

Abstract
One cardinal feature of Huntington's disease (HD) is the degeneration of striatal neurons, whose survival greatly depends on the binding of cortical brain‐derived neurotrophic factor (BDNF) with high‐affinity (TrkB) and low‐affinity neurotrophin receptors [p75 pan‐neurotrophin receptor (p75NTR)]. With a few exceptions, results obtained in HD mouse models demonstrate a reduction in cortical BDNF mRNA and protein, although autopsy data from a limited number of human HD cortices are conflicting. These studies indicate the presence of defects in cortical BDNF gene transcription and transport to striatum. We provide new evidence indicating a significant reduction in BDNF mRNA and protein in the cortex of 20 HD subjects in comparison with 17 controls, which supports the hypothesis of impaired BDNF production in human HD cortex. Analyses of the BDNF isoforms show that transcription from BDNF promoter II and IV is down‐regulated in human HD cortex from an early symptomatic stage. We also found that TrkB mRNA levels are reduced in caudate tissue but not in the cortex, whereas the mRNA levels of T‐Shc (a truncated TrkB isoform) and p75NTR are increased in the caudate. This indicates that, in addition to the reduction in BDNF mRNA, there is also unbalanced neurotrophic receptor signaling in HD.
Keywords: BDNF, TrkB, T‐Sch, p75NTR, Huntington's disease, human brain
INTRODUCTION
Huntington's disease (HD), a dominantly inherited and progressive neurodegenerative disease, is caused by an abnormal expansion of a polyglutamine tract close to the N‐terminus of the huntingtin protein (51). Although mutant huntingtin is ubiquitously expressed (52), and it is known that global and early neuronal dysfunction occurs in the brains of HD patients 44, 45, the loss of neurons in the caudate nucleus and cerebral cortex is a hallmark of the disease (42).
Since the discovery of the HD gene in 1993, genetic and experimental evidence has provided insights into the pathogenesis of HD, and many hypotheses concerning its underlying mechanisms are now being investigated. One that we put forward in 2001 is that brain‐derived neurotrophic factor (BDNF) depletion originating in the cortex may make striatal neurons more vulnerable (57).
BDNF is a small dimeric protein that was discovered in 1982 (7) as the second in a family of molecules with neurotrophic activities. It is primarely synthesised as a 32 kDa precursor known as pro‐BDNF, whose cleavage in the trans‐Golgi network or secretory granules generates a mature 14 kDa biologically active form that is secreted in a basal and activity‐dependent manner (8). Interestingly, more recent findings attribute a functional role to pro‐BDNF at synaptic terminals that triggers intracellular pathways other than mature BDNF or promotes apoptotic cell death 41, 50. BDNF is widely expressed in the adult mammalian central nervous system, but is particularly abundant in the hippocampus and cerebral cortex (34). Its expression increases during brain development, peaks after birth 6, 27, 28, and the fact that it does not seem to decline with age suggests that it plays an essential role in the adult central nervous system 6, 27, 28. BDNF has survival‐ and growth‐promoting activity on a variety of neurons, and in addition to its important role in neuronal development and cell survival, it is also involved in neuronal plasticity as it controls activity‐dependent synaptic transmission (8).
It is known that BDNF is very important for the striatal neurons that die in HD. These require BDNF for their survival and differentiation during development 2, 24, 33, 36, 53, 54, and it also protects them against excitotoxicity, which may be involved in the degeneration observed in HD patients 30, 38, 39, 55. However, striatal neurons do not produce their own BDNF, which is anterogradely transported through the cortical afferents to the point that 95% of the BDNF found in the striatum is of cortical origin 3, 6.
There is now a considerable amount of evidence indicating that defects in cortical BDNF production or its delivery to striatal targets may contribute to the development of HD (56). We have previously reported that, in HD cells and animal models, wild‐type huntingtin acts at the level of the BDNF promoter to facilitate its gene transcription, thus participating in the physiological production of cortical BDNF (57), whereas mutation in huntingtin has a loss‐of‐function effect on BDNF gene transcription. In addition, by acting at the level of a different portion of the BDNF promoter, mutant huntingtin further represses BDNF gene transcription (56). Together, these events cause a net depletion in BDNF mRNA and protein in HD cells and mice. A large number of subsequent experiments using different HD mouse models have further confirmed the evidence of a depletion in cortical BDNF mRNA and protein (56). A different, but not mutually exclusive hypothesis indicates that the same amount of BDNF is produced in the cortex of HD subjects and controls, but its transport from the cortex to the striatum is reduced in the presence of the mutant protein (16).
Post‐mortem assessments of BDNF mRNA and protein in human tissue have led to conflicting findings that are mainly caused by differences in analytical methods. Western blot analyses of the parietal cortex, temporal cortex, hippocampus, caudate and putamen of four grade three HD subjects, indicated that the levels of mature BDNF protein (14 kDa) were between 53% and 82%, less in the caudate and putamen of the HD subjects than in samples from six age‐matched controls, but preserved in the parietal and temporal cortex, and the hippocampus (15). The findings of a second Western blot study of ten HD patients and seven controls by Gauthier et al were in line with these data, showing that mature BDNF protein levels were reduced by about 50% in the striatum of HD patients, but not in the cerebral cortex (16). However, we have previously shown that the levels of BDNF protein (as assessed by enzyme‐linked immunosorbent assay (ELISA), which not only detects the mature form, but also the other processed forms including pro‐BDNF) and BDNF mRNA in the human cortex are consistent with those observed in the various transgenic mouse models of HD: that is, there was a 50% decrease in BDNF levels in the frontoparietal cortex of two subjects with grade 2 and 3 HD in comparison with two age‐matched controls (57).
We have now expanded that work by quantifying BDNF levels in a larger number of samples of post‐mortem cortical tissues from 20 subjects with HD (grades 2–4) and 17 gender‐ and age‐matched controls. BDNF protein and mRNA were respectively evaluated by means of ELISA and quantitative real‐time polymerase chain reaction (PCR) and, as previous data have indicated reduced BDNF promoter exon II and III activity in HD cells and mouse models of HD, we also determined the levels of the BDNF mRNA isoforms whose transcription is specifically driven by the homologous human promoters 21, 57, 58. We conclude that human HD is characterized by a statistically significant reduction in cortical BDNF mRNA (total and different isoforms) and protein.
As the biological effect of BDNF is mediated by the full‐length TrkB receptor in cooperation with the truncated isoforms Trk‐T1 (the dominant truncated isoform expressed in the adult brain), Trk‐T2 8, 41 and T‐Shc (47), we evaluated Trk‐B, Trk‐T1 and T‐Shc mRNA levels in a selection of cortical and striatal tissues from HD and control subjects, and found a reduction in TrkB mRNA levels in the caudate of the HD subjects, and increased expression of T‐Shc. Finally, we also found that p75 pan‐neurotrophin receptor (p75NTR) (13) mRNA levels are increased in the caudate, thus further confirming the dysfunction of Trk‐mediated signaling in the brain of HD subjects.
MATERIALS AND METHODS
Human tissues
The post‐mortem human brain tissues were obtained from the following sources: (i) New York Brain Bank, Columbia University (New York, NY, USA; Controls: T‐168, T‐111, T‐274; HD grade 2: T‐289; grade 4: T‐128, T‐291, T‐178, T‐260, T‐310, T‐329, T‐354); (ii) Harvard Brain Tissue Resource Center (HBTRC) (Belmont, MA, USA; Controls: 6182, 6142, 6002, 5959, 5936, 5919, 5077, 5074, 5021; HD grade 2: 6051, 6121; grade 3: 5570, 5576, 6010, 6183; grade 4: 5507, 6062; (iii) Massachusetts General Hospital (MGH) (Charlestown, MA, USA; Controls: 3688, 3888, 3746, 3899, 3932; HD grade 3: 3176, 3484, 3723, 2866).
