

Abstract
Tau, a major microtubule‐associated protein in brain, forms abnormal fibers in Alzheimer’s disease and several other neurodegenerative diseases. Tau is highly soluble and adopts a natively unfolded structure in solution. In the paired helical filaments of Alzheimer’s disease, small segments of tau adopt a β‐conformation and interact with other tau molecules. In the filament core, the microtubule‐binding repeat region of tau has a cross‐β structure, while the rest of the protein retains its largely unfolded structure and gives rise to the fuzzy coat of the filaments.
TAU—A MICROTUBULE‐ASSOCIATED PROTEIN
The identification of tau protein was closely linked to the discovery of microtubule self‐assembly in the early 1970s. Microtubules represent one of the major cytoskeletal networks responsible for cell shape, mitosis, intracellular transport and other functions. They assemble from the protein tubulin (α‐β‐heterodimers of ∼100 kDa). Previously, tubulin was known as a major “colchicine‐binding protein” in brain (14), but the conditions for assembling this protein into microtubules remained elusive until the work of Weisenberg et al (122). A key observation was that microtubule assembly is regulated by microtubule‐associated proteins (MAPs) and their state of phosphorylation (111); one of these proteins is the “tau factor”, which was first isolated from brain by Kirschner’s group (121). Its major function is the stabilization of microtubules, particularly in axons, and hence its expression is strongly up‐regulated during neuronal development (37) [for reviews, see (20, 30, 43)].
Tau protein is unusual in that it is heat‐stable and acid‐stable, that is, tau is exceptionally well soluble, so that it does not precipitate during boiling and treatment with acids. Its spectral properties are characteristic of a “random coil” protein (25). Electron microscopy of tau gave ambiguous results due to its low contrast (129), but certain techniques of specimen preparation, such as quick‐freeze deep‐etching, revealed a microtubule‐bound “assembly domain” (so‐called, because this domain promotes the assembly of microtubules) and a “projection domain” which protrudes away from the microtubule wall (57). The projection domain represents roughly the N‐terminal half of the molecule and the assembly domain the C‐terminal half (114). High‐resolution shadowing and image reconstructions from unstained microtubules decorated with tau molecules confirmed the largely disordered nature of tau, even when it is bound to the surface of microtubules (105).
Cloning of tau from mouse, cow and human (45, 56, 78) revealed primary sequences and domain compositions (Figure 1). Tau is unusually rich in polar and charged amino acids and has a basic character, except for the initial ∼120 residues, where negative charges predominate. Only five types of residues (G, K, P, S, T) make up half of the sequence. This explains the high solubility and unfolded nature of the protein; however, it renders its abnormal aggregation in Alzheimer’s disease (AD) even more enigmatic. In particular, at first glance, the sequence contains no elements that are particularly amyloidogenic, such as stretches of hydrophobic residues (as in the Aβ peptide) or glutamines (as in the poly‐Q stretches of huntingtin), whose interactions across and along peptide strands favor the formation of stable β‐sheets [for reviews, see (90, 119)]. Tau occurs in a number of isoforms, including “big tau” in the peripheral nervous system (4, 26, 47). There are six major isoforms in the human brain containing 352–441 amino acid residues, which arise from alternative mRNA splicing of exons 2, 3, and 10 of the Tau gene (46). The projection and assembly domains can be separated by chymotryptic cleavage behind Y197 (114), the C‐terminal tail behind D421 can be removed by caspase 3 or 9 (40, 42, 102), a major N‐terminal 17 kDa fragment can be generated by calpain (95), several cleavage sites can be generated by a thrombin‐like activity (5, 71) and cleavage behind E391 in the C‐terminal tail is observed in paired helical filaments (PHFs) (91). Such fragmentation reactions are notable, because smaller fragments of tau have an enhanced tendency to aggregate and could therefore nucleate aggregation within cells.
Figure 1.
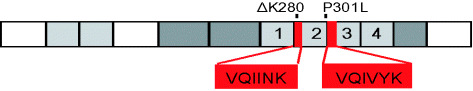
Diagram of tau441, the longest isoform in human brain. The four repeats of 31 or 32 residues each are numbered. The inserts near the N‐terminus and the second repeat can be alternatively spliced, giving rise to six isoforms. The N‐terminal domain up to ∼G120 has an acidic character, the other domains are basic. The left half (residues 1 to ∼200) represents the “projection domain”, the right half the “microtubule assembly domain”. The repeats constitute the core of the microtubule‐binding domain, as well as the core of the paired helical filaments. Two hexapeptide motifs at the beginning of R2 and R3 promote paired helical filament (PHF) aggregation by inducing β‐structure. ΔK280 and P301L are two FTDP‐17 (frontotemporal dementia and parkinsonism linked to chromosome 17) mutations that strongly enhance the rate of PHF aggregation by increasing the propensity for β‐structure.
The C‐terminal half of tau contains three or four semi‐conserved repeats, a feature shared with other MAPs. The general domain structure of tau is similar to that of the neuronal MAP2 and the ubiquitous MAP4; however, they possess a much larger N‐terminal projection domain (21, 82, 94). The size of the projection domain appears to determine the spacing between microtubules in cells (23). Depending on developmentally controlled alternative mRNA splicing, the repeat domain of tau consists of three or four sequences of 31 or 32 residues each: R1 = Q244 − K274, R2 = V275 − S305, R3 = V306 − Q336, R4 = V337 − N368. R2 is encoded by exon 10 of Tau and is absent in the 3‐repeat isoforms. The isoforms can be designated as 0N3R, 1N3R, 2N3R, 0N4R, 1N4R, 2N4R, depending on the number of N‐terminal inserts and C‐terminal repeats. Fetal tau comprises only the shortest form (0N3R), with the other isoforms being added during brain development. In adult human brain, there is a balance of 3R and 4R isoforms, all of which assemble into PHFs (44, 66). This ratio can be perturbed in disease, for example, towards 4R isoforms in some cases of frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP‐17) (38, 60). Unlike humans, rodents express exclusively 4R tau in adult brain (76).
