes were incubated in 200 μl of ChIP direct elution buffer (10 mM Tris-HCl [pH 8.0], 5 mM EDTA, 0.5% SDS, and 300 mM NaCl) at 65°C overnight and were then treated with RNase A and proteinase K, followed by phenol-chloroform extraction. The ethanol-precipitated DNA was subjected to real-time qPCR using the primers 5′-TCTACAGCCTGCGTCCCTCCAA-3′ and 5′-ATGTGCTATCACGTCAGGGCTCC-3′ corresponding to the promoter region of the HOXB9 gene. Real-time qPCR was carried out as described above without a reverse transcription step.
Gene knockout with the CRISPR/Cas9 system.
RNF2KO Huh7OK1 cell lines were established according to the method reported by Fujihara et al. (65). An RNF2-targeting sequence (5′-GAAGAACACCATGACTACAA-3′) was employed for the construction of the pX330 plasmid encoding the guide RNA. The targeting DNA regions were amplified from genomic DNA of Huh7OK1 cells with the primer pair 5′-CAACCACTGAGGATCCAGTGGCTGGGTGATGGGTA-3′ and 5′-TGCCGATATCGAATTCGAGCCAAGATCGTGCCATTGC-3′ and were introduced into pCAG EGxxFP. Both plasmids, pX330 and pCAG EGxxFP, were purchased from Addgene (plasmids 42230 and #50716; Cambridge, MA). Huh7OK1 cells were transfected with these plasmids using Lipofectamine LTX (Thermo Fisher Scientific). GFP-positive cells were isolated using a FACSAria cell sorter (BD Bioscience) at 48 h posttransfection. Single cell clones were established using a colony isolation technique. Deleted regions of each allele of the RNF2 gene were confirmed by direct sequencing. RNF2 mRNA transcribed from each allele in the RNF2KO cell line was expected to lack the sequence AGAATCTATAGCTTCAGTTGGCATTTGCCACCA or TGTTTTACATC. The expression of RNF2 was confirmed by immunoblot analysis.
Statistical analyses.
The measured values are shown as the means ± standard deviations (SDs). The statistical significance of differences in the means was determined with Student’s t test.
ACKNOWLEDGMENTS
We thank T. Wakita, F. V. Chisari, and R. Bartenschlager for providing plasmids and cell lines. We also thank M. Mori for secretarial work and C. Endoh for technical assistance.
This work was supported by Grants-in-Aid from the Japan Agency for Medical Research and Development (20fk0210053h1202, 20fk0310102s0304, 20fk0310105h0004) and scholarship donations from Daiichi Sankyo Company, Ltd.; MSD KK; Otsuka Pharmaceutical Co., Ltd.; Shionogi & Co., Ltd.; and Mitsubishi Tanabe Pharma Co.
We declare that we have no conflicts of interest.
REFERENCES
- 1.WHO. 2017. Global hepatitis report, 2017. http://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/.
