

Abstract
Background
Anti‐neutrophilic cytoplasmic antibodies (ANCA)‐associated vasculitis (AAV) are a group of rare auto‐inflammatory diseases that affects mainly small vessels. AAV includes: granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA). Anti‐cytokine targeted therapy uses biological agents capable of specifically targeting and neutralising cytokine mediators of the inflammatory response.
Objectives
To assess the benefits and harms of anti‐cytokine targeted therapy for adults with AAV.
Search methods
We searched the Cochrane Central Register of Controlled Trials (2019, Issue 7), MEDLINE and Embase up to 16 August 2019. We also examined reference lists of articles, clinical trial registries, websites of regulatory agencies and contacted manufacturers.
Selection criteria
Randomised controlled trials (RCTs) or controlled clinical trials of targeted anti‐cytokine therapy in adults (18 years or older) with AAV compared with placebo, standard therapy or another modality and anti‐cytokine therapy of different type or dose.
Data collection and analysis
We used standard methodological procedures expected by Cochrane.
Main results
We included four RCTs with a total of 440 participants (mean age 48 to 56 years). We analysed the studies in three groups: 1) mepolizumab (300 mg; three separate injections every four weeks for 52 weeks) versus placebo in participants with relapsing or refractory EGPA; 2) belimumab (10 mg/kg on days 0, 14, 28 and every 28 days thereafter until 12 months after the last participant was randomised) or etanercept (25 mg twice a week) with standard therapy (median 25 months) versus placebo with standard therapy (median 19 months) in participants with GPA/MPA; and 3) infliximab (3 mg/kg on days 1 and 14, before the response assessment on day 42) versus rituximab (0.375g/m2 on days 1, 8, 15 and 22) in participants with refractory GPA for up to 12 months. None of the studies were assessed as low risk of bias in all domains: one study did not report randomisation or blinding methods clearly. Three studies were at high risk and one study was at unclear risk of bias for selective outcome reporting.
One trial with 136 participants with relapsing or refractory EGPA compared mepolizumab with placebo during 52 weeks of follow‐up and observed one death in the mepolizumab group (1/68, 1.5%) and none in the placebo group (0/68, 0%) (Peto odds ratio (OR) 7.39, 95% confidence interval (CI) 0.15 to 372.38; low‐certainty evidence). Low‐certainty evidence suggests that more participants in the mepolizumab group had ≥ 24 weeks of accrued remission over 52 weeks compared to placebo (27.9% versus 2.9%; risk ratio (RR) 9.5, 95% CI 2.30 to 39.21), and durable remission within the first 24 weeks sustained until week 52 (19.1% mepolizumab versus 1.5% placebo; RR 13.0, 95% CI 1.75 to 96.63; number needed to treat for an additional beneficial outcome (NNTB) 6, 95% Cl 4 to 13). Mepolizumab probably decreases risk of relapse (55.8% versus 82.4%; RR 0.68, 95% CI 0.53 to 0.86; NNTB 4, 95% CI 3 to 9; moderate‐certainty evidence). There was low‐certainty evidence regarding similar frequency of adverse events (AEs): total AEs (96.9% versus 94.1%; RR 1.03, 95% CI 0.96 to 1.11), serious AEs (17.7% versus 26.5%; RR 0.67, 95% CI 0.35 to 1.28) and withdrawals due to AEs (2.9% versus 1.5%; RR 2.00, 95% CI 0.19 to 21.54). Disease flares were not measured.
Based on two trials with different follow‐up periods (mean of 27 months for etanercept study; up to four years for belimumab study) including people with GPA (n = 263) and a small group of participants with MPA (n = 22) analysed together, we found low‐certainty evidence suggesting that adding an active drug (etanercept or belimumab) to standard therapy does not increase or reduce mortality (3.4% versus 1.4%; Peto OR 2.45, 95% CI 0.55 to 10.97). Etanercept may have little or no effect on remission (92.3% versus 89.5%; RR 0.97, 95% CI 0.89 to 1.07), durable remission (70% versus 75.3%; RR 0.93, 95% CI 0.77 to 1.11; low‐certainty evidence) and disease flares (56% versus 57.1%; RR 0.98, 95% CI 0.76 to 1.27; moderate‐certainty evidence). Low‐certainty evidence suggests that belimumab does not increase or reduce major relapse (1.9% versus 0%; RR 2.94, 95% CI 0.12 to 70.67) or any AE (92.5% versus 82.7%; RR 1.12, 95% CI 0.97 to 1.29). Low‐certainty evidence suggests a similar frequency of serious or severe AEs (47.6% versus 47.6%; RR 1.00, 95% CI 0.80 to 1.27), but more frequent withdrawals due to AEs in the active drug group (11.2%) compared to the placebo group (4.2%), RR 2.66, 95% CI 1.07 to 6.59).
One trial involving 17 participants with refractory GPA compared infliximab versus rituximab added to steroids and cytotoxic agents for 12 months. One participant died in each group (Peto OR 0.88, 95% CI, 0.05 to 15.51; 11% versus 12.5%). We have very low‐certainty evidence for remission (22% versus 50%, RR 0.44, 95% Cl 0.11 to 1.81) and durable remission (11% versus 50%, RR 0.22, 95% CI 0.03 to 1.60), any severe AE (22.3% versus 12.5%; RR 1.78, 95% CI 0.2 to 16.1) and withdrawals due to AEs (0% versus 0%; RR 2.70, 95% CI 0.13 to 58.24). Disease flare/relapse and the frequency of any AE were not reported.
Authors' conclusions
We found four studies but concerns about risk of bias and small sample sizes preclude firm conclusions.
We found moderate‐certainty evidence that in patients with relapsing or refractory EGPA, mepolizumab compared to placebo probably decreases disease relapse and low‐certainty evidence that mepolizumab may increase the probability of accruing at least 24 weeks of disease remission. There were similar frequencies of total and serious AEs in both groups, but the study was too small to reliably assess these outcomes. Mepolizumab may result in little to no difference in mortality. However, there were very few events.
In participants with GPA (and a small subgroup of participants with MPA), etanercept or belimumab may increase the probability of withdrawal due to AEs and may have little to no impact on serious AEs. Etanercept may have little or no impact on durable remission and probably does not reduce disease flare.
Plain language summary
What are the benefits and risks of anti‐cytokine medicines for ANCA‐associated vasculitis?
Why this question is important
The body’s defence (immune) system fights injury or infection by sending white blood cells to surround and protect the affected area. This causes redness and swelling, called inflammation.
Vasculitis is an inflammation of the blood vessels. In vasculitis, instead of reacting to harm, the immune system attacks healthy blood vessels. The reason for this reaction is often unknown.
One rare type of vasculitis is antineutrophil cytoplasmic antibody (ANCA)‐associated vasculitis (AAV). AAV covers three different conditions that are grouped together because they all affect small blood vessels:
‐ MPA: microscopic polyangiitis;
‐ GPA: granulomatosis with polyangiitis; and
‐ EPGA: eosinophilic granulomatosis with polyangiitis.
The areas of the body most commonly affected are kidneys, lungs, joints, ears, nose and nerves. It is important to treat AAV early, to prevent serious damage to these organs.
Currently, the recommended treatment for AAV is to use medicines that control the immune system and medicines against inflammation (steroids). However, this treatment causes serious unwanted effects. Medicines that target cytokines (small molecules that influence the immune system’s reactions) are an alternative option. To evaluate the benefits and risks of anti‐cytokine medicines, we reviewed the evidence from research studies.
How we identified and assessed the evidence
First, we searched for all relevant studies in the medical literature. We then compared the results, and summarised the evidence from all the studies. Finally, we assessed how certain the evidence was. We considered factors such as the way studies were conducted, study sizes, and consistency of findings across studies. Based on our assessments, we categorized the evidence as being of very low, low, moderate or high certainty.
What we found
We found four studies on a total of 440 adults from the USA and Europe. The average age of people ranged between 48 and 56 years. They received treatment for between 2 and 25 months, and were then followed for between 8 weeks and four years. Three studies compared anti‐cytokine medicines (mepolizumab, belimumab and etanercept) to a placebo (fake medicine) and one study compared two different anti‐cytokine medicines (rituximab versus infliximab). Three studies received at least partial funding from pharmaceutical companies.
Mepolizumab versus placebo in people with EGPA that returned after, or did not respond to, initial treatment
Moderate‐certainty evidence indicates that mepolizumab probably reduces the likelihood of the disease returning within a year of treatment.
Low‐certainty evidence suggests that mepolizumab:
‐ may make little or no difference to mortality;
‐ may increase the likelihood of the disease partially or fully disappearing for at least 24 weeks, and may increase the chances of this disappearance lasting for another six months at least;
‐ may make little or no difference to unwanted events, serious unwanted events or withdrawal from studies due to unwanted events.
The impact of mepolizumab on disease flare (worsening) is unknown, as this was not measured.
Etanercept or belimumab versus placebo in GPA and MPA
Moderate‐certainty evidence indicates that etanercept probably makes little or no difference to disease flare.
Low‐certainty evidence suggests that etanercept or belimumab may make little or no difference to:
‐ mortality;
‐ the disease fully disappearing for at least 24 weeks, or disappearance lasting at least another six months after that;
‐ the disease returning strongly;
‐ unwanted events or severe/serious unwanted events
Evidence of low certainty suggests that etanercept or belimumab may slightly increase chances of people withdrawing from studies due to unwanted events.
Infliximab versus rituximab, plus steroids and cytotoxic agents (substances that kill cells), in people with GPA that did not respond to other treatments
The one study we found was too small to assess the differences between treatments (very low‐certainty evidence).
What this means
Mepolizumab probably reduces the likelihood of the disease returning within a year of treatment, and etanercept probably makes little or no difference to disease flare. We are less certain of the other potential benefits or risks of anti‐cytokine medicines because the evidence is of low or very low certainty. Further research is likely to change the findings of this review.
How‐up‐to date is this review?
The evidence in this Cochrane Review is current to August 2019.
Summary of findings
Background
Description of the condition
Vasculitides are a heterogenous group of rare diseases characterised by inflammation of the vessel wall. Infectious, environmental (e.g. drugs), genetic and other factors are probably involved in the pathogenesis of vasculitis (McKinney 2014). The underlying pathology is complex and characteristically involves immune‐mediated inflammation of blood vessel walls, resulting in ischaemic and localised inflammatory injury to tissues in the territory of the affected vessels (Gonzalez‐Gay 2002). Disease subtypes follow the Chapel Hill Consensus Conference (CHCC) nomenclature system, which has been in use since 1994. This document categorises vasculitides on the basis of several aetiological and clinical features, including the diameter of the affected vessels (large, medium, small vessel vasculitis). Major advances in our understanding of the vasculitis pathology, including the importance of ANCA status, were among the main reasons for the CHCC 2012 revision. Additionally, disease eponym names were replaced with descriptive names (e.g. granulomatosis with polyangiitis, instead of Wegener’s granulomatosis) (Jeanette 2013).
According to this nomenclature, ANCA‐associated vasculitis (AAV) predominantly affects small vessels and consists of three entities: granulomatosis with polyangiitis (GPA; Wegener’s granulomatosis), microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA; Churg‐Strauss vasculitis) (Merkel 2015). AAV entities are rare diseases with combined prevalence estimated at between 90 and 144 per million, and with annual incidence of around 20 per million (Cotch 1996; Gibson 2006; Mahr 2004; Watts 2000).
The frequency of ANCA‐associated vasculitis seems to be geographically determined. For example, increased incidence of GPA occurs in northern Europe, whereas greater MPA incidence has been reported in southern Europe (Katsuyama 2014). Some evidence indicates that the incidence is increasing (Watts 2000). Not all AAV conditions have positive ANCA status; this status differs by entity. Approximately 85% to 90% of patients with GPA, 70% of those with MPA and approximately 30% to 40% of patients with EGPA are ANCA‐positive (Mahr 2014; Merkel 2015).
If ANCAs are present in AAV, they are most commonly directed against myeloperoxidase (MPO‐ANCA) or proteinase 3 (PR3‐ANCA) (Harper 2001) and can help in establishing the diagnosis. Increasing evidence suggests that ANCA status determines the disease course and clinical manifestations (Cottin 2017).
Description of the intervention
Treatment recommended for AAV depends on disease severity; induction therapy based on glucocorticosteroids and cyclophosphamide or rituximab is indicated for patients with organ‐threatening disease, particularly of the lungs, kidneys and the nervous system; and glucocorticosteroids and methotrexate or mycophenolate mofetil for those with non‐organ‐threatening AAV (Yates 2016). Cyclophosphamide, which was introduced in the 1970s by Hoffman and Fauci (Kamesh 2002), is a potent, cytotoxic immunosuppressive agent that has dramatically improved survival among patients with some forms of vasculitides. The combination of cyclophosphamide and prednisolone has become the gold standard for treatment of patients with active systemic vasculitis but it is associated with serious adverse effects, including neutropenia, opportunistic infection, premature menopause and increased rates of cancer, most notably, cancer of the bladder (Fauci 1983; Kamesh 2002). Prolonged follow‐up of patients who have received cyclophosphamide reveals increasing over time dose‐dependent rates of bladder cancer (Knight 2004). To overcome these treatment‐related toxicities, strategies for introducing less toxic immunosuppressive agents (e.g. methotrexate or azathioprine after induction therapy with a series of cyclophosphamide infusions) have been used with moderate success (Jayne 2003). However, there remains an unmet need for more effective and less toxic treatments for patients in induction and remission phases of vasculitides. Recently, biological immunotherapies capable of specifically targeting and neutralising cytokine mediators of the inflammatory response have entered clinical practice for a variety of immune‐mediated inflammatory diseases. Among biological therapies, treatment options include anti‐tumour necrosis factor (TNF) agents (infliximab, etanercept, adalimumab) or rituximab, which belongs to the group of anti‐CD20 monoclonal antibodies (Kamesh 2002, Silva‐Fernández 2014). Another Cochrane Review, now in development, is assessing the benefit of rituximab (Riminton 2008). Molecules other than anti‐TNF agents and rituximab, such as abatacept, mepolizumab (an anti‐IL5 antibody) and alemtuzumab (a humanised monoclonal anti‐CD52 antibody) have been used in refractory cases of AAV. These agents hold promise for safer, more effective targeted intervention, and studies in ANCA‐associated vasculitis have commenced. Mepolizumab has been used in a pilot study on EGPA to allow decreasing steroid doses (Kim 2010) and has been fully assessed in a randomised trial called MIRRA (Wechsler 2017).
How the intervention might work
Consideration of the mechanisms underlying chronic inflammatory disease such as vasculitis has revealed a primary, pivotal role of cytokines. Cytokines comprise a diverse family of small molecule mediators of intercellular communication that have essential roles in mobilisation of the inflammatory response. Tumour necrosis factor‐alpha (TNF‐alpha), as one example, has a central role in the downstream production of both pro‐inflammatory and anti‐inflammatory cytokines, which appear to be maintained at a dysfunctional equilibrium in chronic inflammatory disease. TNF‐alpha is among the first of the cytokines to rise in response to bacterial infection (Feldmann 2006). Studies have shown that TNF‐alpha plays a central role in mouse models of renal vasculitis, and the treatment of the affected mice with anti‐TNF‐alpha has improved their outcomes (Feldmann 2006). In another chronic immune‐mediated inflammatory disease, rheumatoid arthritis (RA), the inhibition of TNF‐alpha activity by both monoclonal antibodies and a soluble receptor decoy has been shown to be effective in the management of signs, symptoms and radiographic progression (Chen 2006). Another anti‐cytokine therapy, namely, interleukin‐1 receptor antagonist (IL‐1RA; anakinra), has been shown to provide benefit for patients with RA (Cohen 2002). Future directions and potential therapeutic options include interleukin (IL)‐6 antagonist (B‐cell stimulatory factor‐2; BSF‐2). Several studies have reported that tocilizumab (anti‐interleukin‐6 agent) was successfully used for some systemic diseases such as RA, and its benefit in RA is well established (Berli 2015). Inhibition of pro‐inflammatory cytokines has therefore emerged as an attractive prospect for the management of ANCA‐associated vasculitis.
Why it is important to do this review
ANCA‐associated vasculitis comprises a group of rare systemic diseases. Effective treatment is important because progression of these diseases may be dramatic. Physicians who are responsible for the care of these patients are confronted with an important dilemma: whether to employ conventional cytotoxic immunosuppressive strategies with unsatisfactory primary resistance, relapse and drug‐induced adverse events (AEs); or whether to employ novel biological anti‐cytokine therapies with the potential for greater benefit and tolerability. Further uncertainty is raised by the use of combinations of conventional and novel therapies, and by a rapidly evolving evidence base. Anti‐cytokine therapies represent another treatment option. Systematic reviews can provide some answers.
Use of anti‐cytokine therapy has been reported in patients with ANCA‐associated vasculitides (e.g. Al‐Bishri 2005; Lamprecht 2002; WGET 2005), but no Cochrane systematic review has focused on this type of treatment in this group of patients.
We conducted this review according to the guidelines provided by the Cochrane Musculoskeletal Group Editorial Board (Ghogomu 2014).
Objectives
To assess the benefits and harms of anti‐cytokine targeted therapy for adults with ANCA‐associated vasculitis.
Methods
Criteria for considering studies for this review
Types of studies
We considered randomised controlled trials (RCTs) and controlled clinical trials (CCTs) for inclusion in this review. We planned to include studies reported as abstracts without data in the 'Studies awaiting assessment' category and to contact study authors for additional detailed data, but we have not come across such a study. However, we came across several studies that were registered and completed but did not report their results. In such cases, we attempted to contact the authors for additional information. We applied no restrictions on length of follow‐up or language.
Types of participants
We restricted inclusion in this review to trials that met the following criteria.
All studies primarily concerning ANCA‐associated vasculitis in adult populations (18 years of age or older).
Specific confirmed diagnoses of participants including GPA, EGPA and MPA) .
We excluded patients with other types of vasculitides.
Types of interventions
We considered all randomised controlled comparisons of specifically targeted anti‐cytokine therapy versus placebo, standard therapy or another modality. We considered various types and dosages of anti‐cytokine therapy.
We considered all available anti‐cytokine therapies, such as TNF‐alpha inhibitors (adalimumab, certolizumab, etanercept, golimumab, infliximab), inhibitors of soluble B‐lymphocyte stimulator (BLyS) cytokine (belimumab), interleukin receptor antagonists and interleukin inhibitors (anakinra, basiliximab, benralizumab, brodalumab, canakinumab, clazakizumab, daclizumab, dupilumab, ixekizumab, lebrikizumab, mepolizumab, olokizumab, pitrakinra, reslizumab, rilonacept, sarilumab, secukinumab, siltuximab, sirukumab, tocilizumab, tralokinumab, ustekinumab), as well as anti‐cytokine therapies that will be developed in the future. We did not include interventions that were not specifically directed at cytokines but that may nevertheless alter cytokine expression or function (e.g. corticosteroids).
Types of outcome measures
Major outcomes
Major benefit outcomes included:
mortality;
remission (as defined by study authors, typically as complete absence of disease activity (Merkel 2011) measured by Birmingham Vasculitis Activity Score (BVAS), BVAS/ WG (for GPA) or BVAS v3);
durable remission (defined according to BVAS, BVAS WG or BVAS v3 for at least six months) (WGET 2005); and
disease flare/relapse (as defined by study authors, typically as increased disease activity from a previous low or absent state) (Merkel 2011).
Major harms outcomes included:
total AEs;
serious AEs; and
withdrawals due to AEs.
Minor outcomes
Minor outcomes included:
treatment response (defined as quantifiable improvement in disease activity (Merkel 2011) as assessed by BVAS, BVAS WG or BVAS v3 with cut‐off point as defined by study authors);
health‐related quality of life (as assessed by Short Form (SF)‐36 or other health‐related quality of life measures, including those specific to AAV);
control of asthma/sinonasal disease (as defined by study authors); and
disease damage according to the Vasculitis Damage Index (VDI), the AAV Index of Damage (AVID) or other validated disease damage scores accepted by Outcome Measures in Rheumatology (OMERACT).
Time points
We planned to collect data reported at six months,12 months and more than 12 months, as well as during active treatment and after treatment cessation. Ultimately, we included all time points reported in the studies.
Search methods for identification of studies
We searched all databases from their inception to the 16 August 2019, and we imposed no restriction on language and date of publication.
Electronic searches
We searched the following electronic databases and sources to identify studies (all searches performed on 16 August 2019).
Cochrane Central Register of Controlled Trials (2019, Issue 7).
MEDLINE (OVID).
Embase (OVID).
We searched the following ongoing trial registries (all searches performed on 28 August 2019).
ClinicalTrials.gov (www.ClinicalTrials.gov).
European Trials Register (www.clinicaltrialsregister.eu).
ISRCTN (International Standard Randomised Controlled Trial Number Registry; www.isrctn.com).
WHO (World Health Organization) trials portal (www.who.int/ictrp/en).
For the assessments of AEs, we searched the web sites of regulatory agencies (all searches performed on 3 September 2019), such as the US Food and Drug Administration‐MedWatch (www.fda.gov/Safety/MedWatch/default.htm), the European Medicines Evaluation Agency (www.emea.europa.eu), the Australian Adverse Drug Reactions Bulletin (www.tga.gov.au/adr/aadrb.htm) and the UK Medicines and Healthcare products Regulatory Agency (MHRA) for pharmacovigilance and drug safety updates (www.mhra.gov.uk). We searched each database using basic terms, e.g. ANCA vasculitis, etanercept, mepolizumab, infliximab and belimumab.
See Appendix 1 for the MEDLINE search strategy, Embase search strategy and CENTRAL search strategy.
Searching other resources
We checked the reference lists of all primary studies and review articles for additional references. We searched relevant manufacturers' web sites for trial information. When we identified unpublished trials, we sought contact with relevant authors for further information. We checked www.clinicaltrialresults.org and the web sites of regulatory agencies such as the US Food and Drug Administration‐MedWatch (www.fda.gov) and the European Medicines Evaluation Agency (www.emea.europa.eu) for unpublished data.
We searched for errata and retractions from included studies published in full text on PubMed (www.ncbi.nlm.nih.gov/pubmed) and planned to report in the review the date this was done, but no such cases occurred.
Data collection and analysis
Selection of studies
For study selection, we used Covidence (www.covidence.org), a new tool recommended by Cochrane to facilitate production of systematic reviews. Two review authors (JZ, MK, TMM, MMB, JJ or WS) independently screened titles and abstracts of articles identified by the search to determine their potential for inclusion in the review and coded them as 'yes/maybe' (eligible or potentially eligible/unclear) or 'no.' We retrieved full‐text study reports/publications, and two review authors (JZ, MK, TMM, MMB, JJ or WS) independently screened full texts to identify studies for inclusion and recorded reasons for exclusion of ineligible studies. We resolved disagreements through discussion, or, if required, we consulted a third review author (WS or MMB). We identified and excluded duplicates and collated multiple reports of the same study, so that each study, rather than each report, was the unit of interest in the review. We recorded the selection process in sufficient detail to complete a PRISMA (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses) flow diagram (PRISMA Group) (Moher 2009) and 'Characteristics of excluded studies' tables.
Data extraction and management
If Covidence allowed for adjustment of data extraction forms, we planned to use this tool for data extraction; otherwise, we planned to prepare a data extraction form in Microsoft Excel. Ultimately, we extracted the data to the forms prepared in Microsoft Excel. We recorded study characteristics and outcome data on a data collection form that was piloted on at least one study in the review. We planned that the extraction process would be carried out by one review author and spot‐checked by another one, but to improve the quality of our review we decided to proceed with independent extraction by two review authors (JZ, JJ or MK). We extracted the following study characteristics from the included studies.
Methods: study design, total duration of study, details of any 'run‐in' period, number of study centres and locations, study setting, withdrawals, date of study.
Participants: number of participants included in the study and number of participants in each group, number of participants who completed follow‐up, mean age, age range, sex, disease duration, severity of condition, diagnostic criteria, important ANCA vasculitides‐specific baseline data such as prior treatment, presence of comorbidity.
Interventions: intervention, comparison, dosing regimen, route of administration, concomitant use of steroids, excluded medications, duration of treatment.
Outcomes: major and minor outcomes specified and collected, time points reported.
Characteristics of trial design as outlined in the 'Assessment of risk of bias in included trials' section.
Notes: funding for trial, notable declarations of interest of trial authors.
Each extraction was checked for accuracy against the trial report by an additional review author (MMB, TMM or WS).
Two review authors (JZ, JJ or MK) independently extracted outcome data from the included studies. We extracted numbers of events and participants per treatment group for dichotomous outcomes; and means, standard deviations and numbers of participants per treatment group for continuous outcomes. If reported, we extracted confidence intervals and P values. We planned to note in the 'Characteristics of included studies' table if outcome data were not reported in a useable way and if data were transformed or estimated from a graph. We resolved disagreements by reaching consensus or by involving a third review author (MMB, TMM or WS). One review author (MK, JZ or JJ) transferred data into the Review Manager (RevMan 2014) file. We double‐checked whether that data were entered correctly by comparing data presented in the systematic review with the study reports.
We used results from an intention‐to‐treat analysis, if possible. If a study reported multiple time point measurements, we extracted all time point values and used final values data for analysis. For continuous outcomes, if both final values and change from baseline values were reported for the same outcome, we planned to extract both values and use change values for primary analysis. If investigators reported both adjusted and unadjusted values for the same outcome, we planned to extract both estimates and use adjusted values with the maximum number of covariates.
Assessment of risk of bias in included studies
Two pairs of review authors (JZ, MK, JJ, MMB, TMM or WS) independently assessed the risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). We resolved disagreements by discussion or by consultation with another review author (WS or MMB). We assessed the risk of bias according to the following domains.
Random sequence generation.
Allocation concealment.
Blinding of participants and personnel.
Blinding of outcome assessment.
Incomplete outcome data.
Selective outcome reporting.
Other bias (such as bias related to issues of study design, baseline imbalance, stopping early for benefit, influence of interim results on study conduct, inappropriate administration of co‐interventions and selective reporting of subgroups).
We graded each potential source of bias as high, low or unclear and provided a quote from the study report together with a justification for our judgement in the 'Risk of bias' table. We summarised the risk of bias judgements per outcomes within a study and per outcomes across studies.
When we obtained information on risk of bias related to unpublished data or correspondence with a trialist, we planned to note this in the 'Risk of bias' table.
When considering treatment effects, we took into account the risk of bias for studies that contributed to that outcome.
We presented figures generated by the 'risk of bias' tool to provide summary assessments of the risk of bias.
We considered a study to have low risk of bias if we judged it to be at low risk in all domains for each outcome; otherwise, we considered the study to have a high risk of bias.
Assessment of bias in conducting the systematic review
We conducted the review according to the published protocol and reported deviations from it in the 'Differences between protocol and review' section of the systematic review.
Measures of treatment effect
We analysed dichotomous data as risk ratios (RRs) or Peto odds ratios (OR) when the outcome was a rare event (approximately < 10%) and used 95% confidence intervals (CIs). We planned to analyse continuous data as mean differences (MDs) or standardised mean differences (SMDs), depending on whether the same scale was used to measure the outcome, and 95% CIs. We planned to enter data presented as a scale with a consistent direction of effect across studies.
We planned to back‐translate SMD to a typical scale (e.g. BVAS) by multiplying the SMD by a typical among‐person standard deviation (e.g. standard deviation of the control group at baseline from the most representative trial) (as per Chapter 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2011b)).
If results were reported only as mean differences with standard errors (SEs) or 95% CIs, we planned to pool them using the generic inverse variance method available in RevMan5.
We planned to analyse time‐to‐event data as hazard ratios (HR).
In the 'Effects of intervention' section under 'Results' and in the 'Comments' column of the 'Summary of findings' table, we planned to provide the absolute percentage difference, the relative percentage change from baseline and the number needed to treat for an additional beneficial outcome (NNTB). We provided the NNTB only when the outcome showed a statistically significant difference.
For dichotomous outcomes, such as serious AEs, we planned to calculate the NNTB/number needed to treat for an additional harmful effect (NNTH) from the control group event rate and the risk difference (RD) using the Visual Rx NNT calculator (Cates 2008). When this was not possible, we used RD and 95% CI calculated in RevMan to calculate NNTB with 100/RD formula. We planned to calculate the NNTB/NNTH for continuous measures using the Wells calculator (available at the CMSG Editorial Office; musculoskeletal.cochrane.org).
For dichotomous outcomes, we calculated the absolute RD using the RD statistic in RevMan (RevMan 2014) and expressed the result as a percentage. For continuous outcomes, we planned to calculate the absolute benefit as improvement in the intervention group minus improvement in the control group, in original units, and express this as a percentage.
We calculated the relative percentage change for dichotomous data as the risk ratio ‐ 1 and expressed this as a percentage. For continuous outcomes, we planned to calculate the relative difference in change from baseline as the absolute benefit divided by the baseline mean of the control group.
Unit of analysis issues
When multiple trial arms were reported in a single trial, we planned to include only relevant arms. If two comparisons (e.g. drug A versus placebo and drug B versus placebo) were combined in the same meta‐analysis, we planned to halve the control group to avoid double‐counting.
We did not expect to find cross over studies or cluster RCTs but had we done so, we would have followed the guidance in chapter 16 of the Cochrane Handbook (Higgins 2011b).
Dealing with missing data
