

Abstract
Amyloid‐β (Aβ) is the major component of the insoluble amyloid plaques that accumulate intracerebrally in patients with Alzheimer’s disease (AD). It has been suggested that MDR1‐P‐glycoprotein (ABCB1, P‐gp) plays a substantial role in the elimination of Aβ from the brain. In the present study, MDR1‐transfected LLC cells growing in a polarized cell layer were used to characterize the interaction of Aβ1‐40/1‐42 with P‐gp. In this system, P‐gp‐mediated transport can be followed by the efflux of the fluorescent dye rhodamine‐123, or of Aβ itself from the cells into the apical extracellular space. Aβ significantly decreased the apical efflux of rhodamine‐123, and the transcellular transport of Aβ1‐40 and Aβ1‐42 into the apical chamber could be demonstrated using both ELISA and fluorescence (FITC)‐labeled peptides. This transport was inhibited by a P‐gp modulator. Furthermore, ATP‐dependent, P‐gp‐mediated transport of the fluorescence‐labeled peptides could be demonstrated in isolated, inside‐out membrane vesicles. Our data support the concept that P‐gp is important for the clearance of Aβ from brain, and thus may represent a target protein for the prevention and/or treatment of neurodegenerative disorders such as AD.
INTRODUCTION
The accumulation of the amyloid‐β peptide (Aβ) is one of the characteristic pathological features of Alzheimer’s disease (AD) (4, 21). Cerebral amyloid angiopathy (CAA), which varies in severity among AD cases (25), involves the aggregation of Aβ in the walls of arteries, arterioles and, to a lesser extent, of capillaries and veins (18). Aβ is released by cells into the brain interstitial fluid, where it is eliminated by proteolytic degradation, passive bulk flow and active transport across the blood–brain barrier (BBB) (31, 32). As the latter appears to be a significant pathway for elimination, efforts have been made to identify the mechanisms by which Aβ is transported out of the brain. There is evidence that the low‐density lipoprotein receptor‐related protein (LRP1) is an Aβ‐efflux transporter at the BBB (5, 22). Another candidate is MDR1‐P‐glycoprotein (P‐gp; ABCB1), which is highly expressed at the luminal surface of brain capillary endothelial cells. P‐gp constitutes a major component of the BBB by limiting penetration of various exogenous agents (1, 8, 19). In in vitro experiments, Lam et al provided the first evidence that Aβ may be a substrate for P‐gp (12). On the basis of this hypothesis, we examined the relationship between P‐gp expression and the amount of Aβ deposition in the brains of non‐demented, elderly humans, and found an inverse correlation between vascular P‐gp and the quantity of Aβ‐positive plaques, suggesting that P‐gp might indeed play an important role in the pathogenesis of AD (26). In addition, we found an age‐dependent decrease of P‐gp expression in cases without CAA, and an upregulation of endothelial P‐gp in unaffected vessels of cases with CAA (27). In a series of experiments in genetically modified mice, Cirrito et al recently provided further evidence that the lack of P‐gp expression exacerbates Aβ deposition, and that the activity of this transporter can minimize the development of cerebral Aβ lesions (3).
In the present study, we used LLC cells, a porcine‐derived proximal renal tube epithelial cell line, transfected with human MDR1 (ABCB1) cDNA and maintained in a polarized cell monolayer. This model was used to directly characterize the transcellular transport of the Aβ peptide, and the role of P‐gp in this process. Furthermore, fluorescence‐labeled peptides and isolated, inside‐out membrane vesicles were used to demonstrate a direct, ATP‐dependent transport of these peptides by P‐gp. These in vitro findings indicate a critical function of P‐gp in the regulation of brain Aβ levels, and support the view that dysfunction of this transport protein can promote the emergence of Aβ‐linked proteopathies in humans.
MATERIALS AND METHODS
Chemicals. The 40‐ and 42‐amino acid forms of Aβ (Aβ1‐40 and Aβ1‐42) and fluorescein (FITC)‐conjugated Aβ1‐40/1‐42 were purchased from rPeptide (Athens, GA, USA). Stock solutions of Aβ peptides were prepared according to the manufacturer’s suggestions. FITC‐conjugated Aβ1‐40 and Aβ1‐42 were dissolved in 2 mM NaOH to a final concentration of 200 µM. All four peptide solutions were stored in aliquots at −20°C. The hAmyloid β1–40/42 brain ELISA was obtained from the Genetics Company (Zurich, Switzerland).
[14C]Inulin carboxylic acid (82 MBq/g) was obtained from Biotrend Chemicals (Köln, Germany). Verapamil and rhodamine‐123 (Rh123) were from Sigma (Deisenhofen, Germany). The Rh123 stock solution was prepared by dissolving in DMSO at a final concentration of 2 mM. Cyclosporine A was purchased from Alexis (Grünberg, Germany).
Antibodies. The monoclonal antibodies against human P‐gp, C219 and JSB‐1 were from Alexis (Grünberg, Germany). For immunohistochemistry and immunoblotting, antibodies against LRP were purchased from Calbiochem (Darmstadt, Germany). Alexa Fluor488‐labeled goat anti‐mouse IgG for immunofluorescence microscopy was obtained from Molecular Probes/MoBiTec (Göttingen, Germany).
Cell culture. LLC parental (LLC, clone LLC‐PK1) and stably transfected (LLC‐MDR1) cells were kindly provided by Dr P. Borst (The Netherlands Cancer Institute, Amsterdam), and were cultured in DMEM supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 5% L‐glutamine and 1% non‐essential amino acids at 37°C, 95% humidity and 5% CO2.
