

TO THE EDITOR:
Clonal cytopenia of undetermined significance (CCUS) is defined by the presence of somatic driver mutations/copy number alterations (CNA) in hematopoietic cells, in patients with unexplained cytopenia in 1 or more peripheral blood (PB)–cell lineages, in the absence of overt morphologic dysplasia, excess blasts, and myelodysplastic syndromes (MDS)–defining chromosomal abnormalities.1,2 CCUS is associated with an increased risk for evolution to MDS and/or acute myeloid leukemia (AML), with somatic mutational variant allele frequency (VAF) ≥10%, or carrying ≥2 mutations, being associated with predictive values of 0.86 and 0.88 for diagnosis of a myeloid neoplasm.1 In addition, dominant clone VAF >20% has been associated with a >95% risk of progression to a clinically apparent myeloid neoplasm in 10 years, raising the question as to whether these patients would better be classified as having an early myeloid neoplasm rather than CCUS.3 These data, among others, have led to operational classification criteria for the diagnosis of CCUS that includes the presence of myeloid-relevant somatic mutations with a VAF ≥20%, without overt bone marrow (BM) dysplasia (<10% dysplastic cells).2
MDS are clonal hematopoietic stem cell disorders with clinically relevant morphologic dysplasia in the PB and BM, with <20% blasts and an inherent risk for AML transformation.4 Lower-risk MDS (LR) can be further categorized into morphologic and molecularly annotated entities that have <5% blasts, with lower rates of AML progression but with significant quality of life issues and red blood cell and platelet transfusion dependence (TD).2,4 With the advent of clinical next-generation sequencing (NGS) testing, there are increasing numbers of patients with unexplained cytopenias not meeting criteria for MDS that have somatic mutations/CNA suggestive of CCUS.5 Although these patients are currently not considered to have a hematologic neoplasm, they can be as symptomatic, if not more, in comparison with patients with MDS. However, because of the lack of a formal cancer diagnosis, CCUS patients are often denied MDS-directed therapies or clinical trial enrollment. We carried out this study to compare and contrast the clinical characteristics and survival outcomes between prospectively assessed patients with CCUS at our institution and LR-MDS patients.
CCUS patients were identified from the Mayo Clinic clonal hematopoiesis (CH) clinic, whereas LR-MDS patients were retrospectively identified from our institutional database. For this study, we defined LR-MDS by the 2016 World Health Organization (WHO) criteria for MDS, as long as they were stratified as having very-low-, low-, and intermediate-risk disease by the revised International Prognostic Scoring System (R-IPSS).6 The gold standard criteria applied for dysplasia included the presence of ≥10% dysplastic erythroid and/or granulocytic cells and ≥30% dysplastic megakaryocytes, with micro-megakaryocytes and multinucleated megakaryocytes with separated nuclei being the most reliable dysplastic finding in the megakaryocyte lineage.4 Depending on the extent of dysplasia, further subcategorization into single lineage dysplasia (SLD) and multilineage dysplasia (MLD) was carried out. Although consensus data have suggested using VAF ≥ 20% for the diagnosis of CCUS, in our study, we included patients with a VAF ≥ 2%, based on CH criteria and the fact that formal validation of CCUS consensus criteria are pending.7,8 In our study we further classified CCUS patients into CCUS-HighVAF based on a dominant mutational VAF of ≥20%. In addition, patients with unexplained cytopenias with somatic copy number alerations (SCNA), excluding MDS-defining cytogenetic abnormalities, were also classified as having CCUS.9 Baseline demographics, blood counts, BM morphology, cytogenetics, and NGS results (supplemental Table 1 for NGS panel details) were abstracted. Although the IPSS and R-IPSS prognostication systems have been developed and validated strictly for newly diagnosed MDS patients, given the clinical similarity and lack of a CCUS-specific risk stratification system, in this study, we applied the R-IPSS risk stratification to CCUS patients. Transfusion dependency was defined as requiring at least 1 unit of red cell or platelets every 4 weeks for 2 or more consecutive months. The Mann-Whitney U and Fischer’s exact test were used to compare quantitative and qualitative data in subgroups. Kaplan-Meier overall survival (OS) estimates were used for survival analysis and compared using the log-rank test.