We contacted investigators or study sponsors to verify key study characteristics and to obtain missing numerical outcome data when possible (e.g. when a study was identified as abstract only, or when data were not available for all participants). When this was not possible, and missing data were thought to introduce serious bias, we planned to explore the impact of including such studies in the overall assessment of results by performing a sensitivity analysis. We planned to clearly describe assumptions and imputations required to handle missing data and explore the effect of imputation by performing sensitivity analyses. In all analyses, we included the numbers of participants reported by the authors for particular outcome and we performed sensitivity analyses in the case of studies with missing data.
For dichotomous outcomes (e.g. number of withdrawals due to AEs), we planned to calculate the withdrawal rate by using the number of participants randomised in the group as the denominator.
For continuous outcomes (e.g. mean change in pain score), we planned to calculate the mean MD or the SMD by using the number of participants analysed at that time point. If the number of participants analysed was not presented for each time point, we planned to use the number of randomised participants in each group at baseline.
Where possible, we planned to compute missing SDs from other statistics such as SEs, CIs or P values, according to the methods recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011c). If standard deviations could not be calculated, we planned to impute them (e.g. from other studies in the meta‐analysis) following the advice of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011c).
Assessment of heterogeneity
We assessed the clinical and methodological diversity of the included studies, in terms of participants, interventions, outcomes and study characteristics, to determine whether meta‐analyses were appropriate. We planned to do this by observing data derived from data extraction tables. We planned to assess statistical heterogeneity by visually inspecting forest plots to look for obvious differences in results among studies, and by using I² and Chi² statistical tests.
The Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011) advises that an I² value from 0% to 40% might not represent important heterogeneity; a value from 30% to 60% may represent 'moderate' heterogeneity; a value from 50% to 90% may represent 'substantial' heterogeneity; and a value from 75% to 100% represents 'considerable' heterogeneity. As noted in the Cochrane Handbook for Systematic Reviews of Interventions, we kept in mind that the importance of I² depends on the magnitude and direction of effects, and on strength of the evidence for heterogeneity.
We interpreted the Chi² test with a P value ≤ 0.10 as evidence of statistical heterogeneity.
If we identified substantial heterogeneity, we planned to report this and investigate possible causes by following the recommendations provided in Section 9.6 of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011).
Assessment of reporting biases
If possible, we planned to create and examine a funnel plot to explore possible small study biases. In interpreting funnel plots, we planned to examine different possible reasons for funnel plot asymmetry, as outlined in Section 10.4 of the Cochrane Handbook for Systematic Reviews of Interventions and planned to relate this to review results. If we were able to pool more than 10 trials, we planned to undertake formal statistical tests to investigate funnel plot asymmetry in accordance with the recommendations provided in Section 10.4 of the Cochrane Handbook for Systematic Reviews of Interventions (Sterne 2011).
To assess outcome reporting bias, we checked trial protocols against published reports. For studies published after 1 July 2005, we searched the International Clinical Trials Registry Platform of the World Health Organization (apps.who.int/trialssearch) for the a priori trial protocol. We compared the available protocols with the final results to evaluate whether selective reporting of outcomes occurred.
Data synthesis
We planned to undertake meta‐analysis only when this is meaningful (i.e. when treatments, participants and the underlying clinical question are similar enough for pooling to make sense). For the main analyses, we planned to pool results for the longest follow‐up point available.
We planned to use a random‐effects model and planned to perform a sensitivity analysis based on the fixed‐effect model. However, our analyses included either single studies presented on the forest plots, or there was no statistical heterogeneity. Therefore, sensitivity analysis with a fixed‐effect model would not have been meaningful.
We planned to restrict the primary analysis of self‐reported outcomes in this review to trials at low risk of detection and selection bias. However, there was only one such trial.
'Summary of findings' table
We created a 'Summary of findings' (SoF) table using the following outcomes: mortality, remission, durable remission, disease flare/relapse, total AEs, serious AEs and withdrawals due to AEs. The first SoF table presents the comparison of mepolizumab to placebo for adult patients with EGPA ANCA‐associated vasculitis, followed by the SoF table with comparison of active drug (etanercept or belimumab) and standard therapy with standard therapy and placebo in adult patients with GPA ANCA‐associated vasculitis and SoF table with comparison of infliximab with rituximab in adult patients with refractory GPA ANCA‐associated vasculitis.
Two review authors (JJ and MK) independently assessed the certainty of the evidence. We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness and publication bias) to assess the certainty of a body of evidence as it relates to studies that contribute data to meta‐analyses for prespecified outcomes. We used methods and recommendations described in Sections 8.5 and 8.7, in Chapter 11 and in Chapter 13, Section 13.5 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a; Schünemann 2011a; Schünemann 2011b). We used GRADEpro software to prepare SoF tables (GRADEpro GDT 2015). We justified all decisions to downgrade or upgrade the certainty of studies by using footnotes, and we provided comments to aid the reader's understanding of the review when necessary.
In the 'What happens' column of the SoF table, we provided the absolute percentage difference, the relative percentage change from baseline and the NNTB or the NNTH (but only when the outcome showed a statistically significant difference).
Subgroup analysis and investigation of heterogeneity
If possible, we planned to carry out the following subgroup analyses.
Newly diagnosed or relapsing ‐ as treatment effects may differ in participants receiving treatment for the first time and those receiving repeated treatment.
Different dosage regimens ‐ different doses may show different treatment effects.
Duration of treatment ‐ duration of treatment may also influence the results.
Type of AAV.
ANCA status.
We planned to use the following outcomes in subgroup analyses.
Remission.
Total AEs.
We planned to use the formal test for subgroup interactions in Review Manager (RevMan 2014) and apply caution in interpreting subgroup analyses, as advised in Section 9.6 of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011). We planned to compare the magnitude of effects between subgroups by assessing the overlap of confidence intervals of the summary as estimated. Non‐overlap of confidence intervals indicates statistical significance.
Since the included studies recruited participants with GPA/MPA only or with EGPA only (relapsing or refractory) we decided to summarise the results separately in the population groups (GPA and EGPA) due to their clinical heterogeneity, as combining them would not be clinically meaningful. We did not have enough studies to carry out other subgroup analyses, therefore, we provided the results in subgroups presented by the authors of the analysed studies, if there was stratification for that characteristic at randomisation.
Sensitivity analysis
We planned to carry out the following sensitivity analyses.
Effect of assessing study risk of bias ‐ as adequate allocation concealment and outcome assessor blinding.
Effect of imputing missing data.
Effect of including different types of data (i.e. instead of change value, final value for continuous outcomes; instead of adjusted value, unadjusted value).
However, only one study reported excluding participants from analysis (WGET 2005) and only in the control group. We repeated the analyses that included this study in the scope of the outcomes for the excluded participants, with the assumption that the participants missing in the control group had the best possible outcome.
Since we had planned to perform analyses across all AAV and based on clinical heterogeneity, we decided to pool the data separately for each type of AAV. We performed sensitivity analysis by pooling the different AAV types together for the outcomes for which we had data from more than one study.
Interpreting results and reaching conclusions
We followed the guidelines provided in the Cochrane Handbook for Systematic Reviews of Interventions (see Chapter 12) (Schünemann 2011b) when interpreting results, and we were aware of distinguishing the lack of evidence of effect from the lack of effect. We based our conclusions only on findings from the quantitative or narrative synthesis of the studies included in the studies used in this review. We avoided making recommendations for practice, and our implications for research suggest priorities for future studies while outlining the remaining uncertainties in this area.
Results
Description of studies
Results of the search
We performed our final searches on 16 August 2019. After removing duplicates, we identified 3023 records. We searched trial registries on 28 August 2019 and found 1835 additional records (Figure 1). Of those records, we retrieved 73 and reviewed them in full text. We excluded papers due to wrong study design (43 papers) or wrong intervention (three papers). We excluded two studies that were withdrawn and stopped by the sponsor because they did not enrol any participants (BIANCA‐SC, Eculizumab 2011) and thus published no results. We identified three studies that still await assessment, as there was insufficient information in the databases. Two of them were closed without being completed (NIAID 1999, NIAID 2002); one was terminated due to slow recruitment, and posted no results (ABAVAS 2008).
1.
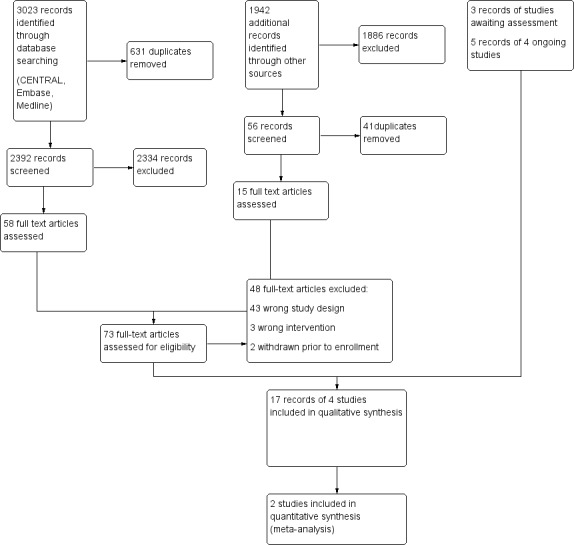
Study flow diagram.
Searching trials registries, we identified four ongoing studies (ABROGATE; ALEVIATE; AAVTCZ; COMBIVAS). The ABROGATE (NCT02108860) and COMBIVAS (NCT03967925) trials are ongoing and are currently recruiting participants. ALEVIATE trial (NCT01405807) has completed recruitment but follow‐up is ongoing. In AAVTCZ trial (JPRN‐UMIN000024574) first enrolment was planned on 1st May 2017.
Searching the databases reporting on AEs, we identified no relevant studies.
Reasons for study exclusions are reported in Characteristics of excluded studies.
We included four studies fulfilling criteria of our review (de Menthon 2011; Wechsler 2017; WGET 2005; BREVAS 2018). These studies were reported in 17 references.
Included studies
The four included studies were described as RCTs. Three were described as double blind (BREVAS 2018; Wechsler 2017; WGET 2005. One study provided no information about blinding (de Menthon 2011). All records were available in English. We extracted data from full‐text publications for all of them, and supplemented these, when available, with data posted in clinicaltrials.gov and clinicaltrialsregister.eu (BREVAS 2018).
We presented details of the methods, participants, interventions and outcome measures in each study in Characteristics of included studies.
Funding
One study was funded from a commercial source (BREVAS 2018). One study was funded from a hospital research program (de Menthon 2011). Two studies had mixed funding from public and commercial sources (Wechsler 2017; WGET 2005).
Participants
The included studies enrolled a total of 440 participants with ANCA vasculitis. The number of participants ranged from 17 (de Menthon 2011) to 181 (WGET 2005). All trials enrolled adult participants (18 years of older). Participants with GPA were included in three studies (de Menthon 2011; WGET 2005; BREVAS 2018). One of those studies also included participants with MPA (BREVAS 2018), but the results of MPA participants were added to the GPA participant analysis. The fourth study focused on participants with relapsing or refractory EGPA (Wechsler 2017). Participants in de Menthon 2011 had refractory disease and failed previous standard treatment.
The mean age of participants was between 48 years (control group of Wechsler 2017) and 56 years (SD 14) (belimumab group of BREVAS 2018). The proportion of females varied between 37% in etanercept group (WGET 2005) to 62% in mepolizumab group (Wechsler 2017). The proportion differed more in de Menthon 2011 study, where there were 12.5% of females in rituximab group and 89% in infliximab group.
Study duration
All studies lasted at least one year. The treatment was maintained up to12 months (de Menthon 2011), 52 weeks (Wechsler 2017), 12 months after the last participant was randomised (BREVAS 2018), and a median of 25 months in etanercept and 19 months in placebo group in WGET 2005 . BREVAS 2018 did not report on mean or median treatment duration. Reported follow‐up differed among the studies: Wechsler 2017 followed participants for 8 weeks, while in de Menthon 2011 study the average follow‐up reached 30.6 months. In WGET 2005 the mean follow‐up for the overall cohort was 27 months, and in BREVAS 2018 study follow‐up was approximately four years.
Location
One study was carried out in the USA (WGET 2005). One was conducted in several French hospitals (de Menthon 2011). Two trials were conducted by international groups in the USA and Europe (BREVAS 2018; Wechsler 2017).
Setting
Two trials were conducted in clinical centres (Wechsler 2017; WGET 2005) and one in hospitals (de Menthon 2011). The setting for one study was not reported (BREVAS 2018).
Interventions
One study compared two pharmacological treatments: rituximab at a dose of 0.375 g/m2 versus infliximab at a dose of 3 mg/kg (de Menthon 2011). Three studies compared active drugs with placebo: belimumab at a dose of 10 mg/kg (BREVAS 2018), mepolizumab 300 mg (Wechsler 2017) and etanercept 25 mg twice a week (WGET 2005).
Major outcome measures
All studies reported mortality as the number of deaths (BREVAS 2018; de Menthon 2011; Wechsler 2017; WGET 2005). Remission was reported in three studies (de Menthon 2011; Wechsler 2017; WGET 2005). One study assessed complete and partial remission, defined as the absence of active vasculitis manifestation (BVAS = 0) and partial regression of the clinical manifestations and a decrease in BVAS by > 50%, respectively (de Menthon 2011). The second study reported remission as the proportion of participants who had remission (BVAS = 0 and the receipt of prednisone at a dose of ≤ 4.0 mg/d over 52 weeks) for a certain period of time (in five categories of remission accrual; however, we described only remission for at least one week and remission for at least 24 weeks); and at weeks 36 and 48 of the study treatment period (Wechsler 2017). The third trial assessed disease remission defined as a BVAS/WG of 0 (WGET 2005).
Three studies also reported durable or sustained remission (de Menthon 2011; Wechsler 2017; WGET 2005), defined as remission for at least six months (WGET 2005); remission achieved within 24 weeks and sustained until week 52 (Wechsler 2017); or persistent remission, defined as remission during a long‐term follow‐up (de Menthon 2011). In BREVAS 2018, all participants were in remission at the beginning of the study. The study authors looked at the maintained remission at one and two years (BVAS = 0).
Minor outcome measures
All trials published data regarding number of participants with confirmed disease relapse/flares (BREVAS 2018; de Menthon 2011; Wechsler 2017; WGET 2005). BREVAS 2018 assessed time to first relapse, which was defined more broadly than vasculitis relapse, i.e. at least one major BVAS item or a minimum total BVAS score of 6 or receiving prohibited medications for any reason, which resulted in treatment failure. The study also reported sensitivity analysis using vasculitis only relapse defined as a minimum total BVAS score of 6, at least one pre‐defined major BVAS item or receiving prohibited medications for vasculitis (BREVAS 2018). The study reported on major relapse, defined as number of participants who experienced at least 1 major BVAS item (BREVAS 2018). Only one study assessed health‐related quality of life change, using scores for the physical and mental health aspects of the SF‐36 (WGET 2005). One study reported the mean score for the VDI at the end of the trial (WGET 2005). Two trials published data regarding disease damage.
AEs were reported in all studies (BREVAS 2018; de Menthon 2011; Wechsler 2017; WGET 2005).
Excluded studies
The most common reason for exclusion (n = 43) was the wrong study design. In three trials, the intervention was inappropriate. Two studies were withdrawn prior to enrolment. Reasons for exclusion are provided in Characteristics of excluded studies.
Risk of bias in included studies
Details for each study are presented in Characteristics of included studies. Figure 2 shows the overall risk of bias in each domain for studies included in this review. The risk of bias by trial can be seen in Figure 3.
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
All included studies were published in full articles. None of the studies was at low risk of bias in all domains.
Random sequence generation
Randomisation methods were not reported in one study (de Menthon 2011). In BREVAS 2018 the authors reported the use of an interactive web‐response system, so we assume that the sequence generation was computer‐based. In the two remaining studies, randomisation was stratified, including generating permuted blocks of varying length (WGET 2005), and centralised computer‐generated permuted‐block schedule (Wechsler 2017).
Allocation
Allocation concealment was at unclear risk of bias in one study (de Menthon 2011) and at low risk of bias in the three remaining studies (BREVAS 2018; Wechsler 2017; WGET 2005).
Blinding
Three studies stated that they were double‐blind, with participants blinded (BREVAS 2018; Wechsler 2017; WGET 2005). One study did not report detailed information about blinding we assessed the risk of bias as unclear (de Menthon 2011). Three studies reported blinding of investigators (BREVAS 2018; Wechsler 2017; WGET 2005). However, in one study the authors highlighted that one potential source of bias in blinding personnel was injection site reactions in the treatment group (WGET 2005). Blinding of outcome assessors was reported in three studies and those trials were judged to be at low risk of bias (BREVAS 2018; Wechsler 2017; WGET 2005). In one study, blinding of outcome assessors was not reported and we assessed the risk of bias as unclear (de Menthon 2011).
Incomplete outcome data
We judged all four studies contributing data to be at a low risk of attrition bias (BREVAS 2018; de Menthon 2011; Wechsler 2017; WGET 2005).
Selective reporting
For one study, no protocol was available, so it was not possible to judge if all outcomes were reported. However, the study reported all important vasculitis outcomes and AEs (de Menthon 2011). In another study, all of its protocol‐specified major and minor outcomes were reported, but other benefit endpoints listed in the protocol were not reported (BREVAS 2018). We judged two studies with protocols to be at high risk of bias, as data on treatment outcomes were reported incompletely (Wechsler 2017; WGET 2005).
Other potential sources of bias
Three studies appeared free of other potential sources of bias (BREVAS 2018; de Menthon 2011; Wechsler 2017; WGET 2005).
Effects of interventions
See: Table 1; Table 2; Table 3
Summary of findings 1. Mepolizumab compared to placebo for adults with EGPA ANCA‐associated vasculitis.
| Mepolizumab compared to placebo for adults with EGPA ANCA‐associated vasculitis | ||||||
| Patient or population: adults (age 18 years and older) with EGPA ANCA‐associated vasculitis Setting: clinical centres Intervention: mepolizumab (300 mg; 3 separate injections every 4 weeks) Comparison: placebo | ||||||
| Outcomes | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | Certainty of the evidence (GRADE) | What happens | ||
| With placebo | With mepolizumab | Difference | ||||
| Mortality follow‐up: 52 weeks Number of participants: 136 (1 RCT) | Peto OR 7.39 (0.15 to 372.38) | 0.0% | 0.0% (0 to 0) | 0.0% fewer (0 fewer to 0 fewer) | ⊕⊕⊝⊝ LOW 1 | During 52 weeks follow‐up, one death was reported in the mepolizumab group and no deaths in placebo group. The low‐certainty evidence suggests that mepolizumab results in little to no difference in mortality. |
| Remission for at least 24 weeks assessed with: BVAS v.3 of 0 (on a scale from 0 to 63) follow‐up: 52 weeks Number of participants: 136 (1 RCT) | RR 9.50 (2.30 to 39.21) | 2.9% | 27.9% (6.8 to 100) | 25.0% more (3.8 more to 112.4 more) | ⊕⊕⊝⊝ LOW 1 | The low‐certainty evidence suggests mepolizumab results in a large increase of the probability of accruing at least 24 weeks of remission over a 52‐week period. NNTB 4, 95% CI 3 to 8 |
| Durable remission within the first 24 weeks sustained until week 52 assessed with: BVAS v.3 of 0 (on a scale from 0 to 63) follow‐up: 52 weeks Number of participants: 136 (1 RCT) | RR 13.00 (1.75 to 96.33) | 1.5% | 19.1% (2.6 to 100) | 17.6% more (1.1 more to 140.2 more) | ⊕⊕⊝⊝ LOW 1 | The low‐certainty evidence suggests that mepolizumab results in a large increase of the probability of durable remission within the first 24 weeks, sustained until week 52. NNTB 6 95% CI 4 to 13 |
| Disease relapse follow‐up: 52 weeks Number of participants: 136 (1 RCT) | RR 0.68 (0.53 to 0.86) | 82.4% | 56.0% (43.6 to 70.8) | 26.4% fewer (38.7 fewer to 11.5 fewer) | ⊕⊕⊕⊝ MODERATE 2 | Mepolizumab probably results in a reduction in disease relapse. NNTB 4, 95% CI 3 to 9 |
| Disease flares | Not measured | |||||
| Any adverse event follow‐up: 52 weeks Number of participants: 136 (1 RCT) | RR 1.03 (0.96 to 1.11) | 94.1% | 96.9% (90.4 to 100) | 2.8% more (3.8 fewer to 10.4 more) | ⊕⊕⊝⊝ LOW 1 | The low‐certainty evidence suggests that mepolizumab does not increase any adverse event. Number of participants reporting similar rates of any AEs in mepolizumab and placebo group (97% vs 94%). |
| Any serious adverse event follow‐up: 52 weeks Number of participants: 136 (1 RCT) | RR 0.67 (0.35 to 1.28) | 26.5% | 17.7% (9.3 to 33.9) | 8.7% fewer (17.2 fewer to 7.4 more) | ⊕⊕⊝⊝ LOW 1 | The low‐certainty evidence suggests that mepolizumab results in little to no difference in any serious adverse event. |
| Any withdrawals due to AEs follow‐up: 52 weeks Number of participants: 136 (1 RCT) | RR 2.00 (0.19 to 21.54) | 1.5% | 2.9% (0.3 to 31.7) | 1.5% more (1.2 fewer to 30.2 more) | ⊕⊕⊝⊝ LOW 1 | The low‐certainty evidence suggests that mepolizumab does not increase any withdrawals due to AEs. The study reported similar number of patients withdrawn from mepolizumab and placebo groups due to AEs. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ANCA: anti‐neutrophilic cytoplasmic antibodies; CI: Confidence interval; BVAS: Birmingham Vasculitis Activity Score; EGPA: eosinophilic granulomatosis with polyangiitis; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1Downgraded by two levels for imprecision: CIs are wide and the number of events was too low to reliably assess optimal information size (OIS); or OIS not met
2Downgraded by one level for imprecision: the number of events was lower than indicated in GRADE guidance
AE ‐ adverse event
ANCA – antineutrophil cytoplasmic antibody
BVAS – Birmingham Vasculitis Activity Score
EGPA – eosinophilic granulomatosis with polyangiitis
NNTB ‐ number needed to treat for an additional beneficial outcome
OR ‐ odds ratio
RCT ‐ randomized controlled trial
RR ‐ relative risk
Summary of findings 2. Active drug (etanercept or belimumab) with standard therapy compared to standard therapy with placebo for adults with GPA ANCA‐associated vasculitis.
| Active drug (etanercept or belimumab) with standard therapy compared to standard therapy with placebo for adults with GPA ANCA‐associated vasculitis | ||||||
| Patient or population: adults (age 18 years and older) with GPA ANCA‐associated vasculitis or microscopic polyangiitis (only a subset of patients in one study, i.e. 7.7% of total population and not analysed separately) Setting: clinical centres in etanercept study and not reported in belimumab study Intervention: active drug (etanercept: 25 mg twice a week for a median of 25 months or belimumab: 10 mg/kg on days 0, 14, 28 and every 28 days thereafter until 12 months after the last participant was randomised) with standard therapy Comparison: standard therapy with placebo | ||||||
| Outcomes | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | Certainty of the evidence (GRADE) | What happens | ||
| With standard therapy and placebo | With active drug (etanercept or belimumab) with standard therapy | Difference | ||||
| Mortality
Number of participants: 285
(2 RCTs) WGET 2005 follow‐up: 27 months; BREVAS 2018 follow‐up: approximately up to 4 years |
Peto OR 2.45 (0.55 to 10.97) | 1.4% | 3.4% (0.8 to 15.3) | 2.0% more (0.6 fewer to 13.9 more) | ⊕⊕⊝⊝ LOW 1 | Five deaths was reported in active drug (etanercept or belimumab) with standard therapy group and two deaths in standard therapy with placebo group. Low‐certainty evidence suggests that active drug (etanercept or belimumab) added to standard therapy when compared with standard therapy does not increase/reduce mortality. |
| Remission (number of patients with remission)
assessed with: BVAS/WG = 0 WGET 2005 follow‐up: 27 months Number of participants: 180 (1 RCT) |
RR 0.97 (0.89 to 1.07) | 92.3% | 89.5% (82.2 to 98.8) | 2.8% fewer (10.2 fewer to 6.5 more) | ⊕⊕⊝⊝ LOW 2, 3 | Low‐certainty evidence suggests that active drug (etanercept or belimumab) added to standard therapy when compared with standard therapy with placebo may have little or no effect on remission. However, any effect is likely to be small. |
| Durable remission assessed with: BVAS/WG = 0 for ≥ 6 months follow‐up: 27 months Number of participants: 174 (1 RCT) | RR 0.93 (0.77 to 1.11) | 75.3% | 70.0% (58 to 83.6) | 5.3% fewer (17.3 fewer to 8.3 more) | ⊕⊕⊝⊝ LOW 1 | Low‐certainty evidence suggests that etanercept added to standard therapy when compared with standard therapy and placebo may have little or no effect on durable remission. |
| Major relapse
assessed with: BVAS (experiencing at least 1 major BVAS item) follow‐up: year 2 week 28 Number of participants: 105 (1 RCT) |
RR 2.94 (0.12 to 70.67) | 0.0% | 0.0% (0 to 0) | 0.0% fewer (0 fewer to 0 fewer) | ⊕⊕⊝⊝ LOW 1 | Low‐certainty evidence suggests that belimumab added to standard therapy when compared with standard therapy and placebo does not increase/reduce major relapse. |
| Disease flare assessed with: BVAS/WG ‐ increase of at least one point in scale follow‐up: 27 months Number of participants: 180 (1 RCT) | RR 0.98 (0.76 to 1.27) | 57.1% | 56.0% (43.4 to 72.6) | 1.1% fewer (13.7 fewer to 15.4 more) | ⊕⊕⊕⊝ MODERATE 3 | Etanercept added to standard therapy when compared with standard therapy with placebo probably does not reduce disease flare. |
| Any adverse event follow‐up: approximately up to 4 years Number of participants: 105 (1 RCT) |
RR 1.12 (0.97 to 1.29) |
82.7% | 92.5% (2.5 to 24) |
9.9% more (2,5 fewer to 24 more) |
⊕⊕⊝⊝ LOW1 | The low‐certainty evidence suggests that belimumab added to standard therapy when compared with standard therapy with placebo may result in little or no difference in any AE. |
| Any severe or serious AE (grade 3, 4 or 5)
assessed with: the National Cancer Institute Toxicity Grading Scale WGET 2005 follow‐up: 27 months; BREVAS 2018 follow‐up: approximately up to 4 years Number of participants: 285 (2 RCTs) |
RR 1.00 (0.80 to 1.27) | 47.6% | 47.6% (38 to 60.4) | 0.0% fewer (9.5 fewer to 12.8 more) | ⊕⊕⊝⊝ LOW 2, 3 | The low certainty evidence suggests that active drug (etanercept or belimumab) added to standard therapy when compared with standard therapy with placebo may result in little or no difference in severe or serious AE (grade 3, 4 or 5). |
| Any withdrawals due to AE
assessed with the National Cancer Institute Toxicity Grading Scale WGET 2005 follow‐up: 27 months; BREVAS 2018 follow‐up: approximately up to 4 years Number of participants: 285 (2 RCTs) |
RR 2.66 (1.07 to 6.59) | 4.2% | 11.2% (4.5 to 27.7) | 7.0% more (0.3 more to 23.5 more) | ⊕⊕⊝⊝ LOW 2, 3 | The low‐certainty evidence suggests that active drug (etanercept or belimumab) added to standard therapy when compared with standard therapy with placebo results in a slight increase in any withdrawals due to AE. NNTH 15, 95% CI 8 to 100 |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ANCA: anti‐neutrophilic cytoplasmic antibodies; CI: Confidence interval;BVAS/WG: Birmingham Vasculitis Activity Score for Wegener's Granulomatosis; GPA: granulomatosis with polyangiitis; RR: Risk ratio; OR: Odds ratio | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1Downgraded by two levels for imprecision: optimal information size (OIS) not achieved; low number of events; confidence intervals very wide or indicating both harm and benefit
2Downgraded by one level due to high risk of selective outcome reporting bias in both studies
3Downgraded by one level for imprecision: OIS criteria are met, but the CIs are very wide; or OIS not achieved, and confidence interval indicates both harm and benefit
AE ‐ adverse event
ANCA – antineutrophil cytoplasmic antibody
BREVAS ‐ Belimumab in Remission of VASculitis
BVAS – Birmingham Vasculitis Activity Score
BVAS/WG – Birmingham Vasculitis Activity Score for granulomatosis with polyangiitis
GPA – granulomatosis with polyangiitis
NNTH ‐ number needed to treat for an additional harmful effect
OR ‐ odds ratio
RCT ‐ randomized controlled trial
RR ‐ relative risk
WGET ‐ Wegener's Granulomatosis Etanercept Trial
Summary of findings 3. Infliximab compared to rituximab for adults with refractory GPA ANCA‐associated vasculitis.
| Infliximab compared to rituximab for adults with refractory GPA ANCA‐associated vasculitis | ||||||
| Patient or population: adults (age 18 years and older) with refractory GPA ANCA‐associated vasculitis Setting: hospitals Intervention: infliximab (3 mg/kg on days 1 and 14, before the response assessment on day 42; further treatment depending on the response) Comparison: rituximab (0.375g/m2 on days 1, 8, 15 and 22 before the response assessment at month 2; further treatment depending on the response) | ||||||
| Outcomes | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | Certainty of the evidence (GRADE) | What happens | ||
| With rituximab | With infliximab | Difference | ||||
| Mortality follow‐up: 12 months Number of participants: 17 (1 RCT) | Peto OR 0.88 (0.05 to 15.51) | 12.5% | 11.0% (0.6 to 193.9) | 1.5% fewer (11.9 fewer to 181.4 more) | ⊕⊝⊝⊝ VERY LOW 1 2 | One death was reported in each group. The evidence is very uncertain about the effect of infliximab when compared to rituximab on mortality. |
| Remission assessed with: BVAS=0 follow‐up: 12 months Number of participants: 17 (1 RCT) | RR 0.44 (0.11 to 1.81) | 50.0% | 22.0% (5.5 to 90.5) | 28.0% fewer (44.5 fewer to 40.5 more) | ⊕⊝⊝⊝ VERY LOW 1 2 | The evidence is very uncertain about the effect of infliximab when compared with rituximab on remission at month 12. |
| Durable remission during additional follow‐up beyond 12 months follow‐up: mean 30.6 months Number of participants: 17 (1 RCT) | RR 0.22 (0.03 to 1.60) | 50.0% | 11.0% (1.5 to 80) | 39.0% fewer (48.5 fewer to 30 more) | ⊕⊝⊝⊝ VERY LOW 1 2 | The evidence is very uncertain about the effect of infliximab when compared with rituximab on durable remission during additional follow‐up beyond 12 months. |
| Disease flare/relapse | This outcome was not reported in included trial. | |||||
| Any adverse event | The total number of any AEs was not reported in the study. | |||||
| Any severe AEs assessed with: the World Health Organization classification (2003) follow‐up: 12 months Number of participants: 17 (1 RCT) | RR 1.78 (0.20 to 16.10) | 12.5% | 22.3% (2.5 to 100) | 9.8% more (10 fewer to 188.8 more) | ⊕⊝⊝⊝ VERY LOW 1 2 | The evidence is very uncertain about the effect of infliximab when compared with rituximab on any severe AEs. |
| Any withdrawals due to AEs assessed with: the World Health Organization classification (2003) follow‐up: 12 months Number of participants: 17 (1 RCT) | RR 2.70 (0.13 to 58.24) | 0.0% | 0.0% (0 to 0) | 0.0% fewer (0 fewer to 0 fewer) | ⊕⊝⊝⊝ VERY LOW 1 2 | The evidence is very uncertain about the effect of infliximab when compared with rituximab on any withdrawals due to AEs. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; GPA: granulomatosis with polyangiitis; RR: Risk ratio; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1Downgraded by one level due to risk of bias – unclear randomisation and concealment, no blinding and unclear selective outcome reporting
2Downgraded by two levels for imprecision: optimal information size (OIS) not met and CI indicates both harm and benefit; and/or low number of events
AE ‐ adverse event
ANCA – antineutrophil cytoplasmic antibody
BVAS – Birmingham Vasculitis Activity Score
GPA – granulomatosis with polyangiitis
OR ‐ odds ratio
RCT ‐ randomized controlled trial
RR ‐ relative risk
The studies included in the review enrolled participants with GPA (three studies: BREVAS 2018; de Menthon 2011; WGET 2005) or EGPA (one study: Wechsler 2017) or MPA (one study: BREVAS 2018). Studies compared a single drug with placebo or with another active drug. The population in BREVAS 2018 was mixed and included mostly participants with GPA (79%), but also with MPA (21%). Therefore, we summarised the results from all studies for the GPA and EGPA populations separately, as well as for comparisons of active drug with placebo and active drug with another active drug.
Comparison 1: Mepolizumab compared to placebo in adults with relapsing or refractory eosinophilic granulomatosis with polyangiitis
Major outcomes
Mortality
In Wechsler 2017, one death was reported in mepolizumab group and no deaths in placebo group (Peto OR 7.39, 95% CI 0.15 to 372.38; Analysis 1.1).
1.1. Analysis.