Preparation of membrane vesicles. Membrane vesicles from LLC and LLC‐MDR1 cells were prepared by hypotonic lysis and differential centrifugation as described previously (11). Membrane vesicles were frozen and stored in liquid nitrogen.
Immunoblot analysis. Membrane vesicles were diluted with sample buffer and incubated at 95°C for 5 min before separation on 7.5% polyacrylamide gels with 50 µg of protein loaded per lane. Immunoblotting was performed with a tank blotting system from BioRad (Munich, Germany) and enhanced chemiluminescence detection (Amersham Biosciences, Freiburg, Germany). P‐gp was detected using the monoclonal antibody C219 at a dilution of 1:500 in Tris‐buffered saline/Tween 20 (250 mM Tris, 1.3 M NaCl, 27 mM KCl, 0.05% Tween 20, pH 7.4). The secondary antibody was a horseradish peroxidase‐conjugated goat anti‐mouse IgG (BioRad) used at a 1:2000 dilution. Immunoblotting for LRP was performed similarly using the monoclonal antibody for LRP at a dilution of 1:500.
Immunofluorescence and confocal laser scanning microscopy. For immunolocalization of P‐gp, LLC‐ and LLC‐MDR1 cells were grown on collagen‐coated glass coverslips (BioCoat, BD Falcon, Heidelberg, Germany), fixed with ethanol for 15 minutes and permeabilized with 0.5% Triton X‐100 in phosphate‐buffered saline (PBS, 137 mM NaCl, 2.7 mM KCl, 8.0 mM Na2HPO4, 1.5 mM KH2PO4, pH 7.4) containing 5% fetal bovine serum for 10 minutes. Cells were incubated with monoclonal antibody C219 at a dilution of 1:50 in PBS at 4°C overnight to stain P‐gp. After three washes with PBS, cells were reincubated with Alexa Fluor488‐labeled goat anti‐mouse IgG (Molecular Probes/MoBiTec, Göttingen, Germany), diluted 1:100 in PBS at room temperature, for 1 h. Images were captured using a confocal scanning unit with a Nikon TE300 microscope and Coherent Innova 70C laser (Chromaphor, Duisburg, Germany). LRP immunofluorescence was performed similarly using an antibody dilution of 1:25.
Transwell transport assays. For transport assays, LLC‐ and LLC‐MDR1 cells were grown on Transwell polyester membrane inserts (12‐mm diameter, 0.4 µm pore size, Corning Costar) at confluence for 3 days, and expression of the recombinant protein was enhanced by treatment with 5 mM sodium butyrate for 24 h. Cells were first washed with transport buffer (142 mM NaCl, 5 mM KCl, 1 mM KH2PO4, 1.2 mM MgSO4, 1.5 mM CaCl2, 5 mM glucose and 12.5 mM HEPES, pH 7.3); subsequently, rhodamine123 (10 µM) and Aβ1‐40/1‐42 (5 µM) were added in transport buffer either to the apical or to the basal compartments. After the times indicated, the fluorescence in the opposite compartments was measured. The intracellular accumulation of fluorescence was determined by lysing the cells with 2 mL of 0.2% sodium dodecyl sulphate in water and measuring the fluorescence in cell lysates.
To study the transcellular transport of FITC‐conjugated Aβ1‐40/1‐42, cells were incubated with 5 µM Aβ1‐40/1‐42 in the basal compartments at 37°C for 1 h. The fluorescence of FITC‐conjugated Aβ1‐40/1‐42 in the apical compartment was measured with a fluorescence photometer (Wallac, Freiburg, Germany) at an excitation wavelength of 485 nm and an emission wavelength of 535 nm. For quantitative determination of Aβ peptides in the apical compartment, the hAmyloid β1‐40/42 Brain ELISA was performed according to the manufacturer’s instructions (The Genetics Company). Cells were incubated for 2 h with 100 nM, 316 nM, 1 µM or 5 µM Aβ1‐40/1‐42 in the basal compartments, respectively. Aβ1‐40 and Aβ1‐42 were detected with rabbit anti‐Aβ1‐40 and rabbit anti‐Aβ1‐42 IgG, respectively. Sites of primary antibody binding were detected with peroxidase‐conjugated donkey anti‐rabbit IgG.
For inhibition studies, the inhibitors verapamil and cyclosporine A were added simultaneously with Rh123 into the basal compartment, and the fluorescence in the apical compartment was measured.
Transcellular leakage was determined by incubating cells with 50 µM [14C]inulin carboxylic acid in the basal compartments and measuring the radioactivity in the apical compartments. For both LLC‐ and LLC‐MDR1 cells, the transcellular leakage was less than 1%/h.
Vesicle transport studies. Transport of 10 µM rhodamine123 or 5 µM FITC‐conjugated Aβ1‐40/1‐42 into membrane vesicles was measured by filtration through a gel matrix. Briefly, membrane vesicles (100 µg of protein) were incubated in the presence of 4 mM ATP, 10 µg/mL creatine kinase and RH123 or FITC‐conjugated Aβ1‐40/1‐42 in an incubation buffer (250 mM sucrose, 10 mM Tris, pH 7.4) in a final volume of 55 µL at 37°C. Aliquots (20 µL) were taken at the indicated time points, diluted in 180 µL ice‐cold incubation buffer and immediately filtered through a Sephadex G50 matrix (Sigma, Deisenhofen, Germany) by centrifugation (450 g, 3 minutes, 4°C). The fluorescence in the flow‐through was measured. In control experiments, ATP was replaced by an equal concentration of 5′‐AMP. ATP‐dependent transport was calculated by subtracting values obtained in the presence of 5′‐AMP from those obtained in the presence of ATP.