A total of 187 patients were included in the study: 75 (40%) with CCUS and 112 (60%) with LR-MDS, with a median age of 66 years (63% male; Table 1). In the CCUS group, 59 (78%) patients had ≥1 somatic mutation, 8 (11%) had clonal cytogenetic abnormalities only, and 8 (11%) patients had both somatic mutations and clonal cytogenetic abnormalities. Sixty-seven (92%) patients had ≥1 somatic mutation, with 45 (61%) having >1 mutation. Common mutations in CCUS included TET2 (30%), SRSF2 (20%), DNMT3A (13%), and ASXL1 (11%) (Figure 1A-B), with 17 (23%) patients being red cell TD and 10 (13%) being platelet TD at diagnosis. Of note, the higher-than-expected TD rates in the CCUS cohort could be secondary to the fact that our institution is a tertiary referral center and that the CH clinic is a specialty referral clinic, introducing an element of referral bias. Based on the R-IPSS stratification system for MDS, 32 (42%), 33 (43%), and 10 (13%) CCUS patients were stratified as having very-low-, low-, and intermediate-risk disease, respectively. In the LR-MDS group, MDS subtypes as defined by the 2016 WHO criteria included 56 (50%) patients with MDS-ring sideroblasts (RS)-SLD, 4 (4%) MDS-SLD, 23 (21%) MDS-MLD, 11 (10%) MDS-EB-1 (excess blasts), 8 (7%) MDS with del(5q), and 10 (9%) MDS-unclassifiable. Ninety-six (86%) LR-MDS patients had ≥1 somatic mutation, whereas 57 (51%) had >1 mutation, with common mutations being SF3B1 67% (95% MDS-RS), TET2 (29%), and DNMT3A (21%). For LR-MDS patients, 49 (44%) were red cell TD, whereas 15 (13%) were platelet TD. On R-IPSS stratification, 49 (44%), 47 (42%), and 16 (14%) MDS patients were stratified as having very-low-, low-, and intermediate-risk disease, respectively.
Table 1.
Clinical characteristics of CCUS and LR-MDS
| Variables | All patients with CCUS and LR-MDS (n = 187) | CCUS (n = 75) | LR-MDS (n = 112) | P |
|---|---|---|---|---|
| Demographics, median (range) | ||||
| Age, y | 66 (20-91) | 65 (20-90) | 67 (37-91) | .796 |
| Sex; males, n (%) | 118 (63.1) | 51 (68) | 67 (59.8) | .282 |
| Blood counts at diagnosis, median (range) | ||||
| HGB, g/dL | 10.6 (5.8-15.9) | 10.8 (6.7-15.9) | 10.4 (5.8-14.4) | .307 |
| MCV, fL | 99.7 (80.4-120.2) | 99.6 (80.4-117.2) | 99.6 (81.5-120.2) | .850 |
| WBC, ×109/L | 4.7 (0.9-13.1) | 4.1 (1.2-13.1) | 5.1 (0.9-13.1) | .002 |
| ANC, ×109/L | 2.5 (0.04-9.4) | 2.1 (0.04-9.4) | 2.7 (0.2-9.4) | .008 |
