

Bacteria adopt different lifestyles depending on their environment and physiological conditions. In Escherichia coli and other enteric bacteria, the transition between the motile and the sessile states is controlled at multiple levels from the regulation of gene expression to the modulation of various processes by the second messenger c-di-GMP as a signaling molecule.
KEYWORDS: transcriptional regulation, motility, repression of flagellar assembly, second messenger
ABSTRACT
PdeL is a transcription regulator and catalytically active c-di-GMP phosphodiesterase (PDE) in Escherichia coli. PdeL has been shown to be a transcription autoregulator, while no other target genes have been identified so far. Here, we show that PdeL represses the transcription of the flagellar class II operon fliFGHIJK and activates sslE, encoding an extracellularly anchored metalloprotease, among additional loci. DNA-binding studies and expression analyses using plasmidic reporters suggest that the regulation of the fliF and sslE promoters by PdeL is direct. Transcription repression of the fliFGHIJK operon, encoding proteins required for the assembly of the flagellar basal body, results in inhibition of motility on soft-agar plates and reduction of flagellum assembly, as shown by fluorescence staining of the flagellar hook protein FlgE. PdeL-mediated repression of motility is independent of its phosphodiesterase activity. Thus, in motility control, the transcription regulator function of PdeL in reducing the number of assembled flagella is apparently epistatic to its phosphodiesterase function, which can indirectly promote the activity of the flagellar motor by lowering the c-di-GMP concentration.
IMPORTANCE Bacteria adopt different lifestyles depending on their environment and physiological conditions. In Escherichia coli and other enteric bacteria, the transition between the motile and the sessile states is controlled at multiple levels from the regulation of gene expression to the modulation of various processes by the second messenger c-di-GMP as a signaling molecule. The significance of our research is in identifying PdeL, a protein of dual function that hydrolyzes c-di-GMP and regulates the transcription of genes, as a repressor of flagellar gene expression and an inhibitor of motility, which adds an additional regulatory switch to the control of motility.
INTRODUCTION
Cyclic di-GMP is a second messenger in bacterial signaling pathways that control the transition between motility and sessility but also the cell cycle, stress responses, and beyond (1–3). Most bacteria encode several diguanylate cyclases (DGCs) and phosphodiesterases (PDEs) to synthesize and hydrolyze c-di-GMP (2), and the Escherichia coli K-12 genome encodes 29 proteins with DGC and PDE domains, of which 13 are enzymatically active PDEs and 12 are enzymatically active DGCs (1, 3, 4).
One of the PDEs of E. coli, PdeL, is both a transcription regulator and a c-di-GMP-specific EAL-type phosphodiesterase (5–7). PdeL carries an N-terminal FixJ/NarL-type DNA-binding domain (DBD) (also referred to as LuxR-type DBD) and a C-terminal EAL-type PDE domain. Such a domain arrangement has been identified only in very few proteins encoded by bacterial genomes (8). FixJ/NarL-type DBDs are present as C-terminal domains in 18 further transcription regulators in E. coli K-12, and in 17 of these transcription regulators, the DBD is fused to a two-component-system-like receiver domain as a regulatory unit. Correspondingly, in the case of PdeL, it was discussed that the EAL-type PDE domain has a regulatory function and that PdeL may represent a trigger enzyme whose activity as a transcriptional regulator is controlled by the binding of c-di-GMP and the enzymatic activity of the PDE domain (9). This regulatory role of the PDE domain seems feasible as the PDE domain exists in a monomer-to-dimer equilibrium with c-di-GMP binding enhancing dimerization (6). Furthermore, the crystal structures revealed structural coupling between dimerization and catalytic activity (6).
With respect to the biological function of PdeL, it has been shown that PdeL variants with enhanced phosphodiesterase activity can suppress a motility defect of pdeH mutants in E. coli K-12 (5). PdeH is the phosphodiesterase that is responsible and required for maintaining low c-di-GMP levels in exponentially growing E. coli cells. A low intracellular c-di-GMP concentration is a prerequisite for motility, as high levels of c-di-GMP result in inhibition of motility by the flagellar motor brake protein YcgR (10–12). With regard to the transcription regulator function of PdeL, it has been shown that it is an autoregulator and that autoregulation is modulated by the intracellular c-di-GMP level (5). These results suggested an intriguing coupling between gene expression and the enzymatic activity of the phosphodiesterase PdeL (5, 9).
In this study, we addressed the transcription regulator function of PdeL. Our data suggest that PdeL represses the transcription of the class II flagellar operon fliFGHIJK, activates sslE, and regulates additional loci. Transcription repression of the fliFGHIJK operon results in inhibition of motility by reducing the number of assembled flagella, independent of the phosphodiesterase activity of PdeL.
RESULTS
PdeL represses a flagellar operon and regulates additional loci.
To characterize the transcription regulator function of PdeL and identify genes regulated by PdeL, we compared transcriptome sequencing (RNA-seq) profiles of transformants of E. coli K-12 ΔpdeL strain T2057 (Table 1) with plasmids carrying Ptac pdeL3×FLAG, Ptac pdeLDBD-cIC-3×FLAG, or a control plasmid (Table 2). Plasmid-directed expression of pdeL3×FLAG was induced by the addition of isopropyl-β-d-thiogalactopyranoside (IPTG) for just 15 min prior to RNA isolation. The RNA-seq data suggest that PdeL3×FLAG represses the class II flagellar operon fliFGHIJK (Table 3). In addition, the data suggest that PdeL3×FLAG activates sslE (yghJ) and regulates additional loci (Table 3; see also Table S1 and Fig. S1 in the supplemental material). The fliFGHIJK operon encodes proteins required for assembling the flagellum basal body MS ring (13, 14). The sslE gene encodes a presumptive outer membrane-anchored, extracellular mucin-degrading metalloprotease. SslE is required for efficient access to small intestinal enterocytes and the optimal delivery of heat-labile toxin in enterotoxigenic E. coli (ETEC) and promotes macrophage activation in neonatal septicemic E. coli (15–17). Other presumably PdeL-repressed loci include genes encoding the synthesis of the enterobacterial common antigen (ECA) (Table 3), a carbohydrate-derived molecule present in the periplasm and outer membrane (18). Furthermore, PdeL3×FLAG triggered the upregulation of the ssrS gene encoding 6S RNA, a component of sigma 70 and sigma S competition for core RNA polymerase (19). The highest changes in transcript levels were observed for the fliFGHIJK operon and sslE (Table 3). Regulation of these loci was also detected in transformants of the ΔpdeL strain with the Ptac pdeLDBD-cIC-3×FLAG plasmid (Fig. S1 and Table S1). The hybrid PdeLDBD-cIC-3×FLAG protein is composed of the DNA-binding domain (DBD) of PdeL and the C-terminal dimerization domain of the phage Lambda repressor cI. Therefore, this result suggests that the changed transcript levels of fliFGHIJK and sslE are attributable to DNA binding by PdeL. Note that the transcript levels were not significantly different in pdeL+ strain BW30270 and its ΔpdeL mutant derivative T2057 (Fig. S1 and Table S1). Taken together the RNA-seq data suggest that plasmid-provided PdeL3×FLAG and PdeLDBD-cIC-3×FLAG repress the flagellar class II fliFGHIJK operon and activate sslE.
TABLE 1.
E. coli K-12 strains
| Strain | Description or genotypea | Reference or construction (reference[s])b |
|---|---|---|
| C41(DE3) | BL21(DE3) derivate for toxic protein expression | 55 |
| Donor strains for transduction | ||
| JW1183-1 | BW25113 ΔycgR784Kan (KEIO collection) | 56 |
| JW1880-1 | BW25113 ΔflhC744Kan (KEIO collection) | 56 |
| M182 stpA::tet | ΔlacIZYA-74 galU galK strA stpA::tet | 57 |
| U171 | U65 ΔpdeHKan | U65 (27)/pKD46 × OA603/OA604 (pKD4) |
| S3754 | MG1655 ΔhnsKan | 58 |
| MG1655 | Wild-type E. coli K-12 flhDC+; motile (no IS1 or IS5 insertion at flhDC) (laboratory storage no. S3837) | CGSC 6300 |
| T2517 | MG1655 flgET224C flhDC+; motile (T224C is an ACA-to-TGC exchange) | No-SCAR recombineering using pKDsgRNA-flgE-T224 (pKES337) and oligonucleotide OA553 |
| T2599 | MG1655 flgET224C IG(flgL-rne)Cm | T2517/pKD46 × OA785/OA786 (pKD3) |
