

Abstract
In Alzheimer's disease (AD) Aβ accumulates because of imbalance between the production of Aβ and its removal from the brain. There is increasing evidence that in most sporadic forms of AD, the accumulation of Aβ is partly, if not in some cases solely, because of defects in its removal—mediated through a combination of diffusion along perivascular extracellular matrix, transport across vessel walls into the blood stream and enzymatic degradation. Multiple enzymes within the central nervous system (CNS) are capable of degrading Aβ. Most are produced by neurons or glia, but some are expressed in the cerebral vasculature, where reduced Aβ‐degrading activity may contribute to the development of cerebral amyloid angiopathy (CAA). Neprilysin and insulin‐degrading enzyme (IDE), which have been most extensively studied, are expressed both neuronally and within the vasculature. The levels of both of these enzymes are reduced in AD although the correlation with enzyme activity is still not entirely clear. Other enzymes shown capable of degrading Aβin vitro or in animal studies include plasmin; endothelin‐converting enzymes ECE‐1 and ‐2; matrix metalloproteinases MMP‐2, ‐3 and ‐9; and angiotensin‐converting enzyme (ACE). The levels of plasmin and plasminogen activators (uPA and tPA) and ECE‐2 are reported to be reduced in AD. Reductions in neprilysin, IDE and plasmin in AD have been associated with possession of APOEε4. We found no change in the level or activity of MMP‐2, ‐3 or ‐9 in AD. The level and activity of ACE are increased, the level being directly related to Aβ plaque load. Up‐regulation of some Aβ‐degrading enzymes may initially compensate for declining activity of others, but as age, genetic factors and diseases such as hypertension and diabetes diminish the effectiveness of other Aβ‐clearance pathways, reductions in the activity of particular Aβ‐degrading enzymes may become critical, leading to the development of AD and CAA.
Keywords: Alzheimer's disease, cerebral amyloid angiopathy, Aβ peptidases, neprilysin, insulin‐degrading enzyme, angiotensin‐converting enzyme, plasmin, endothelin‐converting enzyme, matrix metalloproteases
INTRODUCTION
Aβ accumulation is thought to be central to the pathogenesis of Alzheimer's disease (AD) (62); evidence includes the finding that familial autosomal dominant forms of AD all result from gene mutations that increase either both Aβ1–40 and Aβ1–42, or the ratio of Aβ1–42 : Aβ1–40. The mechanism of Aβ accumulation in cases of late‐onset sporadic AD (LOAD) is less clear. Although some groups have found increased levels or activity of β‐secretase (the rate‐limiting enzyme in Aβ synthesis) in LOAD, other evidence suggests that this is secondary to Aβ accumulation rather than a primary abnormality. The level of Aβ within the brain depends not only on the rate of Aβ production, but also on the rate of its removal through various clearance pathways and by enzyme‐mediated degradation. Several candidate Aβ peptidases are expressed both neuronally and within the cerebral vasculature. This review will focus on studies on human post‐mortem brain tissue of the potential relevance of some of these peptidases to the pathogenesis of AD.
Aβ ACCUMULATION IN AD
Aβ production results from amyloidogenic processing of amyloid precursor protein (APP): the sequential cleavage of APP by β‐ and γ‐secretases 51, 95, 131. The predominant species of Aβ thereby produced are Aβ1–40 and, in lesser amounts, Aβ1–42. Cleavage by α‐secretase of APP within the Aβ segment (non‐amyloidogenic processing) prevents the formation of Aβ24, 79, 84. When present in excess, extracellular Aβ1–42, which is more prone to aggregate than Aβ1–40 (71), tends to precipitate within the brain parenchyma, forming plaques, whereas Aβ1–40 is more likely to reach the cerebral blood vessels and to accumulate within the vascular and perivascular extracellular matrix, leading to cerebral amyloid angiopathy (CAA), present in over 90% of patients with AD.
Excessive accumulation of Aβ in early‐onset familial AD (EOAD) and hereditary CAA, results from increased amyloidogenic processing of APP, often associated with an increased ratio of Aβ1–42 : Aβ1–40 115, 128. This occurs as a result of autosomal dominant mutations in the genes encoding APP (APP), presenilin‐1 (PSEN‐1) or presenilin‐2 (PSEN‐2) (141), or from increased production of APP, in trisomy 21 16, 40 or duplication of the APP locus on chromosome 21 25, 121. APP gene mutations that cause a decrease in the Aβ1–42 : Aβ1–40 ratio (ie, a relative excess of Aβ1–40) usually cause familial CAA(68).
These data constitute strong evidence that Aβ accumulation is central to the pathogenesis of not only AD but also CAA. To date, there is however, little evidence to suggest that an increase in neuronal synthesis of Aβ, or an increase in the overall level of Aβ production via amyloidogenic processing, is responsible for the development of the majority of LOAD and sporadic CAA cases. Several groups have found that the level of β‐secretase‐1 (BACE‐1) activity, the rate‐limiting enzyme in Aβ synthesis, is increased in AD cases compared with controls (reviewed in Stockley and O'Neill (138). Tyler et al (148) observed increased BACE‐1 activity, and a concurrent decrease in α‐secretase activity in tissue homogenates from the temporal cortex in AD. Increased BACE‐1 activity in AD was found to result from a rise in the maximum rate of enzyme activity, mediated by a post‐translational mechanism, rather than a change in BACE‐1 protein levels, which were reduced in AD, especially in cases with advanced disease (139). A study of two strains of mouse transgenic for mutant human APP (one causing early and one late accumulation of Aβ) revealed an increase in the amount of BACE‐1 only after the commencement of Aβ plaque formation (168). Elevated BACE‐1 levels appeared to be restricted to the perimeter pf plaques, and occurred without any change in BACE‐1 mRNA level. These findings argue against a primary role for BACE‐1 in the formation of plaques but suggest that BACE‐1 activation, via a positive feedback loop initiated by parenchymal accumulation of Aβ, could exacerbate Aβ production in AD.
Evidence obtained over the past decade points to deficiencies in Aβ clearance or enzyme‐mediated Aβ degradation as factors potentially responsible for Aβ accumulation, particularly in LOAD and CAA. Intact, soluble Aβ is probably cleared from the brain by several routes: low‐density lipoprotein receptor‐related protein‐1 (LRP‐1)‐mediated transport across vessel walls into the circulation (134), transport of Aβ across the blood–brain barrier, from the abluminal to the luminal side via the P‐glycoprotein (PgP/MDR1/ABCB1) efflux pump 81, 83, 154, and drainage along perivascular basement membranes, possibly into the cerebrospinal fluid (CSF) 114, 158, 159. Aβ can potentially gain access to the CNS from the blood, through binding to receptor for advanced glycation end products (RAGE) in endothelial cells 38, 92. Zlokovic et al recently showed that Aβ within the blood stream binds to soluble LRP‐1; this acts as a peripheral sink, sequestering the circulating Aβ and promoting the passage of soluble Aβ out of CNS (123). The above studies are discussed in detail in accompanying papers in this symposium.
While some soluble Aβ is therefore cleared from the brain still intact, Aβ also undergoes enzymatic degradation 43, 156. Some of the candidate Aβ peptidases can cleave only soluble monomeric Aβ. Others are able to degrade Aβ oligomers and even fibrillar aggregates, that is, forms of Aβ that cannot directly be cleared into the blood or CSF.
CANDIDATE Aβ PEPTIDASES IN THE CNS
Multiple enzymes have been identified that can cleave at either a single or multiple sites within Aβ. The cleavage products are less likely to aggregate and less neurotoxic than Aβ itself. The contributions of these enzymes to the normal homeostasis of Aβ by neurons in vitro and within the CNS of experimental animals have been the subject of a large number of studies. Although some of this information is touched upon below, the emphasis in the present paper is on findings that have been validated by post‐mortem examination of human brain tissue.
Neprilysin
Neprilysin (NEP, also known as neutral endopeptidase‐24.11, EC.3.4.24.11, enkephalinase, neutrophil cluster‐differentiation antigen 10 or common acute lymphoblastic leukemia antigen) is a 90–110 kDa plasma membrane glycoprotein of the neutral zinc metalloendopeptidase family 145, 146, 147. Within the brain, NEP is expressed at pre‐ and post‐synaptic membranes and is involved in the regulation of neuropeptide signaling 13, 57, 135. It is also expressed in the tunica media and endothelium of cortical and leptomeningeal blood vessels 29, 98, where it is involved in the regulation of vascular tone 31, 118.
The first evidence of deficiencies in NEP in AD came from two studies showing that NEP mRNA and protein levels were significantly lower in regions of brain with high plaque burdens, and mRNA levels were lower in hippocampus from AD than control brains despite preservation of MAP‐2 (neuronal) mRNA 4, 165. Subsequent studies yielded inconsistent findings: non‐significant reduction in NEP mRNA in the frontal cortex and hippocampus in AD (29), significant reduction in NEP mRNA in the frontal cortex in AD (122), and no difference in NEP mRNA levels between cases and controls (65).
Antibodies to NEP label neurons within the cerebral cortex (4), particularly pyramidal neurons 29, 98. Carpentier et al (29) reported diminished immunolabeling of hippocampal pyramidal neurons in AD. We found significantly reduced NEP labeling in frontal and temporal neurons in AD and a non‐significant reduction in the amount of NEP in homogenates of frontal cortex (98).
Carpentier et al (29) also noted an inverse relationship between the amounts of Aβ and NEP in the cerebral vasculature. We confirmed this and showed that vessel‐associated NEP is significantly reduced in AD, the reduction being inversely related to CAA severity. In cases of AD with severe CAA, NEP levels were equally reduced in the Aβ‐laden and Aβ‐free vessels (98). The findings indicated that the reduction in cerebrovascular NEP in AD is not simply due to replacement of tunica media by Aβ. Carpentier et al (29) also observed reduced NEP in vessels in which the tunica media had not been destroyed, pointing to global reduction in vessel‐associated NEP as a potential cause of CAA.
Further support for a role for NEP in normally preventing CAA comes from the finding that NEP cannot cleave the abnormal forms of Aβ that result from Dutch, Flemish, Italian or Arctic mutations in APP and which are all associated with severe CAA (142). It is also of note that NEP cleaves monomeric Aβ1–40 more efficiently than it does Aβ1–42 (73). A deficiency in NEP would therefore be expected to decrease the ratio of Aβ1–42 : Aβ1–40, favoring vascular deposition of Aβ.