| AMC, ×109/L | 0.4 (0-3.6) | 0.5 (0.01-3.6) | 0.3 (0-1.1) | .007 |
| PLT, ×109/L | 196 (12-599) | 140 (10-595) | 232 (12-599) | <.001 |
| Myelocytes, % | 0.9 (0-24) | 1.1 (0-24) | 0.7 (0-19) | .324 |
| Metamyelocytes, % | 0.5 (0-11) | 0.5 (0-11) | 0.4 (0-10) | .647 |
| Peripheral blasts, % | 0.04 (0-2) | 0.03 (0-1) | 0.04 (0-2) | .796 |
| Transfusion dependency, n (%)* | ||||
| Red blood cell | 66 (35.3) | 17 (22.6) | 49 (43.7) | .003 |
| Low burden | 32 (48.5) | 9 (53) | 23 (46.9) | |
| High burden | 35 (53) | 9 (53) | 26 (53.1) | .025 |
| Platelets | 24 (12.8) | 9 (12) | 15 (13.4) | .827 |
| Bone marrow characteristics, n (%) | ||||
| Hypercellular | 132 (71) | 43 (57.3) | 89 (83.2) | .001 |
| Hypocellular | 14 (7.8) | 5 (6.9) | 9 (8.4) | .784 |
| Normocellular | 33 (18.3) | 24 (33.3) | 9 (8.4) | <.001 |
| Abnormal MDS flow cytometry | 22 (11.7) | 12 (16) | 10 (8.9) | <.001 |
| BM ring sideroblasts, median (range), % | 16 (0-75) | 2 (0-5) | 24 (0-75) | <.001 |
| BM blasts, median (range), % | 1.1 (0-15) | 0.7 (0-5) | 1.4 (0-15) | .002 |
| Abnormal cytogenetics | 44 (23.5) | 16 (21.3) | 28 (25) | .602 |
| Mutation present on NGS | 163 (87.2) | 67 (89.3) | 96 (85.7) | .251 |
| 1 mutation | 61 (37.4) | 22 (32.8) | 39 (40.6) | .329 |
| >1 mutation | 102 (62.6) | 45 (67.2) | 57 (59.4) | |
| IPSS-R cytogenetic risk category, n (%) | ||||
| Very good | 8 (4.3) | 2 (2.7) | 6 (5.4) | .483 |
| Good | 161 (87) | 63 (86.3) | 98 (87.5) | .826 |
| Intermediate | 12 (6.5) | 5 (6.8) | 7 (6.3) | .98 |
| Poor | 4 (2.2) | 3 (4.1) | 1 (0.9) | .302 |
| Very poor | 0 (0) | 0 (0) | 0 (0) | — |
| NGS analysis, n (%) | ||||
| Epigenetic regulators | ||||
| DNMT3A | 29 (17.4) | 9 (12.7) | 20 (20.8) | .216 |
| IDH1 | 6 (3.6) | 6 (8.5) | 0 (0) | .005 |
| IDH2 | 5 (3) | 2 (2.8) | 3 (3.1) | .65 |
| KDM6A | 1 (0.6) | 1 (1.4) | 0 (0) | .420 |
| TET2 | 49 (29.2) | 21 (29.6) | 28 (28.6) | .87 |
| Chromatin regulators | ||||
| ASXL1 | 23 (13.7) | 9 (12.7) | 14 (14.6) | .822 |
| EZH2 | 1 (0.6) | 0 (0) | 1 (1) | .88 |
| Spliceosome factors | ||||
| SRSF2 | 25 (14.7) | 14 (20) | 11 (11.2) | .132 |
| SF3B1 | 70 (41.2) | 4 (5.6) | 66 (67.4) | <.001 |
| U2AF1 | 14 (8.2) | 8 (11.1) | 6 (6.1) | .266 |
| ZRSR2 | 11 (6.5) | 10 (14.1) | 1 (1) | .001 |
| Cell signaling | ||||
| CBL | 5 (3) | 1 (1.4) | 4 (4.2) | .396 |
| CSF3R | 8 (4.8) | 2 (2.8) | 6 (6.3) | .469 |
| JAK2 | 5 (3) | 3 (4.2) | 2 (2) | .652 |
| KIT | 1 (0.6) | 1 (1.4) | 0 (0) | .420 |
