

Abstract
Background
Lennox‐Gastaut syndrome (LGS) is an age‐specific epilepsy syndrome characterised by multiple seizure types. LGS has a characteristic electroencephalogram, an onset before age eight years, and drug resistance.
This is an updated version of the Cochrane Review published in 2013.
Objectives
To assess the efficacy and tolerability of anti‐seizure medications (ASMs) for LGS.
Search methods
We searched the Cochrane Register of Studies (CRS Web) and MEDLINE (Ovid, 1946 to 28 February 2020) on 2 March 2020. CRS Web includes randomised controlled trials (RCTs) or quasi‐RCTs from the Cochrane Central Register of Controlled Trials (CENTRAL); the Specialised Registers of Cochrane Review Groups, including Cochrane Epilepsy; PubMed; Embase; ClinicalTrials.gov; and the World Health Organization's International Clinical Trials Registry Platform (ICTRP). We imposed no language restrictions. We contacted pharmaceutical companies and colleagues in the field to seek any unpublished or ongoing studies.
Selection criteria
We considered RCTs, including cross‐over trials, of ASMs for LGS in children and adults. We included studies of ASMs used as either monotherapy or as add‐on (adjunctive) therapy. We excluded studies comparing different doses of the same ASM.
Data collection and analysis
We used standard Cochrane methodological procedures, including independent, dual assessment for risk of bias, and applying the GRADE approach to rate the evidence certainty for outcomes.
Main results
We found no trials of ASM monotherapy. The review included 11 trials (1277 participants; approximately 11 weeks to 112 weeks follow‐up after randomisation) using add‐on ASMs for LGS in children, adolescents, and adults.
Two studies compared add‐on cannabidiol (two doses) with add‐on placebo in children, adolescents, and adults. Insufficient information was provided for calculation of different response rate proportions in all seizures. We found high‐certainty evidence that 82 more people per 1000 (confidence interval (CI) 19 more to 350 more) had adverse events (AE) leading to study discontinuation with add‐on cannabidiol, compared to add‐on placebo (two studies; 396 participants; risk ratio (RR) 6.62, 95% CI 1.56 to 28.15).
One study compared add‐on cinromide with add‐on placebo in children and adolescents only. We found very low‐certainty evidence that 35 more people per 1000 (CI 123 fewer to 434 more) had 50% or greater average reduction of overall seizures with add‐on cinromide compared to add‐on placebo (one study; 56 participants; RR 1.15, 95% CI 0.47 to 2.86). This study did not report participants with AE leading to study discontinuation.
One study compared add‐on clobazam (three doses) with add‐on placebo. This study did not report overall seizure cessation or reduction. We found high‐certainty evidence that 106 more people per 1000 (CI 0 more to 538 more) had AE leading to study discontinuation with add‐on clobazam compared to add‐on placebo (one study; 238 participants; RR 4.12, 95% CI 1.01 to 16.87).
One study compared add‐on felbamate with add‐on placebo. No cases of seizure cessation occurred in either regimen during the treatment phase (one study; 73 participants; low‐certainty evidence). There was low‐certainty evidence that 53 more people per 1000 (CI 19 fewer to 716 more) with add‐on felbamate were seizure‐free during an EEG recording at the end of the treatment phase, compared to add‐on placebo (RR 2.92, 95% CI 0.32 to 26.77). The study did not report overall seizure reduction. We found low‐certainty evidence that one fewer person per 1000 (CI 26 fewer to 388 more) with add‐on felbamate had AE leading to study discontinuation compared to add‐on placebo (one study, 73 participants; RR 0.97, 95% CI 0.06 to 14.97).
Two studies compared add‐on lamotrigine with add‐on placebo. Neither study reported overall seizure cessation. We found high‐certainty evidence that 176 more people per 1000 (CI 30 more to 434 more) had ≥ 50% average seizure reduction with add‐on lamotrigine compared to add‐on placebo (one study; 167 participants; RR 2.12, 95% CI 1.19 to 3.76). We found low‐certainty evidence that 40 fewer people per 1000 (CI 68 fewer to 64 more) had AE leading to study‐discontinuation with add‐on lamotrigine compared to add‐on placebo (one study; 169 participants; RR 0.49, 95% CI 0.13 to 1.82).
Two studies compared add‐on rufinamide with add‐on placebo. Neither study reported seizure cessation. We found high‐certainty evidence that 202 more people per 1000 (CI 34 to 567 more) had ≥ 50% average seizure reduction (one study; 138 participants; RR 2.84, 95% CI 1.31 to 6.18). We found low‐certainty evidence that 105 more people per 1000 (CI 17 fewer to 967 more) had AE leading to study discontinuation with add‐on rufinamide compared to add‐on placebo (one study; 59 participants; RR 4.14, 95% CI 0.49 to 34.86). One study compared add‐on rufinamide with another add‐on ASM. This study did not report overall seizure cessation or reduction. We found low‐certainty evidence that three fewer people per 1000 (CI 75 fewer to 715 more) had AE leading to study discontinuation with add‐on rufinamide compared to another add‐on ASM (one study; 37 participants; RR 0.96, 95% CI 0.10 to 9.57).
One study compared add‐on topiramate with add‐on placebo. This study did not report overall seizure cessation. We found low‐certainty evidence for ≥ 75% average seizure reduction with add‐on topiramate (one study; 98 participants; Peto odds ratio (Peto OR) 8.22, 99% CI 0.60 to 112.62) and little or no difference to AE leading to study discontinuation compared to add‐on placebo; no participants experienced AE leading to study discontinuation (one study; 98 participants; low‐certainty evidence).
Authors' conclusions
RCTs of monotherapy and head‐to‐head comparison of add‐on ASMs are currently lacking. However, we found high‐certainty evidence for overall seizure reduction with add‐on lamotrigine and rufinamide, with low‐certainty evidence for AE leading to study discontinuation compared with add‐on placebo or another add‐on ASM. The evidence for other add‐on ASMs for overall seizure cessation or reduction was low to very low with high‐ to low‐certainty evidence for AE leading to study discontinuation.
Future research should consider outcome reporting of overall seizure reduction (applying automated seizure detection devices), impact on development, cognition and behaviour; future research should also investigate age‐specific efficacy of ASMs and target underlying aetiologies.
Plain language summary
Are anti‐seizure medications effective and safe treatments for Lennox‐Gastaut syndrome?
Why is this question important?
Lennox‐Gastaut syndrome (LGS) is a severe type of epilepsy that mainly affects children. The main symptom in LGS is frequent and multiple types of seizures. Seizures are caused by sudden and uncontrolled surges of abnormal electrical activity in the brain. The seizures are difficult to treat with anti‐seizure medications (ASMs). Many different ASMs are given to try and stop the seizures. Two or three ASMs are often given at the same time, which is known as polypharmacy. It is unclear which medications are most effective. Most people with LGS also have learning and behavioural difficulties.
How did we identify and evaluate the evidence?
We searched the medical literature for randomised controlled trials (RCTs) analysing the effects of anti‐seizure medications (ASMs) for treating LGS. We included any RCT that compared ASMs, whether as monotherapy or add‐on (adjunctive) therapy, with placebo (pretend treatment), no treatment or another kind of treatment. We then compared the results of the RCTs we found, and summarised the evidence from all the studies. We rated our confidence in the 'certainty' of evidence, based on factors such as study methods and sizes, and the consistency of findings across studies.
Study characteristics
This review included 11 trials (1277 participants, including children, adolescents and adults). The trials lasted between about 11 weeks and 112 weeks after randomisation. None of the included trials compared one ASM on its own with another treatment. Two trials compared add‐on cannabidiol regimens (cannabis‐based medicine) with add‐on placebo regimens (396 children, adolescents and adults ). One trial compared an add‐on cinromide regimen with an add‐on placebo regimen (56 children and adolescents only). One trial compared an add‐on clobazam regimen with an add‐on placebo regimen (238 participants). One trial compared an add‐on felbamate regimen with an add‐on placebo regimen (73 participants). Two trials compared add‐on lamotrigine regimens with add‐on placebo regimens (186 participants). Two trials compared add‐on rufinamide regimens with add‐on placebo regimens (197 participants). One trial compared an add‐on rufinamide regimen with another ASM regimen (37 participants). One trial compared an add‐on topiramate regimen with an add‐on placebo regimen (98 participants).
Most of the evidence in this review related to people from middle‐ or high‐income countries and, where reported, participants of white ethnicity.
Results and certainty of the evidence
We found high‐certainty evidence that add‐on lamotrigine increased the number of participants with at least 50% reduction in the average number of reported seizures. We also found low‐certainty evidence that add‐on lamotrigine may have reduced the number of participants with adverse events leading to study discontinuation when compared to add‐on placebo.
We found high‐certainty evidence that add‐on rufinamide increased the number of participants with at least 50% reduction in the average number of reported seizures, when compared with add‐on placebo. We also found low‐certainty evidence that add‐on rufinamide may have little or no difference in effect, compared to add‐on placebo or another unspecified ASM, on reduction of the number of participants with adverse events leading to study discontinuation.
Add‐on topiramate may have increased the number of participants with at least 75% reduction in the average number of reported seizures, and probably made little or no difference to the number of adverse events leading to study discontinuation, when compared to add‐on placebo (low‐certainty evidence).
Add‐on felbamate (treatment phase) may have made little or no difference in terms of reported seizure freedom and adverse events leading to study discontinuation when compared to add‐on placebo (low‐certainty evidence). However, we found that when seizures were recorded in a research setting, add‐on felbamate may have increased seizure freedom compared to add‐on placebo (low‐certainty evidence).
We remain uncertain whether other add‐on drug therapies, including cannabidiol, cinromide and clobazam, reduced all types of seizures because this outcome was not reported or had very low‐certainty evidence. We found high‐certainty evidence that add‐on cannabidiol and add‐on clobazam increased the number of participants with adverse events leading to study discontinuation, when compared to add‐on placebo. We did not find any evidence for adverse events leading to study discontinuation in the comparison of add‐on cinromide with add‐on placebo.
The evidence is current to March 2020.
Summary of findings
Background
Description of the condition
This is an updated version of the Cochrane Review published in 2013 (Hancock 2013).
Lennox‐Gastaut Syndrome (LGS) is a severe chronic epilepsy syndrome with onset in early childhood (seizure onset between one and seven years of age, with a peak age of onset at three to five years) (ILAE 1989; ILAE 2020). The disorder commonly persists, with drug‐resistant seizures throughout life (Arzimanoglou 2009).
People with LGS often have ongoing care needs into adulthood because of significant comorbidities, including cognitive, behavioural, and motor impairments. Along with the drug‐resistant seizures, associated severe behavioural and psychiatric disorders can be particularly challenging to manage clinically, as well as by the person living with LGS, their families and caregivers. In addition to the consequences of the underlying aetiology, LGS is regarded as a developmental and epileptic encephalopathy, a concept that refers to the adverse and disruptive impact of frequent epileptiform activity on cerebral activity, even in the absence of clinical seizures.
LGS represents approximately 3% to 5% of all childhood onset epilepsies. In population‐based childhood epilepsy incidence cohorts, only up to 0.9% of patients are identified with LGS when epilepsy is diagnosed; other defining characteristics such as multiple seizure types and electroencephalogram (EEG) features evolve over time (Berg 1999; Berg 2018; Callenbach 1998; Wirrell 2011). The prevalence of LGS was estimated at 26 per 100,000 in a regional US study (Trevathan 1997). This syndrome presents with multiple seizure types and in some people, follows initial presentation with infantile spasms. For three in ten people, LGS may evolve following the initial presentation in infancy with West syndrome (infantile spasms) or Ohtahara syndrome (Cross 2017). However, there are also cases of late‐onset LGS with seizure onset after the age of 10 years, in adolescence and adulthood (Smith 2018).
Underlying aetiologies, identified in 60% to 75% of patients with LGS, are diverse, and include developmental structural brain abnormalities, chromosomal derangements, monogenetic conditions, and less frequently, metabolic disorders and acquired brain insults (e.g. perinatal hypoxic‐ischaemic brain injuries, perinatal CNS infections) (Asadi‐Pooya 2018). The diagnosis of LGS in early childhood can be challenging due to the overlap in clinical presentation with other epileptic syndromes, especially epilepsy with myoclonic‐atonic seizures’ (Cross 2017; Eschbach 2018; Kaminska 1999).
The occurrence of tonic seizures is mandatory for the diagnosis of LGS (ILAE 2020). While atypical absence seizures are the second most characteristic seizure type in LGS, other observed seizure types also include generalised tonic‐clonic, atonic, myoclonic, myoclonic‐atonic, focal seizures, and epileptic spasms (Crespel 2019). The EEG features that form part of the fully evolved electro‐clinical syndrome are slow spike‐wave discharges (< 2.5 Hz) and generalised paroxysmal fast activity in slow wave sleep. Prolonged periods of obtundation or episodes of non‐convulsive status epilepticus are other common epilepsy manifestations interfering with cognitive and developmental function. These episodes and injuries sustained with seizures can have a negative impact on the quality of life as well as increase mortality risk (Autry 2010; Berg 2018).
Description of the intervention
Eligible interventions included any type of anti‐seizure medication (ASM) as either monotherapy or add‐on (adjunctive) therapy.
How the intervention might work
ASMs provide symptomatic treatment with the intent to suppress seizure generation. To date, there is no evidence that ASMs can achieve disease modification and prevent the development of drug‐resistant seizures. Cellular targets for ASMs, in general, include voltage‐gated ion channels, receptors enhancing GABA inhibition or inhibit excitation mediated by glutamate receptors (Rogawski 2016). The anti‐seizure effects of cannabidiol are proposed to be mediated by interaction with diverse molecular targets including G protein‐coupled receptor‐55 (GPR55‐antagonist), transient receptor potential vanilloid 1 (TRPV1‐ activation) channels and adenosine 2A2 receptors (Alves 2020, Lattanzi 2020). Cinromide (3 brono‐N‐ethylcinnamide), an experimental agent, showed its anti‐seizure effect in animal models, through a mechanism of action that is not well understood (Group for the Evaluation of Cinromide 1989). Felbamate (FBM) inhibits glycine‐enhanced N‐methyl‐D‐aspartate (NMDA)‐induced intracellular calcium currents, and at high concentrations, potentiates GABA responses and inhibits excitatory NMDA responses (Shorvon 2010). Clobazam enhances the inhibitory effects of GABA, binding to benzodiazepine receptors at the GABAA ligand‐gated chloride channel complex and boosting chloride conductance through GABA‐regulated channels (Trinka 2015, Brigo 2021). Lamotrigine acts as a use‐dependent blocker of voltage‐sensitive sodium channels, interacts with the open‐channel conformation of voltage‐sensitive sodium channels, interacts at a specific site of the alpha pore‐forming subunit of voltage‐sensitive sodium channels, and inhibits the release of glutamate (Shorvon 2010). Rufinamide acts as a blocker of voltage‐sensitive sodium channels and prevents sodium channels from returning to an activated state, thereby preventing the generation of sustained bursts of high‐frequency action potentials (Shorvon 2010). Topiramate has multiple mechanisms of action: it enhances GABA‐mediated inhibition, inhibits voltage‐dependent sodium channels, enhances potassium channel conduction; it also inhibits L‐type high voltage‐activated calcium channels, decreases glutamate‐mediated excitatory neurotransmission, and can inhibit carbonic anhydrase (Shorvon 2010).
Why it is important to do this review
LGS is complex and one of the most medically refractory epilepsy syndromes. It impacts on individuals' learning, development and mental well‐being, as well as on the psychosocial and socio‐economic situation of families and caregivers. Furthermore, there is evidence to suggest that healthcare and medical treatment costs are higher for people with LGS compared to other types of epilepsy (Pina‐Garza 2017).
Objectives
To assess the efficacy and tolerability of anti‐seizure medications (ASMs) for LGS.
Methods
Criteria for considering studies for this review
Types of studies
We considered randomised controlled trials (RCTs), including cross‐over trials, of ASMs for LGS.
Types of participants
We included children and adults with a diagnosis of LGS. We did not apply age restrictions. As an electrochemical syndrome associated with specific types of epileptic seizures and a characteristic EEG pattern, the diagnosis of LGS was based on clinical criteria. These diagnostic criteria included but were not limited to those provided by the International League Against Epilepsy (ILAE 1989).
Types of interventions
We included any trial that compared ASMs (monotherapy or add‐on therapy) with placebo, no therapy or another therapy. We excluded trials evaluating ketogenic diet, vagus nerve stimulation or other non‐pharmaceutical treatments (including homeopathy or acupuncture) unless they were provided as a co‐intervention with ASMs. In this review update, we also excluded studies comparing different doses of the same drug, which is different to the previous version of this review, although dose comparisons were not pre‐specified in the methods of the review protocol.
Types of outcome measures
If a study reported usable continuous and dichotomous data for quantitative reduction in seizures or other outcomes, we would have reported the dichotomous data as number of participants with a quantitative reduction in the outcome. We included outcome measures reported at any time point. If a study reported multiple time points, we planned to report and meta‐analyse the outcome at the longest follow‐up. However, we found that outcomes reported phases of dose adjustment before and, or after dose maintenance; we retrospectively agreed to prioritise the reporting of results for the treatment period as described in studies.
Primary outcomes
Cessation of all seizures (defined as total cessation of all seizure types within the trial period)
Quantitative reduction of all seizure types (measured as the number of all seizures occurring before treatment was commenced compared with the total number of seizures occurring at the end of the trial period)
Adverse events leading to study discontinuation
Secondary outcomes
Quantitative reduction in the following types of seizures (measured as the number of seizures occurring before treatment was commenced compared with the number occurring at the end of the trial period): absence seizures; tonic seizures; atonic seizures; drop seizures (defined as any seizure type resulting in postural loss); myoclonic seizures; tonic‐clonic seizures; and focal onset seizures
Death (i.e. alive/deceased)
Any adverse events
Search methods for identification of studies
Electronic searches
Searches were run for the original review in March 2003. Subsequent searches were run in February 2009, March 2011, April 2012, October 2012, July 2014, October 2016, and September 2018. For the latest update, we searched the following databases on 2 March 2020. There were no language restrictions.
Cochrane Register of Studies (CRS Web), using the search strategy shown in Appendix 1.
MEDLINE (Ovid), 1946 to 28 February 2020, using the search strategy shown in Appendix 2.
CRS Web includes RCTs or quasi‐RCTs from the Cochrane Central Register of Controlled Trials (CENTRAL); the Specialised Registers of Cochrane Review Groups, including Cochrane Epilepsy; PubMed; Embase; ClinicalTrials.gov; and the World Health Organization's International Clinical Trials Registry Platform (ICTRP).
Searching other resources
One review author (FB) contacted pharmaceutical companies (Eisai, GW Pharmaceuticals, and UCB Pharma) and colleagues in the field on 25 November 2020 in an effort to identify unpublished data.
Data collection and analysis
At least two review authors (from among KJ, FB, CE, and SM) extracted data and resolved any discrepancies by discussion.
Selection of studies
Two review authors (KJ and FB) examined records identified by the search strategy for studies eligible for inclusion. The review authors independently confirmed that studies were RCTs of drug treatment for LGS.
Data extraction and management
At least two review authors (from among KJ, FB, CE, and SM) independently performed data extraction using a specially designed data extraction form. FB and KJ checked and entered data into the Cochrane authoring and statistical software, Review Manager 5 (Review Manager 2020); at least one other review author (CE or SM) checked the data entry.
For each trial, we sought the following information.
Participants
Inclusion criteria
Exclusion criteria
Total number randomised
Baseline imbalances
Withdrawals and exclusions
Age at onset
Age at diagnosis
Age at start of treatment
Sex
Race/ethnicity
Type of seizures
Seizure frequency during the baseline period
Number of background drugs
Co‐morbidities
Interventions
Type of intervention (description, including dose, frequency and route of administration)
Number of participants randomised to each intervention
Duration of baseline period
Duration of treatment period
Co‐interventions (if any)
Compliance
Outcome measures
For each outcome reported in the included studies we extracted the following data:
Outcome name and definition
Time point when each outcome was measured
Time point when each outcome was reported
Validation of the outcome (yes/no/unclear)
Imputation of missing data (e.g. assumptions made for intention‐to‐treat (ITT) analysis)
Assumed risk estimate (e.g. baseline or population risk noted in the Background)
Statistical power (e.g. power and sample size calculation, level of power achieved)
Assessment of risk of bias in included studies
At least two review authors (from among KJ, CE, and FB) independently assessed risk of bias for each of the included trials using the Cochrane 'Risk of bias' tool (Higgins 2011). We assessed risk of bias based on sequence generation, concealment of allocation, methods of blinding, incomplete outcome data, selective reporting, and other types of bias. For each of these categories, the review authors judged the domain to be at 'low', 'high', or 'unclear' risk of bias. We resolved any disagreements by discussion.
Measures of treatment effect
We analysed dichotomous data with a risk ratio (RR) and a 95% confidence interval (CI) or using the Peto odds ratio (Peto OR) and a 99% CI if there were less than 1% events. We analysed continuous data using the mean difference (MD) with a 95% CI where we found data provided as means and standard deviations (SDs).
Unit of analysis issues
For any unit of analysis issues, we planned to deal with them using the guidance in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2020). We planned to analyse randomised cross‐over studies in meta‐analyses, using the results from paired analyses of the first period only to account for carry‐over effect with sequential intervention (Higgins 2020).
For studies in which different doses of the same drug were reported separately, we combined these data into a single treatment group to avoid a duplicative error with multiple‐armed trials.
Dealing with missing data
For this review update, one review author (FB) attempted to contact the study authors of Arzimanoglou 2019 to obtain missing outcome data.
Assessment of heterogeneity
We assessed heterogeneity by visually inspecting forest plots, and by using the Chi² test and I² statistic as follows: 0% to 40% might not be important, 30% to 60% may represent moderate heterogeneity, 50% to 90% may represent substantial heterogeneity, and 75% to 100% indicates considerable heterogeneity (Deeks 2020).
Assessment of reporting biases
We would have used a funnel plot to explore small‐study biases in an outcome if data from sufficient studies (10 or more) had been pooled in a single meta‐analysis.
Data synthesis
We used a random‐effects model on the basis that drug trials assessed different but related intervention effects.
Subgroup analysis and investigation of heterogeneity
We found insufficient evidence to perform subgroup analysis for dosage, timing, and length of treatment.
Sensitivity analysis
No sensitivity analyses were pre‐specified or undertaken.
Summary of findings and assessment of the certainty of the evidence
For this review update, we created 'Summary of findings' tables and used the GRADE approach to evaluate the certainty of the evidence (Schünemann 2020).
We included the following outcomes:
Number of participants free from all seizures.
Number of participants with ≥ 75% reduction in all seizures.
Number of participants with ≥ 50% reduction in all seizures.
Number of participants with adverse events leading to study discontinuation.
Two review authors (from among KJ, FB, and CE) used the GRADE approach to judge the certainty of evidence for outcomes based on five criteria (risk of bias, inconsistency, indirectness, imprecision, and publication bias) (Schunemann 2011). If we had serious concerns regarding one of the five criteria, we downgraded the evidence from 'high quality' by one level; if we had very serious concerns, we downgraded the evidence by two levels. We resolved any discrepancies through discussion and reported our rationale for downgrading evidence in the 'Summary of findings' table footnotes.
We used GRADEpro GDT software (GRADEpro GDT 2015) to record our judgements and to create the 'Summary of findings' tables for each comparison included in the review.
Results
Description of studies
Results of the search
See Figure 1.
1.
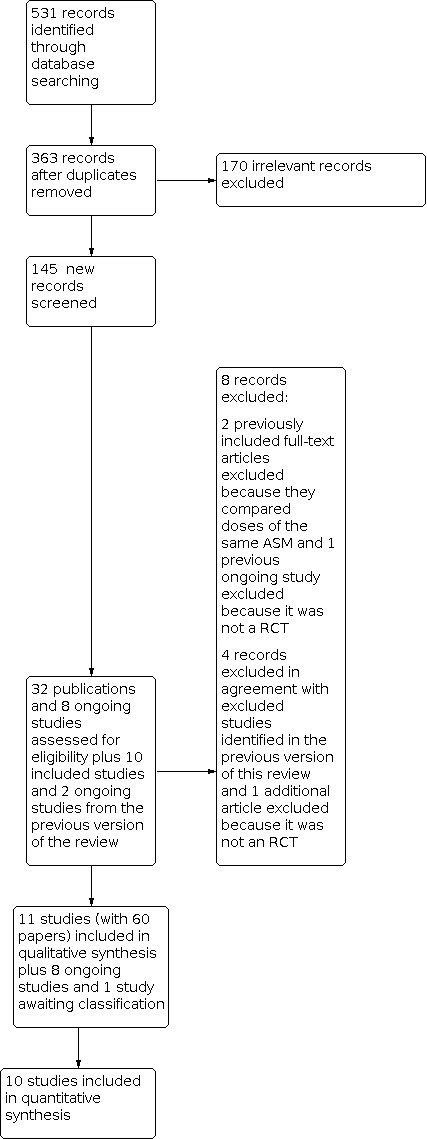
Study flow diagram.
We identified 40 studies, of which 11 met our inclusion criteria (1277 randomised participants). Four studies were published since the previous version of this review; see Characteristics of included studies. We excluded eight studies in total. We excluded two studies from the current review because they compared different doses of the same ASM (Conry 2009; Inanaga 1989), and three other studies were judged not to be RCTs (Oletsky 1996; Perry 2019; Vigevano 1994). We also excluded three studies that were excluded in a previous version of this review because the outcome data were not reported in a usable way (Battaglia 1991; Vajda 1985 ; Vassella 1978); see Characteristics of excluded studies.
We identified eight ongoing studies from ClinicalTrials.gov and the World Health Organization International Clinical Trials Registry Platform (CTRI/2010/091/001449; NCT00004776; NCT01370486; NCT02318537; NCT03355209; NCT03650452; NCT03808935; Wechsler 2017); see Characteristics of ongoing studies. Of these eight trials, one trial of adjunctive rufinamide versus lamotrigine was terminated, and another trial of adjunctive cannabidiol versus placebo had its sponsorship withdrawn. Other adjunctive drug therapies under investigation included topiramate, melatonin, fenfluramine hydrochloride and TAK‐935 versus placebo.
On 25 November 2020, one review author (FB) contacted pharmaceutical companies (Eisai, GW Pharmaceuticals, and UCB Pharma), and colleagues in the field to obtain information on any additional unpublished data. They identified no unpublished data relating to this review. On 4 December 2020, one review author (FB) also contacted the principal investigators of Arzimanoglou 2019 to obtain information on missing data, and the principal investigators of NCT01370486 to obtain information on study completion. We received no response by the time of review completion.
Despite its completion, we listed Ohtahara 2008 in Studies awaiting classification because we were unable to obtain a translation from the Japanese language into English.
Included studies
See Characteristics of included studies.
Two studies published protocols that we considered as part of the evidence synthesis (Devinsky 2018; Thiele 2018). Of the 11 included studies, 10 were randomised and double‐blinded, placebo‐controlled trials (Arzimanoglou 2019; Devinsky 2018; Felbamate Study Group 1993; Glauser 2008; Group for the Evaluation of Cinromide 1989; Motte 1997; Ng 2011; Ohtsuka 2014; Sachdeo 1999; Thiele 2018) and one was a randomised and double‐blinded, cross‐over, placebo‐controlled trial (Eriksson 1998).
Two studies involved children and adolescents only, with ages ranging from one year to 18 years, (Arzimanoglou 2019; Group for the Evaluation of Cinromide 1989) and eight studies involved children, adolescents and adults, with ages ranging from over one year to 55 years (Devinsky 2018; Felbamate Study Group 1993; Glauser 2008; Motte 1997; Ng 2011; Ohtsuka 2014; Sachdeo 1999; Thiele 2018). Another study of children, adolescents and young adults with refractory generalised epilepsy included a subgroup of participants with LGS who were all aged under 18 years (Eriksson 1998).
In ten studies, diagnosis of LGS was reported according to clinical and EEG criteria or the classification of the International League Against Epilepsy (ILAE 1989); (Arzimanoglou 2019; Devinsky 2018; Eriksson 1998; Felbamate Study Group 1993; Glauser 2008; Group for the Evaluation of Cinromide 1989; Ng 2011; Ohtsuka 2014; Sachdeo 1999; Thiele 2018). For one other study, the diagnosis was agreed by an international expert panel of child neurologists (Motte 1997). There was limited reporting of co‐morbidities in the study population but as many as 40% of participants from one study were reported to have underlying causes, such as tuberous sclerosis and cerebral palsy, cerebral dysgenesis, encephalitis and bacterial meningitis (Ohtsuka 2014).
The drug treatment regimens of the included RCTs were as follows: cannabidiol and ASMs (Devinsky 2018; Thiele 2018); cinromide and ASMs (Group for the Evaluation of Cinromide 1989); clobazam and ASMs (Ng 2011); felbamate and ASMs (Felbamate Study Group 1993); lamotrigine and ASMs (Eriksson 1998; Motte 1997); rufinamide and ASMs (Arzimanoglou 2019; Glauser 2008, and Ohtsuka 2014); and topiramate and ASMs (Sachdeo 1999). Aside from ASMs, the two most recent studies of add‐on cannabidiol reported non‐pharmacological co‐interventions including vagus nerve stimulation and ketogenic diet in a subset of participants (Devinsky 2018; Thiele 2018). All studies included a pre‐randomisation, baseline period of four to eight weeks. The follow‐up after randomisation ranged from approximately 11 weeks to 112 weeks, including variable duration of titration and maintenance periods, with or without tapering, and safety follow‐up.
In three studies, the primary outcome for treatment efficacy was overall seizure reduction, which was the main focus of the current review (Eriksson 1998; Felbamate Study Group 1993; Group for the Evaluation of Cinromide 1989). Two studies included primary efficacy outcomes for overall seizure reduction and reduction in drop attacks or tonic‐atonic seizures (Glauser 2008; Sachdeo 1999). Four later studies had a primary efficacy outcome that focused on drop seizure or tonic‐atonic seizure reduction (Devinsky 2018; Ng 2011; Ohtsuka 2014; Thiele 2018). The primary efficacy outcome was major motor seizures in one study (Motte 1997) and behavioural outcomes in another study (Arzimanoglou 2019).
Arzimanoglou 2019
Arzimanoglou 2019 was conducted in 19 centres across Canada, France, Greece, Italy, Poland, and the United States. It involved 37 randomised children (aged one year to less than four years) with inadequately controlled LGS. The participants had LGS with seizures uncontrolled by a fixed dose of one to three concomitant ASMs for a minimum of four weeks before randomisation. The study included an eight‐week pre‐randomisation phase (screening period and baseline visit), followed by a 106‐week randomised phase (including titration and maintenance). Participants were randomised to receive add‐on rufinamide (target maintenance dose: 45 mg/kg/day in two divided doses, given as oral suspension) or any other ASM chosen by the investigator and added to the existing regimen of one to three ASMs. Baseline characteristics were similar in the trial groups although the 'Any other ASM' group had a higher proportion of males. The categories for ethnicity (Hispanic/Latino; non‐Hispanic/Latino) differed from those reported in the 6‐month dataset (White; Black or African‐American). Collectively, the baseline characteristics indicated mostly non‐Hispanic participants (more than 75%) and a majority of white participants (more than 70%) in the study.
Devinsky 2018
Devinsky 2018 was conducted at 30 participating centres (20 in the United States, 5 in Spain, 3 in the United Kingdom, and 2 in France). It involved 225 randomised children and adults (aged 2 to 55 years). They took between one and four ASMs and had at least two drop seizures each week during the baseline period. Participants were randomised to receive add‐on cannabidiol at a dose of either 20 mg/kg/day or 10 mg/kg/day or matching add‐on placebo. The trial included a 4‐week baseline period, a 14‐week treatment period (2 weeks of dose escalation, followed by a maintenance phase of 12 weeks), a tapering period of up to 10 days, and a 4‐week safety follow‐up period after discontinuation of cannabidiol or placebo. Baseline characteristics were similar in the trial groups although a slightly higher proportion of the placebo group also received vagus nerve stimulation as a concomitant intervention (28%) as compared with the 10 and 20mg cannabidiol groups (21% each). The majority of study participants were of white ethnicity (more than 88%).
Eriksson 1998
Eriksson 1998 was conducted in Finland. The study initially involved 30 children (aged over two years), adolescents and two young adults with refractory generalised epilepsy. Twenty of the participants in the open phase had LGS, and all were aged under 18 years. Diagnostic criteria for LGS were based on the classification of the International League Against Epilepsy (ILAE 1989). All included participants experienced more than two seizures per month at baseline. The trial consisted of six phases. There was first an eight‐week baseline phase during which each child was observed on pre‐study medication, followed by an open phase, during which an attempt was made to find the optimal lamotrigine dose for each child. At the end of the open phase, children had been categorised as responders if they showed any improvement (in alertness, behaviour, motor skills, or seizures). The 'responders' (17/27 (13 with LGS); three excluded because of incomplete seizure diaries) were subsequently entered in a double‐blind phase of 12‐week periods during which, for each child, add‐on lamotrigine and placebo tablets were administered in random order. The treatment periods were separated by a three‐week washout phase. This study did not report participants' ethnicity and other characteristics for baseline comparison of treatment groups.
Felbamate Study Group 1993
Felbamate Study Group 1993 involved 73 randomised children and adults (aged 4 years to 36 years) and was conducted in the USA. Included participants had a history of multiple types of seizures and a minimum of 90 atonic seizures or atypical absence seizures per month during an eight‐week pre‐study phase; they took no more than two ASMs at baseline. The trial consisted of a 28‐day baseline period followed by a 14‐day titration phase and a 56‐day maintenance period. The initial dose of add‐on felbamate was 15 mg/kg/day, increased to 30 mg/kg after seven days and to either 14 mg/kg/day or 3600 mg/day (whichever was lower) after 14 days. Baseline characteristics were similar in the trial groups. The study largely involved people of white ethnicity (more than 89%).
Glauser 2008
Glauser 2008 was conducted at 36 sites in nine countries (Belgium, Brazil, Germany, Hungary, Italy, Norway, Poland, Spain, and the United States). It involved 138 randomised children and adults (aged four years to 37 years). Study participants had a history of multiple seizure types, including atypical absence seizures and drop attacks, and a minimum of 90 seizures in the month prior to trial entry. At baseline, included participants took a fixed‐dose regimen of one to three concomitant ASMs. The trial consisted of a 28‐day baseline period at the end of which, patients continuing to meet the study criteria entered an 84‐day double‐blind treatment phase of either add‐on rufinamide or add‐on placebo. This phase consisted of a 14‐day titration period followed by a 70‐day maintenance period. Doses were titrated according to a recommended schedule based on body weight, up to a maximum dose of 45 mg/kg/day.
Baseline characteristics of the two treatment groups were similar, including mostly male participants and with comparable usage of concomitant ASMs. However, a higher proportion of the add‐on placebo group was under 12 years of age, and a higher proportion of the add‐on rufinamide group was over 17 years of age. Both groups largely involved participants of white ethnicity.
Group for the Evaluation of Cinromide 1989
Group for the Evaluation of Cinromide 1989 involved 56 randomised children and adolescents (aged two years to 18 years) and was conducted in the USA. Included participants had seizures for at least six months prior to study entry. At baseline, no individual was receiving more than three marketed antiepileptic drugs and none had previous exposure to cinromide. The trial consisted of a six‐week baseline period, following which participants were randomised to receive either add‐on cinromide or add‐on placebo for a period of 18 weeks. Study medication was initiated at 20 to 40 mg/kg/day, divided into four equal doses. Further increases (to a total daily maximum of 83 to 109 mg/kg) were prescribed at weekly visits if each prior dose was tolerated and seizures continued. Baseline characteristics were comparable between groups, with the majority of study participants being of white ethnicity (more than 92%).
Motte 1997
Motte 1997 was conducted at 43 unspecified sites. The study involved 169 randomised children and young adults (aged three years to 25 years). Included participants experienced more than one type of predominantly generalised seizure for at least one year, and they had seizures at least every other day. They took up to three ASMs at baseline. The trial consisted of a four‐week baseline period during which all recipients received add‐on placebo. Participants were then randomised to receive either add‐on lamotrigine or add‐on placebo for a 16‐week treatment period. Participants were assigned to one of four dosing regimens according to concomitant valproate use and body weight based on paediatric dosing recommendations at that time. The characteristics of the two groups were similar at baseline although the lamotrigine group had a higher proportion of male participants. Both groups largely involved participants of white ethnicity (more than 90%).
Ng 2011
Ng 2011 was conducted at 51 sites in the United States, India, Europe, and Australia. It involved 238 randomised children and adults (aged 2 years to 54 years). They received one to three ASMs with stable dosages for at least 30 days before screening, and they experienced at least two drop seizures per week during the four‐week baseline period. Included participants were randomised to add‐on placebo or one of three doses of add‐on clobazam (0.25, 0.5, and 1.0 mg/kg/day), up to a maximum dosage of 40 mg/day. Treatment included a three‐week titration phase and a 12‐week maintenance phase followed by two to three weeks tapering or continuation in an open‐label extension. Baseline characteristics were comparable between groups, although the medium‐dose clobazam group had a lower mean baseline average weekly drop seizure rate (58.8; SD 119.6) compared with the low‐dose group (98.3; SD 198.5), high‐dose group (94.9; SD 152.2), and placebo group (95.6; SD 168.2). Most study participants in each group were of white ethnicity (57% to 71%).
We included a post‐hoc analysis of Ng 2011 as a subsidiary paper (Paolicchi 2015). This post‐hoc analysis aimed to determine potential drug‐related effects on four behaviour domains of the Child Behaviour Checklist (CBCL): aggressive behaviour, anxious/depressed, attention problems, and somatic complaints (Achenbach 1991).
Ohtsuka 2014
Ohtsuka 2014 included 59 randomised children and adults (aged between 4 years and 30 years), and was conducted in Japan. Included participants experienced at least 90 seizures during the 28 days before the baseline period. They took one to three background ASMs at baseline. The participants were randomised to either add‐on rufinamide (doses titrated according to a predetermined schedule based on body weight, with the target dose maintained during the maintenance period) or add‐on placebo. This study consisted of four phases: a four‐week baseline, a two‐week titration, a 10‐week maintenance, and either a follow‐up visit or entry into an open‐label extension. Baseline characteristics were comparable between groups although there was a relatively higher concomitant use of lamotrigine in the placebo group and relatively higher concomitant use of clobazam in the rufinamide group. Participants' ethnicity was not reported in this study.
Sachdeo 1999
Sachdeo 1999 was conducted at 12 centres in the USA. The study included 98 randomised children and young adults (aged over one year to under 30 years). Included participants experienced seizure types including drop attacks and atypical absence seizures; they had a frequency of at least 60 seizures during the month prior to the baseline phase, while being maintained on one or two standard ASMs. The trial consisted of a baseline phase of four weeks and an 11‐week treatment phase that included three weeks titration and eight weeks maintenance. The participants were titrated up to a dose of 6 mg/kg/day or their maximal tolerated dosage of either add‐on topiramate or add‐on placebo over the first three weeks of the treatment period. Of note, concomitant treatment with felbamate was prohibited part way through the study, due to adverse effects. Baseline characteristics were comparable between groups, although there was a slightly higher proportion of males in the topiramate group. Both groups largely involved participants of white ethnicity (more than 80%).
Thiele 2018
Thiele 2018 was conducted at 24 clinical sites in the USA (17 sites), the Netherlands (one site), and Poland (six sites). It included 171 children and adults (aged two years to 45 years). The study participants experienced more than one type of generalised seizure, including drop seizures, for at least six months. At baseline, they took one to four ASMs, and had at least two drop seizures per week during the four‐week baseline period. Participants were randomised to add‐on cannabidiol 20 mg/kg (given as oral solution) or matching add‐on placebo solution administered orally in two equally divided doses. The study duration was 14 weeks, which included two weeks of dose escalation (starting at a daily dose of 2.5 mg/kg, followed by a maintenance period of 12 weeks), a tapering period of up to 10 days, and a four‐week safety follow‐up period. Baseline characteristics were comparable between groups, although the placebo group had a higher proportion of participants on a ketogenic diet as a concomitant intervention (12% versus 5%). Both groups largely involved participants of white ethnicity (more than 87%).
Excluded studies
See Characteristics of excluded studies.
We excluded eight studies from the search results for this updated review. We excluded two previously included studies that compared different doses of the same ASM (Conry 2009 and Inanaga 1989). Three studies were judged not to be randomised trials (Oletsky 1996; Perry 2019; Vigevano 1994). We also considered studies excluded from the previous version of this review, and agreed on the exclusion of three further studies. We excluded these studies because incomplete reporting for participants with LGS meant that the data were not usable (Battaglia 1991; Vajda 1985 ; Vassella 1978). We did not attempt to contact authors for additional data from these studies, which were published between 29 and 42 years ago. We included one previously excluded study as a subsidiary publication relating to the Felbamate Study Group 1993.
Risk of bias in included studies
We have reported 'Risk of bias' assessments for each study in Characteristics of included studies. Figure 2 summarises the review authors' 'Risk of bias' assessments. Only two included studies of add‐on cannabidiol had published protocols (Devinsky 2018; Thiele 2018). Based on the available information, we identified a high risk of selective reporting bias in five of the 11 RCTs (Devinsky 2018; Eriksson 1998; Felbamate Study Group 1993; Group for the Evaluation of Cinromide 1989; Thiele 2018), three of which were included in the previous version of the review (Eriksson 1998; Felbamate Study Group 1993; Group for the Evaluation of Cinromide 1989). In three of the 11 RCTs, we identified a high risk of bias associated with incomplete outcome data (Arzimanoglou 2019; Eriksson 1998; Group for the Evaluation of Cinromide 1989), two of which were included in the previous version of this review (Eriksson 1998; Group for the Evaluation of Cinromide 1989); the more recent study had a relatively long follow up of two years (Arzimanoglou 2019). We identified a high risk of bias due to the exclusion of 'non‐responders' in the initial open phase of one study (Eriksson 1998).
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
For outcomes included in the 'Summary of findings' tables, we assessed risk of bias at the outcome level. However, we downgraded outcomes for study limitations in one comparison that was stopped prematurely and judged to have a very serious risk of bias due to a combination of selective reporting and incomplete data (Group for the Evaluation of Cinromide 1989).
Allocation
We assessed all eleven included studies to have a low risk of selection bias associated with random sequence generation. In terms of allocation concealment, we judged six of the eleven included studies to have a low risk of selection bias and five studies as having an unclear risk of selection bias.
Blinding
We judged two of the eleven included studies to have a low risk of performance bias and detection bias, and nine studies to have an unclear risk of performance bias and detection bias.
Incomplete outcome data
We judged five of the eleven included studies to have a low risk of attrition bias, three studies as having an unclear risk and three studies as having a high risk of attrition bias.
Selective reporting
We judged six of the eleven included studies to have a low risk of reporting bias and five as having a high risk of reporting bias.
Other potential sources of bias
We did not identify any other potential sources of bias in the eleven included studies.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8
Summary of findings 1. Cannabidiol (10 mg/kg and 20 mg/kg) plus ASMs compared to placebo plus ASMs.
| Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs compared to placebo + ASMs | ||||||
| Patient or population: Lennox‐Gastaut syndrome, any age Setting: multi‐centre, high‐income countries Intervention: cannabidiol (10mg/kg and 20mg/kg) + ASMs Comparison: placebo + ASMs | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo + ASMs | Risk with cannabidiol + ASMs | |||||
| Number of participants free from all seizures follow up: after 14 weeks' treatment (titration and maintenance) | see comment | see comment | not estimable | see comment | see comment | No studies measured this outcome. No participants were free from drop seizures (one study; 225 participants) Analysis 1.1 |
| Number of participants with ≥ 75% reduction in all seizures follow‐up: after 14 weeks' treatment (titration and maintenance) |
see comment | see comment | not estimable | see comment | see comment | No studies measured this outcome. Number of participants with ≥ 75% reduction in drop seizures: RR 3.51 (95% CI 1.24 to 9.92) in favour of the cannabidiol regimen (two studies; 396 participants) Analysis 1.2. |
| Number of participants with ≥ 50% reduction in all seizures follow‐up: after 14 weeks' treatment (titration and maintenance) |
see comment | see comment | not estimable | see comment | see comment | No studies measured this outcome. Number of participants with ≥ 50% reduction in drop seizures: RR 2.12 (95% CI 1.48 to 3.03) in favour of the cannabidiol regimen (two studies; 396 participants; Analysis 1.3). |
| Number of participants with adverse events leading to study discontinuation follow‐up: after 19 weeks | Study population | RR 6.62 (1.56 to 28.15) | 396 (2 RCTs) | ⊕⊕⊕⊕ HIGH | Intervention in Devinsky 2018 involved two doses of cannabidiol (10 mg/kg and 20 mg/kg); 6 of the 7 adverse events leading to study discontinuation occurred in the higher dose group; intervention in Thiele 2018 also involved the higher dose of cannabidiol (20 mg/kg). | |
| 12 per 1000 | 82 per 1000 (19 to 350) | |||||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ASM: anti‐seizure medication; CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
Summary of findings 2. Cinromide plus ASMs compared to placebo plus ASMs.
| Cinromide + ASMs compared to placebo + ASMs | ||||||
| Patient or population: Lennox‐Gastaut syndrome, any age1 Setting: USA Intervention: cinromide + ASMs Comparison: placebo + ASMs | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo + ASMs | Risk with cinromide + ASMs | |||||
| Number of participants free from all seizures follow‐up: after 18 weeks' treatment (titration and maintenance) | Study population | not estimable | 56 (1 RCT) | ⊕⊝⊝⊝ VERY LOW2,3 | ||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Number of participants with ≥ 75% reduction in mean weekly seizures follow‐up: after 18 weeks' treatment (titration and maintenance) | Study population | Peto OR 9.35 (0.45 to 194.96) | 56 (1 RCT) | ⊕⊝⊝⊝ VERY LOW2,4 | Cinromide + ASMs: 3/26 participants; placebo + ASMs: 0/30 participants. |
|
| see comment | see comment | |||||
| Number of participants with ≥ 50% reduction in mean weekly seizures follow‐up: after 18 weeks' treatment (titration and maintenance) | Study population | RR 1.15 (0.47 to 2.86) | 56 (1 RCT) | ⊕⊝⊝⊝ VERY LOW2,4 | ||
| 233 per 1000 | 268 per 1000 (110 to 667) | |||||
| Number of participants with adverse events leading to study discontinuation follow‐up: after 18 weeks |
see comment | see comment | not estimable | see comment | see comment | No studies measured this outcome. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ASM: anti‐seizure medication; CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio; PetoOR: Petoodds ratio. | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1The evidence for this comparison included children and adolescents only.
2Downgraded twice for study limitations because there was a high risk of bias from incomplete data (study terminated prematurely).
3Downgraded twice for imprecision because the study was not powered to detect a between‐group difference in zero event outcomes.
4Downgraded twice for imprecision because the effect estimate has a very wide confidence interval.
Summary of findings 3. Clobazam (low, medium and high doses) plus ASMs compared to placebo plus ASMs.
| Clobazam (low, medium and high doses) + ASMs compared to placebo + ASMs | ||||||
| Patient or population: Lennox‐Gastaut syndrome, any age Setting: multi‐centre, high‐income countries and one middle‐income country Intervention: clobazam (low, medium and high doses) + ASMs Comparison: placebo + ASMs | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo + ASMs | Risk with clobazam + ASMs | |||||
| Number of participants free from all seizures follow‐up: "from the 4‐week baseline period to the 12‐week maintenance period" (with 3 weeks titration) |
see comment | see comment | not estimable | see comment | see comment | No studies measured this outcome. Number of participants free from drop seizures: RR 4.10 (95% CI 1.00 to 16.83) in favour of the clobazam regimen (1 study; 217 participants) Analysis 3.1. |
| Number of participants with ≥ 75% reduction in all seizures follow‐up: "from the 4‐week baseline period to the 12‐week maintenance period" (with 3 weeks titration) |
see comment | see comment | not estimable | see comment | see comment | No studies measured this outcome. |
| Number of participants with ≥ 50% reduction in all seizures follow‐up: "from the 4‐week baseline period to the 12‐week maintenance period" (with 3 weeks titration) |
see comment | see comment | not estimable | see comment | see comment | No studies measured this outcome. |
| Number of participants with adverse events leading to study discontinuation follow‐up: 22 weeks (after baseline, titration, maintenance and tapering) | Study population | RR 4.12 (1.01 to 16.87) | 238 (1 RCT) | ⊕⊕⊕⊕ HIGH | Intervention in Ng 2011 involved three doses of clobazam (low, medium, high) and study authors state that "A dosage related trend was observed for the overall incidence of [adverse events] leading to discontinuation." | |
| 34 per 1000 | 140 per 1000 (34 to 572) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
Summary of findings 4. Felbamate plus ASMs compared to placebo plus ASMs.
| Felbamate + ASMs compared to placebo + ASMs | ||||||
| Patient or population: Lennox‐Gastaut syndrome, any age Setting: USA Intervention: felbamate + ASMs Comparison: placebo + ASMs | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo + ASMs | Risk with felbamate + ASMs | |||||
| Number of participants free from all seizures (recorded by closed‐circuit television and electroencephalography) ‐ follow‐up: after the treatment phase, which "consisted of a 14‐day titration period and a 56‐day maintenance period" | Study population | RR 2.92 (0.32 to 26.77) | 73 (1 RCT) | ⊕⊕⊝⊝ LOW1 | ||
| 28 per 1000 | 81 per 1000 (9 to 744) | |||||
| Number of participants free from all seizures follow‐up: after the treatment phase, which "consisted of a 14‐day titration period and a 56‐day maintenance period" | Study population | not estimable | 73 (1 RCT) | ⊕⊕⊝⊝ LOW2 | ||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Number of participants with ≥ 75% reduction in all seizures follow‐up: after the treatment phase, which "consisted of a 14‐day titration period and a 56‐day maintenance period" |
see comment | see comment | not estimable | see comment | see comment | No studies measured this outcome. |
| Number of participants with ≥ 50% reduction in all seizures follow‐up: after the treatment phase, which "consisted of a 14‐day titration period and a 56‐day maintenance period" |
see comment | see comment | not estimable | see comment | see comment | According to a retrospective analysis "Approximately 50% of patients randomised to FBM obtained at least a 50% reduction in seizure frequency compared with about 15% receiving placebo." |
| Number of participants with adverse events leading to study discontinuation follow up: after 14 weeks | Study population | RR 0.97 (0.06 to 14.97) | 73 (1 RCT) | ⊕⊕⊝⊝ LOW1 | ||
| 28 per 1000 | 27 per 1000 (2 to 416) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ASM: anti‐seizure medication; CI: confidence interval;FBM: felbamate; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1Downgraded twice for imprecision because the effect estimate has a very wide confidence interval.
2Downgraded twice for imprecision because the study was not powered to detect a between‐group difference in zero event outcomes.
Summary of findings 5. Lamotrigine plus ASMs compared to placebo plus ASMs.
| Lamotrigine + ASMs compared to placebo + ASMs | ||||||
| Patient or population: Lennox‐Gastaut syndrome, any age Setting: multi‐centre Intervention: lamotrigine + ASMs Comparison: placebo + ASMs | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo + ASMs | Risk with lamotrigine + ASMs | |||||
| Number of participants free from all seizures follow‐up: after 16 weeks' treatment |
see comment | see comment | not estimable | see comment | see comment | No studies measured this outcome. |
| Number of participants with ≥ 75% reduction in all seizures follow‐up: after 16 weeks' treatment |
see comment | see comment | not estimable | see comment | see comment | No studies measured this outcome. |
| Number of participants with ≥ 50% median reduction in all seizures follow‐up: after 16 weeks' treatment | Study population | RR 2.12 (1.19 to 3.76) | 167 (1 RCT) | ⊕⊕⊕⊕ HIGH | ||
| 157 per 1000 | 333 per 1000 (187 to 591) | |||||
| Number of participants with adverse events leading to study discontinuation follow‐up: after 20 weeks | Study population | RR 0.49 (0.13 to 1.82) | 169 (1 RCT) | ⊕⊕⊝⊝ LOW1 | ||
| 78 per 1000 | 38 per 1000 (10 to 142) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ASM: anti‐seizure medication; CI: confidence interval;RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1Downgraded twice because the effect estimate has a very wide confidence interval.
Summary of findings 6. Rufinamide plus ASMs compared to placebo plus ASMs.
| Rufinamide + ASMs compared to placebo + ASMs | ||||||
| Patient or population: Lennox‐Gastaut syndrome, any age Setting: multi‐centre, high‐income countries and one middle‐income country Intervention: rufinamide + ASMs Comparison: placebo + ASMs | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo + ASMs | Risk with rufinamide + ASMs | |||||
| Number of participants free from all seizures follow‐up: after 84 days/12 weeks' treatment (titration and maintenance) |
see comment | see comment | not estimable | see comment | see comment | No studies measured this outcome. |
| Number of participants with ≥ 75% reduction in all seizures follow‐up: after 84 days/12 weeks' treatment (titration and maintenance) |
see comment | see comment | not estimable | see comment | see comment | No studies measured this outcome. |
| Number of participants with ≥ 50% reduction in all seizures follow‐up: after 84 days/12 weeks' treatment (titration and maintenance) | Study population | RR 2.84 (1.31 to 6.18) | 138 (1 RCT) | ⊕⊕⊕⊕ HIGH | ||
| 109 per 1000 | 311 per 1000 (143 to 676) | |||||
| Number of participants with adverse events leading to study discontinuation follow‐up: after 12 weeks |
33 per 1000 | 138 per 1000 (16 to 1,000) |
RR 4.14 (0.49 to 34.86) | 59 (1 RCT) | ⊕⊕⊝⊝ LOW1 | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ASM: anti‐seizure medication; CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1Downgraded twice because the effect estimate has a very wide confidence interval.
Summary of findings 7. Rufinamide plus ASMs compared to other ASM plus ASMs.
| Rufinamide + ASMs compared to other ASM + ASMs | ||||||
| Patient or population: Lennox‐Gastaut syndrome, any age Setting: multi‐centre, high‐income countries Intervention: rufinamide + ASMs Comparison: placebo + ASMs | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo + ASMs | Risk with rufinamide + ASMs | |||||
| Number of participants free from all seizures follow‐up: after 106 weeks' treatment (titration and maintenance) |
see comments | see comments | not estimable | see comments | see comments | No studies measured this outcome. |
| Number of participants with ≥ 75% reduction in all seizures follow‐up: after 106 weeks' treatment (titration and maintenance) |
see comments | see comments | not estimable | see comments | see comments | No studies measured this outcome. |
| Number of participants with ≥ 50% reduction in all seizures follow‐up: after 106 weeks' treatment (titration and maintenance) |
see comments | see comments | not estimable | see comments | see comments | No studies measured this outcome. |
| Number of participants with treatment‐emergent adverse events leading to study discontinuation follow‐up: after 112 weeks | Study population | RR 0.96 (0.10 to 9.57) | 37 (1 RCT) | ⊕⊕⊝⊝ LOW1 | ||
| 83 per 1000 | 80 per 1000 (8 to 798) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ASM: anti‐seizure medication; CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1Downgraded twice because the effect estimate has a very wide confidence interval.
Summary of findings 8. Topiramate plus ASMs compared to placebo plus ASMs.
| Topiramate + ASMs compared to placebo + ASMs | ||||||
| Patient or population: Lennox‐Gastaut syndrome, any age Setting: multi‐centre, USA Intervention: topiramate + ASMs Comparison: placebo + ASMs | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo + ASMs | Risk with Topiramate+ ASMs | |||||
| Number of participants free from all seizures follow‐up: after 11 weeks' treatment (titration and maintenance) |
see comment | see comment | not estimable | see comment | see comment | No studies measured this outcome. |
| Number of participants with ≥ 75% reduction in all seizures follow‐up: after 11 weeks' treatment (titration and maintenance) | Study population | Peto OR 8.22 (0.60 to 112.62) | 98 (1 RCT) | ⊕⊕⊝⊝ LOW1 | Topiramate + ASMs: 4/48 participants; placebo + ASMs: 0/50 participants. |
|
| see comment | see comment | |||||
| Number of participants with ≥ 50% reduction in all seizures follow‐up: after 11 weeks' treatment (titration and maintenance) |
see comment | see comment | not estimable | see comment | see comment | No studies measured this outcome. |
| Number of participants with adverse events leading to study discontinuation follow‐up: after 11 weeks | Study population | not estimable | 98 (1 RCT) | ⊕⊕⊝⊝ LOW2 | ||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ASM: anti‐seizure medication; CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1Downgraded twice because the effect estimate has a very wide confidence interval.
2Downgraded twice because the study was not powered to detect a between‐group difference in zero event outcomes.
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; and Table 8.
Cannabidiol (10 mg/kg and 20 mg/kg) plus ASMs versus placebo plus ASMs
Two studies contributed data for this comparison (Devinsky 2018; Thiele 2018).
Devinsky 2018 evaluated cannabidiol 10 mg/kg/day and 20 mg/kg/day, whereas Thiele 2018 evaluated only 20 mg/kg/day.
In Devinsky 2018, "A total of 13 patients (6%) discontinued either cannabidiol (11 patients) or placebo (2 patients); in 7 of the 11 patients who discontinued cannabidiol, the treatment was discontinued because of adverse events." In Thiele 2018, "14 patients in the cannabidiol group and one in the placebo group (9%) withdrew from the trial; in nine (60%) of these patients, adverse events were the primary reason for study discontinuation."
Number of participants free from drop seizures during the treatment phase
No participants were free from drop seizures (one study; 225 participants; Analysis 1.1).
1.1. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 1: Number of participants free from drop seizures during the treatment phase
Number of participants with ≥ 75% reduction in drop seizures during the treatment phase
The RR was 3.51 (95% CI 1.24 to 9.92) in favour of the cannabidiol regimen (two studies; 396 participants; Analysis 1.2).
1.2. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 2: Number of participants with ≥ 75% reduction in drop seizures during the treatment phase
Number of participants with ≥ 50% reduction in drop seizures during the treatment phase
The RR was 2.12 (95% CI 1.48 to 3.03) in favour of the cannabidiol regimen (two studies; 396 participants; Analysis 1.3).
1.3. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 3: Number of participants with ≥ 50% reduction in drop seizures during the treatment phase
Number of participants with ≥ 25% reduction in drop seizures during the treatment phase
The RR was 1.45 (95% CI 1.19 to 1.78) in favour of the cannabidiol regimen (two studies; 396 participants; Analysis 1.4).
1.4. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 4: Number of participants with ≥ 25 % reduction in drop seizures during the treatment phase
Number of participants with > 0% to < 25% reduction in drop seizures during the treatment phase
The RR was 1.58 (95% CI 0.85 to 2.93) in favour of the cannabidiol regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 225 participants; Analysis 1.5).
1.5. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 5: Number of participants with > 0% to < 25% reduction in drop seizures during the treatment phase
Number of participants with > 0% to < 25% increase in drop seizures during the treatment phase
The RR was 1.40 (95% CI 0.66 to 3.00) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 225 participants; Analysis 1.6).
1.6. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 6: Number of participants with > 0% to < 25% increase in drop seizures during the treatment phase
Number of participants with > 25% increase in drop seizures during the treatment phase
The RR was 0.71 (95% CI 0.33 to 1.53) in favour of the cannabidiol regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 225 participants; Analysis 1.7).
1.7. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 7: Number of participants with > 25 % increase in drop seizures during the treatment phase
Number of participants with improvement in the patient and caregiver Global Impression of Care scale
The RR was 1.52 (95% CI 1.22 to 1.89) in favour of the cannabidiol regimen (two studies; 392 participants; Analysis 1.8).
1.8. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 8: Number of participants with improvement in the patient and caregiver Global Impression of Care scale
Number of participants free from drop seizures during the maintenance phase
The Peto OR was 7.76 (99% CI 0.75 to 79.85) in favour of the cannabidiol regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 170 participants; Analysis 1.9).
1.9. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 9: Number of participants free from drop seizures during the maintenance phase
Number of participants with ≥ 75% reduction in drop seizures during the maintenance phase
The RR was 2.86 (95% CI 1.28 to 6.40) in favour of the cannabidiol regimen (one study; 170 participants; Analysis 1.10).
1.10. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 10: Number of participants with ≥ 75% reduction in drop seizures during the maintenance phase
Number of participants with ≥ 50% reduction in drop seizures during the maintenance phase
The RR was 1.95 (95% CI 1.25 to 3.05) in favour of the cannabidiol regimen (one study; 170 participants; Analysis 1.11).
1.11. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 11: Number of participants with ≥ 50% reduction in drop seizures during the maintenance phase
Number of participants with ≥ 25% reduction in drop seizures during the maintenance phase
The RR was 1.35 (95% CI 1.02 to 1.78) in favour of the cannabidiol regimen (one study; 170 participants; Analysis 1.12).
1.12. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 12: Number of participants with ≥ 25 % reduction in drop seizures during the maintenance phase
Number of participants with adverse events
The RR was 1.24 (95% CI 1.11 to 1.38) in favour of the placebo regimen (two studies; 396 participants; Analysis 1.13).
1.13. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 13: Number of participants with adverse events
Number of participants with treatment‐related adverse events
The RR was 1.81 (95% CI 1.29 to 2.54) in favour of the placebo regimen (one study; 171 participants; Analysis 1.14).
1.14. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 14: Number of participants with treatment‐related adverse events
Number of participants with serious adverse events
The RR was 4.94 (95% CI 1.76 to 13.85) in favour of the placebo regimen (one study; 171 participants; Analysis 1.15).
1.15. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 15: Number of participants with serious adverse events
Number of participants with adverse events leading to dose reduction
The RR was 5.93 (95% CI 0.73 to 48.22) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 171 participants; Analysis 1.16).
1.16. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 16: Number of participants with adverse events leading to dose reduction
Number of participants with adverse events leading to study discontinuation
The RR was 6.62 (95% CI 1.56 to 28.15) in favour of the placebo regimen (two studies; 396 participants; Analysis 1.17). We assessed the certainty of the evidence for this outcome to be high (Table 1).
1.17. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 17: Number of participants with adverse events leading to study discontinuation
Death
The Peto OR was 7.30 (99% CI 0.04 to 1261.58) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 171 participants; Analysis 1.18).
1.18. Analysis.