| BW30270 and derivatives | ||
| BW30270 | MG1655 rph+ flhDC(IS1); motile (IS1 insertion upstream of the flhDC promoter) (laboratory storage no. S3839) | CGSC 7925 |
| T2055 | BW30270 ΔpdeLKan | BW30270 × T4GT7[U99 ΔpdeLKan] |
| T2057 | BW30270 ΔpdeLFRT | T2055 × pCP20 |
| T2604 | BW30270 flgE+ IG(flgL-rne)Cm | BW30270 × P1vir[IG(flgL-rne)Cm] |
| T2607 | BW30270 flgET224C IG(flgL-rne)Cm | S3839 × P1vir[T2599; cotransduction of flgET224C and IG(flgL-rne)Cm] |
| T2611 | BW30270 flgE+ IG(flgL-rne)FRT | T2604 × pCP20 |
| T2614 | BW30270 flgET224C IG(flgL-rne)FRT | T2607 × pCP20 |
| T1241 and derivatives | ||
| T1241 | BW30270 ilvG+ flhDC(IS1); motile | 59 |
| U98 | T1241 ΔpdeLCm | T1241/pKD46 × OA112/OA115 (pKD3) |
| U99 | T1241 ΔpdeLKan | T1241/pKD46 × OA112/OA115 (pKD4) |
| U102 | T1241 ΔpdeLKan | T1241 × T4GT7[U99 ΔpdeLKan] |
| U106 | T1241 ΔpdeLFRT | U102 × pCP20 |
| U114 | T1241 pdeL3×FLAGKan | T1241/pKD46 × OA274/OA275 (pSUB11) |
| U286 | T1241 pdeL3×FLAG-FRT | U114 × pCP20 |
| U287 | T1241 pdeL3×FLAG-FRT ΔpdeHKan | U286 × P1vir[ΔpdeHKan] |
| U291 | T1241 pdeL3×FLAG-FRT ΔpdeHFRT | U287 × pCP20 |
| Strains for construction of U281 and derivatives | ||
| U53 | T1241 Δ(araC-BAD) Δlac(I-ZYA)FRT Pcp8 araE | 27 |
| U279 | U53 ΔaraFGHCm | U53/pKD46 × OA908/OA909 (pKD3) |
| U281 | U53 ΔaraFGHFRT | U279 × pCP20 |
| U283 | U281 ΔpdeHKan | U281 × P1vir[ΔpdeHKan] |
| U288 | U281 ΔpdeHFRT | U283 × pCP20 |
| U284 | U281 ΔycgRKan | U281 × P1vir[JW1183-1 ΔycgRKan] |
| U289 | U281 ΔycgRFRT | U284 × pCP20 |
| U285 | U281 ΔpdeLKan | U281 × P1vir[U99 ΔpdeLKan] |
| U290 | U281 ΔpdeLFRT | U285 × pCP20 |
| U296 | U281 ΔpdeHFRT ΔpdeLKan | U288 × P1vir[U99 ΔpdeLKan] |
| U303 | U281 ΔpdeHFRT ΔpdeLFRT | U296 × pCP20 |
| U293 | U281 ΔycgRFRT ΔpdeLKan | U289 × P1vir[U99 ΔpdeLKan] |
| U294 | U281 ΔpdeHFRT ΔycgRKan | U288 × P1vir[JW1183-1 ΔycgRKan] |
| U301 | U281 ΔpdeHFRT ΔycgRFRT | U294 × pCP20 |
| U300 | U281 ΔycgRFRT ΔpdeLFRT | U293 × pCP20 |
| U307 | U281 ΔpdeHFRT ΔpdeLFRT ΔycgRKan | U303 × P1vir[JW1183-1 ΔycgRKan] |
| U309 | U281 ΔpdeHFRT ΔpdeLFRT ΔycgRFRT | U307 × pCP20 |
| U298 | U281 IG(fliE-fliF)::[PL fliE FRT-Kan-FRT Ptac fliF] | U281/pKD46 × OA914/OA915 (pKES263) |
| lacZ reporter strains | ||
| U4 | T1241 ΔlacZ | Scarless lacZ deletion constructed using pFDY217 (60, 61) |
| U264 | U4 ΔpdeLCm | U4 × P1vir[U98 ΔpdeLCm] |
| U266 | U4 ΔpdeLFRT | U264 × pCP20 |
| U423 | U4 ΔpdeLFRT ΔflhCKan | U266 × P1vir[T2779 ΔflhCKan] |
| U427 | U4 ΔpdeLFRT ΔflhCFRT | U423 × pCP20 |
| U31 | U4 attB::(Specr PpdeL lacZ) | U4/pLDR8 × pKESL161 |
| U32 | U4 attB::(Specr PpdeL lacZ) ΔhnsKan | U31 × T4GT7[S3754 hnsKan] |
| U33 | U4 attB::(Specr PpdeL lacZ) ΔstpA::tet | U31 × T4GT7[M182 stpA::tet] |
| U34 | U4 attB::(Specr PpdeL lacZ) ΔstpA::tet ΔhnsKan | U33 × T4GT7[S3754 hnsKan] |
| U113 | U4 attB::(Specr PpdeL lacZ) ΔhnsFRT | U32 × pCP20 |
| U226 | U4 attB::(Specr PpdeL lacZ) ΔpdeLCm | U31 × P1vir[U98 ΔpdeLCm] |
| U230 | U4 attB::(Specr PpdeL lacZ) ΔpdeLFRT | U226 × pCP20 |
| U277 | U4 attB::(Specr PpdeL lacZ) (translational fusion) | U4/pLDR8 × pKECY51 |
| U278 | U4 attB::(Specr PpdeL lacZ) (translational fusion) ΔpdeLFRT | U266/pLDR8 × pKECY51 |
IG(flgL-rne)Cm describes the insertion of an FRT-flanked chloramphenicol resistance gene in the intergenic region between flgL and rne. IG(fliE-fliF)::[PL fliE FRT-Kan-FRT Ptac fliF] describes the insertion of a PL-FRT-Kanr-FRT-Ptac cassette in the intergenic region of fliE and fliF, exchanging the native sequence between positions 2013034 and 2013209 (E. coli K-12 MG1655 genome [GenBank accession number U00096.3]).
Strains were constructed by transduction, which is stated as “× phage[donor strain]”; λ Red recombineering, stated as “× PCR primer pair (template),” followed by Flp recombinase-catalyzed deletion of the resistance marker, “× pCP20”; and integration of a PpdeL lacZ fusion at attB, indicated as “× pKESL161” (62).
TABLE 2.
Plasmids
| Plasmid | Feature(s)a | Reference, source, or constructionb |
|---|---|---|
| pBAD30 | araC Para MCS in p15A-ori; Ampr | 63 |
| pCP20 | cI857 PR flp in pSC101-repts; Ampr | 37 |
| pKD3 | FRT-Cmr-FRT in oriRγ; Ampr | 37 |
| pKD4 | FRT-Kanr-FRT in oriRγ; Ampr | 37 |
| pKD46 | araC Para γ-β-exo in pSC101-repts; Ampr | 37 |
| pET-22b(+) | PT7 MCS His6 in pMB1-ori; Ampr | Novagen |
| pEM-Cas9HF1-recA56 | Ptet cas9HF1 PproD recA56 in p15A-ori; Cmr | 41 |
| pKDsgRNA-NT | Ptet sgRNA control Para γ-β-exo in pSC101-repts; Specr | 41 |
| pKDsgRNA-p15A | Ptet sgRNA-p15A Para γ-β-exo in pSC101-repts; Specr | 40 |
| pKEKK7 | PproV proVHA-lacZ in p15A-ori; Kanr Specr attP | 64 |
| pKES263 | FRT-Kanr-FRT-PL in oriRγ; Ampr | 59 |
| pKES268 | PUV5 MCS lacZ in p15A-ori; Kanr Specr attP | 65 |
| pFDY217 | lacI lacOP [ΔlacZ] lacY lacA in pSC-repts; Tetr | 61 |
| pKESK10 | lacIq PUV5 bglG pSC101-ori; Cmr | 61 |
| pKETS24 | lacIq PUV5 MCS in pSC101-ori; Cmr | 66 |
| pKESK22 | lacIq Ptac MCS in p15A-ori; Kanr | 67 |
| pKEDP22 | lacIq Ptac MCS 3×FLAG in pKESK22 | 59 |
| pKESL161 | PpdeL lacZ in p15A-ori; Kanr Specr attP | PpdeL (PCR T929/T930) in pKES268 |
| pKESL162 | lacIq Ptac pdeL in pKESK22 | pdeL (PCR T925/T927) in pKESK22 |
| pKESL165 | lacIq Ptac mVenusc in pKESK22 | mVenus (PCR T146/T368) in pKESK22 |
| pKESL166 | lacIq Ptac pdeL-mVenus in pKESK22 | pdeL (PCR T925/T952) in pKESL165 |
| pKESL209 | lacIq Ptac pdeLHTH5M-mVenus in pKESK22 | pdeLHTH5M (flanking oligonucleotides T925/T972, mut. oligonucleotides OA173/OA174) in pKESL165 |
| pKESL207 | lacIq Ptac pdeL3×FLAG in pKESK22 | pdeL (PCR T925/OA116) in pKEDP22 |
| pKESL201 | lacIq Ptac pdeLDBD-cIC-3×FLAG in pKESK22 | pdeLDBD (PCR T925/OA117) and cIC (PCR OA118/OA121) in pKEDP22 |
| pKECY19 | pdeLHis6 in pET-22b(+); Ampr | pdeL (PCR OA579/OA580) in pET-22b(+) |
| pKECY26 | mNeonGreen in pSC-ori; Cmr | mNeonGreen fragment in pKETS24 |
| pKECY28 | PfliF mNeonGreen in pSC-ori; Cmr | PfliF (PCR OA732/OA733) in pKECY26) |
| pKESL279 | PUV5 mNeonGreen in pSC-ori; Cmr | PUV5 (PCR OA749/OA750) in pKECY26 |
| pKECY44 | araC Para pdeL in pBAD30 | pdeL (PCR T925/T927) in pBAD30 |
| pKECY46 | PsslE lacZ in pSC-ori; Cmr | PsslE (PCR OA777/OA778) in pKECY49 |
| pKECY49 | MCS lacZ in pSC-ori; Cmr | MCS (OA899/OA900) lacZ in pKECY26 |
| pKECY51 | PpdeL lacZ (translational fusion) in p15A-ori; Kanr Specr attP | PpdeL lacZ (pKESL161 XhoI, XbaI) in pKEKK7 |
| pKECY52 | araC Para pdeLHTH5M in pBAD30 | pdeLHTH5M (flanking oligonucleotides T925/T927, mut. oligonucleotides OA173/OA174) in pBAD30 |
| pKECY53 | araC Para pdeLEVL-AAA in pBAD30 | pdeLEVL-AAA (flanking oligonucleotides T925/T927, mut. oligonucleotides OA167/OA168) in pBAD30 |
| pKECY54 | araC Para pdeLDBD-cIC in pBAD30 | pdeLDBD-cIC (flanking oligonucleotides T925/OA905, mut. oligonucleotides OA117/OA118) in pBAD30 |
| pKECY55 | araC Para pdeLF206S in pBAD30 | pdeLF206S (flanking oligonucleotides T925/T927, mut. oligonucleotides OA337/OA338) in pBAD30 |
| pKECY57 | araC Para pdeLG299S in pBAD30 | pdeLG299S (flanking oligonucleotides T925/T927, mut. oligonucleotides OA345/OA346) in pBAD30 |
| pKECY59 | araC Para pdeLD263N in pBAD30 | pdeLD263N (flanking oligonucleotides T925/T927, mut. oligonucleotides OA912/OA913) in pBAD30 |
| pKECY60 | PfliF lacZ in pSC-ori; Cmr (−248 to +63) | PfliF (PCR OA732/OA907) in pKECY49 |
| pKECY78 | lacIq Ptac pdeLDBD(1–105)-cIC(132–236) in pKESK22 | pdeLDBD-cIC (flanking oligonucleotides T925/OA905, mut. oligonucleotides OA117/OA118) in pKESK22 |
| pKECY79 | lacIq Ptac pdeLHTH5M in pKESK22 | pdeLHTH5M (flanking oligonucleotides T925/T927, mut. oligonucleotides OA173/OA174) in pKESK22 |
| pKECY80 | lacIq Ptac pdeLEVL-AAA in pKESK22 | pdeLEVL-AAA (flanking oligonucleotides T925/T927, mut. oligonucleotides OA167/OA168) in pKESK22 |
| pKECY82 | PfliF lacZ in pSC-ori; Cmr (−188 to +63) | PfliF (PCR OB24/OA907) in pKECY49 |
| pKECY83 | PfliF lacZ in pSC-ori; Cmr (−164 to +63) | PfliF (PCR OB25/OA907) in pKECY49 |
| pKECY84 | PfliF lacZ in pSC-ori; Cmr (−142 to +63) | PfliF (PCR OB26/OA907) in pKECY49 |
| pKECY85 | PfliF lacZ in pSC-ori; Cmr (−128 to +63) | PfliF (PCR OB27/OA907) in pKECY49 |
| pKES337 | Ptet sgRNA-flgET224 in KDsgRNA-NT (pKDsgRNA-flgE-T224) | Annealed oligonucleotides OA545/OA546 in pKDsgRNA-NT (BsmBI) |
Features include Cmr (chloramphenicol resistance), Specr (spectinomycin resistance), Kanr (kanamycin resistance), MCS (multiple-cloning site), and pSC-repts (temperature-sensitive replication derivative of pSC101). HTH5M is K54A, S57A, H58A, K61A, and Q62A. EVL-AAA is E143A, V144A, and L145A. Oligonucleotides used for mutagenesis are depicted as mut. oligonucleotides.
