

Abstract
Spinal cord injury (SCI) typically results from sustained trauma to the spinal cord, resulting in loss of neurologic function at the level of the injury. However, activation of various physiological mechanisms secondary to the initial trauma including edema, inflammation, excito‐toxicity, excessive cytokine release and apoptosis may exacerbate the injury and/or retard natural repair mechanisms. Herein, we demonstrate that cytoplasmic extracts prepared from adipose tissue stromal cells (ATSCs) inhibits H2O2‐mediated apoptosis of cultured spinal cord‐derived neural progenitor cells (NPCs) resulting in increased cell survival. The ATSC extracts mediated this effect by decreasing caspase‐3 and c‐Jun–NH2‐terminal kinase (SAPK/JNK) activity, inhibiting cytochrome c release from mitochondria and reducing Bax expression levels in cells. Direct injection of ATSC extracts mixed with Matrigel into the spinal cord immediately after SCI also resulted in reduced apoptotic cell death, astrogliosis and hypo‐myelination but did not reduce the extent of microglia infiltration. Moreover, animals injected with the ATSC extract showed significant functional improvement of hind limbs as measured by the BBB (Basso, Beattie and Bresnahan) scale. Collectively, these studies show a prominent therapeutic effect of ATSC cytoplasmic extracts on SCI principally caused by an inhibition of apoptosis‐mediated cell death, which spares white matter, oligodendrocytes and neurons at the site of injury. The ability of ATSC extracts to prevent secondary pathological events and improve neurologic function after SCI suggests that extracts prepared from autologous cells harvested from SCI patients may have clinical utility.
INTRODUCTION
Traumatic spinal cord injury (SCI) typically causes permanent neurological deficits, which can be largely attributed to retrograde neuronal cell death and failure of surviving neurons to regenerate their severed axons. Landmark experiments conducted over 20 years ago demonstrated that neurologic deficits in SCI are not because of an intrinsic inability of CNS neurons to regenerate, but rather of the formation of a CNS environment that is unfavorable for regeneration (10). Several approaches designed to alter the microenvironment of the injured spinal cord and/or stimulate endogenous repair mechanisms are being actively explored as a means to treat SCI. Importantly, pathological changes that occur because of SCI extend from the onset of trauma to years post injury. During the acute phase, which starts at the moment of the injury and extends over the first few weeks, numerous pathological changes occur that can produce secondary tissue damage. These include hypoxia, excessive release of excitatory neurotransmitters and an increased production of free radicals, which induces damage to lipid‐rich membranes in the spinal cord because of lipid‐peroxidation. A strong inflammatory response also ensues, resulting in infiltration of peripherally derived immune cells such as neutrophils, macrophages and T cells into the injured tissue (29). Infiltrating microglial cells produce various cytotoxic factors including reactive nitrogen species that contribute to secondary damage to the injured spinal cord 25, 30, 36, 42, 47. Other notable physiological changes that may continue up to years following injury include apoptotic cell death, impairment of channel and receptor function, scarring, demyelination and Wallerian degeneration. Each of these processes contributes to deficits in nerve conduction (42).
Several studies have reported that chemical agents including minocycline exhibit some neuroprotective effects following SCI by inhibitory secondary tissue damage 37, 45 but no one product has shown efficacy in both experimental SCI models and clinical trials. Studies conducted over the past two decades have shown that therapeutic strategies aimed at attenuating secondary injury mechanisms, for example, glutamate toxicity, free radical cellular damage, Na+ and Ca2+ influx‐related neuronal death, must be implemented either before or immediately after SCI to be effective and as such may not be amenable for treating human patients 5, 15, 26. In contrast, intravenous administration of methylprednisolone, a potent anti‐inflammatory agent, has been recommended as a standard clinical treatment for SCI based on results from a large‐scale clinical trial (6). However, concerns regarding the interpretation of clinical trial data together with side effects such as increased infection rates, gastrointestinal hemorrhage and respiratory complications have questioned the use of high dose methylprednisolone for treating acute SCI (15). Apoptotic cell death has also been shown to contribute significantly to secondary tissue injury after SCI and as such, pathways regulating apoptosis may also represent important target molecules for therapeutic intervention 11, 24, 27, 39.
Numerous studies have demonstrated that mesenchymal stem cells isolated from human adipose tissue can differentiate into connective tissue cell types 12, 49 as well as neuron‐like cells 18, 19, 33 and as such, may serve as an alternative source of stem/progenitor cells for cell therapy including the treatment of neurological disorders. In this study we evaluated the neuro‐protective effects of cytoplasmic extracts prepared from culture‐expanded adipose tissue stromal cells (ATSCs) following SCI in rats. Our data demonstrate that injection of ATSC extracts directly into the spinal cord immediately after injury suppressed apoptotic cell death, astrogliosis and hypo‐myelination thereby reducing the extent of tissue damage at the site of injury. These changes resulted in significant functional improvement of hind limbs as measured by the BBB (Basso, Beattie and Bresnahan) scale. Animals administered extracts from ATSCs also recovered hind limb reflexes more rapidly and achieved a faster and greater degree of recovery of coordinated hind limb use as compared with untreated, injured rats. Studies conducted in vitro showed that ATSC extract also protected spinal cord‐derived neural progenitor cells (NPCs) from H2O2‐mediated apoptosis by down‐regulating the expression of various apoptotic mediators such as caspase‐3 and Bax as well as inducing expression of survival factors like Bcl2. Therefore, extracts prepared from ATSCs appear to ameliorate secondary tissue damage following SCI by suppressing apoptosis of host cells resident near the site of injury.
MATERIALS AND METHODS
Isolation of spinal cord‐derived neural progenitor cells, adipose‐derived stromal cells (ATSCs) and ATSC cytoplasmic extracts.
NPCs were obtained from the spinal cord of adult rats. Specifically, the complete cervical enlargement (spinal cord level C3 through T1) was obtained by dissection. After removal of the dura, the tissue was minced, washed in sterile phosphate‐buffered saline (PBS) and digested in HBSS supplemented with 0.125% trypsin and 0.01% DNase (Sigma, St Louis, MO, USA) for 30 minutes at 37°C. The dissociated tissue was transferred to culture dishes containing NB medium with B27 supplement, bFGF (20 ng/ml) and EGF (20 ng/ml). Cells were grown as neurospheres for 5 days in petri dishes or on culture dishes coated with poly D‐lysine in a humidified chamber in 5% CO2. NPCs were then plated at 3 × 105 cells/cm2 in DMEM supplemented with 10% FBS (Whittaker, USA) prior to exposure to H2O2. Raw adipose tissue recovered from the abdomen of adult rats was processed according to established methodologies to obtain a stromal vascular function (SVF; Kang et al, 2003, 2004) 18, 19. The SVF was washed extensively with equal volumes of PBS, digested at 37°C for 30 minutes with 0.075% collagenase (Sigma) and then enzyme activity was neutralized by addition of α‐MEM (Life Technologies, Frederick, MA, USA) supplemented with 10% FBS. A high density cell pellet was then obtained by centrifugation at 1200 g for 10 minutes. The pellet was resuspended in red blood cell lysis buffer (Biowhittaker, Walkersville, MD, USA) and incubated at room temperature for 10 minutes. The stromal cell pellet was collected by centrifugation, suspended in α‐MEM containing 10% FBS and the cell suspension was plated at a density of 2 × 105 cells/cm2 and incubated overnight at 37°C in a humidified chamber with 5% CO2. Seventy percent of the media was changed after 48 h and then every fourth day thereafter. ATSCs were then collected after 5 days without further processing. Cytoplasmic extracts were prepared by suspending 2 × 106 cells in NB/B27 minimal media, subjecting them to three to four cycles of rapid freeze/thawing and collecting the supernatant after centrifugation at 15 000 g. The total protein content of each extract was quantified using the Bio‐Rad protein assay kit (Milan, Italy).
Effect of ATSC extracts on NPCs cell survival after H2O2 exposures.
NPCs cultured to 80% confluence were pretreated with 0.6 µg/ml of ATSC cell extract for 8 h followed by 0.05 µg/ml of H2O2 for an additional 6 h. Concentrations of ATSC extract and H2O2 that produced optimal cell survival and cytotoxicity, respectively, were determined empirically. In each experiment, H2O2 was freshly diluted from a 30% stock solution into a small volume of DMEM (1/100th culture volume), incubated at room temperature for 5 minutes and then added directly to the cultured cells followed by light agitation. At the times indicated, cell viability was measured by physically counting cells after staining with Tryphan Blue. Cell viability was also assessed by measuring the ability of cultured cells to reduce (3,4,5‐dimethyl thiazol‐2‐yl) ‐2,5‐diphenyl tetrazolium bromide (MTT, Sigma) to a colored formazan. All viability assays were performed in triplicate and each experiment was repeated at least three times. Raw data from each experiment were analyzed using Student’s t‐test or Fisher test.