An important property of tau is the large number of potential phosphorylation sites [for reviews, see (22, 115)]. This is due to the high frequency of phosphorylatable residues (45 S, 35 T and 5 Y in the largest isoform), combined with the open structure of tau which renders it accessible to many kinases. Many of the sites (up to 17, depending on the isoform) are part of serine‐proline (SP) or threonine‐proline (TP) motifs and represent targets of proline‐directed S/T protein kinases, eg, MAP kinase, glycogen synthase kinase‐3 (GSK‐3β), cyclin‐dependent kinase (cdc) 5, cdc2 and others. Additional sites are targeted by different kinases, including protein kinase A (PKA), protein kinase C, calcium‐calmodulin‐dependent kinase II (CaMKII), serum and glucocorticoid‐dependent kinase (SGK), protein kinase B (PKB), MAP‐microtubule‐dependent kinase (MARK) and SAD kinases. Tyrosine kinases target Y18 [fyn, (79)] and Y394 [abl, (31)]. The lysine‐isoleucine‐glycine‐serine motif (KIGS) or lysine‐cysteine‐glycine‐serine motif (KCGS) motifs in the repeat domain (S262, S293, S324, S356) can be phosphorylated by MARK, PKA, SAD kinases, CaMKII and p70S6K, which strongly reduces the tau‐microtubule interactions (36, 74, 96), [note that phosphorylation at these sites also inhibits tau aggregation, illustrating an analogous role for the repeat domain in the physiological and pathological functions of tau (106)]. A further potent detaching site is phosphoS214, which can be phosphorylated by PKA and other kinases of the AGC group (PKA/PKG/PKC group of protein kinases), and is up‐regulated during mitosis (16, 63). Tau contains one or two cysteines in the repeat domain (C291 in R2, present in 4R isoforms, and C322 in R3), which can be engaged in intra‐ or intermolecular cross‐linking affecting conformation, dimerization and aggregation (108).
The binding of tau to microtubules is mediated through the repeat domain (which binds only weakly by itself but provides specificity for microtubule assembly), in combination with the adjacent proline‐rich flanking domains (which provide efficient targeting to the microtubule surface, according to the “jaws” model (100). In general, 4R tau binds to microtubules more tightly than 3R tau (18, 50, 85). Phosphorylation, especially of the repeat domain, tends to decrease the affinity of tau for microtubules (13, 15). Like soluble tau, microtubule‐bound tau is mostly in a natively unfolded state (Figure 2) and is therefore poorly visible by X‐ray fiber diffraction or (cryo‐) electron microscopy (1, 105). This is in strong contrast to other microtubule‐interacting proteins, such as motor proteins, which show a periodic binding pattern to microtubules, compatible with the tubulin lattice (8 nm axial repeat) (58, 105). Tau appears to bind in an extended fashion to the outer tips of the microtubule protofilaments; consequently, when microtubules are disassembled by low temperature, tau stabilizes the ring‐like disassembly products consisting of tubulin oligomers. Nevertheless, the binding of tau to microtubules is dynamic, as nuclear magnetic resonance (NMR) studies have revealed a high mobility of most residues, even in the bound state (128), and as cellular microtubule‐bound tau is in rapid equilibrium with unbound tau (83, 104). Tau and motor proteins bind to overlapping sites on the outer surface of microtubules and bind in a competitive fashion. This explains why tau can interfere with transport along microtubules, which leads to an inhibition of anterograde axonal transport (109, 113).
Figure 2.

Model of microtubule protofilament with bound kinesin and tau. The protofilament consists of alternating subunits of α‐ and β‐tubulin (∼450 residues each) arranged in a polar fashion. The head domain of kinesin, a microtubule‐dependent motor protein (∼350 residues), has the compact folding typical of most cytoplasmic proteins. By contrast, tau is natively unfolded, its structure is unknown in detail and modeled here as a random chain. Note that tau occupies a much larger volume than kinesin or tubulin.
The conformation of soluble tau is unknown in detail, but is presumably highly variable, as expected for a natively unfolded protein. NMR studies of the repeat domain have revealed little secondary structure, but there are notable motifs of nascent β‐structure near the beginning of R2 and R3 (39, 88). These motifs coincide with the regions involved in PHF assembly (see below). Furthermore, stretches of ∼10 residues after the hexapeptide motifs tend to become helical upon interaction with phospholipid micelles (8), which may mimic the negatively charged microtubule surface. Internal compaction of the repeat domain is evidenced by the relative proximity of fluorescence‐labeled residues, as judged by fluorescence energy transfer. Regarding global folding, fluorescence resonance energy transfer (FRET) studies have pointed to a paperclip‐like folding, resulting in the juxtaposition of the repeat domain with the C‐terminal and N‐terminal ends of the molecule (67, 108) (Figure 3). This doubly folded conformation is reminiscent of the discontinuous epitopes of certain anti‐tau antibodies (Alz50, MC1, TG3), which recognize early stages of AD pathology and are generated by folding of the N‐terminus over the repeat domain (19, 68, 69).
Figure 3.
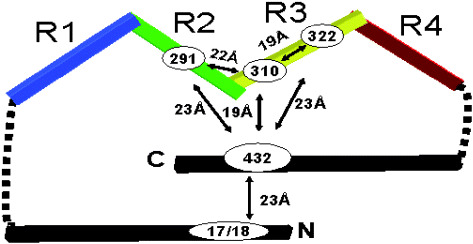
Model of the conformation of tau in solution deduced by fluorescence resonance energy transfer. The molecule shows a paperclip‐like fold which brings the N‐ and C‐terminal ends into the vicinity of the repeat domain. Similar folded conformations are recognized by several antibodies specific for abnormal tau from Alzheimer’s disease brain (eg, Alz‐50, MC1, TG3). Approximate distances between labeled residues are indicated.
Mutations in the Tau gene cause FTDP‐17 (61, 99, 112). At the level of the expressed protein, some mutations perturb the balance of splicing isoforms, mostly in favor of 4R tau (38, 61, 112). These forms bind more tightly to microtubules than 3R forms, which may lead to overstabilization of microtubules and suppression of microtubule dynamics (41). Other mutations alter the protein sequence, mostly in the repeat domain. With regard to tau structure in solution, all mutants investigated so far are similar, in that they have shown the spectroscopic signature of natively unfolded proteins (6). The mutations decrease binding of tau to microtubules, which may lead to microtubule destabilization, although the effects are only moderate (55, 59, 83). In addition, some mutations also increase the propensity of tau to aggregate (49, 89). This is particularly pronounced for mutations that enhance the β‐sheet propensity in the regions of the hexapeptide motifs, for example, ΔK280 and P301L (10).