- 2.Ray RB, Lagging LM, Meyer K, Ray R. 1996. Hepatitis C virus core protein cooperates with ras and transforms primary rat embryo fibroblasts to tumorigenic phenotype. J Virol 70:4438–4443. doi: 10.1128/JVI.70.7.4438-4443.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ray RB, Meyer K, Ray R. 2000. Hepatitis C virus core protein promotes immortalization of primary human hepatocytes. Virology 271:197–204. doi: 10.1006/viro.2000.0295. [DOI] [PubMed] [Google Scholar]
- 4.Shintani Y, Fujie H, Miyoshi H, Tsutsumi T, Tsukamoto K, Kimura S, Moriya K, Koike K. 2004. Hepatitis C virus infection and diabetes: direct involvement of the virus in the development of insulin resistance. Gastroenterology 126:840–848. doi: 10.1053/j.gastro.2003.11.056. [DOI] [PubMed] [Google Scholar]
- 5.Moriya K, Yotsuyanagi H, Shintani Y, Fujie H, Ishibashi K, Matsuura Y, Miyamura T, Koike K. 1997. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J Gen Virol 78:1527–1531. doi: 10.1099/0022-1317-78-7-1527. [DOI] [PubMed] [Google Scholar]
- 6.Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, Matsuura Y, Kimura S, Miyamura T, Koike K. 1998. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med 4:1065–1067. doi: 10.1038/2053. [DOI] [PubMed] [Google Scholar]
- 7.Boldanova T, Suslov A, Heim MH, Necsulea A. 2017. Transcriptional response to hepatitis C virus infection and interferon-alpha treatment in the human liver. EMBO Mol Med 9:816–834. doi: 10.15252/emmm.201607006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin MV, King LY, Chung RT. 2015. Hepatitis C virus-associated cancer. Annu Rev Pathol 10:345–370. doi: 10.1146/annurev-pathol-012414-040323. [DOI] [PubMed] [Google Scholar]
- 9.Nawaz R, Zahid S, Idrees M, Rafique S, Shahid M, Ahad A, Amin I, Almas I, Afzal S. 2017. HCV-induced regulatory alterations of IL-1beta, IL-6, TNF-alpha, and IFN-Upsilon operative, leading liver en-route to non-alcoholic steatohepatitis. Inflamm Res 66:477–486. doi: 10.1007/s00011-017-1029-3. [DOI] [PubMed] [Google Scholar]
- 10.Arora P, Kim EO, Jung JK, Jang KL. 2008. Hepatitis C virus core protein downregulates E-cadherin expression via activation of DNA methyltransferase 1 and 3b. Cancer Lett 261:244–252. doi: 10.1016/j.canlet.2007.11.033. [DOI] [PubMed] [Google Scholar]
- 11.Park SH, Lim JS, Lim SY, Tiwari I, Jang KL. 2011. Hepatitis C virus core protein stimulates cell growth by down-regulating p16 expression via DNA methylation. Cancer Lett 310:61–68. doi: 10.1016/j.canlet.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 12.Duong FH, Christen V, Lin S, Heim MH. 2010. Hepatitis C virus-induced up-regulation of protein phosphatase 2A inhibits histone modification and DNA damage repair. Hepatology 51:741–751. doi: 10.1002/hep.23388. [DOI] [PubMed] [Google Scholar]
- 13.Fischle W, Wang Y, Allis CD. 2003. Histone and chromatin cross-talk. Curr Opin Cell Biol 15:172–183. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 14.Goldknopf IL, Taylor CW, Baum RM, Yeoman LC, Olson MO, Prestayko AW, Busch H. 1975. Isolation and characterization of protein A24, a “histone-like” non-histone chromosomal protein. J Biol Chem 250:7182–7187. [PubMed] [Google Scholar]
- 15.Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y. 2004. Role of histone H2A ubiquitination in Polycomb silencing. Nature 431:873–878. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- 16.Jason LJ, Finn RM, Lindsey G, Ausio J. 2005. Histone H2A ubiquitination does not preclude histone H1 binding, but it facilitates its association with the nucleosome. J Biol Chem 280:4975–4982. doi: 10.1074/jbc.M410203200. [DOI] [PubMed] [Google Scholar]
