

Abstract
The high rate of the sudden infant death syndrome (SIDS) in American Indians in the Northern Plains (3.5/1000) may reflect the high incidence of cigarette smoking and alcohol consumption during pregnancy. Nicotine, a neurotoxic component of cigarettes, and alcohol adversely affect nicotinic receptor binding and subsequent cholinergic development in animals. We measured 3H‐nicotine receptor binding in 16 brainstem nuclei in American Indian SIDS (n = 27) and controls (n = 6). In five nuclei related to cardiorespiratory control, 3H‐nicotinic binding decreased with increasing number of drinks (P < 0.03). There were no differences in binding in SIDS compared with controls, except upon stratification of prenatal exposures. In three mesopontine nuclei critical for arousal there were reductions (P < 0.04) in binding in controls exposed to cigarette smoke compared with controls without exposure; there was no difference between SIDS cases with or without exposure. This study suggests that maternal smoking and alcohol affects 3H‐nicotinic binding in the infant brainstem irrespective of the cause of death. It also suggests that SIDS cases are unable to respond to maternal smoking with the “normal” reduction seen in controls. Future studies are needed to establish the role of adverse prenatal exposures in altered brainstem neurochemistry in SIDS.
INTRODUCTION
Despite the publicity of risk reduction messages (ie, the Back‐to‐Sleep campaign), the sudden infant death syndrome (SIDS) remains the leading cause of postnatal infant mortality in the USA, with an overall rate of 0.53/1000 live births (30). It is defined as the sudden death of an infant less than 1 year of age that remains unexplained after a thorough case investigation, including an autopsy and death scene investigation (49). Of major concern is the wide disparity in SIDS rates among different ethnic and racial groups (30). In the American Indians of Aberdeen Area of the Northern Plains, the population of this study, SIDS is a significant public health problem, with a rate of 3.5/1000, almost seven times the overall US rate (1, 2). Prenatal exposure to maternal cigarette (tobacco) smoke is a major risk factor for SIDS (19, 24, 31). In a recent study of infant mortality and SIDS risk in the Aberdeen Area of the Northern Plains, that is, the Aberdeen Area Infant Mortality Study (AAIMS), 55%–70% of pregnant American Indian women in the Northern Plains were found to smoke during pregnancy compared with the national average of 11% (2). The AAIMS also demonstrated that prenatal exposure to alcohol is a risk factor for SIDS in this population (24), an observation supported in studies of non‐American Indian populations (19, 31). The AAIMS reported an 6.2‐fold increase in SIDS in infants whose mothers drank during the 3 months prior to and after conception, and an 8.2‐fold increase in infants whose mothers binged drank in the first trimester (24). The incidence of maternal drinking during pregnancy overall was approximately 60% (6, 24), compared with the national average of 13% (7)
In the following autopsy study of the AAIMS, we examined the effect of prenatal exposures to cigarette smoke and alcohol upon the distribution and binding of cholinergic nicotinic receptors (nAChRs) in the brainstem of SIDS cases in order to gain insight into the possible role of these exposures in the pathogenesis of abnormal brainstem neurochemistry in SIDS. Nicotine, a major neurotoxin in cigarette smoke, crosses the placental barrier and fetal blood–brain barrier, and binds to the endogenous nAChRs in the fetal brain. The nAChRs are widely expressed in the brain, and are located primarily pre‐synaptically; they are involved in neurotransmitter release, neurite outgrowth and differentiation, and neuronal survival (18, 32, 47). The nAChRs are ligand‐gated ion channels, pentameric in structure, and comprised of homomeric or heteromeric combinations of 12 genes (α2‐10, β2‐4). Each receptor subtype is pharmacologically, anatomically and functionally different (50), for example, the β subunit regulates the rate of agonists/antagonists binding and dissociation (37).
In this study, we chose the radioligand 3H‐nicotine for analysis because it binds with varying affinity to all subunits of the nAChRs; moreover, it binds preferentially to the α4β2 receptor subunit to which nicotine in cigarettes smoke binds (4, 14). Use of this radioligand also allowed us to compare the results from this study with those in a non‐American Indian population that we reported previously (33). In the previous study, we found that 3H‐nicotinic binding was significantly increased in three mesopontine nuclei in controls exposed to smoke prenatally compared with non‐exposed controls, but not in exposed SIDS cases. The affected nuclei were the locus coeruleus (LC) and pontis oralis that are involved in extra‐thalamic and thalamic ascending arousal pathways, respectively, and the parabrachialis lateralis (PBL), involved in the ascending processing of visceral information. The finding of increased binding in these three nuclei in the control cases exposed to smoke suggested to us that the “normative” response to exposure is to increase, that is, up‐regulate, nicotinic binding. This idea is based in part upon considerable animal data indicating up‐regulation of nicotinic binding upon exposure to nicotine in multiple paradigms, including animal models of prenatal exposure (34, 35, 42). The fact that the exposed SIDS cases did not demonstrate this increase suggested to us that SIDS infants are unable to respond in the same way as non‐SIDS cases at the cellular and molecular level, albeit for unknown reasons. Because the sample size was small in this non‐American Indian study, as well as clinical information about exposures was limited, we sought to replicate the mesopontine findings in the autopsy cases of the AAIMS. Indeed, the AAIMS provided the extraordinary opportunity to study the role of prenatal exposures to cigarette smoke and alcohol due to the unprecedented amount of epidemiologic information available to link to brainstem development and SIDS pathology.
In this study, we tested the hypothesis that 3H‐nicotine binding to nicotinic receptors is significantly different (up‐regulated) in mesopontine nuclei between controls exposed to prenatal cigarette smoke compared with unexposed controls, but that this presumably normative response does not occur in exposed SIDS cases, and thus there is no difference in binding between exposed SIDS vs. unexposed SIDS. We were also interested in analyzing the effect of maternal drinking upon 3H‐nicotinic receptor binding in the brainstem in the SIDS cases because of the strong association between maternal drinking with SIDS risk in the AAIMS, in combination with the known adverse effects of alcohol upon nicotinic binding in experimental models (13, 17). We tested the hypothesis that 3H‐nicotine binding is altered in SIDS cases exposed to alcohol during gestation.
MATERIALS AND METHODS
Clinical information.
The AAIMS involved the analysis of 72 infant deaths, 56 (78%) of whom were autopsied. Thirty‐three brains (59% of the autopsied cases) were available for detailed anatomic and neurochemical analysis. SIDS cases were classified according to the National Institute of Child Health and Human Development (NICHD) 1991 definition (49). Control cases were infants that died suddenly but in whom a complete autopsy established an anatomic cause of death. Under the auspice of the AAIMS, information for all SIDS and autopsied control cases was available from maternal interviews, standardized autopsies, death scene investigations and medical chart reviews (24, 27). In the present study, the same SIDS and control cases were examined as in the 5‐HT receptor binding study (4), with the use of adjacent sections for receptor binding to the different radioligands.
3H‐nicotine receptor binding and generation of brainstem autoradiograms.