Cloning was verified by sequencing of the cloned fragments.
The mVenus fragment was amplified from pKES293 (27).
TABLE 3.
PdeL-regulated loci identified by RNA-seq
| Gene ID | Gene or operon | Function and/or description | Fold changea | P value | Reference(s) |
|---|---|---|---|---|---|
| b1937 | fliE | Flagellar basal body and motor assembly proteins; class II locus activated by FlhDC and repressed by CsgD | −2.4 | 5.9E−03 | 68 |
| b1938 | fliF | −5.4 | 1.2E−08 | ||
| b1939 | fliG | −4.2 | 2.6E−06 | ||
| b1940 | fliH | −2.8 | 1.1E−03 | ||
| b1941 | fliI | −2.9 | 3.9E−04 | ||
| b1943 | fliK | −2.5 | 3.8E−03 | ||
| b1044 | ymdA | Uncharacterized; overexpression inhibits biofilm formation; class II locus activated by FlhDC | −2.5 | 3.5E−03 | 30, 69 |
| b3784 | rfe | Enterobacterial common antigen synthesis | −2.5 | 7.7E−04 | 18 |
| b3785 | wzzE | −2.3 | 3.6E−03 | ||
| b3791 | wecE | −2.1 | 7.9E−03 | ||
| b4466 | yghJ-sslE | Secreted mucin-degrading metalloprotease | 4.7 | 2.2E−13 | 15, 17 |
| b1541 | ydfZ | Putative selenoprotein | 2.6 | 2.4E−03 | 32 |
| b2579 | grcA | Stress-induced glycyl radical protein and alternate pyruvate formate-lyase subunit | 2.6 | 2.9E−03 | 70 |
| b2911 | ssrS | 6S RNA | 2.5 | 6.2E−04 | 19 |
| b4595 | yciY | Uncharacterized | 2.4 | 5.6E−03 | 32 |
Values of differential RNA levels of T2057/pKESL207 (Ptac pdeL3×FLAG) versus T2057/pKESK22 (control). The complete RNA-seq data set has been compiled in Table S1 in the supplemental material.
PdeL level and repression of pdeL transcription by H-NS.
To address the difference in the RNA-seq results obtained with plasmidic and chromosomal pdeL, respectively, we studied pdeL expression levels and regulation. To compare the expression levels of chromosomal and plasmidic pdeL, the expression of alleles coding for the PdeL3×FLAG protein was analyzed by Western blotting (Fig. 1A). Chromosomally encoded PdeL3×FLAG was detectable, as shown previously (5, 20), while the Ptac pdeL3×FLAG plasmid in the absence of the inducer (IPTG) directed the synthesis of approximately 10-fold more PdeL3×FLAG (Ptac pdeL3×FLAG carried on plasmid pKESL207 [p15A-ori]). Thus, the Ptac-directed basal level of pdeL3×FLAG expression that is attributable to the leakiness of noninduced Ptac is higher than that of the chromosomal pdeL3×FLAG allele, indicating that the native pdeL gene is expressed at a low level. In experiments addressing the function of the PdeL protein that are described further below, we therefore used plasmids for providing PdeL at a low level. A similar approach has been taken, for example, for characterizing the phosphodiesterase PdeC by using a low-copy-number Ptac pdeC plasmid (21).
FIG 1.

Expression and regulation of pdeL. (A) PdeL3×FLAG levels directed by chromosomal and plasmidic pdeL3×FLAG were compared by Western blotting. Cultures of pdeL+ strain T1241 and isogenic pdeL3×FLAG strain U114 were grown in LB medium for 3 h. In parallel, overnight-grown cultures of transformants of ΔpdeL strain U106 with Ptac pdeL3×FLAG plasmid pKESL207 were grown in LB medium containing kanamycin without the inducer IPTG. Lysates corresponding to bacteria at an OD600 of 0.08 were loaded per lane. For comparison of the level of chromosomally encoded PdeL3×FLAG to the plasmid-directed level of PdeL3×FLAG, a dilution series of the lysate of U106/pKESL207 was loaded, as indicated. Quantified band intensities are depicted below the corresponding lanes. n.d., not detected. (B) PpdeL activity and its regulation by H-NS and StpA were determined using a chromosomal PpdeL lacZ fusion (strain U31) and its isogenic mutant strains U33 (ΔstpA), U113 (Δhns), and U34 (Δhns ΔstpA). Cultures for β-galactosidase assays were inoculated from cultures grown overnight and grown in LB medium to an OD600 of 0.8. wt, wild type. (C) Autoregulation of PpdeL was determined with reporter strain U31 harboring attB::PpdeL lacZ and isogenic ΔpdeL mutant strain U230. PdeL and variants were provided using a Ptac plasmid. No inducer IPTG was added. The following plasmids were used: pKESK22 (+ctrl), pKESL162 (Ptac pdeL) (+PdeL), pKECY78 (Ptac pdeLDBD-cIC) (+DBD-cIC), pKECY79 (Ptac pdeLHTH5M) (+HTH5M), pKECY80 (Ptac pdeLEVL-AAA) (+EVL-AAA) (with replacements of the conserved EVL residues by alanine residues). Cultures for β-galactosidase assays were grown in LB medium containing kanamycin to an OD600 of 0.8. Mean values from at least three independent replicates are shown as bars, and error bars indicate standard deviations.
The expression level of chromosomal pdeL is apparently low, and it was shown previously that H-NS binds the pdeL regulatory region and causes repression (22, 23). H-NS is an abundant nucleoid-associated global repressor and xenogeneic silencer (24, 25). To quantify the repression of pdeL by H-NS, a chromosomal pdeL promoter-lacZ reporter (PpdeL lacZ) was used. The expression level of this transcriptional PpdeL lacZ fusion was low in the wild type and increased approximately 25-fold in the isogenic hns mutant. The deletion of stpA, encoding an H-NS homologue and common H-NS coregulator, had no statistically significant effect (Fig. 1B). These data demonstrate strong repression of the pdeL promoter by H-NS, which is probably the reason why there is no difference between the wild-type and ΔpdeL strains in the RNA-seq data (Table 3) and in the regulation of PpdeL lacZ (Fig. 1C). However, in transformants carrying the Ptac pdeL plasmid (encoding nontagged PdeL), the expression level of PpdeL lacZ was 4-fold increased (Fig. 1C), in agreement with the positive autoregulation reported previously (5). Additional data obtained with PdeL variants suggest that DNA binding by PdeL is essential for positive autoregulation: the substitution of five residues in the DNA recognition helix of the helix-turn-helix motif (HTH5M) of PdeL, mutant PdeLHTH5M, abrogated autoregulation (Fig. 1C). In addition, the hybrid protein PdeLDBD-cIC and PdeLEVL-AAA, carrying a substitution of three alanine residues for the conserved EVL motif of the phosphodiesterase domain, were functional as positive autoregulators, with lower activation by PdeLDBD-cIC (Fig. 1C). Thus, DNA binding but not the phosphodiesterase activity of PdeL is apparently essential for positive autoregulation.
To further explore whether the deletion of the native pdeL gene has any effect on the expression of the PpdeL lacZ fusion at other growth phases, expression was determined along the growth curve in the pdeL+ and ΔpdeL backgrounds. The data show that PpdeL lacZ expression is rather constant and not significantly affected by chromosomally encoded PdeL (Fig. S2). A translational PpdeL lacZ fusion (lacZ fused after the 10th codon of pdeL) likewise directed rather constant β-galactosidase levels, and no significant difference between the pdeL+ and ΔpdeL strains was observed (Fig. S2). These data support the conclusion that the level of chromosomally encoded PdeL is not sufficient for autoregulation, at least in the strain background used and conditions tested here, in contrast to a previous publication (5). In addition, we analyzed whether the deletion of pdeH, encoding the main phosphodiesterase PdeH (5, 20), affects the expression of a chromosomal pdeL3×FLAG allele (Fig. S3). Western blotting shows that PdeL3×FLAG is present throughout growth in both the pdeH+ and ΔpdeH strains, with the protein level decreasing toward the stationary phase (Fig. S3). This decrease has not been observed for the PpdeL lacZ fusions, possibly because β-galactosidase is very stable. Furthermore, no significant differences in the PdeL3×FLAG levels between the pdeL+ and the ΔpdeH strains were observed (Fig. S3), which indicates that the PdeL protein amount is not c-di-GMP regulated.
PdeL regulates the fliF and sslE promoters.
The RNA-seq data identified the fliFGHIJK operon and the sslE gene as loci that were most strongly regulated by PdeL. To analyze their regulation by PdeL, we used transcriptional PfliF mNeonGreen and PsslE lacZ fusions. These were not integrated into the chromosome but were provided on low-copy-number pSC-derived plasmids. PdeL was provided on a plasmid as well since the expression level of the native pdeL gene is low (Fig. 2). The expression and regulation of the PfliF mNeonGreen fusion were determined by flow cytometry. The data show that PfliF directed significant fluorescence in the absence of PdeL, while in the presence of the Ptac pdeL plasmid (noninduced), the expression level was 4-fold reduced, in agreement with the RNA-seq data (Fig. 2A and B). A control plasmid carrying promoterless mNeonGreen directed very low fluorescence, while a constitutive PUV5 promoter directed high fluorescence independent of the presence or absence of PdeL (Fig. 2A and B). These data suggest that PdeL represses the transcription of the plasmidic PfliF mVenus reporter and that the rather low level of PdeL directed by the leaky Ptac promoter is sufficient. In addition, the expression of the plasmidic PsslE lacZ fusion was 7.5-fold upregulated by PdeL (Fig. 2C), which likewise is in congruence with the RNA-seq results. Taken together, these data suggest that transcription initiation at the fliF and sslE promoters is regulated by PdeL.