We found a significant association between NEP labeling and APOE genotype, the only well‐established genetic risk factor for both AD and CAA 30, 33, 53, 129, 140. Possession of APOEε4 was associated with reduced immunolabeling of neurons and vessels for NEP (98). Colinearity of ε4 with the presence of moderate to severe CAA precluded assessment of the independence of this association from NEP levels. However, logistic regression analysis showed low NEP labeling to be a significant independent predictor of moderate to severe CAA. The precise relationship between NEP immunolabeling and enzyme activity, particularly in the vascular tunica media, remains to be established, although the likelihood is that these are directly related.
Correlation between immunolabeling studies and measurements of NEP protein levels in human brain tissue homogenates (usually by densitometry of Western blots) has generally been poor. In most studies NEP levels have not differed significantly between AD cases and controls 65, 98, 122. Wang et al (155) found significantly reduced NEP levels in the mid‐frontal cortex in AD (but not frontotemporal degeneration, suggesting that the reduction in NEP in AD was not simply secondary to neuronal loss). NEP levels correlated directly with brain weight and synaptic protein levels and inversely with plaque counts, formic acid‐extractable Aβ1–40 and Aβ1–42, neurofibrillary tangle counts and phospho‐tau levels. Hellstrom‐Lindahl et al (65) also reported a significant inverse relationship between NEP levels and insoluble Aβ1–40 and Aβ1–42 in the temporal and frontal cortex, in both control and AD brains.
The levels of NEP in post‐mortem brains were shown to decline with age (65). It may be of relevance that the levels of somatostatin, which up‐regulates NEP activity, also fall with age and in AD (124). Somatostatin levels in the frontal cortex were significantly lower in AD cases positive for APOEε4 (61). An APOE‐dependent genetic association between AD and three single nucleotide polymorphism (SNPs) within the somatostatin gene (3q27.3) was reported in a Finnish population (151). The biological link between somatostatin, NEP and APOEε4 merits further exploration.
NEP enzyme activity in CSF is significantly reduced in AD and patients with mild cognitive impairment (MCI) [a prodromal form of cognitive decline that has a high later conversion rate to AD (153)] compared with controls (94). However, when the AD patients were subdivided according to severity of disease, the most severely affected patients showed a significant increase in NEP activity, with levels similar to those in the controls. It is not clear whether NEP activity in the CSF parallels that in brain tissue but these data suggest that (i) CSF measurement of NEP activity has potential as a biomarker of early AD, and (ii) there may be late‐stage up‐regulation of NEP activity (eg, as a response to elevated Aβ). We (98) and others (155) used fluorogenic peptide substrates to measure NEP activity in brain homogenates and found this to be significantly reduced in AD. However, when applied to brain homogenates, the specificity of this method for NEP is questionable (100). We have recently developed immunocapture‐based fluorogenic peptide cleavage assays (100) that allow the measurement of NEP and IDE enzyme activity levels in brain homogenates with much greater specificity (Figure 1).
Figure 1.
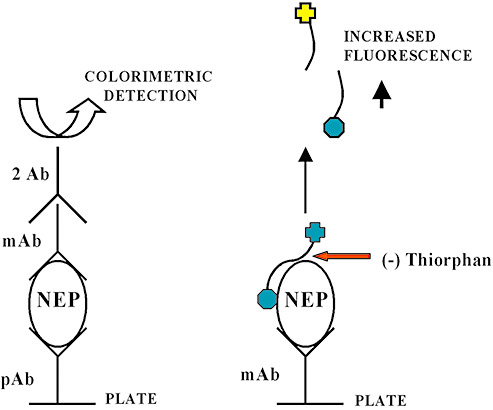
Schematic representation of immunocapture‐based fluorometric enzyme activity assay for the measurement of neprilysin (NEP) and insulin‐degrading (IDE) enzyme activity within brain tissue homogenates. The initial immunocapture phase (similar to a standard indirect sandwich ELISA method), prior to the addition of the fluorogenic substrate, allows the specific measurement of NEP enzyme activity in biological tissues separate from other closely related enzymes. Specificity is demonstrated by almost complete inhibition of fluorogenic peptide substrate cleavage in the presence of thiorphan (200 nM), a NEP‐specific inhibitor. The assay combines high sensitivity and specificity and can be adapted to a 96‐well plate format to permit high‐throughput analysis. ELISA = Enzyme‐linked immunosorbent assay.
Some genetic studies have shown strong associations between polymorphisms in the gene‐encoding NEP (MME) and AD 64, 125, 133 whereas others have found no associations 111, 137. Yamada et al (163) reported that a GT repeat polymorphism in the enhancer/promoter region of NEP was associated with CAA severity.
Insulin‐degrading enzyme
Insulin‐degrading enzyme (IDE) (insulysin, insulinase, EC3.4.24.56) is a zinc metalloendopeptidase that is highly expressed in the liver, testis, muscle and brain (82). It is a single polypeptide with a molecular weight of 110 kDa and is encoded by a gene (IDE) on chromosome 10q23–q25 (2). IDE is predominantly cytosolic 3, 17, 41, with smaller amounts in peroxisomes 8, 103, rough endoplasmic reticulum and plasma membranes 60, 132. A secreted form of IDE is present within extracellular compartments such as CSF (117). IDE degrades a wide range of substrates that share a common amyloidogenic secondary protein structure and include insulin 42, 60, 78, amylin (17), insulin‐like growth factors I and II (101) and Aβ96, 113, 117.
IDE is predominantly expressed neuronally within the brain 18, 32 and has been demonstrated in Aβ plaques and endothelial cells (102). In AD, immunolabeling suggested that neuronal and plaque‐associated IDE was increased in AD (18) but quantitative in situ hybridization demonstrated reduced neuronal IDE mRNA in the dentate granule cells, hilus and CA2‐3 fields of the hippocampus, and Western blot analysis of hippocampal homogenates showed decreased IDE protein levels. The reduction of IDE mRNA and protein in AD was APOEε4 dependent (32). Our immnuolabeling studies have shown that neuronal IDE expression within the pyramidal cells of the hippocampus is highest in CA3/4 neurons and lowest in the CA1 subfield and is significantly reduced in AD cases compared with controls ( Figure 2). In our cohort IDE immunolabeling levels varied with APOE genotype but not significantly, and were lowest in patients with advanced Braak tangle stages and high temporal Aβ plaque load.
Figure 2.
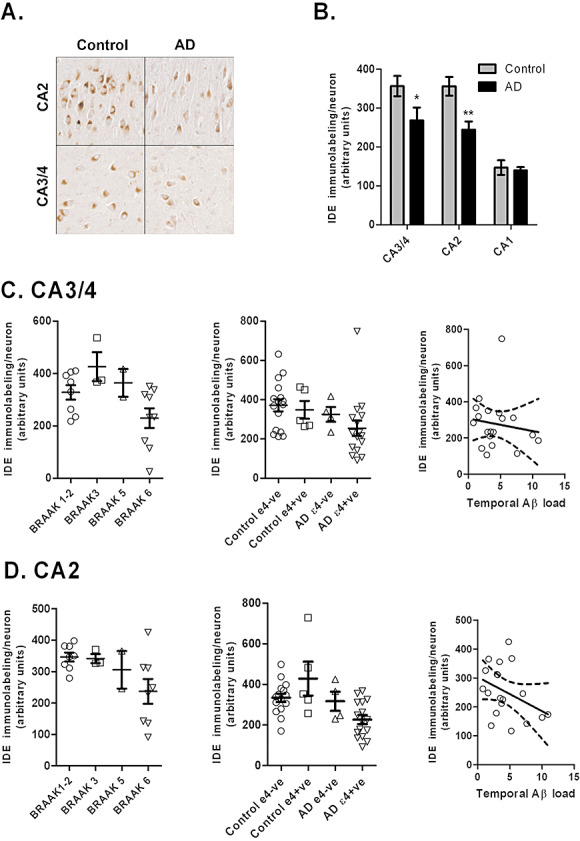
(A) Insulin‐degrading enzyme (IDE) immunolabeling of neurons within the CA3‐4 and CA2 regions of the hippocampus in control cases and Alzheimer's disease (AD). (B) IDE immunolabeling of neurons is significantly reduced in AD cases (n = 20) compared with age‐matched controls (n = 22) (CA3‐4, P < 0.05 and CA2, P < 0.01). Reductions in IDE neuronal labeling in (C) CA3‐4 and (D) CA2 hippocampal regions are inversely related to Braak tangle stage, APOEε4 genotype, and Aβ plaque load in the temporal lobe. The error bars indicate standard error of mean.
Reduced levels of cystolic IDE in AD were reported by Perez et al (113). More recently, Zhao et al (169) demonstrated that hippocampal IDE protein and activity were reduced in MCI and further reduced in AD; in contrast to Perez et al (113), the reductions were within the membrane fraction and not the cytosol. The reduction in membrane‐bound IDE was region‐specific (not seen in the occipital cortex) and correlated inversely with Aβ1–42 load. Region‐specific alterations were also reported by Caccamo et al (26): reduced levels in IDE in the hippocampus and cerebral cortex but increased levels in the cerebellum.
IDE has also been detected immunohistochemically in pericytes, endothelial and cerebrovascular smooth muscle cells 59, 102. IDE was present within occasional vessels in AD but not controls (18). Measurement of IDE levels in vessel‐enriched preparations of brain tissue showed cases with severe CAA to have elevated IDE levels but reduced IDE activity (102), raising the possibility that inactivation or inhibition of IDE activity may contribute to the development of CAA.
There is evidence of genetic linkage of AD to chromosome 10 in the region to which IDE is mapped 19, 104. Several studies observed an association between IDE haplotypes 20, 21, 50, 56, 116, and SNPs within the IDE gene, with AD (152). Other studies 1, 23, 126 have not found significant associations. Genetic variation in close proximity to the IDE gene was found to be associated with clinical disease severity, plaque and neurofibrillary tangle density (116) and plasma Aβ42 levels in AD patients, (50) providing further support for a genetic association with AD. Naturally occurring splice variants of IDE associated with AD were reported to have reduced catalytic activity 54, 55.
Endothelin‐converting enzymes
Endothelin‐converting enzymes ECE‐1 and ECE‐2 (EC 3.4.24.71) are type II integral membrane zinc metalloendopeptidases (144) that are primarily localized to the endothelium throughout the human vasculature (36). They share common catalytic substrates and are responsible for cleaving big endothelins to produce potent vasoconstrictor endothelins 48, 162. In man, four isoforms of ECE‐1 are encoded by a single gene on chromosome 1 (1p36). These differ in their subcellular location: ECE‐1a, 1c and 1d are located predominantly in the plasma membrane 130, 150; ECE‐1b is predominantly intracellular. ECE‐2 is present intracellularly and has an acidic pH optimum in contrast to the neutral pH optimum of ECE‐1 (48).