ASSEMBLY OF TAU INTO PAIRED HELICAL FILAMENTS
The molecular fine structure of PHFs (Figure 4) is unknown in detail, but represents one of the major goals in the field, as it may aid in the development of methods and drugs to prevent aggregation. There was a long gap between Alzheimer’s discovery of neurofibrillary tangles (2) and the identification of PHFs as their basic elements (72, 117). Further time elapsed with attempts to find procedures for the isolation of PHFs (62, 126). One important advance was the image reconstruction of PHFs from negatively stained electron micrographs, which showed each half of the PHF to be composed of three protein densities, with overall dimensions of ∼8 nm × 20 nm (28); subsequent work showed an analogous doubly tripartite structure for “straight filaments”, a minor variant of filament preparations from AD brain (27). These variants depend on details of the charge distribution around the β‐structure forming motifs in the repeat domain at the beginning of R3 and R4 (32). The search for the protein composition of PHFs revealed tau as the major component (17, 29, 45, 53, 75, 80, 92, 125, 127) and molecular cloning elucidated tau sequences from several mammalian species (45, 46, 56, 78). This set the stage for the expression of recombinant tau and the structural and biochemical analysis of tau and PHFs.
Figure 4.
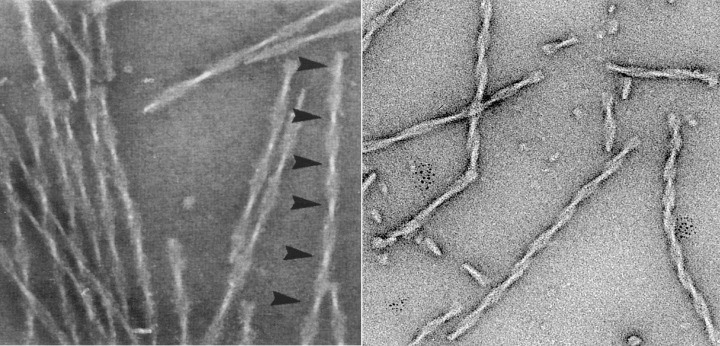
Electron micrographs of paired helical filaments isolated from Alzheimer’s disease brain (left) or assembled in vitro from recombinant tau (repeat domain with pro‐aggregation mutation ΔK280). Note the typical twisted appearance with crossover repeats of ∼80 nm (arrowheads).
One of the difficulties in studying PHF assembly from soluble tau was the high intrinsic solubility resulting from its hydrophilic nature. A further problem was to identify criteria for the in vitro generation of bona fide PHFs. These problems were solved by searching for tau constructs and assembly conditions, including dimerization of the protein by disulfide crosslinking, which accelerates PHF assembly (123). A further important step was the discovery that polyanionic molecules, such as sulphated glycosaminoglycans, RNA, acidic peptides and fatty acid micelles, induce assembly of full‐length tau (48, 70, 97, 124).
The question of whether PHFs can be regarded as “amyloid” was a matter of extended debate. Amyloid is defined structurally as a fibril whose backbone consists of β‐sheets in a “cross‐β” arrangement, recognized by a meridional 0.47 nm reflection in fiber diffraction patterns. On this basis, Kirschner et al (73) proposed a cross‐β structure for both types of fibers found in the brains of AD patients, namely amyloid plaques (made of the Aβ peptide) and neurofibrillary tangles (made of tau). In the case of PHFs, the 0.47 nm reflection was weak, the purity of the preparation was somewhat uncertain and later studies failed to confirm the reflection (107). Instead, other types of reflections were reported, which suggested a non‐amyloid packing of the protein. The puzzle was partially solved with the realization that the aggregation of tau is based on short hexapeptide motifs in the repeat domain (275VQIINK280 and 306VQIVYK311 at the beginning of R2 and R3, (9, 10, 51, 64) (Figure 1). These “aggregation motifs” have a partially hydrophobic character and tend to interact with a cross‐β structure, contributing to the core of PHFs, while the rest of the protein remains largely disordered and makes up the fuzzy coat. In recent years, as a result of improvements in specimen preparation and diffraction techniques, the amyloid nature of tau filaments purified from human brain or assembled from full‐length recombinant protein has been conclusively demonstrated (7, 12). The hexapeptide aggregation motifs coincide with sequences where nascent β‐structure can be detected in soluble tau by NMR spectroscopy, and indeed, this region reveals a very low mobility compared with the fuzzy coat (88, 110). The importance of the hexapeptide motifs is further underscored by proline‐scanning mutagenesis, where single prolines can interrupt β‐structure and inhibit aggregation (“anti‐aggregation” mutants); or, conversely, by mutations that enhance the propensity for β‐structure, such as ΔK280 and P301L, and thus promote aggregation (“pro‐aggregation” mutants) (71).
On the basis of these data, one can anticipate the analysis of PHF structure in several steps. The backbone of PHFs consists of cross‐β structure, and therefore, by analogy with other amyloid fibers (eg, Aβ peptide, yeast prion peptide) (90), it is likely that tau fibers also consist of protofibrils made up of pairs of juxtaposed β‐sheets, interacting axially by hydrogen bonding between their main chain strands and laterally through the sidechains across the sheets. As these residues are near one another their distances can probably be determined by spectroscopic methods. Examples are the recent studies employing site‐directed electron paramagnetic resonance labeling of tau (86, 87), which have concluded that residues near the beginning of R2 (272–289) and R3 (303–320) must lie close to the corresponding residues in neighboring molecules, which could be achieved theoretically by arranging them in successive turns of a beta helix. A higher level of organization will be the arrangement of protofibrils within a PHF. Their number and interactions are currently unknown, but there are several constraints for possible arrangements: (1) The mass‐per‐length of the PHF core, determined by scanning transmission electron microscopy, is about 60–70 kDa/nm, equivalent to roughly 3.5–4.5 repeat domain molecules per nm (11, 125); for variations among PHFs, see (77). By comparison, successive molecules in a cross‐β structure are spaced 0.47 nm apart. This is tantamount to ∼2 molecules per nm, which would allow only ∼2 protofibrils. (2) The cross‐section of the PHF core is about 8 nm × 20 nm. This area is divided up into two halves, each containing three density peaks (and intervening valleys of lower density), so that the effective area is estimated at ∼80 nm2 (27). These features, combined with the typical density of compact protein domains of ∼0.8 kDa/nm3, represent boundary conditions which models of tau folding in PHFs will have to meet. The least well‐defined aspect of the PHF structure is the fuzzy coat (125). PHFs assembled from full‐length tau or from its repeat domain have similar apparent dimensions by electron microscopy, suggesting that the non‐repeat parts, which comprise ∼70% of the protein (roughly residues 1–240, 370–441), make only a small contribution to the images, presumably because they retain their natively unfolded character (7). The extent of the fuzzy coat is best visualized by immunogold labeling where antibody‐binding sites can extend away from the center of the PHF (33). Nevertheless, a substantial fraction of tau molecules in PHFs must have a folded conformation, because PHFs can be immunopurified with antibody MC‐1, whose epitope comprises tau residues near the N‐terminus and within the repeat domain (68).