| KRAS | 3 (1.8) | 2 (2.8) | 1 (1) | .575 |
| MPL | 2 (1.2) | 1 (1.4) | 1 (1) | .46 |
| NRAS | 2 (1.2) | 2 (2.8) | 0 (0) | .175 |
| SH2B3 | 1 (0.6) | 1 (1.4) | 0 (0) | .420 |
| WT1 | 1 (0.6) | 1 (1.4) | 0 (0) | .420 |
| Tumor suppressors | ||||
| ATM | 5 (3) | 5 (7) | 0 (0) | .013 |
| PTEN | 1 (0.6) | 1 (1.4) | 0 (0) | .418 |
| TP53 | 6 (3.5) | 4 (5.6) | 2 (2) | .240 |
| Others | ||||
| BCOR | 5 (3) | 4 (5.6) | 1 (1) | .164 |
| CEBPA | 5 (3) | 0 (0) | 5 (5.2) | .073 |
| CHEK2 | 1 (0.6) | 1 (1.4) | 0 (0) | .420 |
| ETV6 | 1 (0.6) | 0 (0) | 1 (1) | .912 |
| ETNK1 | 1 (0.6) | 0 (0) | 1 (1) | .987 |
| PHF6 | 2 (1.2) | 2 (2.8) | 0 (0) | .175 |
| RAD21 | 1 (0.6) | 1 (1.4) | 0 (0) | .420 |
| RUNX1 | 2 (1.2) | 1 (1.4) | 1 (1) | .654 |
| SETBP1 | 5 (3) | 1 (1.4) | 4 (4) | .400 |
| STAG2 | 3 (1.8) | 3 (4.2) | 0 (0) | .072 |
| IPSS-R risk category; n (%)† | ||||
| Very low | 81 (43.3) | 32 (42.7) | 49 (43.8) | .879 |
| Low | 80 (42.7) | 33 (44) | 47 (42) | .880 |
| Intermediate | 26 (13.9) | 10 (13.3) | 16 (14.3) | .986 |
| High | 0 (0) | 0 (0) | 0 (0) | — |
| Very high | 0 (0) | 0 (0) | 0 (0) | — |
| Treatment type, n (%) | ||||
| None | 87 (47.5) | 58 (77.3) | 29 (26.9) | <.001 |
| Erythropoietin stimulators | 47 (25.7) | 8 (10.7) | 39 (34.8) | <.001 |
| Immune suppression | 13 (7.1) | 2 (2.6) | 11 (10.2) | .077 |
| Hypomethylating agent | 27 (14.8) | 7 (9.3) | 20 (18.5) | .094 |
| Bone marrow transplant | 9 (4.9) | 0 | 9 (8.3) | .011 |
| Clinical course | ||||
| Disease progression, n (%) | 22 (11.8) | 13 (17.3) | 9 (8) | .067 |
| To MDS/chronic myeloid neoplasm | 11 (50) | 10 (76.9) | 1 (11.1) | .008 |
| To AML | 11 (50) | 3 (23.1) | 8 (88.9) | |
| Alive, n (%) | 120 (64.2) | 60 (80) | 60 (53.6) | <.001 |
| Follow-up, median (range, mo) | 32 (0.1-238) | 17 (2-125) | 54 (0.1-238)‡ | <.001 |
HGB, hemoglobin; IPSS-R, International prognostic scoring system revised; MCV, mean corpuscular volume; PLT, platelets; —, lower-risk myelodysplastic syndromes.
Transfusion dependency defined by requirement of at least 1 unit of red cell or platelet transfusion once every 4 weeks for more than 2 consecutive 4-week periods. Low burden defined as 3 to 7 units in a 16-week period; high burden defined as 8+ units in a 16-week period.12
Although IPSS-R was designed for MDS, we also applied it to CCUS patients.
Median OS reached for MDS at 104 months was not reached for CCUS..
Figure 1.