It was not possible to compare the HBTRC samples 4741, 4744, 4751, 4719, 4754, 4797, 4680, 4740 and 4798 used by Gauthier et al, 2004, because of the unavailability of the same tissue.
We analyzed post‐mortem human brain tissue (parietal cortex, Brodmann's area 7) taken from a total of 20 HD patients (Vonsattel's grade 2–4) and 17 gender‐ and age‐matched controls, whose CAG repeat lengths, post‐mortem interval, gender and disease grade are shown in Table 1. The HD cases for which the length of the CAG repeat for the pathological allele was not provided by the brain bank underwent genomic DNA extraction and genotyping for CAG length in the Laboratory of Molecular Genetics of the Division of Biochemistry and Genetics, Neurological Institute Carlo Besta, Milan, in accordance with the published methods for studying trinucleotide repeat diseases (17).
Table 1.
Characteristics of the human brain samples. Parietal cortex (BA7) and caudate have been used. HD samples are all in the pathological range (>35 CAG repeats). Abbreviations: IQR = interquartile range (25th–75th percentile); PMI = post‐mortem interval; HD = Huntington's disease
| Controls | 17 |
| Male | 11 |
| Female | 6 |
| Age | |
| IQR | 15 |
| Median | 63 |
| PMI | |
| IQR | 5 |
| Median | 20 |
| HD | 20 |
| Male | 11 |
| Female | 9 |
| Age | |
| IQR | 15 |
| Median | 54 |
| CAG size | |
| IQR | 3 |
| Median | 45 |
| Grade | |
| Grade 2 | 3 |
| Grade 3 and 4 | 17 |
| PMI | |
| IQR | 10 |
| Median | 19 |
RNA isolation
Total RNA was isolated from 200–300 mg of human tissue using 2 mL of TRIZOL reagent (Invitrogen, Carlsbad, CA, USA), after the tissues had been homogenized in liquid nitrogen with a mortar and pestle. To enhance RNA yield, the samples were precipitated by adding 2 µL of glycogen solution (10 mg/mL) to isopropanol, and incubating them at −80°C overnight. The concentration of RNA was evaluated spectrophotometrically, and its quality was verified by means of agarose gel electrophoresis of 1 µg of each sample. Genomic DNA was digested using 1 U rDNaseI (Applied Biosystems, Foster City, CA, USA) per 1 µg/µL of total RNA and incubation at 37°C for 10 minutes following the manufacturer's instructions. The total RNA was stored in aliquots at −80°C.
RNA retrotranscription
Total RNA (1 µg) was reverse‐transcribed to single‐stranded cDNA using Superscript III RNaseH‐ reverse transcriptase (Invitrogen, Carlsbad, CA, USA) and random primers in a volume of 20 µL, according to the manufacturer's instructions. Two independent reverse transcription (RT) reactions were performed for every RNA stock.
Real‐time PCR for total BDNF and Trk mRNA levels
Because of the large number of samples and their replicates in each PCR plate, different plates were run for BDNF and GAPDH. Three independent PCR analyses were made of each of two independent RT reactions, for a total of six independent measurements.
Using an iCycler Thermal Cycler with a Multicolor Real‐time PCR Detection System (Bio‐Rad, Hercules, CA, USA), all of the reactions were performed in a total volume of 25 µL containing 50 ng cDNA, 50 mM KCl, 20 mM Tris‐HCI, pH 8.4, 0.2 mM dNTPs, iTaq DNA polymerase, 25 units/mL, 3 mM MgCl2, SYBR Green I, 10 nM fluorescein, stabilisers (iQTM SYBR Green Supermix‐Biorad, Hercules, CA, USA), and 0.3 µM of forward and reverse primers. The amplification cycles consisted of an initial denaturing cycle at 95°C for 3 minutes, followed by 45 cycles of 30 s at 95°C, 30 s at 60°C and 30 s at 72°C. Fluorescence was quantified during the 60°C annealing step, and product formation was confirmed by means of melting curve analysis (55°C–94°C). The amounts of target gene mRNA were normalized to the reference gene GAPDH.
The BDNF‐specific primers set up on the BDNF coding sequence (Genbank accession number AF411339) were:
BDNF 5′‐TAACGGCGGCAGACAAAAAGA‐3′;
BDNF 5′‐GAAGTATTGCTTCAGTTGGCCT‐3′.
The obtained amplification product was 101 bp long.
The TrkB‐specific primers were set up referring to Genbank accession number NM_006180.3 for TrkBFL, NM_001007097.1 for Trk‐T‐1, NM_001018066.1 for T‐Shc, and NM_002507.1 for P75NTR.
The primer sequences were:
TrkB 5′‐CAAGACAAGGTGTTGGCCCAG‐3′;
TrkB 5′‐CTGCTCAGGACAGAGGTTATAGC‐3′;
Trk‐T1 5′‐AACCGGTCGGGAACATCTCTC‐3′;
Trk‐T1 5′‐CCCATCCAGTGGGATCTTATG‐3′;
T‐Shc 5′‐CATCTTCTTCGGAAGGTGGCC‐3′;
T‐Shc 5′‐CGGTCTTGGGGGAACCTCTG‐3′;
p75NTR 5′‐TACGGCTACTACCAGGATGAGA‐3′;
p75NTR 5′‐ACCGTGTAATCCAACGGCCA‐3′.
The sizes of the amplification products for TrkB, Trk‐T1, T‐Shc and p75NTR were respectively 255 bp, 158 bp, 143 bp and 265 bp.
The primers used to determine GAPDH expression were:
GAPDH 5′‐ AGCTGAACGGGAAGCTCACT ‐3′;
GAPDH 5′‐ AGGTCCACCACTGACACGTTG ‐3.
The size of the amplified product was 67 bp.
Semi‐quantitative radioactive PCR
Total RNA preparations were used for two independent RT reactions, and duplicate radioactive PCR analyses were made of each RT. The PCRs were performed in a total cDNA volume of 50 µL consisting of 0.25 µg of RNA, 20 mM Tris‐HCl, pH 8.4, 50 mM KCl, 1.5 mM MgCl2, 0.2 mM dNTPs, 1.7 µCi α32PdCTP, 0.4 µM of each primer, and 2 U Taq polymerase (Invitrogen, Carlsbad, CA, USA).
BDNFIIA, IIB and IIC were amplified by means of amplification cycles consisting of an initial denaturing cycle at 94°C for 5 minutes, followed by 30 cycles of 1 minute at 94°C, 45 s at 62°C and 45 s at 72°C. The BDNFIV amplification cycles consisted of an initial denaturing cycle at 94°C for 5 minutes, followed by 30 cycles of 1 minute at 94°C, 45 s at 59°C and 45 s at 72°C.
The primers for the amplification of BDNFII mRNA were:
BDNF II: 5′‐GGGCGATAGGAGTCCATTCAGCAC‐3′,
BDNF IX (set up on BDNF coding region): 5′‐CCAAGCCACCTTGTCCTCGGATG‐3′.
The sizes of the amplification product for BDNF IIA, IIB and IIC were respectively 485 bp, 402 bp and 187 bp.
The primers for the amplification of BDNF IV were:
BDNF IV 5′‐GCTGCAGAACAGAAGGAGTACA‐3′;
BDNF IXa (set up on BDNF coding region): 5′GTCCTCATCCAACAGCTCTTCTATC‐3′. The size of the amplified band was 412 bp.
The same amount of each cDNA was also independently amplified with specific primers for synaptosomal‐associated protein 25 kDa (SNAP‐25), a presynaptic membrane‐associated protein localized in grown cones, axons and presynaptic terminals. The amplification cycles consisted of an initial denaturing cycle at 94°C for 5 minutes, followed by 30 cycles of 30 s at 94°C, 30 s at 57°C and 1 minute at 72°C.