Filamentous tau from AD brain is extensively phosphorylated and there is an ongoing discussion on how phosphorylation and aggregation are related. This issue has been addressed using tau phosphorylated by different kinases and/or pseudophosphorylated forms of tau, where certain residues were exchanged for glutamate [for reviews, see (65, 115)]. These studies have yielded mixed results, perhaps not surprisingly, considering the number of potential phosphorylation sites in tau, the limited specificity of kinase preparations and the heterogeneity of phosphorylation states. In the case of the KXGS motifs in the repeat region, phosphorylation inhibits aggregation rather than promoting it (106). In the present context the important point is that aggregation of tau into bona fide PHFs can be achieved without phosphorylation, demonstrating that phosphorylation does not have a major influence on PHF structure. In cells, phosphorylation of tau can change its properties on at least two levels, namely tau‐microtubule interactions and tau–tau interactions. Phosphorylation tends to decrease the binding of tau to microtubules (35), with phosphorylation of the KXGS motifs and S214 having particularly pronounced effects (63). The result is a decrease in microtubule stability, but more importantly, an increase in the cytosolic pool of tau, which can contribute to aggregation into PHFs.
An important goal of studying PHF assembly is to use the information gained for preventing neuronal degeneration in AD and other neurodegenerative diseases. One of the obstacles in generating cell or animal models has been the high solubility of tau, which means that overexpression of the protein is usually not sufficient to generate neurofibrillary tangles in a reasonable time. This problem can be overcome by using mutations that accelerate the formation of β‐structure, for example, ΔK280 and P301L, by modifying the vicinity of the hexapeptide motifs to allow more extensive β‐conformation (10), or by using multiple FTDP‐17 mutations (120). When these mutations are combined with overexpression in transgenic mice, neurofibrillary tangles develop after ∼1 year (81, 93, 101, 103). Another solution was a mouse model overexpressing wild type 3R and 4R human tau isoforms on a mouse tau knockout background with an excess of 3R over 4R isoforms (3). Such models allow one to test hypotheses on tau‐induced neurodegeneration, such as the question of tau aggregation vs. toxicity, or the relationship between tau and Aβ toxicity. Our tau‐inducible neuronal cell models have indicated that aggregation is toxic and that it can be prevented by switching off tau expression, or by using compounds that inhibit aggregation (71) (Figure 5). So far, a number of different mouse models have been described, resulting in a range of findings that are beyond the scope of this article [for reviews, see (34, 52, 84, 118)]. Furthermore, once the conditions for tau aggregation will be better defined, it will become easier to identify more potent aggregation inhibitors than those currently known (24, 98, 116). Low‐molecular weight compounds have already been effective in preventing tau aggregation in some cell and animal models (54, 71). Thus, there is hope that this search will lead to the development of compounds that will keep the buildup of tau filaments under control in human brain.
Figure 5.
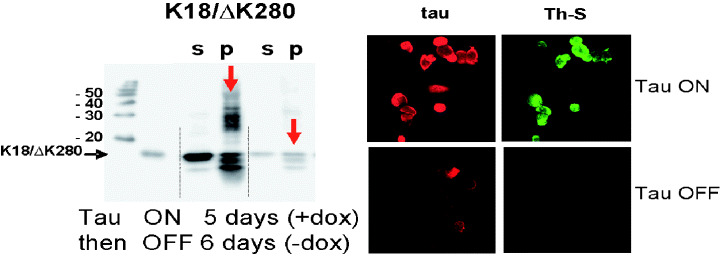
Inducible expression of tau in N2a cells. The panel on the left illustrates that the expression of tau can be switched on by doxycyclin, followed by aggregation (note the high‐molecular weight smear in the gel; s = soluble tau, p = aggregated tau). Expression of tau and aggregates can be reversed by removal of doxycyclin. The panel on the right shows expression of tau (red) and formation of aggregates, as seen by thioflavin S staining (green).
ACKNOWLEDGEMENTS
The authors’ work is supported by the Max‐Planck‐Gesellschaft (MPG), the Deutsche Forschungsgemeinschaft (DFG) and the Institute for the Study on Aging (ISOA). The authors thank Drs. B. Ghetti and M. Goedert for careful reading and constructive comments on the manuscript.
REFERENCES
- 1. Al‐Bassam J, Ozer RS, Safer D, Halpain S, Milligan RA (2002) MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments. J Cell Biol 157:1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alzheimer A (1907) Über eine eigenartige Erkrankung der Hirnrinde. Allg Z Psychia 64:146–148. [Google Scholar]
- 3. Andorfer C, Kress Y, Espinoza M, De Silva R, Tucker KL, Barde YA, Duff K, Davies P (2003) Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem 86:582–590. [DOI] [PubMed] [Google Scholar]
- 4. Andreadis A (2005) Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim Biophys Acta 1739:91–103. [DOI] [PubMed] [Google Scholar]
- 5. Arai T, Guo JP, McGeer PL (2005) Proteolysis of non‐phosphorylated and phosphorylated tau by thrombin. J Biol Chem 280:5145–5153. [DOI] [PubMed] [Google Scholar]
- 6. Barghorn S, Zheng‐Fischhofer Q, Ackmann M, Biernat J, Von Bergen M, Mandelkow E‐M, Mandelkow E (2000) Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry 39:11714–11721. [DOI] [PubMed] [Google Scholar]
- 7. Barghorn S, Davies P, Mandelkow E (2004) Tau paired helical filaments from Alzheimer’s disease brain and assembled in vitro contain beta structure in the core domain. Biochemistry 43:1694–1703. [DOI] [PubMed] [Google Scholar]
- 8. Barré P, Eliezer D (2006) Folding of the repeat domain of tau upon binding to lipid surfaces. J Mol Biol 362:312–326. [DOI] [PubMed] [Google Scholar]
- 9. Von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow E‐M, Mandelkow E (2000) Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif (306‐VQIVYK‐311) forming beta structure. Proc Natl Acad Sci USA 97:5129–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Von Bergen M, Barghorn S, Li L, Marx A, Biernat J, Mandelkow E‐M, Mandelkow E (2001) Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta structure. J Biol Chem 276:48165–48174. [DOI] [PubMed] [Google Scholar]
- 11. Von Bergen M, Barghorn S, Muller SA, Pickhardt M, Biernat J, Mandelkow EM, Davies P, Aebi U, Mandelkow E (2006) The core of tau‐paired helical filaments studied by scanning transmission electron microscopy and limited proteolysis. Biochemistry 45:6446–6457. [DOI] [PubMed] [Google Scholar]