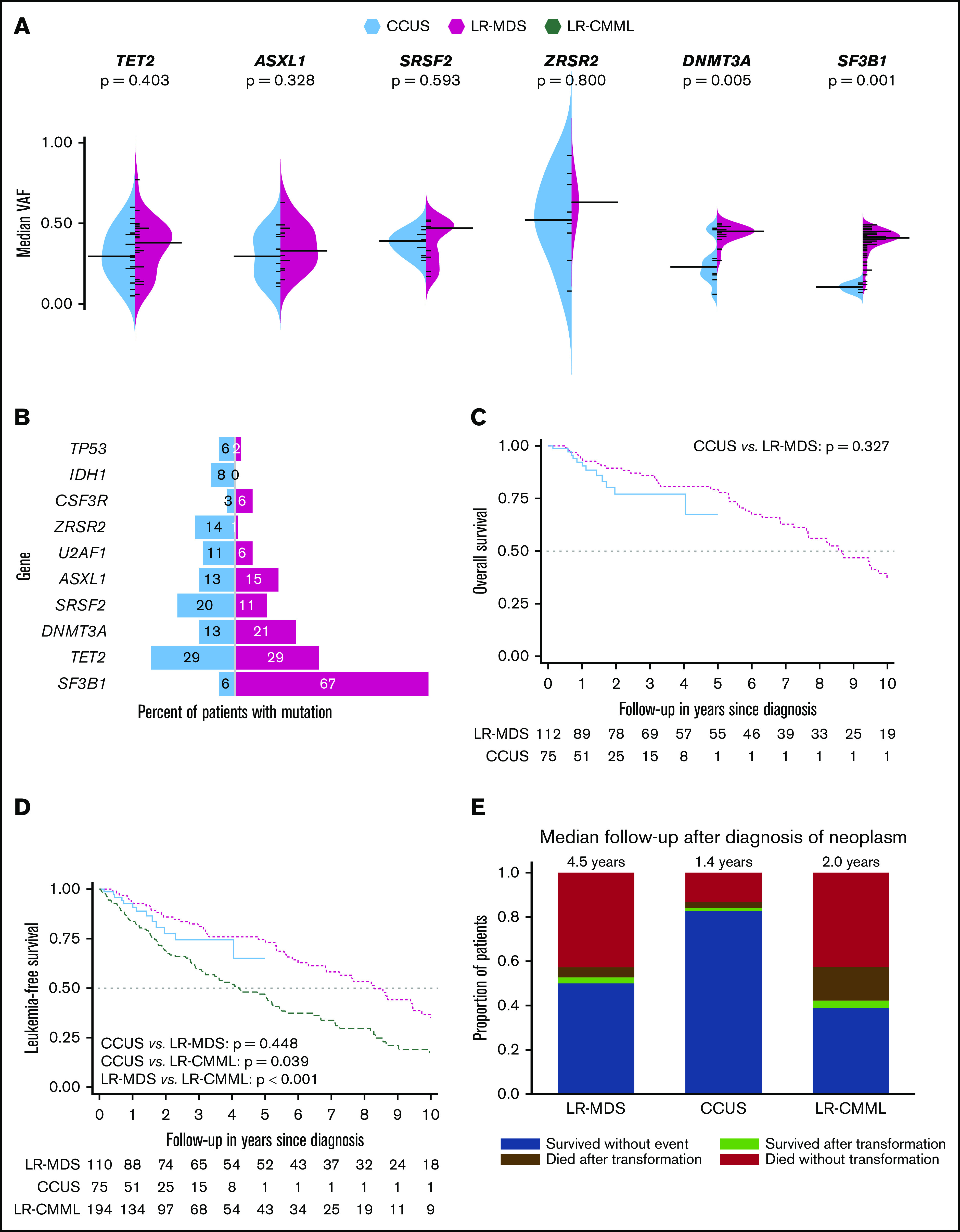
Molecular characteristics and survival outcomes in patients with clonal cytopenias of undetermined significance, lower-risk myelodysplastic syndromes, and lower-risk chronic myelomonocytic leukemia. (A) Split bean plot demonstrating the 6 most common mutations in both CCUS and LR-MDS and their respective VAFs. (B) Bar graph of CCUS and LR-MDS demonstrating the 10 most frequent mutations in all 188 patients and the percentage of patients with that mutation. (C) Kaplan-Meier OS estimate in CCUS and LR-MDS patients, censored at 10 years of follow-up. (D) Kaplan-Meier leukemia-free survival estimate in CCUS, LR-MDS, and LR-CMML patients, censored at 10 years of follow-up. (E) Distribution of survival and progression outcomes in LR-MDS, LR-CMML, and CCUS patients.