Thioflavin T (THT) fibrillogenesis assay. THT is a fluorescent dye exhibiting substantial enhancement in fluorescence intensity upon binding to β‐sheet‐rich (amyloid) fibrils. Because of the correlation between THT fluorescence and the amount of fibrils, a THT fibrillogenesis assay is widely used to follow fibril formation in vitro (14). Here, Aβ1‐40 or Aβ1‐42 peptides (5 µM) were dissolved either in the Transwell assay buffer or the membrane vesicle buffer and incubated at 37°C with (for the membrane vesicle buffer) or without (for the Transwell assay buffer) shaking at 37°C. After 20, 60, 120, 240 and 360 minutes, samples were mixed with THT dissolved in the respective buffers (5 µM final concentration) and assayed for fluorescence at an excitation wavelength of 460 nm, and an emission wavelength of 485 nm. The fluorescence intensity was normalized with the maximal fluorescence value obtained.
RESULTS
Characterization of the cellular model. The expression level of human MDR1 in the transfected and control LLC cells was analyzed by immunoblotting using the monoclonal anti‐P‐gp antibody C219 (Figure 1A). P‐gp was detected in the fully glycosylated form at a molecular mass of about 170 kDa in membranes from LLC‐MDR1 cells, whereas in control cells no signal was obtained.
Figure 1.
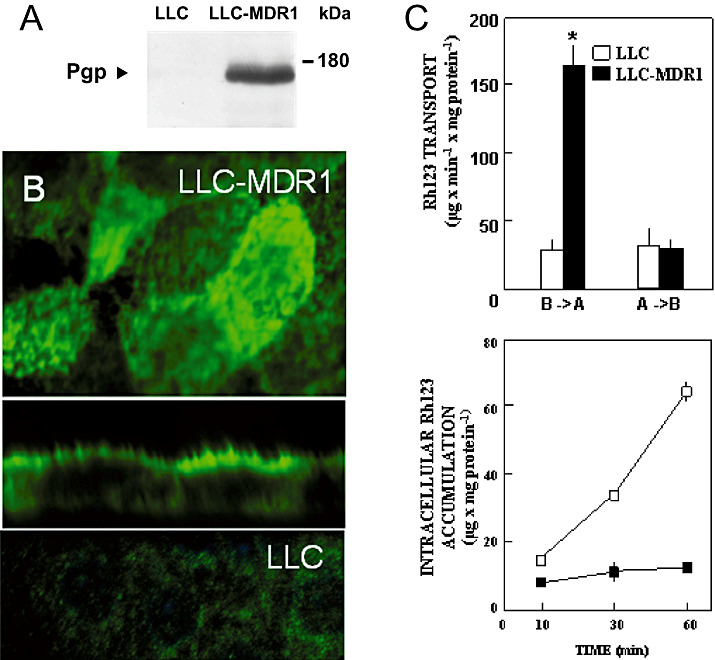
Expression, localization and function of human P‐gp in MDR1‐transfected LLC cells. A. Immunoblot analysis: Membrane vesicles (50 µg protein) from MDR1‐transfected (LLC‐MDR1) and control (LLC) cells were separated by sodium dodecyl sulphate‐polyacrylamide gel electrophoresis as described in the Methods section. Protein levels were analyzed with the C219 monoclonal antibody against P‐gp. B. Confocal laser scanning immunofluorescence microscopy: P‐gp (green fluorescence) was stained with the primary antibody C219 in MDR1‐transfected (upper and middle panel) and control (lower panel) cells. The upper and lower panels show optical sections in the xy‐plane, the middle panel a vertical section in the xz‐plane. C. Transcellular transport (upper panel) and intracellular accumulation (lower panel) of Rh123: LLC (□) and LLC‐MDR1 (▪) cells were grown on Transwell membrane inserts. Upper panel: Rh123 (10 µM) was delivered either to the basal compartments (B→A) or to the apical compartments (A→B). After 60 minutes at 37°C, fluorescence in the opposite compartments was measured. Lower panel: Rh123 (10 µM) was delivered to the basal compartments. At the time points indicated, fluorescence inside the cells was determined. Data represent means ± SD (n = 6). *Significant difference between the two cell lines according to Student’s t‐test (P < 0.05).
The cellular localization of the recombinant transporter in the transfectants was studied by means of confocal laser scanning immunofluorescence microscopy. Using antibody C219, plasma membrane staining could be observed in LLC‐MDR1 cells, and vertically oriented sections showed intense green fluorescence for P‐gp on the apical membrane (Figure 1B). Under the same conditions, no specific staining was observed in non‐transfected, parental LLC cells.
Regarding LRP expression, there were no differences between the LLC cells and the LLC‐MDR1 cells (data not shown).
Transcellular transport of Rh123 is mediated by P‐gp, and is inhibited by Aβ. The function of human P‐gp in the transfected cells was studied by measurement of the transcellular transport of the fluorescent amphiphilic cation rhodamine‐123 (Rh123), a substrate of human P‐gp. LLC and LLC‐MDR1 cells were grown in a confluent, polarized monolayer on Transwell membranes and incubated with Rh123 (10 µM) in the apical or basal compartments. At different time points, the fluorescence that accumulated in the opposite compartment and in the cells was measured. As shown in Figure 1C, there was significant basal‐to‐apical Rh123 transport in LLC‐MDR1 cells, which was markedly higher than apical‐to‐basal transport as well as the basal‐to‐apical transport in the LLC control cells. Permeability of Rh123 in LLC cells was similar for both transport routes. The intracellular accumulation of Rh123 was significantly higher in the LLC cells than in the LLC‐MDR1 cells.