Measurement of intracellular ROS and mitochondrial ATP.
NPCs treated with or without ATSC extracts were plated in glass bottom dishes at a density of 5 × 105 cells/cm2 cultured for 5 days and then treated with 50 µM tert‐butylhydroperoxide alone for 6 h. Cells (5 × 105) were then washed and incubated for 30 minutes at 37°C in media containing 10 µM of DCFDA (5‐(and‐6)‐carboxy‐2′,7′‐dichlorodihydrofluorescein diacetate), a non‐fluorescent compound that freely permeates cells. When inside cells, DCFDA is hydrolyzed to 2′‐7′‐dichlorofluorescin, which yields the fluorescent compound 2′‐7′‐dichlorofluorescin when it interacts with peroxides. Cells were imaged by fluorescent microscopy (Ex/Em = 495/525 nm) at 30 minutes and 1, 3, 5 and 10 h after loading with DCFDA. Adenosine triphosphate (ATP) synthesis was measured by adding treated or untreated NPCs (5 × 105) in 250 µl to 750 µl of substrate buffer (10 mmol L‐malate, 10 mmol L‐pyruvate, 1 mmol ADP, 40 µg/ml digitonin and 0.15 mmol/L adenosine pentaphosphate). Cells were incubated at 37°C for 10 minutes and then at various times 50 µl aliquots of the reaction mixture were withdrawn, quenched in 450 µl of boiling 100 mmol Tris‐HCl, 4 mmol EDTA, pH 7.75 for 2 minutes and further diluted 1/10 in the quenching buffer. The quantity of ATP was measured in a luminometer (Berthold, Detection Systems, Pforzheim, Germany) with the ATP Bioluminescence Assay Kit (Roche Diagnostics, Basel, Switzerland) following the manufacturer’s instructions.
Induction of Spinal Cord Injury (SCI).
Adult female Wistar rats weighing 250 g (∼5 weeks of age) were housed in a controlled environment and provided with standard rodent chow and water. Animal care was in compliance with Korean regulations on protection of animals used for experimental and other scientific purposes. Animals were subjected to traumatic SCI using a modified version of the protocol described by Kang et al (20). Briefly, the spinous processes of T9 and T10 were removed with rongeurs and a laminectomy performed using a dental drill and rongeurs to expose the dorsal spinal cord in anesthetized rats. The dura was incised with microscissors and the dorsal and ventral columns, which included the dorsal and ventral corticospinal tract, were cut by lowering microscissors attached to a sterotaxic arm to a depth of 3.5 mm below the dorsal surface of the spinal cord and cutting twice. Experimental trials were first done to ascertain that this surgical procedure produced a consistent total transaction of the corticospinal tract on both sides.
Semi‐quantitative RT‐PCR.
One week following SCI total cellular RNA was extracted with Trizol (Life Technologies) from spinal cord tissue harvested from a total of four animals per experimental group. Total RNA was reverse transcribed into first strand cDNA using an oligo‐dT primer and then amplified by PCR using the indicated gene specific primers (20 pM). Polymerase chain reactions (PCRs) were performed using an ABI 7700 Prism Sequence Detection System and the SYBER green detection kit (Applied Biosystems, Foster, CA, USA). Primer sequences were designed using Primer Express software (PE‐Applied Biosystems, Warrington, UK) using gene sequences obtained from the GeneBank database. Gene‐specific primers were as follows: GAPDH: 5‐CAT GAC CAC AGT CCA TGC CAT CAC T‐3 and 5‐TGA GGT CCA CCA CCC TGT TGC TGT A‐3, SDF‐1: 5‐TTG CCA GCA CAA AGA CAC TCC‐3 and 5‐CTC CAA AGC AAA CCG AAT ACA G‐3, NGF: 5‐CCA AGG ACG CAG CTT TCT AT‐3 and 5‐CTC CGG TGA GTC CTG TTG AA‐3, PDGFa: 5‐AGC CCA TCC CCG CAG TTT GC‐3 and 5‐GGT TCA GGT TGG AGG TCG CG‐3, MMP2: 5‐ATC TGG TGT CTC CCT TAC GG‐3 and 5‐GTG CAG TGA TGT CCG ACA AC‐3, MMP3: 5‐CCT CTA TGG ACC TCC CAC AGA ATC‐3 and 5‐GTG CCA ATG CCT GGA AAG TTC‐3, TNFR: 5‐CCT GAT TTC CAT CTA CCT CT‐3 and 5‐ACA CTG GAA ATG CGT CTC CAC‐3, TNFa: 5‐CAC GCT CTT TCT GTC TAC TGA‐3 and 5‐GGA CTC CGT GAT GTC TAA GT‐3, CDK4: 5‐TCA GCA CAG TTC GTG AGG T‐3 and 5‐CAG GTA TGT CCG TAG GTC C‐3, RUNX: 5‐CAG CCA GGC CCA GAC CCC AA‐3 and 5‐GTC CAT GCG GCC CGG TGT GC‐3.
Histological analysis of tissues.
Spinal cord tissue was fixed by trans‐cardiac perfusion with 0.1 M PBS followed by 4% paraformaldehyde in 0.1 M phosphate buffer. Spinal cord tissue was then cryo‐protected with 30% sucrose for 48 h, embedded in OCT and cut into 10 µm coronal sections. Spinal cord and sciatic nerve from untreated control animals were processed similarly. Tissue sections were incubated overnight at 4°C with an anti‐ED1 antibody (1:100, Serotec, UK) or an anti‐GFAP antibody (1:1500, DAKO Cytomation, Denmark). Sections were then washed extensively with PBS and then incubated for 30 minutes with either a FITC‐conjugated or Texas Red‐conjugated secondary antibody (1:250, Molecular Probe, USA) and counterstained with Topro‐3 (Molecular Probe) to visualize cell nuclei. Sections were visualized on a Leica TCS sp2 laser scanning microscope (Leica Microsystems, PA, USA). The number of GFAP‐positive and ED‐1 positive cells was determined by counting. Three digital microscopic images at a magnification of 100× were randomly captured from each section from areas around the lesion site. The size of the view field was 0.075 mm2. The number of positively stained cells in the three images was averaged. A total of three sections from each treatment group were examined by an observer blind to the treatment groups and the mean value of cell counting was used to represent one single SCI tissue sample. Results were expressed as the percentage of positive cells per view field ± the standard deviation.
Alternatively, serial 4 µm cryo‐sections of spinal cord tissue harvested at 4 weeks post injury was fixed and stained overnight at 60°C in 0.1% Luxol Fast Blue (Sigma, Oakville, Ontario, Canada) in 95% ethanol and 0.5% acetic acid and then counter stained with 0.05% LiCO3. Staining of nerve cells was also performed using 0.25% of Cresyl Echt Violet (Sigma). For each spinal cord segment, samples were harvested at 500‐µm intervals to yield 20 sections per spinal cord segment. Stained sections were viewed on an Olympus microscope (BH‐2; Olympus, Melville, NY, USA) at 40× magnification and captured with a Magna Fire SP CCD camera (Optronics, Goleta, CA, USA). Image capturing was done using Magna Fire SP 2.1 software (Optronics), and the image processing was done using Photoshop (Adobe System Inc., CA, USA).
In some cases tissues were fixed by trans‐cardiac perfusion with phosphate‐buffered solution (PBS) and then with a 1.0% glutaraldehyde in 0.1 M sodium cacodylate‐HCl (pH 7.4) for 10 minutes at 37°C, washed in 0.1 M sodium cacodylate‐HCl, postfixed in 1.0% osmium tetroxide and 4% tannic acid and embedded in Spurr medium. The tissues were then carefully excised, cut into 1‐mm segments and placed into fresh fixative. Tissues were then washed several times in PBS, postfixed with 1% OsO4 for 2 h at 25°C, dehydrated in graded ethanol solutions, passed through propylene oxide and embedded in Epon 812 resin (Resolution Performance Product, USA). Sections (1 µm) were stained with uranyl acetate and lead citrate‐18 and viewed with an electron microscope (JEM 1200EX‐II, JEOL, Japan).
Preparation of tissue extracts and western blot.