Comparison 1: EGPA ‐ mepolizumab vs placebo, Outcome 1: Mortality
Remission
Wechsler 2017 reported this outcome as accrued weeks of remission over a 52‐week period. Overall, more participants had any remission (for at least one week) in the mepolizumab group compared with the placebo group (RR 2.77, 95% CI 1.62 to 4.74; Relative benefit increase 177%, 95% CI 62 to 374%; RD 33.8%, 95% CI 11.9% to 71.5%; NNTB 3, 95% Cl 2 to 6; Analysis 1.2) and more participants had at least 24 weeks of accrued remission (28% versus 3%; RR 9.5, 95% CI 2.3 to 39.2; Relative benefit increase 850%, 95% CI 130 to 3820%; RD 25%, 95% Cl 3.8% to 112%; NNTB 4, 95% Cl 3 to 8; Analysis 1.2).
1.2. Analysis.

Comparison 1: EGPA ‐ mepolizumab vs placebo, Outcome 2: Remission
In the analysis of accrued weeks of remission over follow‐up, more participants in the mepolizumab group (28%) had ≥ 24 weeks of accrued remission over 52 weeks of follow‐up than in the placebo group (3%) (OR 5.91, 95% CI 2.68 to 13.0).
Durable remission
In Wechsler 2017, durable remission was defined as remission within the first 24 weeks that was sustained until week 52. More participants in the mepolizumab group, compared with control group, experienced durable remission (13 versus 1 participants; RR 13.0, 95% CI 1.75 to 96.33; Relative benefit increase 1200%, 95% CI 75 to 9563%; RD 17.6%, 95% CI 1.1 to 140.2%; NNTB 6, 95% Cl 4 to 13); Analysis 1.3).
1.3. Analysis.

Comparison 1: EGPA ‐ mepolizumab vs placebo, Outcome 3: Durable remission
Disease relapse
Wechsler 2017 reported time to first relapse over 52 weeks of the study, and the number of participants with first EGPA relapse. Fewer participants relapsed in the mepolizumab group than in the placebo group (56% versus 82%; RR 0.68, 95% CI 0.53 to 0.86; RRR 32%, 95% CI 14 to 47%; RD ‐26.4%, 95% CI from ‐38.7 to ‐11.5; NNTB 4, 95% CI 3 to 9; Analysis 1.4). The time to relapse was longer in active treatment, compared with the placebo group (hazard ratio (HR) 0.32, 95% CI 0.21 to 0.5). The study reported details regarding relapses in the mepolizumab and placebo groups in the following categories: any vasculitis relapses (43% versus 65% of participants); any asthma (37% versus 60%); any sinonasal relapses (35% versus 51%); vasculitis only (18% versus 22%); asthma only (19% versus 32%); sinonasal only (6% versus 12%); and any combinations of these.
1.4. Analysis.

Comparison 1: EGPA ‐ mepolizumab vs placebo, Outcome 4: Disease relapse
Total AEs
Wechsler 2017 reported similar rates of participants with any AEs in the mepolizumab and placebo groups (97% versus 94%; RR 1.03, 95% CI 0.96 to 1.11; RD 3%, 95% CI from ‐4 to 1; Analysis 1.5), and also reported any event considered by the investigator to be related to the trial agent (51% versus 35%; RR 1.46, 95% CI 0.98 to 2.17; RD 16%, 95% CI from ‐0.3 to 33; Analysis 1.5). The most common AEs reported in the study were headache (32% versus 18%), nasopharyngitis (18% versus 24%), arthralgia (22% versus 18%), sinusitis (21% versus 16%), and upper respiratory tract infection (21% versus 16%). The number of participants who experienced local injection‐site reactions and systemic reaction was similar in the two groups (15% versus 13% and 6% versus 1% respectively). For serious AEs, see Analysis 1.6.
1.5. Analysis.

Comparison 1: EGPA ‐ mepolizumab vs placebo, Outcome 5: Any AE
1.6. Analysis.

Comparison 1: EGPA ‐ mepolizumab vs placebo, Outcome 6: Any serious AE
Serious and severe AEs
Wechsler 2017 defined all serious AEs as "any untoward medical occurrence that resulted in death, was life‐threatening, resulted in hospitalisation or prolongation of existing hospitalisation, resulted in disability or incapacity, was a congenital anomaly or birth defect, or was indicative of possible drug‐induced liver injury with hyperbilirubinaemia" and specific serious events defined as serious AE which was considered by the investigator as related to the trial agent. The most common serious AE was exacerbation or worsening of asthma (3% of the participants in the mepolizumab group versus 6% of participants in the placebo group).The percentages of participants with any serious AEs or any serious AEs considered to be related to the trial agents were similar in the mepolizumab and placebo groups (18% versus 26%, RR 0.67, 95% CI 0.35 to 1.28; RD ‐8.7, 95% CI from ‐17.2 to 7.4; and 4% versus 4%; RR 1.0, 95% CI 0.21 to 4.78; RD 0, 95% CI from ‐3.5% to 16.7%; Analysis 1.6). One participant in the mepolizumab group died from cardiac arrest during the trial, this participant had a history of coronary artery disease.
Withdrawals due to AEs
Wechsler 2017 reported similar number of participants withdrawn from the mepolizumab and placebo groups due to AEs (3% versus 1%; RR 2.0, 95% CI 0.19 to 21.54; RD 1.5% 95% Cl ‐1.2% to 30.2%; Analysis 1.7).
1.7. Analysis.

Comparison 1: EGPA ‐ mepolizumab vs placebo, Outcome 7: Any withdrawals due to AE
Minor outcomes
Treatment response
Wechsler 2017 did not analyse treatment response as an outcome.
Health‐related quality of life
Wechsler 2017 provided no results of quality of life assessments.
Control of asthma/sinonasal disease
Wechsler 2017 reported changes from baseline in ACQ‐6 and SNOT‐22 scores, but only on graphs. We contacted the study authors to obtain numeric data, without success. To estimate the effect, we used software to read the values from the graph. Values were presented as mean change from baseline in both groups (Analysis 1.8; Analysis 1.9). For ACQ‐6, the change from baseline was similar in both groups; the estimated difference was ‐0.3 (95% CI ‐0.6 to 0.00) (Analysis 1.8). The minimal clinically important difference, as reported by the authors, is 0.5.
1.8. Analysis.

Comparison 1: EGPA ‐ mepolizumab vs placebo, Outcome 8: Control of asthma symptoms with ACQ6 ‐ change from baseline
1.9. Analysis.

Comparison 1: EGPA ‐ mepolizumab vs placebo, Outcome 9: Control of sinonasal symptoms with SNOT22 ‐ change from baseline
For SNOT‐22, the change from baseline was similar in both groups; the estimated difference was ‐4.66 (95% CI ‐10.69 to 1.36) (Analysis 1.9). The minimal clinically important difference, as reported by the authors, is 8.9.
Disease damage
Wechsler 2017 reported data on change from baseline in the VDI in the form of a graph. We contacted the study authors to obtain numeric data, without success. To estimate the effect, we used software to read the values from the graph. Values were presented as mean change from baseline in both groups (Analysis 1.10). The change from baseline was similar in both groups; the estimated difference was ‐0.07 (95% CI ‐0.37 to 0.23) (Analysis 1.8). The minimal clinically important difference has not been determined.
1.10. Analysis.

Comparison 1: EGPA ‐ mepolizumab vs placebo, Outcome 10: Disease damage in VDI ‐ change from baseline
Comparison 2: Etanercept or belimumab compared with placebo in adults with granulomatosis with polyangiitis (GPA) and microscopic polyangiitis
Major outcomes
Mortality
In WGET 2005, four deaths were observed in the group receiving etanercept and two deaths in the group receiving placebo. In BREVAS 2018, one death was observed in the belimumab group and no deaths were observed in the placebo group. When we pooled the results of two studies comparing active drug with placebo, we found similar odds of death in both groups (Peto OR 2.45, 95% CI 0.55 to 10.97; Analysis 2.1).
2.1. Analysis.

Comparison 2: GPA ‐ anticytokine therapy vs control, Outcome 1: Mortality
Remission
WGET 2005 reported remission, defined as a BVAS/WG of 0 at any time during the trial (as a minor outcome), in 80 participants in the etanercept group and 84 participants in the placebo group (RR 0.07, 95% CI 0.89 to 1.07; Analysis 2.2). WGET 2005 stratified participants at randomisation according to the disease severity at baseline (severe or limited) and found similar effect in both groups (HR 0.91, 95% CI 0.63 to 1.32 and HR 0.8, 95% CI 0.44 to 1.43, P for interaction = 0.77).
2.2. Analysis.

Comparison 2: GPA ‐ anticytokine therapy vs control, Outcome 2: Remission
Durable remission
In WGET 2005, sustained remission was defined as BVAS/WG = 0 for ≥ 6 months. The number of participants with sustained remission in the etanercept and placebo groups was similar (62 versus 64 participants; RR 0.93, 95% CI 0.77 to 1.11; RD ‐5.3%, 95% Cl ‐17.3% to 8.3%; Analysis 2.3). The study stratified participants at randomisation according to the disease severity at baseline (severe or limited) and found similar effects in both groups (HR 0.91, 95% CI 0.6 to 1.37; and HR 0.82, 95% CI 0.42 to 1.6; P for interaction = 0.85).
2.3. Analysis.

Comparison 2: GPA ‐ anticytokine therapy vs control, Outcome 3: Durable remission
At baseline, all participants in BREVAS 2018 were in remission. In our analysis, we used the total number of participants included in an intention‐to‐treat (ITT) analysis. BREVAS 2018 reported that, at the year one, week 48 time point, 38 (71.7% of all included in ITT) participants were in remission in the belimumab group and 43 (82.69% of all included in ITT) were in remission in the placebo group (RR 0.87, 95% CI 0.7 to 1.07; calculated using RevMan calculator). At the year two, week 28 time point, 21 (39.62% of all included in ITT) participants were in remission in the belimumab group and 26 (50% of all included in ITT) were in remission in the placebo group (RR 0.79, 95% CI 0.52 to 1.22; calculated using RevMan calculator) .
Disease flare/relapse
WGET 2005 reported that similar percentage of participants in etanercept and placebo groups had no disease flares during the study (42.7% versus 42.9%; RR 0.98, 95% CI 0.76 to 1.27; RD ‐1.1%, 95% Cl ‐13.7% to 15.4%; Analysis 2.5) and the number of any disease flares per 100 person‐years was similar between the groups (66.3 versus 74.1 per 100 person‐years, HR 0.89, 95% CI 0.62 to 1.28) as was the number of severe flares (14.9 versus 12.8 per 100 person‐years, HR 1.05, 95% CI 0.61 to 1.8). Similar percentages of participants who achieved sustained remission relapsed in both groups (30.6% of 62 participants in etanercept versus 32.8% of 64 participants in placebo group).
2.5. Analysis.

Comparison 2: GPA ‐ anticytokine therapy vs control, Outcome 5: No disease flare
BREVAS 2018 reported a similar percentage of participants in the belimumab and placebo groups who experienced vasculitis relapse as the first relapse (11.3% versus 15.4%; RR 0.74, 95% CI 0.27 to 1.97; RD ‐4%, 95% CI from ‐17 to 9; Analysis 2.4) and among participants with relapse the time to first vasculitis relapse was similar in the belimumab and placebo group (median 251 (range 25‐371) days, 105.5 (range 15‐789) days; HR 0.88, 95% CI 0.29 to 2.65). In BREVAS 2018, the first protocol‐specified event that included relapse was defined as a BVAS score of ≥ 6, presence of at least one predefined major BVAS item, or the receipt of prohibited medications for any reason. One participant in the belimumab group had a major relapse during the double‐blind phase of the study. In the placebo group, none of participants experienced major relapse (RR 2.94, 95% CI 0.12 to 70.67; RD 2%, 95% CI from ‐3 to 7; Analysis 2.4).
2.4. Analysis.

Comparison 2: GPA ‐ anticytokine therapy vs control, Outcome 4: Disease relapse
Total AEs
WGET 2005 reported AEs defined as any medical condition in participants who received etanercept and were graded according to the National Cancer Institute Toxicity Grading Scale. The study reported three types of AEs: severe (grade 3), life‐threatening (grade 4) and deaths as fatal AEs (grade 5). For serious or severe AEs, see Analysis 2.8).
2.8. Analysis.

Comparison 2: GPA ‐ anticytokine therapy vs control, Outcome 8: Any severe or serious AE
WGET 2005 reported specific types of AEs such as cytopenias, infections, congestive heart failure and venous thrombotic events. The frequency of cytopenias, infections, venous trombotic events and congestive heart failure was similar in etanercept and placebo group. Similar numbers of participants experienced grade 2 to grade 5 infections (49.4% in each group) and venous thrombotic events (10 events in each group; P = 0.92).
BREVAS 2018 defined AEs as an untoward medical occurrence in a participant, temporally associated with the use of a drug, whether or not it was considered to be related to the medicinal product. In this study, 92.5% participants in belimumab group experienced at least one adverse event, compared to 82.7% participants in the placebo group, at any time during the study (see Analysis 2.6). Most AEs were infections and infestations (56.6% of participants in the belimumab group and 57.7% of participants in the placebo group). The study reported similar percentages of participants in both the belimumab and placebo groups with AEs other than serious (60.38% versus 63.46%; RR 0.95, 95% CI 0.7 to 1.28; RD ‐3.2%, 95% CI from ‐19 to 17.8; Analysis 2.7).
2.6. Analysis.

Comparison 2: GPA ‐ anticytokine therapy vs control, Outcome 6: Any AE
2.7. Analysis.

Comparison 2: GPA ‐ anticytokine therapy vs control, Outcome 7: Any non serious AE
Serious and severe AEs
WGET 2005 provided the definition of severe AEs as adverse event grade 3, life‐threatening events as adverse event grade 4 and fatal AEs as grade 5. The study reported similar percentages of participants with such events in the etanercept and placebo groups (56.2% versus 57.1%), which included deaths (four versus two cases). WGET 2005 did not report serious AEs.
BREVAS 2018 defined serious AEs as AEs that resulted in death, were life‐threatening, required hospitalisation or prolongation of hospitalizations, or resulted in disability or incapacity. There was one death in the belimumab group and no deaths in the placebo group (see Analysis 2.1). The study reported similar percentage of participants with serious AEs in the belimumab and placebo groups (33.96% versus 30.77%). The study reported that there were four cases of any malignancies (including non‐melanoma skin cancer) in the belimumab group (anal cancer, basal cell carcinoma, plasma cell myeloma and one malignancy not specified) with no such events in control group. There was also one case of ischaemic stroke (resulting in death). One participant in the control group was diagnosed with anaemia, one with pancytopenia, and one with sinus bradycardia. One participant in the belimumab group experienced acute kidney injury. Severe AEs were reported in 11 (20.8%) participants in the belimumab group and seven (13.5%) participants in the placebo group. When we pooled the results of both studies (BREVAS 2018; WGET 2005) we found non significant difference between the groups in frequency of serious or severe AEs (RR 1.0, 95% CI 0.8 to 1.27; RD 0, 95% CI from ‐9.5% to 12.8%; Analysis 2.8).
WGET 2005 reported six solid cancers (two cases of mucinous adenocarcinoma of the colon, one metastatic cholangiocarcinoma, one renal‐cell carcinoma, one breast carcinoma, and one liposarcoma), all in the etanercept group (RR 13.29, 95% CI 0.76 to 232.45; RD 0, 95% CI 0 to 0; Analysis 2.9). The rate of cancers in the etanercept and placebo groups were similar (three cases versus four cases of cutaneous basal‐cell or squamous‐cell carcinomas). It is also important to mention AEs during long‐term post‐trial follow‐up. These data were available for 153 participants (85% of the original cohort), with a median follow‐up time of 43 months. During the follow‐up, the rates of cancer were similar in the etanercept and placebo groups (10% versus 7%; RR 1.58, 95% 0.54 to 4.61; RD 3.8%, 95% CI from ‐3% to 23.4%; Analysis 2.9). However, the combined risk of solid malignancy from time of trial enrolment remained higher for the etanercept group (relative risk increase (RRI) 186%, 95% CI 8 to 662; RD 10%, 95% CI 1% to 19%) (WGET 2005).
2.9. Analysis.