In comparison with patients with CCUS, LR-MDS patients were more likely to have higher white blood cell counts (WBC; P = .001), higher absolute neutrophil counts (ANC; P = .005), higher platelet counts (P < .001), be red cell TD (P = .031), have BM RS (P < .001), have higher BM blast% (P = .002), and have SF3B1 mutations (P < .001), whereas CCUS patients were more likely to have higher absolute monocyte counts (AMC; P = .009; Table 1). Mutational VAF for DNMT3A and SF3B1 was higher in LR-MDS in comparison with CCUS (Figure 1A). Importantly, there were no differences with regard to R-IPSS cytogenetic risk groups, R-IPSS prognostic categories, platelet TD, and the distribution of TET2, DNMT3A, SRSF2, ASXL1, and TP53 mutations.
Fifty (83%) of 60 assessable CCUS patients with somatic mutations had at least 1 somatic mutation with VAF ≥20% (CCUS-HighVAF), whereas 58 (97%) had VAF ≥10% (supplemental Table 2). In comparison with CCUS patients with VAF <20%, CCUS-highVAF patients were more likely to have higher AMC (P = .007), less likely to have hypocellular BM (P = .015), more likely to have >1 somatic mutation (76% vs 20%, P = .001), more likely to have TET2 mutations (P = .025), and less likely to have TP53 mutations (P = .013; supplemental Table 3). When CCUS-highVAF patients were compared with LR-MDS patients, they were more likely to have higher AMC (P < .001), lower platelet counts (P < .001), less likely to be RBC TD (P = .036), less likely to have BM RS (P < .001), have lower BM blasts% (P = .001), more likely to have >1 mutation (P = .001), and less likely to have SF3B1 mutations (P < .001) (supplemental Table 4). We also compared the CCUS cohort to 239 WHO-defined lower risk patients with chronic myelomonocytic leukemia (CMML), an MDS/myeloproliferative overlap neoplasm, with risk stratification based on the Mayo Molecular Model.10 In comparison with LR-CMML, CCUS patients were younger in age (P = .003), more anemic (P = .001) and thrombocytopenic (P = .023), had lower WBC, ANC, and AMC (P < .001 for each), had lower PB blasts % (P = .023), and were less likely to have mutations involving TET2 (P = .021), SRSF2 (P < .001), ASXL1 (P = .027), CBL (P = .001), and SETBP1 (P = .010; supplemental Table 5). Of note, CMML arises in the context of age-related clonal hematopoiesis with TET2/SRSF2 mutations skewing hematopoiesis with a monocyte bias and is relatively enriched in ASXL1 (40%), CBL (15%), and SETBP1 (15%) mutations.11
LR-MDS patients had a longer median follow-up (54 vs 16 months), and at last follow-up, 52 (54%) and 12 (16%) deaths have been documented in the LR-MDS and CCUS groups, respectively (5 of 50 in CCUS-HighVAF). Of note, the longer follow-up of the LR-MDS patients in comparison with CCUS patients was in part because of the recent establishment of the CH clinic at our institute (2017) and in part because of the recent creation of the CCUS nosologic category.1,3 Regardless, ongoing follow-up of the CCUS cohort will be very valuable in terms of longer-term outcomes including clonal evolution and OS. CCUS and LR-MDS patients were followed clinically, and BM biopsies were carried out based on clinical need and suspicion to assess for disease progression (no routine/serial biopsies were done in either cohort). Thirteen (18%) CCUS patients progressed to MDS/CMML (n = 10, MDS-8, CMML-2; 6 with CCUS-HighVAF) and AML/blastic plasmacytoid dendritic cell neoplasm (n = 3, AML-2, BPDCN-1; 2 with CCUS-HighVAF) over a median of 16 months, whereas 9 (8%) LR-MDS patients progressed to higher-grade MDS (1) and AML (8), respectively.