In order to study the interference of Aβ1‐40/1‐42 with P‐gp‐mediated transport of Rh123, LLC‐MDR1 cells were incubated with Rh123 (10 µM) in the basolateral compartments, alone (control) or in the presence of Aβ1‐40 or Aβ1‐42. Aβ significantly decreased the apical efflux of Rh123. At a concentration of 5 µM, inhibition observed was similar for Aβ1‐40 (21.5 ± 4.88%) and Aβ1‐42 (21.4 ± 7.35%, mean values ± SD, n = 4), with a half‐maximal inhibition for Aβ1‐42 at about 50 µM.
Transcellular transport of Aβ peptides. We further investigated whether transcellular transport of the Aβ peptide itself can be observed in LLC cells. To this end, we measured Aβ peptides by ELISA in the apical compartments in the inhibition experiments described above. We could detect both Aβ1‐40 and Aβ1‐42 at significantly higher levels in the apical compartments from LLC‐MDR1 cells than from LLC control cells, indicating that the peptides were transported by P‐gp.
For Aβ1‐40, the apical compartment concentrations were significantly higher in LLC‐MDR1 cells at 316 nM (24 ± 1 vs. 1.4 ± 0.7 pg/mL), 1 µM (708 ± 3 vs. 78 ± 18 pg/mL) and 5 µM (850 ± 120 vs. 300 ± 50 pg/mL). The Aβ1‐40 level at 100 nM also was higher (1.8 ± 0.8 vs. 0.6 ± 0.1) but failed to reveal statistical significance (Figure 2A). For Aβ1‐42, the Aβ levels in the apical compartment were below the detection limit for both LLC‐MDR1 and LLC cells at concentrations of 100 nM and 316 nM. For the 1 µM and 5 µM concentrations, significantly higher levels of Aβ1‐42 could be detected in the apical compartment for LLC‐MDR1 cells [77 ± 27 pg/mL (5 µM) and 8.2 ± 0.1 pg/mL (1 µM)], while the concentrations for the LLC cells remained below the detection limit.
Figure 2.
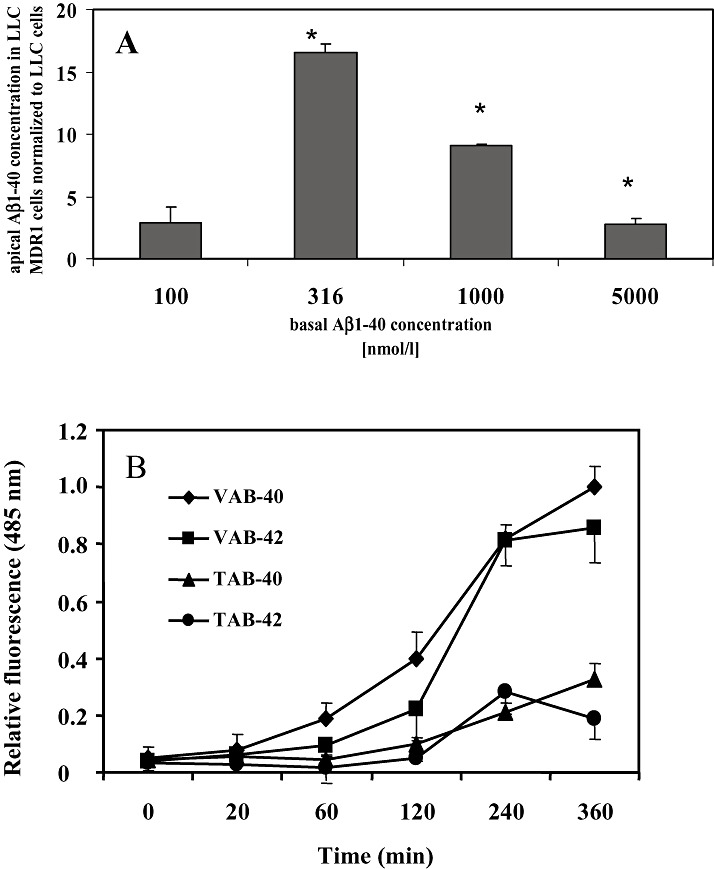
Determination of transcellular transport and fibril formation of Aβ peptides. A. LLC‐MDR1 and LLC control cells were grown on Transwell membrane inserts. After incubation with different concentrations of Aβ1‐40 in the basal compartments at 37°C for 2 h, Aβ peptides were determined in the apical compartments by ELISA. Data are presented as the ratio between LLC‐MDR1 and LLC cells (means ± SD for n = 3). *Significant difference between the two cell lines according to Student’s t‐test (P < 0.05). B. Aβ1‐40 or Aβ1‐42 fibril formation was followed using the thioflavin T assay. Aβ1‐40 or Aβ1‐42 peptides (5 µM) were dissolved either in the Transwell assay (TAB‐40 or TAB‐42) or the membrane vesicle (VAB‐40 or VAB‐42) buffer and incubated at 37°C with (for VAB) or without (for TAB) shaking at 37°C. After the indicated time, samples were mixed with thioflavin T (5 µM final concentration) and the normalized fluorescence intensity determined (excitation 460 nm, emission 485 nm). Data represent means ± SD (n = 3).