Horizontally sectioned blocks of unfixed, hemisected brainstem tissues were frozen, and serially sectioned at 20 µm on a Leitz cryostat and mounted on glass slides. The frozen, unfixed sections were prei‐ncubated with 3.5 nM D,L‐3H‐nicotine (79.5 Ci/mmol, PerkinElmer, Waltham, MA, USA) in 50 nM Tris‐HCL buffer, pH 7.4 and 8 mM CaCl2 for 20 minutes at 25°C (33). Nonspecific binding was determined in a subset of adjacent sections which was incubated in 3.5 nM 3H‐nicotine and 10 µM L‐nicotine bitartrate. To remove unbound ligand, the sections were washed in a series of changes of buffer (1 minute each) followed by 10 s in distilled water. They were dried under a stream of anhydrous air and then exposed to 3H‐sensitive film (LKB Ultrofilm‐3H, Amersham, Piscataway, NJ, USA) for 12 weeks. Films were then developed in a Kodak D‐19 developer (5 minutes), fixed in a Kodak Rapid fixer (5 minutes), washed in running water and hung to dry. A set of 3H standards (Amersham) was included in each X‐ray cassette to permit the conversion of the optical densities of silver grains in autoradiograms to specific activity of tissue‐bound ligand in femtomole per milligram (fmol/mg) of tissue.
Analysis of brainstem autoradiographs.
Receptor binding density (expressed as the specific activity of tissue‐bound ligand) was analyzed in 16 brainstem nuclei of each brainstem specimen (all nuclei were not available in all cases). For each specimen, the selected nuclei were analyzed at eight standardized brainstem levels (two autoradiograms/each nucleus) (26), and their receptors densities were combined and expressed as a mean ± standard error of the mean (SEM). To ascertain tissue boundaries of brainstem nuclei, tissue sections which generated the 3H‐nicotine autoradiographs were stained with hematoxylin‐and‐eosin and compared with the autoradiograph. The brainstem nuclei were determined with reference to the human brainstem atlas of Olszewski and Baxter (36), with the exception of the raphé nuclei, which were classified according to the atlas of Paxinos and Huang (39). The levels and nuclei at which analysis was performed are presented with their atlas number (36) in parentheses: (i) caudal medulla, level of the gracilis (Plate VIII), for analysis of the arcuate nucleus (ARC); (ii) caudal medulla, level of area postrema (Plate X), for analysis of the ARC; (iii) mid‐medulla, level of nucleus of Roller (Plate XII), for analysis of the nucleus of the solitary tract (NTS), hypoglossal nucleus (HG), nucleus centralis medullae oblongata (CEN), principal inferior olive (PO), dorsal accessory olive (DAO), medial accessory olive (MAO), and ARC; (iv) pontomedullary junction (Plate XVIII), for analysis of the ARC; (v) rostral pons, level of nucleus PBL (Plate XXVIII), for analysis of LC, nucleus pontis oralis (PoO), griseum pontis (GRPO), and PBL; (vi) caudal midbrain, level of the decussation of the superior cerebellar peduncle (Plate XXXII), for measurements of interpeduncular nucleus (IPN), nucleus cuneiformis (CUN), raphé dorsalis (RD), inferior colliculus (ICOL), and periaqueductal gray (PAG); (vii) rostral midbrain (Plate XXXVIII) for measurement of the red nucleus (RN).
An MCID imaging system (Interfocus Imaging, Linton, Cambridge, UK) was used to perform quantitative densitometry of autoradiographs. Optical densities were converted to specific activities of tissue bound ligand in fmol/mg tissue with 3H‐standards. Receptor binding density was determined in each brainstem nucleus by digitizing the boundaries of the nucleus upon the color image of the autoradiogram displayed on the computer monitor. There was no detectable nonspecific binding in the tissue autoradiographs. Specific activity data are presented as computer‐generated mosaics with a linear, 15‐step color scale from 0 to 67 fmol/mg tissue. All neurochemical analysis was performed blinded, without knowledge of the diagnosis or age of the case.
Statistical analysis.
For the clinical and autopsy database, group characteristics were compared using Student’s t‐test or the Wilcoxon rank sum test for continuous variables, and Fisher’s exact test for categorical variables. Analysis of covariance (ANCOVA) was used to compare mean nicotine binding by case diagnosis (SIDS vs. control) adjusted for postconceptional age (PCA). ANCOVA was also used to compare cases with a history of maternal smoking/drinking during pregnancy to those without, adjusted for PCA. This analysis was preformed separately for SIDS and control cases, as well as in the combined population. Regression analysis, controlling for PCA, was used to test the effect on nicotine binding of the number of cigarettes/alcoholic drinks during pregnancy in the combined population of SIDS and control cases.
RESULTS
Clinicopathologic information.
Between 1992 and 1996, the period of case accrual in the AAIMS, there were 27 SIDS deaths and six autopsy control (non‐SIDS) cases (total, n = 33). As previously reported, the causes of death in the autopsy control population were: acute respiratory infection (n = 2), congenital heart disease presenting as sudden death (n = 2), meningococcal sepsis (n = 1), and chronic encephalopathy consistent with perinatal hypoxia‐ischemia (n = 1) (27). Detailed clinical and sociodemographic data for the SIDS and autopsy controls in the AAIMS has been published previously (24, 27). Selected demographic data for the dataset in which cases were available for 3H‐nicotine receptor binding in the brainstem are summarized in Table 1, related to the 27 SIDS and six autopsy controls cases reported in previous brainstem AAIMS studies (24, 27). There was no statistically significant difference between mean PCA in SIDS (54.6 ± 13.4 weeks, range 39–90 weeks) and controls (53.8 ± 19.1 weeks, range 39–91 weeks). There was, however, a significant difference in birth weight, with the autopsy controls being on average low (2309 ± 790 g) compared with SIDS cases [3277 ± 405 g (P = 0.03)] (Table 1). In addition, the control cases had significantly lower 1 and 5 minutes Apgar Scores compared with the SIDS cases (P = 0.03) (Table 1). There was no difference in sleep position between SIDS and controls, either “usual placement” or “usually found” after a sleep period (Table 1). The incidence of “usual placement” in the supine position was 40.0% in the SIDS group and 33.3% in the control group (Table 1). There was no statistically significant difference in the incidence of bed sharing, although the incidence was higher in the SIDS group (64.0%) compared with the control group (33.3%).
Table 1.