The primer sequences for the amplification of SNAP‐25 were:
SNAP‐25: 5′CAAATGATGCCCGAGAAAAT‐3′;
SNAP‐25: 5′‐GGAATCAGCCTTCTCCATGA‐ 3′.
The amplification size of the obtained PCR product was 148 bp.
The PCR products were separated by means of non‐denaturing 6% polyacrylamide gel electrophoresis, and visualized by autoradiography.
BDNF IIA, IIB, IIC, IV and SNAP‐25 mRNA levels were calculated using Quantity One software from Bio‐Rad. Different exposure times were used in order to ensure that the autoradiographic bands were in the linear range of intensity.
ELISA
Three independent ELISAs were performed on three different lysates prepared from 150–200 mg of cortical and striatal tissue, with the samples being tested in triplicate in each assay. The tissue lysates were prepared in a lysis buffer consisting of glycerol 10%, 25 mM Tris HCl, pH 7.5, 150 mM NaCl, Triton ×100 1%, 5 mM EDTA, 1 mM EGTA supplemented with 1:100 of Protease Inhibitor Cocktail (Sigma‐Aldrich, St. Louis, MO, USA); 10 µL of lysis buffer were used for each milligram of tissue. The frozen samples were homogenized in ice using a glass dounce homogenizer (15–20 strokes). After sonicating the tissue extract four times at 15 s pulses with the microprobe at 50%–60% output and 60% duty cycle, the samples were acidified, neutralized and centrifuged for 15 minutes at 4°C at maximum Biofuge speed. The supernatants were collected and stored in aliquots at −80°C. The BDNF measurements were made within 1 week of lysate preparation. The samples were assayed for BDNF using an ImmunoAssay System (Promega, Madison, WI, USA) as described by the manufacturer. Total proteins were quantified using Bradford's method.
Statistical analyses and data representation
As BDNF and Trk values do not have a normal distribution, the non‐parametric Mann–Whitney two‐ tailed U‐test was used for the statistical analyses, with significance being set at P < 0.05. The correlations between disease grade or post‐mortem interval (PMI) and the BDNF value were assessed using Spearman's rank correlation coefficient.
Median values were chosen because of the skewed distribution of the data. In each figure, the boundary of the box closest to zero indicates the 25th percentile, the line within the box marks the median or 50th percentile, and the boundary of the box farthest from zero indicates the 75th percentile. When 10 or more samples were analyzed, whiskers above and below the box indicate the 90th and 10th percentiles.
RESULTS
Cortical BDNF protein levels are reduced from the early symptomatic stages of HD
We prepared protein extracts from cortical specimens of 20 HD subjects (neuropathological staging Vonsattel grades 2–4) and 17 age‐ and gender‐matched controls, and measured the level of BDNF protein by means of ELISA. Both pro‐BDNF and mature BDNF can be detected by ELISA but, because of the short life of the pro‐BDNF protein, mature BDNF is always thought to be the predominant BDNF form. To maintain consistency, the analyses were limited to tissue dissected from the superior parietal lobule of the cerebral cortex (Brodmann's area 7: BA7) (Table 1).
Extraction conditions may have a significant effect on the levels of BDNF protein measured in brain tissues (28) and, in this first phase, we followed the same procedure used to extract BDNF from mouse brain (57). The tissues were homogenized with neutral extraction buffers at low salt concentrations (150 mM sodium chloride) containing moderate detergents (Triton ×100 1%), and the protein lysates were treated with acid and then neutralized in order to allow BDNF release from the vesicles, which increases BDNF recovery (28). It is critically important to remember that BDNF protein levels are significantly reduced if the samples are kept for more than 2 weeks at a temperature of −80°C (data not shown), and so we assayed the lysates within 1 week of their preparation. Figure 1A shows the BDNF protein level in each of the 17 healthy controls and 20 HD subjects, which naturally varied in the different samples: in the control group, BDNF protein content in the parietal cortex ranged from 3.4 ± 0.30 pg/mg of tissue lysate to 14.27 ± 2.04 pg/mg; the corresponding figures in the HD group were 2.1 ± 0.28 pg/mg and 13.80 ± 0.28 pg/mg. Such variable values are to be expected in post‐mortem tissue. They may be related to protein degradation, which may depend on the PMI of the selected samples. However, the calculated Spearman rank correlation coefficient did not reveal any significant correlation between PMI and BDNF protein level (see Figure S1A), thus suggesting that the observed variability in BDNF content does not depend on PMI.
Figure 1.
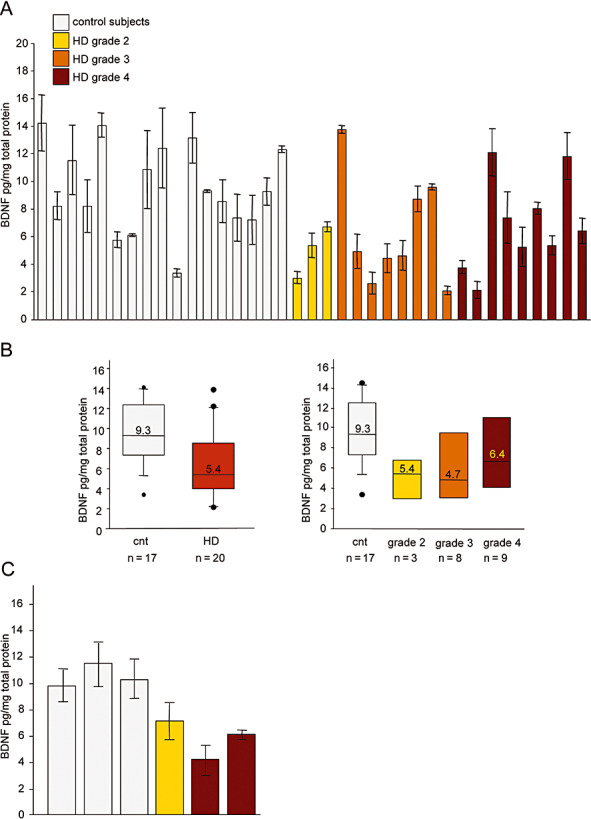
Brain‐derived neurotrophic factor (BDNF) protein levels are reduced in the cortex of Huntington's disease (HD) subjects. A. Cortical BDNF levels were determined by enzyme‐linked immunosorbent assay (ELISA). BDNF protein content was tested in triplicate in each of three independent assays, and so each column represents the mean value ± SD of nine measurements. Control subjects, from left to right: 6182, 6142, 6002, 5959, 5936, 5919, 5077, 5074, 5021,T‐168, T‐111, T‐274, 3688, 3888, 3746, 3899 and 3932; HD subjects, from left to right: 6051, 6121, T‐289, 5576, 6010, 6183, 3176, 2866, 3723, 3484, 5570, 5507, 6062, T‐128, T‐291, T‐178, T‐260, T‐310, T‐329 and T‐354. Statistical analyses made using the non‐parametric Mann–Whitney test revealed a significant reduction in BDNF protein levels in the cortex of the HD subjects (P < 0.05, Mann–Whitney two‐tailed test), and in the grade 2 patients compared with the 17 control subjects (P < 0.01, Mann–Whitney two‐tailed test); Spearman's test showed that there was no progressive reduction in BDNF levels with higher disease grades. B. Median BDNF protein levels in the control and HD groups (left panel), and by HD grade (right panel). The boundary of the box closest to zero indicates the 25th percentile, the line within the box marks the median, and the boundary of the box farthest from zero indicates the 75th percentile. When 10 or more samples were analyzed, whiskers above and below the box indicate the 90th and 10th percentiles. Outliers are indicated as black circles: controls 6182 and 5936 are outliers above the 90th percentile, and control T‐168 beneath the 10th percentile; the grade 3 and 4 HD patients (respectively 5576 and T‐128) fell above the 90th percentile, and the grade 3 and 4 HD patients (5570 and 6062) beneath the 10th percentile. C. Striatal BDNF protein levels as determined by ELISA. Control subjects, from left to right: 5919, 6142, and 6182; HD subjects, from left to right: 6121, 5507 and 5570. Statistical analysis revealed a significant reduction in BDNF protein levels in the caudate of the HD subjects (P < 0.05, Mann–Whitney two‐tailed test).