Spinal cord‐derived NPCs treated with or without ATSC extract and spinal cord tissue dissected from the lesion site from 3–4 animals per group were pooled and then homogenized in 500 µl of lysis buffer (20 mM Tris‐HCl, 150 mM NaCl, 1 mM EDTA, 1% Triton X‐100, 2.5 mM sodium pyrophosphate, 1 mM EGTA, 1 mM glycerophosphate, 1 mM Na3VO4, 1 mM PMSF, pH 7.5). The homogenates were chilled on ice for 15 minutes and then vigorously shaken for a few minutes in the presence of 20 µl of 10% Nonidet P‐40. Lysates were clarified by centrifugation at 15 000 g for 10 minutes and then stored at −80°C. The total protein content of each lysate was quantified using the Bio‐Rad protein assay kit (Milan, Italy). For western blotting, equal amounts (40 µg) of protein extracts in lysis buffer were subjected to 10% SDS‐PAGE analysis and transferred to a nitrocellulose membrane. Anti‐GFAP, anti‐EGFR (1:300, Sigma), anti‐Nestin (1:500, Sigma), anti‐acetyl‐Histone 4 (1:2000, Upstate, USA), anti‐acetyl‐Histone 3 (1:10 000, Upstate), anti cytochrome c (1:500, Cell Signaling, Boston, MA, USA), anti‐Bax (1:500, Cell Signaling), anti‐Bcl 2 (1:500, Cell Signaling), anti‐ED1 (1:200, Serotec), anti‐pSAPK/JNK (1:1000, Cell Signaling), anti‐PLCgamma (1:500, Cell Signaling), anti‐p‐Raf (1:1000, Cell Signaling), anti‐pERK (1:1000, Cell Signaling), anti‐Jak2 (1:1000, Cell Signaling), anti‐pSTAT 3 (1:1000, Cell Signaling), and anti‐β‐Actin (1:500, Sigma) antibodies were incubated with membranes and visualized as described previously (Kang et al, 2006) (20). Relative band intensities were determined by Quality‐one 1‐D analysis software (Bio‐Rad, USA).
Apoptosis assays.
Apoptosis of NPCs and spinal cord tissue was quantified using the deoxynucleotidyl transferase (TdT) In Situ Apoptosis Detector Kit (Roche, USA) according to the manufacturer’s specifications. Briefly, fixed cells and/or tissue sections were incubated at 37°C for 90 minutes in the Terminal deoxynucleotidyl transferase biotin‐duTP Nick (TUNEL) reaction mixture containing TdT, biotinylated dUTP and the TdT reaction buffer. After washing with PBS, samples were incubated at room temperature for 30 minutes with an anti‐horseradish peroxidase antibody conjugated to FITS and then visualized by green fluorescence using a Leica fluorescence microscope (Leica Microsystems). The number of TUNEL‐positive apoptotic cells in each spinal cord specimen was quantified by counting the number of stained cells in three microscopic images (100×) captured around the lesion site. Tissue sections were examined by an observer blind to the treatment groups. The number of positively stained cells in the three images was then averaged. The field of view for each image was 0.075 mm2 and the data were expressed as the percentage of TUNEL‐positive cells per microscopic field. To assess caspase‐3, ‐8 and ‐9 activity, 10 µg of protein isolated from NPCs or injured spinal cord tissues in a total volume of 50 µl was mixed with 50 µl of equilibrated Caspase‐Glo 3/7, 8, or 9 reagents (Promega). After incubating at room temperature for 1 h, luminescence was measured using a TD 20/20 Luminometer (Turner Designs, Sunnyvale, CA, USA). Experimental values were corrected for background and fold‐increase in activity was calculated based on the activity measured in untreated cells. All reactions were performed in triplicate.
Measurement of functional recovery following SCI.
All animals (controls and experimental groups) were placed on a restricted diet to motivate them to perform the various behavioral tests. Behavioral recovery was scored according to the BBB scale, which is composed of 21 different criteria for evaluating a range of hind limb movements from complete paralysis to complete mobility. A BBB score equal to zero represents no hind limb movement and a score equal to twenty represents normal mobility. Statistical analysis was carried out using the Stat View software package (SAS, Cary, NC, USA). Data were reported as the mean ± SEM from five or more independent experiments. Statistically significant differences in functional recovery between groups were calculated by repeated measures of the ANOVA test.
RESULTS
Cytoplasmic extracts from ATSCs block H2O2‐induced expression of apoptotic mediators in cultured NPCs (Figure 1).
Figure 1.
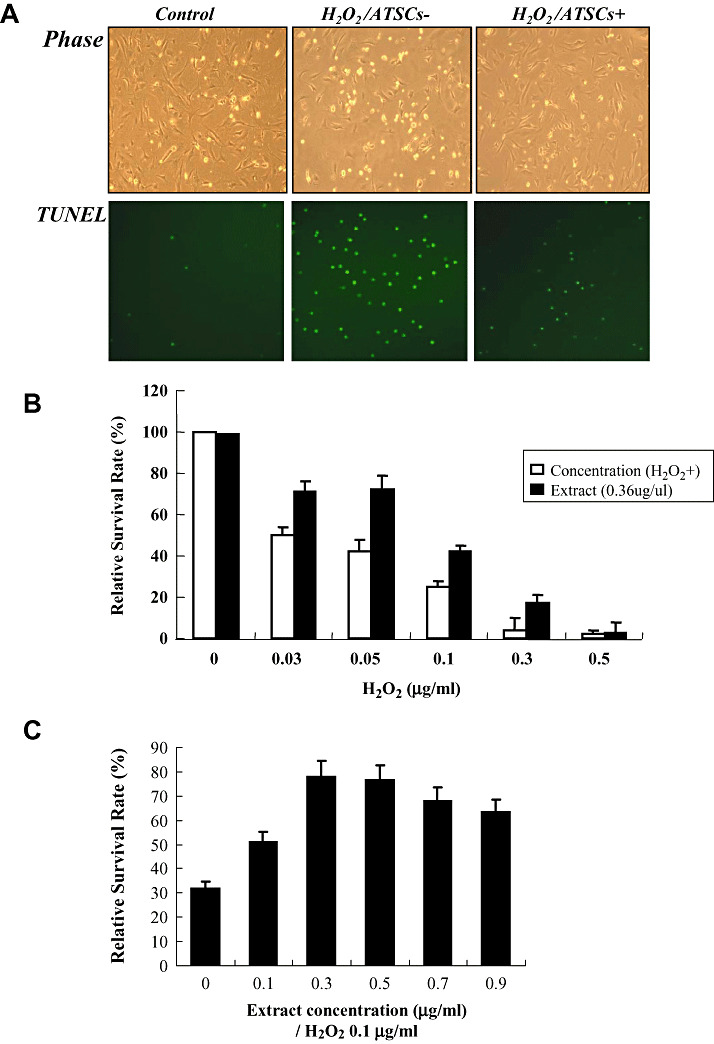
Effect of ATSC extracts on NPC survival after H2O2 exposures. A. Phase contrast and fluorescent micrographs showing the morphology and number of Terminal deoxynucleotidyl transferase biotin‐duTP Nick (TUNEL)‐positive cells in neural progenitor cell (NPC) cultures prior to and after 0.05 µg/ml of H2O2 exposure. Where indicated NPC cultures were pretreated with adipose tissue stromal cell (ATSC) extracts (0.6 µg/ml) for 8 h prior to H2O2 exposure. B. Percentage of NPCs that survived after treatment with the indicated concentrations of H2O2 with or without ATSC extract pretreatment. Experiments were done in triplicate and the mean ± standard deviation are plotted. C. Dose‐dependent effect of ATSC extract on NPC survival in response to exposure to 0.1 µg/ml of H2O2. D. Fluorescent micrographs of NPCs loaded with DCFDA and exposed to 0.05 µg/ml of H2O2 for 5 h. E. Relative caspase‐3 activity and ATP production in cultured NPCs alone, or cells exposed to H2O2 with or without pretreatment with ATSC extracts. Experiments were done in triplicate and the mean ± standard deviation are plotted. F. Western blots showing the expression levels of pSAP/JNK and pERK 1/2 during a time course of H2O2 exposure or cytochrome C, Bax and Bcl2 in NPCs pretreated with or without ATSC extracts. All reactions were performed in triplicate. Numbers indicate relative protein expression levels as determined by densitometry. Data are plotted as the mean ± standard deviation (*P < 0.05; **P < 0.001).
Exposure of spinal cord‐derived NPCs to increasing concentrations of hydrogen peroxide (H2O2) resulted in a progressive and dramatic decrease in cell survival. TUNEL assays confirmed that a majority of cells exposed to H2O2 underwent apoptotic‐mediated cell death (Figure 1A). However, pretreatment of NPCs with cytoplasmic extracts from ATSCs reduced the extent of apoptosis (Figure 1A) resulting in a significant increase in cell survival following exposure to H2O2 (P < 0.001) (Figure 1B). For example, approximately 45% of untreated NPCs survived after exposure to 0.05 µg/ml of H2O2 as compared with 70% of cells pretreated with ATSC extract (Figure 1B). Pretreatment of NPCs with an equal quantity of bovine serum albumin prior to H2O2 exposure had no effect on cell survival (Figure 1B).