In AD, soluble Aβ peptides are assumed to accumulate over time in the cerebral extracellular space and vascular walls and to polymerize into insoluble, β‐sheet‐rich fibrils. To determine if the Aβ peptides are transported mainly in the soluble form and not as high molecular weight, polymerized structures, we used a THT fibrillogenesis assay (14). Figure 2B shows the intensity of THT fluorescence over 6 h under the conditions of our Transwell assay and membrane vesicle transport assay. The data indicate that Aβ40 and Aβ42 form fibrils with a lag time of about 1–2 h. The extent of fibril formation was substantially higher and started earlier under the membrane vesicle assay conditions (incubation in 250 mM sucrose, 10 mM Tris, pH 7.4, shaking at 37°C) than under the Transwell assay conditions (142 mM NaCl, 5 mM KCl, 1 mM KH2PO4, 1.2 mM MgSO4, 1.5 mM CaCl2, 5 mM glucose and 12.5 mM HEPES, pH 7.3, without shaking). However, the maximal incubation times of 20 minutes and 60 minutes in the vesicle assay or Transwell assay, respectively, are within the fibril formation lag time (Figure 2B).
Furthermore, we used FITC‐conjugated Aβ1‐40 and Aβ1‐42 to study the vectorial transport by LLC‐MDR1 and control cells. As shown in Figure 3, cells were incubated with FITC‐conjugated Aβ1‐40 and Aβ1‐42 at a concentration of 5 µM in the apical and basal compartments. A significantly higher basal‐to‐apical transport was observed in the LLC‐MDR1 cells for both peptides (compared with the apical‐to‐basal as well as both transport routes in the control cells). Aβ peptide permeability in LLC control cells was similar in both directions.
Figure 3.
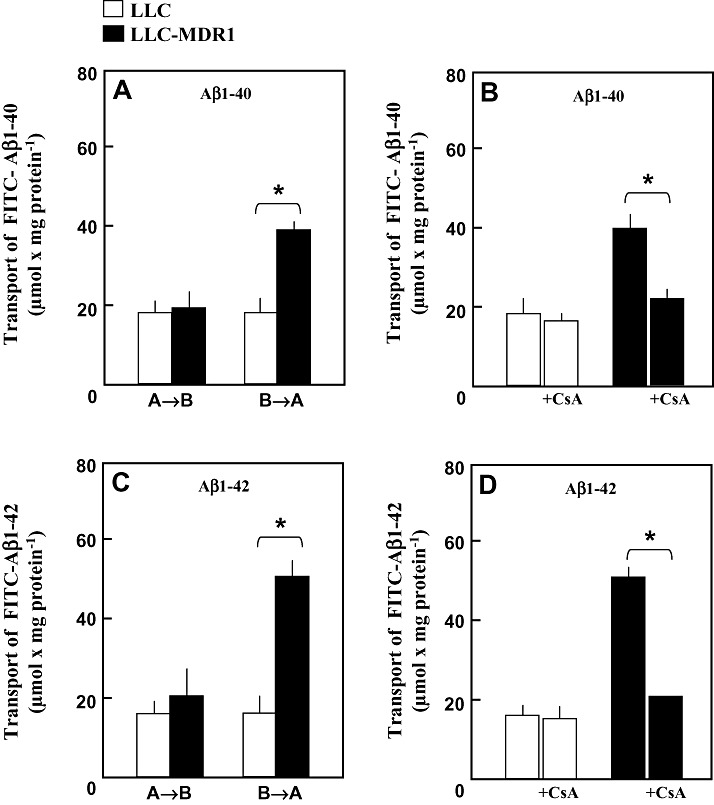
Transcellular Transport of fluorescein‐(FITC‐)‐conjugated Aβ1‐40 and Aβ1‐42. LLC cells (□) and LLC‐MDR1 cells (▪) were grown on Transwell membrane inserts. (A,C) FITC‐Aβ1‐40 and FITC‐Aβ1‐42 (5 µM) were delivered either to the basal compartments (B→A) or to the apical compartments (A→B). After 60 minutes at 37°C, fluorescence in the opposite compartments was measured. (B,D) FITC‐Aβ1‐40 and FITC‐Aβ1‐42 (5 µM) were delivered to the basal compartments in the absence or presence of 10 µM cyclosporine A (+CsA). After 60 minutes at 37°C, fluorescence in the apical compartments was measured. Data represent means ± SD (n = 4). *Significant difference, Student’s t‐test (P < 0.05).
In addition, transcellular transport of FITC‐conjugated Aβ1‐40 and Aβ1‐42 was suppressed to about 40%–50% of control transport in the presence of the P‐gp inhibitor cyclosporine A (Figure 3B,D), further indicating that P‐gp is involved in the basal‐to‐apical transport of Aβ peptides.
Transport studies with membrane vesicles. Inside‐out membrane vesicles were prepared from LLC and LLC‐MDR1 cells. As shown in Figure 4, Rh123 (10 µM) was transported ATP dependently into membrane vesicles from LLC‐MDR1 cells. Net ATP‐dependent transport was calculated by subtracting transport in the presence of 5′‐AMP from that measured in the presence of ATP. In the control cells, rates of ATP‐dependent transport were about 20% of those determined with membrane vesicles from MDR1‐transfected cells. As shown in Figure 4C, Aβ1‐40 and Aβ1‐42 were potent inhibitors of the ATP‐dependent Rh123 transport mediated by P‐gp. At a concentration of 5 µM, both peptides inhibited Rh123 transport to about 25% of control levels, similar to 100 µM verapamil.
Figure 4.