- 17.Zhu P, Zhou W, Wang J, Puc J, Ohgi KA, Erdjument-Bromage H, Tempst P, Glass CK, Rosenfeld MG. 2007. A histone H2A deubiquitinase complex coordinating histone acetylation and H1 dissociation in transcriptional regulation. Mol Cell 27:609–621. doi: 10.1016/j.molcel.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou W, Zhu P, Wang J, Pascual G, Ohgi KA, Lozach J, Glass CK, Rosenfeld MG. 2008. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell 29:69–80. doi: 10.1016/j.molcel.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakagawa T, Kajitani T, Togo S, Masuko N, Ohdan H, Hishikawa Y, Koji T, Matsuyama T, Ikura T, Muramatsu M, Ito T. 2008. Deubiquitylation of histone H2A activates transcriptional initiation via trans-histone cross-talk with H3K4 di- and trimethylation. Genes Dev 22:37–49. doi: 10.1101/gad.1609708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khan AA, Lee AJ, Roh TY. 2015. Polycomb group protein-mediated histone modifications during cell differentiation. Epigenomics 7:75–84. doi: 10.2217/epi.14.61. [DOI] [PubMed] [Google Scholar]
- 21.Lewis EB. 1978. A gene complex controlling segmentation in Drosophila. Nature 276:565–570. doi: 10.1038/276565a0. [DOI] [PubMed] [Google Scholar]
- 22.Holland PW, Booth HA, Bruford EA. 2007. Classification and nomenclature of all human homeobox genes. BMC Biol 5:47. doi: 10.1186/1741-7007-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi Y, Hamada J, Murakawa K, Takada M, Tada M, Nogami I, Hayashi N, Nakamori S, Monden M, Miyamoto M, Katoh H, Moriuchi T. 2004. Expression profiles of 39 HOX genes in normal human adult organs and anaplastic thyroid cancer cell lines by quantitative real-time RT-PCR system. Exp Cell Res 293:144–153. doi: 10.1016/j.yexcr.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 24.Kar SP, Tyrer JP, Li Q, Lawrenson K, Aben KKH, Anton-Culver H, Antonenkova N, Chenevix-Trench G, Baker H, Bandera EV, Bean YT, Beckmann MW, Berchuck A, Bisogna M, Bjorge L, Bogdanova N, Brinton L, Brooks-Wilson A, Butzow R, Campbell I, Carty K, Chang-Claude J, Chen YA, Chen Z, Cook LS, Cramer D, Cunningham JM, Cybulski C, Dansonka-Mieszkowska A, Dennis J, Dicks E, Doherty JA, Dork T, Du Bois A, Durst M, Eccles D, Easton DF, Edwards RP, Ekici AB, Fasching PA, Fridley BL, Gao Y-T, Gentry-Maharaj A, Giles GG, Glasspool R, Goode EL, Goodman MT, Grownwald J, Harrington P, et al. 2015. Network-based integration of GWAS and gene expression identifies a HOX-centric network associated with serous ovarian cancer risk. Cancer Epidemiol Biomarkers Prev 24:1574–1584. doi: 10.1158/1055-9965.EPI-14-1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarkar D, Leung EY, Baguley BC, Finlay GJ, Askarian-Amiri ME. 2015. Epigenetic regulation in human melanoma: past and future. Epigenetics 10:103–121. doi: 10.1080/15592294.2014.1003746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukherjee A, Shrivastava S, Bhanja Chowdhury J, Ray R, Ray RB. 2014. Transcriptional suppression of miR-181c by hepatitis C virus enhances homeobox A1 expression. J Virol 88:7929–7940. doi: 10.1128/JVI.00787-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamagishi T, Hirose S, Kondo T. 2008. Secondary DNA structure formation for Hoxb9 promoter and identification of its specific binding protein. Nucleic Acids Res 36:1965–1975. doi: 10.1093/nar/gkm1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buchwald G, van der Stoop P, Weichenrieder O, Perrakis A, van Lohuizen M, Sixma TK. 2006. Structure and E3-ligase