Immunolabeling of rat (14) and bovine brain (48) has revealed ECE‐1 in neurons, specifically within pyramidal neurons of the hippocampus and layer V of the neocortex (136). A few astrocytes were also labeled (136). In mice, ECE‐2 labeling is largely confined to the brain (164); in the rat brain, ECE‐1 and ECE‐2 mRNA was localized to neurons (105).
Limited information is available on the expression of ECE‐1 and ‐2 in human brain. ECE‐1 immunoreactivity was seen within neurons and their processes in the cerebral cortex (37). We found the strongest labeling for ECE‐1 to be within the cerebrovascular endothelium, whereas ECE‐2 was predominantly neuronal, with strong labeling of hippocampal pyramidal neurons ( Figure 3).
Figure 3.
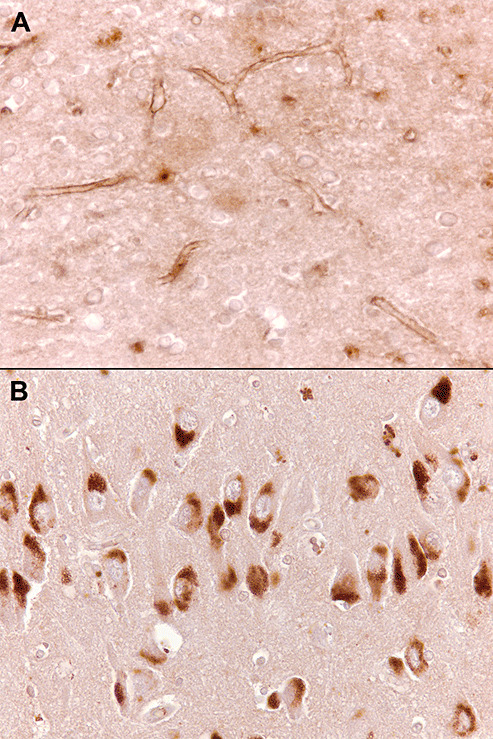
Endothelin‐converting enzyme (ECE‐1) in sections of temporal lobe. A. Immunolabeling ECE‐1 is largely confined to the endothelium. B. Neurons in the CA2 subfield of the hippocampus are strongly immunopositive for ECE‐2.
Although several experimental studies have highlighted a role for ECE‐1 and ECE‐2 in degradation of Aβ44, 45, 46 there are few data from studies on human tissue. A recent microarray study, looking at changes in gene expression in the inferior parietal lobe in LOAD, revealed a highly significant decrease in ECE‐2 gene expression, confirmed by RT‐PCR (157). Immunolabeling showed loss of ECE‐2 from neurons; similar loss was not seen in Parkinson's disease dementia, hippocampal sclerosis or dementia lacking distinctive histological features. Yoshizawa et al (167) found ET‐1 levels in human CSF to be lower in AD patients than controls; it remains to be established whether or not the lower ET‐1 levels are caused by reduced ECE activity.
A SNP in ECE‐1 (chromosome 1p36) was associated with increased hippocampal neuronal ECE‐1 expression and protection from AD in a large case–control study (58). No studies have assessed genetic associations of ECE‐2 with AD.
Angiotensin‐converting enzyme
Angiotensin‐converting enzyme (ACE; peptidyl‐dipeptidase A; EC 3.4.1.5.1) is a membrane‐bound zinc metalloprotease that is widely expressed in the vasculature throughout the body. ACE is encoded by a gene (ACE) on chromosome 17q23.3 and has an important role in the regulation of fluid homeostasis and blood pressure 49, 119. The two major physiological substrates for ACE are angiotensin I (which is converted by ACE to the vasoconstrictor angiotensin II) and bradykinin. Within the human brain, ACE has been detected predominantly in pyramidal neurons in the cortex (layer V) and within the cerebral vasculature 99, 127.
Human genetic studies have provided strong evidence of an association between ACE and AD. Several studies, including a large meta‐analysis, revealed that possession of an insertion (I) polymorphism within intron 16 of ACE is strongly associated with AD risk whereas the deletion (D) variant is protective 47, 75, 76, 77, 90, 107. In a study concerned with the genetic basis of hypertension, serum ACE protein levels were found to be lower in people with the I/I (higher AD risk) genotype (120). These findings have been replicated in human post‐mortem CSF samples from our own cohort; ACE protein levels were significantly decreased in CSF from AD cases compared with controls and were significantly lower in CSF from individuals with the I/I genotype (Figure 4). In vitro studies have shown that ACE can cleave Aβ1–40 and Aβ1–42 66, 69, 110 and converts Aβ1–42 to Aβ1–40 (170). However, Lendon et al (91) found no relationship between ACE genotype and either Aβ load or CAA severity. Other studies have not shown ACE genotype to be associated with vascular dementia or vascular pathology 80, 90.
Figure 4.
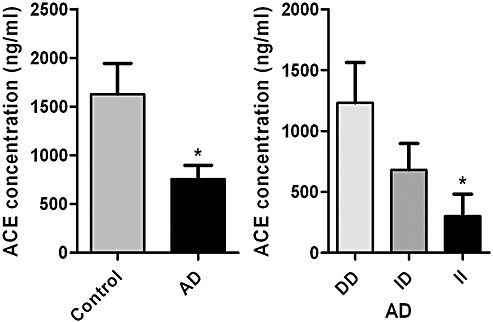
Angiotensin‐converting enzyme (ACE) protein levels in human post‐mortem cerebrospinal fluid (CSF) samples. The bars in the left chart show the mean values and standard error of the mean in Alzheimer's disease (AD) and control samples. In the right chart the levels are subdivided according to ACE (I/D) genotype. ACE levels (measured by sandwich ELISA) were significantly reduced in AD cases (n = 122) compared with controls (n = 20) (P < 0.05). ACE CSF protein levels were significantly lower in AD cases of I/I genotype (n = 29) (ie, the genotype associated with increased risk of AD) than in D/D (protective genotype) (n = 44) (P < 0.05) and lower, but not significantly, in I/D (n = 34) cases. The number of control CSF samples (I/I = 2, I/D = 8, D/D = 7) in this small series was too small for meaningful assessment of the influence of I/D genotype. ACE CSF levels did not vary according to age or gender but showed a weak positive association with post‐mortem delay (which was adjusted for in the statistical analysis). ELISA = Enzyme‐linked immunosorbent assay. *P < 0.05.
The conclusions from animal studies of the effects of ACE on Aβ homeostasis are not entirely clear. Most have found inactivation of the mouse ACE gene or inhibition of ACE in mice not to affect Aβ levels 46, 67 but Zou et al (170) reported that administration of the ACE inhibitor captopril to mice transgenic for the Swedish APP double mutation increased Aβ1–42 deposition.
In post‐mortem brain tissue, ACE protein levels and activity are raised in AD. Savaskan et al (127) described increased immunolabeling of ACE in neurons and blood vessels in the parietal cortex in AD. ACE levels were also raised in hippocampal homogenates (12) and ACE activity increased in the caudate nucleus and cerebral cortex in AD (6). We also found ACE immunolabeling and activity to be elevated in the frontal cortex in AD (99); the activity correlated directly with parenchymal Aβ plaque load, raising the possibility that intracerebral ACE levels are up‐regulated as a response to Aβ accumulation within the brain.
In our series, ACE immunolabeling in frontal and temporal cortex was predominantly perivascular and was significantly increased in moderate to severe CAA. Perivascular ACE co‐localized with the extracellular matrix marker proteins fibronectin and decorin. Angiotensin II (ANG II), the primary cleavage product of ACE, is expressed alongside ACE in the cerebral vasculature (127). The significance of perivascular ACE in CAA is unclear but it may be relevant that we have found strong labeling of blood vessels for ANG II receptor in some patients with CAA (Figure 5). ANG II stimulates the production of ECM, fibronectin in particular 34, 112, and also induces expression of transforming growth factor β (TGF‐β) and its receptors 28, 88. TGF‐β stimulates the synthesis of ECM and is associated with pathological accumulation of ECM in various inflammatory and fibrotic diseases 22, 106 and is elevated in AD (160). Over‐expression of TGF‐β by astrocytes or neurons in transgenic mice resulted in deposition of ECM and accumulation of Aβ perivascularly 149, 161.
Figure 5.
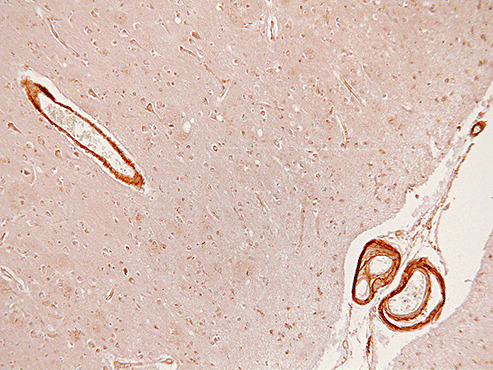
Strong labeling of angiotensin II receptor in arteriolar walls in an Alzheimer's disease (AD) patient with cerebral amyloid angiopathy (CAA).
ACE enzyme activity was increased in the CSF in a series of patients with either MCI or AD compared with controls. ACE activity levels were significantly higher in MCI cases than in AD (63). It remains unclear, however, whether enzyme activity parallels protein levels; another study found that ACE protein levels in CSF did not vary between AD patients and controls (109).
Plasmin
The serine protease, plasmin (EC 3.4.21.7), which is generated from inactive plasminogen, cleaves Aβ at multiple sites (143) and prevents the aggregation of Aβ1–42 into β‐pleated sheets (52). Generation of plasmin from plasminogen results from proteolytic cleavage by either tissue‐type (tPA) or urokinase‐type plasminogen activator (uPA). In vivo data from two separate human APP transgenic mouse models revealed that reduced activity of the tPA system may account for enhanced Aβ levels. In one APP(SI) transgenic mouse model, a decrease in tPA activity (with a corresponding increase in the level of tPA inhibitor) correlated with Aβ accumulation in 14‐month‐old mice (27). Elevated Aβ in another human APP transgenic mouse strain also correlated with increased levels of tPA inhibitor (97). In contrast, in 22‐month‐old transgenic mice, the levels of tPA and uPA activity were increased. Incubation of fibrillar (not soluble) Aβ with primary cortical neuronal cultures caused an increase in tPA and uPA mRNA levels (143). The plasmin system may participate in a feedback mechanism in which fibrillary Aβ induces tPA and uPA synthesis and formation of plasmin which in turn degrades the Aβ.