FIG 2.

PdeL regulates the fliF and sslE promoters. Expression levels of PfliF mNeonGreen and PsslE lacZ fusions were determined by flow cytometry (fluorescence-activated cell sorter [FACS]) and β-galactosidase activity assays, respectively. (A) The activity of PfliF mNeonGreen was tested in the absence (control) and presence of PdeL (+PdeL) of double transformants of ΔpdeL strain T2057. The PfliF mNeonGreen reporter was provided by low-copy-number plasmid pKECY28 (PfliF mNG); a constitutive PlacUV5 mNeonGreen reporter (PUV5 mNG) (provided by pKESL279) and a promoterless mNeonGreen (mNG) promoter (pKECY26) served as controls. Plasmid pKESL162 carries pdeL under the control of Ptac, and parent vector plasmid pKESK22 was used as a control. Cultures were grown in tryptone medium with antibiotics but without the addition of IPTG. Shown are histograms depicting the fluorescence of samples taken after 3 h of growth (OD600 ranging from 0.8 to 1). For each sample, 200,000 bacteria were counted. (B) Mean fluorescence intensities (in arbitrary units) from two independent experiments after 3 h of growth. (C) The activity of the PsslE lacZ fusion was determined in double transformants of ΔpdeL mutant strain U266. The PsslE lacZ reporter was provided by low-copy-number plasmid pKECY46. PdeL was provided under the control of Ptac by pKESL162. Vector plasmid pKESK22 was used as a control in ΔpdeL strain U266 and parental strain U4 (wild type [wt]). Cultures for β-galactosidase assays were grown as described above for panel A. Mean values from at least 3 replicates are shown as bars, and error bars indicate standard deviations.
Next, we analyzed the DNA binding of PdeLHis6 to DNA fragments derived from the fliF and sslE promoter regions. Electrophoretic mobility shift assays (EMSAs) of PfliF fragments revealed DNA binding of PdeLHis6 (Fig. 3). The presumptive location of the PdeLHis6 DNA-binding site overlaps those of the CsgD and FlhD4C2 DNA-binding sites identified previously (26) (Fig. 3A). For the sslE promoter region, specific DNA binding of PdeLHis6 was observed for two fragments, which maps the DNA-binding site 90 bp to 180 bp upstream of the sslE translation start site (Fig. 3B). Although the DNA-binding sites were narrowed down to <100 bp at both loci, no sequence motif could be identified, possibly because the regions are very AT rich (65%). Taking these results together, specific DNA binding of PdeLHis6 to fliF and sslE fragments suggests that PdeL directly regulates transcription at these promoters.
FIG 3.

PdeL specifically binds fliF and sslE DNA fragments. For DNA-binding analyses of PdeLHis6, electrophoretic mobility shift assays (EMSAs) were performed using fliF and sslE promoter DNA fragments. (A) Schematic of the fliF promoter region. Fragments “A” to “F” are depicted as solid lines. Binding sites of CsgD (black) and FlhD4C2 (white) are depicted as boxes. The region of overlapping PdeLHis6-bound fragments is depicted as a gray box. (B) Schematic of the sslE promoter region. An open reading frame (ORF) encompassing 60 codons is indicated (orf60). Fragments “G” to “N” are depicted as solid lines. The gray box depicts the overlapping region of sslE promoter DNA fragments that were PdeLHis6 bound. (C and D) Electrophoretic mobility shift assays with increasing concentrations of purified PdeLHis6 using the fliF (C) and sslE (D) fragments. Oligonucleotides used to amplify DNA fragments A to N are indicated. DNA fragments shifted by PdeLHis6 DNA binding are marked with open arrowheads.
Furthermore, to narrow down the PdeL DNA-binding site at the fliF promoter, the regulation of a deletion series of plasmidic PfliF lacZ fusions was tested as well (Fig. 4). The assays were performed using a Para pdeL plasmid in strain U290, as in this strain, the arabinose regulon is modified to avoid arabinose feedback regulation and thus allows a low level of induction of Para by as little as 2 μM arabinose (27, 28). (Note that the Para pdeL system was also used in motility assays described below.) Repression of PfliF by PdeL was observed for all constructs that carry at least one of the FlhD4C2 DNA-binding sites, while the CsgD DNA-binding site was not required. Thus, the PdeL DNA-binding site presumably overlaps the FlhD4C2 DNA-binding sites (Fig. 4B). Expression analyses in a ΔflhC mutant confirmed that FlhD4C2 is also essential for the activation of PfliF in the plasmidic system (Fig. 4C). The data suggest that PdeL represses the PfliF promoter either directly or by interfering with activation by FlhD4C2.
FIG 4.

The PdeL DNA-binding site overlaps the FlhD4C2 DNA-binding site. (A) Schematic of the PfliF region as shown in Fig. 3. Fragments “−248” to “−128,” depicted as solid lines, represent a deletion series of PfliF lacZ reporters carried on low-copy-number plasmids (pSC derivatives) (Table 2). The presumptive location of the PdeL DNA-binding site is indicated by a dark gray bar, while the light gray bar depicts the region of the PdeL DNA-binding site that was mapped by EMSAs (Fig. 3). The CsgD DNA-binding site maps at positions −178 to −145, and the FlhD4C2 DNA-binding sites map at positions −159 to −143 and −133 to −117 relative to the fliF translation start site. (B) The activity of the plasmidic PfliF lacZ reporters was tested in combination with plasmid pKECY44 carrying pdeL under the control of Para (+pPdeL) or plasmid pBAD30 as a control (−) in ΔlacZ ΔpdeL ΔaraBAD ΔaraFGH strain U290. Cultures for β-galactosidase assays were grown in tryptone medium with antibiotics. l-Arabinose was supplemented to a final concentration of 2 μM 1 h after inoculation. Cultures were harvested at an OD600 of 0.8 (after approximately 5 h of growth). Shown are mean values from four biological replicates, and error bars indicate standard deviations. Statistically significant differences are indicated with asterisks (***, P < 0.001; n.s., not significant). (C) The activity of PfliF lacZ reporters was determined in transformants of ΔpdeL strain U266 and its ΔflhC derivative (U427). Cultures were grown similarly as described above for panel A but without the addition of arabinose. The differences in expression levels are all statistically significant except for PfliF lacZ at position −128.
PdeL inhibits motility.
PdeL represses the class II fliFGHIJK operon encoding proteins required for the assembly of the flagellum basal body. To analyze whether PdeL inhibits motility, we used transformants of strain ΔpdeL U290 and its derivatives with Para pdeL plasmids for gradual induction by arabinose. No difference in motility was detectable for transformants with the empty vector control and the Para pdeL plasmid in the absence of arabinose, in accordance with tight control of Para, while induction with 20 μM arabinose led to reduced motility in the case of Para pdeL (Fig. 5, compare the control and PdeL). Variants of PdeL with substitutions of amino acids in the phosphodiesterase domain had an effect on motility similar to that of wild-type PdeL; i.e., their induction by arabinose caused a reduction of motility (Fig. 5). These PdeL variants include F206S and G299S for enhanced phosphodiesterase activity as well as EVL-AAA and D263N that are inactive as phosphodiesterases (5, 6). In contrast, PdeL variant HTH5M, defective in DNA binding, had the opposite effect: induction by arabinose caused an increase in motility (Fig. 4A). This is in agreement with high flagellar functionality and motility at low c-di-GMP levels (10–12). These data suggest that inhibition of motility by PdeL requires DNA binding, and they show that the DNA-binding mutant PdeLHTH5M is an active phosphodiesterase.
FIG 5.

PdeL inhibits motility by DNA binding. Motility was analyzed on tryptone soft-agar plates (0.25% agar with ampicillin). Shown are scanned images of representative plates that were incubated at 28°C for 8 h. The motilities of transformants of strains U290 (ΔpdeL), U303 (ΔpdeL ΔpdeH), and U309 (ΔpdeL ΔpdeH ΔycgR) with plasmids for the Para-directed expression of PdeL and PdeL variants are shown, as labeled. For induction, 20 μM l-arabinose was added to the soft-agar plates, where indicated. The following plasmids were used: pBAD30 (control), pKECY44 (Para pdeL) (wild type [wt]), pKECY52 (Para pdeLHTH5M) (HTH5M), pKECY55 (Para pdeLF206S) (F206S), pKECY57 (Para pdeLG299S) (G299S), pKECY53 (Para pdeLEVL-AAA) (EVL-AAA), and pKECY59 (Para pdeLD263N) (D263N).
Motility assays in isogenic ΔpdeH and ΔycgR ΔpdeH mutants consolidated the results. Motility is expected to be low in the ΔpdeH mutant lacking the main c-di-GMP phosphodiesterase PdeH but containing the c-di-GMP-dependent flagellar motor brake protein YcgR, while motility is restored in the ΔpdeH ΔycgR double mutants (10–12). The ΔpdeH mutant was not motile, as expected. However, transformants with plasmids encoding the phosphodiesterase mutants F206S and G299S were weakly motile, in agreement with their identification and characterization as PdeL mutants of enhanced phosphodiesterase activity (5). Nevertheless, the induction of mutants F206S and G299S again resulted in the inhibition of the motility of the ΔpdeH strain (Fig. 5). The motility phenotypes of transformants of the ΔycgR ΔpdeH double mutant were similar to that of the wild type, as expected (Fig. 5). Furthermore, the motility of isogenic pdeL+ and ΔpdeL strains in combination with ΔpdeH and ΔycgR was analyzed (Fig. S4), which showed that chromosomal pdeL does not affect motility under the conditions tested, as shown previously (5, 29). Taken together, the motility assays suggest that repression of motility requires DNA binding by PdeL.
To address whether the inhibition of motility by PdeL is attributable to the repression of the fliFGHIJK operon, the fliF promoter was replaced by a constitutive variant of Ptac, and the promoter of the divergent fliE gene was replaced by the constitutive PL promoter (Fig. 6). Note that the promoter replacement resulted in reduced motility per se. Nevertheless, transformants of this Ptac fliF PL fliE strain with the Para pdeL plasmid remained motile irrespective of induction by arabinose (Fig. 6). These data suggest that the inhibition of motility by PdeL involves the repression of the transcription of the fliFGHIJK operon.
FIG 6.

Substitution of fliF and fliE promoters precludes inhibition of motility by PdeL. (A) Strain U298 carries a substitution of the fliE-fliF intergenic region with the fliE promoter replaced by PL and the fliF promoter replaced by Ptac. The fragment used for λ Red-mediated homologous recombination was amplified with oligonucleotides OA914 and OA915. (B) The motility of strain U298 transformed with plasmid pKECY44 (Para pdeL) or vector pBAD30 (control) was assayed on tryptone soft-agar plates with increasing concentrations of l-arabinose, as indicated. Shown are scanned images of representative plates incubated at 28°C for 10 h.