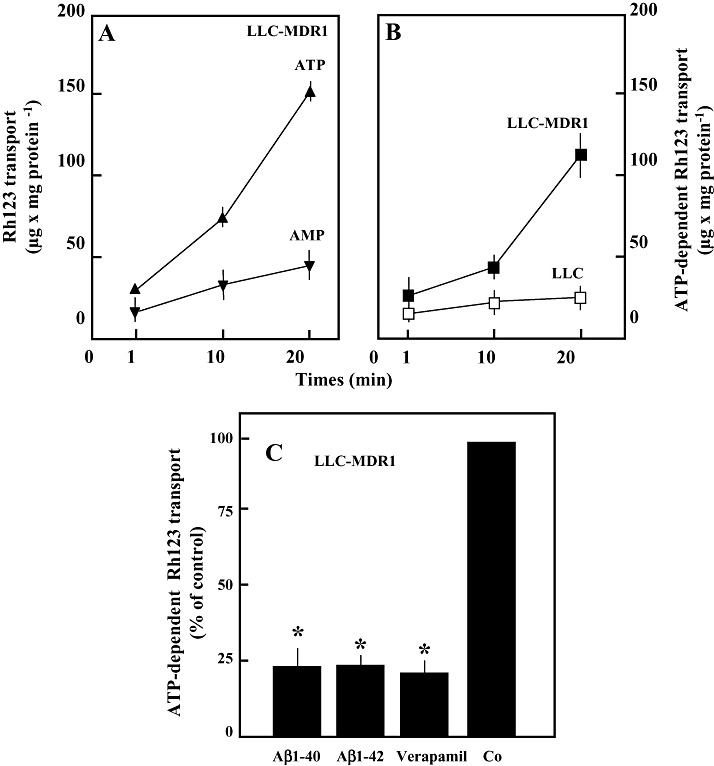
Transport of Rh123 into membrane vesicles from LLC cells and LLC‐MDR1 cells and its inhibition by Aβ peptides. A. Membrane vesicles were incubated with 10 µM Rh123 in the presence of 4 mM ATP (▴) or 4 mM 5′‐AMP (▾). Vesicle‐associated fluorescence was determined by size‐exclusion centrifugation. B. Net ATP‐dependent transport into the vesicles from LLC‐MDR1 (▪) and from LLC (□) cells was calculated by subtracting values obtained in the presence of 5′‐AMP from those in the presence of ATP. C. ATP‐dependent transport (10 µM Rh123) in the presence of 5 µM Aβ1‐40 and Aβ1‐42 and 100 µM verapamil, respectively. Data represent means ± SD (n = 4). *Significant difference from control as determined by Student’s t‐test (P < 0.05).
The membrane vesicles also were used to study the transport of FITC‐conjugated Aβ1‐40 and Aβ1‐42. Both peptides were transported ATP‐dependently into LLC‐MDR1 membrane vesicles (Figure 5). ATP‐dependent transport was detectable in membrane vesicles from LLC control cells, but the transport rate was only about 30% of that measured with LLC‐MDR1 vesicles. Inhibitors of P‐gp markedly decreased the ATP‐dependent uptake of FITC‐conjugated Aβ1‐40 and Aβ1‐42. In the presence of 100 µM verapamil and 10 µM cyclosporine A, uptake was reduced to about 65% of the control levels.
Figure 5.
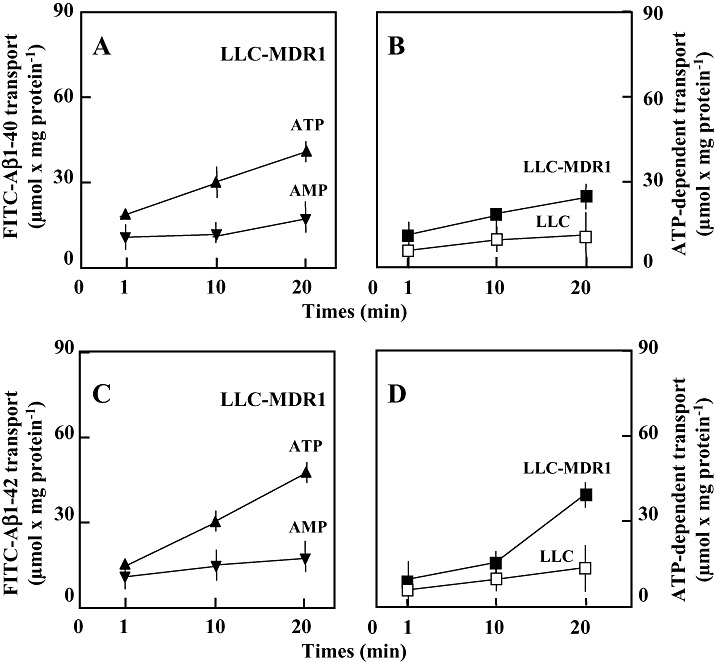
Transport of fluorescein (FITC‐)‐conjugated Aβ1‐40 and Aβ1‐42 into membrane vesicles from LLC‐MDR1 and control cells. (A,C) Membrane vesicles were incubated with 5 µM FITC‐Aβ1‐40 (A) and Aβ1‐42 (C) in the presence of 4 mM ATP (▴) or 4 mM 5′‐AMP (▾). Vesicle‐associated fluorescence was determined by size‐exclusion centrifugation. (B,D) Net ATP‐dependent transport into the vesicles from LLC‐MDR1 (▪) and from LLC (□) cells was calculated by subtracting values obtained in the presence of 5′‐AMP from those obtained in the presence of ATP. Data represent means ± SD (n = 4).
DISCUSSION
Lam et al reported that synthetic Aβ can be transported by P‐gp in vitro in an ATP‐dependent manner (12). Our results are in agreement with these data, and give further evidence for the active involvement of P‐gp in the transmembrane transport of Aβ1‐40 and Aβ1‐42. We used human MDR1‐transfected LLC cells, cultured in a polarized cell monolayer, as an in vitro model of the vascular endothelium at the BBB. This model was used to directly characterize the transcellular transport of the Aβ peptide. In these cells, P‐gp is expressed in the apical membrane, and Aβ transport can be followed by the efflux of the peptide from the cells into the apical compartment. Furthermore, we demonstrated direct ATP‐dependent transport of these peptides by P‐gp using fluorescence‐labeled peptides and isolated, inside‐out membrane vesicles. The THT fibrillogenesis assay indicates that the Aβ peptides are transported mainly in soluble form, and not as polymerized fibrils.