In human post‐mortem tissue, plasmin levels were significantly reduced in the hippocampus and frontal cortex in AD 86, 87. The reduction was confined to APOEε4‐positive cases. The finding that several Aβ‐degrading enzymes (NEP, IDE and plasmin) show a similar relationship between APOE genotype and reduced levels or activity suggests a common pathway through which APOE mediates at least some of its influence on the risk of developing AD and CAA through effects on Aβ degradation.
The PLAU gene which encodes uPA which has been mapped to 10q22 and which lies within the reported region of linkage to AD has not been found to be associated with AD(141).
Matrix metalloproteinases
Matrix metalloproteinases (MMPs) are zinc‐ and calcium‐dependent endopeptidases, several of which are produced by neurons and glial cells. MMP‐2 (EC3.4.24.24), ‐3 (EC 3.4.24B6) and ‐9 (EC 3.4.24.35) all have Aβ‐degrading activity in vitro (10). Although the Aβ‐degrading activity of these MMPs has not yet been explored in vivo, MMP‐9, unlike ECE‐1, NEP and IDE, is capable of cleaving aggregated Aβ‐fibrils (25). Asahina et al (7) reported increased MMP‐9 immunolabeling of neurons in AD, as well as labeling of neurofibrillary tangles, plaques and vessel walls. Backstrom et al (9) found MMP‐2 and ‐9 activity to be elevated in homogenates of hippocampal tissue from AD brains. Several cell types (glial, neuronal and vascular) up‐regulate endogenous MMP‐2 ‐3 and ‐9 expression in response to Aβ stimulation 39, 72, 89.
We looked at MMP‐2, ‐3 and ‐9 levels and activity in the frontal cortex in AD (11). MMP‐2 was detected immunohistochemically in the walls of some blood vessels and scattered white matter glia, MMP‐3 in and around some neurons and within occasional plaques, and MMP‐9 in many neurons. In our series, the level and activity of these three MMPs did not differ significantly between AD and control brains and were not related to Aβ plaque load. Analysis of polymorphisms in the MMP‐3 (‐1171 5A/6A) and MMP‐9 (C‐1562T) genes indicated that the ‐1171 6A MMP‐3 allele (with reduced promoter activity) was associated with AD; the MMP‐9 polymorphism was not.
UP‐REGULATION OF Aβ PEPTIDASES WITH AGE AND IN RESPONSE TO Aβ
IDE levels (85), tPA and uPA activity (143) and MMP‐2 and ‐9 activity (166) were all shown to be increased in aged mice that were transgenic for mutant human APP. Neprilysin levels were also increased, although not significantly, in aged Tg2567 mice (5). Increases in the levels of Aβ peptidases in the transgenic mice seem to parallel increases in Aβ load–somewhat surprisingly given the reduction in levels of these enzymes in AD. An observation in several studies on aged human APP transgenic mice 5, 85, 166 and post‐mortem tissue (29) is the presence of NEP, IDE or MMP‐2‐ or ‐9‐positive astrocytes in close proximity to Aβ plaques, suggesting up‐regulation by Aβ of these enzymes in astrocytes and, possibly, a role for astrocytes in the removal of Aβ, as suggested by Nicoll and Weller (108).
There is in vitro evidence that incubation of neuronal, microglial and vascular smooth muscle cells with fibrillar (not soluble) Aβ or fragments of APP can cause significant elevation in the levels of MMP‐2, ‐3 and ‐9 39, 72, 89, IDE (85) and plasminogen activators (143). These studies, together with others noted earlier, suggest that Aβ‐degrading enzymes are part of a normal regulatory process that act at several stages after Aβ synthesis, up to and including plaque formation and CAA, to limit Aβ accumulation within brain parenchyma and blood vessels (Figure 6). Reduction in the level or activity of an Aβ peptidase may be offset, or at least partially compensated for, by consequent up‐regulation of other Aβ‐degrading enzymes. However, as age, environmental factors, genetic factors, diseases such as hypertension and diabetes impact on the effectiveness of other Aβ‐clearance pathways, impairments in the activity of particular Aβ‐degrading enzymes may become critical (Figure 6).
Figure 6.
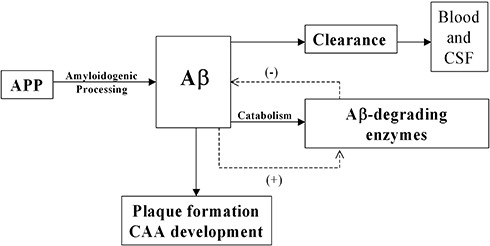
Steady‐state Aβ levels are maintained by a balance between amyloidogenic processing of amyloid precursor protein (APP) and the removal of soluble Aβ by clearance pathways and enzyme‐mediated degradation. Reduced activity of Aβ‐degrading enzymes favors Aβ accumulation. This probably initiates compensatory upregulation (+) of Aβ‐degrading enzymes, increased degradation of Aβ and reduction (−) in its accumulation. However, reduction in the level or activity of Aβ‐degrading enzymes as a result of age, genetic and environmental factors, and a decline in the efficiency of other Aβ clearance pathways, causes excessive accumulation of Aβ, plaque formation and deposition of Aβ within vessel walls.
THERAPEUTIC CONSIDERATIONS
Virus‐mediated delivery of human NEP, in mouse models of AD, has demonstrated the therapeutic potential of exogenous Aβ peptidases for reduction of Aβ levels 67, 70, 93. Most Aβ‐degrading enzymes have multiple physiological roles. NEP and ACE, for example, are involved peripherally in the regulation of blood pressure and vascular tone (118). Drugs that have been developed for the treatment of hypertension and which inhibit NEP, ACE and ECE‐1 15, 35 have the potential to act adversely on the development of AD and this will require careful assessment in animal studies and, if given to patients, close clinical monitoring and post‐mortem evaluation. Current evidence suggests, however, that agents routinely used in the treatment of hypertension, which have the ability to impact on Aβ levels if they penetrate the blood–brain barrier, offer some protection against cognitive decline in MCI and AD [reviewed in Kehoe and Wilcock (74)].
CONCLUSIONS
Aβ‐degrading enzymes are involved in the physiological regulation of Aβ levels in the brain. Altered levels or activity of several of these enzymes have been demonstrated in both AD and CAA and may be influenced by APOE genotype. The multiplicity of enzymes capable of degrading Aβ within the brain and the availability of other pathways of Aβ removal probably limit the immediate impact of deficiency of any particular Aβ‐degrading enzyme. However, as other pathways of Aβ removal decline in efficiency with age or disease, a reduced capacity for enzymatic degradation of Aβ may become critical, resulting in its accumulation within the brain parenchyma and blood vessels and the development of AD and CAA.
ACKNOWLEDGMENTS
This work was supported by Alzheimer's research Trust, the James Tudor Foundation and BRACE (Bristol Research into Alzheimer's and Care of the Elderly).
REFERENCES
- 1. Abraham R, Myers A, Wavrant‐DeVrieze F, Hamshere ML, Thomas HV, Marshall H et al (2001) Substantial linkage disequilibrium across the insulin‐degrading enzyme locus but no association with late‐onset alzheimer's disease. Hum Genet 109:646–652. [DOI] [PubMed] [Google Scholar]
- 2. Affholter JA, Hsieh CL, Francke U, Roth RA (1990) Insulin‐degrading enzyme: stable expression of the human complementary DNA, characterization of its protein product, and chromosomal mapping of the human and mouse genes. Mol Endocrinol 4:1125–1135. [DOI] [PubMed] [Google Scholar]
- 3. Akiyama H, Shii K, Yokono K, Yonezawa K, Sato S, Watanabe K, Baba S (1988) Cellular localization of insulin‐degrading enzyme in rat liver using monoclonal antibodies specific for this enzyme. Biochem Biophys Res Commun 155:914–922. [DOI] [PubMed] [Google Scholar]
- 4. Akiyama H, Kondo H, Ikeda K, Kato M, McGeer PL (2001) Immunohistochemical localization of neprilysin in the human cerebral cortex: inverse association with vulnerability to amyloid beta‐protein (Abeta) deposition. Brain Res 902:277–281. [DOI] [PubMed] [Google Scholar]
- 5. Apelt J, Ach K, Schliebs R (2003) Aging‐related down‐regulation of neprilysin, a putative beta‐amyloid‐degrading enzyme, in transgenic Tg2576 Alzheimer‐like mouse brain is accompanied by an astroglial upregulation in the vicinity of beta‐amyloid plaques. Neurosci Lett 339:183–186. [DOI] [PubMed] [Google Scholar]
- 6. Arregui A, Perry EK, Rossor M, Tomlinson BE (1982) Angiotensin converting enzyme in Alzheimer's disease increased activity in caudate nucleus and cortical areas. J Neurochem 38:1490–1492. [DOI] [PubMed] [Google Scholar]
- 7. Asahina M, Yoshiyama Y, Hattori T (2001) Expression of matrix metalloproteinase‐9 and urinary‐type plasminogen activator in Alzheimer's disease brain. Clin Neuropathol 20:60–63. [PubMed] [Google Scholar]
- 8. Authier F, Cameron PH, Taupin V (1996) Association of insulin‐degrading enzyme with a 70 kDa cytosolic protein in hepatoma cells. Biochem J 319:149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Backstrom JR, Miller CA, Tokes ZA (1992) Characterization of neutral proteinases from Alzheimer‐affected and control brain specimens: identification of calcium‐dependent metalloproteinases from the hippocampus. J Neurochem 58:983–992. [DOI] [PubMed] [Google Scholar]
- 10. Backstrom JR, Lim GP, Cullen MJ, Tokes ZA (1996) Matrix metalloproteinase‐9 (MMP‐9) is synthesized in neurons of the human hippocampus and is capable of degrading the amyloid‐beta peptide (1–40). J Neurosci 16:7910–7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baig S, Kehoe PG, Love S (in press) MMP‐2, ‐3 and ‐9 levels and activity are not related to Abeta load in the frontal cortex in Alzheimer's disease. Neuropathol Appl Neurobiol doi:10.1111/j.1365‐2990.2007.00897.x. [DOI] [PubMed] [Google Scholar]