H2O2 has been shown to increase cellular oxidative stress. Therefore, we measured whether pretreatment with ATSC extracts altered the levels of reactive oxygen species (ROS) in NPCs after exposure to H2O2. Specifically, cultured NPCs were pretreated with varying concentrations of ATSC extract and then exposed to H2O2 (0.1 µg/ml). As shown in Figure 1C, pretreatment with ATSC extract at a dose of 0.3 µg/ml was most effective at preventing neural NPC death induced by H2O2 exposure. In addition, NPCs loaded with the membrane permeable molecule DCFDA exhibited a marked increase in intracellular fluorescence after exposure to H2O2 (Figure 1D). However, this increase in intracellular fluorescence upon H2O2 exposure was essentially abolished by pretreatment of cells with 10 ng/ml ATSCs extract. Importantly, the ATSC extract did not alter ROS production in untreated cells but rather specifically inhibited H2O2‐induced production of ROS (Figure 1D).
H2O2 is also known to trigger activation of caspases (25). Consistent with this finding, we showed that caspase‐3 activity was increased approximately tenfold in NPCs after exposure to H2O2. In contrast, H2O2 exposure of NPCs pretreated with ATSC extract resulted in only a fourfold increase in caspase‐3 activity, which was significantly lower then that measured in non‐treated NPCs (P < 0.001) (Figure 1E). Similarly, H2O2 exposure promoted a significant decrease in ATP production in NPCs (P < 0.05) but pretreatment with ATSC extract reversed this effect to a significant extent (P < 0.05) (Figure 1F). Finally, we also showed that ATSC extracts inhibited the expression of other apoptotic mediators in cells exposed to H2O2. Specifically, H2O2 induced expression of Bax and c‐Jun–NH2‐terminal kinase (SAPK/JNK) and also stimulated release of cytochrome C from mitochondria in NPCs. However, these effects were blunted if NPCs were pretreated with ATSC extracts prior to H2O2 exposure. Moreover, treatment with ATSC extracts also induced expression of specific cell survival factors in NPCs such as Bcl2 and extracellular regulated kinase (ERK) 1 and 2 (Figure 1F). Together, these finding indicate that cytoplasmic extracts prepared from ATSCs promote survival by preventing H2O2‐induced activation of the apoptotic pathway in cells.
Therapeutic application of ATSC extracts in vivo improves motor performance in rats after spinal cord injury (Figure 2).
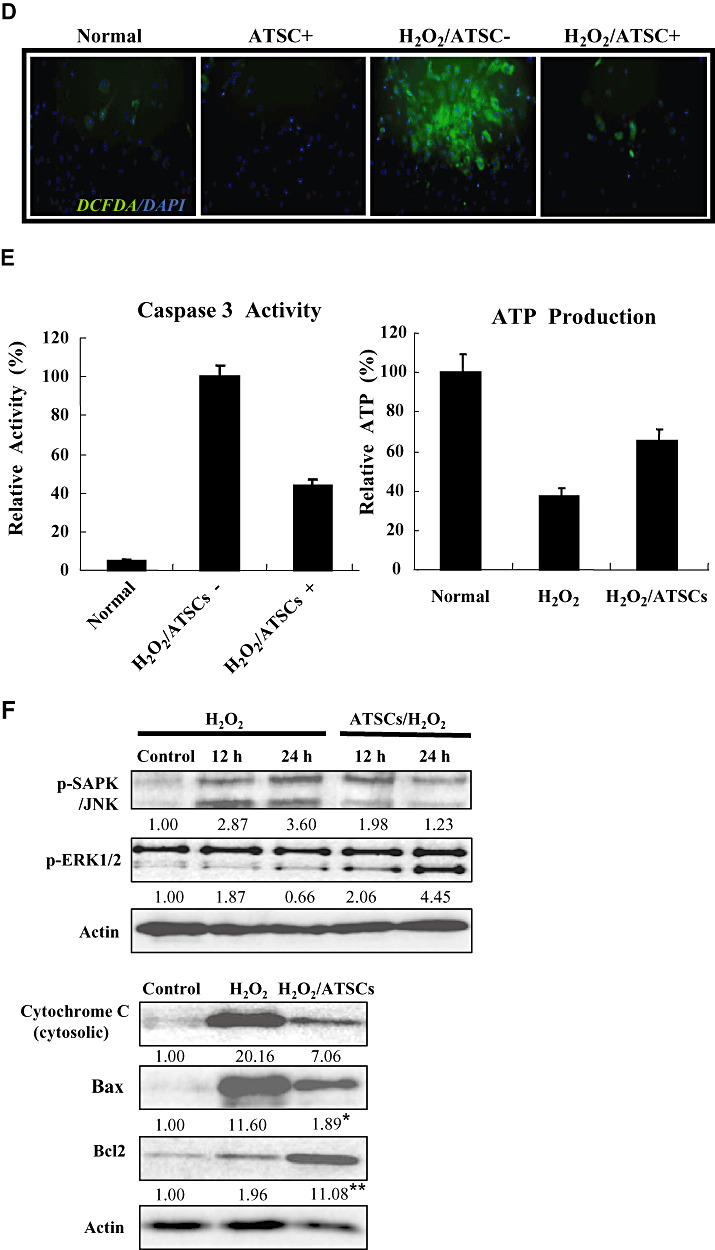
Figure 2.
Locomotor recovery and body weight change after spinal cord injury (SCI) in rats. (A) Hind limb motor function (B) Body weight was evaluated each week up to 6 weeks in uninjured, normal rats and rats subjected to SCI and treated with Matrigel or Matrigel + adipose tissue stromal cell (ATSC) extracts. Animals treated with Matrigel + ATSC extract displayed a significant increase in hind limb motor performance as compared with Matrigel only treated animals (P < 0.05 using the one‐way ANOVA test.). In contrast, animals treated with Matrigel only showed a significant difference in body weight as compared with uninjured animals or those treated with Matrigel + ATSC extract after SCI injury (P < 0.05 using the one way ANOVA test). *Data are the mean ± SD (n = 40). BBB = Basso, Beattie and Bresnahan.
To determine if ATSC extracts exhibit a neuro‐protective effect in vivo, we compared motor function using a modified BBB hind limb locomotor rating scale between uninjured control rats to those administered Matrigel with or without ATSC extract immediately after SCI. As shown in Figure 2A, all animals subjected to SCI exhibited extensive deficits in hind limb function over the first few days post injury as compared with uninjured, control animals, suggesting that all animals experience a similar degree of SCI. However, beginning at the 7th day post injury motor recovery continued to improve in animals administered the ATSC extract as compared with those injected with Matrigel alone. For example, by 3 weeks post SCI, rats administered the ATSC extract consistently supported their weight during plantar stepping, exhibited consistent coordination and followed a predominantly rotated paw position during locomotion and frequent plantar or dorsal stepping. During the same time period injured animals injected only with Matrigel exhibited limited joint locomotion. Additionally, these animals experienced a prominent decrease in body weight over the study period (6 weeks). In contrast, rats treated with the ATSC extract exhibited a gradual increase in body weight over the study period that was not significantly different from uninjured, control animals (Figure 2B).
Effect of ATSCs extract on macrophage and glial cell infiltration/activation within the injured spinal cord (Figure 3).
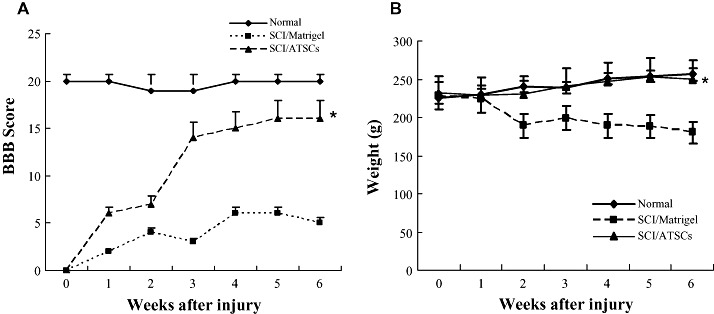
Figure 3.
Macrophage and glial cell infiltration/activation at the site of spinal cord injury. A. Confocal images of spinal cord tissue sections from the lesion epicenter stained with antibodies against GFAP (red), ED‐1 (green) and counter stained with Topro‐3 (blue). The number of GFAP‐positive and ED‐1 positive cells relative to the total number of Topro 3 positive cells was determined by counting and the mean ± the standard deviation plotted. Scale bar = 20 µm. B. Western blot analysis of spinal cord tissue extracts prepared from uninjured, control animals or animals subjected to spinal cord injury (SCI) and treated with Matrigel alone or Matrigel + adipose tissue stromal cell (ATSC) extract. Expression levels of ED1 protein were quantified by densitometry and normalized to that of Actin (*P < 0.05). ED‐1 macrophage and microglial cells (arrows) are indicated. Middle column shows an enlarged picture of control SC2 (Matrigel/SC2; left) and ATSCs extract treated SC2: ATSCs/SC2; right).