To further delineate predictors of clonal evolution, CCUS patients were analyzed using a univariate logistic regression for the binary outcome of progression to myeloid neoplasm vs nonprogression. Once again, within limitation of a small sample size, several variables (supplemental Table 6) were tested, with higher risk cytogenetic groups by R-IPSS (P = .018) and the presence of SF3B1 mutations (P = .014) being statistically associated with clonal evolution/disease progression. We also reviewed the individual prognostic variables contributing to the R-IPSS scores in CCUS and LR-MDS patients (supplemental Table 7) and found that in CCUS patients, 80 (84%) of a cumulative 95 prognostic points were contributed by cytopenias (38% from anemia), whereas in the LR-MDS group, 84 (76%) of a cumulative 110 prognostic points were contributed by cytopenias (55% from anemia). This is explained by the fact that in both categories, karyotypic abnormalities and increased BM blasts were infrequent.
Although hypomethylating agents (HMAs) have not been approved for the management of CCUS, 7 (9%) CCUS patients and 20 (18%) LR-MDS patients treated with HMAs were assessable for response, with 1 patient each in the CCUS group meeting the proposed 2018 International Working Group MDS response criteria for hematologic improvement (HI)–erythroid, HI-platelet, and HI-neutrophil, respectively, in comparison with 4 (20%), 2 (10%), and 1 (5%) patients in the LR-MDS group.12 Three of 8 (37%) CCUS patients and 13 (33%) of 39 LR-MDS patients treated with erythropoiesis-stimulating agents (ESA) met criteria for HI-erythroid.
There was no difference in median OS between CCUS, including CCUS-HighVAF (median OS not reached) vs LR-MDS (median OS, 8.3 years) patients (P = .337; Figure 1C). Within limitations of a shorter follow-up, there was no OS difference between CCUS-HighVAF and CCUS-LowVAF (P = .524) patients or among CCUS patients further stratified by the number of mutations/SCNA (P = .288). Within limitations of a small sample size, we estimated the competing risk for AML progression on all 3 cohorts of patients using the Fine and Gray competing risk model.13 LR-CMML patients were less likely to transform to AML in comparison with LR-MDS patients, whereas the risk in CCUS patients was not significantly different from LR-MDS and LR-CCUS patients (Figure 1D-E).
Increased use of NGS testing has resulted in a higher prevalence of CCUS.5 Because of the lack of a formal neoplastic diagnosis, these patients are often managed with supportive care and are not eligible for MDS-approved therapies and clinical trials. We demonstrate that apart from subtle phenotypic and molecular differences, CCUS patients have overlapping similarities with LR-MDS, including the distribution of common MDS-driver mutations (DNMT3A, TET2, and ASXL1), with the exception of mutant SF3B1, which associates strongly with a diagnosis of MDS-RS.14,15 Although LR-MDS patients were more likely to be red cell TD (44%), 24% of CCUS patients were also red cell TD, with 13% in both groups being platelet TD. We observed higher WBC/ANC and platelet counts in MDS patients in comparison with CCUS patients, largely because of the fact that 50% of the MDS cohort comprised of patients with MDS-RS-SLD, where the dysplasia is largely limited to the erythroid lineage, sparing granulocyte and megakaryocyte development and maturation, and is a limitation of our study. Importantly, although the R-IPSS is currently applicable to MDS patients only, when applied to CCUS patients, risk stratification was similar. In addition, within a short follow-up period, 17% of CCUS patients progressed to MDS/AML, exemplifying that these entities exist in the same spectrum of myeloid neoplasms. Eighty-three percent of CCUS patients had a VAF ≥20%, whereas 91% had a VAF ≥10%, providing credibility to using VAF ≥10% for a diagnosis of CCUS, with further prospective validation needed.