- 12. Berriman J, Serpell LC, Oberg KA, Fink AL, Goedert M, Crowther RA (2003) Tau filaments from human brain and from in vitro assembly of recombinant protein show cross‐beta structure. Proc Natl Acad Sci USA 100:9034–9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Biernat J, Gustke N, Drewes G, Mandelkow E‐M, Mandelkow E (1993) Phosphorylation of serine 262 strongly reduces the binding of tau protein to microtubules: distinction between PHF‐like immunoreactivity and microtubule binding. Neuron 11:153–163. [DOI] [PubMed] [Google Scholar]
- 14. Borisy GG, Taylor EW (1967) The mechanism of action of colchicine. Binding of colchicine‐3H to cellular protein. J Cell Biol 34:525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VMY (1993) Abnormal tau phosphorylation at Ser(396) in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 10:1089–1099. [DOI] [PubMed] [Google Scholar]
- 16. Brandt R, Lee G, Teplow DB, Shalloway D, Abdelghany M (1994) Differential effect of phosphorylation and substrate modulation on tau’s ability to promote microtubule growth and nucleation. J Biol Chem 269:11776–11782. [PubMed] [Google Scholar]
- 17. Brion JP, Passareiro H, Nunez J, Flament‐Durand J (1985) Mise en évidence immunologique de la protéine tau au niveau des lésions de dégénérescence neurofibrillaire de la maladie d’Alzheimer. Arch Biol 95:229–235. [Google Scholar]
- 18. Butner KA, Kirschner MW (1991) Tau‐protein binds to microtubules through a flexible array of distributed weak sites. J Cell Biol 115:717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carmel G, Mager EM, Binder LI, Kuret J (1996) The structural basis of monoclonal‐antibody Alz50’s selectivity for Alzheimers‐disease pathology. J Biol Chem 271:32789–32795. [DOI] [PubMed] [Google Scholar]
- 20. Cassimeris L, Spittle C (2001) Regulation of microtubule‐associated proteins. Int Rev Cytol 210:163–226. [DOI] [PubMed] [Google Scholar]
- 21. Chapin SJ, Bulinski JC (1992. ) Microtubule stabilization by assembly‐promoting microtubule‐associated proteins: a repeat performance. Cell Motil Cytoskeleton 23:236–243. [DOI] [PubMed] [Google Scholar]
- 22. Chen F, David D, Ferrari A, Götz J (2004) Posttranslational modifications of tau—role in human tauopathies and modelling in transgenic animals. Curr Drug Targets 5:503–515. [DOI] [PubMed] [Google Scholar]
- 23. Chen J, Kanai Y, Cowan N, Hirokawa N (1992) Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 360:674–677. [DOI] [PubMed] [Google Scholar]
- 24. Chirita C, Necula M, Kuret J (2004) Ligand‐dependent inhibition and reversal of tau filament formation. Biochemistry 43:2879–2887. [DOI] [PubMed] [Google Scholar]
- 25. Cleveland DW, Hwo SY, Kirschner MW (1977) Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol 116:227–247. [DOI] [PubMed] [Google Scholar]
- 26. Couchie D, Mavilia C, Georgieff I, Liem R, Shelanski M, Nunez J (1992) Primary structure of high molecular weight tau present in the peripheral nervous system. Proc Natl Acad Sci USA 89:4378–4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Crowther RA (1991) Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc Natl Acad Sci USA 88:2288–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crowther RA, Wischik CM (1985) Image reconstruction of the Alzheimer paired helical filament. EMBO J 4:3661–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Delacourte A, Défossez A (1986) Alzheimer’s disease: tau proteins, the promoting factors of microtubule assembly, are major antigenic componente of paired helical filaments. J Neurol Sci 76:173–186. [DOI] [PubMed] [Google Scholar]
- 30. Delacourte A, Buée L (2000) Tau pathology: a marker of neurodegenerative disorders. Curr Opin Neurol 13:371–376. [DOI] [PubMed] [Google Scholar]
- 31. Derkinderen P, Scales TM, Hanger DP, Leung KY, Byers HL, Ward MA, Lenz C, Price C, Bird IN, Perera T, Kellie S, Williamson R, Noble W, Van Etten RA, Leroy K, Brion JP, Reynolds CH, Anderton BH (2005) Tyrosine 394 is phosphorylated in Alzheimer’s paired helical filament tau and in fetal tau with c‐Abl as the candidate tyrosine kinase. J Neurosci 25:6584–6593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DeTure MA, Di Noto L, Purich DL (2002) In vitro assembly of Alzheimer‐like filaments. How a small cluster of charged residues in Tau and MAP2 controls filament morphology. J Biol Chem 277:34755–34759. [DOI] [PubMed] [Google Scholar]
- 33. Dickson D, Ksiezak‐Reding H, Liu W, Davies P, Crowe A, Yen SH (1992) Immunocytochemistry of neurofibrillary tangles with antibodies to subregions of tau protein: identification of hidden and cleaved tau epitopes and a new phosphorylation site. Acta Neuropath 84:596–605. [DOI] [PubMed] [Google Scholar]
- 34. Van Dooren T, Dewachter I, Borghgraef P, Van Leuven F (2005) Transgenic mouse models for APP processing and Alzheimer’s disease: early and late defects. Subcell Biochem 38:45–63. [DOI] [PubMed] [Google Scholar]
- 35. Drechsel DN, Hyman AA, Cobb MH, Kirschner MW (1992) Modulation of the dynamic instability of tubulin assembly by the microtubule‐associated protein tau. Mol Biol Cell 3:1141–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E (1997) MARK, a novel family of protein kinases that phosphorylate microtubule‐associated proteins and trigger microtubule disruption. Cell 89:297–308. [DOI] [PubMed] [Google Scholar]
- 37. Drubin D, Kirschner M (1986) Tau protein function in living cells. J Cell Biol 103:2739–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. D’Souza I, Schellenberg GD (2005) Regulation of tau isoform expression and dementia. Biochim Biophys Acta 1739:104–115. [DOI] [PubMed] [Google Scholar]
- 39. Eliezer D, Barré P, Kobaslija M, Chan D, Li X, Heend L (2005) Residual structure in the repeat domain of tau: echoes of microtubule binding and paired helical filament formation. Biochemistry 44:1026–1036. [DOI] [PubMed] [Google Scholar]
- 40. Fasulo L, Ugolini G, Visintin M, Bradbury A, Brancolini C, Verzillo V, Novak M, Cattaneo A (2000) The neuronal microtubule‐associated protein tau is a substrate for caspase‐3 and an effector of apoptosis. J Neurochem 75:624–633. [DOI] [PubMed] [Google Scholar]
- 41. Feinstein SC, Wilson L (2005) Inability of tau to properly regulate neuronal microtubule dynamics: a loss‐of‐function mechanism by which tau might mediate neuronal cell death. Biochim Biophys Acta 1739:268–279. [DOI] [PubMed] [Google Scholar]