Selected demographic information in the sudden infant death syndrome (SIDS) and control groups.
| Variable | SIDS | Controls | P‐value | ||
|---|---|---|---|---|---|
| n | Mean ± SD or median or % | n | Mean ± SD or median or % | ||
| Male | 25 | 48.0% | 6 | 16.7% | 0.359 |
| Birth weight | 25 | 3277 ± 405 | 6 | 2309 ± 790 | 0.029 |
| Birth length | 25 | 48.6 ± 6.8 | 6 | 46.3 ± 6.8 | 0.461 |
| Gestational age | 22 | 39.0 ± 1.7 | 6 | 35.7 ± 3.8 | 0.087 |
| Gravidity | 25 | 3.0 | 6 | 4.5 | 0.293 |
| 1‐minute Apgar | 25 | 8.0 | 6 | 6.5 | 0.031 |
| 5‐minute Apgar | 25 | 9.0 | 6 | 8.5 | 0.005 |
| Smoking during pregnancy | 25 | 76.0% | 6 | 66.7% | 0.634 |
| Alcohol during pregnancy | 25 | 64.0% | 6 | 33.3% | 0.208 |
| Binge drinking during pregnancy | 25 | 60.0% | 6 | 16.7% | 0.083 |
| Bed sharing | 25 | 64.0% | 6 | 33.3% | 0.208 |
| Usual placement for sleep | 25 | 6 | 1.000 | ||
| Prone | 4 | 16.0% | 1 | 16.7% | |
| Lateral | 11 | 44.0% | 3 | 50.0% | |
| Supine | 10 | 40.0% | 2 | 33.3% | |
| Usually found after sleep period | 25 | 6 | 1.000 | ||
| Prone | 6 | 24.0% | 1 | 16.7% | |
| Lateral | 8 | 32.0% | 2 | 33.3% | |
| Supine | 11 | 44.0% | 3 | 50.0% | |
| Clothing + covers > 5 | 27 | 22.2% | 6 | 33.3% | 0.616 |
A history of maternal smoking and/or alcohol consumption during pregnancy was available in most cases (n = 25 SIDS and n = 6 control cases). Maternal cigarette smoking anytime during pregnancy occurred in 76.0% of SIDS cases compared with 66.7% of control cases, and was not significantly different between the groups (P = 0.634). The consumption of alcohol anytime during pregnancy occurred in 64.0% of SIDS cases and 33.3% of controls, and was not significantly different between the groups. However, 60.0% of SIDS cases and only 16.7% of control cases partook of binge drinking (defined as five or more drinks in one sitting) (Table 1), a marginally significant difference (P = 0.083). There was no difference in the postmortem interval between SIDS and control cases (24.7 vs. 21.6 h), and there was no effect of postmortem interval upon 3H‐nicotine receptor binding in the nuclei assessed (data not shown).
3H‐nicotine receptor binding analysis in the control cases.
The qualitative distribution of 3H‐nicotine binding in the brainstem nuclei of the control (baseline) infants of this American Indian population was essentially identical to that described by us in non‐American Indian control infants (25, 33). In the AAIMS control cases, high binding (>13 fmol/mg tissue) was observed in the PO (highest binding in the brainstem) and MAO in the medulla; in the LC and pontis oralis in the pons, and in the INP and RN in the midbrain (Table 2) (Figure 1). High binding was also observed in the substantia nigria in the midbrain though measurements were not made in this region. 3H‐nicotine binding was visually undetectable in the raphé obscurus and arcuate nucleus in both SIDS and controls; therefore, quantitative measurements were not preformed in either group in these nuclei. We found a significant effect of PCA on 3H‐nicotine binding in all brainstem nuclei analyzed, with a reduction in binding with increasing age (Table 2) (Figure 2). The temporal profiles, however, varied: the slopes ranged from a decrease of 0.12 fmol/mg tissue per week in the NTS to a decrease of 0.35 fmol/mg tissue per week in the RN.
Table 2.
3H‐nicotine binding to nicotine receptors (fmol/mg tissue) in selected brainstem nuclei in SIDS and control groups, adjusted for the effect of postconceptional age (PCA). Abbreviations: NTS = nucleus of the solitary tract; CEN = nucleus centralis medullae oblongata; HG = hypoglossal nucleus; PO = principal inferior olive; DAO = dorsal accessory olive; MAO = medial accessory olive; PBL = parabrachialis lateralis; PoO = nucleus pontis oralis; LC = locus coeruleus; GRPO = griseum pontis; IPN = interpeduncular nucleus; PAG = periaqueductal gray; CUN = nucleus cuneiformis; ICOL = inferior colliculus; RD = raphe dorsalis; RN = red nucleus.
| Site | Age‐adjusted mean ± SE (n) | Diagnosis | Slope | P‐value | |
|---|---|---|---|---|---|
| SIDS | Controls | P‐value | |||
| NTS | 7.0 ± 0.6 (22) | 5.7 ± 1.1 (6) | 0.313 | −0.12 | 0.003 |
| CEN | 9.2 ± 0.8 (23) | 8.4 ± 1.5 (6) | 0.684 | −0.20 | <0.001 |
| HG | 9.0 ± 0.8 (20) | 8.1 ± 1.4 (6) | 0.604 | −0.18 | 0.001 |
| PO | 21.6 ± 1.1 (24) | 20.3 ± 2.1 (6) | 0.593 | −0.26 | <0.001 |
| DAO | 11.8 ± 0.7 (22) | 11.9 ± 1.4 (6) | 0.956 | −0.19 | <0.001 |
| MAO | 14.8 ± 1.0 (22) | 14.5 ± 1.9 (6) | 0.885 | −0.23 | 0.001 |
| PBL | 12.3 ± 1.0 (21) | 11.0 ± 1.9 (6) | 0.551 | −0.24 | 0.001 |
| PoO | 17.1 ± 1.2 (21) | 13.7 ± 2.3 (6) | 0.195 | −0.28 | 0.001 |
| LC | 16.4 ± 1.3 (21) | 13.0 ± 2.3 (6) | 0.212 | −0.27 | 0.002 |
| GRPO | 9.8 ± 0.8 (21) | 8.2 ± 1.5 (6) | 0.367 | −0.14 | 0.009 |
| IPN | 20.1 ± 1.3 (20) | 15.9 ± 2.4 (6) | 0.144 | −0.25 | 0.006 |
| PAG | 13.8 ± 1.2 (18) | 12.8 ± 2.1 (6) | 0.678 | −0.31 | 0.001 |
| CUN | 15.3 ± 1.2 (19) | 12.5 ± 2.2 (6) | 0.274 | −0.29 | 0.001 |
| ICOL | 10.2 ± 0.9 (18) | 8.5 ± 1.6 (5) | 0.378 | −0.21 | 0.002 |
| RD | 15.5 ± 1.2 (17) | 12.6 ± 2.0 (6) | 0.228 | −0.24 | 0.003 |
| RN | 17.5 ± 1.6 (19) | 14.5 ± 2.9 (6) | 0.376 | −0.35 | 0.002 |
Values are expressed as means ± SEM adjusted for PCA. No significant differences in 3H‐nicotinic receptor binding were found between the SIDS and control groups. P < 0.05, significant for either (i) diagnosis of SIDS vs. controls or (ii) the effects of PCA.
Figure 1.
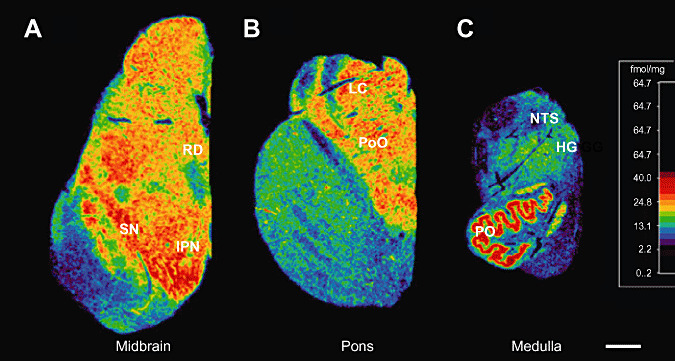
Computer‐generated specific activity mosaics of 3H‐nicotinic binding in the brainstem at the levels of (A) caudal midbrain; (B) mid pons; (C) mid medulla. Tritiated nicotine binding is highest in the principle inferior olive (PO), nucleus pontis oralis (PoO), locus coeruleus (LC), interpeduncular nucleus (IPN) and raphé dorsalis (RD). High binding was also observed in the substantia nigria (SN) though measurements were not made in this region. Brainstem levels standardized to the same scale. Scale bar: 0.4 cm.