Comparison 1: Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs, Outcome 18: Death
Cinromide plus ASMs versus placebo plus ASMs
One study contributed data for this comparison (Group for the Evaluation of Cinromide 1989). This study was terminated prematurely "when it was clear to the sponsor that cinromide was not effective"; only efficacy data collected before study interruption were included in the analysis. This study also reported changes in adjuvant ASM dosage following adverse events for 62% of the cinromide group and 27% of the placebo group.
Number of participants free from all seizures
No participants were free from seizures (one study; 56 participants; Analysis 2.1). We downgraded the certainty of evidence for this outcome twice for study limitations because there was a high risk of bias for incomplete data and twice for imprecision because the study was not powered to detect a between‐group difference in zero event outcomes. Our certainty in the evidence for this outcome was very low (Table 2).
2.1. Analysis.

Comparison 2: Cinromide + ASMs versus placebo + ASMs, Outcome 1: Number of participants free from all seizures
Number of participants with ≥ 75% reduction in mean weekly seizures
The Peto OR was 9.35 (99% CI 0.45 to 194.96) in favour of the cinromide regime, but the CI included the possibility of an effect favouring either treatment regimen (one study; 56 participants; Analysis 2.2). We downgraded the certainty of evidence for this outcome twice for study limitations because there was a high risk of bias for incomplete data and twice for imprecision because the effect estimate has a very wide CI. Our certainty in the evidence for this outcome was very low (Table 2).
2.2. Analysis.

Comparison 2: Cinromide + ASMs versus placebo + ASMs, Outcome 2: Number of participants with ≥ 75% reduction in mean weekly seizures
Number of participants with ≥ 50% reduction in mean weekly seizures
The RR was 1.15 (95% CI 0.47 to 2.86) in favour of the cinromide regime, but the CI included the possibility of an effect favouring either treatment regimen (one study; 56 participants; Analysis 2.3). We downgraded the certainty of evidence for this outcome twice for study limitations because there was a high risk of bias for incomplete data and twice for imprecision because the effect estimate has a very wide CI. Our certainty in the evidence for this outcome was very low (Table 2).
2.3. Analysis.

Comparison 2: Cinromide + ASMs versus placebo + ASMs, Outcome 3: Number of participants with ≥ 50% reduction in mean weekly seizures
Number of participants with ≥ 25% reduction in mean weekly seizures
The RR was 1.07 (95% CI (95% CI 0.59 to 1.91) in favour of the cinromide regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 56 participants; Analysis 2.4).
2.4. Analysis.

Comparison 2: Cinromide + ASMs versus placebo + ASMs, Outcome 4: Number of participants with ≥ 25% reduction in mean weekly seizures
Number of participants with ≥ 0% to < 25% reduction in mean weekly seizures
The RR was 1.15 (95% CI 0.42 to 3.14) in favour of the cinromide regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 56 participants; Analysis 2.5).
2.5. Analysis.

Comparison 2: Cinromide + ASMs versus placebo + ASMs, Outcome 5: Number of participants with ≥ 0% to < 25% reduction in mean weekly seizures
Number of participants with ≥ 0% to < 25% increase in mean weekly seizures
The RR was 0.87 (95% CI 0.21 to 3.52) in favour of the cinromide regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 56 participants; Analysis 2.6).
2.6. Analysis.

Comparison 2: Cinromide + ASMs versus placebo + ASMs, Outcome 6: Number of participants with ≥ 0% to < 25% increase in mean weekly seizures
Number of participants with > 25% increase in mean weekly seizures
The RR was 0.82 (95% CI 0.30 to 2.29) in favour of the cinromide regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 56 participants; Analysis 2.7).
2.7. Analysis.

Comparison 2: Cinromide + ASMs versus placebo + ASMs, Outcome 7: Number of participants with > 25% increase in mean weekly seizures
Number of participants with improvement in global evaluation (at week 12, 18 and 24)
Week 12: the RR was 0.99 (95% CI 0.56 to 1.74) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 56 participants; Analysis 2.8).
2.8. Analysis.

Comparison 2: Cinromide + ASMs versus placebo + ASMs, Outcome 8: Number of participants with improvement in global evaluation
Week 18: the RR was 0.80 (95% CI 0.41 to 1.56) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 56 participants; Analysis 2.8).
Week 24: the RR was 1.28 (95% CI 0.62 to 2.66) in favour of the cinromide regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 56 participants; Analysis 2.8).
Number of participants with no change in global evaluation (at week 12, 18 and 24)
Weeks 12 and 18: the RR was 0.91 (0.50 to 1.64) in favour of the cinromide regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 56 participants; Analysis 2.9).
2.9. Analysis.

Comparison 2: Cinromide + ASMs versus placebo + ASMs, Outcome 9: Number of participants with no change in global evaluation
Week 24: the RR was 1.03 (95% CI 0.46 to 2.27) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 56 participants; Analysis 2.9).
Number of participants with worsening in global evaluation (at week 12, 18 and 24)
Week 12: the RR was 3.46 (95% CI 0.38 to 31.28) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 56 participants; Analysis 2.10).
2.10. Analysis.