To account for the therapeutic effect of ATSC extracts in our SCI model, we first evaluated its effects on microglial cell infiltration/activation and glial scar formation in spinal cord at 1 week post injury. Few ED‐1 immuno‐positive cells were seen within spinal cord tissue of normal, uninjured rats (Figure 3A). As expected, SCI resulted in a significant increase in the number of activated macrophages as evidenced by an increase in the number of ED‐1 immuno‐positive cells (Figure 3A) and expression levels of ED‐1 protein (Figure 3B) at the site of injury (P < 0.05). In addition, significant statistical differences in the number of ED‐1 positive cells and ED‐1 protein levels in all T9‐T10 spinal segments were evident between animals treated with Matrigel alone or that in combination with ATSC extract (Figure 3A,B). Moreover, spinal tissue from injured animals treated with ATSC extract contained ED‐1 positive cells that had a round morphology with blunt processes and were larger in size in comparison to that from Matrigel treated animals (Figure 3A). Therefore, the ATSC extract stimulated invasion of activated macrophages into the site of injury and/or their proliferation. In contrast, evaluation of spinal cord sections 3 mm caudal to the injury epicenter revealed that the relative percentage of GFAP‐positive glial cells in the ventral funiculi, a white matter region with axonal tracts that is important for rat locomotion, increased 20% in tissues from animals treated with Matrigel only after SCI as compared with uninjured controls. In contrast, there were 45% fewer GFAP‐positive glial cells detected in spinal cord from animals treated with Matrigel + ATSC extract after SCI as compared with uninjured normal tissue respectively (Figure 3A). These data were consistent with western blot studies showing a decrease in GFAP and a slight increase in Tuj1 protein expression at the lesion site between ATSC extract treated vs. Matrigel only treated animals (Figure 6A). Spinal tissue from ATSC extract‐treated animals also showed significantly higher levels of Nestin protein expression, a marker of neural stem cells, and lower levels of acetyl‐Histone 3 and 4 expression as compared with that from Matrigel only treated animals, as well (Figure 6A).
Figure 6.
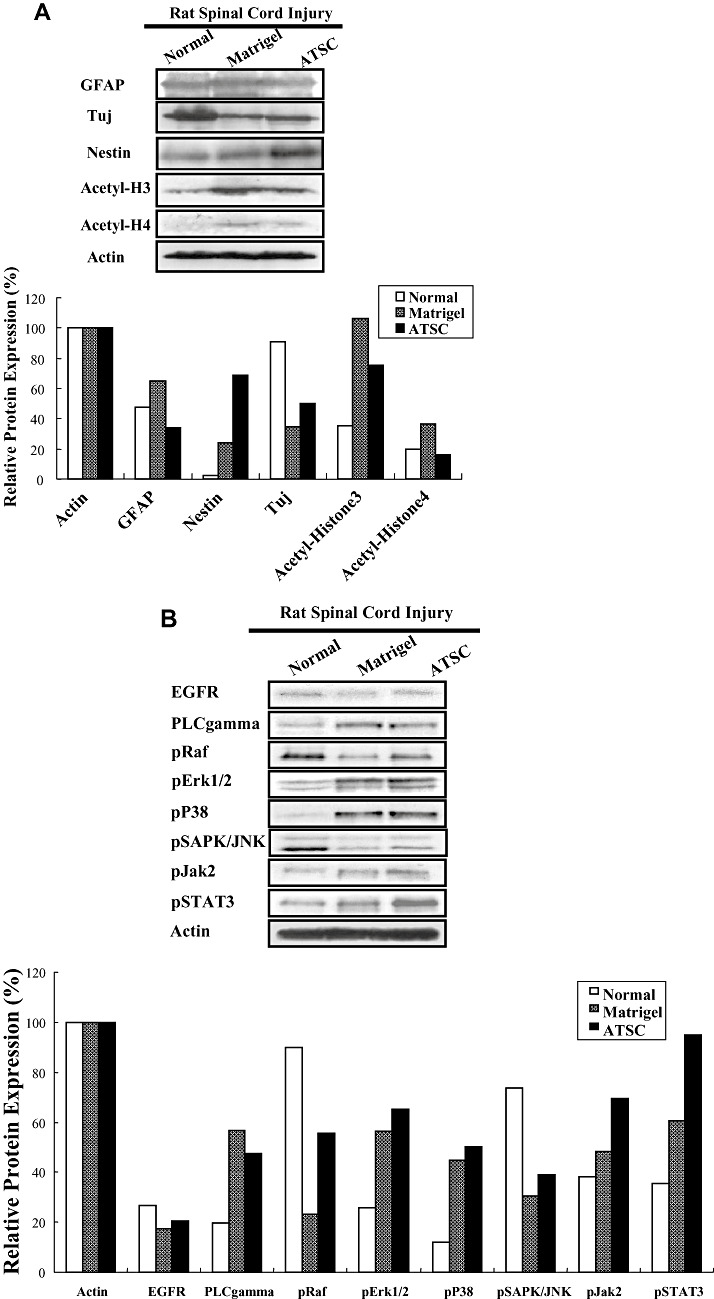
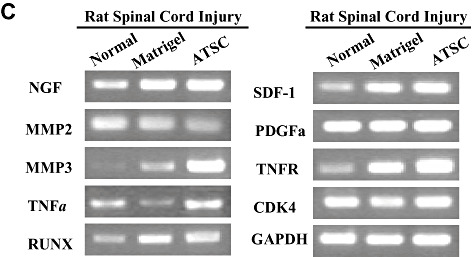
Adipose tissue stromal cell (ATSC) extract alters the expression of signaling molecules regulating cell survival and proliferation within the lesioned spinal cord. A,B. Western blot analysis of spinal cord tissue harvested from uninjured, normal animals or at 1 week post spinal cord injury (SCI) from animals treated with Matrigel or Matrigel + ATSC extract showing expression of (A) neural proteins and acetylated histone H3 and H4 or (B) signaling molecules regulating cell survival and proliferation. Relative protein expression levels were quantified by densitometry. C. Semi‐quantitative RT‐PCR of inflammation related transcripts in spinal cord tissue harvested from uninjured, normal animals or at 1 week post SCI from animals treated with Matrigel or Matrigel + ATSC extract. PCR reactions were performed in triplicate for each tissue sample and then electrophoresed through a 1.5% agarose gel and photographed.
ATSC extract attenuated apoptotic cell death within the lesioned spinal cord (Figure 4).
Figure 4.
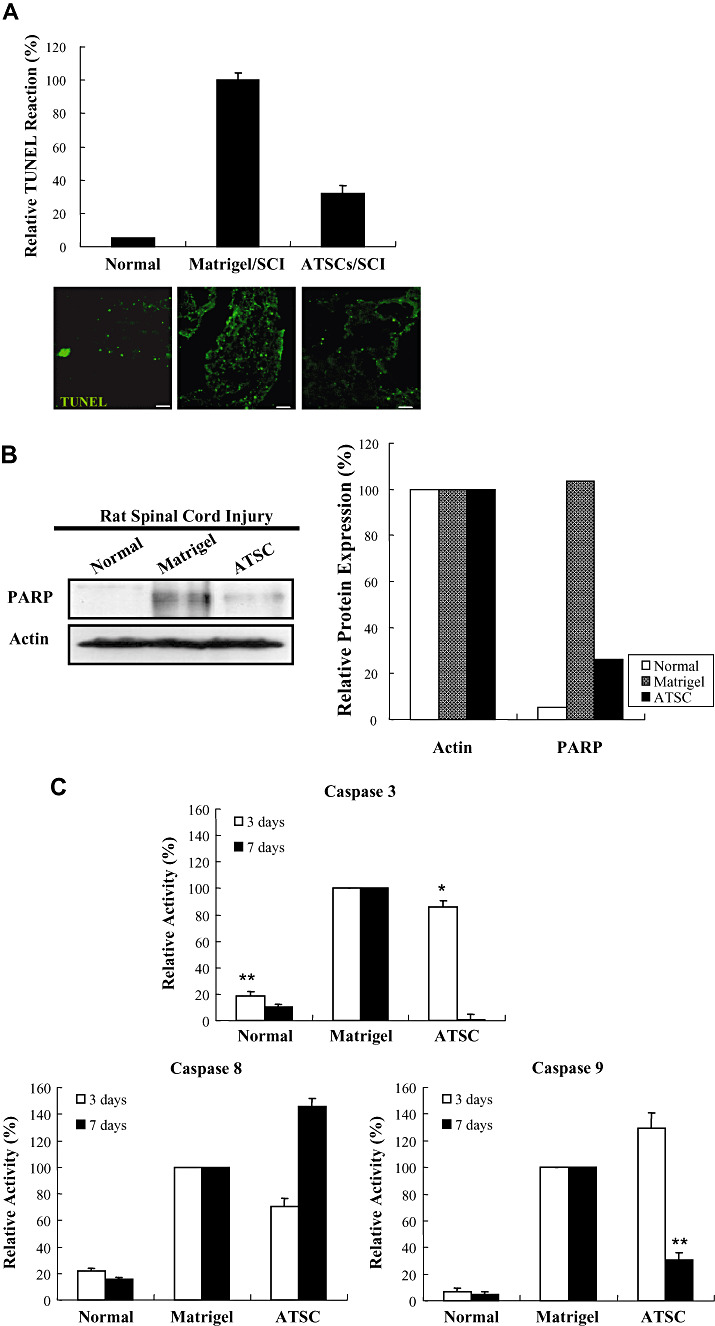
Adipose tissue stromal cell (ATSC) extract down‐regulates indices of apoptosis in spinal cord tissue after traumatic injury. A. The number of Terminal deoxynucleotidyl transferase biotin‐duTP Nick (TUNEL)‐positive cells detected within the spinal cord of uninjured, normal animals and animals treated with Matrigel or Matrigel + ATSC extracts at 1 week post spinal cord injury (SCI). Data plotted are the averaged percentage of TUNEL‐positive cells ± standard deviation from three microscopic images (100×) per experimental group captured around the lesion site. Fluorescent micrographs of representative spinal cord tissue sections are illustrated for each experimental group. TUNEL‐positive cells are represented as green spots. Scale bar = 20 µm. B. Western blot analysis of spinal cord tissue extracts prepared from uninjured, normal animals or animals subjected to SCI and treated with Matrigel alone or Matrigel + ATSC extract. Levels of PARP protein were quantified by densitometry and normalized to that of Actin. C. Caspase‐3, ‐8 and ‐9 activities measured in extracts of spinal cord tissue prepared at 3 and 7 days post injury. Data are presented as a relative activity (%) against protein‐free blank reaction mixture. Data are plotted as the mean ± standard deviation (*P < 0.05; **P < 0.001).