Figure 2.

Developmental profiles of 3H‐nicointe binding in the sudden infant death syndrome (SIDS, closed circles) and control infants (open circle). When SIDS and control data are combined, there is a significant decrease in 3H‐binding with increasing postconceptional age in all nuclei sampled. Illustrated are (A) the raphé dorsalis (P = 0.003) and (B) the locus coeruleus (P = 0.002).
3H‐nicotine receptor binding in SIDS vs. control cases without stratification by history of prenatal exposure to smoking or alcohol.
There were no significant differences in 3H‐nicotine binding in any of the nuclei sampled in SIDS compared with control cases, adjusted for PCA and without stratification by history of adverse prenatal exposures (Table 2). In addition, the relative distribution and developmental profile of 3H‐nicotine receptor binding among the brainstem nuclei was essentially the same as in our previous studies of 3H‐nicotine binding in SIDS and control cases in non‐American Indian populations (25, 33). In both populations, there was a significant effect of age on 3H‐nicotine binding, with decreasing binding with increasing age in both the SIDS and control groups (Table 2) (Figure 2).
3H‐nicotine receptor binding in SIDS and control cases with stratification by history of prenatal exposure to smoking.
We next analyzed 3H‐nicotinic receptor binding in the selected brainstem nuclei in SIDS and control groups stratified by history of maternal smoking status in analysis of covariance models, controlling for PCA. Statistically significant differences were found in the control group in three of the 16 nuclei analyzed: the LC (P = 0.039), PAG (P = 0.005) and RD (P = 0.011), with significantly reduced binding in the control group exposed to maternal smoke in utero (n = 4) compared with the control group without exposure (n = 2) (Figure 3). In contrast, there was no significant difference in binding between SIDS infants exposed to (n = 13) compared with SIDS infants without exposure (n = 4) in these three or any other nuclei analyzed (Figure 3). There were also no significant effects of the average number of cigarettes smoked per day during pregnancy on 3H‐nicotinic receptor binding in SIDS or control cases (Table 3).
Figure 3.
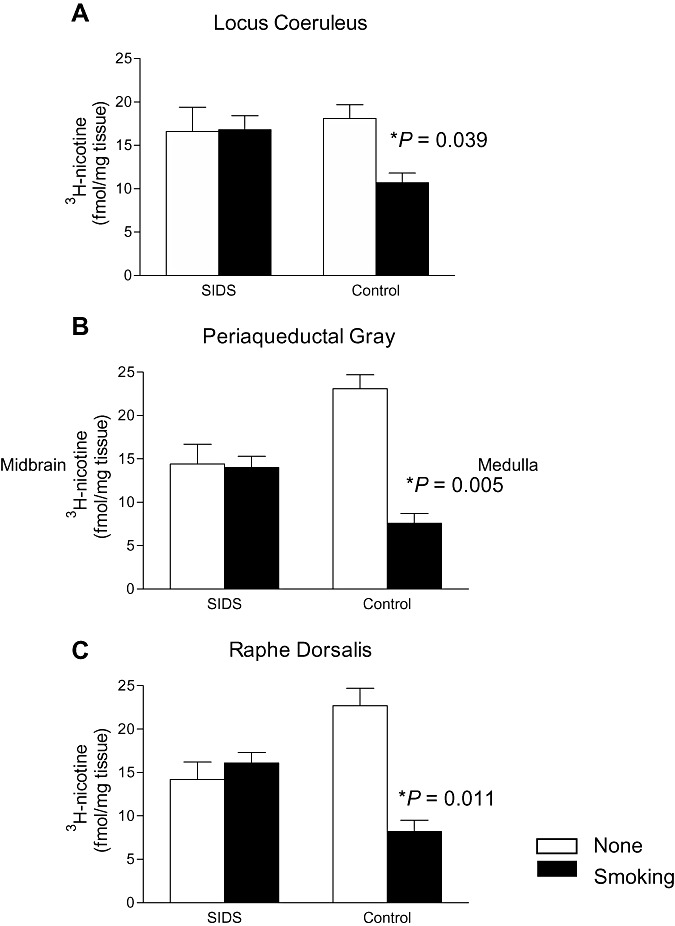
Tritiated‐nicotinic receptor binding in the brainstem nuclei in the sudden infant death syndrome (SIDS) and control groups stratified by history of maternal smoking status and controlling for postconceptional age (mean ± SEM). In the (A) locus coeruleus (P = 0.039), (B) periaqueductal gray (P = 0.005) and (C) raphé dorsalis (P = 0.011), there is a significant reduction in 3H‐nicotinic binding in control infants whose mothers smoked during pregnancy (n = 4) compared with control infants not exposed to maternal smoke (n = 2). Significant differences in binding are not found between SIDS infants exposed to cigarette smoke and SIDS infants not exposed.
Table 3.
Regression of 3H‐nicotinic binding in selected brainstem nuclei in SIDS and control cases combined, by average number of cigarettes (per day) and drinks (per month) during pregnancy, adjusting for postconceptional age.
| n | Average no. of cigarettes per day during pregnancy | Average no. of drinks per month during pregnancy | |||
|---|---|---|---|---|---|
| Slope (SE) | P‐value | Slope (SE) | P‐value | ||
| NTS | 28 | −0.06 (0.11) | 0.608 | −0.09 (0.04) | 0.021 |
| CEN | 28 | −0.08 (0.15) | 0.586 | −0.12 (0.05) | 0.028 |
| HG | 26 | −0.004 (0.14) | 0.976 | −0.11 (0.05) | 0.023 |
| PO | 29 | −0.07 (0.20) | 0.748 | −0.13 (0.07) | 0.076 |
| DO | 27 | 0.08 (0.13) | 0.576 | −0.11 (0.05) | 0.027 |
| MO | 28 | −0.07 (0.19) | 0.707 | −0.15 (0.06) | 0.020 |
| PBL | 26 | −0.13 (0.19) | 0.492 | −0.12 (0.07) | 0.071 |
| POO | 26 | −0.15 (0.23) | 0.524 | −0.10 (0.08) | 0.223 |
| LC | 26 | −0.28 (0.23) | 0.233 | −0.12 (0.08) | 0.176 |
| GRPO | 26 | −0.16 (0.15) | 0.304 | −0.09 (0.05) | 0.126 |
| IPN | 25 | −0.25 (0.25) | 0.317 | −0.06 (0.09) | 0.507 |
| PAG | 23 | −0.16 (0.21) | 0.465 | −0.05 (0.08) | 0.537 |
| CUN | 24 | −0.04 (0.23) | 0.870 | −0.06 (0.08) | 0.439 |
| ICOL | 22 | 0.04 (0.16) | 0.832 | −0.08 (0.06) | 0.149 |
| RD | 22 | −0.15 (0.21) | 0.491 | −0.05 (0.08) | 0.562 |
| RN | 24 | −0.20 (0.29) | 0.501 | −0.12 (0.10) | 0.254 |
Values are expressed as a correlation of 3H‐nicotine binding and (i) the number of cigarettes per day or (ii) the number of drinks per day, adjusting for PCA. In five nuclei there was a significant decrease in 3H‐nicotine binding with increasing numbers of drinks. Data are expressed as means ± SEM. For abbreviations see Table 2.