Our results, and those of other laboratories, suggest that a decrease of P‐gp expression at the BBB diminishes the clearance of Aβ from brain into the blood, thereby promoting the accumulation of the peptide within the brain. The potential role of P‐gp in neurodegeneration is intriguing, as the activity of this transport protein can be modulated by a range of pharmacologic agents (9, 10, 17, 30). Recently, it was reported that the treatment of AD patients with rifampin, one of the most potent inducers of P‐gp, after a period of 3 months led to a notable improvement of cognitive function (15). The mechanism of this effect is unknown, but the possibility that it is mediated by increased, P‐gp‐mediated export of Aβ is worth investigating. In addition to the transport of Aβ out of the brain, P‐gp activity could impede neurodegenerative processes via other constitutive functions of this transport protein. P‐gp generally protects the brain against toxic exogenous compounds. A disturbance of P‐gp expression at the BBB has been hypothesized to be responsible for the enhanced influx of pesticides, leading to a Parkinsonian syndrome (6, 7). Furthermore, P‐gp inhibits the activity of caspases that mediate neuronal apoptosis in neurodegenerative diseases such as AD and Creutzfeldt–Jakob disease (23, 24). In addition to its presence in brain endothelial cells, P‐gp also can be found (to a lesser extent) in astrocytes (20) and microglia (13), and recently was shown to be induced in neurons by epileptic seizures (28, 29). Furthermore, the transporter has been detected in intracellular compartments (2, 16). These findings suggest that the functions of P‐gp in the brain could be more complex than was previously realized, and strengthen the case for P‐gp as a potential target for the development of therapeutic strategies in AD and other neurodegenerative disorders.
CONCLUSION
The present study supports the hypothesis that P‐gp plays a key role in the clearance of Aβ from the brain via the BBB, and therefore might be causally involved in the pathogenesis of cerebral β‐amyloidoses such as AD. As, to date, few data are available that address the relationship between P‐gp and neurodegeneration, further studies are required to shed more light on this issue. This is of major importance, as the activity of P‐gp can be modulated pharmacologically, and thus is a potential target for the prevention or disease‐modifying treatment of CAA and AD.
ACKNOWLEDGMENTS
We thank C. Müller, A. Wolter and I. Geissler for excellent technical assistance.
REFERENCES
- 1. Ambudkar SV, Kim IW, Sauna ZE (2005) The power of the pump: mechanisms of action of P‐glycoprotein (ABCB1). Eur J Pharm Sci 580:1049–1055. [DOI] [PubMed] [Google Scholar]
- 2. Bendayan R, Lee G, Bendayan M (2002) Functional expression and localization of P‐glycoprotein at the blood brain barrier. Microsc Res Tech 57:365–380. [DOI] [PubMed] [Google Scholar]
- 3. Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, Jiang H, Prior JL, Sagare A, Bales KR, Paul SM, Zlokovic BV, Piwnica‐Worms D, Holtzman DM (2005) P‐glycoprotein deficiency at the blood‐brain barrier increases amyloid‐beta deposition in an Alzheimer disease mouse model. J Clin Invest 115:3285–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cummings J (2004) Alzheimer’s disease. N Engl J Med 351:56–67. [DOI] [PubMed] [Google Scholar]
- 5. Deane R, Wu Z, Zlokovic BV (2004) RAGE (yin) versus LRP (yang) balance regulates Alzheimer amyloid beta‐peptide clearance through transport across the blood‐brain barrier. Stroke 35(11 Suppl. 1):2628–2631. [DOI] [PubMed] [Google Scholar]
- 6. Drozdzik M, Bialecka M, Mysliwiec K, Honczarenko K, Stankiewicz J, Sych Z (2003) Polymorphism in the P‐glycoprotein drug transporter MDR1 gene: a possible link between environmental and genetic factors in Parkinson’s disease. Pharmacogenetics 13:259–263. [DOI] [PubMed] [Google Scholar]
- 7. Furuno T, Landi MT, Ceroni M, Caporaso N, Bernucci I, Nappi G, Martignoni E, Schaeffeler E, Eichelbaum M, Schwab M, Zanger UM (2002) Expression polymorphism of the blood‐brain barrier component P‐glycoprotein (MDR1) in relation to Parkinson’s disease. Pharmacogenetics 12:529–534. [DOI] [PubMed] [Google Scholar]
- 8. Gottesman MM, Fojo T, Bates SE (2002) Multidrug resistance in cancer: role of ATP‐dependent transporters. Nat Rev Cancer 2:48–58. [DOI] [PubMed] [Google Scholar]
- 9. Izzo AA (2005) Herb‐drug interactions: an overview of the clinical evidence. Fundam Clin Pharmacol 19:1–16. [DOI] [PubMed] [Google Scholar]
- 10. Johne A, Brockmoeller J, Bauer S, Maurer A, Langheinrich M, Roots I (1999) Pharmacokinetic interaction of digoxin with an herbal extract from St John’s wort (Hypericum perforatum). Clin Pharmacol Ther 66:338–345. [DOI] [PubMed] [Google Scholar]
- 11. Keppler D, Jedlitschky G, Leier I (1998) Transport function and substrate specificity of multidrug resistance protein. Methods Enzymol 292:607–616. [DOI] [PubMed] [Google Scholar]