Comparison 2: Cinromide + ASMs versus placebo + ASMs, Outcome 10: Number of participants with worsening in global evaluation
Week 18: the RR was 4.62 (95% CI 0.55 to 38.74) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 56 participants; Analysis 2.10).
Week 24: the Peto OR was 9.09 (99% CI 0.17 to 475.60) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 56 participants; Analysis 2.11).
2.11. Analysis.

Comparison 2: Cinromide + ASMs versus placebo + ASMs, Outcome 11: Number of participants with worsening in global evaluation ‐ Week 24
Death
No deaths were reported in the study.
Clobazam plus ASMs versus placebo plus ASMs
One study contributed data for this comparison (Ng 2011). According to study authors, "the most common reasons for discontinuing the study were lack of efficacy for placebo‐treated patients and adverse events for clobazam‐treated patients." In the placebo group, 30.5% of participants discontinued the study. Ten participants discontinued treatment due to lack of efficacy, four due to the request of the participant, parent or caregiver, two due to side effects, and two participants were lost to follow up. With clobazam, 13.8% of participants discontinued from the low‐dose group, 27.4% discontinued from the medium‐dose group, and 30.5% discontinued from the high‐dose group. Twenty‐four participants treated with add‐on clobazam discontinued due to side effects, five discontinued due to lack of efficacy, five discontinued due to the request of the participant, parent or caregiver, five discontinued for other reasons such as protocol violations; and four participants were lost to follow up.
A post‐hoc study investigated aggression‐related adverse events that occurred after the first dose of study drug and within 30 days after the last dose of study drug and found no difference between clobazam and placebo (RR 1.89, 95% CI 0.69 to 5.19). Among the 23 participants randomised to rufinamide who experienced aggression‐related adverse events, five had a history of aggression or behavioural problems. Among the four participants randomised to placebo who experienced aggression‐related adverse events, one person had a history of aggression or behavioural problems.
Number of participants free from drop seizures
The RR was 4.10 (95% CI 1.00 to 16.83) in favour of the clobazam regimen (one study; 217 participants; Analysis 3.1).
3.1. Analysis.

Comparison 3: Clobazam (low, medium and high doses) + ASMs versus placebo + ASMs, Outcome 1: Number of participants free from drop seizures
Number of participants with ≥ 75% reduction in drop seizures (from baseline to maintenance phase in average weekly rate)
The RR was 4.04 (95% CI 1.85 to 8.79) in favour of the clobazam regimen (one study; 217 participants; Analysis 3.2).
3.2. Analysis.

Comparison 3: Clobazam (low, medium and high doses) + ASMs versus placebo + ASMs, Outcome 2: Number of participants with ≥ 75 % reduction in drop seizures (from baseline to maintenance phase in average weekly rate)
Number of participants with ≥ 50% reduction in drop seizures (from baseline to maintenance phase in average weekly rate)
The RR was 1.88 (95% CI 1.26 to 2.81) in favour of the clobazam regimen (one study; 217 participants; Analysis 3.3).
3.3. Analysis.

Comparison 3: Clobazam (low, medium and high doses) + ASMs versus placebo + ASMs, Outcome 3: Number of participants with ≥ 50 % reduction in drop seizures (from baseline to maintenance phase in average weekly rate)
Number of participants with ≥ 25% reduction in drop seizures (from baseline to maintenance phase in average weekly rate)
The RR was 1.54 (95% CI 1.17 to 2.03) in favour of the clobazam regimen (one study; 217 participants; Analysis 3.4).
3.4. Analysis.

Comparison 3: Clobazam (low, medium and high doses) + ASMs versus placebo + ASMs, Outcome 4: Number of participants with ≥ 25 % reduction in drop seizures (from baseline to maintenance phase in average weekly rate)
Number of participants with adverse events
The RR was 1.17 (95% CI.097 to 2.03) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 238 participants; Analysis 3.5).
3.5. Analysis.

Comparison 3: Clobazam (low, medium and high doses) + ASMs versus placebo + ASMs, Outcome 5: Number of participants with adverse events
Number of participants with adverse events leading to dose reduction
The RR was 9.23 (95% CI 1.28 to 66.37) in favour of the placebo regimen (one study; 238 participants; Analysis 3.6).
3.6. Analysis.

Comparison 3: Clobazam (low, medium and high doses) + ASMs versus placebo + ASMs, Outcome 6: Number of participants with adverse events leading to dose reduction
Number of participants with aggression‐related adverse events
The RR was 1.89 (95% CI 0.69 to 5.19) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 194 participants; Analysis 3.7).
3.7. Analysis.

Comparison 3: Clobazam (low, medium and high doses) + ASMs versus placebo + ASMs, Outcome 7: Number of participants with aggression‐related adverse events
Number of participants with serious adverse events
The RR was 2.31 (95% CI 0.54 to 9.86) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 238 participants; Analysis 3.8).
3.8. Analysis.

Comparison 3: Clobazam (low, medium and high doses) + ASMs versus placebo + ASMs, Outcome 8: Number of participants with serious adverse events
Number of participants with adverse events leading to study discontinuation
The RR was 4.12 (95% CI 1.01 to 16.87) in favour of the placebo regimen (one study; 238 participants; Analysis 3.9). We assessed the certainty of the evidence for this outcome to be high (Table 3).
3.9. Analysis.

Comparison 3: Clobazam (low, medium and high doses) + ASMs versus placebo + ASMs, Outcome 9: Number of participants with adverse events leading to study discontinuation
Death
There were no deaths in this study (one study; 238 participants; Analysis 3.10).
3.10. Analysis.

Comparison 3: Clobazam (low, medium and high doses) + ASMs versus placebo + ASMs, Outcome 10: Death
Felbamate plus ASMs versus placebo plus ASMs
One study contributed data for this comparison (Felbamate Study Group 1993). One participant had treatment stopped because of somnolence and ataxia in the felbamate group and one because of pancreatitis in the placebo group. According to retrospective analysis, "Approximately 50% of patients randomised to FBM obtained at least a 50% reduction in seizure frequency compared with about 15% receiving placebo" (Jensen 1994).
Number of participants free from all seizures (recorded by closed‐circuit television and electroencephalography)
Treatment phase: the RR was 2.92 (95% CI 0.32 to 26.77) in favour of the felbamate regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 73 participants; Analysis 4.1). We downgraded the certainty of evidence for this outcome twice for imprecision (from high to low) because the effect estimate has a very wide CI (Table 4).
4.1. Analysis.

Comparison 4: Felbamate + ASMs versus placebo + ASMs, Outcome 1: Number of participants free from all seizures (recorded by closed‐circuit television and electroencephalography)
Maintenance phase: the RR was 5.84 (95% CI 0.74 to 46.11) in favour of the felbamate regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 73 participants; Analysis 4.1).
Number of participants free from all seizures
Treatment phase: No participants were free from seizures (one study; 73 participants; Analysis 4.2). We downgraded the certainty of the evidence twice for imprecision (from high to low) because the study was not powered to detect a between‐group difference in zero event outcomes (Table 4).
4.2. Analysis.

Comparison 4: Felbamate + ASMs versus placebo + ASMs, Outcome 2: Number of participants free from all seizures ‐ Treatment phase
Maintenance phase: the RR was 3.89 (95% CI 0.46 to 33.17) in favour of the felbamate regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 73 participants; Analysis 4.3).
4.3. Analysis.

Comparison 4: Felbamate + ASMs versus placebo + ASMs, Outcome 3: Number of participants free from all seizures ‐ Maintenance phase
Number of participants free from atonic seizures
Treatment phase: the Peto OR was 6.43 (99% CI 0.30 to 137.10) in favour of the felbamate regime, but the CI included the possibility of an effect favouring either treatment regimen (one study; 50 participants; Analysis 4.4).
4.4. Analysis.

Comparison 4: Felbamate + ASMs versus placebo + ASMs, Outcome 4: Number of participants free from atonic seizures
Maintenance phase: the Peto OR was 6.99 (99% CI 0.62 to 78.73) in favour of the felbamate regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 50 participants; Analysis 4.4).
Number of participants free from tonic‐clonic seizures
Treatment phase: the RR was 1.63 (95% CI 0.17 to 15.99) in favour of the felbamate regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 29 participants; Analysis 4.5.
4.5. Analysis.

Comparison 4: Felbamate + ASMs versus placebo + ASMs, Outcome 5: Number of participants free from tonic‐clonic seizures
Maintenance phase: the RR was 5.69 (95% CI 0.80 to 40.51) in favour of the felbamate regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 29 participants; Analysis 4.5).
Number of participants with severe side effects
The RR was 2.59 (95% CI 0.75 to 9.01) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 73 participants; Analysis 4.6).
4.6. Analysis.

Comparison 4: Felbamate + ASMs versus placebo + ASMs, Outcome 6: Number of participants with severe side effects
Number of participants with adverse events leading to study discontinuation
The RR was 0.97 (95% CI 0.06 to 14.97) in favour of the felbamate regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 73 participants; Analysis 4.7). We downgraded the certainty of evidence for this outcome twice for imprecision (from high to low) because the effect estimate has a very wide CI (Table 4).
4.7. Analysis.

Comparison 4: Felbamate + ASMs versus placebo + ASMs, Outcome 7: Number of participants with adverse events leading to study discontinuation
Death
No deaths were reported in the study.
Lamotrigine plus ASMs versus placebo plus ASMs
Two studies assessed this comparison (Eriksson 1998; Motte 1997) but only one contributed usable data for this review (Motte 1997).
Eriksson 1998 excluded "non‐responders" involved in an initial open phase of treatment from participating in the randomised study. The study also incompletely reported efficacy and tolerability outcomes for the subgroup of participants with LGS. The study reported that seven of the 20 children with LGS responded to treatment with > 50% seizure reduction, three children with > 75% seizure reduction, and two children became seizure free. However, individual data were only reported for 13 children with LGS, and no data were provided for the baseline number of seizures prior to crossover. In terms of adverse events, the study only reported data for the overall study population with refractory generalised epilepsy.
In this study, seven out of 13 children with LGS entered into the double‐blind phase of the trial showed improvement in the lamotrigine phase compared with the placebo phase, with one child showing a 100% reduction in their seizures. Three participants on lamotrigine had treatment withdrawn (23%), one had deterioration of seizure control, the other two developed a rash. Seven participants receiving placebo had treatment withdrawn (54%), six because of deterioration in seizure control and one other developed a rash.
In Motte 1997, 12% of participants discontinued from the study. "Seven patients in the lamotrigine group stopped treatment early, four because of protocol violations and three because of adverse events. Fourteen patients in the placebo group did not complete the study: seven had adverse events, three had protocol violations, two had a deterioration in the control of seizures, one failed to return for follow‐up, and the parents of one patient withdrew their consent."
Number of participants with ≥ 50% median reduction in all seizures
The RR was 2.12 (95% CI 1.19 to 3.76) in favour of the lamotrigine regimen (one study; 167 participants; Analysis 5.1). We assessed the certainty of the evidence for this outcome to be high (Table 5).
5.1. Analysis.

Comparison 5: Lamotrigine + ASMs versus placebo + ASMs, Outcome 1: Number of participants with ≥ 50% median reduction in all seizures
Number of participants with > 25% to < 50% median reduction in all seizures
The RR was 1.41 (95% CI 0.80 to 2.47) in favour of the lamotrigine regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 167 participants; Analysis 5.2).
5.2. Analysis.

Comparison 5: Lamotrigine + ASMs versus placebo + ASMs, Outcome 2: Number of participants with > 25% to < 50% median reduction in all seizures
Number of participants with either 0 to ≤ 25% median reduction or an increase in all seizures
The RR was 0.61 (95% CI 0.45 to 0.83) in favour of the lamotrigine regimen (one study; 167 participants; Analysis 5.3).
5.3. Analysis.

Comparison 5: Lamotrigine + ASMs versus placebo + ASMs, Outcome 3: Number of participants with either 0 to ≤ 25% median reduction or an increase in all seizures
Number of participants with ≥ 50% median reduction in drop attacks
The RR was 1.66 (95% CI 1.02 to 2.70) in favour of the lamotrigine regimen (one study; 164 participants; Analysis 5.4).
5.4. Analysis.

Comparison 5: Lamotrigine + ASMs versus placebo + ASMs, Outcome 4: Number of participants with ≥ 50% median reduction in drop attacks
Number of participants with > 25% to < 50% median reduction in drop attacks
The RR was 1.61 (95% CI 0.87 to 2.99) in favour of the lamotrigine regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 164 participants; Analysis 5.5).
5.5. Analysis.

Comparison 5: Lamotrigine + ASMs versus placebo + ASMs, Outcome 5: Number of participants with > 25% to < 50% median reduction in drop attacks
Number of participants in with either ≤ 25% median reduction or an increase in drop attacks
The RR was 0.60 (95% CI 0.43 to 0.85) in favour of the lamotrigine regimen (one study; 164 participants; Analysis 5.6).
5.6. Analysis.

Comparison 5: Lamotrigine + ASMs versus placebo + ASMs, Outcome 6: Number of participants with either ≤ 25% median reduction or an increase in the number of drop attacks
Number of participants with ≥ 50% median reduction in tonic‐clonic seizures
The RR was 2.13 (95% CI 1.21 to 3.75) in favour of the lamotrigine regimen (one study; 124 participants; Analysis 5.7.
5.7. Analysis.

Comparison 5: Lamotrigine + ASMs versus placebo + ASMs, Outcome 7: Number of participants with ≥ 50% median reduction in tonic‐clonic seizures
Number of participants with > 25% to < 50% median reduction in tonic‐clonic seizures
The RR was 0.91 (95% CI 0.33 to 2.57) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 124 participants; Analysis 5.8.
5.8. Analysis.

Comparison 5: Lamotrigine + ASMs versus placebo + ASMs, Outcome 8: Number of participants with > 25% to < 50% median reduction in tonic‐clonic seizures
Number of participants with either 0 to ≤ 25% median reduction or an increase in tonic‐clonic seizures
The RR was 0.68 (95% CI 0.49 to 0.93) in favour of the lamotrigine regimen (one study; 124 participants; Analysis 5.9).
5.9. Analysis.

Comparison 5: Lamotrigine + ASMs versus placebo + ASMs, Outcome 9: Number of participants with 0 to ≤ 25% median reduction or an increase in the number of tonic‐clonic seizures
Number of participants with adverse events leading to study discontinuation
The RR was 0.49 (95% CI 0.13 to 1.82) in favour of the lamotrigine regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 169 participants; Analysis 5.10). We downgraded the certainty of evidence for this outcome twice for imprecision because the effect estimate has a very wide CI. Our certainty in the evidence for this outcome was low (Table 5).
5.10. Analysis.

Comparison 5: Lamotrigine + ASMs versus placebo + ASMs, Outcome 10: Number of participants with adverse events leading to study discontinuation
Death
No deaths were reported in the study.
Rufinamide plus ASMs versus placebo plus ASMs
Two studies contributed data for this comparison (Glauser 2008; Ohtsuka 2014). In Glauser 2008, 15 participants (11%) discontinued from the study. "Ten patients in the rufinamide group discontinued therapy during the double‐blind phase because of adverse events (n = 6), unsatisfactory therapeutic effect (n = 3), or withdrawal of consent (n = 1). Five patients in the placebo group did not complete the study because of protocol violations (n = 2), unsatisfactory therapeutic effect (n = 1), administrative problems (n = 1), or withdrawal of consent (n = 1)."
In Ohtsuka 2014, "One patient assigned to the rufinamide group was excluded from the efficacy analysis due to inappropriate diagnosis."
Number of participants with ≥ 50% reduction in all seizures
The RR was 2.84 (95% CI 1.31 to 6.18) in favour of the rufinamide regimen (one study; 138 participants; Analysis 6.1). We assessed the certainty of the evidence for this outcome to be high (Table 6).
6.1. Analysis.

Comparison 6: Rufinamide + ASMs versus placebo + ASMs, Outcome 1: Number of participants with ≥ 50% reduction in all seizures
Number of participants with ≥ 75% reduction in tonic‐atonic seizures
The RR was 10.71 (95% CI 1.46 to 78.39) in favour of the rufinamide regimen (one study; 58 participants; Analysis 6.2).
6.2. Analysis.

Comparison 6: Rufinamide + ASMs versus placebo + ASMs, Outcome 2: Number of participants with ≥ 75 % reduction in tonic‐atonic seizures
Number of participants with ≥ 50% reduction in tonic‐atonic seizures
The RR was 2.70 (95% CI 1.52 to 4.81) in favour of the rufinamide regimen (two studies; 191 participants; Analysis 6.3).
6.3. Analysis.

Comparison 6: Rufinamide + ASMs versus placebo + ASMs, Outcome 3: Number of participants with ≥ 50% reduction in tonic‐atonic seizures
Number of participants with ≥ 25% reduction in tonic‐atonic seizures
The RR was 1.88 (95% CI 0.93 to 3.77) in favour of the rufinamide regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 58 participants; Analysis 6.4).
6.4. Analysis.

Comparison 6: Rufinamide + ASMs versus placebo + ASMs, Outcome 4: Number of participants with ≥ 25% reduction in tonic‐atonic seizures
Number of participants 'unchanged' (< 25% reduction in tonic‐atonic seizures)
The RR was 1.38 (95% CI 0.59 to 3.20) in favour of the rufinamide regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 58 participants; Analysis 6.5).
6.5. Analysis.

Comparison 6: Rufinamide + ASMs versus placebo + ASMs, Outcome 5: Number of participants 'unchanged' (< 25% reduction in tonic‐atonic seizures)
Number of participants with increased tonic‐atonic seizures
The RR was 0.36 (95% CI 0.15 to 0.85) in favour of the rufinamide regimen (one study; 58 participants; Analysis 6.6).
6.6. Analysis.

Comparison 6: Rufinamide + ASMs versus placebo + ASMs, Outcome 6: Number of participants with increased tonic‐atonic seizures
Number of participants with improvement in seizure severity rating
The RR was 1.74 (95% CI 1.13 to 2.68) in favour of the rufinamide regimen (one study; 135 participants; Analysis 6.7).
6.7. Analysis.

Comparison 6: Rufinamide + ASMs versus placebo + ASMs, Outcome 7: Number of participants with improvement in seizure severity rating
Number of participants with adverse events
The RR was 1.13 (95% CI 0.86 to 1.50) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (two studies; 197 participants; Analysis 6.8).
6.8. Analysis.

Comparison 6: Rufinamide + ASMs versus placebo + ASMs, Outcome 8: Number of participants with adverse events
Number of participants with adverse events suspected to be treatment‐related
The RR was 1.27 (95% CI 0.90 to 1.79) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 138 participants; Analysis 6.9).
6.9. Analysis.

Comparison 6: Rufinamide + ASMs versus placebo + ASMs, Outcome 9: Number of participants with adverse events suspected to be treatment‐related
Number of participants with serious adverse events
The RR was 0.86 (95% CI 0.13 to 5.97) in favour of the rufinamide regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 138 participants; Analysis 6.10).
6.10. Analysis.

Comparison 6: Rufinamide + ASMs versus placebo + ASMs, Outcome 10: Number of participants with serious adverse events
Number of participants with adverse events leading to study discontinuation
In Ohtsuka 2014, four of 29 participants in the rufinamide group did not complete the study due to adverse events, and 1 of 30 participants in the placebo group discontinued due to an adverse event. It is unclear if a participant with an inappropriate diagnosis was one of the few withdrawals experiencing adverse events. RR 4.14 (CI 0.49 to 34.86) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 59 participants; Analysis 6.11). We downgraded the certainty of evidence for this outcome twice for imprecision because the effect estimate has a very wide CI. Our certainty in the evidence for this outcome was low (Table 6).
6.11. Analysis.

Comparison 6: Rufinamide + ASMs versus placebo + ASMs, Outcome 11: Number of participants with adverse events leading to study discontinuation
Death
No deaths were reported in the study.
Rufinamide plus ASMs versus other ASM plus ASMs
One study contributed data for this comparison (Arzimanoglou 2019). Ten of 25 people (40%) in the rufinamide group discontinued treatment, primarily due to adverse events (n = 3), patient choice (n = 2), inadequate therapeutic effect (n = 2), or withdrawal of consent (n = 3). Eight of 12 people (67%) in the 'Any other ASM' group discontinued treatment, primarily due to loss to follow‐up (n = 1), patient choice (n = 1), inadequate therapeutic effect (n = 1), withdrawal of consent (n = 4) or other reason (n = 1).
Number of participants with treatment‐emergent adverse events
The RR was 1.06 (95% CI 0.79 to 1.41) in favour of the other ASM regime, but the CI included the possibility of an effect favouring either treatment regimen (one study; 37 participants; Analysis 7.1).
7.1. Analysis.

Comparison 7: Rufinamide + ASMs versus other ASM + ASMs, Outcome 1: Number of participants with treatment‐emergent adverse events
Number of participants with severe treatment‐emergent adverse events
The RR was 0.96 (95% CI 0.20 to 4.53) in favour of the rufinamide regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 37 participants; Analysis 7.2).
7.2. Analysis.

Comparison 7: Rufinamide + ASMs versus other ASM + ASMs, Outcome 2: Number of participants with severe treatment‐emergent adverse events
Number of participants with serious treatment‐emergent adverse events
The RR was 0.96 (95% CI 0.42 to 2.19) in favour of the rufinamide regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 37 participants; Analysis 7.3).
7.3. Analysis.

Comparison 7: Rufinamide + ASMs versus other ASM + ASMs, Outcome 3: Number of participants with serious treatment‐emergent adverse events
Number of participants with treatment‐emergent adverse events leading to study‐drug dose adjustment
Reduction: the Peto OR was 5.91 (99% CI 0.61 to 57.64) in favour of the other ASM regime, but the CI included the possibility of an effect favouring either treatment regimen (one study; 37 participants).
Interruption: the Peto OR was 0.04 (99% CI 0.00 to 2.17) in favour of the rufinamide regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 37 participants; Analysis 7.4).
7.4. Analysis.

Comparison 7: Rufinamide + ASMs versus other ASM + ASMs, Outcome 4: Number of participants with treatment‐emergent adverse events leading to study‐drug dose adjustment
Number of participants with treatment‐emergent adverse events leading to study discontinuation
The RR was 0.96 (95% CI 0.10 to 9.57) in favour of the rufinamide regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 37 participants; Analysis 7.5). We downgraded the certainty of evidence for this outcome twice for imprecision (from high to low) because the effect estimate has a very wide CI (Table 7).
7.5. Analysis.

Comparison 7: Rufinamide + ASMs versus other ASM + ASMs, Outcome 5: Number of participants with treatment‐emergent adverse events leading to study discontinuation
Death
The Peto OR was 4.39 (99% CI 0.02 to 1077.58) in favour of the other ASM regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 37 participants; Analysis 7.6).
7.6. Analysis.

Comparison 7: Rufinamide + ASMs versus other ASM + ASMs, Outcome 6: Death
Child Behaviour Checklist Questionnaire
The MD was 2.60 points (95% CI ‐10.30 to 15.50) in favour of the other ASM regime, but the CI included the possibility of an effect favouring either treatment regimen (one study; 19 participants; Analysis 7.7).
7.7. Analysis.

Comparison 7: Rufinamide + ASMs versus other ASM + ASMs, Outcome 7: Child Behaviour Checklist Questionnaire
Topiramate + ASMs versus placebo + ASMs
One study contributed data for this comparison (Sachdeo 1999). Of the randomised participants, only one person was withdrawn (due to patient choice), from the topiramate group.
Number of participants with ≥ 75% reduction in all seizures
The Peto OR was 8.22 (99% CI 0.60 to 112.62) in favour of the topiramate regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 98 participants; Analysis 8.1). We downgraded the certainty of evidence for this outcome twice for imprecision (from high to low) because the effect estimate has a very wide CI (Table 8).
8.1. Analysis.

Comparison 8: Topiramate + ASMs versus placebo + ASMs, Outcome 1: Number of participants with ≥75% reduction in all seizures
Number of participants free from major seizures (drop attacks and tonic‐clonic seizures)
The RR was 4.08 (95% CI 1.46 to 11.39) in favour of the topiramate regimen (one study; 96 participants; Analysis 8.2).
8.2. Analysis.

Comparison 8: Topiramate + ASMs versus placebo + ASMs, Outcome 2: Number of participants free from major seizures (drop attacks and tonic‐clonic seizures)
Number of participants with ≥ 75% reduction in major seizures (drop attacks and tonic‐clonic seizures)
The RR was 4.35 (95% CI 0.97 to 19.42) in favour of the topiramate regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 96 participants; Analysis 8.3).
8.3. Analysis.

Comparison 8: Topiramate + ASMs versus placebo + ASMs, Outcome 3: Number of participants with ≥ 75% reduction in major seizures (drop attacks and tonic‐clonic seizures)
Number of participants with ≥ 50% reduction in major seizures (drop attacks and tonic‐clonic seizures)
The RR was 4.08 (95% CI 1.46 to 11.39) in favour of the topiramate regimen (one study; 96 participants; Analysis 8.4).
8.4. Analysis.

Comparison 8: Topiramate + ASMs versus placebo + ASMs, Outcome 4: Number of participants with ≥ 50% reduction in major seizures (drop attacks and tonic‐clonic seizures)
Number of participants free from drop attacks
The Peto OR was 8.06 (99% CI 0.05 to 1398.41) in favour of the topiramate regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 96 participants; Analysis 8.5.
8.5. Analysis.

Comparison 8: Topiramate + ASMs versus placebo + ASMs, Outcome 5: Number of participants free from drop attacks
Number of participants with ≥ 75% reduction in drop attacks
The RR was 2.84 (95% CI 0.80 to 10.06) in favour of the topiramate regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 95 participants; Analysis 8.6).
8.6. Analysis.

Comparison 8: Topiramate + ASMs versus placebo + ASMs, Outcome 6: Number of participants with ≥ 75% reduction in drop attacks
Number of participants with ≥ 50% reduction in drop attacks
The RR was 1.98 (95% CI 0.87 to 4.52) in favour of the topiramate regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 95 participants; Analysis 8.7).
8.7. Analysis.

Comparison 8: Topiramate + ASMs versus placebo + ASMs, Outcome 7: Number of participants with ≥ 50% reduction in drop attacks
Number of participants free from drop attacks during the maintenance phase
The RR was 1.33 (95% CI 0.38 to 4.66) in favour of the topiramate regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 95 participants; Analysis 8.8).
8.8. Analysis.

Comparison 8: Topiramate + ASMs versus placebo + ASMs, Outcome 8: Number of participants free from drop attacks during the maintenance phase
Number of participants with severe adverse events
The RR was 2.29 (95% CI 0.86 to 6.11) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 98 participants; Analysis 8.9).
8.9. Analysis.

Comparison 8: Topiramate + ASMs versus placebo + ASMs, Outcome 9: Number of participants with severe adverse events
Number of participants with adverse events leading to dose reduction or temporary discontinuation
The RR was 3.13 (95% CI 0.90 to 10.85) in favour of the placebo regimen but the CI included the possibility of an effect favouring either treatment regimen (one study; 98 participants; Analysis 8.10).
8.10. Analysis.

Comparison 8: Topiramate + ASMs versus placebo + ASMs, Outcome 10: Number of participants with adverse events leading to dose reduction or temporary discontinuation
Number of participants with adverse events leading to study discontinuation
No adverse events led to study discontinuation (one study; 98 participants; Analysis 8.11). We downgraded the certainty of the evidence twice for imprecision (from high to low) because the study was not powered to detect a between‐group difference in zero event outcomes (Table 8).
8.11. Analysis.