3H‐nicotine receptor binding in SIDS and control cases with stratification by history of prenatal exposure to alcohol.
We next analyzed 3H‐nicotinic receptor binding in the selected brainstem nuclei in SIDS and control groups stratified by history of any maternal drinking during pregnancy in analysis of covariance models, controlling for PCA. There was a general trend for reduced nicotinic binding in cases with exposure to maternal drinking, but there were no significant effects in any of the brainstem nuclei sampled, in either the SIDS or control groups (data not shown). The number of drinks per month was calculated from the demographic data as number of days/month drank times number of drinks per day. This number was then averaged over the three trimesters. The number of drinks per month was calculated from the demographic data as number of days/month drank times number of drinks per day. In all brainstem nuclei in the SIDS and control groups combined, 3H‐nicotinic receptor binding decreased with increased average number of drinks per month, with statistical significance achieved in five nuclei (Table 3). These nuclei were the: NTS (P = 0.021), nucleus centralis (P = 0.028), HG (P = 0.023), DAO (P = 0.027) and MAO (P = 0.020) (Figure 4). Models were fit that controlled for the number of cigarettes and the interaction between the number of drinks and number of cigarettes to determine if the effect of drinking was compounded by the effect of smoking. There were no significant interactions, although the slope of the interactions was mostly negative, indicating a trend in the expected direction (data not shown).
Figure 4.
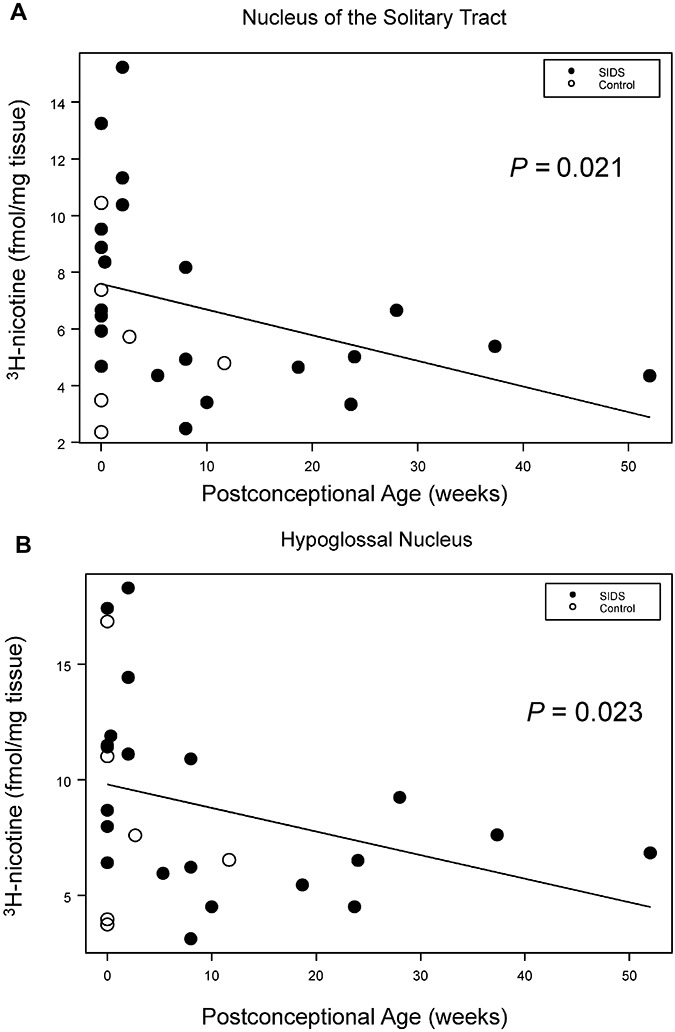
Regression analysis of 3H‐nicotine binding by the average number of alcoholic beverages during pregnancy, illustrated in two of five nuclei with significant changes. (A) Nucleus of the solitary tract (P = 0.021), (B) Hypoglossal nucleus (P = 0.023). sudden infant death syndrome (SIDS, closed circles) and controls (open circles).
DISCUSSION
Maternal cigarette smoking and alcohol consumption during pregnancy are major risk factors for SIDS (24, 31). The high prevalence of these two behaviors in the American Indians of the Northern Plains suggests that they contribute in significant ways to the high SIDS rate among this population. The fundamental mechanism(s) of prenatal exposures to alcohol and tobacco smoke upon the risk for SIDS is unknown, as is the possibility of synergistic effects of cigarette smoke and alcohol upon human brain development. Previously, we found serotonergic (5‐HT) receptor binding abnormalities in the medulla in SIDS infants compared with controls in this same population (27). In the 5‐HT study in this same dataset, 5‐HT receptor binding at the ventral medullary surface (arcuate nucleus) was significantly reduced in infants whose mothers smoked during pregnancy compared with those whose mothers did not (P = 0.01), and was marginally reduced in infants whose mothers drank compared with those whose mother did not (P = 0.07) whether or not they died of SIDS or known causes (controls). These data suggest that alcohol and cigarette smoke each suppresses 5‐HT receptor binding in critical areas related to cardiorespiratory control. In the present study, we postulated in the same high‐risk dataset of American Indian infants that alcohol and cigarette smoke adversely affect the regulation of nicotinic receptor binding in critical brainstem nuclei in SIDS and/or control infants, and that this putative altered nicotinic binding, in addition to altered 5‐HT receptor binding, contributes to the pathogenesis of sudden death. The underlying premises of this study were that endogenous cholinergic nicotine receptors in the human brain bind with exogenous nicotine or interact with alcohol; and that prenatal nicotine exposure causes altered development of the 5‐HT, cholinergic, and other neurotransmitter systems in the brain (9, 29, 43, 44), at least in part via changes in nicotinic receptor binding. When stratified by exposure to maternal cigarette smoking at any time during pregnancy, we found in this study that SIDS infants exposed to prenatal cigarette smoke did not display the same significant reduction in 3H‐nicotine binding in the RD, PAG and LC that the control infants did upon prenatal exposure. We also found a significant correlation between increasing number of drinks and decreasing nicotinic receptor binding. We begin the discussion with consideration of the study’s limitations.
Limitations of the study.