One week after SCI, a dramatic increase in the number of TUNEL‐positive cells was observed in spinal tissue from injured rats as compared with normal, uninjured animals. However, animals injected with Matrigel + ATSC extract showed a marked decrease in the number of TUNEL‐positive cells as compared with Matrigel only treated animals (Figure 4A) (P < 0.001). Therefore, the ATSC extract appeared to prevent apoptotic death of host cells at the site of injury. Western blot analysis also revealed negligible expression levels of Poly (ADP‐ribose) polymerase (PARP) in the cytosolic compartment of spinal cord tissue from normal, uninjured animals. SCI resulted in a dramatic increase in PARP expression within equivalent spinal segments (T9‐T11) from Matrigel injected animals at 7 days post SCI (Figure 4B). PARP expression levels were also significantly greater in injured animals injected with Matrigel + ATSC as compared with uninjured animals (P < 0.001) but were significantly lower as compared with injured animals injected with Matrigel only (P < 0.05) (Figure 4B). Similarly, expression levels of the pro‐apoptotic proteins caspase‐3, ‐8 and ‐9 were dramatically increased within the cytosolic compartment of spinal cord tissue by 3 days post injury in animals subjected to SCI as compared with normal, uninjured animals (Figure 4C). Although expression of these proteins remained elevated at 7 days post SCI in animals injected with Matrigel, caspase‐3 and ‐9 were significantly diminished while caspase‐8 was increased in Matrigel + ATSC extract treated animals (Figure 4C). Because of the fact that caspase‐8 is activated by receptor clustering but caspase‐3 and ‐9 are activated by a transition in mictochondrial membrane permeability, these data indicate that the ATSC extract aided in maintaining the integrity of mitochondrial membranes in cells resident at the site of injury.
ATSC cells extract alleviated hypo‐myelination and neuronal cell death after SCI (Figure 5).
Figure 5.
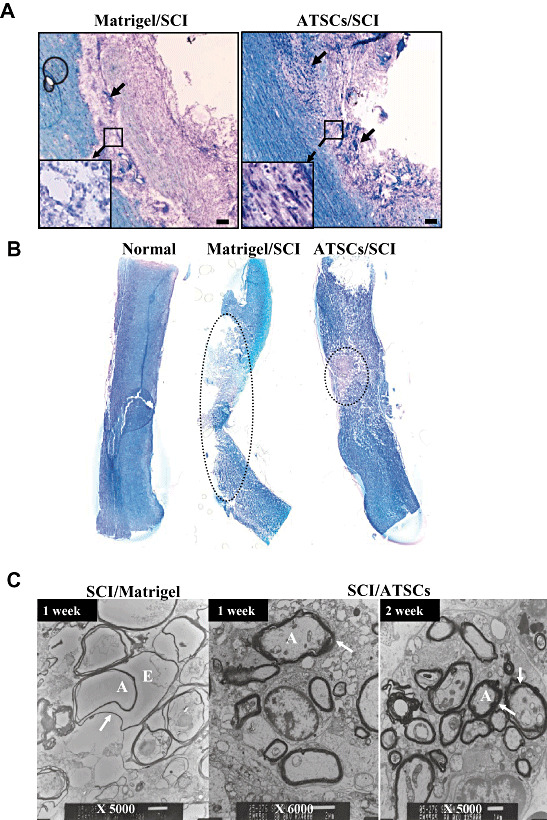
Histo‐pathological evaluation of lesioned spinal cords. Representative micrographs of spinal cord tissue sections at 7 days post injury from animals treated with Matrigel or Matrigel + adipose tissue stromal cell (ATSC) extracts. A. Sections stained with Cresyl Echt Violet and Luxol Fast Blue. Longitudinal dark blue stained cells are nerve cells within the lesion site. Normal and ATSCs extract injected spinal cord tissue was characterized by thick myelin structures across the entire control dimension of the dorsal and the ventral columns and throughout the anterior‐posterior extent of the lesions. Neurons that potentially escaped death (arrows) are indicated. B. Spinal cord tissue sections stained with Cresyl Echt Violet and Luxol Fast Blue. The dotted circle indicates the demyelinated region in the injured spinal cord. Scale bar = 20 µm. C. Transmission electron microscopy of spinal cord tissue harvested from Matrigel only treated animals at 1 week post injury and Matrigel + ATSC extract treated animals at 1 and 2 weeks post injury. Note that myelin sheaths were largely destroyed in the Matrigel only treated animals, while those treated with the ATSC extracts preserved a significant degree of normal myelination (arrow). A = Axon; E = Edema; SCI = spinal cord injury.
Staining of spinal cord tissue from SCI animals treated with Matrigel only revealed weak Luxol Fast Blue (LFB) staining, indicative of neuronal cell death and hypomyelination at the lesion epicenter (Figure 5A). Specifically, staining was completely absent in the epicenter revealing a complete loss of white matter tracts. In contrast, spinal cord tissue prepared from injured animals treated with Matrigel + ATSC cell extract showed significantly greater LFB staining around the lesion site, indicative of enhanced preservation of myelinated fibers and cresyl violet stained nerve cells within the lesion epicenter (Figure 5A). The dotted circle in Figure 5B indicates the demyelinated region (LBF‐negative white zone) in the injured spinal cord, which revealed a protective effect of the ATSC extract. The overall cross section profiles of the lesion epicenters from animals treated with Matrigel only or Matrigel + ATSC extract were comparable. Additionally, there were no significant differences in lesion area size rostral and caudal to the epicenter (data not shown). There are significant differences in overall volume of the spinal cord lesion. However, there was a significant difference in lesion cavity sizes among the three groups, indicating that gray matter tracks at the lesion site might have been preferentially protected by the ATSC extract treatment (Figure 5B). Notably, myelin sheaths surrounding the axons of many neurons in the SCI control group were not compact and exhibited separation of the lamellae and many axons showed vesicular degeneration (Figure 5C).
ATSC extract alters the expression of stress‐induced signaling molecules within the lesioned spinal cord (Figure 6).
We also compared the expression levels of stress‐induced signaling molecules within spinal cord tissue from uninjured animals to SCI animals at 1 week post injury. Western blot analysis revealed that SCI resulted in increased expression of PLC gamma, pERK1/2, p38, pJak2 and pSTAT3 and decreased expression of pRaf and pSAPK/JNK in spinal tissue from Matrigel only treated animals as compared with uninjured controls (Figure 6B). In all cases, treatment with ATSC extract enhanced expression of these signaling molecules in spinal tissue, with the exception of PLC‐gamma. These results suggest that treatment with ATSC extract promotes expression of a signaling programme that may induce proliferation/migration of microglia cells, macrophages and NPCs at the site of tissue injury after SCI. These signaling pathways may also suppress cellular apoptosis by activation of STAT3, which regulates expression of the cell survival factors Bcl2 and Bclxl. Importantly, our data show that treatment with Matrigel + ATSC extract does not inhibit the migration/invasion of macrophages to the site of spinal cord injury (Figures 3A,B). However, application of ATSC extract did significantly reduce the number of GFAP‐positive cells at the site of injury, thereby inhibiting the extent of astrogliosis. Analysis of tissues by semi‐quantitative RT‐PCR further revealed that SCI induces expression of NGF, MMP3, Runx, SDF‐1 and the TNFR and that treatment with ATSC extract further enhanced expression of these factors in spinal cord tissue (Figure 6C). Therefore, ATSC extract appears to mediate functional recovery in spinal cord by inducing a signaling program that promotes proliferation and/or blocks apoptosis of host cells at or near the site of lesion.