Comparison 2: GPA ‐ anticytokine therapy vs control, Outcome 9: Any solid malignancy
Withdrawals due to AEs
WGET 2005 reported nine withdrawals due to AEs in etanercept group and three withdrawals in placebo group. In the BREVAS 2018 publications we identified some discrepancies in the numbers of participants withdrawn from the study. In a figure depicting participant flow, the authors reported seven withdrawals due to AEs in the belimumab group and three withdrawals in placebo group. However, in the results section describing AEs, the authors reported that eight participants in the belimumab group and six participants in the placebo group had AEs leading to study withdrawal. We contacted the authors to clarify this and we received information that the discrepancies were due to different categorisation of AEs in those two study sites, specifically vasculitis flares (classified as lack of efficacy on in the patient flow figure and as AE in the AEs section). The study authors suggested that we use of the number of withdrawals due to AEs presented on the figure depicting patient flow. When we pooled the results from both studies, we observed an increased risk of withdrawal due to AEs in the active drug group as compared with the placebo group (RR 2.66, 95% CI 1.07 to 6.59; RRI 166%, 95% CI 7 to 559%; RD 7%, 95% CI 0.3% to 23.5%; NNTH 15, 95% CI 8 to 100; Analysis 2.10). We used the number of participants withdrawn reported in AEs section of BREVAS study in sensitivity analysis, where the difference became non‐significant (Analysis 4.1).
2.10. Analysis.

Comparison 2: GPA ‐ anticytokine therapy vs control, Outcome 10: Any withdrawals due to AE
4.1. Analysis.

Comparison 4: GPA ‐ sensitivity BREVAS withdrawn due to adverse events, Outcome 1: Any withdrawals due to AE
Minor outcomes
Treatment response
Treatment response was reported in WGET 2005 study as sustained low level of disease activity (BVAS/WG < 3 for at least 6 months) and no significant difference was found between the etanercept and control groups (RR 0.96, 95% CI 0.86 to 1.06; RD ‐4% 95%Cl ‐13% to 5%; Analysis 2.11). The study stratified participants at randomisation according to the disease severity at baseline (severe or limited) and found a similar effect in both groups (HR 0.7, 95%CI 0.48 to 1.03 and 1.24, 95% CI 0.69 to 2.24, P for interaction = 0.11).
2.11. Analysis.

Comparison 2: GPA ‐ anticytokine therapy vs control, Outcome 11: Treatment response
Health‐related quality of life
In WGET 2005, the assessment of health‐related quality of life was based on the SF‐36 questionnaire. The results were only reported as improvements in physical and mental health domains in the etanercept and placebo groups (7.7 and 5.7 versus 8.4 and 8.0).
Control of asthma/sinonasal disease
WGET 2005 and BREVAS 2018 did not report on on this outcome.
Disease damage
WGET 2005 reported a mean score of VDI at baseline and at the end of the trial. The increase in the score was similar in the etanercept and control groups (from 1.6 to 2.0 versus from 1.0 to 1.7, P = 0.5). In all participants the most frequently reported items were hearing loss (25.6%) and proteinuria (> 0.5 g of protein in 24 hours; 18.9%).
BREVAS 2018 reported minimal changes in VDI score in the belimumab group (year one, week 48: 0.1 ± 0.38; year two, week 24: 0.2 ± 0.50) and no change in the placebo group (year one, week 48: 0.0 ± 0.15; year two, week 24: 0.0 ± 0.00).
Comparison 3: Infliximab versus rituximab added to steroids and cytotoxic agents in adults with refractory GPA
Major outcomes
Mortality
In a single study comparing two active drugs, one participant died in each group (de Menthon 2011) during 12 months of the study (Peto OR 0.88, 95% CI 0.05 to 15.51; Analysis 2.1). In additional follow‐up (mean 30.6 months SD 15.4), two more deaths were reported, one in the infliximab group and one in the rituximab group.
Remission
A single study by de Menthon 2011 reported complete remission (CR), defined as BVAS = 0. During 12 months of the study, two participants in the infliximab group had complete remission, while in the rituximab group four participants had complete remission. No beneficial effect in complete remission was found in the case of infliximab in comparison with rituximab (RR 0.44, 95% CI 0.11 to 1.81; RD ‐28%, 95% Cl ‐44.5% to 40.5%; Analysis 2.2). During follow‐up, in the infliximab group, one participant remained in remission. Two participants relapsed, were switched to other treatments and achieved remission. In the rituximab group, three participants remained in remission in the long term. One participant relapsed, achieved remission again and remained in remission.
Durable remission
de Menthon 2011 reported long‐term complete remissions, defined as persistent remission during additional follow‐up beyond 12 months. The study reported one long‐term remission in the infliximab group during 28.9 +/‐ 15.4 months and four long‐term complete remissions during 32.9 +/‐ 16.7 months of follow‐up in rituximab group (RR 0.22, 95% CI 0.03 to 1.60; RD ‐39%, 95% Cl ‐48.5% to 30%; Analysis 2.3).
Disease flare/relapse
de Menthon 2011 reported that during long‐term follow‐up, two out of three participants in remission relapsed, while in rituximab one out of five participants in remission relapsed but achieved complete response again.
Total AEs
de Menthon 2011 reported AEs graded according to the World Health Organization classification (2003), defined AEs as severe, moderate and mild events. Two participants in the infliximab group experienced incidental or mild allergic reactions during infusion. These were one facial erythema and one transient bronchospasm that was described as severe and resulted in withdrawal from the trial. Two participants (one in the infliximab group, one in the rituximab group) experienced severe AEs and died. During the additional long‐term follow‐up in the infliximab group, with mean time of follow‐up 30.6 ± 15.4 months and with all participants available, one participant had infliximab‐related skin rash; one participant experienced hepatitis subsequent to cytomegalovirus infection nine months after the end of the treatment protocol. In the rituximab group, one participant was diagnosed with prostate carcinoma 27 months after the last rituximab infusion. One participant was diagnosed with pancreatic carcinoma two months after the last rituximab infusion, and died three months later.
Serious and severe AEs
de Menthon 2011 reported severe AEs and deaths. There was one severe allergic reaction (transient bronchospasm) in one participant in the infliximab group, and there were two deaths during the study (one in each group) (RR 1.78, 95% CI 0.2 to 16.1; RD 9.8%, 95% Cl ‐10% to 188.8%; Analysis 2.8). Two additional deaths occurred during follow‐up (one in each group). One participant in infliximab group died of invasive aspergillosis at month 2, 60 days after first infusion (severe AE). One participant in the rituximab group experienced sudden death on day 23 of the trial, with no autopsy and no obvious explanation.
Withdrawals due to AEs
de Menthon 2011 reported one withdrawal due to AEs in the infliximab group and did not report withdrawals in the rituximab group (RR 2.7, 95% CI 0.13 to 58.24; RD 0 95% Cl 0% to 0%; Analysis 2.10).
Minor outcomes
Treatment response
de Menthon 2011 reported partial remission (PR), defined as partial regression of the disease and < 50% BVAS (as a part of major outcome). During 12 months of the study, one participant in the infliximab group and one participant in the rituximab group had PR (RR 0.89, 95 CI 0.07 to 12.0; RD ‐1.4% 95%Cl ‐11.6% to 137.5%; Analysis 2.11).
Health‐related quality of life
de Menthon 2011 did not report on quality of life.
Control of asthma/sinonasal disease
de Menthon 2011 did not report on this outcome.
Disease damage
de Menthon 2011 did not report on this outcome.
Sensitivity analysis
We carried out sensitivity analyses for WGET 2005 in Comparison 2, including all randomised and control group participants, assumed to have the best possible outcome. Sensitivity analyses did not change the results of the trial reported for the main analyses (Analysis 3.1; Analysis 3.2; Analysis 3.3; Analysis 3.4; Analysis 3.5; Analysis 3.6; Analysis 3.7).
3.1. Analysis.

Comparison 3: GPA ‐ sensitivity analysis regarding WGET trial, Outcome 1: Mortality
3.2. Analysis.

Comparison 3: GPA ‐ sensitivity analysis regarding WGET trial, Outcome 2: Remission
3.3. Analysis.

Comparison 3: GPA ‐ sensitivity analysis regarding WGET trial, Outcome 3: Durable remission
3.4. Analysis.

Comparison 3: GPA ‐ sensitivity analysis regarding WGET trial, Outcome 4: Disease flare
3.5. Analysis.

Comparison 3: GPA ‐ sensitivity analysis regarding WGET trial, Outcome 5: Any severe or serious AE
3.6. Analysis.

Comparison 3: GPA ‐ sensitivity analysis regarding WGET trial, Outcome 6: Any withdrawals due to AE
3.7. Analysis.

Comparison 3: GPA ‐ sensitivity analysis regarding WGET trial, Outcome 7: Treatment response
Our primary plan outlined in the protocol was to perform analyses across all types of ANCA‐associated vasculitis, but based on clinical heterogeneity as outlined above, we decided to pool the data separately for each type of AAV. We performed sensitivity analysis by pooling the different AAV types together for the outcomes for which we had data from more than one study (Analysis 5.1; Analysis 5.2; Analysis 5.3; Analysis 5.4; Analysis 5.5; Analysis 5.6). The results were similar across AAV‐type and treatment comparisons subgroups for mortality, disease relapse, any serious or severe AEs and withdrawals due to AEs (Analysis 5.1; Analysis 5.4; Analysis 5.5; Analysis 5.6, but the effects differed across AAV‐type and treatment comparisons for any remission and durable remission (Analysis 5.2; Analysis 5.3). This analysis is limited by the low number of studies (single studies in most subgroups).
5.1. Analysis.

Comparison 5: Sensitivity analysis ‐ all AAV together, Outcome 1: Mortality
5.2. Analysis.

Comparison 5: Sensitivity analysis ‐ all AAV together, Outcome 2: Any remission
5.3. Analysis.

Comparison 5: Sensitivity analysis ‐ all AAV together, Outcome 3: Durable remission
5.4. Analysis.

Comparison 5: Sensitivity analysis ‐ all AAV together, Outcome 4: Disease relapse
5.5. Analysis.

Comparison 5: Sensitivity analysis ‐ all AAV together, Outcome 5: Any serious or severe AE
5.6. Analysis.