A major limitation of this study is the small sample size, particularly of autopsy controls. Based upon the robustness of the differences (P < 0.05) between study groups in this study, even with small sample sizes, and the consistency of the nicotine findings in the mesopons with those previously reported by us in a non‐American Indians, however, we believe the findings are valid and warrant further investigation. A second limitation is that the autopsy control cases are likely not representative of living control infants who do not die of SIDS. This limitation is inherent in all autopsy studies of SIDS due to the fact that “normal” infants rarely die. In this study, the controls on average were born prematurely (<37 gestational weeks), and had low birth weight (<2500 g); furthermore, the control group had a high incidence of prenatal exposures to cigarette smoke and alcohol. An additional limitation of the study is the self‐reporting of prenatal exposures by the mother without validation with biomarkers, for example, cotinine measurements in infant blood. In the AAIMS, the design was retrospective, and no measurements of prenatal exposures were available from mothers whose infants subsequently died of SIDS or other causes. A final limitation is that the radioligand selected for use, that is, 3H‐nicotine, does not bind optimally with all receptor subtypes (22), and thus, information about particularly subtypes maybe “masked”. Future analysis using radioligands with different binding selectivity and affinity is needed to exclude all nicotinic receptor deficits are needed.
The effect of maternal smoking upon 3H‐nicotinic binding in the infant brainstem.
The primary finding of this study is that nicotinic receptor binding is reduced in three mesopontine nuclei related to arousal and defense responses in control cases exposed to smoking during pregnancy compared with control cases without exposure, but is not reduced in SIDS cases exposed to smoking. Although definitive conclusions are not possible due to the small sample size of the study, this finding suggests that in response to smoking exposure, 3H‐nicotine binding is down‐regulated in control cases—the “appropriate” or “normal” response—but not in SIDS cases. These data raise the question of a genetic or environmentally acquired defect in the molecular regulation of nicotinic receptor number and/or affinity to nicotine exposure in SIDS. In the previous study reported by us of 3H‐nicotinic binding in a non‐American Indian population of SIDS and controls, we found an increase, as opposed to decrease, in binding in the control cases exposed to smoking, as well as no changes in the SIDS cases (33). The differences in direction of binding changes in the two control population to prenatal exposure to cigarette smoke, that is, increase or decrease in binding, suggests the possibility that the control populations in the two studies are not identical. This lack of consistency may reflect insufficient samples sizes. Yet, the differences may also relate to different perinatal factors in the disparate populations, for example, gestational age, birth weight, the amount and type of exposure to cigarette smoke, and the targeted subunit of the nicotinic receptor.
In both these studies, three mesopontine nuclei were affected: in the non‐American Indian study, they were the LC (3, 5) and pontis oralis (51) (components of thalamic and extra‐thalamic arousal systems, respectively), and PBL (ascending visceral information); in the AAIMS study, the three affected nuclei were the LC (3, 5), PAG (defense‐related region) (12, 21) and RD (ascending 5‐HT arousal system) (41). Interestingly, the LC, which contains the source neurons of the noradrenergic system and has a key role in arousal (5), was affected in both study populations, implicating this nucleus in particular in interactions with maternal cigarette smoke and nicotine in the pathogenesis of arousal deficits associated with prenatal exposure. Experimentally, maternal smoking during pregnancy decreases in the offspring the frequency of spontaneous arousals (23), the arousal response to mild hypoxia (28); and arousals to auditory stimuli (8, 15). In addition, newborn lambs with prenatal nicotine exposure at levels similar to those in smoking humans experience delays in arousal to hypoxia (20). It is unclear if noradrenergic pathways are specifically involved in these arousal deficits upon exposure to prenatal nicotine, but our studies suggest that they are, at least in part, affected.
Based upon experimental studies (48), we expected postnatal nicotinic binding in the human infant brainstem to be up‐regulated in response to prenatal nicotine exposure. In adult postmortem brains of individuals who smoked, protein expression for the different receptor subtypes is also increased (46); in rat exposed brains, nAChR mRNA (16) and receptor binding (34, 35) are increased. With prenatal exposures, Chen et al, however, found a reduction in 125I‐epibatidine (α7 nAChR) binding in the postnatal rat brainstem, with long‐lasting changes in mRNA expression and reduction in neuronal number in the ventral tegmental area (9). These changes in nicotinic binding in prenatal models of exposure are associated with downstream adverse affects upon cholinergic development, and thus, prenatal exposures adversely affect neuronal cell number (10), neuronal cell maturation (40) and synaptogenesis (44). Consequently, the changes in nicotinic binding in response to prenatal exposure in the exposed controls in our two studies cannot be considered “protective”, but rather, reflect the “normal” response to exposure at the cellular and molecular level. The combined American Indian and non‐American Indian data suggest that the LC in particular is “normally” able to alter nicotinic receptor binding levels upon exposure to prenatal cigarette smoke in the control group, but that this capability is not present in exposed SIDS infants, thereby potentially exacerbating the toxic effects of cigarette smoke upon developing ascending arousal pathways—noradrenergic, serotonergic (via the RD), and extra‐thalamic (via the pontis oralis) systems. The reason that the subset of mesopontine nuclei in exposed controls undergo nicotinic binding changes in association with prenatal alcohol exposure and other brainstem nuclei do not is unknown; thus, the vulnerability of these mesopontine nuclei to exposure require further study. Nevertheless, this finding delineates, at least in part, the neuroanatomic substrate of arousal deficits in association with prenatal exposure to cigarette smoke.
The effect of maternal drinking upon 3H‐nicotinic binding in the infant brainstem.
The second finding in this study is that decreasing 3H‐nicotine binding correlates with increasing average number of alcoholic drinks per month in the SIDS and control groups combined, with statistical significance (P < 0.05) reached in five nuclei in the medulla, two of which (NTS and HG) are involved in cardiorespiratory control. Thus, prenatal exposures are not necessarily causative in SIDS, given that they are associated with the same changes in controls, but rather, they may augment the intrinsic abnormality in infants “vulnerable” to SIDS. In this regard, we found significantly reduced 5‐HT1A binding in the NTS and HG in SIDS cases (38) the same sites in which exposure to maternal drinking in pregnancy is associated with altered nicotinic binding in all controls. We speculate that the nicotinic change in the SIDS infant with the 5‐HT1A vulnerability in some way increases the risk for sudden death. The correlation between increasing alcohol intake and decreasing nicotinic binding in infants irrespective of the cause of death suggests that prenatal alcohol exposure interacts with nicotinic receptors to alter the function, as indicated by experimental studies (17, 45). The α7 subunit, for example, is inhibited upon alcohol exposure, and alcohol toxicity is enhanced in α7 knockout mice (11).
CONCLUSION
This study suggests that smoking and alcohol adversely affect 3H‐nicotinic binding in the infant brainstem irrespective of diagnosis. In addition, it suggests that American Indian infants in the Northern Plains that die of SIDS have an intrinsic or acquired defect in regulation of nicotinic receptor binding upon exposure to prenatal cigarette smoke that may exacerbate medullary 5‐HT abnormalities in cardiorespiratory control in the same dataset. The finding now in two independent datasets of changes in postnatal nicotinic binding in controls exposed to prenatal smoke but not in exposed SIDS cases in three mesopontine nuclei is intriguing, despite the small sample sizes in both studies, the difference in the direction (increase/decrease) of the nicotinic binding change, and the affected nuclei (with only the LC involved in both datasets). Nevertheless, the data of these two studies taken together are hypothesis‐generating for experimental studies, as well as for larger human datasets in future research initiatives.