- 42. Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia‐Sierra F, LaPointe N, Miller R, Berry RW, Binder LI, Cryns VL (2003) Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci USA 100:10032–10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Garcia ML, Cleveland DW (2001) Going new places using an old MAP: tau, microtubules and human neurodegenerative disease. Curr Opin Cell Biol 13:41–48. [DOI] [PubMed] [Google Scholar]
- 44. Goedert M, Jakes R (1990) Expression of separate tau isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J 9:4225–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Goedert M, Wischik C, Crowther R, Walker J, Klug A (1988) Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule‐associated protein tau. Proc Natl Acad Sci USA 85:4051–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Goedert M, Spillantini M, Jakes R, Rutherford D, Crowther RA (1989) Multiple isoforms of human microtubule‐associated protein‐tau: sequences and localization in neurofibrillary tangles of Alzheimers‐disease. Neuron 3:519–526. [DOI] [PubMed] [Google Scholar]
- 47. Goedert M, Spillantini MG, Crowther RA (1992) Cloning of a big tau microtubule‐associated protein characteristic of the peripheral nervous system. Proc Natl Acad Sci USA 89:1983–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ Crowther RA (1996) Assembly of microtubule‐associated protein tau into Alzheimer‐like filaments induced by sulphated glycosaminoglycans. Nature 383:550–553. [DOI] [PubMed] [Google Scholar]
- 49. Goedert M, Jakes R, Crowther RA (1999) Effects of frontotemporal dementia FTDP‐17 mutations on heparin‐induced assembly of tau filaments. FEBS Lett 450:306–311. [DOI] [PubMed] [Google Scholar]
- 50. Goode BL, Feinstein SC (1994) Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter‐repeat region of tau. J Cell Biol 124:769–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goux WJ, Kopplin L, Nguyen AD, Leak M, Rutkofsky VD, Shanmuganandam D, Sharma H, Inouye H, Kirschner DA (2004) The formation of straight and twisted filaments from short tau peptides. J Biol Chem 279:26868–26875. [DOI] [PubMed] [Google Scholar]
- 52. Götz J, Ittner LM, Kins S (2006) Do axonal defects in tau and amyloid precursor protein transgenic animals model axonopathy in Alzheimer’s disease? J Neurochem 98:993–1006. [DOI] [PubMed] [Google Scholar]
- 53. Grundke‐Iqbal I, Iqbal K, Tung Y, Quinlan M, Wisniewski H, Binder L (1986) Abnormal phosphorylation of the microtubule‐associated protein tau in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 83:4913–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hall GF, Lee S, Yao J (2002) Neurofibrillary degeneration can be arrested in an in vivo cellular model of human tauopathy by application of a compound which inhibits tau filament formation in vitro. J Mol Neurosci 19:253–260. [DOI] [PubMed] [Google Scholar]
- 55. Hasegawa M, Smith MJ, Goedert M (1998) Tau proteins with FTDP‐17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett 437:207–210. [DOI] [PubMed] [Google Scholar]
- 56. Himmler A, Drechsel D, Kirschner M, Martin D (1989) Tau consists of a set of proteins with repeated C‐terminal microtubule‐binding domains and variable N‐terminal domains. Mol Cell Biol 9:1381–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hirokawa N, Shiomura Y, Okabe S (1988) Tau proteins: the molecular structure and mode of binding to microtubules. J Cell Biol 107:1449–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hoenger A, Sack S, Thormählen M, Marx A, Müller J, Gross H, Mandelkow E (1998) Image reconstructions of microtubules decorated with monomeric and dimeric kinesins: comparison with X‐ray structure and implications for motility. J Cell Biol 141:419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hong M, Zhukareva V, Vogelsberg‐Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM (1998) Mutation‐specific functional impairments in distinct tau isoforms of hereditary FTDP‐17. Science 282:1914–1917. [DOI] [PubMed] [Google Scholar]
- 60. Hutton M (2001) Missense and splice site mutations in tau associated with FTDP‐17: multiple pathogenic mechanisms. Neurology 56:S21–S25. [DOI] [PubMed] [Google Scholar]
- 61. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering‐Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, De Graaff E, Wauters E, Van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, Van Swieten J, Mann D, Lynch T, Heutink P (1998) Association of missense and 5’‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature 393:702–705. [DOI] [PubMed] [Google Scholar]
- 62. Ihara Y, Abraham C, Selkoe DJ (1983) Antibodies to paired helical filaments in Alzheimer’s disease do not recognize normal brain proteins. Nature 304:727–730. [DOI] [PubMed] [Google Scholar]
- 63. Illenberger S, Zheng‐Fischhöfer Q, Preuss U, Stamer K, Baumann K, Trinczek B, Biernat J, Godemann R, Mandelkow E‐M, Mandelkow E (1998) The endogenous and cell‐cycle dependent phosphorylation of tau protein in living cells: implications for Alzheimer’s disease. Mol Biol Cell 9: 1495–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Inouye H, Sharma D, Goux WJ, Kirschner DA (2006) Structure of core domain of fibril‐forming PHF/Tau fragments. Biophys J 90:1774–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Iqbal K, Grundke‐Iqbal I (2005) Metabolic/signal transduction hypothesis of Alzheimer’s disease and other tauopathies. Acta Neuropathol (Berl) 109:25–31. [DOI] [PubMed] [Google Scholar]
- 66. Jakes R, Novak M, Davison M, Wischik CM (1991) Identification of 3‐ and 4‐repeat tau isoforms within the PHF in Alzheimer’s disease. EMBO J 10:2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jeganathan S, Von Bergen M, Brutlach H, Steinhoff H‐J, Mandelkow E (2006) Global hairpin folding of tau in solution. Biochemistry 45:2283–2293. [DOI] [PubMed] [Google Scholar]
- 68. Jicha GA, Bowser R, Kazam IG, Davies P (1997) Alz‐50 and MC‐1, a new monoclonal‐antibody raised to paired helical filaments, recognize conformational epitopes on recombinant‐tau. J Neurosci Res 48:128–132. [DOI] [PubMed] [Google Scholar]