Comparison 8: Topiramate + ASMs versus placebo + ASMs, Outcome 11: Number of participants with adverse events leading to study discontinuation
Death
No deaths were reported in the study.
Discussion
LGS is a complex and severe developmental and epileptic encephalopathy that manifests as multiple seizure types, which often evolve over time. Drop seizures (typically tonic, atonic or myoclonic‐atonic), is the characteristic seizure type. It is a syndrome that is difficult to treat, with many people receiving combination therapy (polypharmacy) but frequently without achieving seizure‐freedom. The aim of this review was to assess the efficacy and tolerability of anti‐seizure medications in the treatment of LGS.
LGS is thought to account for up to 5% of all childhood epilepsies,yet we found only 11 RCTs for the pharmaceutical treatment of this syndrome. There are several possible explanations for this lack of evidence. LGS is primarily a syndrome of childhood; long‐term prognosis is expected to be poor with regard to the cognitive outcome, but seizures tend to become less troublesome into adulthood (Ferlazzo 2010; Vignoli 2017). Preferably, drug treatments would be evaluated earlier in the course of the condition, closer to seizure onset. There has traditionally been reluctance to set up trials in the paediatric age group as it is both difficult and expensive, and continues to be so, despite the early exclusivity clause set out by EU Regulations (European Union 2006). Due to the low incidence of LGS (two cases per 100,000 children), a multicentre collaborative study design to enrol the numbers of individuals is required for sufficient power with consensus on the selection of individuals (diagnostic criteria), drug therapy, and outcome measures (Arzimanoglou 2009). Investigation of adjunctive and especially monotherapy ASM regimens is difficult because LGS characteristics usually evolve over time, including multiple seizure types and typical EEG appearances. As such, many individuals will have already received several ASMs at diagnosis. The natural history of the syndrome shows that the frequency and type of seizures often fluctuate over time, which means the observed improvement or deterioration might not be an effect of the study intervention. Most challenging is the adequate observation and reporting of seizure types with more subtle clinical manifestations, such as atypical absence and myoclonic seizures. This variation may account for the heterogeneous seizure outcome measures applied across the studies included in this review. Some more recent RCTs use pragmatic 'seizure definitions', such as drop seizures with motor manifestations resulting in loss of posture (Devinsky 2018; Ng 2011; Thiele 2018); this approach might be more quantifiable and have more clinical impact. Studies included in this review have applied different seizure outcome measures, with more recent studies focusing on drop seizures, a seizure type that is measurable and is expected to have the most negative clinical impact. Observation and recording of the different seizure types, in particular, those with more subtle clinical manifestations, such as atypical absence and myoclonic seizures, is challenging for carers of participating patients. This factor may be one reason for the variation in seizure outcome measures applied across studies in this review. Furthermore, LGS is an aetiologically heterogeneous condition, and the different aetiologies of LGS could affect the effectiveness outcomes of the different ASMs. For example, selected patients with an underlying monogenetic disorder such as a SCN2A mutation (resulting in gain or loss of sodium channel function) and LGS phenotype and may respond differently to sodium channel‐blocking ASMs (Brunklaus 2020; Wolff 2017).
We found only one trial that included behaviour as a primary outcome domain, even though behaviour and cognition are frequently cited by families as being the most difficult features of the syndrome to accept and manage. The primary focus of most trials of ASMs is on seizure control and it is expected that other types of medications would be indicated for modifying cognitive and behavioural function rather than ASMs.
Recent clinical practice guidelines have recommended the use of some ASMs for the treatment of LGS. The 2004 guidelines of the American Academy of Neurology and American Epilepsy Society for pharmacological management of treatment‐resistant epilepsy (French 2004) found felbamate, lamotrigine, and topiramate to be effective ASMs in treating LGS. The 2018 update of these guidelines (Kanner 2018) listed also rufinamide as an established treatment, reporting that clobazam should be considered to decrease seizure frequency. According to the 2012 National Institute for Health and Care Excellence (NICE) guidelines (NICE 2012), add‐on ASMs that may be considered for children, young people and adults with LGS are rufinamide and topiramate; if they are ineffective or not tolerated, felbamate can be offered. Carbamazepine, gabapentin, oxcarbazepine, pregabalin, tiagabine or vigabatrin should be avoided. According to a 2019 NICE technology appraisal guidance (NICE 2019), cannabidiol with clobazam is recommended as an option for patients aged two years and older with LGS, if their drop seizures are not controlled after 2 or more ASMS, and provided that "the frequency of drop seizures is checked every 6 months, and cannabidiol is stopped if the frequency has not fallen by at least 30% compared with the 6 months before starting treatment."
Summary of main results
We found that the evidence for drug treatment of LGS related specifically to the use of anti‐seizure medications. Some of the included studies involved the treatment of children only, and others involved both children and adults with LGS. All 11 included trials (1277 participants) used ASMs as add‐on therapy and had variable periods of dose adjustment and maintenance; we found no trials of ASMs used as monotherapy and no head‐to‐head comparisons of specific add‐on drugs. We identified one trial registry record for a study that planned to compare add‐on rufinamide with lamotrigine but the study was reported to have been terminated because those supporting the study did not want to proceed with marketing the product.
Three included RCTs reported outcomes for both overall seizure reduction and adverse events leading to study discontinuation. We found high‐certainty evidence that add‐on lamotrigine increased the number of participants with at least 50% reduction in the average number of reported seizures and low‐certainty evidence that add‐on lamotrigine may have reduced the number of participants with adverse events leading to study discontinuation when compared to add‐on placebo. We also found high‐certainty evidence that add‐on rufinamide increased the number of participants with at least 50% reduction in the average number of reported seizures when compared with add‐on placebo and low‐certainty evidence that add‐on rufinamide may have made little or no difference compared to add‐on placebo or another unspecified ASM in terms of reducing the number of participants with adverse events leading to study discontinuation. Add‐on topiramate may have increased the number of participants with at least 75% reduction in the average number of reported seizures, and probably made little or no difference to the number of adverse events leading to study discontinuation when compared to add‐on placebo (low‐certainty evidence). Add‐on felbamate (treatment phase) may have made little or no difference in terms of complete seizure freedom and adverse events leading to study discontinuation when compared to add‐on placebo (low certainty evidence). However, we found that when seizures were recorded under EEG monitoring, add‐on felbamate may have increased seizure freedom compared to add‐on placebo. We remain uncertain whether other add‐on drug therapies, including cannabidiol, cinromide and clobazam, reduced all types of seizures because this outcome was not reported or had very low‐certainty evidence. Despite therapeutic potential for reducing drop seizures, we found high‐certainty evidence that add‐on cannabidiol and add‐on clobazam increased the number of participants with adverse events leading to study discontinuation compared to add‐on placebo. We did not find any evidence for adverse events leading to study discontinuation in the comparison of add‐on cinromide with add‐on placebo.
There were insufficient data to perform subgroup analysis for the pre‐specified variables, dosage, timing, and length of treatment. Most of the evidence included in this review related to people from middle‐ or high‐income countries and, where reported, participants of white ethnicity.
Overall completeness and applicability of evidence
We excluded three studies on the basis that data were not usable. However, we did not attempt to obtain additional data from the authors because the studies were published more than 25 years ago. We acknowledge this as a limitation of the review. In addition, we listed one potentially relevant study as awaiting classification in the absence of a translation into English. We also acknowledge this as a limitation of the review.
All included trials used ASMs as add‐on therapy. Hence, the data on the treatment of LGS from RCTs refer to ASMs given as adjunctive treatments. The earliest trial of an add‐on therapy, cinromide, is not currently used in clinical practice and evidence relates only to patients up to the age of 18 years. We found limited reporting of outcomes for cognition and behaviour. We also found that some of the more recent studies focused on drop seizure reduction as a primary outcome. Our 'Summary of findings' tables focused on overall seizure cessation, reduction and adverse events in accordance with the previous version of this review and because multiple types of seizures are reported in LGS.
Quality of the evidence
For outcomes included in the Summary of Findings table, we found the certainty of evidence to be of moderate‐ to very low‐certainty. We primarily downgraded outcomes because the evidence was not powered to detect a between‐group difference (study limitations). We also downgraded outcomes due to imprecise estimates with wide or very wide CIs. In addition, we downgraded outcomes from one study that was judged to have a very serious risk of bias due to attrition and selective reporting (Group for the Evaluation of Cinromide 1989).
Potential biases in the review process
We made every effort to identify all RCTs on the use of ASMs for LGS through a comprehensive search of the literature, and it is unlikely that we failed to identify large relevant studies. However, despite our efforts, there remains the possibility that we have missed small studies published in the less accessible literature. We also contacted drug companies and experts in the field to obtain information on ongoing trials or unpublished results. Finally, we contacted authors of one included study, published in 2019, to obtain information on missing data, and we contacted trialists to determine the completion of another study. We acknowledge a potential bias in our review process because we did not attempt to contact all study authors for additional data. Unavoidably, some of the authors of this review were familiar with most of the included trials before updating this review. However, data extraction was undertaken blind to the results of the prior version of the review.
Agreements and disagreements with other studies or reviews
We identified a narrative review of drug therapies for LGS that concluded a lack of evidence of the efficacy of any one drug over another. The review reported that drug therapies are difficult to compare even in RCTs, and suggested that non‐medical therapies may be considered after the failure of two to three drugs (Borrelli 2019). We found some evidence in favour of the use of add‐on lamotrigine compared with add‐on placebo in terms of both overall reduction in average reported seizures and adverse events leading to study discontinuation. However, we also found a lack of RCTs assessing the therapeutic potential of monotherapy and head‐to‐head comparison of specific add‐on ASMs. It is beyond the scope of this review to make recommendations on the use of non‐medical therapies for LGS.
We identified several reviews of individual add‐on ASMs for the treatment of LGS. In 2018, a meta‐analysis of add‐on cannabidiol versus add‐on placebo was performed, based on two studies included in this review (Devinsky 2018 and Thiele 2018). In addition to the assessment of different types of seizures, this meta‐analysis also included data for all seizure frequency. The review authors reported that "rates of > 50% reduction of all seizures were also higher among patients randomised to the active drug rather than placebo (37.2% vs 21.2%; RR 1.76 (95% CI 1.07‐2.88); P = 0.025)" (Lattanzi 2018). However, we did not find outcome data reported for greater than 50% reduction in all seizures in Devinsky 2018 or Thiele 2018; these data were not included in our analysis of the evidence.
Previously, an indirect comparison was undertaken of add‐on felbamate, lamotrigine, topiramate, rufinamide and clobazam (Cramer 2013). Although the authors found some evidence in favour of add‐on clobazam, they acknowledged several limitations including overlapping 95% CIs, omission of adverse event outcomes and studies often using dose ranges rather than a specific dosage. The authors of this analysis were also employed by the manufacturer of clobazam. We did not identify publication bias in the included studies in this review but we did find that some study investigators declared financial interests in their study sponsor.
In 2010, one study evaluated the cost effectiveness of rufinamide in the treatment of LGS. Based on data from the UK, this study concluded that, for managing drop seizures, "if society is willing to pay an additional £250 or more for a 1% increase in patients achieving at least a 50% reduction in seizure frequency over 3 years, rufinamide has more than an 80% probability of being cost effective compared with either lamotrigine or topiramate" (Benedict 2010). We did not assess cost‐effectiveness in this review, but recognise a need for up‐to‐date modelling to help inform decision‐making.
Authors' conclusions
Implications for practice.
There is currently a lack of randomised controlled trial (RCT) evidence on the optimal monotherapy for Lennox‐Gastaut syndrome (LGS). However, we found high‐certainty evidence for reduction in average reported seizures with add‐on lamotrigine and rufinamide, and low‐certainty evidence that these treatments may have reduced or made little or no difference to the number of participants with adverse events leading to study discontinuation, when compared with add‐on placebo or another add‐on ASM, respectively. We found very little information on the effects of treatment on development, cognition and behaviour, and incomplete reporting for overall seizure reduction and adverse events leading to study discontinuation across most RCTs of add‐on anti‐seizure medications (ASMs).
Implications for research.
There is a need for high‐quality RCTs to establish the optimal ASM monotherapy for LGS and optimal add‐on treatment through head‐to‐head comparison of add‐on ASMs. We also think more research is necessary across ethnically and geographically diverse populations; outcome reporting needs to include overall seizure cessation and reduction (e.g. recorded under observation if reporting is too difficult), and the impact on developmental progress, cognition, and behaviour. We think future research will need to consider economic evaluation of the different drug treatments for LGS. We also think further studies could explore the efficacy of ASMs within different age groups and aetiologies of LGS.
What's new
| Date | Event | Description |
|---|---|---|
| 7 September 2022 | Amended | Slight amendments made to text in response to feedback |
History
Protocol first published: Issue 4, 2001 Review first published: Issue 3, 2003
| Date | Event | Description |
|---|---|---|
| 2 March 2020 | New citation required and conclusions have changed | Overall the conclusions have expanded since the last version of this review. Conclusions are unchanged for the anti‐seizure medications that were included in the previous version of the review; however the review now includes a few other drugs which were not previously included. |
| 2 March 2020 | New search has been performed | Searches updated 2 March 2020; two new studies have been included. |
| 18 October 2012 | New citation required but conclusions have not changed | Three new studies have been included (Conry 2009; Ng 2011; Glauser 2009). Conclusions remain unchanged. |
| 18 October 2012 | New search has been performed | Searches updated 18 October 2012. |
Acknowledgements
We wish to thank the Cochrane Epilepsy Group for continuous support provided as we prepared this review.
We wish to acknowledge the hard and valuable work that went into the original version of the review by Eleanor Hancock and Helen Cross.
We, and the Cochrane Epilepsy Group, are grateful to the following peer reviewers for their time and comments: Richard Appleton, Myrsini Gianatsi and Danial Sayyad.
We are indebted to the following epilepsy experts, whom we contacted for information about unpublished or ongoing studies: Oriano Mecarelli, Eugen Trinka, Hans Hartmann, Ronit Pressler, Alessandro Patrini (UCB Pharma), Alberto Milan (EISAI), Bruno Ferrò (UCB Pharma), Alessandro Patrini, and Eva Francesca Brognoli (GW Pharmaceuticals).
This review update was supported by the National Institute for Health Research via a Cochrane Incentive Award (NIHR130787) and Cochrane Infrastructure funding to the Epilepsy Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS, or the Department of Health and Social Care.
Appendices
Appendix 1. CRS Web search strategy
#1 MESH DESCRIPTOR Lennox Gastaut Syndrome EXPLODE ALL AND CENTRAL:TARGET
#2 (Lennox Gastaut):AB,KW,MC,MH,TI AND CENTRAL:TARGET
#3 #1 OR #2
Appendix 2. MEDLINE search strategy
The following search strategy includes a modification of the Cochrane Highly Sensitive Search Strategy for identifying randomised trials (Lefebvre 2020).
1. exp Lennox Gastaut Syndrome/ or Lennox Gastaut.tw.
2. (randomised controlled trial or controlled clinical trial or pragmatic clinical trial).pt. or (randomi?ed or placebo or randomly).ab.
3. clinical trials as topic.sh.
4. trial.ti.
5. 2 or 3 or 4
6. exp animals/ not humans.sh.
7. 5 not 6
8. 1 and 7
9. remove duplicates from 8
Data and analyses
Comparison 1. Cannabidiol (10 mg/kg and 20 mg/kg) + ASMs versus placebo + ASMs.
Comparison 2. Cinromide + ASMs versus placebo + ASMs.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Number of participants free from all seizures | 1 | 56 | Peto Odds Ratio (Peto, Fixed, 99% CI) | Not estimable |
| 2.2 Number of participants with ≥ 75% reduction in mean weekly seizures | 1 | 56 | Peto Odds Ratio (Peto, Fixed, 99% CI) | 9.35 [0.45, 194.96] |
| 2.3 Number of participants with ≥ 50% reduction in mean weekly seizures | 1 | 56 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.47, 2.86] |
| 2.4 Number of participants with ≥ 25% reduction in mean weekly seizures | 1 | 56 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.59, 1.91] |
| 2.5 Number of participants with ≥ 0% to < 25% reduction in mean weekly seizures | 1 | 56 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.42, 3.14] |
| 2.6 Number of participants with ≥ 0% to < 25% increase in mean weekly seizures | 1 | 56 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.21, 3.52] |
| 2.7 Number of participants with > 25% increase in mean weekly seizures | 1 | 56 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.30, 2.29] |
| 2.8 Number of participants with improvement in global evaluation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.8.1 Week 12 | 1 | 56 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.56, 1.74] |
| 2.8.2 Week 18 | 1 | 56 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.41, 1.56] |
| 2.8.3 Week 24 | 1 | 56 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.62, 2.66] |
| 2.9 Number of participants with no change in global evaluation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.9.1 Week 12 | 1 | 56 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.50, 1.64] |
| 2.9.2 Week 18 | 1 | 56 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.50, 1.64] |
| 2.9.3 Week 24 | 1 | 56 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.46, 2.27] |
| 2.10 Number of participants with worsening in global evaluation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.10.1 Week 12 | 1 | 56 | Risk Ratio (M‐H, Random, 95% CI) | 3.46 [0.38, 31.28] |
| 2.10.2 Week 18 | 1 | 56 | Risk Ratio (M‐H, Random, 95% CI) | 4.62 [0.55, 38.74] |
| 2.11 Number of participants with worsening in global evaluation ‐ Week 24 | 1 | 56 | Odds Ratio (M‐H, Fixed, 99% CI) | 9.09 [0.17, 475.60] |
Comparison 3. Clobazam (low, medium and high doses) + ASMs versus placebo + ASMs.
Comparison 4. Felbamate + ASMs versus placebo + ASMs.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Number of participants free from all seizures (recorded by closed‐circuit television and electroencephalography) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1.1 Treatment phase | 1 | 73 | Risk Ratio (M‐H, Random, 95% CI) | 2.92 [0.32, 26.77] |
| 4.1.2 Maintenance phase | 1 | 73 | Risk Ratio (M‐H, Random, 95% CI) | 5.84 [0.74, 46.11] |
| 4.2 Number of participants free from all seizures ‐ Treatment phase | 1 | 73 | Odds Ratio (M‐H, Fixed, 99% CI) | Not estimable |
| 4.3 Number of participants free from all seizures ‐ Maintenance phase | 1 | 73 | Risk Ratio (M‐H, Random, 95% CI) | 3.89 [0.46, 33.17] |
| 4.3.1 Maintenance phase | 1 | 73 | Risk Ratio (M‐H, Random, 95% CI) | 3.89 [0.46, 33.17] |
| 4.4 Number of participants free from atonic seizures | 1 | Peto Odds Ratio (Peto, Fixed, 99% CI) | Subtotals only | |
| 4.4.1 Treatment phase | 1 | 50 | Peto Odds Ratio (Peto, Fixed, 99% CI) | 6.43 [0.30, 137.10] |
| 4.4.2 Maintenance phase | 1 | 50 | Peto Odds Ratio (Peto, Fixed, 99% CI) | 6.99 [0.62, 78.73] |
| 4.5 Number of participants free from tonic‐clonic seizures | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.5.1 Treatment phase | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 1.62 [0.17, 15.99] |
| 4.5.2 Maintenance phase | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 5.69 [0.80, 40.51] |
| 4.6 Number of participants with severe side effects | 1 | 73 | Risk Ratio (M‐H, Random, 95% CI) | 2.59 [0.75, 9.01] |
| 4.7 Number of participants with adverse events leading to study discontinuation | 1 | 73 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.06, 14.97] |
Comparison 5. Lamotrigine + ASMs versus placebo + ASMs.
Comparison 6. Rufinamide + ASMs versus placebo + ASMs.
Comparison 7. Rufinamide + ASMs versus other ASM + ASMs.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 7.1 Number of participants with treatment‐emergent adverse events | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.79, 1.41] |
| 7.2 Number of participants with severe treatment‐emergent adverse events | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.20, 4.53] |
| 7.3 Number of participants with serious treatment‐emergent adverse events | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.42, 2.19] |
| 7.4 Number of participants with treatment‐emergent adverse events leading to study‐drug dose adjustment | 1 | Peto Odds Ratio (Peto, Fixed, 99% CI) | Subtotals only | |
| 7.4.1 Reduction | 1 | 37 | Peto Odds Ratio (Peto, Fixed, 99% CI) | 5.91 [0.61, 57.64] |
| 7.4.2 Interruption | 1 | 37 | Peto Odds Ratio (Peto, Fixed, 99% CI) | 0.04 [0.00, 2.17] |
| 7.5 Number of participants with treatment‐emergent adverse events leading to study discontinuation | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.10, 9.57] |
| 7.6 Death | 1 | 37 | Peto Odds Ratio (Peto, Fixed, 99% CI) | 4.39 [0.02, 1077.58] |
| 7.7 Child Behaviour Checklist Questionnaire | 1 | 19 | Mean Difference (IV, Random, 95% CI) | 2.60 [‐10.30, 15.50] |
Comparison 8. Topiramate + ASMs versus placebo + ASMs.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Arzimanoglou 2019.
| Study characteristics | ||
| Methods | Multicentre, randomised, open‐label, Phase III study designed to evaluate the safety, tolerability, pharmacokinetics (PK), and behavioural effects of adjunctive rufinamide in paediatric patients (1 to < 4 years of age) with inadequately controlled seizures associated with LGS. Duration of participation:"106‐week treatment period, including an initial 2‐week titration phase and a 104‐week maintenance phase." This was preceded by 1‐8 weeks screening and baseline, and followed by 2 weeks tapering and a final visit 4 weeks later at 116 weeks. |
|
| Participants |
Duration of participation (from recruitment to last follow‐up):"Eligible patients were 1 to < 4 years of age with a clinical diagnosis of LGS, which might have included presence of a slow background EEG rhythm, slow spike‐wave pattern (< 3 Hz), and/or the presence of polyspikes." Inclusion criteria: • Aged 1 to less than 4 years old; • Clinical diagnosis of LGS, which might include the presence of a slow background EEG rhythm, slow spikes‐waves pattern (< 3 Hz), polyspikes (care should be taken to not include benign myoclonic epilepsy of infancy, atypical benign partial epilepsy [pseudo‐Lennox syndrome], or continuous spike‐waves of slow sleep); • Fixed and documented doses of 1‐3 concomitant regionally approved ASMs for a minimum of 4 weeks prior to randomisation with an inadequate response to treatment; • Consistent seizure documentation (i.e. no uncertainty of the presence of seizures) during the pre‐randomisation phase; • Written informed consent provided by parent(s)/legal representative(s); • Ability to comply with all aspects of the protocol. Exclusion criteria: • Familial short QT syndrome; • Prior treatment with rufinamide within 30 days of baseline visit or discontinuation of rufinamide treatment due to safety issues related to rufinamide; • Evidence of clinically significant disease (e.g. cardiac, respiratory, gastrointestinal, renal disease) that in the opinion of the investigator(s) could affect the patient’s safety or study conduct; • Hypersensitivity to rufinamide and/or triazole, or any of the excipients; • Any history of or concomitant medical condition that, in the opinion of the investigator, would compromise the patient’s ability to safely complete the study; • Scheduled surgery during the projected course of the study. Total number randomised: 37 Baseline imbalances: "The 2 treatment groups were generally well‐balanced for age, weight, and height." However, as with the interim 6‐month data published separately, table 1 suggests a higher proportion of males were in the ‘Any other ASM’ group. The categories for ethnicity are also different to those reported in the 6‐month dataset (Arzimanoglou 2015) Withdrawals and exclusions: 10 of 25 people (40%) in the rufinamide group discontinued and 8 of 12 people (67%) in the 'Any other ASM' group discontinued. Age at onset: Mean, months (SD): rufinamide group 28.3 (10); 'Any other ASM' group 29.8 (9.9) Median, months (min‐max): rufinamide group 28 (12‐46); 'Any other ASM' group 30.5 (13‐47). Age at diagnosis: Mean (SD) time since diagnosis was 19.9 (9.9) months in the rufinamide group and 23.0 (9.5) in the 'Any other ASM' group. Age at start of treatment: Not reported. Sex: M:F (%): rufinamide group 14:11 (56:44); 'Any other ASM' group 10:2 (83.3: 16.7). Race/ethnicity: Hispanic/Latino: Non‐Hispanic or Latino: rufinamide group 5:20 (20:80); 'Any other ASM' group 3:9 (25:75). Type of seizures: Partial (%): rufinamide group 15 (60); 'Any other ASM' group 7 (58.3); Absence (%): rufinamide group 5 (20); 'Any other ASM' group 4 (33.3); Atypical absence (%): rufinamide group 12 (48); 'Any other ASM' group 6 (50); Myoclonic (%): rufinamide group 15 (60); 'Any other ASM' group 10 (83.3); Clonic %): rufinamide group 6 (24); 'Any other ASM' group 4 (33.3); Tonic‐atonic (%): rufinamide group 15 (60); 'Any other ASM' group 8 (66.7); Primary generalised tonic‐clonic (%): rufinamide group 6 (24); 'Any other ASM' group 3 (25); Other: rufinamide group 9 (36); 'Any other ASM' group 1 (8.3); Seizure frequency during the baseline period: not reported. Number of background drugs: "Patients were receiving stable doses of 1‐3 concomitant ASMs for a minimum of 4 weeks prior to randomisation with an inadequate response, and had consistent seizure documents with no uncertainty of the presence of seizures during the pre‐randomisation phase… Among patients in the Safety Analysis Set, 8.1% were taking 1 ASM at baseline, 37.8% were taking 2 ASMs, 45.9% were taking 3 ASMs, 2.7% were taking 4 ASMs, and 5.4% were taking 5 ASMs. At baseline, the most common concomitant ASMs (≥ 20% in either treatment group) were valproic acid, levetiracetam, topiramate, diazepam, vigabatrin, clobazam, and lamotrigine (Table 2). The add‐on ASMs selected by investigators at randomisation for the 'Any other ASM' group were lamotrigine (41.7%), clobazam and topiramate (16.7% each), and phenobarbital, valproic acid, and zonisamide." Severity of illness: Not reported. Comorbidities: Not reported. |
|
| Interventions |
Group name: rufinamide Number randomised to group:25 Description: "…add‐on therapy with rufinamide oral suspension." "The 40 mg/ml oral suspension of rufinamide used in this study has been shown to be bioequivalent to the 400‐mg oral tablet formulation previously used in Study 022… Titration of rufinamide began at a 10 mg/kg/day dose. The dose was increased by 10 mg/kg/day every 3 days to 40 mg/kg/day, at which point the dose was increased by 5mg/kg/day to a target maintenance dose of 45 mg/kg/day, given in 2 equally divided doses. If tolerability issues arose, titration could occur more slowly and/or conclude at a lower maintenance dose. Once the maintenance dose of rufinamide was reached, further dose adjustments were permitted according to the investigator's discretion." Duration of baseline period: Screening and baseline phases lasted 1‐8 weeks in total. Duration of treatment period:"106‐week treatment period, including an initial 2‐week titration phase and a 104‐week maintenance phase." Co‐interventions:"Among patients in the Safety Analysis Set, 8.1% were taking 1 ASM at baseline, 37.8% were taking 2 ASMs, 45.9% were taking 3 ASMs, 2.7% were taking 4 ASMs, and 5.4% were taking 5 ASMs. At baseline, the most common concomitant ASMs (≥ 20% in either treatment group) were valproic acid, levetiracetam, topiramate, diazepam, vigabatrin, clobazam, and lamotrigine." Compliance: "Overall, 18 patients discontinued from the study (rufinamide: n = 10 [40%]; any other ASM: n = 8 [67%])." Rufinamide group: 15/25 (60%) completed. Any other ASM group: 4/12 (33%) completed. Control group name: Any other ASM group Number randomised to group: 12 Description: "Any other approved ASM of the investigator's choice for a 106‐week treatment period, including an initial 2‐week titration phase and a 104‐week maintenance phase" "The administration of other ASMs was undertaken according to the investigator's usual practice by allowing the investigator to add any other approved add‐on ASM of their choice." "Among patients in the Safety Analysis Set, 8.1% were taking 1 ASM at baseline, 37.8% were taking 2 ASMs, 45.9% were taking 3 ASMs, 2.7% were taking 4 ASMs, and 5.4% were taking 5 ASMs. At baseline, the most common concomitant ASMs (≥ 20% in either treatment group) were valproic acid, levetiracetam, topiramate, diazepam, vigabatrin, clobazam, and lamotrigine. The add‐on ASMs selected by investigators at randomisation for the any‐other‐ASM group were lamotrigine (41.7%), clobazam and topiramate (16.7% each), and phenobarbital, valproic acid, and zonisamide (8.3% each)." Duration of baseline period:Screening and baseline phases lasted 1‐8 weeks in total Duration of treatment period:"106‐week treatment period, including an initial 2‐week titration phase and a 104‐week maintenance phase." Compliance:"Overall, 18 patients discontinued from the study (rufinamide: n = 10 [40%]; any other ASM: n = 8 [67%])." Rufinamide group: 15/25 (60%) completed. Any other AED group: 4/12 (33%) completed. |
|
| Outcomes |
Outcome 1: All treatment‐emergent adverse events Time points measured: "An adverse event (AE) was considered treatment‐emergent if the AE was: (a) absent at baseline and emerged during treatment; (b) present at baseline but stopped before treatment and re‐emerged during treatment; or (c) continuous from baseline but became more severe during treatment relative to its pretreatment state." Unit of measurement: Unclear. The definition does not suggest outcome by person although the results table includes % data that suggest outcome reporting by person. Imputation of missing data: Not applicable. Assumed risk estimate:Not reported. Statistical power:"The final Study 303 protocol used a sample size of 75 patients (50 randomised to rufinamide and 25 to any other ASM), to provide 84% power to detect a mean difference of approximately 23 (standard deviation [SD] ¼ 25) in CBCL scores between children with documented psychopathological issues and those without. However, due to the difficulties in enrolling patients in this age group, the sample size was reduced to 37 patients (25 randomised to rufinamide and 12 to any other ASM) during the interim analysis. A sample size of 37 patients provided 72% power. Overall, a total of 24 patients receiving rufinamide and 9 patients receiving any other ASM had CBCL assessments at various times by the end of the study. A sample size of 24 patients had 70% power to detect events with a frequency of 5% and over 90% power to detect events with a frequency of 10%." Outcome 2: Serious treatment‐emergent adverse events Time points measured:See above. Outcome 3: Child Behaviour Checklist (CBLC) questionnaire Time points measured:"In Study 303, the primary efficacy endpoint was the CBCL Total Problems score at the end of the 2‐year treatment period. Change from baseline in CBCL subscores was also assessed as an exploratory endpoint." Time points reported: "The mean and mean change from baseline in CBCL Total Problems scores… (based on the means across Weeks 24, 56, 88, and 106)." Outcome definition:"The CBCL is a 99‐item questionnaire, completed by the patient's parent or guardian, that rates 8 problem areas: emotionally reactive, anxious/depressed, somatic complaints, withdrawn, sleep problems, attention problems, aggressive behavior, and other problems; and additionally produces 3 summary scores: Internalizing, Externalizing, and Total Problems. The Total Problems score is the sum of all problem areas plus 1 additional item and higher scores indicate more problems." Is outcome validated?Unclear: "A more sensitive behavior scale may be required to assess behavior in this patient population in future studies." |
|
| Notes | Study funding sources:The study was funded by Eisai Inc. and all authors were actively or previously working with Eisai Inc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "patients were randomized" "Patients were assigned to treatment groups (rufinamide or any other ASM) using a computer‐generated random allocation sequence." |
| Allocation concealment (selection bias) | Low risk | "Patients were assigned to treatment groups (rufinamide or any other ASM) using a computer‐generated random allocation sequence, which was approved and locked by an independent statistician. Randomization and dose dispensing at each visit was performed centrally by an Interactive Voice Response System." |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not reported. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | There was a high rate of discontinuation from both arms of this study: rufinamide: n = 10 [40%]; any other ASM: n = 8 [67%] yet the Abstract concludes "Long‐term (2 years) adjunctive rufinamide was well tolerated in pediatric patients with LGS." |
| Selective reporting (reporting bias) | Low risk | None identified. |
| Other bias | Low risk | None identified. |
Devinsky 2018.
| Study characteristics | ||
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled trial to assess the efficacy and safety of two doses of cannabidiol, as compared with placebo, added to a regimen of conventional anti‐seizure medication to treat drop seizures in patients with the Lennox–Gastaut syndrome. Parallel RCT "…2:2:1:1 ratio, to receive cannabidiol at a dose of either 20 mg per kilogram of body weight per day (the 20‐mg cannabidiol group) or 10 mg per kilogram per day (the 10‐mg cannabidiol group) or matching placebo." Duration of treatment: "Patients were followed for up to 24 weeks. The trial comprised a 4‐week baseline period, a 14‐week treatment period (2 weeks of dose escalation, followed by 12 weeks of stable dosing [maintenance phase]), a tapering period of up to 10 days, and a 4‐week safety follow‐up period after discontinuation of cannabidiol or placebo." |
|
| Participants |