activity of the Ring-Ring complex of polycomb proteins Bmi1 and Ring1b. EMBO J 25:2465–2474. doi: 10.1038/sj.emboj.7601144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Gottardi A, Pazienza V, Pugnale P, Bruttin F, Rubbia-Brandt L, Juge-Aubry CE, Meier CA, Hadengue A, Negro F. 2006. Peroxisome proliferator-activated receptor-alpha and -gamma mRNA levels are reduced in chronic hepatitis C with steatosis and genotype 3 infection. Aliment Pharmacol Ther 23:107–114. doi: 10.1111/j.1365-2036.2006.02729.x. [DOI] [PubMed] [Google Scholar]
- 30.Fukasawa M, Tanaka Y, Sato S, Ono Y, Nitahara-Kasahara Y, Suzuki T, Miyamura T, Hanada K, Nishijima M. 2006. Enhancement of de novo fatty acid biosynthesis in hepatic cell line Huh7 expressing hepatitis C virus core protein. Biol Pharm Bull 29:1958–1961. doi: 10.1248/bpb.29.1958. [DOI] [PubMed] [Google Scholar]
- 31.Masalova OV, Lesnova EI, Permyakova KY, Samokhvalov EI, Ivanov AV, Kochetkov SN, Kushch AA. 2016. Effect of hepatitis C virus proteins on the production of proinflammatory and profibrotic cytokines in Huh7.5 human hepatoma cells. Mol Biol (Mosk) 50:486–495. (In Russian.) [DOI] [PubMed] [Google Scholar]
- 32.Aizawa S, Okamoto T, Sugiyama Y, Kouwaki T, Ito A, Suzuki T, Ono C, Fukuhara T, Yamamoto M, Okochi M, Hiraga N, Imamura M, Chayama K, Suzuki R, Shoji I, Moriishi K, Moriya K, Koike K, Matsuura Y. 2016. TRC8-dependent degradation of hepatitis C virus immature core protein regulates viral propagation and pathogenesis. Nat Commun 7:11379. doi: 10.1038/ncomms11379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otoguro T, Tanaka T, Kasai H, Yamashita A, Moriishi K. 2016. Inhibitory effect of presenilin inhibitor LY411575 on maturation of hepatitis C virus core protein, production of the viral particle and expression of host proteins involved in pathogenicity. Microbiol Immunol 60:740–753. doi: 10.1111/1348-0421.12448. [DOI] [PubMed] [Google Scholar]
- 34.Zaaroor-Regev D, de Bie P, Scheffner M, Noy T, Shemer R, Heled M, Stein I, Pikarsky E, Ciechanover A. 2010. Regulation of the polycomb protein Ring1B by self-ubiquitination or by E6-AP may have implications to the pathogenesis of Angelman syndrome. Proc Natl Acad Sci U S A 107:6788–6793. doi: 10.1073/pnas.1003108107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mallo M, Alonso CR. 2013. The regulation of Hox gene expression during animal development. Development 140:3951–3963. doi: 10.1242/dev.068346. [DOI] [PubMed] [Google Scholar]
- 36.Cao R, Tsukada Y, Zhang Y. 2005. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell 20:845–854. doi: 10.1016/j.molcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Ben-Saadon R, Zaaroor D, Ziv T, Ciechanover A. 2006. The polycomb protein Ring1B generates self atypical mixed ubiquitin chains required for its in vitro histone H2A ligase activity. Mol Cell 24:701–711. doi: 10.1016/j.molcel.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 38.Karagiannis TC, Kn H, El-Osta A. 2007. Disparity of histone deacetylase inhibition on repair of radiation-induced DNA damage on euchromatin and constitutive heterochromatin compartments. Oncogene 26:3963–3971. doi: 10.1038/sj.onc.1210174. [DOI] [PubMed] [Google Scholar]