PdeL reduces flagellum assembly.
As PdeL represses PfliF and inhibits motility, we tested whether flagellum assembly is inhibited by PdeL. To this end, we constructed a derivative of E. coli K-12 strain BW30270 carrying an flgET224C allele for staining of the flagellar hook protein FlgE by coupling of the cysteine-reactive dye Alexa Fluor 568 C5 maleimide. The flgET224C strain T2614 is motile (Fig. 7A). Transformants of the flgET224C strain with a Ptac pdeL plasmid or the empty vector were grown in LB medium without IPTG, and the numbers of fluorescence-stained flagella were determined by microscopy. Representative microscopy images of individual bacteria with zero to five fluorescent spots presumably representing the flagella and quantitative analysis are shown in Fig. 5B and C. Fluorescent spots were detected for more than 90% of the bacteria transformed with the vector control and the plasmid carrying pdeLHTH5M-mVenus coding for the PdeL DNA-binding mutant. In contrast, no flagella were detected for more than 60% of the bacteria transformed with the Ptac pdeL and Ptac pdeL-mVenus plasmids, while only one or two flagella were stained per bacterium for the remaining 40% of the population (Fig. 7C). Note that the Ptac pdeL-mVenus and Ptac pdeLHTH5M-mVenus plasmids were included in the experiment with the intention to visualize the proteins; however, their expression levels in the absence of IPTG were too low for detection by fluorescence microscopy. We conclude that the significant reduction of motility conferred by leaky Ptac-directed pdeL expression significantly reduces the number of assembled flagella.
FIG 7.

Inhibition of flagellum synthesis by PdeL. The number of flagella per bacterium was determined by coupling the thiol-reactive fluorescent dye Alexa Fluor 568 C5 maleimide to the flagellar hook protein FlgET224C. (A) Motility of wild-type E. coli K-12 strain BW30270, strain T2611 carrying an FRT site in the intergenic region between flgL and the rne allele [IG(flgL-rne)FRT] (strain T2611), and its isogenic flgET224C mutant (strain T2614). Tryptone soft-agar plates were incubated for 4 h at 37°C. OM, outer membrane; IM, inner membrane; PG, peptidoglycan. (B) Representative microscopy images (overlays of transmitted light/phase contrast and red fluorescence [TL+FL]) of transformants of the flgET224C mutant strain T2614 stained with Alexa Fluor 568 C5 maleimide showing 0 to 5 fluorescent spots (in white). Each image represents 5 μm2. (C) Quantitative analysis of the number of fluorescent spots (flagella per bacterium) of transformants of flgET224C mutant strain T2614 with pKESK22 (control), Ptac pdeL provided by pKESL162 (PdeL), Ptac pdeL-mVenus (pKESL166) (PdeL-mV), and Ptac pdeLHTH5M-mVenus (pKESL209) (PdeLHTH5M-mV). Cultures of transformants were used to inoculate 15 ml LB medium with kanamycin to an OD600 of 0.08, and the cultures were grown at 37°C for 3 h and then harvested for flagellum staining and microscopy. Shown are the averaged data from three biological replicates, each based on the evaluation of fluorescent spots of >100 bacteria using ImageJ with the MicrobeJ plug-in.
DISCUSSION
Here, we have shown that PdeL, a transcriptional regulator and c-di-GMP-specific phosphodiesterase in E. coli (5–7), represses the transcription of the flagellar class II fliFGHIJK operon and activates sslE, coding for an extracellularly anchored metalloprotease. The transcriptional repression of the fliFGHIJK operon by PdeL concomitantly causes inhibition of motility and a reduction in flagellum assembly. While PdeL represses motility, a DNA-binding mutant of PdeL enhances motility, as expected for an active c-di-GMP-specific phosphodiesterase. Our data suggest that in motility control, the transcription regulator function of PdeL is epistatic to its function as a c-di-GMP phosphodiesterase.
Regulation of motility by the repression of a flagellar class II operon is unexpected. The control of motility is mediated by a hierarchical regulatory cascade (see reference 30 and references therein). Briefly, the master transcription regulator FlhD4C2 activates class II flagellar genes encoding proteins required for the assembly of the flagellar basal body and hook. The class II fliFGHIJK operon and divergent fliE gene encode flagellum proteins of the basal body and motor complex as well as export proteins important for flagellum assembly (14, 31, 32). Their expression is activated by FlhD4C2 and repressed by CsgD (26, 30, 33). Class II flagellar genes also include fliA, which encodes the alternative RNA polymerase sigma subunit σ28 (FliA), and flgM, encoding the anti-σ28 factor FlgM (14). After the assembly of the basal body and hook, anti-sigma factor FlgM is secreted, and σ28 RNA polymerase directs the transcription of flagellar class III genes, which encode proteins required for the completion of the flagellum (14). The downregulation of flagellar gene expression and flagellum synthesis is achieved by the regulation of FlhD4C2, which is tightly controlled from the transcriptional to posttranslational levels (see reference 30 and references therein). The PdeL-mediated repression of the class II flagellar operon fliFGHIJK adds an additional component to the complex hierarchical control of flagellar gene expression (Fig. 8). PdeL-mediated repression of the fliFGHIJK operon, encoding proteins required at an early step of flagellum assembly, may be a means to prevent the initiation of the assembly of new flagella, while flagellar genes required for later steps remain active, ensuring that flagella whose assembly has been initiated will be completed. Correspondingly, flagellum staining (Fig. 7) revealed a reduction of the number of flagella by PdeL but not complete inhibition.
FIG 8.
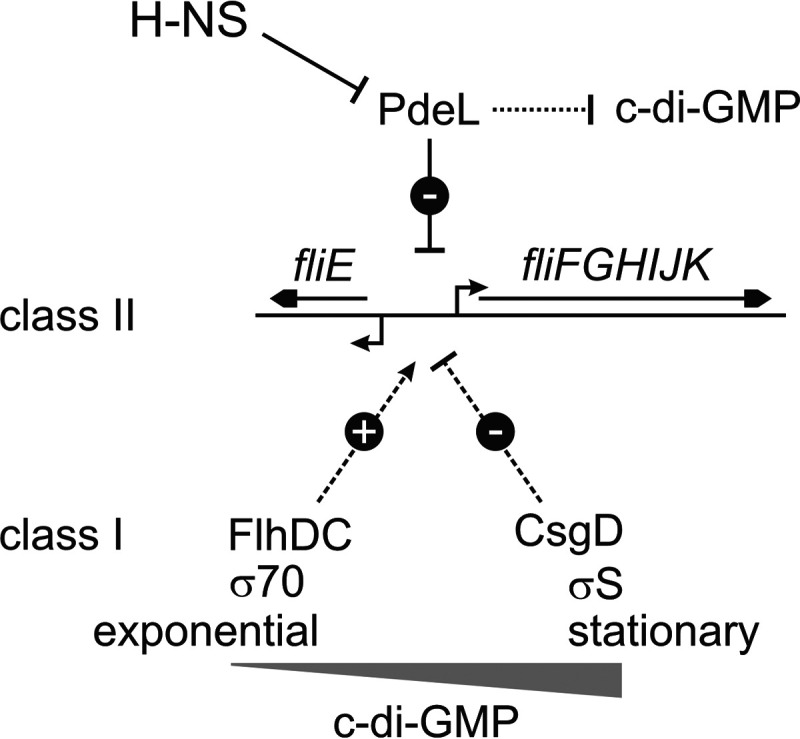
Schematic overview of repression of the fliFGHIJK operon by PdeL. PdeL represses the transcription of the class II flagellar operon fliFGHIJK. The expression of pdeL is repressed by H-NS. In addition, PdeL is a catalytically active c-di-GMP phosphodiesterase. The activation of fliE and fliFGHIJK expression by FlhD4C2 and repression by CsgD integrate flagellar class II gene expression into the regulatory network that controls motility and sessility.
The pdeL gene is strongly repressed by H-NS, and accordingly, its expression level is detectable but very low under standard laboratory growth conditions. Additional data obtained with transcriptional and translational pdeL promoter-lacZ fusions and a chromosomal pdeL3×FLAG allele suggest that PdeL levels are low throughout growth. The low level of expression may explain why ΔpdeL mutants have no phenotype with respect to either regulating transcription or affecting c-di-GMP levels (5, 29; this work). This result corresponds to data that demonstrated that only PdeL variants with enhanced phosphodiesterase activity that are expressed at a 4-fold-higher level than wild-type pdeL suppress a motility defect of the ΔpdeH mutant, while wild-type pdeL does not (5). As shown here, the brief induction of pdeL in the RNA-seq analysis as well as the low-level plasmid-directed pdeL expression conferred transcription regulation of fliFGHIJK and sslE as well as positive autoregulation of pdeL transcription and significantly reduced flagellum assembly and inhibited motility. Considering the 25-fold repression of pdeL by H-NS and the glycolytic-flux-dependent repression of pdeL by the transcriptional regulator Cra (34), there should be a physiological condition under which the native pdeL gene directs the synthesis of PdeL levels that are sufficient for it being functional. It is also possible that an additional positive transcription regulator is required to overcome H-NS-mediated repression of pdeL in vivo, which has not yet been addressed.
Taking these results together, PdeL has a dual activity as a transcriptional regulator and a c-di-GMP-specific phosphodiesterase. Our result that the reduction in the number of assembled flagella is attributable to the transcription regulator function of PdeL reveals that yet another role of the biological function of PdeL needs to be considered.
MATERIALS AND METHODS
Bacterial strains, media, and plasmids.
E. coli K-12 strains and their construction are summarized in Table 1. Strains were constructed by transduction using T4GT7 (35) or P1vir (36), by Lambda Red-mediated chromosomal recombineering (37, 38), and by scarless Cas9-assisted recombineering (no-SCAR) (39, 40). Plasmids are listed in Table 2, and oligonucleotides used for the construction of strains and plasmids are listed in Table S2 in the supplemental material. Bacteria were cultivated in LB medium (10 g/liter Bacto tryptone, 5 g/liter Bacto yeast extract, and 5 g/liter NaCl), tryptone medium (10 g/liter Bacto tryptone, 5 g/liter NaCl), SOB medium (20 g/liter Bacto tryptone, 5 g/liter Bacto yeast extract, 0.5 g/liter NaCl, 2.5 mM KCl, 10 mM MgCl2 [pH 7.0]), or SOC medium (SOB medium with 0.4% glucose). For plates, 15 g/liter Bacto agar was added. Antibiotics were added to final concentrations of 50 μg/ml ampicillin (Amp), 15 μg/ml chloramphenicol (Cm), and 15 μg/ml kanamycin (Kan), as required; IPTG and l-arabinose were added as described below.