DISCUSSION
Traumatic injury to the spinal cord often results in permanent injury because of the limited capacity of the nervous system to regenerate lost neurons and/or repair severed nervous connections. Moreover, because of the complex organization of the spinal cord, which conveys both sensory and motor information, SCI leads to a wide range of functional deficits (34). Although the majority of spinal cord injuries are incomplete, which enables some motor/sensory information to be transmitted; patients still experience severe neurological deficits and/or malfunction of other organ systems. Subsequent to traumatic injury, neuronal and glial cell loss is apparent within and surrounding the lesion epicenter within 4 h and axonal loss within the spinal cord becomes apparent within 48 h (13). The initial injury then instigates a progressive wave of secondary physiological responses including edema, ischemia, inflammation, excitotoxicity, disturbances to ionic homeostasis, excessive cytokine release and cellular apoptosis that exacerbate the injury. Typically, a fluid‐filled cyst then arises and persists in the chronic state. Cavitation is also presented rostral and caudal to the injury epicenter (26). The latter leads to the destruction of axonal tracks left intact after the initial trauma, further impeding functional recovery after SCI (43). A therapeutic window is thought to exist between the onset of injury and the initiation of secondary physiological responses. Accordingly, a variety of pharmacological agents targeting these secondary pathological events have been evaluated and in some cases shown to possess neuroprotective or neuro‐regenerative properties in animal models of SCI, thereby providing a basis for their use in clinical trials 7, 8, 16, 21, 23.
Our studies show that administration of cytoplasmic extracts directly into the spinal cord following traumatic injury significantly improved recovery of hind limb function in rats as measured using a modified BBB motor performance test. Functional deficits in hind limb performance in our model of lower thoracic SCI are largely caused by loss of white matter axonal tracts 4, 44 and to a lesser degree the death of motor neurons related to trunk stability (14). The white matter degeneration is principally caused by the initial injury (mechanical lesion) while secondary processes such as Na+ influx‐mediated intra axonal Ca2+ accumulation leads to protease activation and degradation of the surrounding connective tissue 41, 44. Apoptosis of surviving oligodendrocytes further leads to demyelination of the surviving axons 2, 48. Our results indicate that ATSC extracts are therapeutically beneficial in that they prevent apoptosis of host cells (neurons, oligodendrocytes and glial cells) after SCI thereby maintaining a larger volume of white matter at the injury site and reducing the overall size of the lesion. Consequently, the preservation of white matter together with increased survival of ventral horn motor neurons results in a significant enhancement in hind limb function. We also noted a discernable reduction in the number of reactive astrocytes in the spared white matter. This likely also contributed to recovery since reactive astrocytes secrete inhibitory molecules such as neurocan and trabecular meshwork‐inducible glucocorticoid response protein (TIGR) that restrict repair/regeneration of motor neurons 3, 17. Moreover, astroglial scaring is also known to prevent axonal regeneration (17).
Our biochemical studies provide direct evidence that ATSC extracts promote functional recovery by diminishing several indices of the injury‐mediated pathological changes associated with SCI. For example, we showed that treatment with the ATSC extract limited cellular apoptosis at the injury site as evidenced by decreased expression of PARP and caspase‐3 and ‐9 together with a reduction in the number of TUNEL‐positive cells. Caspases are known to be important intracellular signaling molecules that promote apoptosis after spinal cord injury. Moreover, among members of the caspase family, caspase‐3 is regarded as the principal effector of apoptosis 1, 32. Caspase‐3 is known to directly cleave PARP, which blocks DNA repair but also preserves the cellular pools of ATP that may be necessary for the execution of the cell death programme (9). PARP activity is believed to contribute to neuronal cell death in a variety of neurological conditions (38) including traumatic brain injury (23) and SCI (36) as a result of energy failure or the modification of the activity of various proteins by poly ADP‐ribosylation (9). At present, it remains unclear if ATSC extracts exert their inhibitor effect on apoptosis at the level of cell surface receptor activation, cell signaling or transcriptional activation (6, 4). However, our in vitro biochemical studies revealed that ATSC extracts promote discernable changes in expressed levels of survival and proliferation promoting signaling molecules including Bcl2, ERK1/2, p38, pJak2, PLCgamma and pSTAT3 in NPCs (Figure 6B). Other studies have indicated that alterations in these signaling pathways have neuroprotective effects in brain injury models 46, 48. Furthermore, it is well established that Jak/STAT3 and PLC gamma/Raf/Erk1/2 signaling is induced by cytokines or other factors that promote cell survival and inhibit apoptosis (31). P38MAPK and SAPK/JNK are stress‐induced signaling pathway molecules that can be involved in cytokine production and cell survival, differentiation, growth and apoptosis (22).
Inflammation is a universal defense and reparative response to tissue injury. It appears that the inflammatory and immunological response to injury within the CNS is qualitatively and quantitatively different than that which is occurring in other tissues (35). Activation of both resident and recruited immune cells appears to be the cellular source of cytokines induced following SCI, which promote inflammation 3, 40. Damage to the cord also results in extensive proliferation of cells in and around the epicenter, many of which are microglia and macrophages. This sequence of events is well‐documented for Wallerian degeneration and demyelinating disease 16, 21, 28. In our study, ATSC extract did not attenuate macrophage and microglial cell infiltration into the lesion site but did protect neural cells from ED1‐positive immune cell‐mediated signaling that promotes apoptosis (3, 4, 5). Therefore, the foremost protective effect of ATSC extracts in SCI is the suppression of acute secondary apoptotic cell death. The latter results in the preservation of white matter and neurons around the lesion site, resulting in improved functional recovery (2, 5). Therefore, ATSC extracts may provide a powerful autoplastic therapy for neurodegenerative conditions in humans.
ACKNOWLEDGEMENTS
This work was supported by the 21st Century Frontier/Stem Cell Research Committee (SC3130).