Comparison 5: Sensitivity analysis ‐ all AAV together, Outcome 6: Any withdrawals due to AE
Discussion
Summary of main results
The aim of this review was to provide an overview of the benefits and harms of anti‐cytokine targeted therapies in patients with ANCA‐associated vasculitis. The review identified four RCTs investigating the effects of anti‐cytokine interventions on mortality, remission, durable remission, disease flare, any AEs, any serious AEs, and any withdrawals due to AEs. Due to differences in studied populations and treatments, the results were not pooled together, but were analysed in three comparisons: 1) mepolizumab versus placebo in relapsing or refractory EGPA; 2) etanercept/belimumab added to standard therapy versus standard therapy with placebo in GPA (with 8% participants with MPA); and 3) infliximab versus rituximab in refractory GPA.
A single trial of mepolizumab compared with placebo in relapsing or refractory EGPA showed that the use of experimental treatment probably decreases the risk of disease relapse (moderate‐certainty evidence) and may increase the probability of accruing at least 24 weeks of remission and durable remission (low‐certainty evidence). However, due to low‐certainty evidence and a low number of events there is uncertainty regarding the effect of mepolizumab on mortality. The evidence on any AEs, any severe adverse effects and any withdrawals due to adverse effects was limited, as the study included a low number of participants.
We found low‐certainty evidence that in patients with GPA (with 8% participants with MPA), etanercept or belimumab added to standard therapy as compared with standard therapy alone may increase the probability of withdrawal due to AE; etanercept or belimumab may have little or no impact on serious AEs; etanercept may have little or no impact on durable remission, and belimumab may have little or no impact on major relapse. Moderate‐certainty evidence suggests little or no improvement in disease flare with the use of etanercept. Low‐certainty evidence suggests that active drug (etanercept or belimumab) added to standard therapy, when compared with standard therapy, does not increase or reduce mortality.
Due to very low‐certainty evidence for all outcomes, the effects of infliximab as compared to rituximab in the management of patients with refractory GPA is uncertain.
Overall, based on obtained data, there is low‐ to moderate‐certainty evidence of the clinically relevant effect of one specific anti‐cytokine, mepolizumab, in one form of ANCA‐associated vasculitis (relapsing or refractory EGPA), with uncertain side effect risk; but none for any other anti‐cytokines in any vasculitis.
Overall completeness and applicability of evidence
It should be highlighted that only four studies were included in the review, of which three had major limitations and one was conducted in a small number of participants. This is a significant limitation of the available data. Three additional studies were planned, but were withdrawn prior to enrolment or closed without being completed, and do not add evidence to our systematic review. We have also identified four ongoing studies that may add evidence in future updates of the review.
The majority of included studies (three of the four included) were conducted mainly in participants with GPA. However, one of these studies included a small group of participants with refractory disease, so it had to be analysed separately and its results may not be applicable to patients with non‐refractory disease. The remaining two studies included mainly participants with GPA, but also a small subgroup of participants with MPA, who comprised 8% of the overall pooled population from those two studies. Therefore, the results of these three studies, mainly in participants with GPA, may not be applicable to patients with MPA or other form of ANCA‐associated vasculitis, such as EGPA.
One study was conducted in participants with EGPA. The authors reported that compared to placebo in relapsing or refractory EGPA participants, mepolizumab resulted in higher rates of remission (including durable remission) and fewer disease relapse events. The results of this study may not be applicable to patients with other forms of ANCA‐associated vasculitis, such as GPA and MPA.
The studies included in the review assessed all or most of the major outcomes defined by this review, but they included small numbers of patients and for several outcomes the confidence intervals were wide and imprecise. Wechsler 2017 showed the beneficial effect of mepolizumab in relapsing or refractory EGPA, which maybe considered clinically relevant. However, the risk of side effects is uncertain, as the number of patients in the study was not large enough to assess safety.
None of the studies included in this systematic review address potential benefit in lowering the mortality in ANCA‐associated vasculitis, as the number of death events was too low to draw any conclusions.
To summarise, the studies identified by this review addressed questions for single drugs in single groups of patients. There are still many gaps in evidence, suggesting the need for further studies. The effects of interventions in patients with other forms of ANCA‐associated vasculitis are not sufficiently covered by current evidence. Further investigation of safety of these interventions is also needed.
Quality of the evidence
All the included studies were described as double‐blind, randomised placebo‐controlled trials. Two studies were judged to be at low risk of bias in all domains except selective reporting, which was judged to be at high risk of bias (Wechsler 2017; WGET 2005). One study was judged to be at unclear risk of bias for random sequence generation, high risk of bias for blinding, unclear risk of bias for allocation and low risk of bias for incomplete outcome data (de Menthon 2011). Additionally for this study, we had concern about risk of bias in selective outcome reporting, as there was no protocol available (de Menthon 2011). One study did not provide information about all outcomes specified in the protocol (BREVAS 2018). Three studies appeared free of other potential sources of bias. The studies enrolled different types of patients. Patients with GPA were included in three studies (BREVAS 2018; de Menthon 2011; WGET 2005). However, BREVAS 2018 included a mixed population, with the majority of participants with GPA and one fifth of the participants with MPA, analysed together. Another study enrolled participants with relapsing or refractory EGPA (Wechsler 2017). For this reason, we analysed these separately. However, all of the analyses provided imprecise results with wide CIs. Publication bias was not assessed due to the low number of studies. For the comparison of mepolizumab and placebo in patients with EGPA, we judged the certainty of the evidence for the following outcomes to be low: mortality, remission accrued for at least 24 weeks, durable remission, any AE, any serious AEs and any withdrawals due to AEs. We judged the cetertainty of evidence to be moderate for disease relapse. The certainty of evidence was downgraded due to imprecision. For the comparison of etanercept/belimumab and placebo in patients with GPA/MPA, we judged the certainty of evidence to be low for mortality, remission, durable remission, major relapse, any severe or serious AEs, and any withdrawals due to AEs. We judged the certainty of evidence to be moderate for disease flare. The certainty of evidence was downgraded due to risk of bias (high risk of selective outcome reporting in both studies) and/or for imprecision. For the comparison of infliximab with rituximab, the certainty of evidence was very low for all outcomes. It was downgraded for risk of bias in the study and imprecision.
Potential biases in the review process
We conducted an extensive search to identify relevant studies. In addition to a comprehensive database search we sought information from other sources, such as registries of clinical trials, the websites of regulatory agencies, pharmacovigilance agencies and drug safety updates. We checked for additional data in all primary studies, review articles and relevant manufacturer websites. We searched PubMed for errata or retractions from included studies published in full‐text. Study selection on the basis of titles and abstracts and on the basis of full texts, data extraction and risk of bias assessments were done by two review authors independently and additionally checked by a senior review author to reduce chance of errors, while our GRADE assessment was done by a junior review author and checked by two senior review authors.
Agreements and disagreements with other studies or reviews
This Cochrane Review stays in agreement with other literature reviews in this topic (Silva‐Fernandez 2014, Lutalo 2015; Souza 2017), pointing out that there is insufficient evidence regarding use most of biological treatments in ANCA‐associated vasculitis. However, Silva‐Fernandez 2014 had broader scope and addressed all types of vasculitis and all types of biological therapies. Silva‐Fernandez 2014 was not focused only on evidence from RCTs, but also included previously published systematic reviews, RCTs, cohort studies and case series. That review had its last search in April 2014 and included two RCTs that we also identified. Silva‐Fernandez 2014 concluded that etanercept is not effective in maintaining remission in patients with GPA and that large RCTs on biological therapies in systemic vasculitis are needed. Lutalo 2015 was not described as a systematic review. It aimed to summarise both the clinical trials and clinical practice regarding use of biological therapies in the treatment of ANCA‐associated vasculitis. They searched literature up to November 2014, described three of the RCTs that were included in our review (BREVAS 2018 was still ongoing at the time). Lutalo 2015 concluded that drugs belonging to anti‐TNF‐α agents are not recommended in inducing remission in patients with ANCA‐associated vasculitis and that the data for other biological agents (including those assessed in our review) is limited. The guidelines of the Brazilian Society of Rheumatology, based on a systematic review with literature searched up to October 2016, recommended not to use etanercept in the induction of remission in patients with ANCA‐associated vasculitis (Souza 2017). Our review adds to the evidence from previous reviews the results of two studies on anti‐cytokine agents: the results of BREVAS 2018 in patients with GPA and the results of Wechsler 2017 on mepolizumab in relapsing or refractory EGPA.
Authors' conclusions
Implications for practice.
We found only four studies, of which three had major limitations and one was conducted in a small number of patients. There is thus limited evidence for drawing conclusions about the benefit and risk of anti‐cytokine therapy in ANCA‐associated vasculitis. In patients with relapsing or refractory EGPA, compared to placebo, we found moderate‐certainty evidence that mepolizumab probably decreases disease relapse, and low‐certainty evidence that mepolizumab may increase disease remission. There was low‐certainty evidence on the similar frequency of AEs and serious AEs in both groups. The effect of mepolizumab as compared with placebo is uncertain, due to low‐certainty evidence.
We found low‐certainty evidence that etanercept/belimumab may increase the probability of withdrawal due to AEs, and low‐certainty evidence that etanercept/belimumab may have little or no impact on durable remission, major relapse and serious AEs. We found moderate‐certainty evidence of no improvement in disease flare. Low‐certainty evidence suggests that active drug (etanercept or belimumab) added to standard therapy when compared with standard therapy does not increase or reduce mortality.
Due to very low‐certainty evidence for all outcomes, the effects of infliximab, as compared to rituximab, in the management of patients with refractory GPA, is uncertain.
Implications for research.
Future research should be adequately powered and should ensure proper adherence to treatment to assess the effects of the intervention on clinically important outcomes in patients with ANCA‐associated vasculitis (AVV). It should also be reported properly, which will enable meaningful conclusions regarding the long‐term benefits and harms of anti‐cytokine targeted therapy. Due to the high heterogeneity of enrolled cases, especially concerning the uneven population of GPA and MPA in BREVAS 2018, further investigations on belimumab benefits and harms are justified. Due to limited evidence on the safety of mepolizumab, further investigations are justified.
History
Protocol first published: Issue 1, 2010 Review first published: Issue 9, 2020
| Date | Event | Description |
|---|---|---|
| 6 January 2016 | Amended | New review authors added, protocol updated in accordance with newer classification of vasculitis and Cochrane MECIR standards |
| 11 November 2009 | Amended | CMSG ID C162‐P |
Acknowledgements
We are grateful to Mr. Ireneusz Korfel for advice regarding design of the search strategy.
Appendices
Appendix 1. MEDLINE search strategy
Granulomatosis with Polyangiitis.sh.
(Polyangiitis adj2 Granulomatosis).tw.
(Wegener$ adj2 Granulomatosis).tw.
Microscopic Polyangiitis.sh.
Anti‐Neutrophil Cytoplasmic Antibody‐Associated Vasculitis.sh. or (anca associated vasculitis).mp.
Eosinophilic granulomatosis with polyangiitis.tw.
Microscopic Polyangiitis.tw.
Eosinophilic Granulomatous Vasculiti$.tw.
EGPA.tw.
Churg‐Strauss Syndrome.sh.
Churg‐Strauss Syndrome.tw.
Churg‐Strauss vasculitis.tw.
Allergic Granulomato$.tw.
Allergic Angiiti$.tw.
Allergic Granulomato$ Angiiti$.tw.
GPA.tw.
MPA.tw.
1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 or 14 or 15 or 16 or 17
randomized controlled trial.pt.
randomized controlled trials.mp.
random allocation.mp.
controlled clinical trial.pt.
controlled clinical trials.mp.
randomized.ab.
clinical trials.mp.
(clinical trial or clinical trial phase i or clinical trial phase ii or clinical trial phase iii or clinical trial phase iv).pt.
placebo.ab.
drug therapy.fs.
randomly.ab.
(random$ or RCT or RCTs).tw.
(controlled adj5 (trial$ or stud$)).tw.
(clinical$ adj5 trial$).tw.
((control or treatment or experiment$ or intervention) adj5 (group$ or subject$ or patient$)).tw.
(quasi‐random$ or quasi random$ or pseudo‐random$ or pseud or random$).tw.
((singl$ or doubl$ or tripl$ or trebl$) adj5 (blind$ or mask$)).tw.
placebo$.tw.
trial$.ab.
groups.ab.
animals.sh. not (humans.sh. and animals.sh.)
Tumor Necrosis Factors.sh.
Tumo?r Necrosis Factor‐alpha antagonist$.tw.
anti‐tumo?r necrosis factor$.tw.
Tumo?r Necrosis Factor‐alpha inhibitor$.tw.
anti‐tnf.tw.
antibodies monoclonal.sh.
Interleukin$ receptor$ antagonist$.tw.
Receptors, Interleukin.sh.
Interleukin$ antagonist$.tw.
Interleukin$ inhibitor$.tw.
Interleukin 1 Receptor Antagonist Protein.sh.
(Interleukin‐$ adj2 antagonist$).tw.
(Interleukin‐$ adj2 antagonist$).nm.
(IL‐$ adj2 antagonist$).tw.
(IL‐$ adj2 antagonist$).nm.
(Etanercept or Enbrel or TNF Receptor Type II IgG Fusion Protein or tnr001OR Recombinant Human Dimeric TNF Receptor Type II IgG Fusion Protein or TNFR‐Fc Fusion Protein or TNFR‐Fc Fusion Protein or TNFR‐Fc Fusion Protein or TNR 001 or TNR‐001).nm. or (Etanercept or Enbrel or TNF Receptor Type II IgG Fusion Protein or tnr001OR Recombinant Human Dimeric TNF Receptor Type II IgG Fusion Protein or TNFR‐Fc Fusion Protein or TNFR‐Fc Fusion Protein or TNFR‐Fc Fusion Protein or TNR 001 or TNR‐001).tw.
(Infliximab or Remicade or Remsima or MAb cA2 or Monoclonal Antibody cA2 or CT‐P13).tw. or (Infliximab or Remicade or Remsima or MAb cA2 or Monoclonal Antibody cA2 or CT‐P13).nm.
(Adalimumab or Humira or D2E7 Antibody).tw. or (Adalimumab or Humira or D2E7 Antibody).nm.
(Golimumab or Simponi or Belimumab).tw. or (Golimumab or Simponi or Belimumab).nm.
(Certolizumab pegol or Cimzia or Cimzias or CDP870 or CDP870s or CDP 870 or Certolizumab Pegols).tw. or (Certolizumab pegol or Cimzia or Cimzias or CDP870 or CDP870s or CDP 870 or Certolizumab Pegols).nm.
(Anakinra or Kineret or Urine‐Derived IL1 Inhibitor or IL1 Febrile Inhibitor or Urine IL‐1 Inhibitor or IL‐1Ra or Antril or recombinant interleukin 1 receptor antagonist or recombinant interleukin 1 receptor blocker).nm. or (Anakinra or Kineret or Urine‐Derived IL1 Inhibitor or IL1 Febrile Inhibitor or Urine IL‐1 Inhibitor or IL‐1Ra or Antril or recombinant interleukin 1 receptor antagonist or recombinant interleukin 1 receptor blocker).tw.
(Rilonacept or interleukin‐1 Trap or Arcalyst).nm. or (Rilonacept or interleukin‐1 Trap or Arcalyst).tw.
(canakinumab or Ilaris or Novartis Pharma or ACZ 885 or ACZ‐885 or ACZ885 or anti‐interleukin‐1beta monoclonal antibody).nm. or (canakinumab or Ilaris or Novartis Pharma or ACZ 885 or ACZ‐885 or ACZ885 or anti‐interleukin‐1beta monoclonal antibody).tw.
(Tocilizumab or Atlizumab or Actemra or monoclonal antibody, MRA or il ‐6 or anti‐IL‐6 or anti‐interluekin‐6 or interleukin 6).nm. or (Tocilizumab or Atlizumab or Actemra or monoclonal antibody, MRA or il ‐6 or anti‐IL‐6 or anti‐interluekin‐6 or interleukin 6).tw.
(Siltuximab or Sylvant or CNTO 328 monoclonal antibody or cClB8 monoclonal antibody or monoclonal antibody CNTO 328 or monoclonal antibody CNTO‐328 or monoclonal antibody CNTO328).nm. or (Siltuximab or Sylvant or CNTO 328 monoclonal antibody or cClB8 monoclonal antibody or monoclonal antibody CNTO 328 or monoclonal antibody CNTO‐328 or monoclonal antibody CNTO328).tw.
(Daclizumab or Zenapax or dacliximab or dacluzimab or Ro 24‐7375 or Ro‐24‐7375 or Zinbryta).nm. or (Daclizumab or Zenapax or dacliximab or dacluzimab or Ro 24‐7375 or Ro‐24‐7375 or Zinbryta).tw.
(Basiliximab or Simultec or Simulect or CHI 621 or SDZ CHI 621).nm. or (Basiliximab or Simultec or Simulect or CHI 621 or SDZ CHI 621).tw.
(Mepolizumab or Nucala or SB‐240563 or SB24056 or Bosatria).nm. or (Mepolizumab or Nucala or SB‐240563 or SB24056 or Bosatria).tw.
(Reslizumab or Cinquil or Cinqair or sch 55700 or sch‐55700 or DCP‐835 or DCP835).nm. or (Reslizumab or Cinquil or Cinqair or sch 55700 or sch‐55700 or DCP‐835 or DCP835).tw.
(Benralizumab or medi 563).nm. or (Benralizumab or medi 563).tw.
(Tralokinumab or cat 354).nm. or (Tralokinumab or cat 354).tw.
(Lebrikizumab il ‐13 or anti‐IL‐13 or anti‐interluekin‐13 or interleukin 13).nm. or (Lebrikizumab il ‐13 or anti‐IL‐13 or anti‐interluekin‐13 or interleukin 13).tw.
(Pitrakinra or binetrakin).nm. or (Pitrakinra or binetrakin).tw.
(Dupilumab or REGN668).nm. or (Dupilumab or REGN668).tw.
(Ustekinumab or Stelara or CNTO 1275 or CNTO‐1275).nm. or (Ustekinumab or Stelara or CNTO 1275 or CNTO‐1275).tw.
(Ixekizumab or Taltz or LY‐2439821 or LY2439821 or il ‐17 or anti‐IL‐17 or anti‐interluekin‐17 or interleukin 17).nm. or (Ixekizumab or Taltz or LY‐2439821 or LY2439821 or il ‐17 or anti‐IL‐17 or anti‐interluekin‐17 or interleukin 17).tw.
Sarilumab.nm. or Sarilumab.tw
Olokizumab.nm. or Olokizumab.tw
Sirukumab.nm. or Sirukumab.tw
(Clazakizumab or ALD518 or BMS‐945429).nm. or (Clazakizumab or ALD518 or BMS‐945429).tw
(Secukinumab or Cosentyx or AIN 457 or AIN‐457 or AIN457).nm or (Secukinumab or Cosentyx or AIN 457 or AIN‐457 or AIN457).tw
(Brodalumab or AMG‐827).nm. or (Brodalumab or AMG‐827).tw
(interleukin‐2 or interleukin 2 or IL‐2 or IL2 or IL 2 or Il‐2 antagonist or Il‐2 receptor antagonist).nm. or (interleukin‐2 or interleukin 2 or IL‐2 or IL2 or IL 2 or Il‐2 antagonist or Il‐2 receptor antagonist).tw
(interleukin‐3 or interleukin 3 or IL‐3 or IL3 or IL 3 or Il‐3 antagonist or Il‐3 receptor antagonist).nm. or (interleukin‐3 or interleukin 3 or IL‐3 or IL3 or IL 3 or Il‐3 antagonist or Il‐3 receptor antagonist).tw
(Il‐4 antagonist or Il‐4 receptor antagonist or Il‐4 receptor or interleukin 4 or interleukin‐4 or Il‐4 or Il 4).nm. or (Il‐4 antagonist or Il‐4 receptor antagonist or Il‐4 receptor or interleukin 4 or interleukin‐4 or Il‐4 or Il 4).tw
(interleukin‐7 or interleukin 7 or IL‐7 or IL7 or IL 7 or Il‐7 antagonist or Il‐7 receptor antagonist).nm. or (interleukin‐7 or interleukin 7 or IL‐7 or IL7 or IL 7 or Il‐7 antagonist or Il‐7 receptor antagonist).tw
(interleukin‐8 or interleukin 8 or IL‐8 or IL8 or IL 8 or Il‐8 antagonist or Il‐8 receptor antagonist).nm. or (interleukin‐8 or interleukin 8 or IL‐8 or IL8 or IL 8 or Il‐8 antagonist or Il‐8 receptor antagonist).tw
(interleukin‐9 or interleukin 9 or IL‐9 or IL9 or IL 9 or Il‐9 antagonist or Il‐9 receptor antagonist).nm. or (interleukin‐9 or interleukin 9 or IL‐9 or IL9 or IL 9 or Il‐9 antagonist or Il‐9 receptor antagonist).tw
(interleukin‐10 or interleukin 10 or IL‐10 or IL10 or IL 10 or Il‐10 antagonist or Il‐10 receptor antagonist).nm. or (interleukin‐10 or interleukin 10 or IL‐10 or IL10 or IL 10 or Il‐10 antagonist or Il‐10 receptor antagonist).tw
(interleukin‐11 or interleukin 11 or IL‐11 or IL11 or IL 11 or Il‐11 antagonist or Il‐11 receptor antagonist).nm. or (interleukin‐11 or interleukin 11 or IL‐11 or IL11 or IL 11 or Il‐11 antagonist or Il‐11 receptor antagonist).tw
(interleukin‐12 or interleukin 12 or IL‐12 or IL12 or IL 12 or Il‐12 antagonist or Il‐12 receptor antagonist).nm. or (interleukin‐12 or interleukin 12 or IL‐12 or IL12 or IL 12 or Il‐12 antagonist or Il‐12 receptor antagonist).tw
(interleukin‐14 or interleukin 14 or IL‐14 or IL14 or IL 14 or Il‐14 antagonist or Il‐14 receptor antagonist).nm. or (interleukin‐14 or interleukin 14 or IL‐14 or IL14 or IL 14 or Il‐14 antagonist or Il‐14 receptor antagonist).tw
(interleukin‐15 or interleukin 15 or IL‐15 or IL15 or IL 15 or Il‐15 antagonist or Il‐15 receptor antagonist).nm. or (interleukin‐15 or interleukin 15 or IL‐15 or IL15 or IL 15 or Il‐15 antagonist or Il‐15 receptor antagonist).tw
(interleukin‐16 or interleukin 16 or IL‐16 or IL16 or IL 16 or Il‐16 antagonist or Il‐16 receptor antagonist).nm. or (interleukin‐16 or interleukin 16 or IL‐16 or IL16 or IL 16 or Il‐16 antagonist or Il‐16 receptor antagonist).tw
(interleukin‐18 or interleukin 18 or IL‐18 or IL18 or IL 18 or Il‐18 antagonist or Il‐18 receptor antagonist).nm. or (interleukin‐18 or interleukin 18 or IL‐18 or IL18 or IL 18 or Il‐18 antagonist or Il‐18 receptor antagonist).tw
(interleukin‐19 or interleukin 19 or IL‐19 or IL19 or IL 19 or Il‐19 antagonist or Il‐19 receptor antagonist).nm. or (interleukin‐19 or interleukin 19 or IL‐19 or IL19 or IL 19 or Il‐19 antagonist or Il‐19 receptor antagonist).tw
(interleukin‐20 or interleukin 20 or IL‐20 or IL20 or IL 20 or Il‐20 antagonist or Il‐20 receptor antagonist).nm. or (interleukin‐20 or interleukin 20 or IL‐20 or IL20 or IL 29 or Il‐20antagonist or Il‐20 receptor antagonist).tw
(interleukin‐21 or interleukin 21 or IL‐21 or IL21 or IL 21 or Il‐21 antagonist or Il‐21 receptor antagonist).nm. or (interleukin‐21 or interleukin 21 or IL‐21 or IL21 or IL 21 or Il‐21 antagonist or Il‐21 receptor antagonist).tw
(interleukin‐22 or interleukin 22 or IL‐22 or IL22 or IL 22 or Il‐22 antagonist or Il‐22 receptor antagonist).nm. or (interleukin‐22 or interleukin 22 or IL‐22 or IL22 or IL 22 or Il‐22 antagonist or Il‐22 receptor antagonist).tw
(interleukin‐23 or interleukin 23 or IL‐23 or IL23 or IL 23 or Il‐23 antagonist or Il‐23 receptor antagonist).nm. or (interleukin‐23 or interleukin 23 or IL‐23 or IL23 or IL 23 or Il‐23 antagonist or Il‐23 receptor antagonist).tw
(interleukin‐24 or interleukin 24 or IL‐24 or IL24 or IL 24 or Il‐24 antagonist or Il‐24 receptor antagonist).nm. or (interleukin‐24 or interleukin 24 or IL‐24 or IL24 or IL 24 or Il‐24 antagonist or Il‐24 receptor antagonist).tw
(interleukin‐25 or interleukin 25 or IL‐25 or IL25 or IL 25 or Il‐25 antagonist or Il‐25 receptor antagonist).nm. or (interleukin‐25 or interleukin 25 or IL‐25 or IL25 or IL 25 or Il‐25 antagonist or Il‐25 receptor antagonist).tw
(interleukin‐26 or interleukin 26 or IL‐26 or IL26 or IL 26 or Il‐26 antagonist or Il‐26 receptor antagonist).nm. or (interleukin‐26 or interleukin 26 or IL‐26 or IL26 or IL 26 or Il‐26 antagonist or Il‐26 receptor antagonist).tw
(interleukin‐27 or interleukin 27 or IL‐27 or IL27 or IL 27 or Il‐27 antagonist or Il‐27 receptor antagonist).nm. or (interleukin‐27 or interleukin 27 or IL‐27 or IL27 or IL 27 or Il‐27 antagonist or Il‐27 receptor antagonist).tw
(interleukin‐28 or interleukin 28 or IL‐28 or IL28 or IL 28 or Il‐28 antagonist or Il‐28 receptor antagonist).nm. or (interleukin‐28 or interleukin 28 or IL‐28 or IL28 or IL 28 or Il‐28 antagonist or Il‐28 receptor antagonist).tw
(interleukin‐29 or interleukin 29 or IL‐29 or IL29 or IL 29 or Il‐29 antagonist or Il‐29 receptor antagonist).nm. or (interleukin‐29 or interleukin 29 or IL‐29 or IL29 or IL 29 or Il‐29 antagonist or Il‐29 receptor antagonist).tw
(interleukin‐30 or interleukin 30 or IL‐30 or IL30 or IL 30 or Il‐30 antagonist or Il‐30 receptor antagonist).nm. or (interleukin‐30 or interleukin 30 or IL‐30 or IL30 or IL 30 or Il‐30 antagonist or Il‐30 receptor antagonist).tw
(interleukin‐31 or interleukin 31 or IL‐31 or IL31 or IL 31 or Il‐31 antagonist or Il‐31 receptor antagonist).nm. or (interleukin‐31 or interleukin 31 or IL‐31 or IL31 or IL 31 or Il‐31 antagonist or Il‐31 receptor antagonist).tw
(interleukin‐32 or interleukin 32 or IL‐32 or IL32 or IL 32 or Il‐32 antagonist or Il‐32 receptor antagonist).nm. or (interleukin‐32 or interleukin 32 or IL‐32 or IL32 or IL 32 or Il‐32 antagonist or Il‐32 receptor antagonist).tw
(interleukin‐33 or interleukin 33 or IL‐33 or IL33 or IL 33 or Il‐33 antagonist or Il‐33 receptor antagonist).nm. or (interleukin‐33 or interleukin 33 or IL‐33 or IL33 or IL 33 or Il‐33 antagonist or Il‐33 receptor antagonist).tw
40 or 41 or 42 or 43 or 44 or 45 or 46 or 47 or 48 or 49 or 50 or 51 or 52 or 53 or 54 or 55 or 56 or 57 or 58 or 59 or 60 or 61 or 62 or 63 or 64 or 65 or 66 or 67 or 68 or 69 or 70 or 71 or 72 or 73 or 74 or 75 or 76 or 77 or 78 or 79 or 80 or 81 or 82 or 83 or 84 or 85 or 86 or 87 or 88 or 89 or 90 or 91 or 92 or 93 or 94 or 95 or 96 or 97 or 98 or 99 or 100 or 101 or 102 or 103 or 104 or 105 or 106 or 107 or 108 or 109
19 or 20 or 21 or 22 or 23 or 24 or 25 or 26 or 27 or 28 or 29 or 30 or 31 or 32 or 33 or 34 or 35 or 36 or 37 or 38
111 not 39
18 and 112 and 110
Appendix 2. Embase search strategy
'granulomatosis'/exp OR granulomatosis:ab,ti
'wegener granulomatosis'/exp OR wegener AND granulomatosis
'microscopic polyangiitis'/exp OR 'microscopic polyangiitis':ti,ab
'churg strauss syndrome'/exp OR 'churg strauss syndrome':ti,ab
'vasculitis'/exp OR vasculitis:ti,ab OR 'anca associated vasculitis'
'allergic granulomato* angiiti*':ti,ab
1 OR 2 OR 3 OR 4 OR 5 OR 6
random*
factorial* OR crossover* OR cross AND over* OR 'cross over*' OR placebo* OR doubl* AND adj5 AND blind* OR singl* AND adj5 AND blind* OR assign* OR allocat* OR volunteer*
'crossover procedure' OR 'double blind' AND procedure OR randomized AND controlled AND trial OR 'single blind' AND procedure
group*:ti,ab
trial*:ab
8 OR 9 OR 10 OR 11 OR 12
'cytokine'/exp OR cytokine:ti,ab
'interleukin'/exp OR interleukin:ti,ab
'tumor necrosis factor':ti,ab
'cytokine receptor'/exp OR 'cytokine receptor':ti,ab
'tumor necrosis factor inhibitor'/exp
'tumor necrosis factor inhibitor'/exp OR 'tumor necrosis factor inhibitor':ti,ab
'etanercept'/exp OR etanercept:ti,ab OR enbrel:ti,ab OR (tnf AND receptor AND type AND ii AND igg AND fusion AND protein) OR tnr001 OR (recombinant AND human AND dimerictnf AND receptor AND type AND ii AND igg AND fusion AND protein) OR ('tnfr fc' AND fusion AND protein) OR (tnr AND 001) OR 'tnr 001' OR 'belimumab'/exp OR belimumab:ti,ab