- 12. Barnes NM, Cheng CH, Costall B, Naylor RJ, Williams TJ, Wischik CM (1991) Angiotensin converting enzyme density is increased in temporal cortex from patients with Alzheimer's disease. Eur J Pharmacol 200:289–292. [DOI] [PubMed] [Google Scholar]
- 13. Barnes K, Turner AJ, Kenny AJ (1992) Membrane localization of endopeptidase‐24.11 and peptidyl dipeptidase a (angiotensin converting enzyme) in the pig brain: a study using subcellular fractionation and electron microscopic immunocytochemistry. J Neurochem 58:2088–2096. [DOI] [PubMed] [Google Scholar]
- 14. Barnes K, Walkden BJ, Wilkinson TC, Turner AJ (1997) Expression of endothelin‐converting enzyme in both neuroblastoma and glial cell lines and its localization in rat hippocampus. J Neurochem 68:570–577. [DOI] [PubMed] [Google Scholar]
- 15. Battistini B, Daull P, Jeng AY (2005) CGS 35601, a triple inhibitor of angiotensin converting enzyme, neutral endopeptidase and endothelin converting enzyme. Cardiovasc Drug Rev 23:317–330. [DOI] [PubMed] [Google Scholar]
- 16. Belza MG, Urich H (1986) Cerebral amyloid angiopathy in Down's syndrome. Clin Neuropathol 5:257–260. [PubMed] [Google Scholar]
- 17. Bennett RG, Duckworth WC, Hamel FG (2000) Degradation of amylin by insulin‐degrading enzyme. J Biol Chem 275:36621–36625. [DOI] [PubMed] [Google Scholar]
- 18. Bernstein HG, Ansorge S, Riederer P, Reiser M, Frolich L, Bogerts B (1999) Insulin‐degrading enzyme in the Alzheimer's disease brain: prominent localization in neurons and senile plaques. Neurosci Lett 263:161–164. [DOI] [PubMed] [Google Scholar]
- 19. Bertram L, Blacker D, Mullin K, Keeney D, Jones J, Basu S et al (2000) Evidence for genetic linkage of Alzheimer's disease to chromosome 10q. Science 290:2302–2303. [DOI] [PubMed] [Google Scholar]
- 20. Bjork BF, Katzov H, Kehoe P, Fratiglioni L, Winblad B, Prince JA, Graff C (2007) Positive association between risk for late‐onset alzheimer disease and genetic variation in IDE. Neurobiol Aging 28:1374–1380. [DOI] [PubMed] [Google Scholar]
- 21. Blomqvist ME, Silburn PA, Buchanan DD, Andreasen N, Blennow K, Pedersen NL et al (2004) Sequence variation in the proximity of IDE may impact age at onset of both Parkinson disease and Alzheimer disease. Neurogenetics 5:115–119. [DOI] [PubMed] [Google Scholar]
- 22. Border WA, Ruoslahti E (1990) Transforming growth factor‐beta 1 induces extracellular matrix formation in glomerulonephritis. Cell Differ Dev 32:425–431. [DOI] [PubMed] [Google Scholar]
- 23. Boussaha M, Hannequin D, Verpillat P, Brice A, Frebourg T, Campion D (2002) Polymorphisms of insulin degrading enzyme gene are not associated with Alzheimer's disease. Neurosci Lett 329:121–123. [DOI] [PubMed] [Google Scholar]
- 24. Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ et al (1998) Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha‐secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem 273:27765–27767. [DOI] [PubMed] [Google Scholar]
- 25. Cabrejo L, Guyant‐Marechal L, Laquerriere A, Vercelletto M, De la Fourniere F, Thomas‐Anterion C et al (2006) Phenotype associated with APP duplication in five families. Brain 129:2966–2976. [DOI] [PubMed] [Google Scholar]
- 26. Caccamo A, Oddo S, Sugarman MC, Akbari Y, LaFerla FM (2005) Age‐ and region‐dependent alterations in Abeta‐degrading enzymes: implications for Abeta‐induced disorders. Neurobiol Aging 26:645–654. [DOI] [PubMed] [Google Scholar]
- 27. Cacquevel M, Launay S, Castel H, Benchenane K, Cheenne S, Buee L et al (2007) Ageing and amyloid‐beta peptide deposition contribute to an impaired brain tissue plasminogen activator activity by different mechanisms. Neurobiol Dis 27:164–173. [DOI] [PubMed] [Google Scholar]
- 28. Campbell SE, Katwa LC (1997) Angiotensin II stimulated expression of transforming growth factor‐beta1 in cardiac fibroblasts and myofibroblasts. J Mol Cell Cardiol 29:1947–1958. [DOI] [PubMed] [Google Scholar]
- 29. Carpentier M, Robitaille Y, DesGroseillers L, Boileau G, Marcinkiewicz M (2002) Declining expression of neprilysin in Alzheimer disease vasculature: possible involvement in cerebral amyloid angiopathy. J Neuropathol Exp Neurol 61:849–856. [DOI] [PubMed] [Google Scholar]
- 30. Chalmers K, Wilcock GK, Love S (2003) APOE epsilon 4 influences the pathological phenotype of Alzheimer's disease by favouring cerebrovascular over parenchymal accumulation of a beta protein. Neuropathol Appl Neurobiol 29:231–238. [DOI] [PubMed] [Google Scholar]
- 31. Chen HH, Burnett JC Jr (1999) The natriuretic peptides in heart failure: diagnostic and therapeutic potentials. Proc Assoc Am Physicians 111:406–416. [DOI] [PubMed] [Google Scholar]
- 32. Cook DG, Leverenz JB, McMillan PJ, Kulstad JJ, Ericksen S, Roth RA et al (2003) Reduced hippocampal insulin‐degrading enzyme in late‐onset alzheimer's disease is associated with the apolipoprotein E‐epsilon4 allele. Am J Pathol 162:313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW et al (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261:921–923. [DOI] [PubMed] [Google Scholar]
- 34. Crawford DC, Chobanian AV, Brecher P (1994) Angiotensin II induces fibronectin expression associated with cardiac fibrosis in the rat. Circ Res 74:727–739. [DOI] [PubMed] [Google Scholar]
- 35. Daull P, Lepage R, Benrezzak O, Cayer J, Beaudoin M, Belleville K et al (2006) The first preclinical pharmacotoxicological safety assessment of CGS 35601, a triple vasopeptidase inhibitor, in chronically instrumented, conscious, and unrestrained spontaneously hypertensive rats. Drug Chem Toxicol 29:183–202. [DOI] [PubMed] [Google Scholar]
- 36. Davenport AP, Kuc RE, Mockridge JW (1998) Endothelin‐converting enzyme in the human vasculature: evidence for differential conversion of big endothelin‐3 by endothelial and smooth‐muscle cells. J Cardiovasc Pharmacol 31(Suppl. 1):S1–S3. [DOI] [PubMed] [Google Scholar]
- 37. Davenport AP, Kuc RE, Plumpton C, Mockridge JW, Barker PJ, Huskisson NS (1998) Endothelin‐converting enzyme in human tissues. Histochem J 30:359–374. [Google Scholar]
- 38. Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E et al (2003) RAGE mediates amyloid‐beta peptide transport across the blood‐brain barrier and accumulation in brain. Nat Med 9:907–913. [DOI] [PubMed] [Google Scholar]
- 39. Deb S, Zhang JW, Gottschall PE (1999) Activated isoforms of MMP‐2 are induced in U87 human glioma cells in response to beta‐amyloid peptide. J Neurosci Res 55:44–53. [DOI] [PubMed] [Google Scholar]
- 40. Donahue JE, Khurana JS, Adelman LS (1998) Intracerebral hemorrhage in two patients with Down's syndrome and cerebral amyloid angiopathy. Acta Neuropathol 95:213–216. [DOI] [PubMed] [Google Scholar]
- 41. Duckworth WC, Bennett RG, Hamel FG (1994) A direct inhibitory effect of insulin on a cytosolic proteolytic complex containing insulin‐degrading enzyme and multicatalytic proteinase. J Biol Chem 269:24575–24580. [PubMed] [Google Scholar]
- 42. Duckworth WC, Bennett RG, Hamel FG (1998) Insulin degradation: progress and potential. Endocr Rev 19:608–624. [DOI] [PubMed] [Google Scholar]
- 43. Eckman EA, Eckman CB (2005) Abeta‐degrading enzymes: modulators of Alzheimer's disease pathogenesis and targets for therapeutic intervention. Biochem Soc Trans 33:1101–1105. [DOI] [PubMed] [Google Scholar]
- 44. Eckman EA, Reed DK, Eckman CB (2001) Degradation of the Alzheimer's amyloid beta peptide by endothelin‐converting enzyme. J Biol Chem 276:24540–24548. [DOI] [PubMed] [Google Scholar]
- 45. Eckman EA, Watson M, Marlow L, Sambamurti K, Eckman CB (2003) Alzheimer's disease beta‐amyloid peptide is increased in mice deficient in endothelin‐converting enzyme. J Biol Chem 278:2081–2084. [DOI] [PubMed] [Google Scholar]
- 46. Eckman EA, Adams SK, Troendle FJ, Stodola BA, Kahn MA, Fauq AH et al (2006) Regulation of steady‐state beta‐amyloid levels in the brain by neprilysin and endothelin‐converting enzyme but not angiotensin‐converting enzyme. J Biol Chem 281:30471–30478. [DOI] [PubMed] [Google Scholar]
- 47. Elkins JS, Douglas VC, Johnston SC (2004) Alzheimer disease risk and genetic variation in ACE: a meta‐analysis. Neurology 62:363–368. [DOI] [PubMed] [Google Scholar]
- 48. Emoto N, Yanagisawa M (1995) Endothelin‐converting enzyme‐2 is a membrane‐bound, phosphoramidon‐sensitive metalloprotease with acidic pH optimum. J Biol Chem 270:15262–15268. [DOI] [PubMed] [Google Scholar]
- 49. Erdos EG, Skidgel RA (1987) The angiotensin I‐converting enzyme. Lab Invest 56:345–348. [PubMed] [Google Scholar]
- 50. Ertekin‐Taner N, Allen M, Fadale D, Scanlin L, Younkin L, Petersen RC et al (2004) Genetic variants in a haplotype block spanning IDE are significantly associated with plasma Abeta42 levels and risk for Alzheimer disease. Hum Mutat 23:334–342. [DOI] [PubMed] [Google Scholar]
- 51. Evin G, Weidemann A (2002) Biogenesis and metabolism of Alzheimer's disease Abeta amyloid peptides. Peptides 23:1285–1297. [DOI] [PubMed] [Google Scholar]