As most of the BDNF values in the control and HD groups did not fit a normal distribution curve, the non‐parametric Mann–Whitney U‐test was used to determine the statistical significance of BDNF protein content between the two cohorts. This analysis revealed that, in comparison with the 17 control subjects, BDNF protein was significantly reduced in the HD group (P < 0.05, two‐tailed test), particularly in the three grade two patients (P < 0.01). The fact that there were no significant differences between the different grades suggests that, at least in our cases, there is no correlation between cortical BDNF protein levels and disease stage (as also shown by Spearman's test). As the CAG repeat length was very similar among the HD patients (see Table 1), it was not possible to correlate BDNF levels with CAG length. Figure 1B shows median cortical BDNF protein levels in the controls and HD patients, which better represent the average value of not normally distributed data, and also reduce the skewing caused by outliers.
The ELISA‐determined BDNF levels in our control human cortex samples were reasonably similar to some values previously reported by others 22, 26, but much lower than those observed in mice, which are in the range of ng/mg of protein lysate 28, 57. In the same ELISA experiments, we tested BDNF protein content in mouse cortical lysates as a control, and found levels of 5–10 ng/mg of protein lysate, thus excluding the possibility that the lower levels in human brain were caused by differences in assay sensitivity. Mouse brain tissues can be immediately processed after dissection or stored in liquid nitrogen, which is clearly not possible in the case of human post‐mortem material. Protein degradation and possible differences in the activity of the enzymes controlling BDNF cleavage cannot be excluded, and may lead to an underestimate of BDNF protein content, as well as the observed and expected variability. Finally, a different protein extraction protocol may be required to ensure greater BDNF recovery, but we could not test different protocols because of the limited amount of tissue available. However, we conclude that HD subjects have significantly lower cortical BDNF protein levels than controls and, importantly, our findings show that this reduction occurs at an early symptomatic stage (grade 2).
As striatal neurons are the primary sites of degeneration in HD, and BDNF of cortical origin is vital for their survival, we measured BDNF protein in post‐mortem striatum extracts from controls and HD patients (grade 3 and 4) whose cortical BDNF content had been previously analyzed. Caudate samples are the most requested by HD researchers, and therefore only very small amounts are available from brain banks. We obtained caudate tissues from eight controls and eight HD subjects, but use ELISA in the case of only three HD subjects (grades 2 and 4) and three controls (in most cases, the amount of post‐mortem caudate was not enough for protein lysate preparation and subsequent ELISA). Although derived from a small number of subjects, our data show that the BDNF protein content was significantly reduced in the presence of the HD mutation (P < 0.05, Mann–Whitney two‐tailed test; see Figure 1C), and suggest that the clearly demonstrated reduction in cortical BDNF leads to reduced striatal BDNF levels.
BDNF gene transcription is affected in the cortex of HD subjects
The human BDNF gene contains ten non‐coding and one major coding exon 29, 40. Transcription is initiated at BDNF exon I, II, III, IV, V, VI, VII or IX, and the splice donor site of each of these exon sequences is spliced to the major coding exon acceptor site, thus leading to a remarkable heterogeneity in BDNF expression patterns.
To verify whether the decreased cortical levels of BDNF protein in HD subjects are caused by reduced BDNF gene transcription, total BDNF mRNA levels were evaluated in the same tissue samples as those tested by ELISA.
The RNA was extracted, and the samples were treated with DNase to eliminate genomic DNA contamination. The quality controls included spectrophotometer analyses at 260 and 280 nm (an A260:A280 ratio of >1.8 was considered an acceptable indicator of RNA purity), and routinely performed agarose gel electrophoresis using RNA stained with ethidium bromide in order to identify ribosomal bands (28S and 18S) as an indicator of RNA integrity (see Figure S2). The quality of the mRNA in the tissue samples was also evaluated by examining the constancy of GAPDH expression, and quantitative real‐time PCR experiments showed no quantitative differences in GAPDH mRNA between the HD patients and controls. Subsequently, the same cDNA samples were analyzed for their total BDNF mRNA content using primers recognizing the downstream coding exons present in all BDNF transcripts.
Figure 2A shows total BDNF mRNA levels in HD and control subjects, normalized to GAPDH mRNA content. There was no correlation between BDNF mRNA level and PMI (see Figure S1B). In line with the ELISA data, BDNF mRNA levels were significantly lower than those observed in the 17 control subjects (P < 0.001, Mann–Whitney two‐tailed test), although the extent of the reduction did not correlate with disease stage (according to Spearman's test). Figure 2B shows the median total BDNF mRNA levels in the control and HD groups. These data confirm the reduction in cortical BDNF in the absence of a correlation between BDNF mRNA levels and disease grade, as in the case of BDNF protein levels.
Figure 2.
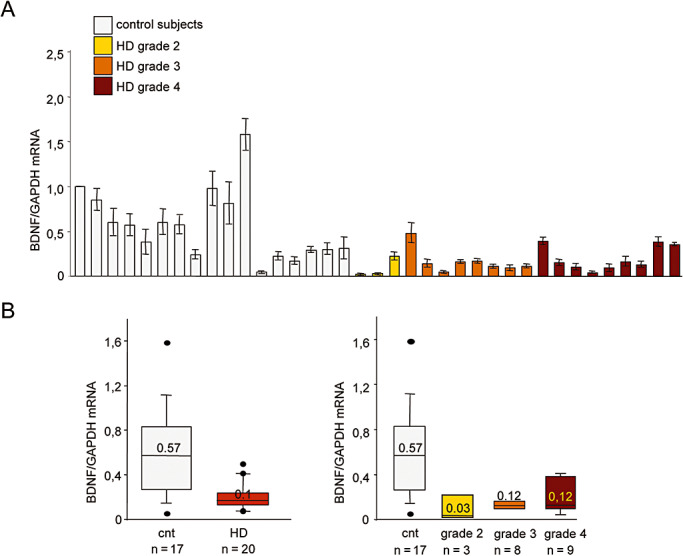
Total brain‐derived neurotrophic factor (BDNF) mRNA levels are reduced in the cortex of Huntington's disease (HD) subjects. A. Total BDNF mRNA levels were determined by means of quantitative real‐time polymerase chain reaction (PCR) and normalized to the level of GAPDH mRNA. The values are the averages ± SD of six independent PCR experiments. Control subjects, from left to right: T‐274, 6182, 6142, 6002, 5959, 5936, 5919, 5077, 5074, 5021, T‐168, T‐111, 3688, 3888, 3746, 3899 and 3932; HD subjects, from left to right: 6051, 6121, T‐289, 5576, 6010, 6183, 3176, 2866, 3723, 3484, 5570, 5507, 6062, T‐128, T‐291, T‐178, T‐260, T‐310, T‐329 and T‐354. BDNF mRNA levels were significantly reduced in the cortex of the HD subjects (P < 0.001, Mann–Whitney two‐tailed test), but did not significantly decrease with increasing disease grade (Spearman's test). B. Left panel: median BDNF mRNA levels in the control and HD groups. Right panel: median BDNF mRNA levels at different disease stages. The boundary of the box closest to zero indicates the 25th percentile, the line within the box marks the median, and the boundary of the box farthest from zero indicates the 75th percentile. When 10 or more samples were analyzed, whiskers above and below the box indicate the 90th and 10th percentiles. Outliers are indicated as black circles. Left panel: control T‐168 fell above the 90th percentile, and control T‐111 beneath the 10th percentile. The grade 3 and 4 HD patients (respectively 5576 and 5507) fell above the 90th percentile, and the grade 2 patients 6051 and 6121 fell beneath the 10th percentile.