ACKNOWLEDGMENTS
We would like to extend our appreciation to the many families who participated in this study. We appreciate the assistance provided by the Aberdeen Area Tribal Chairman’s Health Board, the 10 participating Tribal Communities, the Office of Epidemiology, Aberdeen Area Indian Health Service, and the Prenatal Infant Mortality Review Committee.
The National Institutes for Child Health and Human Development, US Centers for Disease Controls and Prevention and Indian Health Service funded the Aberdeen Area Indian Health Service (AAIMS) through interagency agreements. The neuropathologic analysis in the AAIMS was supported by subcontract NICHD‐CRMC‐92‐05 (HCK), the CJ Martin Overseas Fellowship (NHMRC) and the First Candle/SIDS Alliance (JRD).
Disclaimer: The content of this publication does not necessarily reflect the views or policies of the U.S Department of Health and Human Services or the Indian Health Service, nor does the mention of trade names, commercial products, or organizations imply endorsement by the USA
REFERENCES
- 1. Indian Health Service (1998) Regional Differences in Indian Health 1997. US Department of Health and Human Services, Public Health Service, Indian Health Service: Rockville, MD. [Google Scholar]
- 2. Indian Health Service (2000) Regional Differences in Indian Health 1998–1999. Indian Health Service: Rockville, MD. [Google Scholar]
- 3. Aston‐Jones G, Rajkowski J, Cohen J (1999) Role of locus coeruleus in attention and behavioral flexibility. Biol Psychiatry 46:1309–1320. [DOI] [PubMed] [Google Scholar]
- 4. Benwell ME, Balfour DJ, Anderson JM (1988) Evidence that tobacco smoking increases the density of (‐)‐[3H]nicotine binding sites in human brain. J Neurochem 50:1243–1247. [DOI] [PubMed] [Google Scholar]
- 5. Bouret S, Sara SJ (2005) Network reset: a simplified overarching theory of locus coeruleus noradrenaline function. Trends Neurosci 28:574–582. [DOI] [PubMed] [Google Scholar]
- 6. Bull LB, Kvigne VL, Leonardson GR, Lacina L, Welty TK (1999) Validation of a self‐administered questionnaire to screen for prenatal alcohol use in Northern Plains Indian women. Am J Prev Med 16:240–243. [PubMed] [Google Scholar]
- 7. Centers of Disease Control and Prevention (2002) Alcohol use among women of childbearing age‐United States. Morb Mortal Wkly Rep 51:273–276. [PubMed] [Google Scholar]
- 8. Chang AB, Wilson SJ, Masters IB, Yuill M, Williams J, Williams G, Hubbard M (2003) Altered arousal response in infants exposed to cigarette smoke. Arch Dis Child 88:30–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen H, Parker SL, Matta SG, Sharp BM (2005) Gestational nicotine exposure reduces nicotinic cholinergic receptor (nAChR) expression in dopaminergic brain regions of adolescent rats. Eur J Neurosci 22:380–388. [DOI] [PubMed] [Google Scholar]
- 10. Chen WJ, King KA, Lee RE, Sedtal CS, Smith AM (2006) Effects of nicotine exposure during prenatal or perinatal period on cell numbers in adult rat hippocampus and cerebellum: a stereology study. Life Sci 79:2221–2227. [DOI] [PubMed] [Google Scholar]
- 11. De Fiebre NC, De Fiebre CM (2005) alpha7 Nicotinic acetylcholine receptor knockout selectively enhances ethanol‐, but not beta‐amyloid‐induced neurotoxicity. Neurosci Lett 373:42–47. [DOI] [PubMed] [Google Scholar]
- 12. De Oca BM, DeCola JP, Maren S, Fanselow MS (1998) Distinct regions of the periaqueductal gray are involved in the acquisition and expression of defensive responses. J Neurosci 18:3426–3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dohrman DP, Reiter CK (2003) Ethanol modulates nicotine‐induced upregulation of nAChRs. Brain Res 975:90–98. [DOI] [PubMed] [Google Scholar]
- 14. Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ (1992) A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha 4 and beta 2 subunits and is up‐regulated by chronic nicotine treatment. Mol Pharmacol 41:31–37. [PubMed] [Google Scholar]
- 15. Franco P, Groswasser J, Hassid S, Lanquart JP, Scaillet S, Kahn A (1999) Prenatal exposure to cigarette smoking is associated with a decrease in arousal in infants. J Pediatr 135:34–38. [DOI] [PubMed] [Google Scholar]
- 16. Frank MG, Srere H, Ledezma C, O’Hara B, Heller HC (2001) Prenatal nicotine alters vigilance states and AchR gene expression in the neonatal rat: implications for SIDS. Am J Physiol Regul Integr Comp Physiol 280:R1134–R1140. [DOI] [PubMed] [Google Scholar]
- 17. Gorbounova O, Svensson A‐L, Jonsson P, Mousavi M, Miao H, Hellstrom‐Lindahl E, Nordberg A (1998) Chronic ethanol treatment decreases [3H]epibatidine and [3H]nicotine binding and differentially regulates mRNA levels of nicotinic acetylcholine receptor subunits expressed in M10 and SH‐SY5Y neuroblastoma cells. J Neurochem 70:1134–1142. [DOI] [PubMed] [Google Scholar]
- 18. Gotti C, Clementi F (2004) Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol 74:363–396. [DOI] [PubMed] [Google Scholar]
- 19. Gunn AJ, Gunn TR, Mitchell EA (2000) Clinical review article: is changing the sleep environment enough? Current recommendations for SIDS. Sleep Med Rev 4:453–469. [DOI] [PubMed] [Google Scholar]
- 20. Hafstrom O, Milerad J, Sundell HW (2002) Prenatal nicotine exposure blunts the cardiorespiratory response to hypoxia in lambs. Am J Respir Crit Care Med 166:1544–1549. [DOI] [PubMed] [Google Scholar]
- 21. Hayward LF, Castellanos M, Davenport PW (2004) Parabrachial neurons mediate dorsal periaqueductal gray evoked respiratory responses in the rat. J Appl Physiol 96:1146–1154. [DOI] [PubMed] [Google Scholar]
- 22. Hellstrom‐Lindahl E, Court JA (2000) Nicotinic acetylcholine receptors during prenatal development and brain pathology in human aging. Behav Brain Res 113:159–168. [DOI] [PubMed] [Google Scholar]
- 23. Horne RS, Ferens D, Watts AM, Vitkovic J, Lacey B, Andrew S, Cranage SM, Chau B, Greaves R, Adamson TM (2002) Effects of maternal tobacco smoking, sleeping position, and sleep state on arousal in healthy term infants. Arch Dis Child Fetal Neonatal Ed 87:F100–F105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iyasu S, Randall LL, Welty TK, Hsia J, Kinney HC, Mandell F, McClain M, Randall B, Habbe D, Wilson H, Willinger M (2002) Risk factors for sudden infant death syndrome among northern plains, Indians. JAMA 288:2717–2723. [DOI] [PubMed] [Google Scholar]