REFERENCES
- 1. Armstrond RC, Aja TJ, Hoand KD, Gaur S, Bai X, Alnemri ES, Litwack G, Karanewsky DS, Fritz LC, Tomaselli KJ (1997) Activation of the CED3/ICE‐related protease‐CPP32 in cerebellar granule neurons undergoing apoptosis but not necrosis. J Neurosci 17:553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM (2002) The neonatal brain against hypoxic‐ischemic injury. Ann Neurol 52:54–61. [DOI] [PubMed] [Google Scholar]
- 3. Bareyre FM, Schwab ME (2003) Inflammation, degeneration and regeneration in the injured spinal cord: insights from DNA microarrays. TRENDS Neurosci 26:555–563. [DOI] [PubMed] [Google Scholar]
- 4. Blight AR (1983) Cellular morphology of chronic spinal cord injury in the cat: analysis of myelinated axons by line‐sampling. Neuroscience 10:521–543. [DOI] [PubMed] [Google Scholar]
- 5. Blight AR (2002) Miracles and molecules—progress in spinal cord repair. Nat Neurosci Suppl 5:1051–1054. [DOI] [PubMed] [Google Scholar]
- 6. Bracken MB, Shepard MJ, Collins WF, Holford TR, Young W, Baskin DS, Eisenberg HM, Flamm E, Leo‐Summers L, Maroon PH (1990) A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal‐cord injury. Results of the Second National Acute Spinal Cord Injury Study. N Engl J Med 322:1405–1411. [DOI] [PubMed] [Google Scholar]
- 7. Bruck W, Friede RL (1990) Anti‐macrophage CR3 antibody blocks myelin phagocytosis by macrophages in vitro . Acta Neuropathol 80:415–418. [DOI] [PubMed] [Google Scholar]
- 8. Bruck W, Friede RL (1990) The role of complement in myelin phagocytosis during PNS Wallerian degeneration. J Neurol Sci 103:182–187. [DOI] [PubMed] [Google Scholar]
- 9. Burkle A (2001) Physiology and pathophysiology of poly(ADP‐ribosyl)ation. Bioessays 23:795–806. [DOI] [PubMed] [Google Scholar]
- 10. David S, Aguayo AJ (1981) Axonal elongation into peripheral nervous system “bridges” after central nervous system injury in adult rats. Science 214:931–933. [DOI] [PubMed] [Google Scholar]
- 11. Emery E, Aldana P, Bunge MB, Puckett W, Srinivasan A, Kean RW, Bethea J, Levi AD (1998) Apoptosis after traumatic human spinal cord injury. J Neurosurg 89:911–920. [DOI] [PubMed] [Google Scholar]
- 12. Erickson GR, Gimble JM, Franklin DM, Rice HE, Awad H, Guilak F (2002) Chondrogenic potential of adipose tissue‐derived stromal cells in vitro and in vivo . Biochem Biophys Res Commun 290:763–769. [DOI] [PubMed] [Google Scholar]
- 13. Grossman SD, Rosenberg LJ, Wrathall JR (2001) Temporal‐spatial pattern of acute neuronal and glial loss after spinal cord contusion. Exp Neurol 168:273–282. [DOI] [PubMed] [Google Scholar]
- 14. Holstege G (1991) Descending motor pathways and the spinal motor system: limbic and non‐limbic components. Prog Brain Res 87:307–421. [DOI] [PubMed] [Google Scholar]
- 15. Hurlbert RJ (2001) The role of steroids in acute spinal cord injury: an evidence‐based analysis. Spine 26:S39–S46. [DOI] [PubMed] [Google Scholar]
- 16. Jander S, Lausberg F, Stoll G (2001) Differential recruitment of CD8+ macrophages during Wallerian degeneration in the peripheral and central nervous system. Brain Pathol 11:27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jones LL, Margolis RU, Tuszynski MH (2003) The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp Neurol 182:399–411. [DOI] [PubMed] [Google Scholar]
- 18. Kang SK, Lee DH, Bae YC, Kim HK, Baik SY, Jung JS (2003) Improvement of neurological deficits by intracerebral transplantation of human adipose tissue‐derived stromal cells after cerebral ischemia in rats. Exp Neurol 183:355–366. [DOI] [PubMed] [Google Scholar]
- 19. Kang SK, Putnam LA, Ylostalo J, Popescu IR, Dufour J, Belousov A, Bruce AB (2004) Neurogenesis of Rhesus adipose stromal cells. J Cell Sci 117:4289–4299. [DOI] [PubMed] [Google Scholar]
- 20. Kang SK, So HW, Moon YS, Kim CH (2006) Proteomics analysis of injured spinal cord tissue protein using 2 D‐E and MALDI‐TOF Mass. Porteomics 6:2797–2812. [DOI] [PubMed] [Google Scholar]
- 21. Kiefer R, Kieseier BC, Stoll G, Hartung HP (2001) The role of macrophages in immune‐mediated damage to the peripheral nervous system. Prog Neurobiol 64:109–127. [DOI] [PubMed] [Google Scholar]
- 22. Kyriakis JM, Avruch J (2001) Mammalian mitogen‐activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81:807–886. [DOI] [PubMed] [Google Scholar]
- 23. LaPlaca MC, Zhang J, Raghupathi R, Li JH, Smith F, Bareyre FM, Snyder SH, Graham DI, McIntosh TK (2001) Pharmacologic inhibition of poly(ADP‐ribose) polymerase is neuroprotective following traumatic brain injury in rats. J Neurotrauma 18:369–376. [DOI] [PubMed] [Google Scholar]
- 24. Li M, Ona VO, Chen M, Kaul M, Tenneti L, Zhang X, Stieg PE, Lipton SA, Friedlander RM (2000) Functional role and therapeutic implications of neuronal caspase‐1 and ‐3 in a mouse model of traumatic spinal cord injury. Neuroscience 99:333–342. [DOI] [PubMed] [Google Scholar]
- 25. Liu D, Ling X, Wen J, Liu J (2000) The role of reactive nitrogen species in secondary spinal cord injury: formation of nitric oxide, peroxynitrite, and nitrated protein. J Neurochem 75:2144–2154. [DOI] [PubMed] [Google Scholar]
- 26. McTigue DM, Tani M, Krivacic K, Chernosky A, Kelner GS, Maciejewski D, Maki R, Ransohoff RM, Stokes BT (1998) Selective chemokine mRNA accumulation in the rat spinal cord after contusion injury. J Neurosci Res 53:368–376. [DOI] [PubMed] [Google Scholar]
- 27. McTigue DM, Popovich PG, Jakeman LB, Stokes BT (2002) Strategies for spinal cord injury repair. Prog Brain Res 128:3–8. [DOI] [PubMed] [Google Scholar]
- 28. Moalem G, Monsonego A, Shani Y, Cohen IR, Schwartz M (1999) Differential T cell response in central and peripheral nerve injury: connection with immune privilege. FASEB J 13:1207–1217. [DOI] [PubMed] [Google Scholar]
- 29. Omura T, Omura K, Sano M, Sawada T, Hasegawa T, Nagano A (2005) Spatiotemporal quantification of recruit and resident macrophages after crush nerve injury utilizing immunohistochemistry. Brain Res 1057:29–36. [DOI] [PubMed] [Google Scholar]
- 30. Popovich PG, Wei P, Stokes BT (1997) Cellular inflammatory response after spinal cord injury in Sprague‐Dawley and Lewis rats. J Comp Neurol 377:443–464. [DOI] [PubMed] [Google Scholar]
- 31. Roux PP, Blenis J (2004) ERK and P38 MAPK‐activated protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68:320–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ruemmele FM, Dionne S, Levy E, Seidman EG (1999) TNFα‐induced ICE‐6 cell apoptosis requires activation of ICE caspases whereas complete inhibition of the caspase leads to necrotic cell death. Biochem Biophys Res Commun 260:159–166. [DOI] [PubMed] [Google Scholar]
- 33. Safford KM, Hicok KC, Safford SD, Halvorsen YD, Wilkison WO, Gimble JM, Rice HE (2002) Neurogenic differentiation of murine and human adipose‐derived stromal cells. Biochem Biophys Res Commun 294:371–379. [DOI] [PubMed] [Google Scholar]
- 34. Schwab ME, Bartholdi D (1996) Degeneration and regeneration of axons in the lesioned spinal cord. Physiol Rev 76:319–370. [DOI] [PubMed] [Google Scholar]
- 35. Schwartz M, Moalem G, Leibowitz‐Amit R, Cohen IR (1999) Innate and adaptive immune responses can be beneficial for CNS repair. Trends Neurosci 22:295–299. [DOI] [PubMed] [Google Scholar]
- 36. Scott GS, Jakeman LB, Stokes BT, Szabo C (1999) Peroxynitrite production and activation of poly (adenosine diphosphate‐ribose) synthetase in spinal cord injury. Ann Neurol 45:120–124. [PubMed] [Google Scholar]
- 37. Scott GS, Cuzzocrea S, Genovese T, Koprowski H, Hooper DC (2005) Uric acid protects against secondary damage after spinal cord injury. PNAS 102:3483–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Skaper SD (2003) Poly(ADP‐Ribose) polymerase‐1 in acute neuronal death and inflammation: a strategy for neuroprotection. Ann NY Acad Sci 993:217–228. [DOI] [PubMed] [Google Scholar]
- 39. Springer JE, Azbill RD, Knapp PE (1999) Activation of the caspase‐3 apoptotic cascade in traumatic spinal cord injury. Nat Med 5:943–946. [DOI] [PubMed] [Google Scholar]
- 40. Sribnick EA, Wingrave JM, Matzelle DD, Wilford GG, Ray SK, Banik NL (2005) Estrogen attenuated markers of inflammation and decreased lesion volume in acute spinal cord injury in rats. J Neurosci Res 82:283–293. [DOI] [PubMed] [Google Scholar]
- 41. Stys PK, Waxman SG, Ransom BR (1992) Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na(+)‐Ca2+ exchanger. J Neurosci 12:430–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taoka Y, Okajima K (1998) Spinal cord injury in the rat. Prog Neurobiol 56:341–358. [DOI] [PubMed] [Google Scholar]
- 43. Tator CH, Fehlings MG (1991) Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J Neurosurg 75:15–26. [DOI] [PubMed] [Google Scholar]
- 44. Teng YD, Wrathall JR (1997) Local blockade of sodium channels by tetrodotoxin ameliorates tissue loss and long‐term functional deficits resulting from experimental spinal cord injury. J Neurosci 17:4359–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Teng YD, Choi H, Onario RC, Zhu S, Desilets FC, Lan S, Woodard EJ, Snyder EY, Eichler ME, Friedlander RM (2004) Minocycline inhibits contusion‐triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. PNAS 101:3071–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J (2001) Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci 21:2580–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu J, Kim GM, Chen S, Yan P, Ahmed SH, Ku G, Beckman JS, Xu XM, Hsu CY (2001) iNOS and nitrotyrosine expression after spinal cord injury. J Neurotrauma 18:523–532. [DOI] [PubMed] [Google Scholar]
- 48. Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J (1998) Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. PNAS 95:15769–15774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, Benhaim P, Lorenz HP, Hedrick MH (2001) Multilineage cells from human adipose tissue: implications for cell‐based therapies. Tissue Eng 7:211–228. [DOI] [PubMed] [Google Scholar]