- 52. Exley C, Korchazhkina OV (2001) Plasmin cleaves Abeta42 in vitro and prevents its aggregation into beta‐pleated sheet structures. Neuroreport 12:2967–2970. [DOI] [PubMed] [Google Scholar]
- 53. Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R et al (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta‐analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278:1349–1356. [PubMed] [Google Scholar]
- 54. Farris W, Mansourian S, Leissring MA, Eckman EA, Bertram L, Eckman CB, Tanzi RE, Selkoe DJ (2004) Partial loss‐of‐function mutations in insulin‐degrading enzyme that induce diabetes also impair degradation of amyloid beta‐protein. Am J Pathol 164:1425–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Farris W, Leissring MA, Hemming ML, Chang AY, Selkoe DJ (2005) Alternative splicing of human insulin‐degrading enzyme yields a novel isoform with a decreased ability to degrade insulin and amyloid beta‐protein. Biochemistry 44:6513–6525. [DOI] [PubMed] [Google Scholar]
- 56. Feuk L, McCarthy S, Andersson B, Prince JA, Brookes AJ (2005) Mutation screening of a haplotype block around the insulin degrading enzyme gene and association with Alzheimer's disease. Am J Med Genet B Neuropsychiatr Genet 136:69–71. [DOI] [PubMed] [Google Scholar]
- 57. Fukami S, Watanabe K, Iwata N, Haraoka J, Lu B, Gerard NP et al (2002) Abeta‐degrading endopeptidase, neprilysin, in mouse brain: synaptic and axonal localization inversely correlating with Abeta pathology. Neurosci Res 43:39–56. [DOI] [PubMed] [Google Scholar]
- 58. Funalot B, Ouimet T, Claperon A, Fallet C, Delacourte A, Epelbaum J et al (2004) Endothelin‐converting enzyme‐1 is expressed in human cerebral cortex and protects against Alzheimer's disease. Mol Psychiatry 9:1059, 1122–1128. [DOI] [PubMed] [Google Scholar]
- 59. Gao W, Eisenhauer PB, Conn K, Lynch JA, Wells JM, Ullman MD et al (2004) Insulin degrading enzyme is expressed in the human cerebrovascular endothelium and in cultured human cerebrovascular endothelial cells. Neurosci Lett 371:6–11. [DOI] [PubMed] [Google Scholar]
- 60. Goldfine ID, Williams JA, Bailey AC, Wong KY, Iwamoto Y, Yokono K et al (1984) Degradation of insulin by isolated mouse pancreatic acini. Evidence for cell surface protease activity. Diabetes 33:64–72. [DOI] [PubMed] [Google Scholar]
- 61. Grouselle D, Winsky‐Sommerer R, David JP, Delacourte A, Dournaud P, Epelbaum J (1998) Loss of somatostatin‐like immunoreactivity in the frontal cortex of Alzheimer patients carrying the apolipoprotein epsilon 4 allele. Neurosci Lett 255:21–24. [DOI] [PubMed] [Google Scholar]
- 62. Hardy J (1997) Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci 20:154–159. [DOI] [PubMed] [Google Scholar]
- 63. He M, Ohrui T, Maruyama M, Tomita N, Nakayama K, Higuchi M et al (2006) ACE activity in CSF of patients with mild cognitive impairment and Alzheimer disease. Neurology 67:1309–1310. [DOI] [PubMed] [Google Scholar]
- 64. Helisalmi S, Hiltunen M, Vepsalainen S, Iivonen S, Mannermaa A, Lehtovirta M et al (2004) Polymorphisms in neprilysin gene affect the risk of Alzheimer's disease in Finnish patients. J Neurol Neurosurg Psychiatry 75:1746–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hellström‐Lindahl E, Ravid R, Nordberg A (2006) Age‐dependent decline of neprilysin in Alzheimer's disease and normal brain: Inverse correlation with Abeta levels. Neurobiol Aging 29:210–221. [DOI] [PubMed] [Google Scholar]
- 66. Hemming ML, Selkoe DJ (2005) Amyloid beta‐protein is degraded by cellular angiotensin‐converting enzyme (ACE) and elevated by an ACE inhibitor. J Biol Chem 280:37644–37650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hemming ML, Selkoe DJ, Farris W (2007) Effects of prolonged angiotensin‐converting enzyme inhibitor treatment on amyloid beta‐protein metabolism in mouse models of Alzheimer disease. Neurobiol Dis 26:273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Herzig MC, Van Nostrand WE, Jucker M (2006) Mechanism of cerebral beta‐amyloid angiopathy: murine and cellular models. Brain Pathol 16:40–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hu J, Igarashi A, Kamata M, Nakagawa H (2001) Angiotensin‐converting enzyme degrades Alzheimer amyloid beta‐peptide (A beta); retards a beta aggregation, deposition, fibril formation; and inhibits cytotoxicity. J Biol Chem 276:47863–47868. [DOI] [PubMed] [Google Scholar]
- 70. Iwata N, Mizukami H, Shirotani K, Takaki Y, Muramatsu S, Lu B et al (2004) Presynaptic localization of neprilysin contributes to efficient clearance of amyloid‐beta peptide in mouse brain. J Neurosci 24:991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jarrett JT, Berger EP, Lansbury PT Jr (1993) The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry 32:4693–4697. [DOI] [PubMed] [Google Scholar]
- 72. Jung SS, Zhang W, Van Nostrand WE (2003) Pathogenic a beta induces the expression and activation of matrix metalloproteinase‐2 in human cerebrovascular smooth muscle cells. J Neurochem 85:1208–1215. [DOI] [PubMed] [Google Scholar]
- 73. Kanemitsu H, Tomiyama T, Mori H (2003) Human neprilysin is capable of degrading amyloid beta peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci Lett 350:113–116. [DOI] [PubMed] [Google Scholar]
- 74. Kehoe PG, Wilcock GK (2007) Is inhibition of the renin‐angiotensin system a new treatment option for Alzheimer's disease? Lancet Neurol 6:373–378. [DOI] [PubMed] [Google Scholar]
- 75. Kehoe PG, Russ C, McIlory S, Williams H, Holmans P, Holmes C et al (1999) Variation in DCP1, encoding ACE, is associated with susceptibility to Alzheimer disease. Nat Genet 21:71–72. [DOI] [PubMed] [Google Scholar]
- 76. Kehoe PG, Katzov H, Feuk L, Bennet AM, Johansson B, Wiman B et al (2003) Haplotypes extending across ACE are associated with Alzheimer's disease. Hum Mol Genet 12:859–867. [DOI] [PubMed] [Google Scholar]
- 77. Kehoe PG, Katzov H, Andreasen N, Gatz M, Wilcock GK, Cairns NJ et al (2004) Common variants of ACE contribute to variable age‐at‐onset of Alzheimer's disease. Hum Genet 114:478–483. [DOI] [PubMed] [Google Scholar]
- 78. Kirschner RJ, Goldberg AL (1983) A high molecular weight metalloendoprotease from the cytosol of mammalian cells. J Biol Chem 258:967–976. [PubMed] [Google Scholar]
- 79. Koike H, Tomioka S, Sorimachi H, Saido TC, Maruyama K, Okuyama A et al (1999) Membrane‐anchored metalloprotease MDC9 has an alpha‐secretase activity responsible for processing the amyloid precursor protein. Biochem J 343:371–375. [PMC free article] [PubMed] [Google Scholar]
- 80. Kolsch H, Jessen F, Freymann N, Kreis M, Hentschel F, Maier W, Heun R (2005) ACE I/D polymorphism is a risk factor of Alzheimer's disease but not of vascular dementia. Neurosci Lett 377:37–39. [DOI] [PubMed] [Google Scholar]
- 81. Kuhnke D, Jedlitschky G, Grube M, Krohn M, Jucker M, Mosyagin I et al (2007) MDR1‐P‐Glycoprotein (ABCB1) Mediates Transport of Alzheimer's amyloid‐beta peptides–implications for the mechanisms of Abeta clearance at the blood‐brain barrier. Brain Pathol 17:347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kuo WL, Montag AG, Rosner MR (1993) Insulin‐degrading enzyme is differentially expressed and developmentally regulated in various rat tissues. Endocrinology 132:604–611. [DOI] [PubMed] [Google Scholar]
- 83. Lam FC, Liu R, Lu P, Shapiro AB, Renoir JM, Sharom FJ, Reiner PB (2001) beta‐Amyloid efflux mediated by p‐glycoprotein. J Neurochem 76:1121–1128. [DOI] [PubMed] [Google Scholar]
- 84. Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M et al (1999) Constitutive and regulated alpha‐secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA 96:3922–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Leal MC, Dorfman VB, Gamba AF, Frangione B, Wisniewski T, Castano EM et al (2006) Plaque‐associated overexpression of insulin‐degrading enzyme in the cerebral cortex of aged transgenic tg2576 mice with Alzheimer pathology. J Neuropathol Exp Neurol 65:976–987. [DOI] [PubMed] [Google Scholar]
- 86. Ledesma MD, Da Silva JS, Crassaerts K, Delacourte A, De Strooper B, Dotti CG (2000) Brain plasmin enhances APP alpha‐cleavage and Abeta degradation and is reduced in Alzheimer's disease brains. EMBO Rep 1:530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ledesma MD, Abad‐Rodriguez J, Galvan C, Biondi E, Navarro P, Delacourte A et al (2003) Raft disorganization leads to reduced plasmin activity in Alzheimer's disease brains. EMBO Rep 4:1190–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lee AA, Dillmann WH, McCulloch AD, Villarreal FJ (1995) Angiotensin II stimulates the autocrine production of transforming growth factor‐beta 1 in adult rat cardiac fibroblasts. J Mol Cell Cardiol 27:2347–2357. [DOI] [PubMed] [Google Scholar]
- 89. Lee JM, Yin KJ, Hsin I, Chen S, Fryer JD, Holtzman DM et al (2003) Matrix metalloproteinase‐9 and spontaneous hemorrhage in an animal model of cerebral amyloid angiopathy. Ann Neurol 54:379–382. [DOI] [PubMed] [Google Scholar]
- 90. Lehmann DJ, Cortina‐Borja M, Warden DR, Smith AD, Sleegers K, Prince JA et al (2005) Large meta‐analysis establishes the ACE insertion‐deletion polymorphism as a marker of Alzheimer's disease. Am J Epidemiol 162:305–317. [DOI] [PubMed] [Google Scholar]