Transcription from BDNF promoter exons II and IV is affected in the cortex of HD subjects
Our earlier studies of mouse models of HD have shown that wild‐type but not mutant huntingtin stimulates BDNF gene transcription by acting at the level of BDNF promoter II, which contains the RE1/NRSE silencer element and whose activity is reduced by wild‐type huntingtin 56, 57, 58, 59, 61. In addition, mutant huntingtin has a toxic effect and reduces transcription from BDNF exon III and IV promoters 56, 57.
We therefore tested whether BDNF gene transcription from the homolog BDNF exon II, IV and V promoters in humans is decreased in the cortex of HD subjects, concentrating on the well‐characterized exon II promoter, which also contains the RE1/NRSE silencer (RE1 ID: hum26143, according to http://bioinformatics.leeds.ac.uk/RE1db_mkII/) (25), and the exon IV promoter, which contains regulatory elements that recognize calcium‐responsive transcription factor, cAMP/calcium‐responsive element binding proteins, and methyl‐CpG binding protein 2 12, 31, 49.
We selected samples from 10 subjects with HD (grades 3 and 4) and 10 gender‐ and age‐matched controls, and determined their BDNF II and IV mRNA levels by means of semiquantitative radioactive reverse transcription PCR. The presence of three splice donor sites in human BDNF exon II leads to the production of the three BDNF isoforms, BDNF IIA, IIB and IIC. These were all detectable in all of the subjects, but their levels were lower in the cerebral cortex of the HD patients, thus suggesting that transcription from BDNF promoter exon II is affected in HD ( Figure 3A). SNAP‐25, which is involved in neurotransmitter exocytosis in neurons, was amplified as a housekeeping gene. A previous study has found reduced SNAP‐25 mRNA levels in the prefrontal association cortex of HD subjects (23), but the levels were unaffected in our parietal cortex specimens. We then quantitatively evaluated BDNF II mRNA content in four independent PCR reactions, densitometrically analyzed the three amplified BDNF isoforms and the SNAP‐25 band, and determined the ratio between the sum of the absolute values of BDNF II and SNAP‐25 mRNA in each sample. BDNF II mRNA levels were significantly reduced in the HD patients (P < 0.01, Mann–Whitney two‐tailed test), with no significant difference between the grade 3 and grade 4 patients. Similar results were obtained when BDNF II mRNA was normalized over GAPDH content (data not shown). Figure 3B (left panel) shows the median BDNF II mRNA levels in the control and HD subjects.
Figure 3.
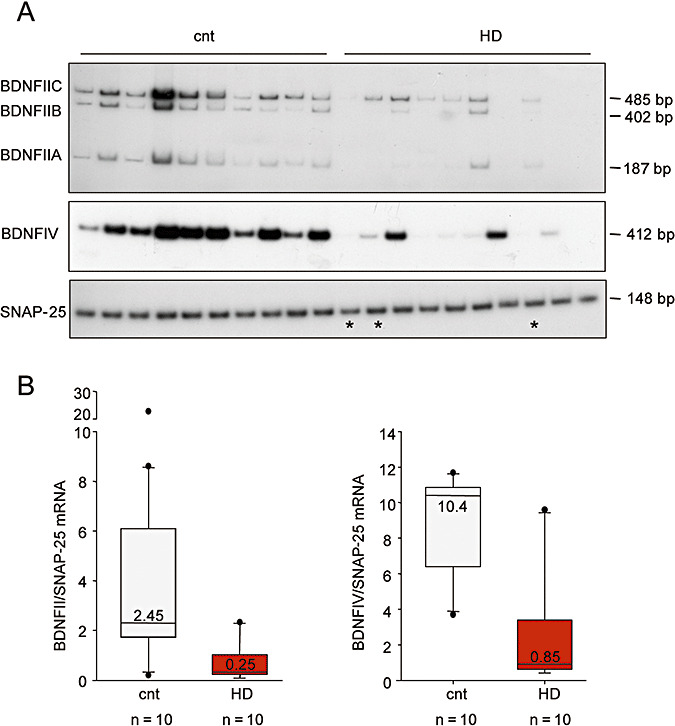
Expression of brain‐derived neurotrophic factor (BDNF) isoforms in the cortex of Huntington's disease (HD) subjects. A. BDNF II and IV mRNA levels were determined by means of semiquantitative radioactive reverse transcription polymerase chain reaction (RT‐PCR) in 10 controls and 10 subjects with grade 3 (n = 7) and 4 HD (n = 3) (* indicates grade 4 subjects). The presence of three splice donor sites in human BDNF exon II leads to the production of three BDNF isoforms: BDNF mRNA IIA, IIB and IIC. Synaptosome‐associated protein 25 kDa (SNAP‐25) was amplified as a housekeeping gene in addition to GAPDH (data not shown). The data come from one of four independent PCR experiments. Control subjects, from left to right: T‐274, 6182, 6142, 6002, 5959, 5936, 5919, 5077, 5074 and 5021; HD subjects, from left to right: T‐291, T‐310, 3176, 2866, 3723, 3484, 5570, 6062, 6010 and 6183. B. Quantification of BDNF II (left panel) and IV mRNA (right panel). Each of the amplified bands was densitometrically analyzed, and the BDNF level expressed as an absolute value normalized to the absolute value of SNAP‐25. The data were calculated from four independent PCR experiments. BDNF II and IV mRNA were significantly reduced in the HD patients (P < 0.01, Mann–Whitney two‐tailed test). BDNF mRNA levels are shown as the median value within each group. Similar results were obtained when the data were normalized to GAPDH content. Outliers are indicated as black circles. Left panel: control patients 6182 and 6002 fell above the 90th percentile, and control 5919 beneath the 10th percentile. Grade 3 HD patient 3484 fell above the 90th percentile. Right panel: control patient 6002 fell above the 90th percentile, and control T‐274 beneath the 10th percentile. Grade 3 HD patient 3176 fell above the 90th percentile.
Figure 3A shows that BDNF IV mRNA levels were also lower in the HD subjects than in the controls (P < 0.01, Mann–Whitney two‐tailed test). Similar results were obtained when BDNF IV mRNA was normalized over GAPDH content (data not shown). Figure 3B (right panel) shows the median BDNF IV mRNA levels in the controls and HD patients.
Trk B and p75NTR mRNA levels in the brain of HD subjects
Cortical BDNF is released at the axon terminals and captured by striatal neurons after binding to TrkB receptor tyrosine kinase 3, 8, 35. Although most of the functions attributed to BDNF are associated with full‐length TrkB, the brain also contains two truncated isoforms that lack a large part of the intracellular domain (Trk‐T1 and Trk‐T2) and do not show any protein tyrosine kinase activity 8, 32, 35. The suggested roles of truncated receptors include growth and development, and the negative modulation of TrkB expression and function (thus decreasing BDNF‐mediated signaling). They can also regulate BDNF storage and release by glial cells (8). A novel truncated Trk isoform that is preferentially expressed in neurons and localized on the plasma membrane has been identified more recently, which lacks the tyrosine kinase domain but has an Shc binding site and is therefore known as T‐Shc 20, 47. In addition, BDNF binds to the p75NTR, which is mainly expressed during early neuronal development, but may be re‐expressed in adult brain under various pathological conditions (13).
In order to investigate the role of TrkB in HD, we evaluated its levels, and those of the highly expressed truncated Trk‐T1 and T‐Shc isoforms in the caudate nucleus and cerebral cortex of eight subjects with HD (grades 2, 3 and 4) and eight age‐matched controls. As a previous study of four cortical and striatal specimens taken from HD subjects (18) found a reduction in TrkB protein levels, we used quantitative real‐time PCR to investigate whether this reduction may be caused by decreased TrkB mRNA levels.