Mutants of flgE were constructed by no-SCAR (39–41). Briefly, E. coli K-12 strain MG1655 (CGSC 6300) (Table 1) was doubly transformed with plasmids carrying high-fidelity cas9 (pEM-cas9HF1-recA56) and a single guide RNA (sgRNA) targeting flgE (sgRNA-flgET224) (pKES337). Double transformants were selected at 28°C on LB plates containing chloramphenicol (20 μg/ml) and spectinomycin (Spec) (50 μg/ml). The efficiency of Cas9-HF1/sgRNA-flgET224-targeted killing was evaluated by spotting 10 μl of 10−2 to 10−7 serial dilutions on LB plates containing chloramphenicol and spectinomycin without and with anhydrotetracycline (aTC) (100 ng/ml) for inducing the transcription of casHF1 and sgRNA-flgET224. For the mutagenesis of flgET224, electrocompetent bacteria of these double transformants were prepared from cultures that were grown in SOB medium with chloramphenicol and spectinomycin containing 10 mM l-arabinose. Electrocompetent bacteria (40 μl) were mixed with 500 pmol of the flgET224C-specific recombineering oligonucleotide OA553 (Table S2); after electroporation, 1 ml SOC medium was added, and the bacteria were incubated for 1 h at 28°C. To isolate the flgET224C mutants and assess the efficiency of mutagenesis, 100 μl was plated on LB plates containing chloramphenicol, spectinomycin, and aTC that were incubated at 28°C, and in parallel, 10 μl of 10−1 to 10−6 serial dilutions was spotted on the same selective plates. Colonies were restreaked, followed by PCR screening using the flgET224C-specific primer OA575 and primer OA547. Of the positive clones, the flgE fragment was PCR amplified (primers OA547 and OA548) and sequenced. Clones carrying the flgET224C mutation were cured for the temperature-sensitive sgRNA-flgET224C plasmid (pKES337) by streaking on LB plates containing chloramphenicol and incubation at 42°C. Subsequently, plasmid pEM-Cas9HF1-recA56 was cured as described previously (39) by transformation with pKDsgRNA-p15a, followed by the induction of cas9HF1 and the p15A-specific sgRNA with aTC for 2 h in liquid culture (LB medium with spectinomycin and aTC) and the selection of cured clones on LB plates containing spectinomycin and aTC at 28°C. Clones successfully cured of pEM-Cas9HF1-recA56 were streaked on LB plates at 42°C to cure the temperature-sensitive plasmid pKDsgRNA-p15A. The flgET224C mutant that was cured of all plasmids was stored as strain T2517. Subsequently, for the cotransduction of the flgET224C allele, a pKD3-derived FRT (Flp recombination target)-Cm-FRT fragment was inserted in the intergenic region between flgL and rne (IG_flgL-rne::KD3-Cmr), which maps 8 kb downstream of flgE, yielding strain T2599.
Expression analyses and standard molecular techniques.
Standard molecular methods were performed as described previously (42). For expression analyses of promoter-lacZ fusions, β-galactosidase assays were performed in at least 3 biological replicates, as described previously (36). Mean β-galactosidase activities determined for transcriptional and translational PpdeL lacZ fusions along the growth curve were normalized relative to the total protein amount per 1 optical density at 600 nm (OD600) of bacteria since the OD600 depends on the size of the bacteria, which varies during growth. In particular, the protein amount per 1 OD600 was determined using the Pierce bicinchoninic acid (BCA) protein assay (Thermo Scientific, Germany) for one replicate of each strain per time point, and the average total protein content per time point was used for normalizing the mean β-galactosidase activities.
Fluorescence microscopy.
For fluorescence staining of flagella, the thiol-reactive fluorescent dye Alexa Fluor 568 C5 maleimide (Thermo Fisher Scientific, Germany) was used to label the flagellar hook protein FlgET224C in strain T2614 (BW30270 flgET224C), similar to what was described previously for Bacillus subtilis (43, 44). For flagellum staining, 1 ml of the culture was harvested by centrifugation at 8,000 relative centrifugal force (rcf) for 1 min. Bacterial cell pellets were washed 3 times with phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 [pH 7.4]) prior to labeling. Next, the bacterial cell pellet was resuspended in 200 μl PBS containing 5 μl Alexa Fluor 568 C5 maleimide, which was prepared by dissolving 1 mg of Alexa Fluor 568 C5 maleimide in 200 μl of dimethyl sulfoxide. The samples were incubated for 20 min in the dark at room temperature. The dye was removed by washing the bacteria three times with PBS. Finally, the bacterial cell pellet was resuspended in 100 μl PBS and used for microscopy on 1% agarose pads. The Alexa Fluor 568 C5 maleimide dye was excited at 577 nm for 500 ms. Image acquisition was performed using a Zeiss Axio Imager.M2 microscope with an EC Plan-Neofluar 100×/1.30-numerical-aperture oil Ph3 M27 objective. Images were captured and processed using ZEN 2012 software (Carl Zeiss Microscopy GmbH, Germany). The number of flagella was analyzed using ImageJ and the MicrobeJ plug-in (45, 46).
Motility assay.
Motility was assayed on tryptone soft-agar plates (0.25% Bacto agar on petri dishes with a 94-mm lid radius) (47) that were supplemented with antibiotics and l-arabinose, as indicated in the figure legends. Petri dishes with a 60-mm lid radius were used to assay the motility of strains carrying an frt site in the intergenic region between flgE and rne used for labeling of the flagellar hook protein FlgET224C. The soft-agar plates were freshly poured and allowed to cool for no longer than 1 h at room temperature before spotting 4 μl of a densely grown culture onto the center of the plate. The plates were incubated at 28°C for 8 h unless otherwise specified in the figure legends, followed by scanning of the plates for image acquisition. For better visibility, the color of the images was transformed to grayscale, and the contrast was enhanced using CorelDRAW 2019 software.
RNA isolation and RNA-seq.
For RNA-seq, total RNA was isolated from transformants of E. coli strain BW30270 with plasmid pKESK22 (control) and from strain T2057 with plasmids pKESK22 (control), pKESL207 (Ptac pdeL3×FLAG), and pKESL201 (Ptac pdeLDBD-cIC-3×FLAG). These transformants were inoculated to an OD600 of 0.08 in 10 ml LB medium supplemented with kanamycin and grown for 1 h to an OD600 of approximately 0.3 at 37°C with shaking. Next, Ptac-directed transcription was induced by adding IPTG to a final concentration of 1 mM. After 15 min of additional growth, 2-ml samples of the cultures were harvested by adding them to RNAprotect bacterial reagent (Qiagen, Germany), followed by total RNA isolation using the RNeasy minikit (Qiagen, Germany), with on-column DNA digestion by DNase I treatment, according to the manufacturer’s instructions (Qiagen, Germany). The integrity of the RNA was validated by separating 0.25 μg of RNA on 8% denaturing acrylamide gels (7 M urea, 45 mM Tris base, 45 mM boric acid, 1 mM EDTA, 8% acrylamide-bisacrylamide [19:1, wt/wt]) and visual inspection of the 23S rRNA and 16S rRNA bands. The preparation of RNA-seq libraries was performed according to methods described previously (48), with minor adaptations. The depletion of processed RNAs and rRNAs was performed by treatment with Terminator 5′-phosphate-dependent exonuclease (TEX) (Biozym, Germany). In brief, 7 μg of total RNA was digested with 7 U of TEX for 60 min at 30°C. The reaction was stopped by the addition of EDTA-NaOH (pH 8.0) to a final concentration of 5 mM, followed by extraction with phenol-chloroform-isoamyl alcohol (25:24:1, vol/vol/vol) and ethanol precipitation by the addition of 3 volumes of ethanol–3 M Na-acetate (pH 6.5) (30:1, vol/vol). Subsequently, the 5′ triphosphates of the RNAs were removed by treatment with 20 U of RppH (New England BioLabs [NEB]) for 60 min at 37°C. The RNA was extracted with phenol-chloroform-isoamyl alcohol and ethanol precipitated, as described above. The synthesis of cDNA libraries was performed by the Cologne Center for Genomics (CCG) (http://portal.ccg.uni-koeln.de/ccg/) using an Illumina TruSeq RNA library kit according to the manufacturer’s instructions. Three biological replicates of each strain were sequenced, with 5 million reads each, using an Illumina HiSeq 4000 instrument.
For data analysis, the sequencing raw data files were uploaded to the public Galaxy Web platform (https://usegalaxy.org/) (49). First, raw sequencing reads were uploaded in FASTQ file format and aligned to the E. coli K-12 MG1655 genome (GenBank assembly accession number GCA_000005845.2) using Bowtie2 (50) with default settings for paired-end libraries. These generated BAM files containing the aligned reads were used to calculate individual gene abundances. For this, HTSeq (51) was used in union mode for stranded data sets with a minimum alignment quality of 10. Tabular lists of gene abundances for each sample were used for differential expression analysis using DEseq2 (52) with default settings. In addition, the aligned reads compiled in the BAM files of the individual samples were inspected using IGV (Integrated Genome Viewer) (53).
Western blotting.
For comparison of chromosomal and plasmid-directed levels of PdeL, strain U114 (T1241 pdeL3×FLAG-Kan) coding for epitope-tagged PdeL3×FLAG under the control of its native promoter was used (Table 1). The plasmid-directed expression of Ptac pdeL3×FLAG (pKESL207) was tested in transformants of strain U106 (T1241 ΔpdeL). Parental strain T1241 carrying the wild-type pdeL allele was used as a negative control. Cultures grown overnight were used to inoculate 20 ml of LB medium (containing kanamycin for U106/pKESL207) to an OD600 of 0.08 and grown for 3 h at 37°C. Bacteria from a 500-μl culture sample were pelleted for 1 min at 13,000 rpm. For the detection of chromosomal pdeL3×FLAG, strain U286 (T1241 pdeL3×FLAG-FRT) and its isogenic ΔpdeH mutant strain U291 (T1241 pdeL3×FLAG-FRT ΔpdeHFRT) were used. Cultures grown overnight were used to inoculate 100 ml LB medium and grown for 10 h at 37°C. Every hour, bacteria from a 1-ml culture sample were pelleted for 1 min at 13,000 rpm.
SDS-PAGE and Western blot procedures were performed as described previously (42). Per lane, cells at an OD600 of 0.08 were loaded, and equal loading was verified by 2,2,2-trichloroethane staining of total protein (54). Epitope-tagged PdeL3×FLAG was detected using primary antibody anti-FLAG M2 from mouse (diluted 1:4,000) (catalogue number F3165; Sigma) and Alexa Fluor 680 fluorescent dye-labeled secondary anti-mouse antibody from goat (diluted 1:10,000) (catalogue number A21057; Thermo Scientific). Quantification of protein bands was performed with Odyssey V3.0 software.