Population description:"Patients with Lennox–Gastaut syndrome were eligible for inclusion in the trial if they were between 2 and 55 years of age; had an electroencephalogram that showed a pattern of slow (< 3.0 Hz) spike‐and‐wave complexes, which is characteristic of the disorder; and had at least two types of generalised seizures, including drop seizures, for at least 6 months. A drop seizure was defined as an epileptic seizure (atonic, tonic, or tonic–clonic) involving the entire body, trunk, or head that leads or could lead to a fall, injury, or slumping in a chair." Inclusion criteria: See above population description. Also, "Eligible patients were taking between one and four antiepileptic drugs and had at least two drop seizures each week during the baseline period. All medication doses and non‐pharmacologic interventions for epilepsy (including ketogenic diet and vagus nerve stimulation) had to be stable in the 4 weeks before screening and throughout the trial." Exclusion criteria: "unstable medical conditions during the 4 weeks before screening, a history of alcohol or substance abuse, use of recreational or medicinal cannabis in the previous 3 months, use of corticotropins in the previous 6 months, or current use of felbamate for less than 1 year." Total number randomised: 225 Baseline imbalances: "Baseline characteristics were similar in the trial groups" It is noted that a slightly higher proportion of the placebo group also received vagus nerve stimulation as a concomitant intervention (28%) as compared with the 10 and 20 mg cannabidiol groups (21% each). Withdrawals and exclusions: "A total of 13 patients (6%) discontinued either cannabidiol (11 patients) or placebo (2 patients); in 7 of the 11 patients who discontinued cannabidiol, the treatment was discontinued because of adverse events." Age at onset:Age, mean (SD): placebo group, 15.3 (9.3); 10‐mg cannabidiol group, 15.4 (9.5); 20‐mg cannabidiol group, 16 (10.8). Age at diagnosis:Not reported. Age at start of treatment:Not reported. Sex: Males (%): placebo group, 44 (58); 10‐mg cannabidiol group, 40 (55); 20‐mg cannabidiol group, 45 (59). Race/ethnicity: White (%): placebo group, 69 (90.8); 10‐mg cannabidiol group, 62 (84.9); 20‐mg cannabidiol group, 67 (88.2). Other (Includes Black/African American, Asian, and ‘Other’ (verbatim terms included Biracial, Hispanic, Indian, and Latino‐Hispanic)) (%): placebo group, 7 (9.2); 10‐mg cannabidiol group, 11 (15.1); 20‐mg cannabidiol group, 9 (11.8). Type of seizures: Drop seizures; Total seizures; Non‐drop seizures; Seizure frequency during the baseline period; Median number of seizures during the 28‐day baseline period (interquartile range): Drop seizures: placebo group, 80.3 (47.8 to 148); 10 mg cannabidiol, 86.9 (40.6 to 190); 20‐mg cannabidiol, 85.5 (38.3 to 161.5) Total seizures (all types combined): placebo group, 180.6 (90.4 to 431.3); 10‐mg cannabidiol, 165 (81.3 to 359); 20‐mg cannabidiol, 174.3 (82.7 to 392.4) Non‐drop seizures: placebo group, 78 (22 to 216); 10‐mg cannabidiol, 95.7 (14 to 280); 20‐mg cannabidiol, 93.7 (22.2 to 278.4) Number of background drugs: Median number of previous antiepileptic drugs (range): Placebo group, 6 (1‐22) 10‐mg cannabidiol, 6 (0‐21) 20‐mg cannabidiol, 6 (1‐18) Severity of illness:Not reported Comorbidities: Not reported |
|
| Interventions |
Intervention group 1: 20‐mg cannabidiol Number randomised to group:76 Description: "…to receive cannabidiol at a dose of either 20 mg per kilogram of body weight per day… The active treatment was a plant‐derived pharmaceutical formulation of purified cannabidiol oral solution (100 mg per milliliter). Cannabidiol and the matching placebo solution (excipients alone) were provided in identical 100‐ml amber glass bottles. The cannabidiol or placebo was administered orally twice daily in equally divided doses starting at 2.5 mg per kilogram per day and increasing by 2.5 to 5.0 mg per kilogram every other day until the target dose was reached." Duration of baseline period:28 days; "Patients began a 4‐week baseline period after screening" Duration of treatment period:14 weeks; "The trial comprised…a 14‐week treatment period (2 weeks of dose escalation, followed by 12 weeks of stable dosing [maintenance phase]), a tapering period of up to 10 days, and a 4‐week safety follow‐up period after discontinuation of cannabidiol or placebo" Co‐interventions: Vagus nerve stimulation; Ketogenic diet; "medication doses and non‐pharmacologic interventions for epilepsy (including ketogenic diet and vagus nerve stimulation) had to be stable in the 4 weeks before screening and throughout the trial." Intervention group 2: 10‐mg cannabidiol Number randomised to group:73 Description: "…to receive cannabidiol at a dose of…10 mg per kilogram per day…The active treatment was a plant‐derived pharmaceutical formulation of purified cannabidiol oral solution (100 mg per milliliter). Cannabidiol and the matching placebo solution (excipients alone) were provided in identical 100‐ml amber glass bottles. The cannabidiol or placebo was administered orally twice daily in equally divided doses starting at 2.5 mg per kilogram per day and increasing by 2.5 to 5.0 mg per kilogram every other day until the target dose was reached." Duration of baseline period:28 days; "Patients began a 4‐week baseline period after screening" Duration of treatment period:14 weeks; "The trial comprised…a 14‐week treatment period (2 weeks of dose escalation, followed by 12 weeks of stable dosing [maintenance phase]), a tapering period of up to 10 days, and a 4‐week safety follow‐up period after discontinuation of cannabidiol or placebo" Control group: Placebo Description: "…matching placebo administered at a volume equivalent to that for either the 20‐mg cannabidiol dose or the 10‐mg dose. The active treatment was a plant‐derived pharmaceutical formulation of purified cannabidiol oral solution (100 mg per milliliter). Cannabidiol and the matching placebo solution (excipients alone) were provided in identical 100‐ml amber glass bottles. The cannabidiol or placebo was administered orally twice daily in equally divided doses starting at 2.5 mg per kilogram per day and increasing by 2.5 to 5.0 mg per kilogram every other day until the target dose was reached." "Placebo oral solution contains the excipients sesame oil and anhydrous ethanol with added sweetener (sucralose) and strawberry flavoring" Duration of baseline period:28 days; "Patients began a 4‐week baseline period after screening" Duration of treatment period:14 weeks; "The trial comprised…a 14‐week treatment period (2 weeks of dose escalation, followed by 12 weeks of stable dosing [maintenance phase]), a tapering period of up to 10 days, and a 4‐week safety follow‐up period after discontinuation of cannabidiol or placebo." |
|
| Outcomes |
Outcome 1: percentage change from baseline in the frequency of drop seizures (average per 28 days) during the treatment period Time points measured:Average per 28 days during the treatment period – 14 weeks Time points reported:Across the duration of the treatment period Outcome definition:"Patients or their caregivers were trained to record, using an interactive voice‐response system, the number and type of seizures, including drop seizures, that occurred each day. They also recorded in paper diaries cannabidiol or placebo use, use of concomitant medications, and adverse events that occurred during the treatment and follow‐up periods." Imputation of missing data:"Sensitivity analyses were performed for the primary and key secondary outcomes, including one in which missing data from the days that were not reported in the interactive response system were imputed as the highest number of seizures for each patient according to the last observation carried forward, the next observation carried backward, and the mean number of daily seizures during the treatment period (calculated from non missing data)" For the primary outcome, "The results of the sensitivity analyses, including those performed to account for missing data, were consistent with the results of the primary analysis" Statistical power:"On the basis of previously reported placebo effects on seizure rates in other trials involving patients with the Lennox–Gastaut syndrome and allowing for a slightly greater placebo effect because of a higher expectation of effect with cannabidiol than with other agents, we assumed that the patients assigned to receive placebo would have a mean 18% reduction from baseline in drop‐seizure frequency and that patients assigned to receive cannabidiol would have a mean 50% reduction. We calculated that 50 patients per trial group would provide 80% power to detect a 32 percentage‐point difference between the cannabidiol group and the placebo group at a two‐tailed significance level of 5%. Because of the rapid recruitment after notification of pending closure of recruitment, more patients underwent randomisation than originally planned." Outcome 2: "percentage of patients who had at least a 50% reduction from baseline in drop‐seizure frequency" Time points measured:Not pre‐specified in methods; "over the 12‐week, double‐blind maintenance period" (as reported in protocol). Time points reported:Across the duration of the treatment period. Outcome definition:"Patients or their caregivers were trained to record, using an interactive voice‐response system, the number and type of seizures, including drop seizures, that occurred each day. They also recorded in paper diaries cannabidiol or placebo use, use of concomitant medications, and adverse events that occurred during the treatment and follow‐up periods." Imputation of missing data:"Sensitivity analyses were performed for the primary and key secondary outcomes, including one in which missing data from the days that were not reported in the interactive response system were imputed as the highest number of seizures for each patient according to the last observation carried forward, the next observation carried backward, and the mean number of daily seizures during the treatment period (calculated from non missing data)" "The direction of these findings was consistent in the sensitivity analyses" Outcome 3: "the percentage change from baseline in the frequency of all types of seizures (total seizures)" Time points measured:Not pre‐specified in methods; Average per week and "during the Weeks 1–4, 5–8, and 9–12" for the outcome relating to drop seizures (as reported in protocol) Time points reported:"per 28 days during the treatment period" Outcome definition:"Patients or their caregivers were trained to record, using an interactive voice‐response system, the number and type of seizures, including drop seizures, that occurred each day. They also recorded in paper diaries cannabidiol or placebo use, use of concomitant medications, and adverse events that occurred during the treatment and follow‐up periods." Imputation of missing data:"Sensitivity analyses were performed for the primary and key secondary outcomes, including one in which missing data from the days that were not reported in the interactive response system were imputed as the highest number of seizures for each patient according to the last observation carried forward, the next observation carried backward, and the mean number of daily seizures during the treatment period (calculated from non missing data)" "The direction of these findings was consistent in the sensitivity analyses." Outcome 4: "the Patient or Caregiver Global Impression of Change from baseline in overall condition" Time points measured:From baseline and "the last visit." Time points reported: Change from baseline to the last visit. Outcome definition:"…assessed on a 7‐point scale that included three categories of improvement (slightly improved, much improved, or very much improved), three categories of worsening (slightly worse, much worse, or very much worse), and an option of 'no change'." Outcome 5: Adverse events ‘adverse events’ were not specifically identified as an outcome measure in the methods of the full report. Time points measured:Not specified in methods or results of full report. Time points reported: Not specified in the full report. Outcome definition:"Any new unfavorable/unintended signs/symptoms (including abnormal laboratory findings), or diagnosis or worsening of a pre‐existing condition, which is present following screening (Visit 1) and the post treatment, safety follow‐up visit (Visit 10), which may or may not be considered to be related to the IMP. Any event that is the result of a study procedure must be recorded as an AE." (as reported in the protocol). |
|
| Notes |
Study funding sources:"Supported by GW Pharmaceuticals" Conflicts of interest:All completed disclosure forms by authors identified relevant conflicts of interest outside of the submitted work. Other: Inclusion and exclusion criteria in the full report are not fully consistent with those reported in the protocol. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A computer‐generated block randomisation schedule, with block sizes of six, was produced by an independent statistician and held at a central location." |
| Allocation concealment (selection bias) | Low risk | "An interactive voice‐response or Web‐based response system was used to randomly assign the patients, in a 2:2:1:1 ratio, to receive cannabidiol at a dose of either 20 mg per kilogram of body weight per day (the 20‐mg cannabidiol group) or 10 mg per kilogram per day (the 10‐mg cannabidiol group) or matching placebo administered at a volume equivalent to that for either the 20‐mg cannabidiol dose or the 10‐mg dose (the placebo group)." |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | The abstract and introduction refer to double blinding and the methods refer to a "matching placebo" but provide no further information on blinding. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The abstract and introduction refer to double blinding and the methods refer to a "matching placebo" but provide no further information on blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The abstract and introduction refer to double blinding and the methods refer to a "matching placebo" but provide no further information on blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | "For patients who withdrew from the trial, data up to the time of withdrawal were included in the outcome analyses, and no imputation for missing data was performed." The number of dropouts (13/225) was relatively low overall although the proportion was slightly higher from the cannabidiol groups (11/49) than placebo (2/76). |
| Selective reporting (reporting bias) | High risk | A number of secondary outcomes referred to in the methods and protocol were reported in the full report as using descriptive statistics only but analyses are available in the supplementary paper. "The type 1 error was not controlled in the analysis of the other secondary outcomes. Therefore, only descriptive statistics and 95% confidence are presented in Table S3 in the Supplementary Appendix." |
| Other bias | Low risk | None identified. |
Eriksson 1998.
| Study characteristics | ||
| Methods | Cross‐over RCT with lamotrigine (LTG) in children and young adults with intractable generalised epilepsy. Baseline: 8 weeks Open phase: 2‐12 months Washout: 3 weeks Period 1: 12 weeks Washout: 3 weeks Period 2: 12 weeks Total: 46 to 86 weeks |
|
| Participants |
Population description:"All children and adolescents older than 2 years with refractory or intractable generalized epilepsy referred to the Department of Child Neurology, Karolinska Hospital, were eligible for the trial if they had more than two seizures per month." Inclusion criteria:"All children and adolescents older than 2 years with refractory or intractable generalized epilepsy referred to the Department of Child Neurology, Karolinska Hospital, were eligible for the trial if they had more than two seizures per month. 'Refractory' was defined as 'not seizure free' despite consecutive treatment attempts with at least three conventional ASMs in therapeutic doses giving adequate recommended high plasma concentrations verified by therapeutic drug monitoring (TDM). 'Generalized' was defined according to the ILAE’s classification and required a scalp EEG showing generalized or multifocal epileptiform activity." Exclusion criteria: "the presence of liver, renal, or progressive neurologic disease, or the diagnosis of focal epilepsy." Total number randomised:17 with refractory generalised epilepsy (based on Table 1, 13/17 had LGS). Baseline imbalances: Not reported. Withdrawals and exclusions:"Three children were excluded from the analyses because of incomplete seizure diaries (patients 11, 28, and 30). One child was withdrawn during the double‐blind phase for family reasons (patient 21), and the family of another (patient 14) refused to carry through because the child became seizure free after the first phase of the double‐blind period. Both children had received LTG." Age at onset: Mean: 9.3 (SD 3.9) Age at diagnosis:Not reported. Age at start of treatment: Not reported. Sex: Not reported. Race/ethnicity: Not reported. Type of seizures: Data reported individually at baseline, pre‐randomisation. Across participants with LGS, seizure types included: Tonic‐atonic seizures (n = 20/20) Myoclonic seizures (n = 20/20) Atypical absences (n = 20/20) Tonic‐clonic seizures (n = 13/20) Other seizure types (n = 2/20) Seizure frequency during the baseline period: Median = 96 (range 27 to 315) Number of background drugs:Data reported individually: 1‐3 drugs in the patients with LGS Comorbidities: Not reported. |
|
| Interventions |
Group: Lamotrigine Number randomised to group:N = 17 with refractory generalised epilepsy (based on Table 1, 13/17 had LGS). Description: "During the open phase, LTG was added to the current treatment, starting with 1 mg/kg/day divided into two daily doses and increased by 1 mg/kg/day every 2 weeks until a clinical response or side effects were observed. However, for children taking sodium valproate (VPA) and no enzyme‐inducing ASM, the initial dose of LTG was < 0.5m g/kg/day, and the dose increments were 0.5 mg/kg/day at intervals as described. LTG was administered either as 12.5‐mg capsules or as 25‐ or 50‐mg tablets supplied by Wellcome. The optimal LTG dose (i.e. the dose that produced the best clinical response for individual patients, was determined after a mean period of 5 months (range, 2‐12 months). At the end of the open phase, each child was classified as a responder or a nonresponder. Children with a ≥ 50% reduction in seizure frequency or reduction in the severity of seizures or both, or definite improvements in behavior or motor skills or both, were classified as responders and went on to the double‐blind phase" "Double‐blind phase: two 12‐week periods during which the previously determined individual dose of LTG for each child and identical placebo tablets were administered in random order." Washout phase: "The treatment periods were separated by a 3‐week washout phase." Duration of baseline period:8 weeks Duration of treatment period:12 weeks Co‐interventions: Existing antiepileptic medications (AEDs) Compliance:Not reported. Group name: placebo Number randomised to group:N = 17 with refractory generalised epilepsy (based on Table 1, 13/17 had LGS). Description:See above. "During the course of the study, the patients were evaluated every 2 weeks during a 1‐day visit to our clinic for children with epilepsy. The evaluation was based on an interview of the parents and, if possible, the child, a 4‐h observation of the child by a trained nurse, and a physical examination. Each child’s condition was classified as improved, unchanged, or deteriorated. The LTG dose was increased during the open phase if the child was classified as improved or unchanged, but not seizure free. The dose was reduced if the child’s overall clinical status showed deterioration. The overall clinical status was assessed by the same investigator (A‐S.E.) at all follow‐up visits. At entry into the study, each child’s usual ASMs were in steady state and were kept constant throughout the study and within the pre study range, as measured by plasma drug levels." Duration of baseline period:8 weeks Duration of treatment period:12 weeks Co‐interventions:Existing antiepileptic medications (ASMs). Compliance:Not reported. |
|
| Outcomes |
Outcome 1: Percentage reduction in average monthly seizure frequency Time points measured:Appears to be the duration of the treatment phase but it is unclear if the baseline is the washout period. Time points reported:Appears to be the duration of the treatment phase but it is unclear if the baseline is the washout period. Outcome definition: Not reported but it is noted that atypical absences and myoclonic events were excluded from outcome assessments. Is outcome/tool validated? Unclear Imputation of missing data: No ITT reported. Assumed risk estimate: Not reported. Power: Not reported; small sample size but cross‐over study. Outcome 2: Severity of seizures Imputation of missing data: Not reported. Time points reported:Not reported. Outcome definition: Not reported. Is outcome/tool validated?Unclear Imputation of missing data:Not reported. Assumed risk estimate:Not reported. Power: Not reported. Outcome 3: Functional status of the patients Time points measured: Not reported. Time points reported:Not reported. Outcome definition: Not reported. Is outcome/tool validated? Unclear Imputation of missing data: Not reported. Assumed risk estimate: Not reported. Power:Not reported. Outcome 4: Frequency of adverse events Time points measured:Appears to be the duration of the treatment phase. Time points reported:Appears to be the duration of the treatment phase. Is outcome/tool validated?Not applicable. Imputation of missing data: No ITT analysis reported. Assumed risk estimate: Not reported. Power: Not reported. |
|
| Notes |
Study funding sources:This study was supported by grants from the Barnavård Society, the Margarethahemmet Foundation, and Wellcome Sweden AB. Possible conflict of interests:Not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Described as "random" and having a "randomisation code." |
| Allocation concealment (selection bias) | Unclear risk | Concealment of allocation unclear (not reported). |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Although the study is described as "double blind" and "identical placebo tablets were administered," there is no clarification of who was blinded. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Although the study is described as "double blind" and "identical placebo tablets were administered," there is no clarification of who was blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Although the study is described as "double blind" and "identical placebo tablets were administered," there is no clarification of who was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "Non‐responders" in the open phase were not included in the RCT; individual data presented incompletely for participants with LGS. |
| Selective reporting (reporting bias) | High risk | Some outcomes reported in the Methods were not reported in the results for the RCT. |
| Other bias | Low risk | None identified. |
Felbamate Study Group 1993.
| Study characteristics | ||
| Methods | Parallel RCT aimed to evaluate the efficacy and safety of felbamate in patients with Lennox‐Gastaut syndrome. Duration of participation: Eight‐week pre‐study screening phase then a 28‐day baseline (during which participants received their usual antiepileptic therapies) followed by 70 days with additional felbamate or placebo. |
|
| Participants |
Population description:Patients with the Lennox‐Gastaut syndrome ‐ a severe epileptic encephalopathy characterised by the onset of multiple types of seizures during childhood. Inclusion criteria: "a history of multiple types of seizures and a minimum of 90 atonic seizures (seizures characterised by a sufficient change in posture to cause a fall from the sitting or standing position) or atypical absence seizures per month during an eight‐week pre study screening phase, were taking no more than two antiepileptic drugs, had no evidence of progressive central nervous system lesions on magnetic resonance imaging or computed tomography, weighed at least 11.3kg, and had a slow spike‐wave complex (≤2.5 Hz) on electroencephalography." Exclusion criteria:"Female patients were excluded if they were pregnant or were not using adequate contraception. Patients with a history of identifiable progressive neurologic disorders, anoxic episodes within the past year, poor compliance with past antiepileptic therapy, recent drug or alcohol abuse, a major medical illness, or previous suicide attempts were excluded. Patients who had recently received corticotropin, were following ketogenic diets, or had inadequate supervision by parents (or guardians) were also excluded." Total number randomised:73 Baseline imbalances: "The felbamate and placebo groups were comparable with respect to demographic and pretreatment characteristics." It is noted from Table 1 that both groups largely involved white participants (> 89%). Withdrawals and exclusions: "One patient in the felbamate group withdrew from the trial…because of somnolence and ataxia, and one patient in the placebo group withdrew from the trial…because of pancreatitis." Age at onset: Mean age, years at baseline (range) Felbamate: 12 (4‐24) Placebo: 14 (4‐36) Age at diagnosis: Not reported. Age at start if treatment: Not reported. Sex: Felbamate (M:F): 27:10 Placebo (M:F): 24:12 Race/ethnicity: Felbamate: White – 33; Black ‐ 2; Other ‐ 2 Placebo: White – 33; Black – 1; Other ‐ 2 Type of seizures:Atonic, Total and Tonic‐clonic recorded in the baseline demographics. Seizure frequency during the baseline period: Felbamate atonic: 370 Placebo atonic: 228 Felbamate Total: 1617 Placebo Total: 716 Felbamate tonic‐clonic: 9 Placebo tonic‐clonic: 6 Number of background drugs: Reported across the patient lifetime. No more than two antiepileptic drugs at baseline according to the trial eligibility criteria. Comorbidities: Not reported. |
|
| Interventions |
Group: Felbamate Number randomised to group:37 Description:"The treatment phase consisted of a 14‐day titration period and a 56‐day maintenance period. The initial dose of felbamate was 15 mg per kilogram of body weight per day; the daily dose was increased to 30 mg per kilogram after 7 days and to the maximal dose after 14 days. The maximal dose was either 45 mg per kilogram per day or 3600 mg per day, whichever represented the lower dose. During the maintenance period (study days 43 to 98), the patients continued to receive the maximal tolerated dose of the study drug…Felbamate (200 mg) and placebo were prepared in identical‐appearing capsules. The doses of felbamate were established on the basis of body weight and were given four times daily." Duration of baseline period: "a 28‐day base‐line phase." Duration of treatment period: 70 days Co‐interventions:Usual antiepileptic medications. Group: Placebo Number randomised to group:36 Description: "The treatment phase consisted of a 14‐day titration period and a 56‐day maintenance period…Felbamate (200 mg) and placebo were prepared in identical‐appearing capsules…To maintain the double‐blind conditions, the number of capsules taken by patients in the placebo group at each dosage level was based on body weight." Duration of baseline period:"a 28‐day base‐line phase." Duration of treatment period:70 days Co‐interventions: Usual antiepileptic medications. |
|
| Outcomes |
Outcome 1: Frequency of seizures during periods of video monitoring Time points measured:"study days 42, 49, 70, and 98" Time points reported:Not reported. Outcome definition:Not reported. Is outcome/tool validated?Unclear Imputation of missing data: "All patients were included in the efficacy analyses." Assumed risk estimate: Not applicable. Power: Not reported. Outcome 2: "patient’s status" Time points measured:Not reported. Time points reported:Not reported. Outcome definition: Not reported. Is outcome/tool validated?Unclear Imputation of missing data: "All patients were included in the efficacy analyses." Assumed risk estimate:Not applicable. Power: Not reported. Outcome 3: Parental counts of total seizures Time points measured:Not reported. Time points reported: Not reported. Outcome definition:Not reported. Is outcome/tool validated? Unclear Imputation of missing data:"All patients were included in the efficacy analyses." Assumed risk estimate:Not applicable. Power: Not reported. Outcome 4: Parental counts of generalised tonic‐clonic seizures Time points measured: Not reported. Time points reported: Not reported. Outcome definition: Not reported. Is outcome/tool validated?Unclear Imputation of missing data: "All patients were included in the efficacy analyses." Assumed risk estimate: Not applicable. Power:Not reported. Outcome 5: Adverse symptoms Time points measured:Not reported. Time points reported: Not reported. Outcome definition: Not reported. Is outcome/tool validated?Unclear Imputation of missing data: "All patients were included in the efficacy analyses." Assumed risk estimate: Not applicable. Power: Not reported. |
|
| Notes |
Study funding sources:Supported by a grant from Wallace Laboratories, Division of Carter‐Wallace, Inc., Cranbury, N.J., and a grant (M01‐RR‐00865) from the Public Health Service. Possible conflicts of interest:At least one author in the study group was from the grant provider, Wallace Laboratories. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The patients were then randomly assigned in blocks of two to receive either felbamate or placebo by a separate computer‐generated randomisation schedule at each study center." |
| Allocation concealment (selection bias) | Low risk | "computer‐generated randomisation schedule" |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Though the trial is described as "double‐blind," it is unclear who was blinded. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Though the trial is described as "double‐blind," it is unclear who was blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Though the trial is described as "double‐blind," it is unclear who was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Although only one person from each group was reported to have withdrawn during the study and a total of 73 people were studied for treatment efficacy, it is not specified how many people were randomised in total. |
| Selective reporting (reporting bias) | High risk | Incomplete reporting on one of the study’s primary outcomes specified in the methods – patient status. Reporting also appears to be inconsistent between methods and results for other outcomes. |
| Other bias | Low risk | None identified. |
Glauser 2008.
| Study characteristics | ||
| Methods | Parallel RCT aimed to evaluate the efficacy and tolerability of rufinamide adjunctive therapy in patients with Lennox–Gastaut syndrome. Duration of participation: "This study consisted of a 28‐day baseline period followed by an 84‐day, double‐blind, placebo‐controlled, parallel‐group treatment period." |
|
| Participants |
Population description:"Patients diagnosed with Lennox–Gastaut syndrome based on the International League Against Epilepsy classification." Inclusion criteria: "Patients 4 to 30 years of age were eligible if they had a history of multiple seizure types, which had to include atypical absence seizures and drop attacks (i.e. tonic–atonic or astatic seizures); a minimum of 90 seizures in the month before the 28‐day baseline period; an EEG within 6 months of study entry demonstrating a pattern of slow spike‐and‐wave complexes (< 2.5 Hz); a weight of at least 18 kg; a fixed‐dose regimen of one to three concomitant antiepileptic drugs during the baseline period; and a CT scan or MRI study confirming the absence of a progressive lesion." Exclusion criteria: "Patients were ineligible if they were receiving more than three antiepileptic drugs; were pregnant or not using adequate contraception; had a correctable etiology of their seizures (active infection, neoplasm, metabolic disturbance); had a history of generalized tonic– clonic status epilepticus within 30 days before baseline; or had a history of any clinically significant non neurologic medical condition." Total number randomised:138 (reported also as 139) Baseline imbalances: "The baseline characteristics of the two treatment groups were similar, with a preponderance of males… The two treatment groups had a comparable distribution of concomitant antiepileptic drugs with valproic acid, lamotrigine, and topiramate the most frequently used." On review of Table 2, the demographics suggest that 10% more of the placebo group were under 12 years of age compared with the rufinamide group, and just over 10% more of the placebo group were aged 17 years or over compared with the rufinamide group. Both groups largely involved white participants. In terms of concomitant ASMs used by > 10% of patients, Table 2 suggests just over 10% lower use of lamotrigine in the placebo group as compared with the rufinamide group. Withdrawals and exclusions: "Ten patients in the rufinamide group discontinued therapy during the double‐blind phase because of adverse events (n = 6), unsatisfactory therapeutic effect (n = 3), or withdrawal of consent (n = 1). Five patients in the placebo group did not complete the study because of protocol violations (n = 2), unsatisfactory therapeutic effect (n = 1), administrative problems (n = 1), or withdrawal of consent (n = 1)." Age at onset: Age, median years (range) Rufinamide: 13 (4‐35) Placebo: 10.5 (4‐37) Age at diagnosis: Not reported. Age at start of treatment: Not reported. Sex:n (%) Rufinamide: male, 46 (62.2); female, 28 (37.8) Placebo: male, 40 (62.5); female, 24 (37.5) Race/ethnicity:n (%) Rufinamide: White, 62 (83.8); Black, 6 (8.1); Other, 6 (8.1) Placebo: White, 53 (82.8); Black, 4 (6.3); Other, 7 (10.9) Type of seizures:Includes absence and atypical absence seizure, tonic seizures, atonic seizures, myoclonic seizures, tonic‐clonic seizures and partial seizures. Seizure frequency during the baseline period: Baseline Rufinamide median frequency (range): 290.0 (48.0–53,760.0) Baseline Placebo median frequency (range): 205.0 (21.0–109,714.0) Number of background drugs: Not reported – Table 2 only reports concomitant ASMs used by > 10% of patients. Comorbidities:Not reported. |
|
| Interventions |
Group name: Rufinamide Number randomised to group:74 Description: "At the end of the 28‐day baseline period, patients continuing to meet the study criteria entered the 84‐day double‐blind treatment phase. This phase consisted of a 14‐day titration period followed by a 70‐day maintenance period." "Rufinamide was supplied as 100‐, 200‐, and 400‐mg tablets with corresponding matching placebo tablets. Doses were titrated according to a recommended schedule based on weight up to a maximum dose of approximately 45 mg/kg per day (table 1). The dose of medication at the end of the titration period (day 14) was the dose that the patient was to receive during the entire maintenance period." Duration of baseline period:28 days Duration of treatment period:84 days Co‐interventions: 1‐3 other antiepileptic drugs. Compliance: Not reported. Group name: Placebo Number randomised to group: 64 Description: "At the end of the 28‐day baseline period, patients continuing to meet the study criteria entered the 84‐day double‐blind treatment phase. This phase consisted of a 14‐day titration period followed by a 70‐day maintenance period." "Rufinamide was supplied as 100‐, 200‐, and 400‐mg tablets with corresponding matching placebo tablets. Doses were titrated according to a recommended schedule based on weight up to a maximum dose of approximately 45 mg/kg per day (table 1). The dose of medication at the end of the titration period (day 14) was the dose that the patient was to receive during the entire maintenance period." Duration of baseline period:28 days Duration of treatment period:84 days Co‐interventions: 1‐3 other antiepileptic drugs. Compliance: Not reported. |
|
| Outcomes |
Outcome 1: Percent change in total seizure frequency Time points measured:"per 28 days in the double‐blind phase relative to the baseline phase." Time points reported:Per 28 days relative to baseline. Outcome definition:Not reported. Is outcome/tool validated?Unclear Imputation of missing data: "All efficacy analyses were based on the intent‐to‐treat population consisting of all randomised patients who received double‐blind study drug." Assumed risk estimate:Not applicable. Power:"The study’s sample size was determined assuming that adjunctive rufinamide therapy would result in a reduction in seizure frequency 22.5% greater than that of placebo adjunctive therapy. With approximately 64 randomised patients per treatment group, the study had a statistical power of 91.3% at a two‐sided significance level of 0.025 to detect this difference." Outcome 2: Percent change in tonic–atonic (i.e. the sum of tonic and atonic) seizure frequency Time points measured:"per 28 days in the double‐blind phase relative to the baseline phase." Time points reported:per 28 days relative to baseline. Outcome definition:Not reported. Is outcome/tool validated?Unclear Imputation of missing data: "All efficacy analyses were based on the intent‐to‐treat population consisting of all randomised patients who received double‐blind study drug." Assumed risk estimate:Not applicable. Power: See outcome 1. Outcome 3: Seizure severity rating from the global evaluation of the patient’s condition Time points measured:Not reported. Time points reported: At the end of the double‐blind phase. Is outcome/tool validated? Unclear Imputation of missing data:"All efficacy analyses were based on the intent‐to‐treat population consisting of all randomised patients who received double‐blind study drug." Assumed risk estimate: Not applicable. Power:See outcome 1. Outcome 4: Response to treatment – percentage of patients with ≥ 50% reduction in seizure frequency Time points measured:During the double‐blind phase relative to baseline. Time points reported: Per 28 days relative to baseline. Outcome definition: Not reported. Is outcome/tool validated? Unclear Imputation of missing data:"All efficacy analyses were based on the intent‐to‐treat population consisting of all randomised patients who received double‐blind study drug." Assumed risk estimate: Not applicable. Power: See outcome 1. Outcome 5: Percentage change in seizure frequency for each seizure subtype other than tonic–atonic seizures Time points measured: "per 28 days in the double‐blind phase relative to the baseline phase." Time points reported: "per 28 days in the double‐blind phase relative to the baseline phase." Outcome definition: Not reported. Is outcome/tool validated? Unclear Imputation of missing data:"All efficacy analyses were based on the intent‐to‐treat population consisting of all randomised patients who received double‐blind study drug." Assumed risk estimate: Not applicable. Power: See outcome 1. Outcome 6: Parental/guardian global evaluation of seizure severity and the patient’s condition Time points measured:The end of the double‐blind phase. Time points reported:At the end of the double‐blind phase. Outcome definition:Global evaluation was "composed of alertness, interaction with environment, daily activity performance, responsiveness to verbal requests, and seizure severity." Is outcome/tool validated? Unclear Imputation of missing data:"All efficacy analyses were based on the intent‐to‐treat population consisting of all randomised patients who received double‐blind study drug." Assumed risk estimate: Not applicable. Power: See outcome 1. Outcome 7: Adverse events (incidence; incidence of serious adverse events; drug‐related adverse events; withdrawal from the study because of adverse events; adverse events reported by 10% or more of patients in either group) Time points measured:Not reported Time points reported:Not reported Outcome definition: Adverse events reported in multiple ways in the results section. Is outcome/tool validated? Unclear Imputation of missing data:"All efficacy analyses were based on the intent‐to‐treat population consisting of all randomised patients who received double‐blind study drug." Assumed risk estimate: Not applicable. Power: See outcome 1. |