- 39.Machida K, McNamara G, Cheng KT, Huang J, Wang CH, Comai L, Ou JH, Lai MM. 2010. Hepatitis C virus inhibits DNA damage repair through reactive oxygen and nitrogen species and by interfering with the ATM-NBS1/Mre11/Rad50 DNA repair pathway in monocytes and hepatocytes. J Immunol 185:6985–6998. doi: 10.4049/jimmunol.1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, Shafer A, Neri F, Lee K, Kutyavin T, Stehling-Sun S, Johnson AK, Canfield TK, Giste E, Diegel M, Bates D, Hansen RS, Neph S, Sabo PJ, Heimfeld S, Raubitschek A, Ziegler S, Cotsapas C, Sotoodehnia N, Glass I, Sunyaev SR, Kaul R, Stamatoyannopoulos JA. 2012. Systematic localization of common disease-associated variation in regulatory DNA. Science 337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li B, Huang Q, Wei GH. 2019. The role of HOX transcription factors in cancer predisposition and progression. Cancers (Basel) 11:528. doi: 10.3390/cancers11040528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Joo MK, Park JJ, Chun HJ. 2016. Impact of homeobox genes in gastrointestinal cancer. World J Gastroenterol 22:8247–8256. doi: 10.3748/wjg.v22.i37.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Faber J, Krivtsov AV, Stubbs MC, Wright R, Davis TN, van den Heuvel-Eibrink M, Zwaan CM, Kung AL, Armstrong SA. 2009. HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood 113:2375–2385. doi: 10.1182/blood-2007-09-113597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rauch T, Wang ZD, Zhang XM, Zhong XY, Wu XW, Lau SK, Kernstine KH, Riggs AD, Pfeifer GP. 2007. Homeobox gene methylation in lung cancer studied by genome-wide analysis with a microarray-based methylated CpG island recovery assay. Proc Natl Acad Sci U S A 104:5527–5532. doi: 10.1073/pnas.0701059104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Z, Dahiya S, Provencher H, Muir B, Carney E, Coser K, Shioda T, Ma XJ, Sgroi DC. 2007. The prognostic biomarkers HOXB13, IL17BR, and CHDH are regulated by estrogen in breast cancer. Clin Cancer Res 13:6327–6334. doi: 10.1158/1078-0432.CCR-07-0310. [DOI] [PubMed] [Google Scholar]
- 46.Jung CY, Kim RS, Lee SJ, Wang CH, Jeng MH. 2004. HOXB13 homeodomain protein suppresses the growth of prostate cancer cells by the negative regulation of T-cell factor 4. Cancer Res 64:3046–3051. doi: 10.1158/0008-5472.can-03-2614. [DOI] [PubMed] [Google Scholar]
- 47.Hamdane N, Juhling F, Crouchet E, El Saghire H, Thumann C, Oudot MA, Bandiera S, Saviano A, Ponsolles C, Suarez AAR, Li S, Fujiwara N, Ono A, Davidson I, Bardeesy N, Schmidl C, Bock C, Schuster C, Lupberger J, Habersetzer F, Doffoel M, Piardi T, Sommacale D, Imamura M, Uchida T, Ohdan H, Aikata H, Chayama K, Boldanova T, Pessaux P, Fuchs BC, Hoshida Y, Zeisel MB, Duong FHT, Baumert TF. 2019. HCV-induced epigenetic changes associated with liver cancer risk persist after sustained virologic response. Gastroenterology 156:2313–2329.e7. doi: 10.1053/j.gastro.2019.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kalb R, Latwiel S, Baymaz HI, Jansen PWTC, Muller CW, Vermeulen M, Muller J. 2014. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat Struct Mol Biol 21:569–571. doi: 10.1038/nsmb.2833. [DOI] [PubMed] [Google Scholar]
- 49.Chan ST, Lee J, Narula M, Ou JJ. 2016. Suppression of host innate immune response by hepatitis C virus via induction of autophagic degradation of TRAF6. J Virol 90:10928–10935. doi: 10.1128/JVI.01365-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matsui C, Deng L, Minami N, Abe T, Koike K, Shoji I. 2018. Hepatitis C virus NS5A protein promotes the lysosomal degradation of hepatocyte nuclear factor 1alpha via chaperone-mediated autophagy. J Virol 92. doi: 10.1128/JVI.00639-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stevenson NJ, Bourke NM, Ryan EJ, Binder M, Fanning L, Johnston JA, Hegarty JE, Long A, O’Farrelly C. 2013. Hepatitis C virus targets the interferon-alpha JAK/STAT pathway by promoting proteasomal degradation in immune cells and hepatocytes. FEBS Lett 587:1571–1578. doi: 10.1016/j.febslet.2013.03.041. [DOI] [PubMed] [Google Scholar]