'infliximab'/exp OR infliximab:ti,ab OR remicade:ti,ab OR remsima:ti,ab OR (mab AND ca2) OR (monoclonal AND antibody AND ca2) OR 'ct p13'
'adalimumab'/exp OR adalimumab:ti,ab OR humira:ti,ab OR (d2e7 AND antibody)
'monoclonal antibody'/exp OR 'monoclonal antibody':ti,ab
golimumab:ti,ab OR simponi:ti,ab OR 'golimumab'/exp
'certolizumab pegol*':ti,ab OR cimzia*:ti,ab OR cdp870 OR cdp870s OR 'cdp 870' OR 'certolizumab'/exp
'anakinra'/exp OR anakinra:ti,ab OR kineret:ti,ab OR ('urine derived':ti,ab AND il1:ti,ab AND inhibitor:ti,ab) OR (il1:ti,ab AND febrile:ti,ab AND inhibitor:ti,ab) OR (urine:ti,ab AND 'il 1':ti,ab AND inhibitor:ti,ab) OR 'il 1ra':ti,ab OR antril:ti,ab OR (recombinant:ti,ab AND interleukin:ti,ab AND 1:ti,ab AND receptor:ti,ab AND antagonist:ti,ab) OR (recombinant:ti,ab AND interleukin:ti,ab AND 1:ti,ab AND receptor:ti,ab AND blocker:ti,ab)
rilonacept:ti,ab OR ('interleukin 1':ti,ab AND trap:ti,ab) OR arcalyst:ti,ab
'rilonacept'/exp
canakinumab:ti,ab OR ilaris:ti,ab OR (acz AND 885) OR 'acz 885' OR acz885 OR ('anti interleukin 1beta':ti,ab AND monoclonal:ti,ab AND antibody:ti,ab)
'canakinumab'/exp
tocilizumab:ti,ab OR atlizumab:ti,ab OR actemra:ti,ab OR (monoclonal:ti,ab AND antibody,:ti,ab AND mra:ti,ab) OR (il:ti,ab AND ‐6:ti,ab) OR 'anti il 6':ti,ab OR 'anti interluekin 6':ti,ab OR 'interleukin 6':ti,ab
'tocilizumab'/exp
siltuximab:ti,ab OR sylvant:ti,ab OR (cnto AND 328 AND monoclonal AND antibody) OR (cclb8 AND monoclonal AND antibody) OR (monoclonal AND antibody AND 'cnto 328') OR (monoclonal AND antibody AND cnto328)
'siltuximab'/exp
daclizumab:ti,ab OR zenapax:ti,ab OR dacliximab:ti,ab OR dacluzimab:ti,ab OR (ro AND '24 7375') OR 'ro 24 7375' OR zinbryta:ti,ab
'daclizumab'/exp
basiliximab:ti,ab OR simultec:ti,ab OR (chi AND 621) OR (sdz AND chi AND 621)
'basiliximab'/exp
mepolizumab:ti,ab OR nucala:ti,ab OR 'sb 240563' OR sb24056 OR bosatria
'mepolizumab'/exp
reslizumab:ti,ab OR cinquil:ti,ab OR cinqair:ti,ab OR (sch AND 55700) OR 'sch 55700' OR 'dcp 835' OR dcp835
'reslizumab'/exp
benralizumab:ti,ab OR (medi AND 563)
'benralizumab'/exp
tralokinumab:ti,ab OR (cat AND 354)
'tralokinumab'/exp
lebrikizumab:ti,ab OR 'il 13'/exp OR 'il 13':ti,ab OR (anti AND il AND 13) OR (anti AND interluekin AND 13) OR (interleukin AND 13)
'lebrikizumab'/exp
pitrakinra:ti,ab OR binetrakin:ti,ab
'pitrakinra'/exp
dupilumab:ti,ab OR regn668
'dupilumab'/exp
ustekinumab:ti,ab OR stelara:ti,ab OR (cnto AND 1275) OR 'cnto 1275'
'ustekinumab'/exp
ixekizumab:ti,ab OR taltz:ti,ab OR 'ly 2439821' OR ly2439821 OR (il:ti,ab AND 17:ti,ab) OR 'il 17'/exp OR 'anti il 17' OR 'anti interluekin 17':ti,ab OR 'interleukin 17':ti,ab OR 'interleukin 17'/exp
'ixekizumab'/exp
'sarilumab'/exp OR sarilumab:ti,ab
'olokizumab'/exp OR olokizumab:ti,ab
'sirukumab'/exp OR sirukumab:ti,ab
clazakizumab:ti,ab OR ald518 OR 'bms 945429'
'clazakizumab'/exp
secukinumab:ti,ab OR cosentyx:ti,ab OR (ain AND 457) OR 'ain 457' OR ain457
'secukinumab'/exp
'brodalumab'/exp OR brodalumab:ti,ab OR 'amg 827'
14 OR 15 OR 16 OR 17 OR 18 OR 19 OR 20 OR 21 OR 22 OR 23 OR 24 OR 25 OR 26 OR 27 OR 28 OR 29 OR 30 OR 31 OR 32 OR 33 OR 34 OR 35 OR 36 OR 37 OR 38 OR 39 OR 40 OR 41 OR 42 OR 43 OR 44 OR 45 OR 46 OR 47 OR 48 OR 49 OR 50 OR 51 OR 52 OR 53 OR 54 OR 55 OR 56 OR 57 OR 58 OR 59 OR 60 OR 61 OR 62 OR 63 OR 64
'human'/exp OR human*
7 AND 13 AND 65 AND 66
67 AND [embase]/lim NOT [medline]/lim
Appendix 3. CENTRAL search strategy
random* or trial* crossover* or 'cross over*' or double near blind* or singl* near blind or assign* or allocat*
MeSH descriptor: [Double‐Blind Method] explode all trees
MeSH descriptor: [Randomized Controlled Trial] explode all trees
MeSH descriptor: [Single‐Blind Method] explode all trees
MeSH descriptor: [Cross‐Over Studies] explode all trees
1 or 2 or 3 or 4 or 5
granulomatosis or 'wegener granulomatos*' or 'microscopic polyangi*' or 'churg strauss syndrome' or 'allergic granulomato* angiiti*'
MeSH descriptor: [Anti‐Neutrophil Cytoplasmic Antibody‐Associated Vasculitis] explode all trees
MeSH descriptor: [Granulomatosis with Polyangiitis] explode all trees
MeSH descriptor: [Churg‐Strauss Syndrome] explode all trees
7 or 8 or 9 or 10
Tumor Necrosis Factor‐alpha' and (antagonist* or inhibitor*)
'Tumor Necrosis Factor‐alpha receptor*' and (antagonist* or inhibitor*)
Interleukin* and (antagonist* and inhibitor*)
MeSH descriptor: [Tumor Necrosis Factor‐alpha] explode all trees
MeSH descriptor: [Interleukins] explode all trees
12 or 13 or 14 or 15 or 16
Etanercept or Enbrel or 'TNF Receptor Type II IgG Fusion Protein' or tnr001 or 'Recombinant Human Dimeric TNF Receptor Type II IgG Fusion Protein' or 'TNFR‐Fc Fusion Protein' or 'TNFR‐Fc Fusion Protein' or 'TNFR‐Fc Fusion Protein'
MeSH descriptor: [Etanercept] explode all trees
18 or 19
Infliximab or Remicade or Remsima or 'MAb cA2' or 'Monoclonal Antibody cA2' or CT‐P13
MeSH descriptor: [Infliximab] explode all trees
21 or 22
Adalimumab or Humira or 'D2E7 Antibody'
MeSH descriptor: [Adalimumab] explode all trees
24 or 25
Golimumab or Simponi
Certolizumab pegol* or Cimzia* or CDP870*
MeSH descriptor: [Certolizumab Pegol] explode all trees
28 or 29
Anakinra or Kineret or 'Urine‐Derived IL1 Inhibitor' or 'IL1 Febrile Inhibitor' or 'Urine IL‐1 Inhibitor' or IL‐1Ra or Antril or 'recombinant interleukin 1 receptor antagonist' or 'recombinant interleukin 1 receptor blocker'
MeSH descriptor: [Interleukin 1 Receptor Antagonist Protein] explode all trees
31 or 32
Rilonaept or 'interleukin‐1 Trap' or Arcalyst
Canakimumab or Ilaris or 'Novartis Pharma' or 'ACZ 885' or 'anti‐interleukin‐1beta monoclonal antibody'
Tocilizumab or Atlizumab or Actemra
Siltuximab or Sylvant or 'CNTO 328 monoclonal antibody' or 'cClB8 monoclonal antibody'
Daclizumab or Zenapax or dacliximab or dacluzimab or 'Ro 24‐7375'
Basiliximab or Simultec or 'CHI 621' or 'SDZ CHI 621'
Mepolizumab or Nucala or 'SB‐240563' or Bosatria
Reslizumab or Cinquil or 'sch 55700'
Benralizumab or 'medi 563'
Tralokinumab or 'cat 354'
Lebrikizumab
Pitrakinra or binetrakin
Dupilumab or 'REGN 668'
Ustekinumab or Stelara or 'CNTO 1275'
MeSH descriptor: [Ustekinumab] explode all trees
47 or 48
Ixekizumab or taltz or 'ly 2439821'
sarilumab or belimumab
olokizumab
sirukumab or clazakizumab or secukinumab or brodalumab or 'amg 827'
17 or 20 or 23 or 26 or 27 or 30 or 33 or 34 or 35 or 36 or 37 or 38 or 39 or 40 or 41 or 42 or 43 or 44 or 45 or 46 or 49 or 50 or 51 or 52 or 53
6 and 11 and 54
Data and analyses
Comparison 1. EGPA ‐ mepolizumab vs placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Mortality | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Subtotals only | |
| 1.2 Remission | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.2.1 Any remission (for at least 1 week) | 1 | 136 | Risk Ratio (M‐H, Random, 95% CI) | 2.77 [1.62, 4.74] |
| 1.2.2 Remission for at least 24 weeks | 1 | 136 | Risk Ratio (M‐H, Random, 95% CI) | 9.50 [2.30, 39.21] |
| 1.3 Durable remission | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.4 Disease relapse | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.5 Any AE | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.5.1 Any event | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.5.2 Any event related to trial agent | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.6 Any serious AE | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.6.1 Any serious event | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.6.2 Any serious event related to trial agent | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.7 Any withdrawals due to AE | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.8 Control of asthma symptoms with ACQ6 ‐ change from baseline | 1 | 136 | Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.60, 0.00] |
| 1.9 Control of sinonasal symptoms with SNOT22 ‐ change from baseline | 1 | 136 | Mean Difference (IV, Random, 95% CI) | ‐4.66 [‐10.69, 1.36] |
| 1.10 Disease damage in VDI ‐ change from baseline | 1 | 136 | Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.37, 0.23] |
Comparison 2. GPA ‐ anticytokine therapy vs control.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Mortality | 3 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Subtotals only | |
| 2.1.1 Active drug vs placebo | 2 | 285 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.45 [0.55, 10.97] |
| 2.1.2 Infliximab vs rituximab | 1 | 17 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.88 [0.05, 15.51] |
| 2.2 Remission | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.2.1 Etanercept vs placebo, remission at any time during study | 1 | 180 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.89, 1.07] |
| 2.2.2 Infliximab vs rituximab | 1 | 17 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.11, 1.81] |
| 2.3 Durable remission | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.3.1 Etanercept vs placebo | 1 | 174 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.77, 1.11] |
| 2.3.2 Infliximab vs rituximab long term | 1 | 17 | Risk Ratio (M‐H, Random, 95% CI) | 0.22 [0.03, 1.60] |
| 2.4 Disease relapse | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.4.1 Any relapse ‐ belimumab vs placebo | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.27, 1.97] |
| 2.4.2 Major relapse ‐ belimumab vs placebo | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 2.94 [0.12, 70.67] |
| 2.5 No disease flare | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.5.1 Etanercept vs placebo | 1 | 180 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.76, 1.27] |
| 2.6 Any AE | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.97, 1.29] |
| 2.7 Any non serious AE | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.70, 1.28] |
| 2.7.1 Belimumab vs placebo | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.70, 1.28] |
| 2.8 Any severe or serious AE | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.8.1 Active drug vs placebo | 2 | 285 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.80, 1.27] |
| 2.8.2 Infliximab vs rituximab | 1 | 17 | Risk Ratio (M‐H, Random, 95% CI) | 1.78 [0.20, 16.10] |
| 2.9 Any solid malignancy | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.9.1 During trial and follow up | 1 | 180 | Risk Ratio (M‐H, Random, 95% CI) | 2.86 [1.08, 7.62] |
| 2.9.2 During trial | 1 | 180 | Risk Ratio (M‐H, Random, 95% CI) | 13.29 [0.76, 232.45] |
| 2.9.3 During follow up | 1 | 153 | Risk Ratio (M‐H, Random, 95% CI) | 1.58 [0.54, 4.61] |
| 2.10 Any withdrawals due to AE | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.10.1 Active drug vs placebo | 2 | 285 | Risk Ratio (M‐H, Random, 95% CI) | 2.66 [1.07, 6.59] |
| 2.10.2 Infliximab vs rituximab | 1 | 17 | Risk Ratio (M‐H, Random, 95% CI) | 2.70 [0.13, 58.24] |
| 2.11 Treatment response | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.11.1 Sustained low level of disease activity (Etanercept vs placebo) | 1 | 174 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.86, 1.06] |
| 2.11.2 Partial remission (infliximab vs rituximab) | 1 | 17 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.07, 12.00] |
Comparison 3. GPA ‐ sensitivity analysis regarding WGET trial.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Mortality | 2 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Subtotals only | |
| 3.1.1 Active drug vs placebo | 2 | 286 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.47 [0.55, 11.07] |
| 3.2 Remission | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.2.1 Etanercept vs placebo | 1 | 181 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.90, 1.08] |
| 3.3 Durable remission | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.3.1 Etanercept vs placebo | 1 | 181 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.83, 1.21] |
| 3.4 Disease flare | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.4.1 Etanercept vs placebo | 1 | 181 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.77, 1.28] |
| 3.5 Any severe or serious AE | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.5.1 Active drug vs placebo | 2 | 286 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.80, 1.28] |
| 3.6 Any withdrawals due to AE | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.6.1 Active drug vs placebo | 2 | 286 | Risk Ratio (M‐H, Random, 95% CI) | 2.67 [1.08, 6.63] |
| 3.7 Treatment response | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.7.1 Sustained low level of disease activity (Etanercept vs placebo) | 1 | 181 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.92, 1.17] |
Comparison 4. GPA ‐ sensitivity BREVAS withdrawn due to adverse events.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Any withdrawals due to AE | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1.1 Active drug vs placebo | 2 | 286 | Risk Ratio (M‐H, Random, 95% CI) | 1.83 [0.80, 4.19] |
Comparison 5. Sensitivity analysis ‐ all AAV together.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Mortality | 4 | 438 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.25 [0.64, 7.93] |
| 5.1.1 EGPA | 1 | 136 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 7.39 [0.15, 372.38] |
| 5.1.2 GPA active drug vs placebo | 2 | 285 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.45 [0.55, 10.97] |
| 5.1.3 GPA infliximab vs rituximab | 1 | 17 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.88 [0.05, 15.51] |
| 5.2 Any remission | 3 | 333 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.36, 3.76] |
| 5.2.1 EGPA | 1 | 136 | Risk Ratio (M‐H, Random, 95% CI) | 2.77 [1.62, 4.74] |
| 5.2.2 GPA active drug vs placebo | 1 | 180 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.89, 1.07] |
| 5.2.3 GPA infliximab vs rituximab | 1 | 17 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.11, 1.81] |
| 5.3 Durable remission | 3 | 327 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [0.20, 8.61] |
| 5.3.1 EGPA | 1 | 136 | Risk Ratio (M‐H, Random, 95% CI) | 13.00 [1.75, 96.63] |
| 5.3.2 GPA active drug vs placebo | 1 | 174 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.77, 1.11] |
| 5.3.3 GPA infliximab vs rituximab | 1 | 17 | Risk Ratio (M‐H, Random, 95% CI) | 0.22 [0.03, 1.60] |
| 5.4 Disease relapse | 2 | 241 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.54, 0.86] |
| 5.4.1 EGPA | 1 | 136 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.53, 0.86] |
| 5.4.2 GPA active drug vs placebo | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.27, 1.97] |
| 5.5 Any serious or severe AE | 4 | 438 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.78, 1.20] |
| 5.5.1 EGPA | 1 | 136 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.35, 1.28] |
| 5.5.2 GPA active drug vs placebo | 2 | 285 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.80, 1.27] |
| 5.5.3 Infliximab vs rituximab | 1 | 17 | Risk Ratio (M‐H, Random, 95% CI) | 1.78 [0.20, 16.10] |
| 5.6 Any withdrawals due to AE | 4 | 438 | Risk Ratio (M‐H, Random, 95% CI) | 2.57 [1.13, 5.83] |
| 5.6.1 EGPA | 1 | 136 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.19, 21.54] |
| 5.6.2 GPA active drug vs placebo | 2 | 285 | Risk Ratio (M‐H, Random, 95% CI) | 2.66 [1.07, 6.59] |
| 5.6.3 GPA infliximab vs rituximab | 1 | 17 | Risk Ratio (M‐H, Random, 95% CI) | 2.70 [0.13, 58.24] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
BREVAS 2018.
| Study characteristics | ||
| Methods |
Study design: double‐blind parallel RCT (initially planned as phase 3 trial) Location: Australia, Belgium, Canada, Czech Republic, Germany, Ireland, Italy, Mexico, Peru, Poland, Russian Federation, Spain, Switzerland, United Kingdom, United States Setting: Not reported Number of centres: 37 Time frame of the study: 20 March 2013 through 06 February 2017 Duration of follow‐up: approximately up to 4 years |
|
| Participants |
Inclusion criteria:
Exclusion criteria:
Total number of participants: 106 participants randomised, Belimumab group N = 53, Placebo group N = 52, N = 1 participant randomised in error Characteristics: GPA 79%, MPA 21% (The results of MPA patients were added to the GPA patient analysis) Mean (SD) age: Belimumab 56 ± 14, Placebo 54 ± 14 Female sex: Belimumab 49.1%, Placebo 48.1% CrCl value: NR Disease duration (time since diagnosis, median(SD)): Belimumab 2.3 years (range 0.3‐14.9), placebo 1.3 years (0.01‐20.6) Age at onset of symptoms: NR Race/ethnic group: Belimumab: African American/African Heritage 1, American Indian or Alaskan Native 6, Central/South Asian Heritage 0, White 46. Placebo: African American/African Heritage 1, American Indian or Alaskan Native 5, Central/South Asian Heritage 2, White 44 Disease diagnosed at the enrolment of the study: NR Disease assessment index BVAS (mean(SD)): NR Damage index: NR Global assessment: NR Quality of life: NR Disease severity (limited vs severe): NR Organ involvement: NR Prior treatment: NR ANCA type: anti‐PR3 77%, anti‐MPO 23% |
|
| Interventions |
Belimumab group: Belimumab 10 mg/kg administered intravenously over 1 hour, on days 0, 14, 28 and every 28 days thereafter until 12 months after the last participant was randomised (In Belgium‐ all participants received belimumab at dose 10mg/day every 28 days until Week 24, and a final evaluation was at Week 28). Placebo group: Placebo administered intravenously over 1 hour on days 0, 14, 28 and every 28 days thereafter until 12 months after the last participant was randomised. (In Belgium‐only open‐label extension, all participants received belimumab 10mg/day every 28 days until Week 24, and a final evaluation was at Week 28). Mean or median treatment duration not reported. Description of treatment and concomitant treatment: All participants received oral azathioprine at a target dose of 2 mg/kg/day. |
|
| Outcomes |
Major outcomes:
Minor outcomes:
Other outcomes:
At double‐blind Week 48 of year 1 and double‐blind Week 28 (specified in protocol) of year 2 and by visit;
|
|
| Notes |
Discontinuation: 32 (9.52%) participants (12 participants from placebo group and 20 participant from belimumab group): 10 (9.52%) due to AEs, 11 (10.48%) lack of efficacy, 1 (0,95%) protocol violation, 2 (1.9%) other‐study closed/terminated, 5 (4.76%) physician decision, 3 (2.86%) consent withdrawal. Funding: GlaxoSmithKline The study was initially planned as phase 3 trial with planned enrolment of approximately 300, then it ws changed to exploratory trial with enrolment of approximately 100 patients, the design changed from "event‐driven" to "fixed completion” specified as 12 months after the randomisation of the last patient. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A total of 164 participants were screened and 106 were enrolled and randomised in a 1:1 ratio to receive placebo or belimumab" ‐ information from EudraCT and clinicaltrials.gov. Authors reported the use of interactive web response system so it was assumed that the sequence generation was computed based. |
| Allocation concealment (selection bias) | Low risk | Central randomisation: Use of interactive web response system |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Masking: Quadruple (Participant, Care Provider, Investigator, Outcomes Assessor)" ‐ information from EudraCT and clinicaltrials.gov. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Masking: Quadruple (Participant, Care Provider, Investigator, Outcomes Assessor)" ‐ information from EudraCT and clinicaltrials.gov. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT was performed (all randomised participants who received at least one dose of study drug) and all patients were included in primary analysis, no missing outcome data identified, all reasons of withdrawn were described. |
| Selective reporting (reporting bias) | High risk | Full protocol available; not all outcomes specified in protocol were reported (see in the description of outcomes). |
| Other bias | Low risk | No other biases were identified. |
de Menthon 2011.
| Study characteristics | ||
| Methods |
Study design: double‐blinded parallel RCT Location: France: Dijon, Nancy, Lyon, Bordeaux, Rennes, Caen, Nantes, Reims, Rouen, Paris Setting: Hospitals Number of centres: Not reported Time frame of the study: June 2004 through June 2007 Duration of follow‐up mean (SD): 30.6 ± 15.4 months (28.9 ± 15.4 months for the infliximab group and 32.9 ± 16.7 months for the rituximab group) |
|
| Participants |
Inclusion criteria: Wegeners granulomatosis according to Chapel Hill criteria, refractory disease despite optimal treatment: failure to respond to steroids and several immunosuppressants (alone or combination), among the latter ‐ at least pulses and then oral cyclophosphamide. Exclusion criteria: not reported Total number of participants: 17 participants randomised, Infliximab group N=9, Rituximab group N=8 Characteristics: Mean (SD) age: Infliximab 52.9 ± 17, Rituximab 51.4 ± 15 Female sex: Infliximab 88.9%, Rituximab 12.5% CrCl value: Not reported Disease duration (time since diagnosis, mean(SD)): Infliximab 74.8 ± 70 months, Rituximab 76.3 ± 47 months Age at onset of symptoms: Not reported Race/ethnic group: Not reported Disease diagnosed at the enrolment of the study: Not reported Disease assessment index BVAS (mean(SD)): Infliximab 13.1 ± 5.5 Rituximab 12.6 ± 7 Damage index: Not reported Global assessment: Not reported Quality of life: Not reported Disease severity (limited vs severe): 100% refractory Organ involvement: Infliximab: Diagnosis: Poor General Condition 5 (56%), Ear nose and throat involvement 8 (89%), lung nodules 9 (100%), purpura 2 (22%), arthralgias 3 (33%), scleritis 3 (33%), mononeuritis multiplex 3 (33%), rapidly progressing glomerulonephritis 4 (44%) Inclusion: Poor General Condition 6 (67%), Ear nose and throat involvement 6 (67%), lung nodules 8 (89%), mononeuritis multiplex 1 (11%), rapidly progressing glomerulonephritis 2 (22%), renal insufficiency 1 (11%) Rituximab: Diagnosis: Poor General Condition 6 (75%), Ear nose and throat involvement 4 (50%), lung nodules 5 (62.5%), purpura 1 (12.5%), arthralgias 4 (50%), mononeuritis multiplex 2 (25%), rapidly progressing glomerulonephritis 2 (25%), tracheal stenosis 2 (25%), pericarditis 1 (12.5%), orchitis 1 (12.5%), subcutaneus nodules (12.5%), retroocular pseudotumour 1 (12.5%) Inclusion: Poor General Condition 5 (67.5%), Ear nose and throat involvement 5 (67.5%), lung nodules 4 (50%), mononeuritis multiplex 2 (25%), rapidly progressing glomerulonephritis 2 (25%),pleural effusion 1 (12.5%), tracheal stenosis 2 (25%), subcutaneus nodules 1 (12.5%), retroocular pseudotumour 1 (12.5%) Prior treatment: Not reported ANCA positive in immunofluorescence (14 c‐ANCA and 1 p‐ANCA) at inclusion: infliximab: 6, Rituximab: 7 |
|
| Interventions |
Infliximab group: The initial IV dose (3 mg/kg) administered on days 1 and 14 and the response was assessed on day 42. In case of a complete remission, the dose maintained for the next 6 months. In case of a partial remission or no response ‐ the dose increased to 5 mg/kg, and the therapeutic response re‐evaluated 4 weeks later (day 73). In case of a complete remission after dose increase ‐ the dose unchanged for the rest of the study (14 infusions). In case of no response on day 73 ‐ infliximab stopped. Rituximab group: Administered IV (0.375g/m2) on days 1, 8, 15 and 22. In case of a partial or complete remission at month 2, the same dose maintained for subsequent infusions (months 4, 8 and 12). In case of no response at month 2 ‐ rituximab stopped. Description of treatment and concomitant treatment: Infliximab: One patient (12.5%) was taking corticosteroid (CS) at inclusion, with mean ± SD doses of 19.4 ± 11.8 mg/day for the infliximab group, 3 patients did not receive oral cyclophosphamide (CYC) after failure of IV CYC (cytopenia, haemorrhagic cystitis and high cumulated doses of CYC) Rituximab: One patient was taking CS at inclusion 41.4 ± 28 mg/day for the rituximab group (P = 0.34), 1 patient did not receive oral CYC after IV CYC failure because of high cumulated doses of CYC. |
|
| Outcomes |
Major outcomes:
Minor outcomes:
|
|
| Notes |
Discontinuation: 10 (58.82%) participants (7 participants from infliximab group and 3 participant from rituximab group): 7 (41.17%) due to progressive disease, 1 (5.88%) due to AE, 2 (11.76%) died, Funding: This trial was supported by a grant from the Programme Hospitalier de Recherche Clinique (PHRC 2003 n. P020931). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "After inclusion, patients were randomly assigned to receive either infliximab or rituximab". Authors do not describe methods of sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | Method of concealment is not described. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information how participants and personnel were blinded and the outcome is likely to be influenced by lack or insufficient of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information how participants and personnel were blinded and the outcome is likely to be influenced by lack or insufficient of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing outcome data. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available, so insufficient information to judge if all outcomes reported; outcomes that are of interest in the review have been reported. |
| Other bias | Low risk | No other biases were identified. |
Wechsler 2017.
| Study characteristics | ||
| Methods |
Study design: double‐blinded parallel RCT Location: USA: Boston, Denver, Cleveland, Salt Lake City. Italy: Pisa, Firenze, Milano. France: Montpellier, Marseille, Bron, Paris, Suresnes. UK: Portsmouth, Leicester. Spain: Barcelona. Belgium: Bruxelles. Canada: Hamilton, Toronto. Germany: Kirchheim Gunter Teck, Neumünster, Hessen, Thueringen, Kiel, Freiburg. Japan: Miyagi, Kanagawa. Setting: clinical centres Number of centres: 31 Time frame of the study: February 2014 through June 2015 Duration of follow‐up: continued until September 2016 (8 weeks) |
|
| Participants |
Inclusion criteria:
Exclusion criteria:
Total number of participants: 136 participants randomised, Mepolizumab group n = 68, Placebo group n = 68 Characteristics: Mean age: Mepolizumab 49 ± 12 Placebo 48 ± 14 Female sex: Mepolizumab 42 (62%); Placebo 38 (56%) BMI mean (SD): Mepolizumab 27.5 (4.4); Placebo 28.2 (5.7) CrCl value: not reported Disease duration (time since diagnosis; mean (SD)): Mepolizumab 5.2 (4.4) years; Placebo 5.9 (4.9) years Age at onset of symptoms: not reported Race/ethnic group (%): Mepolizumab white 89.7% other 10.3% Placebo white 94.1% other 5.9% Disease diagnosed at enrolment to the study: not reported Disease assessment BVAS > 0 Mepolizumab 37 (54%) Placebo 48 (71%) VDI mean (SD): Mepolizumab 4.7 (3.4) Placebo 4.4 (2.8) Global assessment: NR Quality of life assessment: NR Disease severity (limited /severe): NR Organ Involvement n (%): Mepolizumab Asthma and eosinophilia 68 (100%), Neuropathy (mono or poly) 32 (47%), Pulmonary infiltrates (non‐fixed) 50 (74%), sinonasal abnormality 64 (94%), cardiomyopathy 13 (19%), glomerulonephritis 1 (1%), alveolar haemorrhage 3 (4%), palpable purpura 9 (13%), biopsy evidence 25 (37%) Placebo Asthma and eosinophilia 68 (100%), Neuropathy (mono or poly) 24 (35%), Pulmonary infiltrates (non fixed) 48 (71%), sinonasal abnormality 64 (94%), cardiomyopathy 7 (10%), glomerulonephritis 0 (0%), alveolar haemorrhage 1 (1%), palpable purpura 8 (12%), biopsy evidence 31 (46%) Prior treatment: Mepolizumab: Prednisolone or prednisone dose — mg/day median (range) 12 (7.5‐40) Immunosuppressive therapy at baseline 41 (60%) Placebo: Prednisolone or prednisone dose — mg/day median 11 (7.5‐50) Immunosuppressive therapy at baseline number (%): 31 (46%) Patients with ANCA laboratory results number (%): Mepolizumab: ANCA positive (MPO or PR3) 13 (19) Placebo: ANCA positive (MPO or PR3) 13 (19) |