|
| Notes |
Study funding sources:Sponsored by Eisai Pharmaceutical and conducted by Novartis Pharmaceutical. Possible conflict of interests:Study investigators declared receipt of personal compensation (honorarium) and grants from the study sponsor. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Eligible patients were randomly assigned in blocks of four to receive either rufinamide or matching placebo in addition to their standard antiepileptic drug regimens. Patients were randomised at the country/center level and were assigned sequential numbers at each site during the first visit." |
| Allocation concealment (selection bias) | Unclear risk | Not reported. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Trial described as "double‐blind" but it is unclear who was blinded. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Trial described as "double‐blind" but it is unclear who was blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Trial described as "double‐blind" but it is unclear who was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Seizure severity rating reported incompletely but ITT analysis reported and there was a comparable rate of withdrawal: 14% from the rufinamide group and 8% from placebo. |
| Selective reporting (reporting bias) | Low risk | None identified. |
| Other bias | Low risk | None identified. |
Group for the Evaluation of Cinromide 1989.
| Study characteristics | ||
| Methods | Parallel RCT aimed to investigate efficacy of adjunctive cinromide in patients with Lennox‐Gastaut syndrome. Duration of participation: 24 weeks. |
|
| Participants |
Population description:See inclusion criteria. Inclusion criteria: "1. History of multiple seizure types with seizure onset during the 1st decade, at least 40 clinically recognisable atonic, myoclonic, tonic and/or absence seizures every 2 weeks during the 6 weeks before the study entry, those presenting with absence seizures had at least a history of atonic, tonic, or myoclonic seizures. Other seizure types may have been present, but patients with predominantly partial seizures were not accepted. 2. Predominantly generalised (definitely bilateral), slow (1‐2.5 hertz), spike – and – wave discharges were demonstrated on EEG recorded during the 3 months before study entry ; additional abnormalities were noted, patients whose spike‐and‐wave discharges were typically secondary to discharges from a single focus were not accepted. 3. Patients aged 2‐18 years. 4. A patients must have had seizures for at least 6 months and could be receiving no more than 3 marketed antiepileptic drugs (acceptable plasma levels must have been measured at least twice during the 4 months before the study entry). 5. Parents or guardians, will give informed consent for patient participation problem ahead to demonstrate the ability to Minister medication as instructed and to maintain satisfactory seizure counts as required by the study protocol. Exclusion criteria: 1. History of tonic or tonic‐clonic status epilepticus within the previous 6 months or history of prolonged (> 1hr) episodes of non Conti friable seizure activity of any type more than once every 2 weeks during the previous 3 months. 2. An anoxic episode requiring resuscitation during the previous year. 3. A progressive CNS lesion or progressive have Patrick, cardiovascular, her mother to poetic, renal, pulmonary, gastrointestinal, ophthalmologic or endocrine disease. 4. A predisposing condition which might interfere with absorption, distribution are excretion of drugs. 5. Severe side effects from pre study medications. 6. An antiepileptic drug added 2 or removed from their medication regimen within the previous 4 weeks (3 months in the case of valproate) or a change since the most recent pre study plasma level measurements, or if they had been maintained on the ketogenic diet within the previous 2 weeks or treated with ACTH or corticosteroid within the previous 4 weeks. 7. Previous exposure to cinromide or, 8. Such severe retardation or other impairment that safety and efficacy evaluation would have been particularly unreliable or difficult to perform." Total number randomised: 56 Baseline imbalances: None identified. Age at onset:Not reported. Age at diagnosis:Not reported. Age at start of treatment: Mean (SD), years Cinromide: 7.38 (3.65); Placebo 7.93 (4.87) Sex: 34 male, 22 female Race/ethnicity: (number of participants) Cinromide: White (24); Black (1); Other (1) Placebo: White (28); Black (1); Other (0) Type of seizures:All seizure types Seizure frequency during the baseline period: (number of participants) Cinromide: one (1), two (9), three (9), four or more (7) Placebo: one (4), two (12), three (10), four or more (4) Number of background drugs:(number of participants) Cinromide: one (3), two (9), three (14) Placebo: one (5), two (13), three (12) Comorbidities:Not reported. |
|
| Interventions |
Group: Cinromide Number randomised to group:26 Description:Study medication (cinromide or placebo) was initiated at 20 to 40 milligram/kg/day, divided into 4 equal doses. Further increase in (to a total daily maximum of 83 ‐ 109 milligram/kg) were prescribed at weekly visits according to fix dosing schedule, if each prior dose was well tolerated and seizures continued. The objective was to achieve an optimal dose during the 1st 6 treatment weeks and maintain this dose during at least 12 treatment weeks. Doses and regimens of prior antiepileptic drugs were not to be altered during baseline. Doses of these drugs could be changed during treatment periods if plasma drug concentration exceeded specified limits above and below baseline means. Additional antiepileptic medication was permitted on an acute (≤24 h) basis for treatment of status epilepticus without necessarily this qualifying a patient from further study. Duration of baseline period: 6 weeks Duration of treatment period:18 weeks Co‐interventions: Not reported. Compliance:Not reported. Group: Placebo Number randomised to group:30 Description:Not reported. Duration of baseline period: 6 weeks Duration of treatment period:18 weeks Co‐interventions:Not reported. Compliance: Not reported. |
|
| Outcomes |
Outcome 1: mean weekly seizure frequency (for total seizures and by seizure type) Time points measured:At baseline and during treatment phase. Time points reported: At baseline and during treatment phase. Outcome definition:See above. Is outcome/tool validated?Unclear Imputation of missing data:No intention to treat or modified intention to treat analyses applied. Assumed risk estimate:Not reported. Power:Not reported. Outcome 2: Global evaluation Time points measured:After 6, 12 and 18 treatment weeks compared with previous 6 weeks during baseline. Time points reported:After 6, 12 and 18 treatment weeks compared with previous 6 weeks during baseline. Outcome definition:'relevant clinical factors including seizure frequency and severity, signs and symptoms, our general function ability describing patients as mildly, moderately or severely improved/deteriorated'. Is outcome/tool validated? Unclear Imputation of missing data:No intention to treat or modified intention to treat analyses applied. Assumed risk estimate:Not reported. Power: Not reported. |
|
| Notes |
Study funding sources:Not reported. Possible conflicts of interest:Not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "assigned randomly" |
| Allocation concealment (selection bias) | Unclear risk | Unclear concealment of allocation (not reported). |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "Double‐blind" but unclear who was blinded. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Double‐blind" but unclear who was blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Double‐blind" but unclear who was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 73 patients entered the study and 17 patients had incomplete outcome data because of premature termination. Outcomes only reported for the remaining 56 patients (76% of initially enrolled patients). |
| Selective reporting (reporting bias) | High risk | Study terminated prematurely when it was clear to the sponsor that cinromide was not effective; only efficacy data collected before study interruption were included in the analysis. |
| Other bias | Low risk | None identified. |
Motte 1997.
| Study characteristics | ||
| Methods | Parallel RCT aimed to evaluate lamotrigine in patients with the Lennox–Gastaut syndrome. Duration of participation:4 week baseline period followed by 16 week treatment period. |
|
| Participants |
Population description:Children and young adults fulfilling diagnostic criteria for the Lennox‐Gastaut syndrome. Inclusion criteria:"The diagnostic criteria for the Lennox–Gastaut syndrome in this study were agreed on by an international expert panel of child neurologists. Patients 3 to 25 years of age were eligible if they had had more than one type of predominantly generalized seizure, including tonic–clonic seizures and drop attacks (atonic, tonic, or major myoclonic), for at least one year; they were younger than 11 years at the onset of epilepsy; they had seizures at least every other day or with a similar average frequency; and they had intellectual impairment or a clinical impression of intellectual deterioration (on the basis of results of developmental assessments or IQ tests) and a recent electroencephalogram demonstrating an abnormal background and a pattern of slow spike‐and‐wave complexes (< 2.5 Hz). If focal abnormalities occurred during electroencephalography, they occurred concurrently with a slow spike‐and‐wave pattern and were not considered by the investigator to be the most important electroencephalographic abnormality." Exclusion criteria: "Patients were ineligible if they had a progressive neurodegenerative disorder, they were receiving more than three antiepileptic drugs, or they weighed less than 15 kg and were taking valproate." Total no. randomised:169 Baseline imbalances: "The characteristics of the two groups were similar, although the lamotrigine group had a higher proportion of male subjects (P = 0.02)." Both groups had a high majority (> 90%) of participants who were white. Withdrawals and exclusions:"Seven patients in the lamotrigine group stopped treatment early, four because of protocol violations and three because of adverse events. Fourteen patients in the placebo group did not complete the study: seven had adverse events, three had protocol violations, two had a deterioration in the control of seizures, one failed to return for follow‐up, and the parents of one patient withdrew their consent." Age at onset: Age at baseline, mean years ±SD Lamotrigine: 9.6 ± 5.2 Placebo: 10.9 ± 5.9 Age at diagnosis:Not reported. Age at start of treatment:Not reported. Sex:Male sex, n (%) Lamotrigine: 54 (68) Placebo: 45 (50) Females, n (%) Lamotrigine: 25 (32) Placebo: 45 (50) Race/ethnicity: White, n (%) Lamotrigine: 74 (94) Placebo: 84 (93) Black, n (%) Lamotrigine: 3 (4) Placebo: 3 (3) Other, n (%) Lamotrigine: 2 (3) Placebo: 3 (3) Type of seizures:Baseline ‐ All major seizures; All drop attacks; All tonic‐clonic seizures; Atypical absence seizures. Seizure frequency during the baseline period: Baseline ‐ All major seizures, seizures/week (range) Lamotrigine: 16.4 (3.1 to 249.4) Placebo: 13.5 (1.5 to 592.8) Number of background drugs: Concomitant treatment with valproate specified in table 3, page 4. However, it was an exclusion criterion for participants to take more than three antiepileptic drugs. Comorbidities:Not reported. "Approximately 40 percent of the patients in each group had a history of infantile spasms, and approximately 25 percent of each group had a history of status epilepticus." |
|
| Interventions |
Group: Lamotrigine Number randomised to group:79 Description:"During a four‐week single‐blind baseline period (the physician investigators were not blinded), all patients received placebo. Their eligibility for the study was confirmed, and their standard antiepileptic‐drug regimens were continued. Eligible patients were then randomly assigned in a double‐blind fashion to receive either lamotrigine or placebo. The randomization was not stratified according to site or country. Because of the ability of valproate to inhibit the clearance of lamotrigine and increase plasma lamotrigine concentrations, patients were assigned to one of four dosing regimens according to concomitant valproate use and body weight (≤25 kg or >25 kg)(Table 1). The 16‐week treatment period comprised a 6‐week period in which the dose of active drug or placebo was increased, 2 weeks in which the dose was fixed, and an additional 8 weeks during which the fixed dose could be increased during week 8 or 12 to no more than the maximal allowable daily dose if seizures were still occurring (100 to 200 mg for patients concomitantly receiving valproate and 300 to 400 mg for patients who were not receiving valproate) (Table 1). At the end of the treatment period, the study drug was gradually discontinued in a double‐blind manner by reducing the dose to 50 percent for two weeks and then to 25 percent for a further two weeks. The study drug was also discontinued gradually in patients who stopped treatment prematurely." Duration of baseline period:4 weeks Duration of treatment period:16 weeks Co‐interventions: "patients’ standard anti‐seizure medications regimens." Compliance:"Of the 179 patients who entered the 4‐week placebo base‐line period, 10 were not enrolled in the subsequent 16‐week treatment phase." Unclear – some participants were ruled out before the treatment phase due to a lack of compliance with the study procedures. These individuals were not included in the number of people randomised. Subsequently, some participants were also excluded due to protocol violations. Group: Placebo No. randomised to group:90 Description: See above. Duration of baseline period:4 weeks Duration of treatment period:16 weeks Co‐interventions:"patients’ standard antiepileptic‐drug regimens." Compliance: See above. |
|
| Outcomes |
Outcome 1: Number of all major motor seizures Time points measured:"in daily diaries throughout the base‐line and treatment period." Time points reported: "from base line during treatment weeks 1 through 16.' Outcome definition: "drop attacks and tonic‐clonic seizures." Is outcome/tool validated?Unclear Imputation of missing data: Interpreted as per protocol not ITT analysis. Assumed risk estimate:Not reported. Power: Not reported. Outcome 2: Adverse events Time points measured:"Clinicians obtained information on the occurrence of adverse events…from the patients’ parents or guardians during clinic visits during treatment weeks 2, 4, 8, 12, 16, and 20." Time points reported:"from base line during treatment weeks 1 through 16." Outcome definition: "defined as any undesirable effects, irrespective of the relation to the administration of the study drug." Is outcome/tool validated? Unclear Imputation of missing data:Interpreted as per protocol not ITT analysis. "All safety analyses were based on the 169 patients who received at least one dose of study drug and had analyzable data." Assumed risk estimate:Not reported. Power:Not reported. |
|
| Notes |
Study funding sources:"Funded by Glaxo Wellcome" Possible conflicts of interest: Not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Eligible patients were then randomly assigned..." |
| Allocation concealment (selection bias) | Unclear risk | Concealment of allocation unclear (not reported). |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Although the trial is reported to be "double‐blind," it is not specified who is blinded. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Although the trial is reported to be "double‐blind," it is not specified who is blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Although the trial is reported to be "double‐blind," it is not specified who is blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None identified. |
| Selective reporting (reporting bias) | Low risk | None identified. |
| Other bias | Low risk | None identified. |
Ng 2011.
| Study characteristics | ||
| Methods | RCT, aimed to evaluate efficacy and safety of clobazam, a 1,5‐benzodiazepine, as adjunctive therapy for Lennox‐Gastaut syndrome (LGS). Duration of participation:"Study consisted of 4‐week baseline, 3‐week titration, and 12‐week maintenance phases, followed by a 2‐ or 3‐week taper or continuation in an open‐label extension.' |
|
| Participants |
Population description:"Patients aged 2–60 years weighing ≥12.5 kg were eligible to participate in the CONTAIN trial if they had onset of LGS before 11 years of age. A clinical diagnosis of LGS was evidenced by ≥ 1 type of generalized seizure (including drop seizures) for ≥ 6 months and a previous EEG report documenting generalized, slow spike‐and‐wave (< 2.5 Hz) patterns." Study inclusion and exclusion criteria are provided in a separate, online appendix on the Neurology website at www.neurology.org Total number randomised:238 Baseline imbalances: Table 1 appears to show a comparatively lower mean baseline average weekly drop seizure rate in the medium dose of clobazam group (58.8; SD 119.6) as compared with low (98.3; SD 198.5) and high dose groups (94.9; SD 152.2) and placebo group (95.6; SD 168.2). "Demographics and baseline characteristics were comparable between groups (table 1) and were similar between the safety and mITT populations. Mean age was 12.4 years (range, 2–54 years), and the majority (60.5%) were male." Withdrawals and exclusions: "At the time of the [protocol] amendment, 81 patients had enrolled. Of these, 29 (36%) discontinued, 26 before the amendment went into effect and 3 after…" Following the amendment, 157 patients enrolled, of which 32 (20%) discontinued." "The most common reasons for discontinuing the study were lack of efficacy for placebo‐treated patients and adverse events (AEs) for clobazam‐treated patients. All 238 randomised patients were included in the safety population. The mITT population excluded 21 patients who did not have 1 daily seizure measurement during the maintenance period. Thus, efficacy analyses included 217 patients (57 for placebo and 53, 58, and 49 for the low‐, medium‐, and high‐dosage clobazam groups)." Premature discontinuation from placebo group: n = 18 (30.5%). Reasons included lack of efficacy (10), patient/parent/caregiver request (4), AE (2), loss to follow‐up (2); Premature discontinuation from low‐dose clobazam: n = 8 (13.8%). Reasons included AE (4) , loss to follow‐up (2), protocol violation (1), lack of efficacy (1); Premature discontinuation from medium‐dose clobazam: n = 17 (27.4%). Reasons included AE (8), lack of efficacy (4), patient/parent/caregiver request (2), protocol violation (2), termination of study or withdrawal of patient by study sponsor (1); Premature discontinuation from high‐dose clobazam: n = 18 (30.5%). Reasons included AE (12), patient/parent/caregiver request (3), loss to follow‐up (2), other (1). Age at onset:Not reported. Age at diagnosis: Not reported. Age at start of treatment: Age, mean (SD) at baseline, years Placebo: 13 (9.2) Low‐dose clobazam: 10.9 (7.2) Medium‐dose clobazam: 14.1 (10.4) High‐dose clobazam: 11.7 (8.5 Sex: Male, n (%) Placebo: 38 (64.4) Low‐dose clobazam: 36 (62.1) Medium‐dose clobazam: 36 (58.1) High‐dose clobazam: 34 (57.6) Race/ethnicity: Race, n (%) Placebo: White 42 (71.2); Asian 13 (22); Black 3 (5.1); Other 1 (1.7) Low‐dose clobazam: White 33 (56.9); Asian 16 (27.6); Black 8 (13.8); Other 1 (1.7) Medium‐dose clobazam: White 35 (56.5); Asian 16 (25.8); Black 9 (14.5); Other 2 (3.2) High‐dose clobazam: White 37 (62.7); Asian 16 (27.1); Black 5 (8.5); Other 1 (1.7) Type of seizures:Not reported. Seizure frequency during the baseline period: Not reported. Number of background drugs: Not reported. "Approximately 50% of all patients were receiving concomitant valproic acid, valproate semisodium, or valproate sodium." Severity of illness:Not reported. Comorbidities: Not reported. Other relevant sociodemographics: Not reported. |
|
| Interventions |
Intervention group 1: Clobazam (low ‐ 0.25 mg/kg/day; medium ‐ 0.5 mg/kg/day; high ‐ 1.0 mg/kg/day) Number randomised to group:Total n = 179 (58 + 62 +59) Description:1) placebo 2) low‐dosage clobazam: target of 0.25 mg/kg/day (maximum, 10 mg/day) 3) medium‐dosage clobazam: target of 0.5 mg/kg/day (maximum, 20 mg/day) or 4) high dosage clobazam: target of 1.0 mg/kg/day (maximum, 40 mg/day).' Clobazam 5‐mg tablets and matching placebo tablets were supplied. During titration, clobazam 5 mg/day or 10 mg/day or placebo (in divided doses) was initiated, and dosage was increased per schedule every 7 days until the assigned target dosage was attained. At any time beginning with week 1 during titration, investigators could decrease daily dosages by a single tablet (placebo or clobazam 5 mg/day) if patients developed any signs or symptoms representing difficulty tolerating study drug.' Duration of baseline period: 4 weeks baseline. Duration of treatment period: "3‐week titration, and 12‐week maintenance periods, followed by either continuation in an open‐label study or a 2‐ or 3‐week taper period, depending on weight, with a follow‐up visit 1 week after last dose." Co‐interventions: At baseline, "Approximately 50% of all patients were receiving concomitant valproic acid, valproate semisodium, or valproate sodium." Compliance:Not reported. Group name: Placebo Number randomised to group:59 Description: "…matching placebo tablets were supplied. During titration... placebo (in divided doses) was initiated, and dosage was increased per schedule every 7 days until the assigned target dosage was attained. At any time beginning with week 1 during titration, investigators could decrease daily dosages by a single tablet (placebo or clobazam 5 mg/day) if patients developed any signs or symptoms representing difficulty tolerating study drug." Duration of baseline period:4 weeks baseline Duration of treatment period:"3‐week titration, and 12‐week maintenance periods, followed by either continuation in an open‐label study or a 2‐ or 3‐week taper period, depending on weight, with a follow‐up visit 1 week after last dose." Co‐interventions: At baseline, "Approximately 50% of all patients were receiving concomitant valproic acid, valproate semisodium, or valproate sodium." Compliance:Not reported. |
|
| Outcomes |
Outcome 1: Percentage decrease in average weekly rate of drop seizures Time points measured:"from the 4‐week baseline period to the 12‐week maintenance period." Time points reported: "from baseline to maintenance period." Outcome definition:"Drop seizures were recorded by patients’ parents/caregivers in daily seizure diaries. Drop seizure was defined as a drop attack or spell involving the entire body, trunk, or head that led to a fall, injury, slumping in a chair, or the patient’s head hitting a surface or that could have led to a fall or injury, depending on the patient’s position at the time of the attack or spell. Drop seizures were recorded as a single seizure (occurring ≥15 minutes before and after the next seizure) or cluster of seizures (≥ 2 drop seizures with < 5 minutes between any 2 consecutive seizures). For clusters, an exact number of drop seizures or a seizure range (10–20 drop seizures or >20 drop seizures) could have been recorded." Is outcome/tool validated?Unclear Imputation of missing data:"The mITT population excluded 21 patients who did not have ≥ 1 daily seizure measurement during the maintenance period. Thus, efficacy analyses included 217 patients (57 for placebo and 53, 58, and 49 for the low‐, medium‐, and high‐dosage clobazam groups)." Assumed risk estimate:Not reported. Statistical power: "This study was not designed nor powered to evaluate the effect of clobazam on non drop seizures. Although most patients had a history of non drop seizures, patients were not required to have had a minimum number of non drop seizures during the baseline period to be randomised. As a result, the sample size for non drop seizure analysis was smaller than for drop seizure analysis." Outcome 2: "Percentage decrease in average weekly rate of non drop seizures (classified according to International League Against Epilepsy guidelines)" Time points measured:"from baseline to maintenance period." Time points reported: Baseline to maintenance period. Outcome definition: "Of note, tonic clonic seizures that did not result in drop attacks were counted as non drop seizures." Is outcome/tool validated?Unclear Imputation of missing data: See above . Assumed risk estimate: Not reported. Power:See above Outcome 3: Total seizures (drop and non‐drop) Time points measured:See above. Time points reported:See above. Outcome definition:See above. Is outcome/tool validated? See above. Imputation of missing data:See above. Assumed risk estimate: See above. Power:See above. Outcome 4: Responder rates Time points measured: "from baseline to maintenance period." Time point reported: "from baseline to maintenance period." Outcome definition:Percentages with ≥ 25%, ≥ 50%, ≥ 75%, and 100% decreases in drop seizures. Is outcome/tool validated?Unclear Imputation of missing data:See above. Assumed risk estimate:Not reported. Power:See above. Outcome 5: "physicians’ and caregivers’ global evaluations of the patients’ overall changes in symptoms over time" Time points measured:Not reported. Time points reported: "from baseline to week 15/end of treatment." Outcome definition:"using a 7‐point Likert scale, with 1 = very much improved and 7 = very much worse." Is outcome/tool validated?Unclear Imputation of missing data: See above. Assumed risk estimate:Not reported. Power:See above. Outcome 6: Adverse events Time points measured:Not reported. Time points reported:Not reported. Outcome definition:Not reported. Imputation of missing data: Not applicable. "All 238 randomised patients were included in the safety population." Assumed risk estimate: Not reported. Power:See above. |
|
| Notes |
Study funding sources:"This study was funded by Lundbeck Inc. (Deerfield, IL). Manuscript preparation, including editing and formatting the manuscript, incorporating author comments, preparing tables and figures, and coordinating submission requirements, was provided by Robin L. Stromberg, PhD, of JK Associates, Inc. (Conshohocken, PA), and Michael A. Nissen, ELS, of Lundbeck Inc. This support was funded by Lundbeck." Possible conflicts of interest:Study investigators declared receipt of financial support from the study sponsor and several contributors were employed by Lundbeck or had a consultant or advisory role with the sponsor. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "patients were stratified by weight (12.5 kg to ≤ 30 kg, > 30 kg) randomly assigned (through central randomization via interactive voice response system)." |
| Allocation concealment (selection bias) | Low risk | Central randomisation via interactive voice response system. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blinded trial but it is unclear who was blinded. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blinded trial but it is unclear who was blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blinded trial but it is unclear who was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was a high rate of premature discontinuation from the placebo group, moderate and high‐dose clobazam groups." |
| Selective reporting (reporting bias) | Low risk | None identified. |
| Other bias | Low risk | None identified. |
Ohtsuka 2014.
| Study characteristics | ||
| Methods | Randomised, double‐blind, placebo‐controlled trial to evaluate the efficacy, safety, and pharmacokinetics of rufinamide as an adjunctive therapy for patients with LGS. Parallel RCT Duration of participation: "This study consisted of the following four periods: a 4‐week baseline, a 2‐week titration, a 10‐week maintenance, and either a follow‐up visit or entry into an open‐label extension." |
|
| Participants |
Population description:"Eligible patients were aged 4‐30 years, weighing 15 kg or more. LGS was diagnosed based on a history of tonic and/or atonic seizures and atypical absence seizures and slow spike‐and‐wave complex patterns on EEG within 6 months before the baseline period." Inclusion criteria:"The patients should have experienced at least 90 seizures during the 28 days before the baseline period." Exclusion criteria:"Patients were excluded from the study if they experienced tonic—clonic status epilepticus during the baseline period. Patients were also excluded if they had other clinically severe diseases or electrocardiographic/laboratory abnormalities." Total number randomised: 59 Baseline imbalances:"Patients in both groups had similar demographic and baseline characteristics"; no imbalances identified although there appeared to be relatively higher concomitant use of lamotrigine in the placebo group and relatively higher concomitant use of clobazam in the rufinamide group. Withdrawals and exclusions:"One patient assigned to the rufinamide group was excluded from the efficacy analysis due to inappropriate diagnosis." Age at onset:Mean age (SD), year Rufinamide: 16 (7.1) Placebo: 13.9 (6.1) Age at diagnosis:Mean time since LGS diagnosis (SD), year Rufinamide: 10.5 (7.1) Placebo: 9.3 (5.8) Age at start of treatment: Not reported. Sex:Male, (%) Rufinamide: 17/28 (60.7) Placebo: 19 (63.3) Race/ethnicity:Not reported. Type of seizures: Multiple seizure types Rufinamide: 19 (67.9) Placebo: 24 (80) Tonic‐atonic seizure only Rufinamide: 9 (32.1) Placebo: 6 (20) Seizure frequency during the baseline period:Median (range) per 28 days Rufinamide: 253 (95.4 to 22,499.4) Placebo: 296.7 (63 to 5759.7) Number of background drugs:n (%) One concomitant ASM Rufinamide: 2 (7.1) Placebo: 1 (3.3) Two concomitant ASMs Rufinamide: 3 (10.7) Placebo: 9 (30) Three concomitant ASMs Rufinamide: 23 (82.1) Placebo: 20 (66.7) Severity of illness: Not reported. Comorbidities:"Approximately 40% of patients had underlying causes: for example, tuberous sclerosis and cerebral palsy in five patients each; cerebral dysgenesis, encephalitis and bacterial meningitis in three patients each." Underlying causes:n (%) Rufinamide: 12 (42.9) Placebo: 13 (43.3) Transition from West syndrome, n (%) Rufinamide: 15 (53.6) Placebo: 15 (50) |
|
| Interventions |
Intervention group 1: Rufinamide Number randomised to group:29 Description:"Rufinamide was supplied as 100‐ and 200‐mg tablets…Doses were titrated according to a predetermined schedule based on body weight (Table 1). The corresponding target dose was maintained during the maintenance period. One‐step dose reduction was allowed only when investigators judged it necessary due to safety concerns. A rescue treatment (e.g. intravenous injection or rectal suppository of diazepam) was permitted for transient seizure aggravation including status epilepticus. One to three ASMs were allowed to be administered concomitantly, but they had to remain unchanged throughout the trial." Duration of baseline period:4‐week baseline. Duration of treatment period:"…a 2‐week titration, a 10‐week maintenance, and either a follow‐up visit or entry into an open‐label extension…" Co‐interventions:1‐3 concomitant ASMs. "Transient seizure aggravations were observed in 13 (22.0%) of the 59 patients [eight (27.6%) in the rufinamide group and five (16.7%) in the placebo group]. All events required rescue medication, and all medications actually used were diazepam suppositories." Control group: Placebo Number randomised to group: 30 Description:See above. Duration of baseline period:4‐week baseline. Duration of treatment period:"…a 2‐week titration, a 10‐week maintenance, and either a follow‐up visit or entry into an open‐label extension…" Co‐interventions:1‐3 concomitant ASMs; see above. |
|
| Outcomes |
Outcome 1: Percent change in the frequency of tonic‐atonic seizures per 28 days Time points measured:See below. Time points reported: See below. Outcome definition:"Percent change in seizure frequency was defined as [(D − B)/B] × 100, where D and B were the seizure frequencies per 28 days in the double‐blind period and the baseline period, respectively. The double‐blind period included the titration and the maintenance periods." Imputation of missing data:"One patient assigned to the rufinamide group was excluded from the efficacy analysis due to inappropriate diagnosis." Statistical power:"…As the result of this simulation, a sample size of 23 patients in each group was considered sufficient for a >80% power to detect a significant difference in per‐cent change in the tonic—atonic seizure frequency between the rufinamide group and the placebo group. Considering the possibility of early discontinuations, we set the target number of patients to 50 in total." Outcome 2: Percent change in total seizure frequency Outcome definition:See above. Imputation of missing data: See above. Outcome 3: 50% responder rate for the tonic‐atonic seizure frequency Outcome definition: See above. Imputation of missing data:See above. Outcome 4: "percent change in the frequency of seizures other than tonic‐atonic seizures" Outcome definition: See above. Imputation of missing data:See above. Outcome 5: "clinical global impression of the patient’s condition" Time points measured:"At Day 84" Time points reported:Not reported. Outcome definition: "clinical global impression of the patient’s condition, including seizure status, was evaluated by investigators at Day 84 using a 7‐point scale (markedly improved, improved, slightly improved, unchanged, slightly worsened, worsened, and markedly worsened)." Imputation of missing data:See above. Outcome 6: Number of patients who experienced adverse events Time points measured:Not reported. Time points reported: Not reported. Outcome definition:Not reported. Imputation of missing data:See above. Four of the included 29 people in the rufinamide group did not complete the study due to adverse events, and 1 of 39 included people in the placebo group discontinued due to an adverse event. However, one of the 29 participants in the rufinamide group was identified as having an inappropriate diagnosis and it is unclear whether this participant was one of the four who discontinued due to adverse events. |
|
| Notes |
Study funding sources:"Eisai Co., Ltd. (Tokyo, Japan) was solely responsible for the design and conduct of the study as well as the collection, management, and analysis of the data. This study was also supported by grant from Japanese government which was specifically aimed to resolve "Drug Lag" problem in Japan." Possible conflicts of interest:The first author served as a consultant for the study sponsor and another was an employee. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "a multicenter, randomised, double‐blind, placebo‐controlled trial." |
| Allocation concealment (selection bias) | Unclear risk | Not reported. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "we conducted a randomised, double‐blind, placebo‐controlled trial" but no detail is provided on who was blinded. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "We conducted a randomised, double‐blind, placebo‐controlled trial" but no detail is provided on who was blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "We conducted a randomised, double‐blind, placebo‐controlled trial" but no detail is provided on who was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | "One patient assigned to the rufinamide group was excluded from the efficacy analysis due to inappropriate diagnosis" – although the omission of one person’s data may be a low risk of bias judgement, this exclusion only refers to the efficacy data and not the safety data. Also, the methods section reports that 4 of 29 people discontinued from rufinamide, and 1 person from placebo but they appear to be included in analyses without a description of assumptions for ITT analysis. |
| Selective reporting (reporting bias) | Low risk | None identified. |
| Other bias | Low risk | None identified. |
Sachdeo 1999.
| Study characteristics | ||
| Methods | Parallalel RCT aimed to evaluate the safety and efficacy of topiramate as adjunctive therapy for Lennox–Gastaut syndrome. Duration of participation:"The study was composed of a baseline phase of 4 weeks and an 11‐week double‐blind treatment phase (a 3‐week dose titration period followed by an 8‐week maintenance period)." |
|
| Participants |