- 91. Lendon CL, Thaker U, Harris JM, McDonagh AM, Lambert JC, Chartier‐Harlin MC et al (2002) The angiotensin 1‐converting enzyme insertion (I)/deletion (D) polymorphism does not influence the extent of amyloid or tau pathology in patients with sporadic Alzheimer's disease. Neurosci Lett 328:314–318. [DOI] [PubMed] [Google Scholar]
- 92. Mackic JB, Stins M, McComb JG, Calero M, Ghiso J, Kim KS et al (1998) Human blood‐brain barrier receptors for Alzheimer's amyloid‐beta 1‐ 40. Asymmetrical binding, endocytosis, and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J Clin Invest 102:734–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Marr RA, Rockenstein E, Mukherjee A, Kindy MS, Hersh LB, Gage FH et al (2003) Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J Neurosci 23:1992–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Maruyama M, Higuchi M, Takaki Y, Matsuba Y, Tanji H, Nemoto M et al (2005) Cerebrospinal fluid neprilysin is reduced in prodromal Alzheimer's disease. Ann Neurol 57:832–842. [DOI] [PubMed] [Google Scholar]
- 95. Mattson MP (2004) Pathways towards and away from Alzheimer's disease. Nature 430:631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. McDermott JR, Gibson AM (1997) Degradation of Alzheimer's beta‐amyloid protein by human and rat brain peptidases: involvement of insulin‐degrading enzyme. Neurochem Res 22:49–56. [DOI] [PubMed] [Google Scholar]
- 97. Melchor JP, Pawlak R, Strickland S (2003) The tissue plasminogen activator‐plasminogen proteolytic cascade accelerates amyloid‐beta (Abeta) degradation and inhibits Abeta‐induced neurodegeneration. J Neurosci 23:8867–8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Miners JS, Van Helmond Z, Chalmers K, Wilcock G, Love S, Kehoe PG (2006) Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy. J Neuropathol Exp Neurol 65:1012–1021. [DOI] [PubMed] [Google Scholar]
- 99. Miners JS, Ashby E, Van Helmond Z, Chalmers KA, Palmer LE, Love S, Kehoe PG (in press) Angiotensin‐converting enzyme (ACE) levels and activity in Alzheimer's disease, and relationship of perivascular ACE‐1 to cerebral amyloid angiopathy. Neuropathol Appl Neurobiol doi:10.1111/j.1365‐2990.2007.00885.x. [DOI] [PubMed] [Google Scholar]
- 100. Miners JS, Verbeek MM, Rikkert MO, Kehoe PG, Love S (2008) Immunocapture‐based fluorometric assay for the measurement of neprilysin‐specific enzyme activity in brain tissue homogenates and cerebrospinal fluid. J Neurosci Methods 167:229–236. [DOI] [PubMed] [Google Scholar]
- 101. Misbin RI, Almira EC (1989) Degradation of insulin and insulin‐like growth factors by enzyme purified from human erythrocytes. Comparison of degradation products observed with A14‐ and B26‐[125I]monoiodoinsulin. Diabetes 38:152–158. [DOI] [PubMed] [Google Scholar]
- 102. Morelli L, Llovera RE, Mathov I, Lue LF, Frangione B, Ghiso J, Castano EM (2004) Insulin‐degrading enzyme in brain microvessels: proteolysis of amyloid {beta} vasculotropic variants and reduced activity in cerebral amyloid angiopathy. J Biol Chem 279:56004–56013. [DOI] [PubMed] [Google Scholar]
- 103. Morita M, Kurochkin IV, Motojima K, Goto S, Takano T, Okamura S et al (2000) Insulin‐degrading enzyme exists inside of rat liver peroxisomes and degrades oxidized proteins. Cell Struct Funct 25:309–315. [DOI] [PubMed] [Google Scholar]
- 104. Myers A, Holmans P, Marshall H, Kwon J, Meyer D, Ramic D et al (2000) Susceptibility locus for Alzheimer's disease on chromosome 10. Science 290:2304–2305. [DOI] [PubMed] [Google Scholar]
- 105. Nakagomi S, Kiryu‐Seo S, Kiyama H (2000) Endothelin‐converting enzymes and endothelin receptor B messenger RNAs are expressed in different neural cell species and these messenger RNAs are coordinately induced in neurons and astrocytes respectively following nerve injury. Neuroscience 101:441–449. [DOI] [PubMed] [Google Scholar]
- 106. Nakamura T, Miller D, Ruoslahti E, Border WA (1992) Production of extracellular matrix by glomerular epithelial cells is regulated by transforming growth factor‐beta 1. Kidney Int 41:1213–1221. [DOI] [PubMed] [Google Scholar]
- 107. Narain Y, Yip A, Murphy T, Brayne C, Easton D, Evans JG et al (2000) The ACE gene and Alzheimer's disease susceptibility. J Med Genet 37:695–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Nicoll JA, Weller RO (2003) A new role for astrocytes: beta‐amyloid homeostasis and degradation. Trends Mol Med 9:281–282. [DOI] [PubMed] [Google Scholar]
- 109. Nielsen HM, Londos E, Minthon L, Janciauskiene SM (2007) Soluble adhesion molecules and angiotensin‐converting enzyme in dementia. Neurobiol Dis 26:27–35. [DOI] [PubMed] [Google Scholar]
- 110. Oba R, Igarashi A, Kamata M, Nagata K, Takano S, Nakagawa H (2005) The N‐terminal active centre of human angiotensin‐converting enzyme degrades Alzheimer amyloid beta‐peptide. Eur J Neurosci 21:733–740. [DOI] [PubMed] [Google Scholar]
- 111. Oda M, Morino H, Maruyama H, Terasawa H, Izumi Y, Torii T et al (2002) Dinucleotide repeat polymorphisms in the neprilysin gene are not associated with sporadic Alzheimer's disease. Neurosci Lett 320:105–107. [DOI] [PubMed] [Google Scholar]
- 112. Ohrui T, Matsui T, Yamaya M, Arai H, Ebihara S, Maruyama M, Sasaki H (2004) Angiotensin‐converting enzyme inhibitors and incidence of Alzheimer's disease in Japan. J Am Geriatr Soc 52:649–650. [DOI] [PubMed] [Google Scholar]
- 113. Perez A, Morelli L, Cresto JC, Castano EM (2000) Degradation of soluble amyloid beta‐peptides 1–40, 1–42, and the Dutch variant 1–40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochem Res 25:247–255. [DOI] [PubMed] [Google Scholar]
- 114. Preston SD, Steart PV, Wilkinson A, Nicoll JA, Weller RO (2003) Capillary and arterial cerebral amyloid angiopathy in Alzheimer's disease: defining the perivascular route for the elimination of amyloid beta from the human brain. Neuropathol Appl Neurobiol 29:106–117. [DOI] [PubMed] [Google Scholar]
- 115. Price DL, Tanzi RE, Borchelt DR, Sisodia SS (1998) Alzheimer's disease: genetic studies and transgenic models. Annu Rev Genet 32:461–493. [DOI] [PubMed] [Google Scholar]
- 116. Prince JA, Feuk L, Gu HF, Johansson B, Gatz M, Blennow K, Brookes AJ (2003) Genetic variation in a haplotype block spanning IDE influences Alzheimer disease. Hum Mutat 22:363–371. [DOI] [PubMed] [Google Scholar]
- 117. Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB et al (1998) Insulin‐degrading enzyme regulates extracellular levels of amyloid beta‐protein by degradation. J Biol Chem 273:32730–32738. [DOI] [PubMed] [Google Scholar]
- 118. Quaschning T (2005) Vasopeptidase inhibition for blood pressure control: emerging experience. Curr Pharm Des 11:3293–3299. [DOI] [PubMed] [Google Scholar]
- 119. Reid IA (1992) Interactions between ANG II, sympathetic nervous system, and baroreceptor reflexes in regulation of blood pressure. Am J Physiol 262:E763–E778. [DOI] [PubMed] [Google Scholar]
- 120. Rigat B, Hubert C, Alhenc‐Gelas F, Cambien F, Corvol P, Soubrier F (1990) An insertion/deletion polymorphism in the angiotensin I‐converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest 86:1343–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Rovelet‐Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A et al (2006) APP locus duplication causes autosomal dominant early‐onset alzheimer disease with cerebral amyloid angiopathy. Nat Genet 38:24–26. [DOI] [PubMed] [Google Scholar]
- 122. Russo R, Borghi R, Markesbery W, Tabaton M, Piccini A (2005) Neprylisin decreases uniformly in Alzheimer's disease and in normal aging. FEBS Lett 579:6027–6030. [DOI] [PubMed] [Google Scholar]
- 123. Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R et al (2007) Clearance of amyloid‐beta by circulating lipoprotein receptors. Nat Med 13:1029–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Saito T, Iwata N, Tsubuki S, Takaki Y, Takano J, Huang SM et al (2005) Somatostatin regulates brain amyloid beta peptide Abeta42 through modulation of proteolytic degradation. Nat Med 11:434–439. [DOI] [PubMed] [Google Scholar]
- 125. Sakai A, Ujike H, Nakata K, Takehisa Y, Imamura T, Uchida N et al (2004) Association of the Neprilysin gene with susceptibility to late‐onset alzheimer's disease. Dement Geriatr Cogn Disord 17:164–169. [DOI] [PubMed] [Google Scholar]
- 126. Sakai A, Ujike H, Nakata K, Takehisa Y, Imamura T, Uchida N et al (2004) No association between the insulin degrading enzyme gene and Alzheimer's disease in a Japanese population. Am J Med Genet B Neuropsychiatr Genet 125:87–91. [DOI] [PubMed] [Google Scholar]
- 127. Savaskan E, Hock C, Olivieri G, Bruttel S, Rosenberg C, Hulette C, Muller‐Spahn F (2001) Cortical alterations of angiotensin converting enzyme, angiotensin II and AT1 receptor in Alzheimer's dementia. Neurobiol Aging 22:541–546. [DOI] [PubMed] [Google Scholar]
- 128. Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N et al (1996) Secreted amyloid beta‐protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med 2:864–870. [DOI] [PubMed] [Google Scholar]
- 129. Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH et al (1993) Increased amyloid beta‐peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late‐onset alzheimer disease. Proc Natl Acad Sci USA 90:9649–9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Schweizer A, Valdenaire O, Nelbock P, Deuschle U, Dumas Milne Edwards JB, Stumpf JG, Loffler BM (1997) Human endothelin‐converting enzyme (ECE‐1): three isoforms with distinct subcellular localizations. Biochem J 328:871–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Selkoe DJ (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81:741–766. [DOI] [PubMed] [Google Scholar]