- 12. Lam FC, Liu R, Lu P, Shapiro AB, Renoir JM, Sharom FJ, Reiner PB (2001) beta‐Amyloid efflux mediated by p‐glycoprotein. J Neurochem 76:1121–1128. [DOI] [PubMed] [Google Scholar]
- 13. Lee G, Schlichter L, Bendayan M, Bendayan R (2001) Functional expression of P‐glycoprotein in rat brain microglia. J Pharmacol Exp Ther 299:204–212. [PubMed] [Google Scholar]
- 14. LeVine H 3rd (1999) Quantification of beta‐sheet amyloid fibril structures with thioflavin T. Methods Enzymol 309:274–284. [DOI] [PubMed] [Google Scholar]
- 15. Loeb MB, Molloy DW, Smieja M, Standish T, Goldsmith CH, Mahony J, Smith S, Borrie M, Decoteau E, Davidson W, McDougall A, Gnarpe J, O’Donnell M, Chernesky M (2004) A randomized, controlled trial of doxycycline and rifampin for patients with Alzheimer’s disease. J Am Geriatr Soc 52:381–387. [DOI] [PubMed] [Google Scholar]
- 16. Rajagopal A, Simon SM (2003) Subcellular localization and activity of multidrug resistance proteins. Mol Biol Cell 14:3389–3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rautio J, Humphreys JE, Webster LO, Balakrishnan A, Keogh JP, Kunta JR, Serabjit‐Singh CJ, Polli JW (2006) In vitro P‐glycoprotein inhibition assays for assessment of clinical drug interaction potential of new drug candidates: a recommendation for probe substrates. Drug Metab Dispos 34:786–792. [DOI] [PubMed] [Google Scholar]
- 18. Revesz T, Holton JL, Lashley T, Plant G, Rostagno A, Ghiso J, Frangione B (2002) Sporadic and familial cerebral amyloid angiopathies. Brain Pathol 12:343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schinkel AH, Smit JJ, Van Tellingen O, Beijnen JH, Wagenaar E, Van DeemTer L, Mol CA, Van der Valk MA, Robanus‐Maandag EC, Te Riele HP, Berns AJM, Borst P (1994) Disruption of the mouse mdr1a P‐glycoprotein gene leads to a deficiency in the blood‐brain barrier and to increased sensitivity to drugs. Cell 77:491–502. [DOI] [PubMed] [Google Scholar]
- 20. Schlachetzki F, Pardridge WM (2003) P‐glycoprotein and caveolin‐1alpha in endothelium and astrocytes of primate brain. Neuroreport 14:2041–2046. [DOI] [PubMed] [Google Scholar]
- 21. Selkoe DJ (1999) Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 399:A23–A31. [DOI] [PubMed] [Google Scholar]
- 22. Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV (2000) Clearance of Alzheimer’s amyloid‐ß 1‐40 peptide from brain by LDL receptor‐related protein‐1 at the blood‐brain barrier. J Clin Invest 106:1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Smyth MJ, Krasovskis E, Sutton VR, Johnstone RW (1998) The drug efflux protein, P‐glycoprotein, additionally protects drug‐resistant tumor cells from multiple forms of caspase‐dependent apoptosis. Proc Natl Acad Sci USA 95:7024–7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tainton KM, Smyth MJ, Jackson JT, Tanner JE, Cerruti L, Jane SM, Darcy PK, Johnstone RW (2004) Mutational analysis of P‐glycoprotein: suppression of caspase activation in the absence of ATP‐dependent drug efflux. Cell Death Differ 11:1028–1037. [DOI] [PubMed] [Google Scholar]
- 25. Vinters VH (1987) Cerebral amyloid angiopathy: a critical review. Stroke 18:311–324. [DOI] [PubMed] [Google Scholar]
- 26. Vogelgesang S, Cascorbi I, Schroeder E, Pahnke J, Kroemer HK, Siegmund W, Kunert‐Keil C, Walker LC, Warzok RW (2002) Deposition of Alzheimer’s beta‐amyloid is inversely correlated with P‐glycoprotein expression in the brains of elderly non‐demented humans. Pharmacogenetics 12:535–541. [DOI] [PubMed] [Google Scholar]
- 27. Vogelgesang S, Warzok RW, Cascorbi I, Kunert‐Keil C, Schroeder E, Kroemer HK, Siegmund W, Walker LC, Pahnke J (2004) The role of P‐glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 1:121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Volk HA, Burkhardt K, Potschka H, Chen J, Becker A, Loscher W (2004) Neuronal expression of the drug efflux transporter P‐glycoprotein in the rat hippocampus after limbic seizures. Neuroscience 123:751–759. [DOI] [PubMed] [Google Scholar]
- 29. Volk H, Potschka H, Loscher W (2005) Immunohistochemical localization of P‐glycoprotein in rat brain and detection of its increased expression by seizures are sensitive to fixation and staining variables. J Histochem Cytochem 53:517–531. [DOI] [PubMed] [Google Scholar]
- 30. Westphal K, Weinbrenner A, Gießmann T, Stuhr M, Franke G, Zschiesche M, Oertel R, Terhaag B, Kroemer HK, Siegmund W (2000) Oral bioavailability of digoxin is enhanced by talinolol: evidence for involvement of intestinal P‐glycoprotein. Clin Pharmacol Ther 68:6–12. [DOI] [PubMed] [Google Scholar]
- 31. Zlokovic BV, Yamada S, Holtzman D, Ghiso J, Frangione B (2000) Clearance of amyloid beta‐peptide from brain: transport or metabolism? Nat Med 6:718–719. [DOI] [PubMed] [Google Scholar]
- 32. Zlokovic BV, Deane R, Sallstrom J, Chow N, Miano JM (2005) Neurovascular pathways and Alzheimer amyloid beta‐peptide. Brain Pathol 15:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]