Population description: "Lennox–Gastaut syndrome is a severe childhood epilepsy syndrome characterized by multiple seizure types and a specific abnormal EEG pattern. Mental retardation or regression is common, but may not always be present at the onset of the disease. The typical EEG pattern consists of generalized, slow spike‐and‐wave discharges often accompanied by other multifocal abnormalities. Although a number of seizure types can occur, those most commonly associated with this syndrome are tonic, atonic, and absence seizures. Because it is difficult at times for parents to differentiate tonic from atonic seizures, these types of seizures are combined in drug studies and called drop attacks. Lennox–Gastaut syndrome typically develops between 1 and 8 years of age, although its greatest frequency of initial onset occurs before 5 years of age. The long‐term prognosis of Lennox–Gastaut syndrome is frequently poor, with deteriorating mental function and persistently high rates of seizures." Inclusion criteria: "Male and female patients older than 1 year but younger than 30 years and weighing at least 11.5 kg were eligible for this study. Female subjects had to be either premenarchal or practicing an acceptable method of birth control. Eligible patients had to have a prior EEG tracing showing a slow spike‐and‐wave pattern and seizure types including drop attacks (i.e. tonic‐atonic seizures) and either a history of or active atypical absence seizures. Other seizure types could include tonic‐clonic, myoclonic, and partial‐onset seizures. Seizures were classified according to the International Classification of Epilepsies and Epileptic Seizures. Patients were required to have at least 60 seizures (of all eligible types) during the month before entering the baseline phase while being maintained on one or two standard AEDs." Exclusion criteria:"Patients were excluded from study participation if they had a history of recent significant cardiovascular, respiratory, hepatic, renal, gastrointestinal, or hematologic illness, or malignancy; seizures due to progressive disease; documented status epilepticus within 3 months of baseline; drug or alcohol abuse; a psychiatric or mood disorder requiring medication or electroconvulsant therapy within 6 months of baseline; poor compliance with therapy; anoxic episodes requiring resuscitation within 1 year before the study; or nephrolithiasis. Additional exclusion criteria included treatment with an experimental drug or use of an experimental device within 60 days of baseline; use of acetazolamide or zonisamide within 60 days of baseline; treatment with a ketogenic diet or adrenocorticotropic hormone within 6 months before the study; use of benzodiazepines on more than an occasional basis (unless used as a concomitant AED); presence of clinically significant EKG abnormalities; or history of an inability to take medication or maintain a seizure calendar, independently or with assistance. By protocol amendment, after approximately 50% of the patients had been enrolled, felbamate was prohibited because of its association with aplastic anemia." Total number randomised: 98 Baseline imbalances:"The topiramate and placebo treatment groups were comparable both demographically and in terms of seizure and medication history." There appeared to be a slightly higher proportion of males in the topiramate group. Both groups largely involved white participants (>80%) Withdrawals and exclusions: Of the randomised participants, only one person was withdrawn due to patient choice, from the topiramate group. Age at onset:Baseline mean age, years (SD) Topiramate: 11.2 (6.2) Placebo: 11.2 (7.7) Age at diagnosis:Not reported. Age at start of treatment: Not reported. Sex: Male/Female Topiramate: 28/20 Placebo: 25/25 Race/ethnicity: Topiramate: White, 39; African American, 4; Other, 5 Placebo: White, 46; African American, 3; Other, 1 Type of seizures:Drop attacks (tonic‐atonic seizures), Major seizures (drop attacks and tonic‐clonic seizures), All types of seizures. Seizure frequency during the baseline period:All types of seizures, n (range). Topiramate: 267 (13 to 3795) Number of background drugs: One ‐ 19 Two ‐ 27 Three – 2 Placebo: One – 20 Two – 29 Three ‐ 1 Comorbidities: Not reported but exclusion criteria specified some co‐morbidities. |
|
| Interventions |
Group: Topiramate Number randomised to group:48 Description: "The study was composed of a baseline phase of 4 weeks and an 11‐week double‐blind treatment phase (a 3‐week dose titration period followed by an 8‐week maintenance period)." "During the 4‐week baseline period, patients received a constant dose of one or two AEDs. Patients who qualified for entry into the double‐blind treatment period were randomised at each center to receive either topiramate or matching placebo tablets while maintaining their background dose(s) of AEDs. The titration period was divided into three 1‐week intervals. During the first week, study drug dosage was approximately 1 mg/kg/d administered twice daily in equal doses. Dosage was increased to approximately 3 mg/kg/d during the second week and to a target dose of 6 mg/kg/d during the third week. If the patient did not tolerate the titration schedule, the rate of titration was either maintained or reduced at the discretion of the investigator. Patients were then followed for an 8‐week maintenance period on 6 mg/kg/d or their maximal tolerated dosage. Dosages could be reduced to whatever level was appropriate in terms of adverse events at any time during the trial. Rechallenges were permitted if dosages of study drug were temporarily discontinued." Duration of baseline period:4 weeks Duration of treatment period:11 weeks, including titration and maintenance periods. Co‐interventions:Background dose(s) of ASMs but with a number of exclusion criteria for study participation. Note that felbamate was prohibited part way through the study due to adverse effects. Compliance: Only one participant was withdrawn – patient choice. Group: Placebo Number randomised to group:50 Description: "The study was composed of a baseline phase of 4 weeks and an 11‐week double‐blind treatment phase (a 3‐week dose titration period followed by an 8‐week maintenance period)." "During the 4‐week baseline period, patients received a constant dose of one or two ASMs. Patients who qualified for entry into the double‐blind treatment period were randomised at each center to receive either topiramate or matching placebo tablets while maintaining their background dose(s) of AEDs. The titration period was divided into three 1‐week intervals. During the first week, study drug dosage was approximately 1 mg/kg/d administered twice daily in equal doses. Dosage was increased to approximately 3 mg/kg/d during the second week and to a target dose of 6 mg/kg/d during the third week. If the patient did not tolerate the titration schedule, the rate of titration was either maintained or reduced at the discretion of the investigator. Patients were then followed for an 8‐week maintenance period on 6 mg/kg/d or their maximal tolerated dosage. Dosages could be reduced to whatever level was appropriate in terms of adverse events at any time during the trial. Rechallenges were permitted if dosages of study drug were temporarily discontinued." Duration of baseline period: 4 weeks Duration of treatment period:11 weeks, including titration and maintenance periods. Co‐interventions: Background dose(s) of ASMs but with a number of exclusion criteria for study participation. Note that felbamate was prohibited part way through the study due to adverse effects. Compliance:No participant was withdrawn. |
|
| Outcomes |
Outcome 1: Percentage reduction in the average (median) monthly (28‐day) seizure rate for all seizure types Time points measured:Monthly throughout the double‐blind phase from baseline. Time points reported: From baseline during the double‐blind phase. Outcome definition: See above. Is outcome/tool validated? Unclear Imputation of missing data:Not applicable. Assumed risk estimate: Not reported. Power: "A sample size of 40 patients per treatment group was determined based on the detection of a 30% difference between groups in percentage reduction from baseline in seizure rates. The calculation of sample size assumed a type I error level of 5%, a power of 80%, and an SD of 70%. As the study population approached 80, investigators were notified to stop enrolment. At that time, a number of patients had been screened for study entry, and it was decided that it would not be ethical to deny entry to these patients. Thus, the actual sample size exceeded the planned sample size." Outcome 2: Percentage reduction in drop attacks (tonic‐atonic seizures) Time points measured:Monthly throughout the double‐blind phase from baseline. Time points reported: From baseline during the double‐blind phase. Is outcome/tool validated? Unclear Imputation of missing data: Not applicable. Assumed risk estimate:Not reported. Power: See above. Outcome 3: Percentage reduction in the average (median) monthly (28‐day) rate of major seizures (drop seizures and tonic‐clonic seizures) Time points measured:Monthly throughout the double‐blind phase from baseline. Time points reported:From baseline during the double‐blind phase. Outcome definition: See above. Is outcome/tool validated? Unclear Imputation of missing data: Not applicable. Assumed risk estimate:Not reported. Power: See above. Outcome 4: Percentage of treatment responders, defined as a ≥ 50%, ≥ 75% or 100% reduction from baseline for drop attacks, major seizures and all seizures Time points measured:Monthly throughout the double‐blind phase from baseline. Time points reported: From baseline during the double‐blind phase. Outcome definition: See above. Is outcome/tool validated? Unclear Imputation of missing data: Not applicable. Assumed risk estimate:Not reported. Power: See above. Outcome 5: Parental global evaluation of seizure severity Time points measured: "relative to baseline was rated at the final double‐blind visit." Time points reported:"during the double‐blind phase relative to baseline." Outcome definition: "Parental global evaluation of seizure severity improvement relative to baseline was rated…using a seven‐point scale, as either very much improved, much improved, minimally improved, no change, minimally worse, much worse, or very much worse." Is outcome/tool validated? Unclear Imputation of missing data: Not applicable. Assumed risk estimate:Not reported. Power: See above. Outcome 6: Parental global evaluation of mental status Time points measured: "relative to baseline was recorded at the final visit of the double‐blind treatment phase." Time points reported:"As evaluated by parents or guardians at the end of the double‐blind treatment." Outcome definition:"On a seven‐point scale, the patient’s improvement in level of alertness, level of interaction with the environment, ability (or the caregiver’s ability) to perform activities of daily living, and responsiveness to verbal requests were recorded as either very much improved, much improved, minimally improved, no change, minimally worse, much worse, or very much worse." Is outcome/tool validated? Unclear Imputation of missing data: Not applicable. Assumed risk estimate:Not reported. Power: See above. Outcome 7: Adverse events / Treatment‐emergent adverse events Time points measured:"Adverse events were reported at each study visit." Time points reported: Not reported. Outcome definition: "Treatment‐emergent adverse events, defined as those events that occurred for the first time or worsened after treatment, were summarized by treatment group." Is outcome/tool validated? Unclear Imputation of missing data: Not applicable. Assumed risk estimate:Not reported. Power: See above. |
|
| Notes |
Study funding sources:"Funded by the R.W. Johnson Pharmaceutical Research Institute and PHS grant no. M01‐RR00865." Possible conflicts of interest:Author(s) based at the above Institute. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomised." |
| Allocation concealment (selection bias) | Low risk | "Allocation was by random permuted blocks of four and was stratified by center." |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "During the double‐blind phase, investigators, patients, study monitors, and observers remained blinded to codes." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "During the double‐blind phase, investigators, patients, study monitors, and observers remained blinded to codes." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "During the double‐blind phase, investigators, patients, study monitors, and observers remained blinded to codes." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only one person was withdrawn of 98 randomised participants. |
| Selective reporting (reporting bias) | Low risk | None identified. |
| Other bias | Low risk | None identified. |
Thiele 2018.
| Study characteristics | ||
| Methods | The GWPCARE4 study was designed to assess the efficacy and safety of cannabidiol compared with placebo as add‐on therapy to existing antiepileptic drugs for the treatment of seizures associated with Lennox‐Gastaut syndrome in children and adults." Parallel RCT Duration of participation:"All patients received treatment for 14 weeks, which included 2 weeks of dose escalation (starting at a daily dose of 2·5 mg/kg, followed by 12 weeks of stable dosing [maintenance]), a tapering period of up to 10 days, and a 4‐week safety follow‐up period (appendix)." |
|
| Participants |
Population description:"Eligible patients were aged between 2 and 55 years, with a clinical diagnosis of Lennox‐Gastaut syndrome (including documented history of slow [< 3·0 Hz] spike‐and‐wave electroencephalograms), and evidence of more than one type of generalised seizure, including drop seizures, for at least 6 months. The definition of Lennox‐Gastaut syndrome chosen for this trial was the same as has been used in other multicentre trials." Inclusion criteria:"Eligible patients were aged between 2 and 55 years, with a clinical diagnosis of Lennox‐Gastaut syndrome (including documented history of slow [< 3·0 Hz] spike‐and‐wave electroencephalograms), and evidence of more than one type of generalised seizure, including drop seizures, for at least 6 months. The definition of Lennox‐Gastaut syndrome chosen for this trial was the same as has been used in other multicentre trials. Patients who were refractory (i.e. inadequately managed on at least two antiepileptic drugs, inclusive of previous and current treatments), were taking one to four antiepileptic drugs, and had at least two drop seizures per week during the 4‐week baseline period were eligible. Patients in whom all medications and interventions for epilepsy (including ketogenic diet and vagus nerve stimulation) were stable for 4 weeks before screening were included." Exclusion criteria:"Patients who had a clinically significant unstable illness (other than epilepsy) in the 4 weeks before screening or randomisation, had a history of alcohol or substance misuse, were recreational or medicinal cannabis users, had taken corticotrophins in the previous 6 months, or who had been taking felbamate for less than 1 year before screening were excluded. Patients with a positive urine tetrahydrocannabinol screen at the beginning of the study, and female patients who were pregnant, lactating, or planning pregnancy during or within 3 months of completing the trial were also ineligible." Total number randomised:171 Baseline imbalances:"Patient demographics and baseline characteristics were similar across the two treatment groups (table 1)." No imbalances identified in table 1 but the placebo group had a higher proportion of participants on a ketogenic diet as a concomitant intervention (12% versus 5%). Withdrawals and exclusions:"14 patients in the cannabidiol group and one in the placebo group withdrew from the trial; in nine (60%) of these patients, adverse events were the primary reason for study discontinuation." Age at onset:Mean age, years (SD) Cannabidiol: 15.5 (8.7) Placebo: 15.3 (9.8) Age, years (range) Cannabidiol: 14.2 (2.7 to 39) Placebo: 13.3 (2.8 to 45.1) Age at diagnosis:Not reported. Age at start of treatment:Not reported. Sex:Male: Female (%) Cannabidiol: 45:41 (52:48) Placebo: 43:42 (51:49) Race/ethnicity:White (%) Cannabidiol: 75 (87) Placebo: 79 (93) Other *(%) Cannabidiol: 11 (13) Placebo: 6 (7) *"Includes patients who identified as black or African American, Asian, Hispanic, Latino, and Arabian" Type of seizures:Monthly frequency of drop seizures, total seizures and non‐drop seizures reported at baseline. Seizure frequency during the baseline period: Drop seizures, median (IQR) Cannabidiol: 71.4 (27 to 156) Placebo: 74.7 (47.3 to 144) Total seizures, median (IQR) Cannabidiol: 144.6 (72 to 385.7) Placebo: 176.7 (68.6 to 359.5) Non‐drop seizures, median (IQR) Cannabidiol: 94 (19.8 to 311) Placebo: 85 (20.5 to 220) Number of background drugs: Previous ASMs per patient, median (range) Cannabidiol: 6 (1‐18) Placebo: 6 (0‐28) Concomitant AEDs per patient, median (range) Cannabidiol: 3 (1‐5) Placebo: 3 (1‐4) Severity of illness: Not reported. Comorbidities: Not reported. |
|
| Interventions |
Intervention group 1: Add‐on cannabidiol Number randomised to group:86 Description:"Patients received 20 mg/kg of a pharmaceutical formulation of purified cannabidiol (100 mg/mL, GW Pharmaceuticals (Cambridge, UK) in oral solution daily, or matching placebo solution. Cannabidiol or placebo was administered orally in two equally divided doses (morning and evening) for 14 weeks. The 20 mg/kg dose of cannabidiol was approved by an independent data safety monitoring committee (DSMC), who reviewed data from a dose‐ranging safety and pharmacokinetic evaluation22 of three doses of cannabidiol (5, 10, and 20 mg/kg daily) in Dravet syndrome and identified 20 mg/kg per day as a safe dose without unacceptable side‐effects." "All patients received treatment for 14 weeks, which included 2 weeks of dose escalation (starting at a daily dose of 2·5 mg/kg, followed by 12 weeks of stable dosing [maintenance]), a tapering period of up to 10 days, and a 4‐week safety follow‐up period (appendix)." Duration of baseline period:"4‐week baseline period"/ "4‐week screening period." Duration of treatment period:"All patients received treatment for 14 weeks." Co‐interventions:Ketogenic diet in 5% of cannabidiol group and 12% of placebo group; Vagus nerve stimulation 30% of cannabidiol group and 29% of placebo group; Concomitant ASMs (including clobazam, valproate, lamotrigine, levetiracetam and rufinamide) Supplementary appendix details concomitant ASMs in table S3 but the list excludes rescue medications. Control group: Placebo Number randomised to group:85 Description:See above. Duration of baseline period:"4‐week baseline period"/ "4‐week screening period." Duration of treatment period:"All patients received treatment for 14 weeks." Co‐interventions:See above. |
|
| Outcomes |
Outcome 1: "the percentage change in monthly frequency of drop seizures from baseline" Note that the protocol in the Supplementary appendix specifies this as a "mean" percentage change although the statistical analysis of the full report refers to a "median" which is understood to be "Because the seizure data had a non‐normal distribution." Time points measured:"measured during the 14‐week treatment period." Time points reported: "over the 14‐week treatment period." Outcome definition:"1 month was defined as 28 days. A drop seizure was defined as an attack or spell (atonic, tonic, or tonic‐clonic) involving the entire body, trunk, or head that led or could have led to a fall, injury, slumping in a chair, or hitting the patient’s head on a surface. The functional definition of drop seizure used in this trial was reviewed and approved by the US Food and Drug Administration, the European Medicines Agency, and an independent committee of experts from the Epilepsy Study Consortium, and was similar to that used in a previous clobazam trial.9 All seizure types or descriptions given by each patient were confirmed by the Epilepsy Study Consortium." Imputation of missing data:"The primary endpoint was analysed in the intention‐to‐treat analysis dataset, which included all randomly assigned patients who received at least one dose of cannabidiol or placebo and had post‐baseline efficacy data…Analyses using the per‐protocol analysis set were additionally performed for the primary and key secondary endpoints only." Statistical power: 'Thus, a sample size of 100 patients (50 per group) was calculated to provide 80% power with a two‐tailed significance level of 0·05." Outcome 2: "the proportion of patients in each treatment group that achieved a reduction of 50% or more in monthly frequency of drop seizures" Time points measured:Assumed to be the same as for Outcome 1, over the 14‐week treatment period. Time points reported:During the treatment period. Outcome definition:See Outcome 1 for definition of drop seizures; one month defined as 28 days. Imputation of missing data:"Secondary endpoints were also analysed in the intention‐to‐treat dataset, apart from seizure reduction during the maintenance period and patient or caregiver GIC, which were analysed in all patients who had post‐baseline efficacy data for those endpoints. Analyses using the per‐protocol analysis set were additionally performed for the primary and key secondary endpoints only." Outcome 3: "percentage change in total seizure frequency from baseline during the treatment period (i.e. sum of all individual seizure subtypes reported)" Time points measured:Assumed to be the same as for Outcome 1, over the 14‐week treatment period. Time points reported: Over the 14‐week period. Outcome definition:See Outcome 1 for definition of drop seizures; one month defined as 28 days. Outcome 4: "change from baseline in patient and caregiver global impression of change (GIC)" Time points measured:"at the end of treatment." Time points reported:"At their last visit to the clinic…compared with baseline." Outcome definition: Not reported in the full report. See Supplementary appendix: "The S/CGICSD, as appropriate, comprises a question to be rated on a three‐point scale for each seizure subtype… The markers are: Average duration of seizures has decreased; Average duration of seizures has stayed the same; Average duration of seizures has increased. The patient or their caregiver will be asked to assess the average duration of the patient’s seizures at Visit 2 (i.e. prior to commencement of IMP) as a memory aid for subsequent visits. If the main caregiver is not available at the appropriate visit then this information can be captured over the telephone, ideally on the day of the visit or otherwise within three days." Imputation of missing data:See above. Outcome 5: "Responder analysis (i.e. the proportion of patients who achieved a ≥ 25%, ≥ 50%, ≥ 75%, or 100% reduction in drop seizures from baseline)" Time points measured:Assumed to be the same as for Outcome 1, over the 14‐week treatment period. Time points reported: "…during the treatment period (A) and the maintenance period alone (B)." Outcome definition: See Outcome 1 for definition of drop seizures. Imputation of missing data:See Outcome 2 for details of ITT and per protocol analysis. Outcome 6: "percentage change in the frequency of non‐drop, convulsive (tonic‐clonic, tonic, clonic, or atonic seizures), non‐convulsive (myoclonic, countable focal, other focal, or absence seizures), and individual seizure types" Time points measured: Assumed to be the same as for Outcome 1, over the 14‐week treatment period. Time points reported: During the 14‐week treatment period and during the 12‐week maintenance period alone. Outcome definition: Not reported. Imputation of missing data: See Outcome 2 for details of ITT and per protocol analysis. Outcome 7: "the proportion of patients with adverse events" Time points measured:Assumed to be the same as for Outcome 1, over the 14‐week treatment period. Time points reported:Not reported. Outcome definition:"measured by the investigators using standard severity measures (i.e. mild, moderate, or severe)." Imputation of missing data: "The safety analyses included all randomised patients who received at least one dose of the study drug." Outcome 8: "Columbia Suicide Severity Rating Scale (C‐SSRS) scores" Time points measured: Not reported. Time points reported:Not reported. Outcome definition:Not defined in the full report. See the Supplementary appendix, protocol: "The definitions of behavioral suicidal events used in this scale are based on those used in the Columbia Suicide History Form. Questions are asked on suicidal behavior, suicidal ideation and intensity of ideation. At screening (Visit 1), questions will be in relation to lifetime experiences (Baseline). Questioning at all subsequent visits will be in relation to the last assessment (Since Last Visit). The C‐SSRS will be used for patients 19 years of age and above. The C‐SSRS Children’s will be used for patients six to 18 years of age. The C‐SSRS is to be administered by the investigator or his/her qualified designee at every visit as indicated in the Schedule of Assessments (APPENDIX 1); "qualified designee" is defined as physician, osteopath, nurse practitioner, clinical psychologist or physician’s assistant, who is licensed and has completed the C‐SSRS training within the last two years. The survey should be completed by the same assessor, where possible, throughout the study. Assessments will be conducted only if patients are of an appropriate age (six years of age and older) and capable of understanding and answering the questions, in the investigator’s opinion." Imputation of missing data:"The safety analyses included all randomised patients who received at least one dose of the study drug." Outcome 9: "frequency of episodes of status epilepticus" Time points measured: Assumed to be the same as for Outcome 1, over the 14‐week treatment period. Time points reported: Not reported. Outcome definition: Not defined. Imputation of missing data:"The safety analyses included all randomised patients who received at least one dose of the study drug." |
|
| Notes |
Study funding sources:"This study was funded by GW Pharmaceuticals (Cambridge, UK). Greenwich Biosciences (US subsidiary of GW Pharmaceuticals) also provided financial support for medical writing." Possible conflicts of interest: Study investigators declared receipt of grants from the study sponsor, employment by the study sponsor and ownership of shares or stock options with the study sponsor. Other:It is noted that the inclusion and exclusion criteria in the full report are not fully consistent with those reported in the protocol. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "We did a randomised, double‐blind, placebo‐controlled, phase 3 trial…At visit 1, each patient was assigned a unique number via an interactive voice response system (IVRS) and then at visit 2 the IVRS was used to randomly assign eligible participants to treatment in a 1:1 ratio. The randomisation schedule was produced by an independent statistician, and was stratified by age group." |
| Allocation concealment (selection bias) | Low risk | The randomisation schedule was produced by an independent statistician. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | "We did a randomised, double‐blind, placebo‐controlled, phase 3 trial…Both cannabidiol and placebo were provided in identical 100 mL amber glass bottles and could not be distinguished visually. GW Pharmaceuticals manufactured and supplied the study drug. All patients, caregivers, investigators, and individuals assessing data were masked to group assignment." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "We did a randomised, double‐blind, placebo‐controlled, phase 3 trial… Both cannabidiol and placebo were provided in identical 100 mL amber glass bottles and could not be distinguished visually. GW Pharmaceuticals manufactured and supplied the study drug. All patients, caregivers, investigators, and individuals assessing data were masked to group assignment." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "We did a randomised, double‐blind, placebo‐controlled, phase 3 trial… Both cannabidiol and placebo were provided in identical 100 mL amber glass bottles and could not be distinguished visually. GW Pharmaceuticals manufactured and supplied the study drug. All patients, caregivers, investigators, and individuals assessing data were masked to group assignment." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The statistical analysis clearly outlined ITT and per protocol approaches for the outcomes. "All 15 patients who withdrew early from the trial were excluded from the per‐protocol analysis set (used in sensitivity analyses)." |
| Selective reporting (reporting bias) | High risk | "Analyses for the other secondary endpoints are presented in the appendix, with the exception of the Cannabis Withdrawal Scale, number of hospital admissions, and cognitive function, for which insufficient data was collected." |
| Other bias | Low risk | None identified. |
AED: antiepileptic drug; ASM: anti‐seizure medication; EEG: electroencephalogram; ITT: Intention‐to‐treat; LGS: Lennox‐Gastaut syndrome; RCT: randomised controlled trial.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Battaglia 1991 | Identified for exclusion in the previous version of this review and agreed for exclusion in the review update. This cross‐over study included a subset of participants with Lennox‐Gastaut syndrome but the data were not usable because individual case data were reported incompletely. Review authors did not attempt to seek additional data from this study, which was published over 29 years ago. |
| Conry 2009 | Excluded from this review update because it compared different doses of the same ASM. |
| Inanaga 1989 | Excluded from this review update because it compared different doses of the same ASM. |
| Oletsky 1996 | Excluded from this review update because it was not a randomised trial. |
| Perry 2019 | Excluded from this review update because it was not a randomised controlled trial. |
| Vajda 1985 | Identified for exclusion in the previous version of this review and agreed for exclusion in the review update. This study abstract reported a subset of participants with LGS but with no results provided separately for them. Review authors did not attempt to seek additional data from this study, which was published over 35 years ago. |
| Vassella 1978 | Identified for exclusion in the previous version of this review and agreed for exclusion in the review update. The abstract for this cross‐over study does not report results separately for the two periods of the study. Previous review authors were able to access a translation (from German into English) of the full article but data were not usable because the reporting of results was unclear. Review authors did not attempt to seek additional data from this study, which was published over 42 years ago. |
| Vigevano 1994 | Full text unobtainable but judged not to be a RCT based on the abstract. |
ASM: anti‐seizure medication; LGS: Lennox‐Gastaut syndrome; RCT: randomised controlled trial.
Characteristics of studies awaiting classification [ordered by study ID]
Ohtahara 2008.
| Methods | RCT |
| Participants | 63 participants in subgroup with Lennox‐Gastaut syndrome |
| Interventions | Lamotrigine vs zonisamide |
| Outcomes | Reduction in drop attacks and all seizure types |
| Notes | No English translation available |
Characteristics of ongoing studies [ordered by study ID]
CTRI/2010/091/001449.
| Study name | A Comparative, randomised, Open label, Multicentric, Prospective Clinical Study to Evaluate the Efficacy, Safety and Tolerability of Rufinamide Tablet Vs. Lamotrigine (Adjunctive Therapy) in the treatment of seizures associated with Lennox‐Gastaut syndrome. |
| Methods | RCT |
| Participants | People with Lennox‐Gastaut syndrome |
| Interventions | Adjunctive rufinamide versus adjunctive lamotrigine |
| Outcomes | Seizure frequency; global assessment of efficacy and tolerability; improvement in signs and symptoms of headache, dizziness, fatigue, nausea, sleepiness |
| Starting date | Not applicable |
| Contact information | Dr Shailesh Singh, Vice President Medical & Regulatory Affairs. Email: shailesh.singh@ajantapharma.com |
| Notes | Study terminated. "It was decided by Ajanta Management that they would not be keen to proceed with marketing of this product. Thus the proposed Clinical Trial was not initiated." |
NCT00004776.
| Study name | Phase III randomised, Double‐Blind, Placebo‐Controlled Study of Oral Topiramate for Lennox‐Gastaut Syndrome |
| Methods | RCT |
| Participants | People with Lennox‐Gastaut syndrome |
| Interventions | Topiramate versus placebo ‐ no further details reported |
| Outcomes | No details reported |
| Starting date | November 1993 |
| Contact information | Not reported |
| Notes | Recruitment completed |
NCT01370486.
| Study name | Melatonin Versus Placebo in the Lennox‐Gastaut Syndrome: Neurophysiological and Neuropsychological Effects |
| Methods | Randomised, placebo‐controlled crossover trial |
| Participants | People with Lennox‐Gastaut syndrome |
| Interventions | Melatonin versus placebo |
| Outcomes | Diminution of at least 50% of the nocturnal interictal discharges and tonic seizures with the melatonin treatment; augmentation of at least 15% of the amount of deep slow sleep with the melatonin treatment |
| Starting date | August 2011 |
| Contact information | Dr Giovanni B. Foletti. Principal Investigator. Telephone ++ 41 21 821 46 46. Email: NCT01370486, 25/10, Melatonin Versus Placebo in the Lennox‐Gastaut Syndrome: Neurophysiological and Neuropsychological Effects" type="EXTERNAL">giovanni.foletti@ilavigny.ch |
| Notes | Recruitment status unknown |
NCT02318537.
| Study name | Cannabidiol Oral Solution as an Adjunctive Therapy for Treatment of Participants With Inadequately Controlled Lennox‐Gastaut Syndrome |
| Methods | RCT |
| Participants | People with inadequately controlled Lennox‐Gastaut syndrome |
| Interventions | Adjunctive cannabidiol versus placebo |
| Outcomes | Percent change from baseline in the frequency of motor seizures involving the trunk or extremities [tonic, atonic, generalized tonic‐clonic (GTC), focal seizures with motor components (FSMC)] ; Percent change from baseline in severity of motor seizures involving the trunk or extremities (tonic, clonic, GTC, FSMC); Percent change from baseline in frequency of all seizure activity independent of seizure type; Percent change from baseline in the severity of all seizure activity independent of seizure type; Percent change from baseline in the duration of all seizure activity independent of seizure type; Change from baseline in parent(s)/caregiver(s) Clinical Global Impressions of Improvement (CGI‐I); Change from baseline in Investigator CGI‐I ; Change from baseline in parent(s)/caregiver(s) Clinical Global Impressions of Severity (CGI‐S); Change from baseline in Investigator CGI‐S |
| Starting date | Not applicable |
| Contact information | Not reported |
| Notes | 'Withdrawn (Sponsor elected not to continue with study)'. |
NCT03355209.
| Study name | A Study to Investigate the Efficacy and Safety of ZX008 (Fenfluramine Hydrochloride) as an Adjunctive Therapy in Children and Adults With Lennox‐Gastaut Syndrome |
| Methods | This is a multicenter, double‐blind, parallel‐group, placebo controlled study to evaluate the effect of adjunctive ZX008 (Fenfluramine Hydrochloride) for the treatment of uncontrolled seizures in children and adults with LGS. |
| Participants | It will include patients with LGS aged between 2 and 35 years, receiving at least 1 concomitant ASM and up to 4 concomitant antiepileptic treatments. |
| Interventions | Subjects will be randomised to receive 1 of 2 doses of ZX008 (0.2 mg/kg/day or 0.8 mg/kg/day given as oral solution) or placebo. In the subsequent open‐Label phase ZX008 is supplied as an oral solution. |
| Outcomes | The main outcome will be the change from baseline in frequency of seizures that result in drops in subjects receiving ZX008 compared to placebo [Time Frame: Up to 20 weeks maintenance and taper period]. Secondary outcome will be safety and tolerability. |
| Starting date | 27 November 2017 |
| Contact information | |
| Notes | Contact: ZX008 Clinical Trials Information Desk |
NCT03650452.
| Study name | A Phase 2, Multicenter, randomised, Double‐blind, Placebo‐controlled Study to Evaluate the Efficacy, Safety, and Tolerability of TAK‐935 (OV935) as an Adjunctive Therapy in Pediatric Patients With Developmental and/or Epileptic Encephalopathies (ELEKTRA) |
| Methods | This randomised, double‐blind study will assess the effects of TAK‐935 (OV935), compared to placebo, on efficacy, safety, and tolerability in paediatric patients with Dravet syndrome or LGS. |
| Participants | Pediatric patients with Dravet syndrome or LGS. |
| Interventions | The study will consist of a screening Period and Treatment Period (8‐week Dose Optimization Period and 12‐week Maintenance Period). The overall time to patients in this study is approximately 30 weeks. This randomised trial will be followed by an open‐label extension study. |
| Outcomes | Following outcomes will be evaluated: (1) percent change from baseline in frequency of all seizures (convulsive and drop) per 28 days in patients receiving TAK‐935 (OV935) as compared to placebo during the Maintenance Period; (2) percent change from baseline in all seizures (convulsive and drop) in patients receiving TAK‐935 (OV935) as compared to placebo during Treatment Period [Time Frame: up to 20 weeks]; (3) percent change from baseline in frequency of convulsive seizures in Dravet patients and drop seizures in Lennox Gastaut syndrome (LGS) patients, respectively, receiving TAK‐935 (OV935) as compared to placebo during Maintenance Period [Time Frame: up to 12 weeks]; (4) percentage of patients receiving TAK‐935 (OV935) as compared to placebo in the Dravet Syndrome Stratum and LGS Stratum, respectively, considered treatment responders throughout Maintenance Period [Time Frame: up to 12 weeks]; (5) Change in Clinician's and Caregiver's Clinical Global Impression of Severity and Change (CGI‐S/C) [Time Frame: up to 20 weeks]; and (6) Correlation of TAK‐935 concentration and plasma 24S‐hydroxycholesterol (24HC) levels [Time Frame: up to 20 weeks]. |
| Starting date | 8 August 2018 |
| Contact information | TakedaOvid Therapeutics Inc. |
| Notes | This multi‐centre trial will be conducted worldwide and will enrol approximately 126 patients. |
NCT03808935.
| Study name | Cannabis Extract in Refractory Epilepsy Study (CERES) |
| Methods | Phase III, double‐blind, randomised, placebo‐controlled, parallel‐group trial, followed by an open phase aimed at evaluating the efficacy and safety of adjunctive cannabidiol for the treatment of patients with LGS and other epileptic syndromes. |
| Participants | It will included adult patients (18 years of age and older) with drug‐resistant epilepsy, including patients with Dravet and LGS, and patients with frequent convulsive seizures. |
| Interventions | Patients will be randomised to receive cannabidiol at a maximum total daily dose of approximately 300 mg versus placebo. |
| Outcomes | Following outcomes will be assessed: Frequency of seizures; side effects; blood levels of ASMs, CBD, THC, and liver enzymes; impact on cognition and quality of life; genetics. |
| Starting date | January 10, 2019 |
| Contact information | The Epilepsy Research Program of the Ontario Brain Institute |
| Notes | It is estimated that a total of 80 participants (40 assigned to treatment and 40 to control group) will be recruited. |