|
| Interventions |
Mepolizumab group: mepolizumab 300 mg ‐ 3 separate injections (100 mg each) every 4 weeks, in addition to standard care for 52 weeks. Placebo group: subcutaneously, placebo 0.9% sodium chloride 300 mg every 4 weeks, in addition to standard care for 52 weeks Description of treatment and concomitant treatment: Participants taking glucocorticosteroids during treatment period: Mepolizumab: Prednisone 47 (69%) Prednisolone 22 (32%) Methylprednisolone sodium succinate 6 (9%) Methylprednisolone 4 (6%) Dexamethasone 1 (1%) Hydrocortisone sodium succinate 1 (1%) Hydrocortisone 1 (1%) Triamcinolone acetonide 1 (1%) Placebo: Prednisone 49 (72%) Prednisolone 22 (32%) Methylprednisolone sodium succinate 4 (6%) Methylprednisolone 4 (6%) Dexamethasone 3 (4%) Hydrocortisone sodium succinate 2 (3%) Triamcinolone 1 (1%) |
|
| Outcomes |
Major outcomes:
Minor outcomes:
Other outcomes:
|
|
| Notes |
Discontinuation: 14 (10.29%) participants (9 participants from placebo group and 9 participant from mepolizumab group): 2 (1.47%) due to AEs, 3 (2.21%) lack of efficacy, 3 (2.21%) met protocol‐defined stopping criteria, 1 (0.74%) physician decision, 5 (3.68%) withdrawal from the trial. Funding: Supported by grants from GlaxoSmithKline (115921) and the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (U01AI097073), and by the Division of Intramural Research, NIAID, National Institutes of Health. Ongoing study: Mepolizumab Long‐term Access Programme for Subjects who Participated in Study MEA115921. Placebo‐controlled Study of Mepolizumab in the Treatment of Eosinophilic Granulomatosis with Polyangiitis in Subjects Receiving Standard‐of‐care Therapy (ID: MEA116841, ClinicalTrials.gov Identifier: NCT03298061). A Phase III, multi‐centre, multinational, non‐randomised, open with one arm: mepolizumab 100mg SC. Estimated Primary Completion Date: September 7, 2018 (Final data collection date for major outcome measure) Authors contacted for the data reported only on the graphs and not usable for the reporting in the systematic review ‐ information that the results will be posted on clinicaltrials.gov, but not posted yet. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization was performed with the use of a centralized computer–generated, permuted‐block schedule, stratified according to three subgroups: participation in a mechanistic–biomarker substudy in the United States, recruitment in Japan, and the remainder of recruited participants." |
| Allocation concealment (selection bias) | Low risk | "The randomization schedule was generated using the GSK validated randomization software RandAll. Equal numbers of subjects were allocated to each treatment." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The randomization schedule was sent as a signed, hard copy, controlled document, marked as private, for the attention of the unblinded qualified designee at each centre or sent as a GSK Secure email to the attention of the unblinded qualified designee. Clinicians who were treating and evaluating patients were unaware of the preparation of the trial agents, the trial‐group assignments, and the white‐cell counts and white‐cell differential counts for the duration of the trial." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Procedures must be in place to ensure the blind is maintained by any site staff involved in administration of the drug or clinical care or assessment of the subject, and by the subject themselves." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT was performed and all patients were included in primary analysis, no missing outcome data identified. |
| Selective reporting (reporting bias) | High risk | Not all of the study’s pre‐specified major outcomes have been reported in the publication such as SF‐36 score or WPAI index. ACQ‐6 and SNOT‐22 score were shown on Figures (and read from them). |
| Other bias | Low risk | No other biases was identified. |
WGET 2005.
| Study characteristics | ||
| Methods |
Study design: double‐blinded parallel RCT Location: USA: Michigan, San Francisco, Rochester, Maltimore, Durham, Cleveland, Boston, New York Setting: clinical centres Number of centres: 8 Time frame of the study: June 2000 through September 2003 Duration of follow‐up mean: 27 months |
|
| Participants |
Inclusion criteria:
Exclusion criteria:
Total number of participants: 181 participants randomised. Etanercept group: 89 Placebo group: 91 Characteristics:
|
|
| Interventions | All participants were followed for 12 months after randomization of the last participant. Etanercept group: 25 mg twice a week, subcutaneous; for 25 months (median value) Placebo group: lyophilised powder containing 40 mg of mannitol, 10 mg of sucrose, and 1,2 mg of tris(hydroxymethyl)methylamine twice a week, subcutaneous; for 19 months (median value) Concominant treatment for both groups: ‐ Patients with severe GPA received cyclophosphamide + glucocorticoids at the time of enrolment. Those with limited GPA receive methotrexate and glucocorticoids. After control of the patients’ disease, the standard medications are tapered according to regimens consistent with patient safety to reduce the risk of medication‐associated morbidity and to test the benefit of etanercept in sustaining disease remissions. ‐ All patients received prophylaxis against pneumocystis infection and osteoporosis. |
|
| Outcomes |
Major outcome:
Minor outcomes:
Other outcomes:
|
|
| Notes |
Discontinuation: 69 (38.12%) participants (34 participants from placebo group and 35 participant from etanercept group): 12 (6.63%) due to AEs, 4 (2.21%) died, 20 (11.94%) had treatment failure, 9 (4.97%) physician decision, 18 (9.94%) participant decision, 6 (3.31%) other reasons. Funded by:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Treatment assignments are generated in permuted blocks of varying lengths." |
| Allocation concealment (selection bias) | Low risk | Central allocation. "Patients are assigned to one of six experimental medication “bins”: A1, B2, C3, D4, E5, or F6" (information obtained from the author of the study) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants receive medicinal dosage from the same bin. "The packaging of etanercept and placebo into identical vials". Blinding of study personnel ensured. "One potential source of unmasking among clinical personnel is the occurrence of injection site reactions. Injection site reactions have been reported in both the placebo‐ and etanercept‐assigned groups in other trials, albeit more commonly among etanercept‐assigned patients." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The board is masked to the specific treatment assignment of each group. In the evaluation of all data regarding the harm and benefit, the board receives data relating to treatment group (i.e., either etanercept or placebo) labelled as either treatment “A” or “B.” |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis, however 7% of patients in control group were excluded from primary analysis because lost to follow‐up and 0% were excluded in etanercept group. Including those patients would not have clinically relevant impact on the intervention effect estimate for major outcome |
| Selective reporting (reporting bias) | High risk | One or more outcomes of interest in the review are reported incompletely so that they cannot be entered in a meta‐analysis. |
| Other bias | Low risk | No other biases were identified. |
AE – adverse event
ANCA – antineutrophil cytoplasmic antibody
anti‐MPO – anti‐myeloperoxidase
anti‐PR3 – Anti‐proteinase 3 ‐
BMI – body mass index
BREVAS‐ Belimumab in Remission of VASculitis
BVAS – Birmingham Vasculitis Activity Score
BVAS/WG – Birmingham Vasculitis Activity Score for granulomatosis with polyangiitis
CrCl – creatinine clearance
EGPA – eosinophilic granulomatosis with polyangiitis
EudraCT ‐ European Union Drug Regulating Authorities Clinical Trials Database
EULAR – European League against Rheumatism
GPA – granulomatosis with polyangiitis
HBcAb – hepatitis B core antibody
HBsAg – hepatitis B Surface antigen
HIV ‐ human immunodeficiency virus
ITT – intention to treat
IV – intravenous
MPA – microscopic polyangiitis
NR – not reported
QTc(F) – QT interval corrected using Fridericia formula
RCT – randomized controlled trial
SC – subcutaneous
SD – standard deviation
VDI – Vasculitis Damage Index
WGET ‐ Wegener's Granulomatosis Etanercept Trial
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Anwar 2017 | Wrong study design ‐ not RCT (editorial) |
| Berti 2017 | Wrong study design ‐ not RCT (non‐systematic review) |
| BIANCA‐SC | Withdrawn prior to enrolment |
| Booth 2002 | Wrong study design ‐ not RCT (letter) |
| Booth 2004 | Wrong study design ‐ not RCT (open‐label, multi‐centre, prospective trial, not randomised); patients in both study subgroups received infliximab |
| Bosch 2007 | Wrong study design ‐ not RCT (systematic review) |
| Cattran 2011 | Wrong intervention ‐ mycophenolate mofetil vs azathioprine, not anti‐cytokine drugs was assessed |
| Eculizumab 2011 | Withdrawn (the study failed to enrol any patient and sponsor wished to stop) |
| Fraser 2016 | Wrong study design ‐ not RCT |
| Guillevin 2005 | Wrong study design ‐ not RCT (editorial) |
| Herrmann 2012 | Wrong study design ‐ not RCT; single arm study |
| Jayne 2015 | Wrong study design ‐ not RCT (non‐systematic review) |
| Kahn 2010 | Wrong study design ‐ not RCT (letter to the editor) |
| Kontkanen 2010 | Wrong study design ‐ not RCT (letter to the editor) |
| Krishna 2017 | Wrong study design ‐ not RCT (non‐systematic review) |
| Lamprecht 2004 | Wrong study design ‐ not RCT (letter) |
| Lamprecht 2005 | Wrong study design ‐ not RCT (letter to the editor) |
| Lau 2011 | Wrong study design ‐ not RCT (case report) |
| Lee 2008 | Wrong study design ‐ not RCT (non‐systematic review) |
| Lutalo 2015 | Wrong study design ‐ not RCT (non‐systematic review) |
| MEPOCHUSS 2006 | Wrong study design, not RCT, single arm study |
| Merkel 2013 | Wrong study design ‐ not RCT (non‐systematic review) |
| Moosig 2011 | Wrong study design ‐ not RCT (clinical trial, phase II, letter) |
| Moosig 2013 | Wrong study design ‐ not RCT (non‐systematic review) |
| Mukhtyar 2011 | Wrong study design ‐ not RCT |
| Mukhtyar 2016 | Wrong study design ‐ not RCT (abstract of recommendation) |
| Murgia 2014 | Wrong study design ‐ not RCT (non‐systematic review) |
| Niles 2017 | Wrong study design ‐ not RCT (prospective, observational safety study) |
| Puéchal 2017 | Wrong study design ‐ not RCT (non‐systematic review) |
| Rhee 2018 | Wrong study design ‐ post hoc analysis |
| Rheumatology Annual Meeting 2013 | Wrong study design ‐ not RCT |
| Rozin 2003 | Wrong study design ‐ not RCT (letter to the editor) |
| Ruppert 2008 | Wrong study design ‐ not RCT (letter to the editor) |
| Saadoun 2016 | Wrong study design ‐ not RCT (editorial) |
| Sakai 2015 | Wrong study design ‐ not RCT (case report) |
| Samson 2013 | Wrong study design ‐ not RCT, mixed participants with eosinophilic granulomatosis with polyangiitis, polyarteritis nodosa and microscopic polyangiitis from prospective studies |
| Samson 2014 | Wrong intervention ‐ intervention included azathioprine versus cyclophosphamide, not anti‐cytokine drugs |
| Scherer 2006 | Wrong study design ‐ not RCT (non‐systematic review) |
| Seror 2010 | Wrong intervention ‐ intervention included methotrexate versus azathioprine, not anti‐cytokine drugs. Results from pre‐randomisation phase of the prospective multicentre randomised open‐label Wegener’s Granulomatosis‐Entretien (WEGENT) trial |
| Silva 2012 | Wrong study design ‐ not RCT |
| Silva 2013 | Wrong study design ‐ not RCT |
| Silva‐Fernandez 2014 | Wrong study design ‐ not RCT (systematic review which include systematic reviews and meta‐analysis, clinical trials, cohort studies, and case series) |
| Singer 2017 | Wrong study design ‐ not RCT (non‐systematic review) |
| Solans 2008 | Wrong study design ‐ not RCT (editorial) |
| Spina 2009 | Wrong study design ‐ not RCT (letter which describes case report) |
| Tanaka 2012 | Wrong study design ‐ not RCT (non‐systematic review) |
| Watts 2010 | Wrong study design ‐ not RCT (preliminary results of randomised, placebo‐controlled trial of rituximab therapy) |
| Westman 2013 | Wrong study design ‐ not RCT (conference abstract of non‐systematic review) |
BIANCA‐SC ‐ A Study of the Efficacy, Safety, and Tolerability of Blisibimod in Addition to Methotrexate During Induction of Remission in Subjects With ANCA‐Associated Small Vessel Vasculitis
MEPOCHUSS ‐ Safety and Efficacy Study of Mepolizumab in Churg Strauss Syndrome
RCT – randomized controlled trial
Characteristics of studies awaiting classification [ordered by study ID]
ABAVAS 2008.
| Methods | A phase III, multi‐centre, randomised, double‐blind, placebo‐controlled trial with two arms: 1. arm ‐ abatacept 250 mg intravenous 2. arm ‐ placebo |
| Participants |
Inclusion criteria:
Exclusion criteria:
abatacept any time investigational drug within 28 days (or >5 terminal half‐lives) currently treated with biological agent methotrexate within 3 months rituximab, anti‐TNF therapy, or IL‐1 receptor antagonists within last year cyclophosphamide within last six months. |
| Interventions | Arm 1: Abatacept 500 mg for patients under 60kg 750mg for patients 60‐100kg 1g for patients > 100kg given as IV infusion over 30 minutes at day 0, 14, 28 and then monthly for a further 11 months 914 infusions in total) Arm 2: saline placebo only IV |
| Outcomes | Major outcome:
Minor outcomes:
|
| Notes | Contact person: Alan Salama Imperial College London ClinicalTrials.gov Identifier: NCT00482066 EudraCT number 2006‐001859‐35 |
NIAID 1999.
| Methods | No information available; study data were never published |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes |
NIAID 2002.
| Methods | No information available; study data were never published |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes |
AAV – Anti‐neutrophilic cytoplasmic antibodies (ANCA)‐associated vasculitis
ABAVAS ‐ Abatacept in ANCA Associated Vasculitis
ANCA – antineutrophil cytoplasmic antibody
anti‐MPO – anti‐myeloperoxidase
anti‐PR3 – Anti‐proteinase 3 ‐
BVAS – Birmingham Vasculitis Activity Score
ELISA – enzyme‐bound immunosorbent analysis
HIV – human immunodeficiency virus
IV – intravenous
MCP‐1 – monocyte chemoattractant protein‐1
NIAID – National Institute of Allergy and Infectious Diseases
Characteristics of ongoing studies [ordered by study ID]
AAVTCZ.
| Study name | Clinical trial of tocilizumab versus cyclophosphamide for microscopic polyangiitis and granulomatosis with polyangiitis |
| Methods | A phase II, randomised, open ‐ but assessors are blinded, controlled trial with two arms: 1. arm: intravenous tocilizumab (TCZ) plus high dose glucocorticoids 2. arm: intravenous cyclophosphamide (IVCY) plus high dose glucocorticoids |
| Participants |
Inclusion criteria:
Exclusion criteria:
|
| Interventions | Arm 1 Week 0‐16: tocilizumab (8 mg/kg) IV every 2 weeks. Week 20 and 24: tocilizumab (8 mg/kg) IV every 4 weeks. If no response (a participant does not achieve BVAS v3=0) at week 16, tocilizumab continued every 2 weeks until week 24. Week 28‐52: If a complete remission achieved at week 24, tocilizumab (8 mg/kg) continued IV every 4 weeks until week 48. Arm 2 Week 0‐24: cyclophosphamide (15 mg/kg, doses modified for renal dysfunction) IV (IVCY) every 4 weeks (at least 3 times and up to 6 times). 4 weeks after the last dose of IVCY to week 52: If a complete remission achieved azathioprine orally every day until week 52. Prednisolone (PSL) prescribed by the same schedule to both treatment groups. Week 0‐24: Oral PSL at a dose of 0.8 mg/kg/day for the first 4 weeks, then tapered according to the prefixed schedule. Week 25‐52: Continued oral PSL at a dose of 7.5mg per day. |
| Outcomes | Major outcome measures:
Minor outcome measures:
|
| Starting date | 1 May 2018 (date of first enrolment) |
| Contact information | Masayoshi Harigai ‐ Tokyo women's medical university Institute of Rheumatology harigai.masayoshi@twmu.ac.jp UMIN000024574 |
| Notes |
ABROGATE.
| Study name | Abatacept (CTLA4‐Ig) for the Treatment of Relapsing, Non‐Severe, Granulomatosis With Polyangiitis (Wegener's) (ABROGATE) |
| Methods | A phase III, multi‐centre, randomised, double‐blind, placebo‐controlled trial with two arms: 1. arm: 125 mg abatacept 2. arm: placebo administered by subcutaneous injection once a week. |
| Participants | Inclusion criteria:
Exclusion criteria: 1. Presence of involvement that meets the criteria for severe disease. 2. Treatment prior to screening with: Cyclophosphamide within 3 months, Methylprednisolone 1000 mg within 28 days, Prednisone/prednisolone > 30 mg/day for > 28 days. 3. Begininng or increasing the dose of the immunosuppressive agent used for the disease maintenance (methotrexate, azathioprine, mycophenolate) within 3 months before the study. 4. Active infection (including chronic infection). 5. Pregnant or nursing. 6. HIV, hepatitis C positive status, or a positive hepatitis B surface antigen. 7. Inability to comply with study guidelines. 8. Cytopenia: platelet count below 100,000/mm3, white blood cell count below 3,000/mm3 (3 x 109/L), absolute neutrophil count below 1500/mm3, haemoglobin below 8.5 g/dL. 9. Chronic renal insufficiency with a creatinine clearance of <= 20 ml/min. 10. Known current use of illegal drugs. 11. Other uncontrolled disease (co‐morbidity) that could interfere with fulfilling the study requirements or substantially increase the risk of study procedures. 12. Any previous or current history of cancer except properly treated basal cell or squamous cell carcinomas of the skin, or cervical carcinoma in situ treated or excised in a curative procedure. 13. Investigational agent or device within 30 days before the study or 5 half lives of the drug (whichever is longer). 14. A live vaccination <3 months before enrolment. 15. Current active tuberculosis (clinical, radiographic, or laboratory evidence). 16. Any history of active tuberculosis within the past 3 years and >3 years ago if proper treatment not documented. 17. Latent tuberculosis not properly treated or currently treated with isoniazid or other therapy recommended in the guidelines for up to 4 weeks before the study. Participants with a positive tuberculosis screening test indicative of latent tuberculosis can be eligible for the study if there is no evidence of current tuberculosis on chest X‐ray at screening and they are receiving treatment with isoniazid or other therapy for latent tuberculosis recommended in the guidelines (e.g., CDC) for at least 4 weeks before the study and complete this treatment according to the guidelines. 20. History of herpes zoster resolved <2 months before the study. 21. Treatment with: Rituximab or any other biologic agent depleting B cell within the last 6 months Alemtuzumab or anti‐thymocyte globulin within the past 12 months. Intravenous immunoglobulin or plasma exchange within the last 3 months. Infliximab, etanercept, adalimumab, tocilizumab, or any other biologics within the last 3 months or 5 half lives of the agent (whichever is longer). |
| Interventions | Arm 1 abatacept 125 mg administered by subcutaneous injection once a week for at least 12 months. Participants may be removed from treatment earlier due to a disease relapse, disease worsening, or if they have not achieved remission by treatment month 6. Arm 2 blinded placebo administered by subcutaneous injection once a week for at least 12 months. Participants may be removed from treatment earlier due to a disease relapse, disease worsening, or if they have not achieved remission by treatment month 6. |
| Outcomes | Major outcome measures:
Minor outcome measures:
|
| Starting date | April 2015 |
| Contact information | Cristina Burroughs 1‐888‐772‐8315 abrogate@epi.usf.edu |
| Notes | Estimated Primary Completion Date: August 2019 (Final data collection date for major outcome measure) ClinicalTrials.gov Identifier: NCT02108860 |
ALEVIATE.
| Study name | Alemtuzumab for ANCA Associated Refractory Vasculitis ‐ a Study of Safety and Efficacy |
| Methods | A open label, randomised, multi‐centre study with two arms: 1. arm: Alemtuzumab ‐ high dose (60mg) 2. arm: Alemtuzumab ‐ low dose (30mg) |
| Participants |
Inclusion criteria:
Exclusion criteria:
Any alemtuzumab Within the past 3 months: IVIg, infliximab, etanercept, adalimumab, abatacept, anti‐thymocyte globulin or plasma exchange Within the past 6 months: rituximab. 7. Required intensive care unit treatment. 8. Active infection with HIV, hepatitis B or hepatitis C or other infection which requires parenteral or long‐term oral antibiotics. 9. History of idiopathic thrombocytopenic purpura or platelet count below 50,000 x 106/l at screening. 10. Pregnancy or nursing or not adequate contraception in pre‐menopausal women. 11. Any condition which can be associated with detrimental effect of the study to the participant. 12. Any other multisystem autoimmune disease, such as Churg Strauss angiitis, systemic lupus erythematosus,anti‐glomerular basement membrane disease and cryoglobulinaemia. 13. Any previous or current history of cancer (other than resected basal cell carcinoma). |
| Interventions | Arm 1: Alemtuzumab (Campath 1H)‐ high dose (60mg) ‐ Alemtuzumab will be administered on Day 1 and Day 2 at 0 and 6 months. Arm 2: Alemtuzumab (Campath 1H)‐ low dose (30mg) ‐ Alemtuzumab will be administered on Day 1 and Day 2 at 0 and 6 months. |
| Outcomes | Major outcome:
Minor outcomes:
|
| Starting date | February 2011 |
| Contact information | David Jayne FMedSci Professor of Clinical Autoimmunity Department of Medicine School of Clinical Medicine University of Cambridge CB2 2QQ Tel 01223 325039 dj106@cam.ac.uk |
| Notes | We received information that the trial has completed recruitment but follow‐up is ongoing. |
COMBIVAS.
| Study name | A Randomised, Double Blind, Controlled Mechanistic Study of Rituximab and Belimumab Combination Therapy in PR3 ANCA‐associated Vasculitis |
| Methods | A phase II randomised, double‐blind, multi‐centre study with two arms: 1. arm: 200 mg belimumab with rituximab 2. arm: rituximab with placebo administered by subcutaneous injection once a week |
| Participants | nclusion Criteria:
Exclusion Criteria: At screening:
Any B cell targeted therapy within 364 days, Cyclophosphamide within 180 days, Any steroid injection within 60 days (unless provided during or 14 days before screening period) Methylprednisolone (IV) >1.5mg between 14 days prior to screening and Day 1 (including Day 1). 8. Prednisolone on average >10mg/day (or equivalent) orally during 30 days prior to screening. |
| Interventions | Arm 1 Belimumab (Benlysta) 200 mg administered by subcutaneous injection once a week for 12 months co‐administration of rituximab Arm 2 Placebo administered by subcutaneus injection once a week for 12 months. co‐administration of rituximab |
| Outcomes | Major outcome measures:
Minor outcomes measures:
|
| Starting date | February 2019 |
| Contact information | Kim Mynard 01223 349350 kim.mynard@addenbrookes.nhs.uk Principal Investigator: Rachel Jones |
| Notes | Estimated Primary Completion Date: February 2022 (Final data collection date for major outcome measure) ClinicalTrials.gov Identifier: NCT03967925 |
AAV – Anti‐neutrophilic cytoplasmic antibodies (ANCA)‐associated vasculitis
AAVTCZ ‐ Clinical trial of tocilizumab versus cyclophosphamide for microscopic polyangiitis and granulomatosis with polyangiitis
ABROGATE ‐ Abatacept for the Treatment of Relapsing, Non‐Severe, Granulomatosis With Polyangiitis
ACR – American College of Rheumatology
AE – adverse event
ALEVIATE ‐ Alemtuzumab for ANCA Associated Refractory Vasculitis
ANCA – antineutrophil cytoplasmic antibody
anti‐MPO – anti‐myeloperoxidase
anti‐PR3 – Anti‐proteinase 3 ‐
BVAS – Birmingham Vasculitis Activity Score
BVAS/WG – Birmingham Vasculitis Activity Score for granulomatosis with polyangiitis
CDC – Centre for Disease Control
COMBIVAS ‐ A Randomised, Double Blind, Controlled Mechanistic Study of Rituximab and Belimumab Combination Therapy in PR3 ANCA‐associated Vasculitis
CS – corticosteroid
DNA – deoxyribonucleic acid
eGFR – Estimated glomerular filtration rate
EGPA – eosinophilic granulomatosis with polyangiitis
ELISA – enzyme‐bound immunosorbent analysis
EQ5D – The EuroQol (European Quality of Life) Five Dimension Scale
EUVAS ‐ European Vasculitis Study Group
GPA – granulomatosis with polyangiitis
HB ‐ hepatitis B
HBV ‐ hepatitis B virus
HIV – human immunodeficiency virus
IV – intravenous
IVCY – intravenous cyclophosphamide
Kg – kilogram
MCP‐1 – monocyte chemoattractant protein‐1
MPA – microscopic polyangiitis
PSL ‐ prednisolone
SAE – serious adverse event
SF‐36 – Short Form‐36
TCZ – tocilizumab
Differences between protocol and review
In the protocol we indicated that type of AAV will be a subject of subgroup analysis. However, we decided not to pool the results from two types of AAV together (i.e. GPA and EGPA) due to their clinical heterogeneity, as it would not be clinically meaningful to combine the results and derive conclusions for all types of AAV on the basis of studies targeted to two separate populations of AAV (i.e. not mixed).
Contributions of authors
MMB, TMM and WS developed the concept of the study; JZ, MK and MMB developed the search strategy. All review authors contributed to preparation of the protocol and agreed upon this final version of the review.
We thank the authors of the previous version of this protocol for their contributions.
Sources of support
Internal sources
Jagiellonian University Medical College, Poland
External sources
No sources of support supplied
Declarations of interest
We are not aware of any direct conflicts of interest.
TMM, JZ, and JDJ declared no conflicts of interest.
MMB receives honoraria (as a freelancer) from a company that works for pharmaceutical companies.
MK received honoraria for articles with topics not related to the topic of the review from a publishing company which does work for several pharmaceutical companies.
WS participates in a trial sponsored by a pharmaceutical company that is not related to vasculitis but to non‐cardiac surgery.
New
References
References to studies included in this review
BREVAS 2018 {published data only}
- Jayne D, Blockmans D, Luqmani R, Ji B, Green Y, et al. AB0692 Belimumab in combination with azathioprine for remission maintenance in granulomatosis with polyangiitis and microscopic polyangiitis: effect on biomarkers. In: Annals of the Rheumatic Diseases. Vol. 77. 2018:1488.
- Jayne D, Blockmans D, Luqmani R, Moiseev S, Green Y, et al. Efficacy and Safety of Belimumab and Azathioprine for Maintenance of Remission in Antineutrophil Cytoplasmic Antibody-Associated Vasculitis: A Randomized Controlled Study. Arthritis Rheumatol 2019;71(6):952‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayne D, Blockmans D, Luqmani, R, Ji B, Green Y, Hall L, et al. Efficacy and safety of belimumab in combination with azathioprine for remission maintenance in granulomatosis with polyangiitis and microscopic polyangiitis: A multicenter randomized, placebo-controlled study.. In: Arthritis and Rheumatology. Vol. 69. 2017.