Protein purification.
For the purification of C-terminally six-histidine-tagged PdeL (PdeLHis6), strain C41(DE3) was transformed with plasmid pKECY19 carrying the pdeLHis6 gene under the control of the T7 promoter (Table 2). For protein purification, cultures grown overnight were used to inoculate 0.5 liter of LB medium containing 50 μg/ml ampicillin to an OD600 of 0.08, and the cultures were grown for 2 h at 37°C to an OD600 of 1.0. Protein expression was then induced by adding IPTG to a final concentration of 200 μM, and the cultures were grown for an additional 2 h at 28°C. Bacteria were harvested by centrifugation, washed in resuspension buffer (50 mM Tris-HCl [pH 8.0], 250 mM KCl, 20 mM imidazole), and pelleted again for storage at −80°C. Lysate preparation was performed at 4°C; the cell pellets were resuspended in lysis buffer (4 ml/g of bacterial cells) (50 mM Tris-HCl [pH 8.0], 250 mM KCl, 20 mM imidazole, 20 μg/ml DNase I [5,000 U/ml] [New England BioLabs, USA]) and lysed by sonication (40% amplitude for 4 min with a 2-s pulse/2-s pause) (Sonics Vibracell VCX750 high-volume ultrasonic cell disrupter). The lysate was cleared by centrifugation (38,000 rcf for 60 min) (Sorvall SS34 rotor). Next, the supernatant was loaded onto a 1-ml HisTrap HP column using an Äkta fast protein liquid chromatography (FPLC) system (GE Healthcare, Germany). The column was washed with wash buffer (50 mM Tris-HCl [pH 8.0], 250 mM KCl) with increasing imidazole concentrations (20 mM, 40 mM, and 60 mM), with at least 15 column volumes at each step. The proteins were eluted with elution buffer (20 mM Tris-HCl [pH 8.0], 250 mM KCl, 250 mM imidazole), and 1-ml fractions were collected and analyzed by SDS-PAGE. Protein concentrations were measured by using the Qubit fluorometric quantitation system (Invitrogen, Germany). Aliquots of the proteins were stored at −80°C.
Electrophoretic mobility shift assays.
For DNA-binding analyses by electrophoretic mobility shift assays (EMSAs), DNA fragments were amplified by PCR using the primers indicated in Fig. 3 and Table S2. DNA-binding reactions of purified PdeLHis6 were set up on ice in 10 μl binding buffer (20 mM Tris-HCl [pH 8], 100 mM KCl, 2 mM dithiothreitol [DTT], 10% glycerol, and 10 mM CaCl2) with 20 nM DNA fragment and PdeLHis6 protein at the concentrations specified in the figure legends. The samples were incubated at 30°C for 20 min and then loaded onto an 8% polyacrylamide gel (acrylamide-bisacrylamide [29:1] in 0.5× TBE [45 mM Tris base, 45 mM boric acid, 1 mM EDTA]). The gel was run for 10 min at 50 V, followed by 45 min at 200 V. For documentation, the gel was stained with ethidium bromide (0.5 μg/ml in 0.5× TBE) for 20 min.
Flow cytometry.
Expression analyses of green fluorescent mNeonGreen reporter gene fusions were performed by flow cytometry. Briefly, 30 ml of tryptone medium was inoculated from cultures grown overnight to an OD600 of 0.08 and grown at 37°C. Samples of the cultures (500 μl) were taken after 3 h of growth at an OD600 of 0.8. Fluorescence detection measurement of 200,000 cells was conducted using a BD FACSCalibur instrument and CellQuest (BD Biosciences, USA). The fluorescence of mNeonGreen was excited with a blue laser at 488 nm (530/30-nm-band-pass filter). Data processing and statistical analyses were performed using Flowing software (http://flowingsoftware.btk.fi). The recorded events were gated for the background of the growth medium.
Data availability.
Raw sequencing files (FASTQ file format) and gene abundance tables (TXT file format) have been deposited and are available in NCBI’s Gene Expression Omnibus under accession number GSE136273.
Supplementary Material
ACKNOWLEDGMENTS
RNA sequencing was performed by the Cologne Center for Genomics (CCG) (http://portal.ccg.uni-koeln.de/ccg/). We thank Prerana Wagle, CECAD Bioinformatics Facility, University of Cologne, for support in data analyses; Hannes Bredderman, Vanessa Holtwick, and Bilge Ipek Meriç for plasmids and strains; and Gert Bange and Patricia Bedrunka, Marburg, Germany, for suggestion of the flgE mutant.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Jenal U, Reinders A, Lori C. 2017. Cyclic di-GMP: second messenger extraordinaire. Nat Rev Microbiol 15:271–284. doi: 10.1038/nrmicro.2016.190. [DOI] [PubMed] [Google Scholar]
- 2.Römling U, Galperin MY, Gomelsky M. 2013. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev 77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hengge R. 2009. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol 7:263–273. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- 4.Hengge R, Galperin MY, Ghigo J-M, Gomelsky M, Green J, Hughes KT, Jenal U, Landini P. 2016. Systematic nomenclature for GGDEF and EAL domain-containing cyclic di-GMP turnover proteins of Escherichia coli. J Bacteriol 198:7–11. doi: 10.1128/JB.00424-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reinders A, Hee C-S, Ozaki S, Mazur A, Boehm A, Schirmer T, Jenal U. 2016. Expression and genetic activation of cyclic di-GMP-specific phosphodiesterases in Escherichia coli. J Bacteriol 198:448–462. doi: 10.1128/JB.00604-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sundriyal A, Massa C, Samoray D, Zehender F, Sharpe T, Jenal U, Schirmer T. 2014. Inherent regulation of EAL domain-catalyzed hydrolysis of second messenger c-di-GMP. J Biol Chem 289:6978–6990. doi: 10.1074/jbc.M113.516195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt AJ, Ryjenkov DA, Gomelsky M. 2005. The ubiquitous protein domain EAL is a cyclic diguanylate-specific phosphodiesterase: enzymatically active and inactive EAL domains. J Bacteriol 187:4774–4781. doi: 10.1128/JB.187.14.4774-4781.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.UniProt Consortium. 2017. UniProt: the universal protein knowledgebase. Nucleic Acids Res 45:D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hengge R. 2016. Trigger phosphodiesterases as a novel class of c-di-GMP effector proteins. Philos Trans R Soc Lond B Biol Sci 371:20150498. doi: 10.1098/rstb.2015.0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paul K, Nieto V, Carlquist WC, Blair DF, Harshey RM. 2010. The c-di-GMP binding protein YcgR controls flagellar motor direction and speed to affect chemotaxis by a “backstop brake” mechanism. Mol Cell 38:128–139. doi: 10.1016/j.molcel.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boehm A, Kaiser M, Li H, Spangler C, Kasper CA, Ackermann M, Kaever V, Sourjik V, Roth V, Jenal U. 2010. Second messenger-mediated adjustment of bacterial swimming velocity. Cell 141:107–116. doi: 10.1016/j.cell.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 12.Fang X, Gomelsky M. 2010. A post-translational, c-di-GMP-dependent mechanism regulating flagellar motility. Mol Microbiol 76:1295–1305. doi: 10.1111/j.1365-2958.2010.07179.x. [DOI] [PubMed] [Google Scholar]
- 13.Chevance FF, Hughes KT. 2008. Coordinating assembly of a bacterial macromolecular machine. Nat Rev Microbiol 6:455–465. doi: 10.1038/nrmicro1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Macnab RM. 2003. How bacteria assemble flagella. Annu Rev Microbiol 57:77–100. doi: 10.1146/annurev.micro.57.030502.090832. [DOI] [PubMed] [Google Scholar]
- 15.Luo Q, Kumar P, Vickers TJ, Sheikh A, Lewis WG, Rasko DA, Sistrunk J, Fleckenstein JM. 2014. Enterotoxigenic Escherichia coli secretes a highly conserved mucin-degrading metalloprotease to effectively engage intestinal epithelial cells. Infect Immun 82:509–521. doi: 10.1128/IAI.01106-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nesta B, Valeri M, Spagnuolo A, Rosini R, Mora M, Donato P, Alteri CJ, Del Vecchio M, Buccato S, Pezzicoli A, Bertoldi I, Buzzigoli L, Tuscano G, Falduto M, Rippa V, Ashhab Y, Bensi G, Fontana MR, Seib KL, Mobley HLT, Pizza M, Soriani M, Serino L. 2014. SslE elicits functional antibodies that impair in vitro mucinase activity and in vivo colonization by both intestinal and extraintestinal Escherichia coli strains. PLoS Pathog 10:e1004124. doi: 10.1371/journal.ppat.1004124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tapader R, Bose D, Dutta P, Das S, Pal A. 2018. SslE (YghJ), a cell-associated and secreted lipoprotein of neonatal septicemic Escherichia coli, induces Toll-like receptor 2-dependent macrophage activation and proinflammation through NF-κB and MAP kinase signaling. Infect Immun 86:e00399-18. doi: 10.1128/IAI.00399-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitchell AM, Srikumar T, Silhavy TJ. 2018. Cyclic enterobacterial common antigen maintains the outer membrane permeability barrier of Escherichia coli in a manner controlled by YhdP. mBio 9:e01321-18. doi: 10.1128/mBio.01321-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cavanagh AT, Wassarman KM. 2014. 6S RNA, a global regulator of transcription in Escherichia coli, Bacillus subtilis, and beyond. Annu Rev Microbiol 68:45–60. doi: 10.1146/annurev-micro-092611-150135. [DOI] [PubMed] [Google Scholar]
- 20.Sarenko O, Klauck G, Wilke FM, Pfiffer V, Richter AM, Herbst S, Kaever V, Hengge R. 2017. More than enzymes that make or break cyclic di-GMP—local signaling in the interactome of GGDEF/EAL domain proteins of Escherichia coli. mBio 8:e01639-17. doi: 10.1128/mBio.01639-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herbst S, Lorkowski M, Sarenko O, Nguyen TKL, Jaenicke T, Hengge R. 2018. Transmembrane redox control and proteolysis of PdeC, a novel type of c‐di‐GMP phosphodiesterase. EMBO J 37:e97825. doi: 10.15252/embj.201797825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uyar E, Kurokawa K, Yoshimura M, Ishikawa S, Ogasawara N, Oshima T. 2009. Differential binding profiles of StpA in wild-type and hns mutant cells: a comparative analysis of cooperative partners by chromatin immunoprecipitation-microarray analysis. J Bacteriol 191:2388–2391. doi: 10.1128/JB.01594-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rangarajan AA, Schnetz K. 2018. Interference of transcription across H-NS binding sites and repression by H-NS. Mol Microbiol 108:226–239. doi: 10.1111/mmi.13926. [DOI] [PubMed] [Google Scholar]