Despite the expected variability in the control and HD populations, Figure 4A shows that the levels of full‐length TrkB mRNA normalized to GAPDH content were significantly lower in the caudate nucleus, but not in the cerebral cortex of the HD patients (P < 0.01, Mann–Whitney two‐tailed test).
Figure 4.
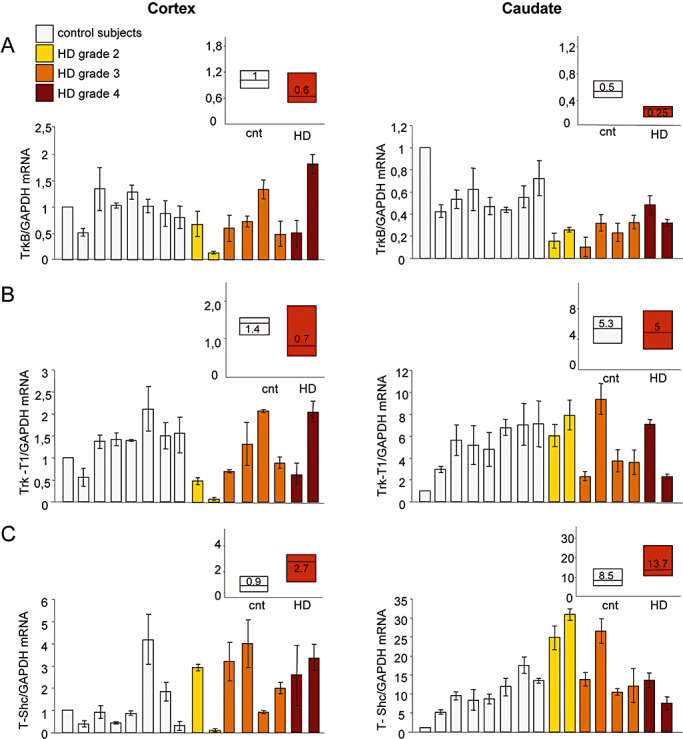
BDNF receptors mRNA levels in the cortex and caudate of Huntington's disease (HD) subjects. TrkB, Trk‐T1 and T‐Shc mRNA levels were determined by means of quantitative real‐time polymerase chain reaction (PCR) in the cortex and caudate of eight controls and eight HD subjects. Trk mRNA was normalized to the level of GAPDH mRNA. The values are the averages ± SD of six independent PCR experiments. Control subjects, from left to right: 6182, 6142, 6002, 5959, 5936, 5919, 5074 and 5021; HD subjects, from left to right: 6051, 6121, 5570, 5576, 6010, 6183, 5507 and 6062. A. TrkB mRNA levels were significantly reduced in the caudate of the HD subjects (P < 0.01, Mann–Whitney two‐tailed test), but not in the cortex. The decreased levels did not correlate with disease grade (Spearman's test). B. The levels of Trk‐T1 mRNA were not significantly lower in the cortex or caudate of the HD subjects. C. T‐Shc mRNA levels were significantly increased in the cortex and caudate of the HD subjects (P < 0.05, Mann–Whitney two‐tailed test). A, B and C respectively show median TrkB, Trk‐T1 and T‐Shc mRNA levels in the control and HD subjects. The boundary of the box closest to zero indicates the 25th percentile, the line within the box marks the median, and the boundary of the box farthest from zero indicates the 75th percentile.
The levels of truncated Trk‐T1 receptors in HD caudate nucleus and cortex were unaltered (Figure 4B), but there was a significant increase in T‐Shc mRNA in both brain areas (P < 0.05, Mann–Whitney two‐tailed test) (Figure 4C). In vitro cell line studies have shown that the transport of TrkB to the cell membrane is influenced by interactions between T‐Shc and TrkB (20), and so the increased levels of T‐Shc in the caudate may reflect an attempt to relocalize functional TrkB to the plasma membrane and/or indicate additional defects in TrkB signaling insofar as it is known that T‐Shc can negatively modulate TrkB function 20, 47.
Finally, we assessed p75NTR mRNA levels in the same tissues and, as shown in Figure 5A, found that they were significantly increased in the caudate of the HD subjects (P < 0.01, Mann–Whitney two‐tailed test). There were no differences in the cortical specimens of the HD and control subjects, thus suggesting that the increased level of p75NTR mRNA may be specific to the caudate. Figure 5B shows the median cortical and striatal p75NTR mRNA levels in the controls and HD patients.
Figure 5.
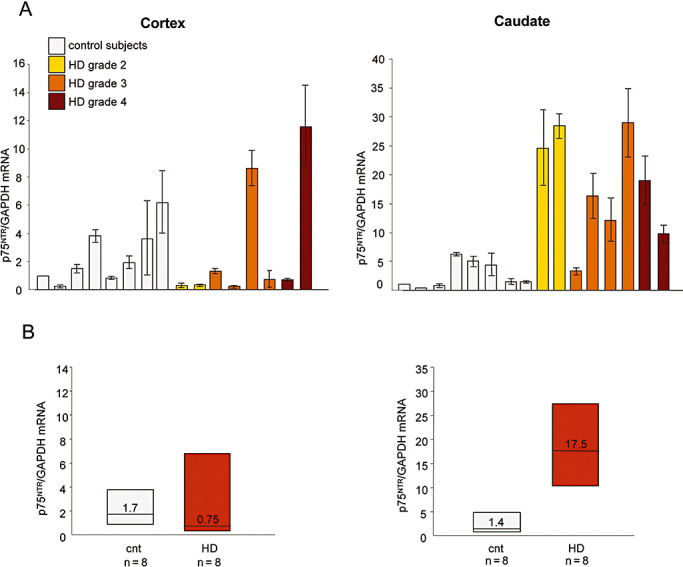
p75 pan‐neurotrophin receptor (p75NTR) mRNA levels in the cortex and caudate of Huntington's disease (HD) subjects. A. p75NTR mRNA levels were determined by quantitative real‐time polymerase chain reaction (PCR) in the cortex and caudate of the eight controls and eight HD subjects shown in Figure 4, and normalized to the level of GAPDH mRNA. The values are the averages ± SD of six independent PCR experiments. p75NTR mRNA levels were significantly higher in the caudate of the HD patients (P < 0.01, Mann–Whitney two‐tailed test), but not in the cerebral cortex. B. Median p75NTR mRNA level in control and HD subjects. The boundary of the box closest to zero indicates the 25th percentile, the line within the box marks the median, and the boundary of the box farthest from zero indicates the 75th percentile.
DISCUSSION
We report reduced BDNF protein levels in the largest collection of post‐mortem samples of human cerebral cortices so far analyzed, and a reduction in total BDNF mRNA levels due to decreased transcription by BDNF exon II and exon IV promoters.
These findings are consistent with the large amount of data obtained from various mouse models of the disease (56) showing a reproducible and progressive reduction in cortical BDNF mRNA and protein levels (59). In our human samples, we found that the level of cortical BDNF is reduced from the early symptomatic stage of HD, but did not seem to correlate with disease progression.
Analysis of the potential correlation between BDNF levels and CAG length was precluded by the over‐similarity of the CAG repeat sizes in this study. The only published data concerning such a correlation comes from an in vitro study showing that cells transfected with an huntingtin exon 1 cDNA construct bearing 47 or 72 CAG repeats led to a similar reduction in BDNF content (10).
We also found that cortical BDNF levels vary within the normal and HD population. We first associated this variability with between‐subject differences in the PMI, but these did not affect the statistical outcome of a significant reduction in BDNF levels in subjects with HD.