- NCT01663623. A Phase 3, Multi-Center, Multinational, Randomized, Double-Blind, Study to Evaluate the Efficacy and Safety of Belimumab (HGS1006) in Combination With Azathioprine for the Maintenance of Remission in Wegener's Granulomatosis and Microscopic Polyangiitis. clinicaltrials.gov/ct2/show/study/NCT01663623?term=BREVAS&rank=1 (first received 13 August 2012).
de Menthon 2011 {published data only}
- Menthon M, Cohen P, Pagnoux C, Buchler M, Sibilia J, Détrée F, et al. Infliximab or rituximab for refractory Wegener's granulomatosis: long-term follow up. A prospective randomised multicentre study on 17 patients. Clinical and Experimental Rheumatology 2011;29(1 suppl 64):S63-71. [PubMed] [Google Scholar]
Wechsler 2017 {published data only}
- Krishna R, West FM, Zappetti D. Is Anti-IL 5 therapy effective in eosinophilic granulomatosis with polyangiitis? Clinical Pulmonary Medicine 2017;24(6):267-8. [Google Scholar]
- Smith R. Mepolizumab for the treatment of eosinophilic granulomatosis with polyangiitis. Rheumatology 2017;56:iii15. [Google Scholar]
- Steinfeld J, Bradford ES, Brown J, Mallett S, Yancey SW, Wechsler ME. Mepolizumab for the treatment of patients with eosinophilic granulomatosis with polyangiitis: Post-hoc results of a phase iii randomized, placebo-controlled trial. In: Arthritis Rheumatol. Vol. 69. 2017.
- Steinfeld J, Mallett S, Brown J, Bradford E S, Yancey S, Wechsler M. Eosinophilic Granulomatosis with Polyangiitis Patient Characteristics and Clinical Response to Mepolizumab Compared to Placebo. American Journal of Respiratory and Critical Care Medicine 2018;197:A3012. [Google Scholar]
- Steinfeld J, Bradford ES, Brown J, Mallett S, Yancey SW, Akuthota P, et al. Evaluation of clinical benefit from treatment with mepolizumab for patients with eosinophilic granulomatosis with polyangiitis. J Allergy Clin Immunol 2019 Jun;143(6):2170-2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler ME, Akuthota P, Jayne D, Khoury P, Klion A, Langford C, et al. Mepolizumab for the treatment of patients with eosinophilic granulomatosis with polyangiitis: A phase iii randomized, placebo-controlled trial. American Journal of Respiratory and Critical Care Medicine 2017;195:107. [Google Scholar]
- Wechsler ME, Akuthota P, Jayne D, Khoury P, Klion A, Langford CA, Merkel PA, Moosig F, Specks U, Cid MC, Luqmani R, Brown J, Mallett S, Philipson R, Yancey SW, Steinfeld J, Weller PF, Gleich GJ, EGPA Mepolizumab Study Team. Mepolizumab or Placebo for Eosinophilic Granulomatosis with Polyangiitis. The New England Journal of Medicine 2017;376(20):1921-1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
WGET 2005 {published data only}
- Performance of the Birmingham Vasculitis Activity Score for Wegener's Granulomatosis (BVAS/WG) in a randomized clinical trial. Cleveland Clinic Journal of Medicine 2002;69:SII-SSI.
- Seo P, Min YI, Holbrook JT, Hoffman GS, Merkel PA, Spiera R, et al. Damage caused by Wegener's granulomatosis and its treatment: prospective data from the Wegener's Granulomatosis Etanercept Trial (WGET). Arthritis and Rheumatism 2005;52(7):2168-78. [DOI] [PubMed] [Google Scholar]
- Silva F, Seo P, Schroeder DR, Stone JH, Merkel PA, Hoffman GS, et al. Solid malignancies among etanercept-treated patients with granulomatosis with polyangiitis (Wegener’s) long-term follow-up of a multicenter longitudinal cohort. Arthritis & Rheumatology 2011;63(8):2495-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JH, Holbrook JT, Marriott MA, Tibbs AK, Sejismundo LP, Min YI, et al. Solid malignancies among patients in the Wegener's Granulomatosis Etanercept Trial. Arthritis and Rheumatism 2006;54(5):1608-18. [DOI] [PubMed] [Google Scholar]
- WGET Research Group. Design of the Wegener's Granulomatosis Etanercept Trial (WGET). Controlled Clinical Trials 2002;23(4):450-68. [DOI] [PubMed] [Google Scholar]
- Wegener's Granulomatosis Etanercept Trial Research, Group. Etanercept plus standard therapy for Wegener's granulomatosis. The New England Journal of Medicine 2005;352(4):351-61. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Anwar 2017 {published data only}
- Anwar S, Karim MY. Update on systemic vasculitides. Journal of Clinical Pathology 2017;70:476-82. [DOI] [PubMed] [Google Scholar]
Berti 2017 {published and unpublished data}
- Berti A, Specks U, Keogh KA, Cornec D. Current and future treatment options for eosinophilic granulomatosis with polyangiitis. Current Treatment Options in Rheumatology 2017;3(4):193–206. [Google Scholar]
BIANCA‐SC {unpublished data only}
- BIANCA-SC: A Study of the Efficacy, Safety, and Tolerability of Blisibimod in Addition to Methotrexate During Induction of Remission in Subjects With ANCA-Associated Small Vessel Vasculitis (BIANCA-SC). Available from: https://clinicaltrials.gov/ct2/show/NCT01598857 2012.
Booth 2002 {published data only}
- Booth AD, Jefferson HJ, Ayliffe W, Andrews PA, Jayne DR. Safety and efficacy of TNFalpha blockade in relapsing vasculitis. Annals of the Rheumatic Diseases 2002;61(6):559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Booth 2004 {published data only}
- Booth A, Harper L, Hammad T, Bacon P, Griffith M, Levy J, et al. Prospective study of TNFalpha blockade with infliximab in anti-neutrophil cytoplasmic antibody-associated systemic vasculitis. Journal of the American Society of Nephrology 2004;15(3):717-21. [DOI] [PubMed] [Google Scholar]
Bosch 2007 {published data only}
- Bosch X, Guilabert A, Espinosa G, Mirapeix E. Treatment of antineutrophil cytoplasmic antibody associated vasculitis: a systematic review. JAMA 2007;298(6):655-69. [DOI] [PubMed] [Google Scholar]
Cattran 2011 {published data only}
- Cattran DC, Hladunewich MA. Maintenance immunosuppression in antineutrophil cytoplasmic antibody - associated vasculitis. American Journal of Kidney Diseases 2011;57(6):818-21. [DOI] [PubMed] [Google Scholar]
Eculizumab 2011 {published data only}
- Targeting Complement Activation in Antineutrophil Cytoplasmic Autoantibodies (ANCA)-Vasculitis. Clinicaltrials.gov February 23, 2017. [https://www.clinicaltrials.gov/ct2/show/NCT01275287?id=NCT01275287&rank=1]
Fraser 2016 {published data only}
- Fraser D, Boyle S, Amft N. Perianal Crohn Disease after Treatment with Rituximab for Active Granulomatosis with Polyangiitis. The Journal of Rheumatology 2016;43(12):2199-2200. [DOI] [PubMed] [Google Scholar]
Guillevin 2005 {published data only}
- Guillevin L. Are anti-TNFAb indicated in systemic necrotizing vasculitides? [Faut-il prescrire des anti-TNF dans les vascularites necrosantes?]. La Revue de Médecine Interne 2005;26(10):769-70. [DOI] [PubMed] [Google Scholar]
Herrmann 2012 {published data only}
- Herrmann K, Gross WL, Moosig F. Extended follow-up after stopping mepolizumab in relapsing/refractory Churg-Strauss syndrome. Clinical and Experimental Rheumatology 2012;30(1 Suppl 70):S62-5. [PubMed] [Google Scholar]
Jayne 2015 {published data only}
- Jayne D, Rasmussen N. Twenty-five years of European Union collaboration in ANCA-associated vasculitis research. Nephrology Dialysis Transplantation 2015;30:i1-7. [DOI] [PubMed] [Google Scholar]
Kahn 2010 {published data only}
- Kahn JE, Grandpeix-Guyodo C, Marroun I, Catherinot E, Mellot F, Roufosse F, et al. Sustained response to mepolizumab in refractory Churg-Strauss syndrome. The Journal of Allergy and Clinical Immunology 2010;125(1):267-70. [DOI] [PubMed] [Google Scholar]
Kontkanen 2010 {published data only}
- Kontkanen M, Paimela L, Kaarniranta K. Regression of necrotizing scleritis in Wegener's granulomatosis after infliximab treatment. Acta Ophthalmologica 2010;88(3):e96-7. [DOI] [PubMed] [Google Scholar]
Krishna 2017 {published data only}
- Krishna R, West FM Zappetti D. Is Anti-IL 5 therapy effective in eosinophilic granulomatosis with polyangiitis? Clinical Pulmonary Medicine 2017;24(6):267-268. [Google Scholar]
Lamprecht 2004 {published data only}
- Lamprecht P, Gross WL. A brief history of Wegener's granulomatosis: on limited, localized, and generalized forms of the disease: comment on the article by the Wegener's Granulomatosis Etanercept Trial Research Group. Arthritis and Rheumatism 2004;50(1):334-6. [DOI] [PubMed] [Google Scholar]
Lamprecht 2005 {published data only}
- Lamprecht P, Voswinkel J, Gross WL. TNF-inhibitors in Wegener's granulomatosis. Kidney & Blood Pressure Research 2005;28(1):62. [DOI] [PubMed] [Google Scholar]
Lau 2011 {published data only}
- Lau MT, Cooper W, Bye PT, Yan K. Difficult asthma and Churg-Strauss-like syndrome: a cautionary tale. Respirology 2011;16(1):180-1. [DOI] [PubMed] [Google Scholar]
Lee 2008 {published data only}
- Lee RW, D'Cruz David P. Novel therapies for anti-neutrophil cytoplasmic antibody-associated vasculitis. Drugs 2008;68(6):747-70. [DOI] [PubMed] [Google Scholar]
Lutalo 2015 {published data only}
- Lutalo MK, D'Cruz David P. Biological drugs in ANCA-associated vasculitis. International Immunopharmacology 2015;27(2):209-12. [DOI] [PubMed] [Google Scholar]
MEPOCHUSS 2006 {published data only}
- 2006-001791-20. A Phase II, single center open label, prospective trial to evaluate the efficacy and safety of mepolizumab for patients with refractory or relapsing Churg Strauss Syndrome. clinicaltrialsregister.eu/ctr-search/trial/2006-001791-20/DE/ (first received 2 January 2008).
Merkel 2013 {published data only}
- Merkel PA. The future of international clinical trials in vasculitis. Presse Medicale 2013;42(4):637-41. [DOI] [PubMed] [Google Scholar]
Moosig 2011 {published data only}
- Moosig F, Gross WL, Herrmann K, Bremer JP, Hellmich B. Targeting interleukin-5 in refractory and relapsing Churg-Strauss syndrome. Annals of Internal Medicine 2011;155(5):341-3. [DOI] [PubMed] [Google Scholar]
Moosig 2013 {published data only}
- Moosig F. Eosinophilic granulomatosis with polyangiitis: future therapies. Presse Medicale 2013;42(4 Pt 2):510-2. [DOI] [PubMed] [Google Scholar]
Mukhtyar 2011 {published data only}
- Mukhtyar C. Clinical trials in ANCA associated vasculitis. Current Immunology Reviews 2011;7(4):358-87. [Google Scholar]
Mukhtyar 2016 {published data only}
- Mukhtyar C. EULAR/ERA-EDTA recommendations for the management of ANCA-associated vasculitis. Annals of the Rheumatic Diseases 2016;75:24. [DOI] [PubMed] [Google Scholar]
Murgia 2014 {published data only}
- Murgia G, Firinu D, Manconi PE, Del Giacco SR. Biologics for ANCA-associated vasculitis. Inflammation & Allergy Drug Targets 2014;13(4):275-87. [DOI] [PubMed] [Google Scholar]
Niles 2017 {published data only}
- Niles J, Allen N, Block JA, Koening CL, Langford CA, Abril A, Lee AS, Merkel PA, Mertz LE, Monach PA, Moreland LW, Nachman PH, Peikert T, Spiera RF, Wallace D, Erblang F, Cascino MD, Duncombe P, Malik V, Brunetta P. Safety following initiation of rituximab in granulomatosis with polyangiitis (GPA) or microscopic polyangiitis (MPA): Interim analysis of the rituximab in ANCA-associated vasculitis registry (RAVER). In: Annals of the Rheumatic Diseases. 2017:318.
Puéchal 2017 {published data only}
- Puéchal X. Biologic treatment prospects in eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Revue du Rhumatisme Monographies 2017;84(3):256-61. [Google Scholar]
Rhee 2018 {published data only}
- Rhee RL, Davis JC, Ding L, Fervenza FC, Hoffman GS, Kallenberg CGM, Langford CA, McCune WJ, Monach PA, Seo P, Spiera R, St Clair EW, Specks U, Stone JH, Merkel PA. The utility of urinalysis in determining the risk of renal relapse in ANCA-associated vasculitis. Clinical Journal of the American Society of Nephrology 2018;13(2):251-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rheumatology Annual Meeting 2013 {published data only}
- British Society for Rheumatology and British Health Professionals. Rheumatology 2013 - British Society for Rheumatology and British Health Professionals in Rheumatology Annual Meeting 2013. In: Rheumatology. Vol. 52. 2013:Suppl 1:i1-173. [PubMed]
Rozin 2003 {published data only}
- Rozin A. Infliximab efficiency in refractory Wegener's granulomatosis. Rheumatology 2003;42(9):1124-6. [DOI] [PubMed] [Google Scholar]
Ruppert 2008 {published data only}
- Ruppert AM, Averous G, Stanciu D, Deroide N, Riehm S, Poindron V, et al. Development of Churg-Strauss syndrome with controlled asthma during omalizumab treatment. The Journal of Allergy and Clinical Immunology 2008;121(1):253-4. [DOI] [PubMed] [Google Scholar]
Saadoun 2016 {published data only}
- Saadoun D, Cacoub P. Moving towards new treatments for vasculitis. Expert Opinion on Emerging Drugs 2016;21(3):239-42. [DOI] [PubMed] [Google Scholar]
Sakai 2015 {published data only}
- Sakai R, Shibata A, Chino K, Kondo T, Okuyama A, Takei H, et al. Corticosteroid- and cyclophosphamide-free treatment of anti-neutrophil cytoplasmic antibody-associated vasculitis using tocilizumab. Modern Rheumatology 2015;25(5):810-1. [DOI] [PubMed] [Google Scholar]
Samson 2013 {published data only}
- Samson M, Puéchal X, Devilliers H, Ribi C, Cohen P, Bienvenu B, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) (EGPA), polyarteritis nodosa (PAN) and microscopic polyangiitis (MPA) with no initial Five-Factor Score-defined poor-prognosis factors (FFS = 0): Baseline factors associated with cytotoxic agent and immunomodulator prescription. Presse Medicale 2013;42(4):666. [Google Scholar]
Samson 2014 {published data only}
Scherer 2006 {published data only}
- Scherer HU, Burmester GR. Biologicals in the treatment of rheumatic diseases [Biologika in der Therapie rheumatischer Erkrankungen]. Deutsche Medizinische Wochenschrift 2006;131(41):2279-85. [DOI] [PubMed] [Google Scholar]
Seror 2010 {published data only}
- Seror R, Pagnoux C, Ruivard M, Landru I, Wahl D, Riviere S, et al. Treatment strategies and outcome of induction-refractory Wegener's granulomatosis or microscopic polyangiitis: analysis of 32 patients with first-line induction-refractory disease in the WEGENT trial. Annals of the Rheumatic Diseases 2010;69(12):2125-30. [DOI] [PubMed]
Silva 2012 {published data only}
- Silva F, Cisternas M, Specks Ul. TNF-alpha blocker therapy and solid malignancy risk in ANCA-associated vasculitis. Current Rheumatology Reports 2012;14(6):501-08. [DOI] [PubMed] [Google Scholar]
Silva 2013 {published data only}
- Silva L, Blanco R, Martínez-Taboada V, Loza E, Pego JM, Rúa-Figueroa I, et al. AB0761 TNF inhibitors for the treatment of ANCA associated vasculitis: A systematic review. In: Annals of the Rheumatic Disease. Vol. 71. 2013:682.
Silva‐Fernandez 2014 {published data only}
- Silva-Fernandez L, Loza E, Martinez-Taboada VM, Blanco R, Rua-Figueroa I, Pego-Reigosa JM, et al. Biological therapy for systemic vasculitis: a systematic review. Seminars in Arthritis and Rheumatism 2014;43(4):542-57. [DOI] [PubMed] [Google Scholar]
Singer 2017 {published data only}
- Singer O, McCune WJ. Update on maintenance therapy for granulomatosis with polyangiitis and microscopic polyangiitis. Current Opinion in Rheumatology 2017;29(3):248-53. [DOI] [PubMed] [Google Scholar]
Solans 2008 {published data only}
- Solans Laque R, Bosch Gil JA. Anti-TNF alpha agents in vasculitis [Farmacos antifactor de necrosis tumoral en las vasculitis sistemicas.]. Medicina Clínica 2008;130(3):93-4. [DOI] [PubMed] [Google Scholar]
Spina 2009 {published data only}
- Spina MF, Miadonna AA. Role of omalizumab and steroids in Churg-Strauss syndrome. Journal of Allergy and Clinical Immunology 2009;124(3):600-1. [DOI] [PubMed] [Google Scholar]
Tanaka 2012 {published data only}
- Tanaka T, Ogata A, Shima Y, Narazaki M, Kumanogoh A, Kishimoto T. Therapeutic implications of tocilizumab, a humanized anti-interleukin-6 receptor antibody, for various immune-mediated diseases: An update review. Current Rheumatology Reviews 2012;8(3):209-26. [Google Scholar]
Watts 2010 {published data only}
- Watts RA. Vasculitis syndromes: Inducing remission in ANCA-positive vasculitis: time to RAVE? Nature reviews. Rheumatology 2010;6(3):127-128. [DOI] [PubMed] [Google Scholar]
Westman 2013 {published data only}
- Westman K. SP0067 Long term outcomes in systemic vasculitis. In: Annals of the Rheumatic Disease. Vol. 71. 2013:17.
References to studies awaiting assessment
ABAVAS 2008 {published data only}
- NCT00482066. A Pilot Study Examining the Effect of Abatacept in ANCA Associated Vasculitis. clinicaltrials.gov/ct2/show/NCT00482066 (first received 4 June 2007).
NIAID 1999 {published data only}
- NCT00001901. Phase I/II Trial of TNFR:Fc (Etanercept) in Patients With Wegener's Granulomatosis. clinicaltrials.gov/ct2/show/NCT00001901 (first received 4 November 1999).
NIAID 2002 {published data only}
- NCT00040248. A Randomized Trial Examining the Use of Daclizumab in Wegener's Granulomatosis. clinicaltrials.gov/ct2/show/NCT00040248 (first received 24 June 2002).
References to ongoing studies
AAVTCZ {published data only}
- JPRN-UMIN000024574. Clinical trial of tocilizumab versus cyclophosphamide for microscopic polyangiitis and granulomatosis with polyangiitis. apps.who.int/trialsearch/Trial2.aspx?TrialID=JPRN-UMIN000024574 (first received 1 May 2018).
ABROGATE {published data only}
- NCT02108860. Abatacept (CTLA4-Ig) for the Treatment of Relapsing, Non-Severe, Granulomatosis With Polyangiitis (Wegener's) (ABROGATE). clinicaltrials.gov/ct2/show/NCT02108860 (first received 9 April 2014).
ALEVIATE {published data only}
- Gopaluni S, Goymer D, McClure M, Cahill H, Broadhurst E, Smith, R, et al. Alemtuzumab for relapsing and refractory primary systemic vasculitis - A trial of efficacy and safety (aleviate): A randomised open-label phase ii clinical trial. In: Rheumatology. Vol. 58 Suppl 2. 2019:kez063.026.
- NCT01405807. Alemtuzumab for ANCA Associated Refractory Vasculitis - a Study of Safety and Efficacy. clinicaltrials.gov/ct2/show/NCT01405807?id=NCT01405807&rank=1 (first received 29 July 2011).
COMBIVAS {published data only}
- NCT03967925. Rituximab and Belimumab Combination Therapy in PR3 COMBIVAS. https://clinicaltrials.gov/ct2/show/NCT03967925?cond=ANCA+Associated+Vasculitis&draw=2&rank=12.
Additional references
Al‐Bishri 2005
- Al-Bishri J, le Riche N, Pope JE. Refractory polyarteritis nodosa successfully treated with infliximab. Journal of Rheumatology 2005;32(7):1371-3. [PubMed] [Google Scholar]
Berli 2015
- Berti A, Cavalli G, Campochiaro C, Guglielmi B, Baldissera E, Cappio S, et al. Interleukin-6 in ANCA-associated vasculitis: rationale for successful treatment with tocilizumab. Seminars in Arthritis and Rheumatism 2015 Aug;45(1):48-54. [DOI] [PubMed] [Google Scholar]
Cates 2008 [Computer program]
- Visual Rx. Version 3. Dr. Christopher Cates EBM web site, 2008. Available at nntonline.net.
Chen 2006
- Chen YF, Jobanputra P, Barton P, Jowett S, Bryan S, Clark W, et al. A systematic review of the effectiveness of adalimumab, etanercept and infliximab for the treatment of rheumatoid arthritis in adults and an economic evaluation of their cost-effectiveness. Health Technology Assessment 2006;10(42):1-229. [DOI] [PubMed] [Google Scholar]
Cohen 2002
- Cohen S, Hurd E, Cush J, Schiff M, Weinblatt ME, Moreland LW, et al. Treatment of rheumatoid arthritis with anakinra, a recombinant human interleukin-1 receptor antagonist, in combination with methotrexate: results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled trial [see comment]. Arthritis & Rheumatism 2002;46(3):614-24. [DOI] [PubMed] [Google Scholar]
Cotch 1996
- Cotch MF, Hoffman GS, Yerg DE, Kaufman GI, Targonski P, Kaslow RA. The epidemiology of Wegener's granulomatosis. Estimates of the five-year period prevalence, annual mortality, and geographic disease distribution from population-based data sources. Arthritis & Rheumatism 1996;39(1):87-92. [DOI] [PubMed] [Google Scholar]
Cottin 2017
- Cottin V, Bel E, Bottero P, Dalhoff K, Humbert M, Lazor R, et al. Revisiting the systemic vasculitis in eosinophilic granulomatosis with polyangiitis (Churg-Strauss): a study of 157 patients by the Groupe d'Etudes et de Recherchesur les Maladies Orphelines Pulmonaires and the European Respiratory SocietyTaskforce on eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Autoimmunity Reviews 2017;16(1):1-9. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG. Chapter 9. Analysing data and undertaking meta-analyses. In: Higgins JPT, Green S editor(s) Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Fauci 1983
- Fauci AS, Haynes BF, Katz P, Wolff SM. Wegener's granulomatosis: prospective clinical and therapeutic experience with 85 patients for 21 years. Annals of Internal Medicine 1983;98:76-85. [DOI] [PubMed] [Google Scholar]
Feldmann 2006
- Feldmann M, Pusey CD. Is there a role for TNF-alpha in anti-neutrophil cytoplasmic antibody-associated vasculitis? Lessons from other chronic inflammatory diseases. Journal of the American Society of Nephrology 2006;17(5):1243-52. [DOI] [PubMed] [Google Scholar]
Ghogomu 2014
- Ghogomu EA, Maxwell LJ, Buchbinder R, Rader T, Pardo Pardo J, et al. Updated method guidelines for Cochrane musculoskeletal group systematic reviews and metaanalyses. Journal of Rheumatology 2014;41(2):194-205. [DOI] [PubMed] [Google Scholar]
Gibson 2006
- Gibson A, Stamp LK, Chapman PT, O'Donnell JL. The epidemiology of Wegener's granulomatosis and microscopic polyangiitis in a Southern Hemisphere region. Rheumatology 2006;45(5):624-8. [DOI] [PubMed] [Google Scholar]
Gonzalez‐Gay 2002
- Gonzalez-Gay MA, Garcia-Porrua C. Systemic vasculitides. Best Practice & Research in Clinical Rheumatology 2002;16(5):833-45. [DOI] [PubMed] [Google Scholar]
GRADEpro GDT 2015 [Computer program]
- McMaster University (developed by Evidence Prime, Inc.) GRADEpro GDT. Version accessed 13 June 2018. Hamilton (ON): McMaster University (developed by Evidence Prime, Inc.), 2015. Available at gradepro.org.
Harper 2001
- Harper L, Radford D, Plant T. IgG from myeloperoxidase–antineutrophil cytoplasmic antibody–positive patients stimulates greater activation of primed neutrophils than IgG from proteinase 3–antineutrophil cytoplasmic antibody–positive patients. Arthritis & Rheumatology 2001;44(4):921-30. [DOI] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Altman DG, Sterne JAC editor(s). Chapter 8. Assessing risk of bias in included studies. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from cochrane.handbook.org.
Higgins 2011b
- Higgins JPT, Deeks JJ, Altman DG (editors). Chapter 16: Special topics in statistics. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org.
Higgins 2011c
- Higgins JPT, Deeks JJ (editors). Chapter 7: Selecting studies and collecting data. Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011).
Jayne 2003
- Jayne D, Rasmussen N, Andrassy K, Bacon P, Tervaert JWC, Dadoniene J, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. New England Journal of Medicine 2003;349(1):36-44. [DOI] [PubMed] [Google Scholar]
Jeanette 2013
- Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis & Rheumatism 2013;65(1):1-11. [DOI] [PubMed] [Google Scholar]
Kamesh 2002
- Kamesh L, Harper L, Savage COS. ANCA-positive vasculitis. Journal of the American Society of Nephrology 2002;13:1953-60. [DOI] [PubMed] [Google Scholar]
Katsuyama 2014
- Katsuyama T, Sada KE, Makino H. Current concept and epidemiology of systemic vasculitides. Allergology International 2014;63(4):505-13. [DOI] [PubMed] [Google Scholar]
Kim 2010
- Kim S, Marigowda G, Oren E, Israel E, Wechsler ME. Mepolizumab as a steroid-sparing treatment option in patients with Churg-Strauss syndrome. Journal of Allergy and Clinical Immunology 2010;125(6):1336-43. [DOI] [PubMed] [Google Scholar]
Knight 2004
- Knight A, Askling J, Granath F, Sparen P, Ekbom A. Urinary bladder cancer in Wegener's granulomatosis: risks and relation to cyclophosphamide. Annals of the Rheumatic Diseases 2004;63(10):1307-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lamprecht 2002
- Lamprecht P, Voswinkel J, Lilienthal T, Nolle B, Heller M, Gross WL, et al. Effectiveness of TNF-alpha blockade with infliximab in refractory Wegener's granulomatosis. Rheumatology 2002;41(11):1303-7. [DOI] [PubMed] [Google Scholar]
Mahr 2004
- Mahr A, Guillevin L, Poissonnet M, Segolene A. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener's granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: a capture-recapture estimate. Arthritis & Rheumatism 2004;51(1):92-9. [DOI] [PubMed] [Google Scholar]
Mahr 2014
- Mahr A, Moosig F, Neumann T, Szczeklik W, Taillé C, Vaglio A, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): evolutions in classification, etiopathogenesis, assessment and management. Current Opinion in Rheumatology 2014;26(1):16-23. [DOI] [PubMed] [Google Scholar]
McKinney 2014
- McKinney EF, Willcocks LC, Broecker V, Smith KGC. The immunopathology of ANCA-associated vasculitis. Seminars in Immunopathology 2014;36(4):461-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
Merkel 2011
- Merkel PA, Aydin SZ, Boers M, Direskeneli H, Herlyn K, Seo P, et al. The OMERACT core set of outcome measures for use in clinical trials of ANCA-associated vasculitis. Journal of Rheumatology 2011;38(7):1480-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Merkel 2015
- Merkel PA, Matteson EL, Ramirez Curtis M. Overview of and approach to the vasculitides in adults. In: Post TW (editor). UpToDate, Waltham, MA. September 22, 2015.
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG. PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ 2009;21(339):b2535. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Riminton 2008
- Riminton S. Lymphocyte directed biologic therapies for primary systemic necrotizing vasculitis (registered title). Available from:cochrane.org/title/lymphocyte-directed-biologic-therapies-for-primary-systemic-necrotizing-vasculitis 2008 (Accessed 23 March 2017).
Schünemann 2011a
- Schünemann HJ, Oxman AD, Higgins JPT, Vist GE, Glasziou P, Guyatt GH. Chapter 11. Presenting results and ‘Summary of findings' tables. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Schünemann 2011b
- Schünemann H, Oxman AD, Vist GE, Higgins JBT, Deeks JJ, Glasziou P, Guyatt GH. Chapter 12. Interpreting results and drawing conclusions.In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011.. Available from handbook.cochrane.org.
Silva‐Fernández 2014
- Silva-Fernández L, Loza E, Martínez-Taboada VM, Blanco R, Rúa-Figueroa I, Pego-Reigosa JM, et al. Biological therapy for systemic vasculitis: a systematic review. Seminars in Arthritis and Rheumatism 2014;43(4):542-57. [DOI] [PubMed] [Google Scholar]
Souza 2017
- Souza AWS, Calich AL, Mariz HA, Ochtrop MLG, Bacchiega ABS, Ferreira GA, Rêgo J, Perez MO, Pereira RMR, Bernardo WM, Levy RA. Recommendations of the Brazilian Society of Rheumatology for the induction therapy of ANCA-associated vasculitis. Rev Bras Reumatol Engl Ed. 2017;57 Suppl 2:484-496. [DOI] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JAC, Egger M, Moher D editor(s). Chapter 10. Addressing reporting biases. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Watts 2000
- Watts RA, Lane SE, Bentham G, Scott DG. Epidemiology of systemic vasculitis: a ten-year study in the United Kingdom. Arthritis & Rheumatism 2000;43(2):414-9. [DOI] [PubMed] [Google Scholar]
Yates 2016
- Yates M, Watts RA, Bajema IM, Cid MC, Crestani B, Hauser T, et al. EULAR/ERA-EDTA recommendations for the management of ANCA-associated vasculitis. Annals of Rheumatic Diseases 2016;75(9):1583-94. [DOI] [PubMed] [Google Scholar]