- 132. Seta K, Hayashi T, Sugawara A, Kasuno K, Watanabe S, Sumi Y et al (1998) Atrial natriuretic peptide as a preload depressor in acute renal failure secondary to congestive heart failure. Ren Fail 20:717–723. [DOI] [PubMed] [Google Scholar]
- 133. Shi J, Zhang S, Tang M, Ma C, Zhao J, Li T et al (2005) Mutation screening and association study of the neprilysin gene in sporadic Alzheimer's disease in Chinese persons. J Gerontol A Biol Sci Med Sci 60:301–306. [DOI] [PubMed] [Google Scholar]
- 134. Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B et al (2000) Clearance of Alzheimer's amyloid‐ss(1–40) peptide from brain by LDL receptor‐related protein‐1 at the blood‐brain barrier. J Clin Invest 106:1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Skidgel RA, Erdos EG (2004) Angiotensin converting enzyme (ACE) and neprilysin hydrolyze neuropeptides: a brief history, the beginning and follow‐ups to early studies. Peptides 25:521–525. [DOI] [PubMed] [Google Scholar]
- 136. Sluck JM, Lin RC, Katolik LI, Jeng AY, Lehmann JC (1999) Endothelin converting enzyme‐1‐, endothelin‐1‐, and endothelin‐3‐like immunoreactivity in the rat brain. Neuroscience 91:1483–1497. [DOI] [PubMed] [Google Scholar]
- 137. Sodeyama N, Mizusawa H, Yamada M, Itoh Y, Otomo E, Matsushita M (2001) Lack of association of neprilysin polymorphism with Alzheimer's disease and Alzheimer's disease‐type neuropathological changes. J Neurol Neurosurg Psychiatry 71:817–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Stockley JH, O'Neill C (2007) The proteins BACE1 and BACE2 and beta‐secretase activity in normal and Alzheimer's disease brain. Biochem Soc Trans 35:574–576. [DOI] [PubMed] [Google Scholar]
- 139. Stockley JH, Ravid R, O'Neill C (2006) Altered beta‐secretase enzyme kinetics and levels of both BACE1 and BACE2 in the Alzheimer's disease brain. FEBS Lett 580:6550–6560. [DOI] [PubMed] [Google Scholar]
- 140. Strittmatter WJ, Saunders AM, Schmechel D, Pericak‐Vance M, Enghild J, Salvesen GS, Roses AD (1993) Apolipoprotein E: high‐avidity binding to beta‐amyloid and increased frequency of type 4 allele in late‐onset familial Alzheimer disease. Proc Natl Acad Sci USA 90:1977–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Tanzi RE, Bertram L (2005) Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell 120:545–555. [DOI] [PubMed] [Google Scholar]
- 142. Tsubuki S, Takaki Y, Saido TC (2003) Dutch, Flemish, Italian, and Arctic mutations of APP and resistance of Abeta to physiologically relevant proteolytic degradation. Lancet 361:1957–1958. [DOI] [PubMed] [Google Scholar]
- 143. Tucker HM, Kihiko‐Ehmann M, Wright S, Rydel RE, Estus S (2000) Tissue plasminogen activator requires plasminogen to modulate amyloid‐beta neurotoxicity and deposition. J Neurochem 75:2172–2177. [DOI] [PubMed] [Google Scholar]
- 144. Turner AJ, Murphy LJ (1996) Molecular pharmacology of endothelin converting enzymes. Biochem Pharmacol 51:91–102. [DOI] [PubMed] [Google Scholar]
- 145. Turner AJ, Tanzawa K (1997) Mammalian membrane metallopeptidases: nEP, ECE, KELL, and PEX. FASEB J 11:355–364. [DOI] [PubMed] [Google Scholar]
- 146. Turner AJ, Brown CD, Carson JA, Barnes K (2000) The neprilysin family in health and disease. Adv Exp Med Biol 477:229–240. [DOI] [PubMed] [Google Scholar]
- 147. Turner AJ, Isaac RE, Coates D (2001) The neprilysin (NEP) family of zinc metalloendopeptidases: genomics and function. Bioessays 23:261–269. [DOI] [PubMed] [Google Scholar]
- 148. Tyler SJ, Dawbarn D, Wilcock GK, Allen SJ (2002) alpha‐ and beta‐secretase: profound changes in Alzheimer's disease. Biochem Biophys Res Commun 299:373–376. [DOI] [PubMed] [Google Scholar]
- 149. Ueberham U, Ueberham E, Bruckner MK, Seeger G, Gartner U, Gruschka H et al (2005) Inducible neuronal expression of transgenic TGF‐beta1 in vivo: dissection of short‐term and long‐term effects. Eur J Neurosci 22:50–64. [DOI] [PubMed] [Google Scholar]
- 150. Valdenaire O, Lepailleur‐Enouf D, Egidy G, Thouard A, Barret A, Vranckx R et al (1999) A fourth isoform of endothelin‐converting enzyme (ECE‐1) is generated from an additional promoter molecular cloning and characterization. Eur J Biochem 264:341–349. [DOI] [PubMed] [Google Scholar]
- 151. Vepsäläinen S, Helisalmi S, Koivisto AM, Tapaninen T, Hiltunen M, Soininen H (2007) Somatostatin genetic variants modify the risk for Alzheimer's disease among Finnish patients. J Neurol 254:1432–1459. [DOI] [PubMed] [Google Scholar]
- 152. Vepsalainen S, Parkinson M, Helisalmi S, Mannermaa A, Soininen H, Tanzi RE et al (2007) Insulin‐degrading enzyme is genetically associated with Alzheimer's disease in the Finnish population. J Med Genet 44:606–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Visser PJ, Verhey FR (2008) Mild cognitive impairment as predictor for Alzheimer's disease in clinical practice: effect of age and diagnostic criteria. Psychol Med 38:113–122. [DOI] [PubMed] [Google Scholar]
- 154. Vogelgesang S, Cascorbi I, Schroeder E, Pahnke J, Kroemer HK, Siegmund W et al (2002) Deposition of Alzheimer's beta‐amyloid is inversely correlated with P‐glycoprotein expression in the brains of elderly non‐demented humans. Pharmacogenetics 12:535–541. [DOI] [PubMed] [Google Scholar]
- 155. Wang DS, Lipton RB, Katz MJ, Davies P, Buschke H, Kuslansky G et al (2005) Decreased neprilysin immunoreactivity in Alzheimer disease, but not in pathological aging. J Neuropathol Exp Neurol 64:378–385. [DOI] [PubMed] [Google Scholar]
- 156. Wang DS, Dickson DW, Malter JS (2006) beta‐Amyloid Degradation and Alzheimer's disease. J Biomed Biotechnol 2006:58406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Weeraratna AT, Kalehua A, Deleon I, Bertak D, Maher G, Wade MS et al (2007) Alterations in immunological and neurological gene expression patterns in Alzheimer's disease tissues. Exp Cell Res 313:450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE (1998) Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer's disease. Am J Pathol 153:725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Weller RO, Massey A, Kuo YM, Roher AE (2000) Cerebral amyloid angiopathy: accumulation of a beta in interstitial fluid drainage pathways in Alzheimer's disease. Ann NY Acad Sci 903:110–117. [DOI] [PubMed] [Google Scholar]
- 160. Wyss‐Coray T, Masliah E, Mallory M, McConlogue L, Johnson‐Wood K, Lin C, Mucke L (1997) Amyloidogenic role of cytokine TGF‐beta1 in transgenic mice and in Alzheimer's disease. Nature 389:603–606. [DOI] [PubMed] [Google Scholar]
- 161. Wyss‐Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L et al (2001) TGF‐beta1 promotes microglial amyloid‐beta clearance and reduces plaque burden in transgenic mice. Nat Med 7:612–618. [DOI] [PubMed] [Google Scholar]
- 162. Xu D, Emoto N, Giaid A, Slaughter C, Kaw S, DeWit D, Yanagisawa M (1994) ECE‐1: a membrane‐bound metalloprotease that catalyzes the proteolytic activation of big endothelin‐1. Cell 78:473–485. [DOI] [PubMed] [Google Scholar]
- 163. Yamada M, Sodeyama N, Itoh Y, Takahashi A, Otomo E, Matsushita M, Mizusawa H (2003) Association of neprilysin polymorphism with cerebral amyloid angiopathy. J Neurol Neurosurg Psychiatry 74:749–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Yanagisawa H, Hammer RE, Richardson JA, Emoto N, Williams SC, Takeda S et al (2000) Disruption of ECE‐1 and ECE‐2 reveals a role for endothelin‐converting enzyme‐2 in murine cardiac development. J Clin Invest 105:1373–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Yasojima K, Akiyama H, McGeer EG, McGeer PL (2001) Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of beta‐amyloid peptide. Neurosci Lett 297:97–100. [DOI] [PubMed] [Google Scholar]
- 166. Yin KJ, Cirrito JR, Yan P, Hu X, Xiao Q, Pan X et al (2006) Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid‐beta peptide catabolism. J Neurosci 26:10939–10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Yoshizawa T, Iwamoto H, Mizusawa H, Suzuki N, Matsumoto H, Kanazawa I (1992) Cerebrospinal fluid endothelin‐1 in Alzheimer's disease and senile dementia of Alzheimer type. Neuropeptides 22:85–88. [DOI] [PubMed] [Google Scholar]
- 168. Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O'Connor T et al (2007) Beta‐site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J Neurosci 27:3639–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Zhao Z, Xiang Z, Haroutunian V, Buxbaum JD, Stetka B, Pasinetti GM (2007) Insulin degrading enzyme activity selectively decreases in the hippocampal formation of cases at high risk to develop Alzheimer's disease. Neurobiol Aging 28:824–830. [DOI] [PubMed] [Google Scholar]
- 170. Zou K, Yamaguchi H, Akatsu H, Sakamoto T, Ko M, Mizoguchi K et al (2007) Angiotensin‐converting enzyme converts amyloid beta‐protein 1–42 (Abeta(1–42)) to Abeta(1–40), and its inhibition enhances brain Abeta deposition. J Neurosci 27:8628–8635. [DOI] [PMC free article] [PubMed] [Google Scholar]