Wechsler 2017.
| Study name | Study of Perampanel as Adjunctive Treatment for Inadequately Controlled Seizures Associated With Lennox‐Gastaut Syndrome |
| Methods | Ongoing multicentre, double‐blind, randomised, placebo‐controlled trial of adjunctive perampanel in LGS patients >=2 years of age with inadequately controlled seizures. |
| Participants | It will include patients aged between 2 and 11 years, with at least 2 drop seizures during baseline and currently being treated with stable doses of 1‐3 antiepileptic drugs. |
| Interventions | Patients will receive perampanel 2‐8 mg/day in a single daily dose. The study will include a Core Phase (Pre‐randomisation screening and Baseline [4‐8 weeks]; Randomisation [6‐week Titration; 12‐week Maintenance; 4‐week Follow‐up]) and Open‐Label Extension Phase (52‐week Conversion/Maintenance; 4‐week Follow‐up). |
| Outcomes | The primary endpoint is change in drop seizure (tonic, atonic, myoclonic) frequency per 28 days: other outcomes include reductions in total and non‐drop seizures per 28 days, safety and perampanel pharmacokinetics; quality of life, long‐term efficacy and resource utilisation. |
| Starting date | 13 December 2016 |
| Contact information | Contact: Eisai Medical Information |
| Notes | It is estimated that approximately 142 patients will be randomised to receive adjunctive perampanel or placebo. |
Differences between protocol and review
Compared to the review protocol and the previous version of this review, we rewrote the introduction section to provide a more updated background on the topic based on recent advances in the literature. Furthermore, we modified the methods sections to meet the current Methodological Expectations of Cochrane Intervention Reviews (MECIR) standards, as follows:
We differentiated between primary and secondary outcomes. Among primary outcomes, we analysed adverse events leading to study discontinuation. We specified how we dealt with studies reporting both median quantitative reduction and the number of participants with a quantitative reduction in an outcome. We also specified that we included outcome measures reported at any time point, and if a study reported multiple time points, we planned to prioritise reporting and meta‐analysis of the outcome at the longest follow‐up. However, we found that outcomes reported phases of dose adjustment before and, or after dose maintenance; we retrospectively agreed to prioritise the reporting of results for the treatment period as described in studies.
We specified in more detail which data we planned to extract, through dual processes, and by whom.
Two authors independently assessed risk of bias for each of the included trials using the Cochrane 'Risk of bias' tool (Higgins 2011).
We used the GRADE approach to evaluate the certainty of evidence for the selected primary outcomes (Schünemann 2020). We presented the certainty of evidence for primary outcomes in 'Summary of findings' tables.
We excluded studies comparing different doses of the same ASM. As such, we excluded Conry 2009 and Inanaga 1989, which were included in the previous version of the review although the methods did not pre‐specify dose comparisons.Ohtahara 2008
We removed two previously included tables: 'Methodological quality of included studies' and 'Participant characteristics'; we provided relevant data in the Included studies tables.
We updated information on the methods used as a measurement of treatment effect for continuous variables, including use of Peto OR where there was < 1% events and we also provided rationale for using a random‐effect model in data synthesis.
We updated the Unit of analysis issues and revised the 'Assessment of reporting biases' section.
We updated the 'Subgroup and sensitivity analysis', specifying that we did not consider comparisons between doses of the same ASM.
We updated the 'Summarising and interpreting results' section.
Contributions of authors
FB: initial screening; data extraction; risk of bias assessment; data entry and analysis; GRADE assessment; drafting and revision of review update.
KJ: initial screening; data extraction; risk of bias assessment; data entry and analysis; GRADE assessment and creation of Summary of findings tables; drafting and revision of review update.
CE: data extraction; risk of bias assessment, drafting and revision of review update.
SM: data extraction; risk of bias assessment, drafting and revision of review update.
Sources of support
Internal sources
No sources of support provided
External sources
National Institute for Health and Care Research, UK
Declarations of interest
FB: Francesco Brigo received travel support and accommodation by Lusofarmaco to attend the annual Congress of the Italian Chapter of ILAE; he received fees for speaking from Lusofarmaco.
KJ: is employed as a NIHR Network Support Fellow for the Cochrane Mental Health and Neuroscience Network and Cochrane Acute and Emergency Care Network.
CE: none known.
SM: received travel support and accommodation by Eisai to attend the 2018 American Epilepsy Society Annual Meeting.
Edited (no change to conclusions)
References
References to studies included in this review
Arzimanoglou 2019 {published data only}
- Arzimanoglou A, Ferreira J, Satlin A, Olhaye O, Kumar D, Dhadda S, et al. Evaluation of long-term safety, tolerability, and behavioral outcomes with adjunctive rufinamide in pediatric patients (≥1 to <4 years old) with Lennox-Gastaut syndrome: final results from randomized study 303. European Journal of Paediatric Neurology 2019;23(1):126-35. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Arzimanoglou A, Ferreira J, Satlin A, Olhaye O, Kumar D, Dhadda S, et al. Safety and cognitive development effects of rufinamide in paediatric patients with Lennox-Gastaut syndrome (LGS): study 303 final results. Developmental Medicine and Child Neurology 2017;59(Suppl 4):135-6, Abstract no: 242. [DOI: 10.1111/dmcn.13623] [DOI] [Google Scholar]
- Arzimanoglou A, Ferreira JA, Satlin A, Mendes S, Williams B, Critchley D, et al. Safety and pharmacokinetic profile of rufinamide in pediatric patients aged less than 4 years with Lennox-Gastaut syndrome: an interim analysis from a multicenter, randomized, active-controlled, open-label study. European Journal of Paediatric Neurology 2016;20(3):393‐402. [DOI: 10.1016/j.ejpn.2015.12.015] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Auvin S, Williams B, McMurray R, Kumar D, Perdomo C, Malhotra M. Novel seizure outcomes in patients with Lennox-Gastaut syndrome: post hoc analysis of seizure-free days in rufinamide Study 303. Epilepsia Open 2019;4(2):275‐80. [DOI: 10.1002/epi4.12314] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auvin S, Williams B, McMurray R, Kumar D, Perdomo C, Malhotra M. Post hoc analysis of rufinamide study 303: seizure-free days in patients with Lennox-Gastaut Syndrome (LGS). Neurology 2018;90(15 Suppl):Abstract no: P1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Devinsky 2018 {published data only}
- Devinsky O, Patel AD, Cross JH, Villanueva V, Wirrell EC, Privitera M, et al. Effect of cannabidiol on drop seizures in the Lennox-Gastaut syndrome. New England Journal of Medicine 2018;378(20):1888-97. [DOI: 10.1056/NEJMoa1714631] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Patel A, Devinsky O, Cross JH, Villanueva V, Wirrell E, VanLandingham K, et al. Cannabidiol (CBD) significantly reduces drop seizure frequency in Lennox-Gastaut syndrome (LGS): results of a dose-ranging, multi-center, randomized, double-blind, placebo-controlled trial (GWPCARE3). Neurology 2017;89(8):e100. [DOI: 10.1212/wnl.0000000000004380] [DOI] [Google Scholar]
- Wirrell E, Devinsky O, Patel A, Zuberi S, Cross J, Villanueva V, et al. Cannabidiol (CBD) significantly reduces drop and total seizure frequency in Lennox Gastaut Syndrome (LGS): results of a dose ranging, multicenter, randomized, double blind, placebo controlled trial (GWPCARE3). Annals of Neurology 2017;82(S21):S279-80, Abstract no: 22. [Google Scholar]
- Zuberi S, Devinsky O, Patel A, Cross JH, Villanueva V, Wirrell EC, et al. Cannabidiol (CBD) significantly reduces drop and total seizure frequency in Lennox-Gastaut syndrome (LGS): results of a dose-ranging, multi-centre, randomised, double-blind, placebo-controlled trial (GWPCARE3). Epilepsia 2017;58(Suppl 5):S13-4, Abstract no: 0026. [DOI: 10.1111/epi.13944] [DOI] [Google Scholar]
Eriksson 1998 {published data only}
- Eriksson AS, Nergardh A, Boreus L, Knutsson E. Double-blind cross-over study with lamotrigine in children with Lennox-Gastaut syndrome and other types of generalized intractable epilepsy. Epilepsia 1995;36(Suppl 3):S110-1. [Google Scholar]
- Eriksson AS, Nergardh A, Hoppu K. The efficacy of lamotrigine in children and adolescents with refractory generalised epilepsy: a randomised, double blind, crossover study. Epilepsia 1998;39(5):495-501. [PMID: ] [DOI] [PubMed] [Google Scholar]
Felbamate Study Group 1993 {published data only}
- Dodson WE. Felbamate in the treatment of Lennox-Gastaut syndrome: results of a 12-month open-label study following a randomized clinical trial. Epilepsia 1993;34(Suppl 7):S18-24. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Felbamate Study Group in Lennox Gastaut syndrome. Efficacy of felbamate in childhood epileptic encephalopathy (Lennox-Gastaut syndrome). New England Journal of Medicine 1993;328(1):29-33. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Garofalo EA, Olson LD, Sackellares JC, Childs J, Tornow MA. Felbamate for the treatment of Lennox-Gastaut syndrome. Epilepsia 1990;31(5):619. [Google Scholar]
- Ritter F, Dreifuss FE, Sackellares JC, Shields D, French J, Dodson WE, et al. A double-blind trial of felbamate in Lennox-Gastaut syndrome. Epilepsia 1991;32(Suppl 3):13. [Google Scholar]
Glauser 2008 {published and unpublished data}
- Glauser T, Kluger G, Krauss G, Arroyo S. Effects of rufinamide on the frequency of different seizure types in patients with Lennox-Gastaut syndrome: a long-term study. Epilepsia 2007;48(Suppl 7):156, abstract no: p415. [Google Scholar]
- Glauser T, Kluger G, Sachdeo R, Krauss G, Perdomo C, Arroyo S. Open-label extension study of the efficacy and safety of rufinamide adjunctive therapy in patients with Lennox-Gastaut syndrome. Epilepsia 2005;46(Suppl 6):408, abstract no: p1356. [Google Scholar]
- Glauser T, Kluger G, Sachdeo R, Krauss G, Perdomo C, Arroyo S. Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology 2008;70(21):1950-8. [DOI: 10.1212/01.wnl.0000303813.95800.0d] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Glauser T, Kluger G, Sachdeo R, Krauss G, Perdomo C, Arroyo S. Short-term and long-term efficacy and safety of rufinamide as adjunctive therapy in patients with inadequately controlled Lennox-Gastaut syndrome. Annals of Neurology 2005;58(Suppl 9):S92, abstract no: M15. [Google Scholar]
- Glauser T, Kluger G, Sachedo R, Krauss G, Perdomo C, Arroyo S. Efficacy and safety of rufinamide adjunctive therapy in patients with Lennox-Gastaut syndrome (LGS): a multicenter, randomized, double-blind, placebo-controlled, parallel trial. Neurology 2005;64(10):1826, abstract no: LBS.001. [Google Scholar]
- Glauser T, Santana B V, Wang W, Narurkar M. Early and sustained response to rufinamide as adjunctive therapy for seizures associated with Lennox Gastaut syndrome. Epilepsia 2009;50(Suppl 11):261, abstract no: 2.229. [Google Scholar]
- Kluger G, Glauser T, Krauss G, Seeruthun R, Perdomo C, Arroyo S. Adjunctive rufinamide in Lennox-Gastaut syndrome: a long-term, open-label extension study. Acta Neurologica Scandinavica 2010;122(3):202-8. [DOI: 10.1111/j.1600-0404.2010.01334.x] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Kluger G, Glauser T, Sachdeo R, Krauss G, Perdomo C, Arroyo S. Short-term and long-term efficacy and safety of rufinamide as adjunctive therapy in patients with inadequately controlled Lennox-Gastaut syndrome. Epilepsia 2006;47(Suppl 3):139, abstract no: p537. [Google Scholar]
- Krauss GL, Glauser T, Kluger G, Arroyo S. Long-term safety of rufinamide in patients with Lennox-Gastaut syndrome. Epilepsia 2007;48(Suppl 6):359, abstract no: 3.303. [Google Scholar]
- McMurray R, Striano P. Treatment of adults with Lennox-Gastaut syndrome: further analysis of efficacy and safety/tolerability of rufinamide. Neurology & Therapy 2016;5(1):35-43. [DOI: 10.1007/s40120-016-0041-9] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striano P, McMurray R. Rufinamide as adjunctive treatment for adults with Lennox-Gastaut syndrome: subgroup analysis from a phase III trial. Epilepsia 2015;56(Suppl 1):211, Abstract no: p0860. [DOI: 10.1111/epi.13241] [DOI] [Google Scholar]
Group for the Evaluation of Cinromide 1989 {published data only}
- Group for the Evaluation of Cinromide in the Lennox-Gastaut syndrome. Double-blind, placebo-controlled evaluation of cinromide in patients with the Lennox-Gastaut syndrome. Epilepsia 1989;30(4):422-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Motte 1997 {published data only}
- Billard C, Motte J, Arvidsson D, Trevathan E, Talladai M, Campos J, et al. Double-blind, placebo-controlled evaluation of the safety and efficacy of lamotrigine (Lamictal) for the treatment of patients with a clinical diagnosis of Lennox-Gastaut syndrome. Epilepsia 1996;37(Suppl 4):92. [Google Scholar]
- Gallagher J, Mullens L, Rigdon G, Donahue R. Clinician and caregiver assessments of global response to therapy strongly agree in a double-blind, placebo-controlled trial of lamotrigine in the Lennox-Gastaut syndrome. Neurology 1997;48(3):A109. [Google Scholar]
- Motte J, Trevathan E, Arvidsson JF, Barrera MN, Mullens EL, Manasco P. Lamotrigine for generalised seizures associated with the Lennox-Gastaut syndrome. Lamictal Lennox-Gastaut Study Group. New England Journal of Medicine 1997;337(25):1807-12. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Mullens L, Gallagher J, Manasco P, for the Lamictal Lennox-Gastaut Study Group. Improved neurological function accompanies effective control of the Lennox-Gastaut syndrome with Lamictal: results of a multinational, placebo-controlled trial. Epilepsia 1996;37(Suppl 5):163, abstract no: 6.47. [Google Scholar]
- Trevathan E, Motte J, Arvidsson J, Manasco P, Mullens L. Safety and tolerability of adjunctive Lamictal® for the treatment of the Lennox-Gastaut syndrome: results of a multinational, double-blind, placebo-controlled trial. Epilepsia 1996;37(Suppl 5):202, abstract no: I.3. [Google Scholar]
Ng 2011 {published data only}
- Chung SS, Gidal BE, Lemming OM, Karnik-Henry M, Hackler E, Tolbert D et al. Combination AED treatment with clobazam in patients with Lennox–Gastaut syndrome: post hoc analyses of the CONTAIN study. Neurology 2018;90(15 Suppl):Abstract no: P4.264. [Google Scholar]
- Conry J, Ng Y, Collins S, Bradt J, Stolle J, Tracy K, et al. Safety and efficacy of clobazam, a novel 1,5-benzodiazepine, in the treatment of Lennox-Gastaut syndrome. Epilepsia 2007;48(Suppl 6):360-1, Abstract no: 3.305. [Google Scholar]
- Conry J, Ng Y, Drummond R, Stolle J, Sagar S. Efficacy and safety of clobazam in the treatment of seizures associated with Lennox Gastaut syndrome: results of a phase III trial. In: 64th Annual Meeting of the American Epilepsy Society; 2010 December 3-7; San Antonio, TX. www.aesnet.org/go/publications/aes-abstracts/abstract-search/mode/display/st/clobazam/sy/2010/sb/All/id/, (accessed 28 April 2012):Abstract no: 1.283.
- Conry J, Ng YT, Peng G, Lee D, Isojarvi J. Efficacy and safety of clobazam for LGS patients who completed all 15 weeks of the phase III CONTAIN trial. Epilepsy Currents 2014;14(Suppl 1):391-2, Abstract no: 3.204. [Google Scholar]
- Conry JA, Ng YT, Drummond R, Stolle J, Owen JR, Weinberg MA. Efficacy and safety of clobazam for seizures associated with Lennox-Gastaut syndrome (LGS): results of a phase III study. Annals of Neurology 2011;70(S15):S113, Abstract no: 14. [DOI: 10.1002/ana.22572] [DOI] [Google Scholar]
- Isojarvi J, Lee D, Peng G, Sperling MR. Clobazam-treated patients with Lennox-Gastaut syndrome experienced fewer seizure-related injuries than placebo patients during trial OV-1012. Epilepsia 2016;57(6):e113-6. [DOI: 10.1111/epi.13388] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isojarvi J, Lee D, Renfroe JB. Somnolence and sedation were transient AES for most patients receiving clobazam during a phase III study in Lennox-Gastaut syndrome (LGS). Annals of Neurology 2013;74(Suppl 17):S75, Abstract no: M2224. [DOI: 10.1002/ana.24068] [DOI] [Google Scholar]
- Isojarvi J, Lee D. Response to clobazam in relationship to baseline seizure frequency. Neurology 2013;80(7 Suppl):Abstract no: P07.176. [Google Scholar]
- Lee D, Rosenfeld W, Isojarvi J. Use of rescue medications by Lennox-Gastaut (LGS) patients treated with clobazam. Annals of Neurology 2013;74(Suppl 17):S72, Abstract no. M2211. [DOI: ] [Google Scholar]
- Ng YT, Conry JA, Drummond R, Stolle J, Weinberg MA, OV-1012 Study Investigators. Randomized, phase III study results of clobazam in Lennox-Gastaut syndrome. Neurology 2011;77(15):1473-81. [DOI: 10.1212/WNL.0b013e318232de76] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicchi J, Isojarvi J, Lee D. Aggression in LGS patients treated with clobazam during the CONTAIN trial. Epilepsy Currents 2013;13(Suppl 1):266-7, Abstract no: 2.13. [Google Scholar]
- Paolicchi JM, Ross G, Lee D, Drummond R, Isojarvi J. Clobazam and aggression-related adverse events in pediatric patients with Lennox-Gastaut syndrome. Pediatric Neurology 2015;53(4):338-42. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Rosenfeld W, Isojarvi J, Nichol K, Lee D. Response to clobazam among benzodiazepine-experienced LGS patients during the CONTAIN trial. Epilepsy Currents 2014;14(Suppl 1):392, Abstract no: 3.205. [Google Scholar]
- Rosenfeld W, Lee D, Isojarvi J. Use of rescue medications by Lennox-Gastaut (LGS) patients treated with clobazam during the CONTAIN trial. Neurology 2013;80(7 Suppl):Abstract no: P01.046. [Google Scholar]
Ohtsuka 2014 {published data only}
- Ohtsuka Y, Yoshinaga H, Shirasaka Y, Takayama R, Takano H, Iyoda K. Rufinamide as an adjunctive therapy for Lennox-Gastaut syndrome: a randomized double-blind placebo-controlled trial in Japan. Epilepsy Research 2014;108(9):1627-36. [PMID: ] [DOI] [PubMed] [Google Scholar]
Sachdeo 1999 {published data only}
- Glauser TA, Levisohn PM, Ritter F, Sachdeo RC. Topiramate in Lennox-Gastaut syndrome: open-label treatment of patients completing a randomized controlled trial. Topiramate YL Study Group. Epilepsia 2000;41(Suppl 1):S86-90. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Glauser TA, Sachdeo RC, Ritter FJ, Reife R, Lim P, Topiramate YL Study Group. A double-blind trial of topiramate in Lennox-Gastaut syndrome (LGS). Epilepsia 1997;38(Suppl 3):131. [Google Scholar]
- Ritter FJ, Glauser TA, Sachdeo RC, Shu-Chen W. Long-term experience with topiramate in Lennox-Gastaut syndrome. Epilepsia 1998;39(Suppl 2):2-3. [Google Scholar]
- Ritter FJ, Sachdeo RC, Glauser TA, Topiramate YL Study Group. Topiramate as open-label adjunctive therapy in Lennox-Gastaut syndrome. Epilepsia 1997;38(Suppl 3):94. [Google Scholar]
- Ritter FJ, Topiramate YL Study Group. Open-label treatment of Lennox-Gastaut syndrome with topiramate. Epilepsia 1997;38(Suppl 3):37. [DOI] [PubMed] [Google Scholar]
- Sachdeo RC, Glauser TA, Ritter F, Reife R, Lim P, Pledger G. A double-blind, randomized trial of topiramate in Lennox-Gastaut syndrome. Topiramate YL Study Group. Neurology 1999;52(9):1882-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Sachdeo S, Sachdeo RC, Kugler S. An open label evaluation of topiramate in Lennox-Gastaut syndrome. Epilepsia 1996;37(Suppl 5):112, abstract no: 4.89. [Google Scholar]
Thiele 2018 {published data only}
- French J, Thiele E, Mazurkiewicz-Beldzinska M, Benbadis S, Marsh E, Joshi C, et al. Cannabidiol (CBD) significantly reduces drop seizure frequency in Lennox-Gastaut syndrome (LGS): results of a multi-center, randomized, double-blind, placebo controlled trial (GWPCARE4). Neurology 2017;88(16 Suppl):Abstract no: S21.001. [Google Scholar]
- Joshi C, Thiele E, Marsh E, French J. Treatment with cannabidiol (CBD) significantly reduces drop and total seizure frequency in Lennox-Gastaut Syndrome (LGS): results of a multicenter, randomized, double-blind, placebo controlled trial (GWPCARE4). Annals of Neurology 2017;82(S21):S293, Abstract no: 42. [Google Scholar]
- Mazurkiewicz-Beldzinska M, Thiele EA, Benbadis S, Marsh ED, Joshi C, French JA, et al. Treatment with cannabidiol (CBD) significantly reduces drop seizure frequency in Lennox-Gastaut syndrome (LGS): results of a multi-centre, randomised, double-blind, placebo-controlled trial (GWPCARE4). Epilepsia 2017;58(Suppl 5):S55, Abstract no: p0238. [DOI: 10.1111/epi.13944] [DOI] [Google Scholar]
- Thiele EA, Marsh ED, French JA, Mazurkiewicz-Beldzinska M, Benbadis SR, Joshi C, et al. Cannabidiol in patients with seizures associated with Lennox-Gastaut syndrome (GWPCARE4): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2018;391(10125):1085-96. [PMID: ] [DOI] [PubMed] [Google Scholar]
- Thiele EA, Mazurkiewicz-Beldzinska M, Benbadis S, Marsh ED, Joshi C, French JA et al. Treatment with cannabidiol (CBD) significantly reduces drop seizure frequency in Lennox Gastaut Syndrome (LGS): results of a multi-center, randomized, double-blind, placebo-controlled trial (GWPCARE4). Neurotherapeutics 2017;14(3):824-5. [DOI: 10.1007/s13311-017-0543-x] [DOI] [Google Scholar]
References to studies excluded from this review
Battaglia 1991 {published data only}
- Battaglia A, Ferrari AR, Guerrini R. Double-blind placebo-controlled trial of flunarizine as add-on therapy in refractory childhood epilepsy. Brain and Development 1991;13(4):217-22. [PMID: ] [DOI] [PubMed] [Google Scholar]
Conry 2009 {published data only}
- Conry JA, Ng YT, Paolicchi JM, Kernitsky L, Mitchell WG, Ritter FJ, et al. Clobazam in the treatment of Lennox-Gastaut syndrome. Epilepsia 2009;50(5):1158-66. [PMID: ] [DOI] [PubMed] [Google Scholar]
Inanaga 1989 {published data only}
- Inanaga K, Kumashiro H, Fukyama Y, Ohtahara S, Shirouzu M. Clinical study of oral administration of DN-1417, a TRH analog, in patients with intractable epilepsy. Epilepsia 1989;30(4):438-45. [PMID: ] [DOI] [PubMed] [Google Scholar]
Oletsky 1996 {published data only}
- Oletsky H, Kelley K, Stertz B, Reeves-Tyer P, Flamini R, Malow B, et al. The efficacy of felbamate as add-on therapy to valproic acid in the Lennox-Gastaut syndrome. Epilepsia 1996;37(Suppl 5):155, Abstract no: 6.13. [DOI] [PubMed] [Google Scholar]
Perry 2019 {published data only}
- Perry MS. Don't fear the reefer - evidence mounts for plant-based cannabidiol as treatment for epilepsy. Epilepsy Currents 2019;19(2):93-5. [DOI: 10.1177/1535759719835671] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Vajda 1985 {published data only}
- Vajda FJ, Bladin PF, Parsons BJ. Clinical experience with clobazam: a new 1,5 benzodiazepine in the treatment of refractory epilepsy. Clinical and Experimental Neurology 1985;21:177-82. [PMID: ] [PubMed] [Google Scholar]
Vassella 1978 {published data only}
- Vassella F, Rudeberg A, Da Silva V, Pavlincova E. Double-blind study on the anticonvulsive effect of phenobarbital and valproate in the Lennox syndrome [Doppletblind-Untersuchung uber die antikonvulsive Wirkung von Phenobarbital und Valproat beim Lennox-Syndrom]. Schweizerische Medizinische Wochenschrift 1978;108(19):713-6. [PMID: ] [PubMed] [Google Scholar]
Vigevano 1994 {published data only}
- Vigevano F, Cilio MR, Antonini L. Clinical experience with Felbamate in paediatric age. In: Bollettino - Lega Italiana Contro L'Epilessia. Vol. 86-87. 1994:63-6. [DOI: 10.1016/s0920-1211(01)00290-x] [DOI]
References to studies awaiting assessment
Ohtahara 2008 {published data only}
- Ohtahara S, Linuma K, Fujiwara T, Yamatogi Y. Single-blind and controlled comparative study of lamotrigine with zonisamide for refractory pediatric epilepsy. Journal of the Japan Epilepsy Society 2008;25(4):425-40. [DOI: 10.3805/jjes.25.425] [DOI] [Google Scholar]
References to ongoing studies
CTRI/2010/091/001449 {published data only}
- CTRI/2010/091/001449. A comparative, randomized, open label, multicentric, prospective clinical study to evaluate the efficacy, safety and tolerability of rufinamide tablet vs. lamotrigine (adjunctive therapy) in the treatment of seizures associated with Lennox-Gastaut syndrome. http://www.ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=2237 2011.
NCT00004776 {published data only}
- NCT00004776. Phase III randomized, double-blind, placebo-controlled study of oral topiramate for Lennox-Gastaut syndrome. http://ClinicalTrials.gov/show/NCT00004776 1993.
NCT01370486 {published data only}
- NCT01370486. Melatonin versus placebo in the Lennox-Gastaut syndrome: neurophysiological and neuropsychological effects. http://ClinicalTrials.gov/show/NCT01370486 2011.
NCT02318537 {published data only}
- NCT02318537. Cannabidiol oral solution as an adjunctive therapy for treatment of subjects with inadequately controlled Lennox-Gastaut syndrome. https://ClinicalTrials.gov/show/NCT02318537 2016.
NCT03355209 {unpublished data only}
- NCT03355209. A study to investigate the efficacy and safety of ZX008 (fenfluramine hydrochloride) as an adjunctive therapy in children and adults with Lennox-Gastaut syndrome. https://clinicaltrials.gov/show/NCT03355209 (first received November 28, 2017).
NCT03650452 {published data only}
- NCT03650452. A phase 2, multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy, safety, and tolerability of TAK-935 (OV935) as an adjunctive therapy in pediatric patients with developmental and/or epileptic encephalopathies. https://ClinicalTrials.gov/show/NCT03650452 2018.
NCT03808935 {unpublished data only}
- NCT03808935. Cannabis extract in refractory epilepsy study. https://clinicaltrials.gov/show/NCT03808935 2019.
Wechsler 2017 {unpublished data only}
- Wechsler RT, Popli G, Dimos J, Ferreira J, Bibbiani F, Laurenza A, et al. Design and methods of study 338: a multicenter, double-blind, randomized, placebo-controlled trial of perampanel as adjunctive treatment in subjects >=2 years of age with inadequately controlled seizures associated with Lennox-Gastaut syndrome. Epilepsia 2017;58(Suppl 5):S157-8, Abstract no: p0891. [DOI: 10.1111/epi.13944] [DOI] [Google Scholar]
- Wechsler RT, Popli G, Dimos J, Ferreira J, Bibbiani F, Laurenza A, et al. Design and methods of study 338: a multicentre, double-blind, randomised, placebo-controlled trial of perampanel as adjunctive treatment in patients >=2 years of age with inadequately controlled seizures associated with Lennox-Gastaut syndrome. Developmental Medicine and Child Neurology 2017;59(Suppl 4):121-2, Abstract no: 207. [DOI: 10.1111/dmcn.13623] [DOI] [Google Scholar]
Additional references
Achenbach 1991
- Achenbach TM. Manual for the Child Behavior Checklist/4–18 and 1991 Profile. Department of Psychiatry. University of Vermont, Burlington, VT, 1991. [Google Scholar]
Alves 2020
- Alves P, Amaral C, Teixeira N, Correia-da-Silva G. Cannabis sativa: much more beyond Δ 9-tetrahydrocannabinol. Pharmacological Research 2020;157:104822. [DOI: 10.1016/j.phrs.2020.104822] [PMID: ] [DOI] [PubMed] [Google Scholar]
Arzimanoglou 2009
- Arzimanoglou A, French J, Blume WT, Cross J, Ernst JP, Feucht M, et al. Lennox Gastaut syndrome: a consensus approach on diagnosis, assessment, management and trial methodology. Lancet Neurology 2009;8(1):82-93. [PMID: ] [DOI] [PubMed] [Google Scholar]
Asadi‐Pooya 2018
- Asadi-Pooya AA. Lennox-Gastaut syndrome: a comprehensive review. Neurological Sciences 2018;39(3):403-14. [PMID: ] [DOI] [PubMed] [Google Scholar]
Autry 2010
- Autry AR, Trevathan E, Van Naarden Braun K, Yeargin-Allsopp M. Increased risk of death among children with Lennox-Gastaut syndrome and infantile spasms. Journal of Child Neurology 2010;25(4):441-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Benedict 2010
- Benedict A, Verdian L, Maclaine G. The cost effectiveness of rufinamide in the treatment of Lennox-Gastaut syndrome in the UK. Pharmacoeconomics 2010;28(3):185-99 [Erratum in: Pharmacoeconomics 2011;29(12):1014]. [DOI: 10.2165/11313640-000000000-00000] [PMID: ] [DOI] [PubMed] [Google Scholar]
Berg 1999
- Berg AT, Shinnar S, Levy SR, Testa FM. Newly diagnosed epilepsy in children: presentation at diagnosis. Epilepsia 1999;40(4):445-52. [PMID: ] [DOI] [PubMed] [Google Scholar]
Berg 2018
- Berg AT, Levy SR, Testa FM. Evolution and course of early life developmental encephalopathic epilepsies: focus on Lennox-Gastaut syndrome. Epilepsia 2018;59(11):2096-105. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Borrelli 2019
- Borrelli S, El Tahry R. Therapeutic approach to Lennox-Gastaut syndrome: a systematic review. Acta Neurololgica Belgica 2019;119(3):315-24. [PMID: ] [DOI] [PubMed] [Google Scholar]
Brigo 2021
- Brigo F, Lattanzi S. Anticonvulsant agents: benzodiazepines (clobazam, clonazepam, diazepam, lorazepam, midazolam). In: Riederer P, Laux G, Nagatsu T, Le W, Riederer C, editors(s). NeuroPsychopharmacotherapy. Springer International Publishing, 2021. [ISBN: 978-3-030-62060-8] [Google Scholar]
Brunklaus 2020
- Brunklaus A, Du J, Steckler F, Ghanty II, Johannesen KM, Fenger CD, et al. Biological concepts in human sodium channel epilepsies and their relevance in clinical practice. Epilepsia 2020;61(3):387-99. [DOI: 10.1111/epi.16438] [DOI] [PubMed] [Google Scholar]
Callenbach 1998
- Callenbach PM, Geerts AT, Arts WF, Donselaar CA, Peters AC, Stroink H, et al. Familial occurrence of epilepsy in children with newly diagnosed multiple seizures: Dutch Study of Epilepsy in Childhood. Epilepsia 1998;39(3):331-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Cramer 2013
- Cramer JA, Sapin C, François C. Indirect comparison of clobazam and other therapies for Lennox-Gastaut syndrome. Acta Neurologica Scandinavica 2013;128(2):91-9. [DOI: 10.1111/ane.12086] [PMID: ] [DOI] [PubMed] [Google Scholar]
Crespel 2019
- Crespel A, Gelisse P, Macorig G, Nikanorova M, Ferlazzo E, Genton P. Lennox-Gastaut syndrome. In: Bureau M, Genton P, Delgado-Escueta A, Dravet C, Guerrini R, Tassinari CA, et al, editors(s). Epileptic Syndromes in Infancy, Childhood and Adolescence. 6th edition. Paris: John Libbey Eurotext, 2019. [ISBN: 978-2742015726] [Google Scholar]
Cross 2017
- Cross JH, Auvin S, Falip M, Striano P, Arzimanoglou A. Expert opinion on the management of Lennox-Gastaut syndrome: treatment algorithms and practical considerations. Frontiers in Neurology 2017;8:505. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2020
- Deeks JJ, Higgins JPT, Altman DG (editors). Chapter 10: Analysing data and undertaking meta-analyses. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.1 (updated September 2020). Cochrane, 2020. Available from www.training.cochrane.org/handbook.
Eschbach 2018
- Eschbach K, Moss A, Joshi C, Angione K, Smith G, Dempsey A, et al. Diagnosis switching and outcomes in a cohort of patients with potential epilepsy with myoclonic-atonic seizures. Epilepsy Research 2018;147:95-101. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
European Union 2006
- The European Parliament and the Council of the European Union. Regulation (EC) No 1901/2006 of the European Council and of the Council of 12th December 2006 on medicinal products for paediatric use and amending Regulation (EEC) No 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation (EC) No 726/2004. www.emea.europa.eu/htms/human/paediatrics/regulation.htm 2006.
Ferlazzo 2010
- Ferlazzo E, Nikanorova M, Italiano D, Bureau M, Dravet C, Calarese T, et al. Lennox-Gastaut syndrome in adulthood: clinical and EEG features. Epilepsy Research 2010;89(2-3):271-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
French 2004
- French JA, Kanner AM, Bautista J, Abou-Khalil B, Browne T, Harden CL, et al. Efficacy and tolerability of the new antiepileptic drugs I: treatment of new onset epilepsy: report of the Therapeutics and Technology Assessment Subcommittee and Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2004;62(8):1252-60. [PMID: ] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT , Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from training.cochrane.org/handbook/archive/v5.1/.
Higgins 2020
- Higgins JP, Eldridge S, Li T (editors). Chapter 23: Including variants on randomized trials. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.1 (updated September 2020). Cochrane, 2020. Available from training.cochrane.org/handbook.
ILAE 1989
- International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia 1989;30(4):389-99. [DOI] [PubMed] [Google Scholar]
ILAE 2020
- International League Against Epilepsy. Lennox Gastaut syndrome. https://www.epilepsydiagnosis.org/syndrome/lgs-overview.html (accessed 7 December 2020).
Jensen 1994
- Jensen PK. Felbamate in the treatment of Lennox-Gastaut syndrome. Epilepsia 1994;35(Suppl 5):S54-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kaminska 1999
- Kaminska A, Ickowicz A, Plouin P, Bru MF, Dellatolas G, Dulac O. Delineation of cryptogenic Lennox-Gastaut syndrome and myoclonic astatic epilepsy using multiple correspondence analysis. Epilepsy Research 1999;36(1):15-29. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kanner 2018
- Kanner AM, Ashman E, Gloss D, Harden C, Bourgeois B, Bautista JF, et al. Practice guideline update summary: efficacy and tolerability of the new antiepileptic drugs II: treatment-resistant epilepsy: report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2018;91(2):82-90. [PMID: ] [DOI] [PubMed] [Google Scholar]
Lattanzi 2018
- Lattanzi S, Brigo F, Cagnetti C, Trinka E, Silvestrini M. Efficacy and safety of adjunctive cannabidiol in patients with Lennox-Gastaut syndrome: a systematic review and meta-analysis. CNS Drugs 2018;32(10):905-16. [DOI: 10.1007/s40263-018-0558-9] [PMID: ] [DOI] [PubMed] [Google Scholar]
Lattanzi 2020
- Lattanzi S, Trinka E, Striano P, Zaccara G, Del Giovane C, Nardone R et al. Cannabidiol efficacy and clobazam status: a systematic review and meta-analysis. Epilepsia 2020;61(6):1090–8. [DOI] [PubMed] [Google Scholar]
Lefebvre 2020
- Lefebvre C, Glanville J, Briscoe S, Littlewood A, Marshall C, Metzendorf M-I, et al. Technical Supplement to Chapter 4: Searching for and selecting studies. In: Higgins JPT, Thomas J, Chandler J, Cumpston MS, Li T, Page MJ, Welch VA (eds). Cochrane Handbook for Systematic Reviews of Interventions Version 6.1 (updated September 2020). Cochrane, 2020. Available from www.training.cochrane.org/handbook.
NICE 2012
- National Institute for Health and Care Excellence (NICE). Epilepsies: diagnosis and management. Clinical guideline [CG137]. Available from: https://www.nice.org.uk/guidance/cg137 (accessed 19/02/2021) 11 January 2012. [PubMed]
NICE 2019
- National Institute for Health and Care Excellence (NICE). Cannabidiol with clobazam for treating seizures associated with Lennox–Gastaut syndrome. Technology appraisal guidance [TA615]. Available from: https://www.nice.org.uk/guidance/ta615 (accessed 19/02/2021) 18 December 2019.
Paolicchi 2015
- Paolicchi JM, Ross G, Lee D, Drummond R, Isojarvi J. Clobazam and aggression-related adverse events in pediatric patients with Lennox-Gastaut syndrome. Pediatric Neurology 2015;53(4):338-42. [PMID: ] [DOI] [PubMed] [Google Scholar]
Pina‐Garza 2017
- Piña-Garza JE, Montouris GD, Vekeman F, Cheng WY, Tuttle E, Giguere-Duval P, et al. Assessment of treatment patterns and healthcare costs associated with probable Lennox-Gastaut syndrome. Epilepsy & Behavior 2017;73:46-50. [PMID: ] [DOI] [PubMed] [Google Scholar]
Review Manager 2020 [Computer program]
- Review Manager 5 (RevMan 5). Version 5.4.1. Cochrane, 2020.
Rogawski 2016
- Rogawski MA, Löscher W, Rho JM. Mechanisms of action of antiseizure drugs and the ketogenic diet. Cold Spring Harbor Perspectives in Medicine 2016;6(5):a022780. [DOI: 10.1101/cshperspect.a022780] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2020
- Schünemann HJ, Vist GE, Higgins JPT, Santesso N, Deeks JJ, Glasziou P, et al. Chapter 15: Interpreting results and drawing conclusions. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.1 (updated September 2020). Cochrane, 2020. Available from www.training.cochrane.org/handbook.
Shorvon 2010
- Shorvon SD. Handbook of Epilepsy Treatment. Wiley-Blackwell, 2010. [Google Scholar]
Smith 2018
- Smith KM, Britton JW, Cascino GD. Late-onset Lennox-Gastaut syndrome: diagnostic evaluation and outcome. Neurology. Clinical Practice 2018;8(5):397-402. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Trevathan 1997
- Trevathan E, Murphy CC, Yeargin-Allsopp M. Prevalence and descriptive epidemiology of Lennox-Gastaut syndrome among Atlanta children. Epilepsia 1997;38(12):1283-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Trinka 2015
- Trinka E, Brigo F. Benzodiazepines used in the treatment of epilepsy. In: Treatment of epilepsies. 4th edition. Oxford: Blackwell Publishing, 2015. [Google Scholar]
Vignoli 2017
- Vignoli A, Oggioni G, De Maria G, Peron A, Savini MN, Zambrelli E, et al. Lennox-Gastaut syndrome in adulthood: long-term clinical follow-up of 38 patients and analysis of their recorded seizures. Epilepsy & Behavior 2017;77:73-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Wirrell 2011
- Wirrell EC, Grossardt BR, Wong-Kisiel LC, Nickels KC. Incidence and classification of new-onset epilepsy and epilepsy syndromes in children in Olmsted County, Minnesota from 1980 to 2004: a population-based study. Epilepsy Research 2011;95(1-2):110-8. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wolff 2017
- Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, Gardella E, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017;140(5):1316-36. [DOI: 10.1093/brain/awx054] [PMID: ] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Hancock 2003
- Hancock EC, Cross HJ. Treatment of Lennox‐Gastaut syndrome. Cochrane Database of Systematic Reviews 2003, Issue 3. Art. No: CD003277. [DOI: 10.1002/14651858.CD003277] [DOI] [PubMed] [Google Scholar]
Hancock 2009
- Hancock EC, Cross HJ. Treatment of Lennox‐Gastaut syndrome. Cochrane Database of Systematic Reviews 2009, Issue 3. Art. No: CD003277. [DOI: 10.1002/14651858.CD003277.pub2] [DOI] [PubMed] [Google Scholar]
Hancock 2013
- Hancock EC, Cross JH. Treatment of Lennox‐Gastaut syndrome. Cochrane Database of Systematic Reviews 2013, Issue 2. Art. No: CD003277. [DOI: 10.1002/14651858.CD003277.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]