- 25. Kinney HC, O’Donnell TJ, Kriger P, White WF (1993) Early developmental changes in [3H]nicotine binding in the human brainstem. Neuroscience 55:1127–1138. [DOI] [PubMed] [Google Scholar]
- 26. Kinney HC, Filiano JJ, White WF (2001) Medullary serotonergic network deficiency in the sudden infant death syndrome: review of a 15‐year study of a single dataset. J Neuropathol Exp Neurol 60:228–247. [DOI] [PubMed] [Google Scholar]
- 27. Kinney HC, Randall LL, Sleeper LA, Willinger M, Belliveau RA, Zec N, Rava LA, Dominici L, Iyasu S, Randall B, Habbe D, Wilson H, Mandell F, McClain M, Welty TK (2003) Serotonergic brainstem abnormalities in Northern Plains Indians with the sudden infant death syndrome. J Neuropathol Exp Neurol 62:1178–1191. [DOI] [PubMed] [Google Scholar]
- 28. Lewis KW, Bosque EM (1995) Deficient hypoxia awakening response in infants of smoking mothers: possible relationship to sudden infant death syndrome. J Pediatr 127:691–699. [DOI] [PubMed] [Google Scholar]
- 29. Luo Z, Costy‐Bennett S, Fregosi RF (2004) Prenatal nicotine exposure increases the strength of GABA(A) receptor‐mediated inhibition of respiratory rhythm in neonatal rats. J Physiol 561:387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mathews TJ, MacDorman MF (2006) Infant mortality statistics from the 2003 period linked birth/infant death data set. Natl Vital Stat Rep 54:1–29. [PubMed] [Google Scholar]
- 31. Matturri L, Ottaviani G, Lavezzi AM (2006) Maternal smoking and sudden infant death syndrome: epidemiological study related to pathology. Virchows Arch 449:697–706. [DOI] [PubMed] [Google Scholar]
- 32. Mihailescu S, Palomero‐Rivero M, Meade‐Huerta P, Maza‐Flores A, Drucker‐Colin R (1998) Effects of nicotine and mecamylamine on rat dorsal raphe neurons. Eur J Pharmacol 360:31–36. [DOI] [PubMed] [Google Scholar]
- 33. Nachmanoff DB, Panigrahy A, Filiano JJ, Mandell F, Sleeper LA, Valdes‐Dapena M, Krous HF, White WF, Kinney HC (1998) Brainstem 3H‐nicotine receptor binding in the sudden infant death syndrome. J Neuropathol Exp Neurol 57:1018–1025. [DOI] [PubMed] [Google Scholar]
- 34. Narayanan U, Birru S, Vaglenova J, Breese CR (2002) Nicotinic receptor expression following nicotine exposure via maternal milk. Neuroreport 13:961–963. [DOI] [PubMed] [Google Scholar]
- 35. Nguyen HN, Rasmussen BA, Perry DC (2004) Binding and functional activity of nicotinic cholinergic receptors in selected rat brain regions are increased following long‐term but not short‐term nicotine treatment. J Neurochem 90:40–49. [DOI] [PubMed] [Google Scholar]
- 36. Olszewski J, Baxter D (1982) Atlas of the Human Brainstem. Karger: Basel. [Google Scholar]
- 37. Papke RL (1993) The kinetic properties of neuronal nicotinic receptors: genetic basis of functional diversity. Prog Neurobiol 41:509–531. [DOI] [PubMed] [Google Scholar]
- 38. Paterson DS, Trachtenberg FL, Thompson EG, Belliveau RA, Beggs AH, Darnall R, Chadwick AE, Krous HF, Kinney HC (2006) Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. JAMA 296:2124–2132. [DOI] [PubMed] [Google Scholar]
- 39. Paxinos G, Huang X‐F (1995) Atlas of the Human Brainstem. Academic Press, Inc.: San Diego. [Google Scholar]
- 40. Roy TS, Seidler FJ, Slotkin TA (2002) Prenatal nicotine exposure evokes alterations of cell structure in hippocampus and somatosensory cortex. J Pharmacol Exp Ther 300:124–133. [DOI] [PubMed] [Google Scholar]
- 41. Schenberg LC, Lovick TA (1995) Attenuation of the midbrain‐evoked defense reaction by selective stimulation of medullary raphe neurons in rats. Am J Physiol 269:R1378–R1389. [DOI] [PubMed] [Google Scholar]
- 42. Slotkin TA, Pinkerton KE, Auman JT, Qiao D, Seidler FJ (2002) Perinatal exposure to environmental tobacco smoke upregulates nicotinic cholinergic receptors in monkey brain. Brain Res Dev Brain Res 133:175–179. [DOI] [PubMed] [Google Scholar]
- 43. Slotkin TA, Southard MC, Adam SJ, Cousins MM, Seidler FJ (2004) Alpha7 nicotinic acetylcholine receptors targeted by cholinergic developmental neurotoxicants: nicotine and chlorpyrifos. Brain Res Bull 64:227–235. [DOI] [PubMed] [Google Scholar]
- 44. Slotkin TA, Tate CA, Cousins MM, Seidler FJ (2006) Prenatal nicotine exposure alters the responses to subsequent nicotine administration and withdrawal in adolescence: serotonin receptors and cell signaling. Neuropsychopharmacology 31:2462–2475. [DOI] [PubMed] [Google Scholar]
- 45. Soderpalm B, Ericson M, Olausson P, Blomqvist O, Engel JA (2000) Nicotinic mechanisms involved in the dopamine activating and reinforcing properties of ethanol. Behav Brain Res 113:85–96. [DOI] [PubMed] [Google Scholar]
- 46. Teaktong T, Graham AJ, Johnson M, Court JA, Perry EK (2003) Selective changes in nicotinic acetylcholine receptor subtypes related to tobacco smoking: an immunohistochemical study. Neuropathol Appl Neurobiol 30:243–254. [DOI] [PubMed] [Google Scholar]
- 47. Torrao AS, Britto LR (2002) Neurotransmitter regulation of neural development: acetylcholine and nicotinic receptors. An Acad Bras Cienc 74:453–461. [DOI] [PubMed] [Google Scholar]
- 48. Wang H, Sun X (2005) Desensitized nicotinic receptors in brain. Brain Res Brain Res Rev 48:420–437. [DOI] [PubMed] [Google Scholar]
- 49. Willinger M, James LS, Catz C (1991) Defining the sudden infant death syndrome (SIDS): deliberations of an expert panel convened by the National Institute of Child Health and Human Development. Pediat Pathol 11:677–684. [DOI] [PubMed] [Google Scholar]
- 50. Wu J, Liu Q, Yu K, Hu J, Kuo YP, Segerberg M, St John PA, Lukas RJ (2006) Roles of nicotinic acetylcholine receptor beta subunits in function of human alpha4‐containing nicotinic receptors. J Physiol 576:103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xi MC, Morales FR, Chase MH (2004) Interactions between GABAergic and cholinergic processes in the nucleus pontis oralis: neuronal mechanisms controlling active (rapid eye movement) sleep and wakefulness. J Neurosci 24:10670–10678. [DOI] [PMC free article] [PubMed] [Google Scholar]