- 24.Shen BA, Landick R. 2019. Transcription of bacterial chromatin. J Mol Biol 431:4040–4066. doi: 10.1016/j.jmb.2019.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh K, Milstein JN, Navarre WW. 2016. Xenogeneic silencing and its impact on bacterial genomes. Annu Rev Microbiol 70:199–213. doi: 10.1146/annurev-micro-102215-095301. [DOI] [PubMed] [Google Scholar]
- 26.Ogasawara H, Yamamoto K, Ishihama A. 2011. Role of the biofilm master regulator CsgD in cross-regulation between biofilm formation and flagellar synthesis. J Bacteriol 193:2587–2597. doi: 10.1128/JB.01468-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Breddermann H, Schnetz K. 2016. Correlation of antagonistic regulation of leuO transcription with the cellular levels of BglJ-RcsB and LeuO in Escherichia coli. Front Cell Infect Microbiol 6:106. doi: 10.3389/fcimb.2016.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khlebnikov A, Risa Ø, Skaug T, Carrier TA, Keasling JD. 2000. Regulatable arabinose-inducible gene expression system with consistent control in all cells of a culture. J Bacteriol 182:7029–7034. doi: 10.1128/jb.182.24.7029-7034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heroven AK, Böhme K, Dersch P. 2012. The Csr/Rsm system of Yersinia and related pathogens: a post-transcriptional strategy for managing virulence. RNA Biol 9:379–391. doi: 10.4161/rna.19333. [DOI] [PubMed] [Google Scholar]
- 30.Fitzgerald DM, Bonocora RP, Wade JT. 2014. Comprehensive mapping of the Escherichia coli flagellar regulatory network. PLoS Genet 10:e1004649. doi: 10.1371/journal.pgen.1004649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berg HC. 2003. The rotary motor of bacterial flagella. Annu Rev Biochem 72:19–54. doi: 10.1146/annurev.biochem.72.121801.161737. [DOI] [PubMed] [Google Scholar]
- 32.Keseler IM, Mackie A, Santos-Zavaleta A, Billington R, Bonavides-Martínez C, Caspi R, Fulcher C, Gama-Castro S, Kothari A, Krummenacker M, Latendresse M, Muñiz-Rascado L, Ong Q, Paley S, Peralta-Gil M, Subhraveti P, Velázquez-Ramírez DA, Weaver D, Collado-Vides J, Paulsen I, Karp PD. 2017. The EcoCyc database: reflecting new knowledge about Escherichia coli K-12. Nucleic Acids Res 45:D543–D550. doi: 10.1093/nar/gkw1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stafford GP, Ogi T, Hughes C. 2005. Binding and transcriptional activation of non-flagellar genes by the Escherichia coli flagellar master regulator FlhD2C2. Microbiology (Reading) 151:1779–1788. doi: 10.1099/mic.0.27879-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimada T, Fujita N, Maeda M, Ishihama A. 2005. Systematic search for the Cra-binding promoters using genomic SELEX system. Genes Cells 10:907–918. doi: 10.1111/j.1365-2443.2005.00888.x. [DOI] [PubMed] [Google Scholar]
- 35.Wilson GG, Young KYK, Edlin GJ, Konigsberg W. 1979. High-frequency generalised transduction by bacteriophage T4. Nature 280:80–82. doi: 10.1038/280080a0. [DOI] [PubMed] [Google Scholar]
- 36.Miller JH. 1992. A short course in bacterial genetics. A laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 37.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uzzau S, Figueroa-Bossi N, Rubino S, Bossi L. 2001. Epitope tagging of chromosomal genes in Salmonella. Proc Natl Acad Sci U S A 98:15264–15269. doi: 10.1073/pnas.261348198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reisch CR, Prather KLJ. 2017. Scarless Cas9 assisted recombineering (no-SCAR) in Escherichia coli, an easy-to-use system for genome editing. Curr Protoc Mol Biol 117:31.8.1–31.8.20. doi: 10.1002/cpmb.29. [DOI] [PubMed] [Google Scholar]
- 40.Reisch CR, Prather KLJ. 2015. The no-SCAR (scarless Cas9 assisted recombineering) system for genome editing in Escherichia coli. Sci Rep 5:15096. doi: 10.1038/srep15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moreb EA, Hoover B, Yaseen A, Valyasevi N, Roecker Z, Menacho-Melgar R, Lynch MD. 2017. Managing the SOS response for enhanced CRISPR-Cas-based recombineering in E. coli through transient inhibition of host RecA activity. ACS Synth Biol 6:2209–2218. doi: 10.1021/acssynbio.7b00174. [DOI] [PubMed] [Google Scholar]
- 42.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 43.Courtney CR, Cozy LM, Kearns DB. 2012. Molecular characterization of the flagellar hook in Bacillus subtilis. J Bacteriol 194:4619–4629. doi: 10.1128/JB.00444-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Najafi J, Shaebani MR, John T, Altegoer F, Bange G, Wagner C. 2018. Flagellar number governs bacterial spreading and transport efficiency. Sci Adv 4:eaar6425. doi: 10.1126/sciadv.aar6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ducret A, Quardokus EM, Brun YV. 2016. MicrobeJ, a tool for high throughput bacterial cell detection and quantitative analysis. Nat Microbiol 1:16077. doi: 10.1038/nmicrobiol.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kearns DB. 2010. A field guide to bacterial swarming motility. Nat Rev Microbiol 8:634–644. doi: 10.1038/nrmicro2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borries A, Vogel J, Sharma CM. 2012. Differential RNA sequencing (dRNA-Seq): deep-sequencing-based analysis of primary transcriptomes, p 109–121. In Harbers M, Kahl G (ed), Tag-based next generation sequencing. Wiley-VCH Verlag GmbH & Co KGaA, Weinheim, Germany. [Google Scholar]
- 49.Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Eberhard C, Grüning B, Guerler A, Hillman-Jackson J, Von Kuster G, Rasche E, Soranzo N, Turaga N, Taylor J, Nekrutenko A, Goecks J. 2016. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res 44:W3–W10. doi: 10.1093/nar/gkw343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ladner CL, Yang J, Turner RJ, Edwards RA. 2004. Visible fluorescent detection of proteins in polyacrylamide gels without staining. Anal Biochem 326:13–20. doi: 10.1016/j.ab.2003.10.047. [DOI] [PubMed] [Google Scholar]
- 55.Miroux B, Walker JE. 1996. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol 260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 56.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang A, Rimsky S, Reaban ME, Buc H, Belfort M. 1996. Escherichia coli protein analogs StpA and H-NS: regulatory loops, similar and disparate effects on nucleic acids dynamics. EMBO J 15:1340–1349. doi: 10.1002/j.1460-2075.1996.tb00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stratmann T, Pul Ü, Wurm R, Wagner R, Schnetz K. 2012. RcsB-BglJ activates the Escherichia coli leuO gene, encoding an H-NS antagonist and pleiotropic regulator of virulence determinants. Mol Microbiol 83:1109–1123. doi: 10.1111/j.1365-2958.2012.07993.x. [DOI] [PubMed] [Google Scholar]
- 59.Pannen D, Fabisch M, Gausling L, Schnetz K. 2016. Interaction of the RcsB response regulator with auxiliary transcription regulators in Escherichia coli. J Biol Chem 291:2357–2370. doi: 10.1074/jbc.M115.696815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hamilton CM, Aldea M, Washburn BK, Babitzke P, Kushner SR. 1989. New method for generating deletions and gene replacements in Escherichia coli. J Bacteriol 171:4617–4622. doi: 10.1128/jb.171.9.4617-4622.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dole S, Kühn S, Schnetz K. 2002. Post-transcriptional enhancement of Escherichia coli bgl operon silencing by limitation of BglG-mediated antitermination at low transcription rates. Mol Microbiol 43:217–226. doi: 10.1046/j.1365-2958.2002.02734.x. [DOI] [PubMed] [Google Scholar]
- 62.Diederich L, Rasmussen LJ, Messer W. 1992. New cloning vectors for integration into the lambda attachment site attB of the Escherichia coli chromosome. Plasmid 28:14–24. doi: 10.1016/0147-619x(92)90032-6. [DOI] [PubMed] [Google Scholar]
- 63.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kavalchuk K, Madhusudan S, Schnetz K. 2012. RNase III initiates rapid degradation of proU mRNA upon hypo-osmotic stress in Escherichia coli. RNA Biol 9:98–109. doi: 10.4161/rna.9.1.18228. [DOI] [PubMed] [Google Scholar]
- 65.Salscheider SL, Jahn A, Schnetz K. 2014. Transcriptional regulation by BglJ-RcsB, a pleiotropic heteromeric activator in Escherichia coli. Nucleic Acids Res 42:2999–3008. doi: 10.1093/nar/gkt1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fragel SM, Montada A, Heermann R, Baumann U, Schacherl M, Schnetz K. 2019. Characterization of the pleiotropic LysR-type transcription regulator LeuO of Escherichia coli. Nucleic Acids Res 47:7363–7379. doi: 10.1093/nar/gkz506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Venkatesh GR, Kembou Koungni FC, Paukner A, Stratmann T, Blissenbach B, Schnetz K. 2010. BglJ-RcsB heterodimers relieve repression of the Escherichia coli bgl operon by H-NS. J Bacteriol 192:6456–6464. doi: 10.1128/JB.00807-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Subramanian S, Kearns DB. 2019. Functional regulators of bacterial flagella. Annu Rev Microbiol 73:225–246. doi: 10.1146/annurev-micro-020518-115725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim M, Kim K. 2017. Stress-responsively modulated ymdAB-clsC operon plays a role in biofilm formation and apramycin susceptibility in Escherichia coli. FEMS Microbiol Lett 364:fnx114. doi: 10.1093/femsle/fnx114. [DOI] [PubMed] [Google Scholar]
- 70.Wagner AFV, Schultz S, Bomke J, Pils T, Lehmann WD, Knappe J. 2001. YfiD of Escherichia coli and Y06I of bacteriophage T4 as autonomous glycyl radical cofactors reconstituting the catalytic center of oxygen-fragmented pyruvate formate-lyase. Biochem Biophys Res Commun 285:456–462. doi: 10.1006/bbrc.2001.5186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing files (FASTQ file format) and gene abundance tables (TXT file format) have been deposited and are available in NCBI’s Gene Expression Omnibus under accession number GSE136273.