This study is limited by the length of follow up for CCUS (16 months) compared with LR-MDS patients (54 months) and the fact that there could be a selection bias in detection of CCUS patients who have significant cytopenias and comorbidities, because they are more likely to seek care. In addition, given that 50% of our LR-MDS patients had MDS-RS-SLD, where SF3B1 is a dominant mutation and where BM dysplasia is largely restricted to the erythroid lineage, this could certainly influence phenotypic/molecular comparisons between LR-MDS and CCUS patients. However, despite these limitations, we demonstrate the evident clinical and molecular overlap between CCUS and LR-MDS patients and strongly urge the academic community to consider including CCUS, especially CCUS-HighVAF (≥20%), as a MDS subtype in the next iteration of the WHO classification system. In addition, given the lack of a prognostication system, we also urge the MDS consortium to consider assessing CCUS-HighVAF patients as they develop the IPSS-Molecular model. These steps will ensure better care for CCUS patients and importantly provide access to enrollment in MDS-directed clinical trials.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
Informed consent from the patient was obtained to share relevant data as outlined in the manuscript, including genetic information.
The authors thank the Mayo Clinic Center for Individualized Medicine for sponsoring the clonal hematopoiesis clinic and the Henry J. Predolin leukemia fund for developing the myelodysplastic syndrome/acute myeloid leukemia biorepository.
Footnotes
All raw sequencing data will be made available on e-mail request to the corresponding author at patnaik.mrinal@mayo.edu. Individual variant details are provided in the supplemental files.
Contribution: M. Li and M.M.P. helped with design of the study and data analysis; A.F. and T.L. helped with cytogenetic studies, NGS studies, and supplemental data; M.B. helped with statistical analysis and figure design; and all authors helped with the final draft of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Mrinal M. Patnaik, Division of Hematology, Mayo Clinic, 200 First St SW, Rochester, MN 55905; e-mail: patnaik.mrinal@mayo.edu.
References
- 1.Malcovati L, Gallì A, Travaglino E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood. 2017;129(25):3371-3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cazzola M. Myelodysplastic syndromes. N Engl J Med. 2020;383(14):1358-1374. [DOI] [PubMed] [Google Scholar]
- 3.Gondek LP, DeZern AE. Assessing clonal haematopoiesis: clinical burdens and benefits of diagnosing myelodysplastic syndrome precursor states. Lancet Haematol. 2020;7(1):e73-e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia [published correction appears in Blood. 2016;128(3):462-463]. Blood. 2016;127(20):2391-2405. [DOI] [PubMed] [Google Scholar]
- 5.Mangaonkar AA, Ferrer A, Pinto E Vairo F, et al. Clinical applications and utility of a precision medicine approach for patients with unexplained cytopenias. Mayo Clin Proc. 2019;94(9):1753-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454-2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488-2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377(2):111-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.In: Swerdlow S, Camp E, Harris NL, eds., et al. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues, Lyon, France: International Agency for Research on Cancer; 2008: [Google Scholar]
- 10.Patnaik MM, Itzykson R, Lasho TL, et al. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two-center study of 466 patients. Leukemia. 2014;28(11):2206-2212. [DOI] [PubMed] [Google Scholar]
- 11.Patnaik MM, Lasho T. Myelodysplastic syndrome/myeloproliferative neoplasm overlap syndromes: a focused review. Hematology Am Soc Hematol Educ Program. 2020;2020:460-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Platzbecker U, Fenaux P, Adès L, et al. Proposals for revised IWG 2018 hematological response criteria in patients with MDS included in clinical trials. Blood. 2019;133(10):1020-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Austin PC, Fine JP. Practical recommendations for reporting Fine-Gray model analyses for competing risk data. Stat Med. 2017;36(27):4391-4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Makishima H, Yoshizato T, Yoshida K, et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet. 2017;49(2):204-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patnaik MM, Lasho TL, Hodnefield JM, et al. SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood. 2012;119(2):569-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.