Previously published data indicate that differences in BDNF protein and mRNA levels may be caused by individual variations in life style. BDNF levels significantly increase in mice on regimens of dietary restriction (14) or physical exercise favored by environmental enrichment (46). Moreover, BDNF expression undergoes diurnal oscillations (9). Animal models have also shown that antidepressants affect BDNF gene transcription in brain (11) in an exon‐ and tissue‐specific manner. In particular, selective serotoninergic or noradrenegic reuptake inhibitors increase BDNF mRNA in different brain areas, whereas neuroleptics and antipsychotic drugs down‐regulate BDNF gene transcription 4, 5, 11. As HD patients are normally exposed to these drugs, some of the variability may have been caused by the drugs taken by the individual subjects but, as no data are available concerning the drugs administered to the cases examined in this study, we could not seek any correlation with pharmacological treatments.
On the basis of many previous animal studies and our new human data, we conclude that reduced BDNF promoter activity in human cortex is a feature of HD, and reduces the amount of BDNF produced in the cortex and transported from the cortex to the striatum. It is important to note that this occurs from the early symptomatic stages, thus suggesting that there may be a large window for therapeutical interventions aimed at restoring BDNF levels 56, 59, 60.
Our findings differ from those of an early study showing unaltered BDNF protein levels in the cortex and reduced BDNF levels in the striatum of four subjects with HD compared with six age‐matched control subjects (15) and, more recently, Gauthier et al observed reduced BDNF protein levels in the striatum, but not in the cerebral cortex of ten HD subjects and seven age‐matched controls. On the basis of these findings and in vitro experiments showing reduced BDNF vesicle transport in HD cells, it has been suggested that the lower BDNF levels in the striatum may also be caused by reduced transport from the cerebral cortex to striatal targets (16). However, it is likely that the differences in the results are caused by the diversity of the considered samples and the different methods of analyzing BDNF protein. Most strikingly, we observed considerable between‐sample variability that was not found by Gauthier et al, probably because they studied a more homogenous collection of tissue specimens. Ferrer and Gauthier determined BDNF protein levels by means of Western blotting, which exclusively detects mature BDNF, but is less sensitive and has less quantitative power than ELISA. As our study uses ELISA, which measures both the mature and pro‐BDNF, it is possible that the lower levels of cortical BDNF protein may be caused by reductions in both forms, although we think the latter represents a very minor fraction as its half‐life is very short because of its fast processing to mature BDNF. Finally, it is worth noting that our finding of reduced cortical BDNF mRNA and protein levels does not exclude the hypothesis of impaired vesicle trafficking along the cortical afferents, and both could contribute to reducing the delivery of BDNF to striatal neurons.
Given the extensive evidence linking BDNF to HD, the BDNF gene has been tested as a potential modifier of age at HD onset caused by the presence of the BDNF Val66Met polymorphism influencing BDNF vesicle transport (56) but, despite an initial report of a correlation (1), subsequent studies have failed to reveal any significant effect (56). As the Val66Met polymorphism affects BDNF transport from the Golgi region to appropriate secretory granules, and not its transcription or biological activity, it is possible that the lack of an association indicates that the defect in BDNF transport has no impact on age at disease onset, although it may still have an effect on disease progression. The possibility that a defect in BDNF transcriptional activity is present pre‐symptomatically and may therefore affect age at onset and/or disease progression remains open.
Taken together, our data suggest that strategies aimed at increasing striatal BDNF levels may be beneficial in HD 56, 60. However, whether or how a neuron responds will depend on the expression of its high affinity TrkB receptor. We found that the levels of TrkB mRNA (which activates the intracellular signaling pathways leading to cell survival and differentiation following BDNF binding) are significantly decreased in the caudate but not in the cortex of HD subjects. On the contrary, the mRNA levels of Trk‐T1 (a truncated TrkB isoform highly expressed in the brain) were not significantly affected in either the cortical or striatal specimens, which is in line with the findings of previous studies of other pathological conditions (48). We also found that T‐Shc mRNA levels were increased in both the cortex and the caudate of HD subjects. This last receptor, which is mainly expressed in neurons and preferentially located on the plasma membrane, can be involved in TrkB localization on the plasma membrane, but can also act as a negative regulator of TrkB signaling in the brain 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, and so increased T‐Shc mRNA indicates a receptor imbalance. More recent findings indicate that unprocessed neurotrophin can bind to p75NTR, a member of the tumor necrosis factor receptor superfamily. Pro‐BDNF binding to p75NTR is linked to several intracellular signal transduction pathways that mediates biological actions other than those of the Trk receptors, and p75NTR is involved in events promoting cell migration, differentiation and axon growth 13, 41. A number of reports indicate that its expression is dramatically increased under neurodegenerative conditions: for example, increased p75NTR levels have been found in the basal nucleus of patients with Alzheimer's disease or temporal lobe epilepsy 19, 37. In line with this, our data showing that p75NTR mRNA levels are increased in the caudate but not the cortex of HD subjects reflect what is known in other neurodegenerative diseases. It has also been reported that p75NTR binds pro‐BDNF (an immature form of the neurotrophin), recruits sortilin co‐receptors and leads to apoptosis (50). This suggests that higher p75NTR levels may increase apoptotic cell death in a diseased brain. As p75NTR is highly expressed in glial cells (37), we hypothesize that increased levels of striatal p75NTR may be associated with the reactive gliosis often found in neurodegenerative diseases.
In conclusion, our data indicate that there is a clear reduction in BDNF mRNA and protein levels in the cortex of subjects with HD, thus suggesting that the administration of BDNF may be beneficial. In line with this, it has been shown that depleted BDNF in HD transgenic mice worsens the phenotype, whereas BDNF administration improves the neuropathological and behavioral phenotype (10). Although our finding that TrkB, T‐Shc and p75NTR levels were severely affected in the same specimens is a warning that BDNF administration alone may not be sufficient, it is known to benefit mice with decreased striatum levels of TrkB receptor (10). This suggests that BDNF may have a neuroprotective function even in the presence of lower levels of TrkB, or that the activation of a limited amount of TrkB may be enough to improve the behavioral phenotype in mice.
We believe that our results strongly support beginning a systematic effort to evaluate the impact of increasing BDNF levels in HD mice (56). The discovery that RE1/NRSE is a target of normal huntingtin function on the BDNF promoter, and the elucidation of the mechanism through which this protein stimulates BDNF gene transcription 60, 61, have led to the development of a cell‐based drug screening system and the identification of small molecules capable of increasing endogenous BDNF production in HD cells and the transcription from other RE1/NRSE neuronal genes (43). It is hoped that small molecules capable of passing the blood‐brain barrier and activating BDNF and RE1/NRSE‐controlled gene transcription will become available in the future and prove useful in HD.
Supporting information
Figure S1. Correlation between post‐mortem interval (PMI) and (A) brain‐derived neurotrophic factor (BDNF) protein and (B) mRNA level in control and Huntington’s disease patients. No correlation between BDNF level and PMI was observed, according to Spearman test.
Figure S2. A selection of RNA used in this study.
Supporting info item
Supporting info item
ACKNOWLEDGMENTS
We are very grateful to Professor Alberto Clivio, Department of Preclinical Science, Faculty of Medicine, University of Milan, for his generous hospitality and collaboration, and to Cinzia Gellera and Cateria Mariotti, Besta Neurological Institute, Milan, for their help in determining CAG size.
This study was funded by grants from the Huntington's Disease Society of America Coalition for the Cure (USA), High Q Foundation (USA), Telethon (Italy), Fondazione Cariplo (Italy), Ministero dell’Istruzione dell’Università e della Ricerca (Italy), NeuroNE (European Union's 6th Framework Programme), to E. Cattaneo. E. Cattaneo and M.E. MacDonald are members of the Huntingtin Function Team, Coalition for the Cure (Huntington's Disease Society of America).