- 52.Xue B, Yang D, Wang J, Xu Y, Wang X, Qin Y, Tian R, Chen S, Xie Q, Liu N, Zhu H. 2016. ISG12a restricts hepatitis C virus infection through the ubiquitination-dependent degradation pathway. J Virol 90:6832–6845. doi: 10.1128/JVI.00352-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Osna NA, White RL, Krutik VM, Wang T, Weinman SA, Donohue TM Jr. 2008. Proteasome activation by hepatitis C core protein is reversed by ethanol-induced oxidative stress. Gastroenterology 134:2144–2152. doi: 10.1053/j.gastro.2008.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, Bell GW, Otte AP, Vidal M, Gifford DK, Young RA, Jaenisch R. 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 55.Brookes E, de Santiago I, Hebenstreit D, Morris KJ, Carroll T, Xie SQ, Stock JK, Heidemann M, Eick D, Nozaki N, Kimura H, Ragoussis J, Teichmann SA, Pombo A. 2012. Polycomb associates genome-wide with a specific RNA polymerase II variant, and regulates metabolic genes in ESCs. Cell Stem Cell 10:157–170. doi: 10.1016/j.stem.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okamoto T, Omori H, Kaname Y, Abe T, Nishimura Y, Suzuki T, Miyamura T, Yoshimori T, Moriishi K, Matsuura Y. 2008. A single-amino-acid mutation in hepatitis C virus NS5A disrupting FKBP8 interaction impairs viral replication. J Virol 82:3480–3489. doi: 10.1128/JVI.02253-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhong J, Gastaminza P, Cheng GF, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kasai H, Kawakami K, Yokoe H, Yoshimura K, Matsuda M, Yasumoto J, Maekawa S, Yamashita A, Tanaka T, Ikeda M, Kato N, Okamoto T, Matsuura Y, Sakamoto N, Enomoto N, Takeda S, Fujii H, Tsubuki M, Kusunoki M, Moriishi K. 2015. Involvement of FKBP6 in hepatitis C virus replication. Sci Rep 5:16699. doi: 10.1038/srep16699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moriishi K, Shoji I, Mori Y, Suzuki R, Suzuki T, Kataoka C, Matsuura Y. 2010. Involvement of PA28gamma in the propagation of hepatitis C virus. Hepatology 52:411–420. doi: 10.1002/hep.23680. [DOI] [PubMed] [Google Scholar]
- 61.Kao SH, Wang WL, Chen CY, Chang YL, Wu YY, Wang YT, Wang SP, Nesvizhskii AI, Chen YJ, Hong TM, Yang PC. 2015. Analysis of protein stability by the cycloheximide chase assay. Bio Protoc 5:e1734. doi: 10.21769/BioProtoc.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okamoto T, Nishimura Y, Ichimura T, Suzuki K, Miyamura T, Suzuki T, Moriishi K, Matsuura Y. 2006. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J 25:5015–5025. doi: 10.1038/sj.emboj.7601367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kikuchi K, Kasai H, Watanabe A, Lai AY, Kondo M. 2008. IL-7 specifies B cell fate at the common lymphoid progenitor to pre-proB transition stage by maintaining early B cell factor expression. J Immunol 181:383–392. doi: 10.4049/jimmunol.181.1.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sakurai N, Inamochi Y, Inoue T, Hariya N, Kawamura M, Yamada M, Dey A, Nishiyama A, Kubota T, Ozato K, Goda T, Mochizuki K. 2017. BRD4 regulates adiponectin gene induction by recruiting the P-TEFb complex to the transcribed region of the gene. Sci Rep 7:11962. doi: 10.1038/s41598-017-12342-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fujihara Y, Ikawa M. 2014. CRISPR/Cas9-based genome editing in mice by single plasmid injection. Methods Enzymol 546:319–336. doi: 10.1016/B978-0-12-801185-0.00015-5. [DOI] [PubMed] [Google Scholar]