- 69. Jicha GA, Berenfeld B, Davies P (1999) Sequence requirements for formation of conformational variants of tau similar to those found in Alzheimer’s disease. J Neurosci Res 55:713–723. [DOI] [PubMed] [Google Scholar]
- 70. Kampers T, Friedhoff P, Biernat J, Mandelkow E‐M, Mandelkow E (1996) RNA stimulates aggregation of microtubule‐associated protein tau into Alzheimer‐like paired helical filaments. FEBS Lett 399:344–349. [DOI] [PubMed] [Google Scholar]
- 71. Khlistunova I, Biernat J, Wang Y‐P, Pickhardt M, Von Bergen M, Gazova Z, Mandelkow E, Mandelkow E‐M (2006) Inducible expression of tau in cell models of Alzheimer’s disease: aggregation is toxic to cells but can be rescued by inhibitor drugs. J Biol Chem 281:1205–1214. [DOI] [PubMed] [Google Scholar]
- 72. Kidd M (1963) Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature 197:192–193. [DOI] [PubMed] [Google Scholar]
- 73. Kirschner DA, Abraham C, Selkoe DJ (1986) X‐ray diffraction from intraneural paired helical filaments and extraneural amyloid fibers in Alzheimer disease indicates cross‐beta conformation. Proc Natl Acad Sci USA 83:503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kishi M, Pan YA, Crump JG, Sanes JR (2005) Mammalian SAD kinases are required for neuronal polarization. Science 307:929–932. [DOI] [PubMed] [Google Scholar]
- 75. Kosik KS, Joachim CL, Selkoe DJ (1986) Microtubule‐associated protein tau is a major antigenic component of paired helical filaments in Alzheimer’s disease. Proc Natl Acad Sci USA 83:4044–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kosik KS, Orecchio LD, Bakalis S, Neve RL (1989) Developmentally regulated expression of specific tau sequences. Neuron 2:1389–1397. [DOI] [PubMed] [Google Scholar]
- 77. Ksiezak‐Reding H, Wall JS (2005) Characterization of paired helical filaments by scanning transmission electron microscopy. Microsc Res Tech 67:126–140. [DOI] [PubMed] [Google Scholar]
- 78. Lee G, Cowan N, Kirschner M (1988) The primary structure and heterogeneity of tau protein from mouse brain. Science 239:285–288. [DOI] [PubMed] [Google Scholar]
- 79. Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, Do LH, Andreadis A, Van Hoesen G, Ksiezak‐Reding H (2004) Phosphorylation of tau by fyn: implications for Alzheimer’s disease. J Neurosci 24:2304–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lee VMY, Balin BJ, Otvos L, Trojanowski JQ (1991) A68—a major subunit of paired helical filaments and derivatized forms of normal tau. Science 251:675–678. [DOI] [PubMed] [Google Scholar]
- 81. Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn‐Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M (2000) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25:402–405. [DOI] [PubMed] [Google Scholar]
- 82. Lewis SA, Wang D, Cowan NJ (1988) Microtubule‐associated protein MAP2 shares a microtubule binding motif with tau protein. Science 242:936–939. [DOI] [PubMed] [Google Scholar]
- 83. Lu M, Kosik KS (2001) Competition for microtubule‐binding with dual expression of tau missense and splice isoforms. Mol Biol Cell 12:171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. McGowan E, Eriksen J, Hutton M (2006) A decade of modeling Alzheimer’s disease in transgenic mice. Trends Genet 22:281–289. [DOI] [PubMed] [Google Scholar]
- 85. Makrides V, Massie MR, Feinstein SC, Lew J (2004) Evidence for two distinct binding sites for tau on microtubules. Proc Natl Acad Sci USA 101:6746–6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Margittai M, Langen R (2004) Template‐assisted filament growth by parallel stacking of tau. Proc Natl Acad Sci USA 101:10278–10283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Margittai M, Langen R (2006) Side chain‐dependent stacking modulates tau filament structure. J Biol Chem 281:37820–37827. [DOI] [PubMed] [Google Scholar]
- 88. Mukrasch MD, Biernat J, Von Bergen M, Griesinger C, Mandelkow E, Zweckstetter M (2005) Sites of tau important for aggregation populate beta structure and bind to microtubules and polyanions. J Biol Chem 280:24978–24986. [DOI] [PubMed] [Google Scholar]
- 89. Nacharaju P, Lewis J, Easson C, Yen S, Hackett J, Hutton M, Yen SH (1999) Accelerated filament formation from tau protein with specific FTDP‐17 mutations. FEBS Lett 447:195–199. [DOI] [PubMed] [Google Scholar]
- 90. Nelson R, Eisenberg D (2006) Recent atomic models of amyloid fibril structure. Curr Opin Struct Biol 16:260–265. [DOI] [PubMed] [Google Scholar]
- 91. Novak M, Jakes R, Edwards P, Milstein C, Wischik C (1991) Difference between the tau protein of Alzheimer paired helical filament core and normal tau revealed by epitope analysis of monoclonal antibodies 423 and 7.51. Proc Natl Acad Sci USA 88:5837–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Nukina N, Ihara Y (1986) One of the antigenic determinants of paired helical filaments is related to tau protein. J Biochem (Tokyo) 99:1541–1544. [DOI] [PubMed] [Google Scholar]
- 93. Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple‐transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39:409–421. [DOI] [PubMed] [Google Scholar]
- 94. Olmsted JB (1991) Non‐motor microtubule‐associated proteins. Curr Opin Cell Biol 3:52–58. [DOI] [PubMed] [Google Scholar]
- 95. Park SY, Ferreira A (2005) The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta‐amyloid‐induced neurodegeneration. J Neurosci 25:5365–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Pei JJ, An WL, Zhou XW, Nishimura T, Norberg J, Benedikz E, Götz J, Winblad B (2006) P70 S6 kinase mediates tau phosphorylation and synthesis. FEBS Lett 580:107–114. [DOI] [PubMed] [Google Scholar]
- 97. Perez M, Valpuesta JM, Medina M, Degarcini EM, Avila J (1996) Polymerization of tau into filaments in the presence of heparin: the minimal sequence required for tau‐tau‐interaction. J Neurochem 67:1183–1190. [DOI] [PubMed] [Google Scholar]
- 98. Pickhardt M, Gazova Z, Von Bergen M, Khlistunova I, Wang Y‐P, Hascher A, Mandelkow E‐M, Biernat J, Mandelkow E (2005) Anthraquinones inhibit tau aggregation and dissolve Alzheimer paired helical filaments in vitro and in cells. J Biol Chem 280:3628–3635. [DOI] [PubMed] [Google Scholar]
- 99. Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43:815–825. [DOI] [PubMed] [Google Scholar]
- 100. Preuss U, Biernat J, Mandelkow E‐M, Mandelkow E (1997) The “jaws” model of tau‐microtubule interaction examined in CHO cells. J Cell Sci 110:789–800. [DOI] [PubMed] [Google Scholar]
- 101. Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, Guimaraes A, Yue M, Lewis J, Carlson G, Hutton M, Ashe KH (2005) Age‐dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci 25:10637–10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Rissman RA, Poon WW, Blurton‐Jones M, Oddo S, Torp R, Vitek MP, LaFerla FM, Rohn TT, Cotman CW (2004) Caspase‐cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest 114:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sahara N, Lewis J, DeTure M, McGowan E, Dickson DW, Hutton M, Yen SH (2002) Assembly of tau in transgenic animals expressing P301L tau: alteration of phosphorylation and solubility. J Neurochem 83:1498–1508. [DOI] [PubMed] [Google Scholar]
- 104. Samsonov A, Yu JZ, Rasenick M, Popov SV (2004) Tau interaction with microtubules in vivo. J Cell Sci 117:6129–6141. [DOI] [PubMed] [Google Scholar]
- 105. Santarella RA, Skiniotis G, Goldie KN, Tittmann P, Gross H, Mandelkow EM, Mandelkow E, Hoenger A (2004) Surface‐decoration of microtubules by human tau. J Mol Biol 339:539–553. [DOI] [PubMed] [Google Scholar]
- 106. Schneider A, Biernat J, Von Bergen M, Mandelkow E, Mandelkow E‐M (1999) Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 38:3549–3558. [DOI] [PubMed] [Google Scholar]
- 107. Schweers O, Schönbrunn E, Marx A, Mandelkow E (1994) Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta structure. J Biol Chem 269: 24290–24297. [PubMed] [Google Scholar]
- 108. Schweers O, Mandelkow E‐M, Biernat J, Mandelkow E (1995) Oxidation of cysteine 322 in the repeat domain of microtubule‐associated protein tau controls the assembly of Alzheimer paired helical filaments. Proc Natl Acad Sci USA 92:8463–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Seitz A, Kojima H, Oiwa K, Mandelkow E‐M, Song Y‐H, Mandelkow E (2002) Single‐molecule investigation of the interference between kinesin and Tau on microtubules. EMBO J 21:4896–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sillen A, Leroy A, Wieruszeski JM, Loyens A, Beauvillain JC, Buee L, Landrieu I, Lippens G (2005) Regions of tau implicated in the paired helical fragment core as defined by NMR. Chembiochem 6:1849–1856. [DOI] [PubMed] [Google Scholar]
- 111. Sloboda RD, Rudolph SA, Rosenbaum JL, Greengard P (1975) Cyclic AMP‐dependent endogenous phosphorylation of a microtubule‐associated protein. Proc Natl Acad Sci USA 72:177–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 95:7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow E‐M (2002) Tau blocks traffic of organelles, neurofilaments, and APP‐vesicles in neurons and enhances oxidative stress. J Cell Biol 156:1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Steiner B, Mandelkow E‐M, Biernat J, Gustke N, Meyer HE, Schmidt B, Mieskes G, Söling HD, Drechsel D, Kirschner MW, Goedert M, Mandelkow E (1990) Phosphorylation of microtubule‐associated protein tau: identification of the site for Ca++‐calmodulin dependent kinase and relationship with tau phosphorylation in Alzheimer tangles. EMBO J 9:3539–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Stoothoff WH, Johnson GV (2005) Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta 1739:280–297. [DOI] [PubMed] [Google Scholar]
- 116. Taniguchi S, Suzuki N, Masuda M, Hisanaga S, Iwatsubo T, Goedert M, Hasegawa M (2005) Inhibition of heparin‐induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J Biol Chem 280:7614–7623. [DOI] [PubMed] [Google Scholar]
- 117. Terry RD (1963) The fine structure of neurofibrillary tangles in Alzheimer’s disease. J Neuropathol Exp Neurol 22:629–642. [DOI] [PubMed] [Google Scholar]
- 118. Terwel D, Dewachter I, Van Leuven F (2002) Axonal transport, tau protein, and neurodegeneration in Alzheimer’s disease. Neuromolecular Med 2:151–165. [DOI] [PubMed] [Google Scholar]
- 119. Tycko R (2004) Progress towards a molecular‐level structural understanding of amyloid fibrils. Curr Opin Struct Biol 14:96–103. [DOI] [PubMed] [Google Scholar]
- 120. Vogelsberg‐Ragaglia V, Bruce J, Richter‐Landsberg C, Zhang B, Hong M, Trojanowski JQ, Lee VM (2000) Distinct FTDP‐17 missense mutations in tau produce tau aggregates and other pathological phenotypes in transfected CHO cells. Mol Biol Cell 11:4093–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW (1975) A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA 72:1858–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Weisenberg RC (1972) Microtubule formation in vitro in solutions containing low calcium concentrations. Science 177:1104–1105. [DOI] [PubMed] [Google Scholar]
- 123. Wille H, Drewes G, Biernat J, Mandelkow E‐M, Mandelkow E (1992) Alzheimer‐like paired helical filaments and antiparallel dimers formed from microtubule‐associated protein tau in vitro. J Cell Biol 118:573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Wilson D, Binder LI (1997) Free fatty acids stimulate the polymerization of tau and amyloid Abeta peptides: in vitro evidence for a common effector of pathogenesis in Alzheimer’s disease. Am J Pathol 150:2181–2195. [PMC free article] [PubMed] [Google Scholar]
- 125. Wischik CM, Novak M, Thogersen HC, Edwards PC, Runswick MJ, Jakes R, Walker JE, Milstein C, Roth M, Klug A (1988) Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci USA 85:4506–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wisniewski HM, Merz PA, Iqbal K (1984) Ultrastructure of paired helical filaments of Alzheimer’s neurofibrillary tangle. J Neuropathol Exp Neurol 43:643–656. [DOI] [PubMed] [Google Scholar]
- 127. Wood JG, Mirra SS, Pollock NJ, Binder LI (1986) Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the microtubule‐associated protein tau. Proc Natl Acad Sci USA 83:4040–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Woody RW, Clark DC, Roberts GC, Martin SR, Bayley PM (1983) Molecular flexibility in microtubule proteins: proton nuclear magnetic resonance characterization. Biochemistry 22: 2186–2192. [DOI] [PubMed] [Google Scholar]
- 129. Zingsheim HP, Herzog W, Weber K (1979) Differences in surface morphology of microtubules reconstituted from pure brain tubulin using two different microtubule‐associated proteins: the high molecular weight MAP 2 proteins and tau proteins. Eur J Cell Biol 19:175–183. [PubMed] [Google Scholar]