REFERENCES
- 1. Alberch J, Lopez M, Badenas C, Carrasco JL, Mila M, Munoz E, Canals JM (2005) Association between BDNF val66met polymorphism and age at onset in Huntington disease. Neurology 65:964–965. [DOI] [PubMed] [Google Scholar]
- 2. Alcantara S, Frisen J, Del Rio JA, Soriano E, Barbacid M, Silos‐Santiago I (1997) TrkB signaling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy‐induced cell death. J Neurosci 17:3623–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL et al (1997) Anterograde transport of brain‐derived neurotrophic factor and its role in the brain. Nature 389:856–860. [DOI] [PubMed] [Google Scholar]
- 4. Angelucci F, Aloe L, Iannitelli A, Gruber SH, Mathe AA (2005) Effect of chronic olanzapine treatment on nerve growth factor and brain‐derived neurotrophic factor in the rat brain. Eur Neuropsychopharmacol 15:311–317. [DOI] [PubMed] [Google Scholar]
- 5. Bai O, Chlan‐Fourney J, Bowen R, Keegan D, Li XM (2003) Expression of brain‐derived neurotrophic factor mRNA in rat hippocampus after treatment with antipsychotic drugs. J Neurosci Res 71:127–131. [DOI] [PubMed] [Google Scholar]
- 6. Baquet ZC, Gorski JA, Jones KR (2004) Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain‐derived neurotrophic factor. J Neurosci 24:4250–4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barde YA, Edgar D, Thoenen H (1982) Purification of a new neurotrophic factor from mammalian brain. EMBO J 1:549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Binder DK, Scharfman HE (2004) Brain‐derived neurotrophic factor. Growth Factors 22:123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bova R, Micheli MR, Qualadrucci P, Zucconi GG (1998) BDNF and trkB mRNAs oscillate in rat brain during the light‐dark cycle. Brain Res Mol Brain Res 57:321–324. [DOI] [PubMed] [Google Scholar]
- 10. Canals JM, Pineda JR, Torres‐Peraza JF, Bosch M, Martin‐Ibanez R, Munoz MT et al (2004) Brain‐derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington's disease. J Neurosci 24:7727–7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Castren E, Voikar V, Rantamaki T (2007) Role of neurotrophic factors in depression. Curr Opin Pharmacol 7:18–21. [DOI] [PubMed] [Google Scholar]
- 12. Chen WG, West AE, Tao X, Corfas G, Szentirmay MN, Sawadogo M et al (2003) Upstream stimulatory factors are mediators of Ca2+‐responsive transcription in neurons. J Neurosci 23:2572–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dechant G, Barde YA (2002) The neurotrophin receptor p75(NTR): novel functions and implications for diseases of the nervous system. Nat Neurosci 5:1131–1136. [DOI] [PubMed] [Google Scholar]
- 14. Duan W, Guo Z, Jiang H, Ware M, Li XJ, Mattson MP (2003) Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc Natl Acad Sci USA 100:2911–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferrer I, Goutan E, Marin C, Rey MJ, Ribalta T (2000) Brain‐derived neurotrophic factor in Huntington disease. Brain Res 866:257–261. [DOI] [PubMed] [Google Scholar]
- 16. Gauthier LR, Charrin BC, Borrell‐Pages M, Dompierre JP, Rangone H, Cordelieres FP et al (2004) Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118:127–138. [DOI] [PubMed] [Google Scholar]
- 17. Gellera C, Meoni C, Castellotti B, Zappacosta B, Girotti F, Taroni F, DiDonato S (1996) Errors in Huntington disease diagnostic test caused by trinucleotide deletion in the IT15 gene. Am J Hum Genet 59:475–477. [PMC free article] [PubMed] [Google Scholar]
- 18. Gines S, Bosch M, Marco S, Gavalda N, Diaz‐Hernandez M, Lucas JJ et al (2006) Reduced expression of the TrkB receptor in Huntington's disease mouse models and in human brain. Eur J Neurosci 23:649–658. [DOI] [PubMed] [Google Scholar]
- 19. Ginsberg SD, Che S, Wuu J, Counts SE, Mufson EJ (2006) Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer's disease. J Neurochem 97:475–487. [DOI] [PubMed] [Google Scholar]
- 20. Haapasalo A, Sipola I, Larsson K, Akerman KE, Stoilov P, Stamm S et al (2002) Regulation of TRKB surface expression by brain‐derived neurotrophic factor and truncated TRKB isoforms. J Biol Chem 277:43160–43167. [DOI] [PubMed] [Google Scholar]
- 21. Hermel E, Gafni J, Propp SS, Leavitt BR, Wellington CL, Young JE et al (2004) Specific caspase interactions and amplification are involved in selective neuronal vulnerability in Huntington's disease. Cell Death Differ 11:424–438. [DOI] [PubMed] [Google Scholar]
- 22. Hock C, Heese K, Hulette C, Rosenberg C, Otten U (2000) Region‐specific neurotrophin imbalances in Alzheimer disease: decreased levels of brain‐derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch Neurol 57:846–851. [DOI] [PubMed] [Google Scholar]
- 23. Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G et al (2006) Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet 15:965–977. [DOI] [PubMed] [Google Scholar]
- 24. Ivkovic S, Ehrlich ME (1999) Expression of the striatal DARPP‐32/ARPP‐21 phenotype in GABAergic neurons requires neurotrophins in vivo and in vitro. J Neurosci 19:5409–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Johnson R, Gamblin RJ, Ooi L, Bruce AW, Donaldson IJ, Westhead DR et al (2006) Identification of the REST regulon reveals extensive transposable element‐mediated binding site duplication. Nucleic Acids Res 34:3862–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Karege F, Vaudan G, Schwald M, Perroud N, La Harpe R (2005) Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Brain Res Mol Brain Res 136:29–37. [DOI] [PubMed] [Google Scholar]
- 27. Katoh‐Semba R, Takeuchi IK, Semba R, Kato K (1997) Distribution of brain‐derived neurotrophic factor in rats and its changes with development in the brain. J Neurochem 69:34–42. [DOI] [PubMed] [Google Scholar]
- 28. Kolbeck R, Bartke I, Eberle W, Barde YA (1999) Brain‐derived neurotrophic factor levels in the nervous system of wild‐type and neurotrophin gene mutant mice. J Neurochem 72:1930–1938. [DOI] [PubMed] [Google Scholar]
- 29. Liu QR, Walther D, Drgon T, Polesskaya O, Lesnick TG, Strain KJ et al (2005) Human brain derived neurotrophic factor (BDNF) genes, splicing patterns, and assessments of associations with substance abuse and Parkinson's disease. Am J Med Genet B Neuropsychiatr Genet 134:93–103. [DOI] [PubMed] [Google Scholar]
- 30. Martinez‐Serrano A, Bjorklund A (1996) Protection of the neostriatum against excitotoxic damage by neurotrophin‐producing, genetically modified neural stem cells. J Neurosci 16:4604–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y et al (2003) DNA methylation‐related chromatin remodeling in activity‐dependent BDNF gene regulation. Science 302:890–893. [DOI] [PubMed] [Google Scholar]
- 32. Middlemas DS, Lindberg RA, Hunter T (1991) trkB, a neural receptor protein‐tyrosine kinase: evidence for a full‐length and two truncated receptors. Mol Cell Biol 11:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mizuno K, Carnahan J, Nawa H (1994) Brain‐derived neurotrophic factor promotes differentiation of striatal GABAergic neurons. Dev Biol 165:243–256. [DOI] [PubMed] [Google Scholar]
- 34. Murer MG, Yan Q, Raisman‐Vozari R (2001) Brain‐derived neurotrophic factor in the control human brain, and in Alzheimer's disease and Parkinson's disease. Prog Neurobiol 63:71–124. [DOI] [PubMed] [Google Scholar]
- 35. Nagappan G, Lu B (2005) Activity‐dependent modulation of the BDNF receptor TrkB: mechanisms and implications. Trends Neurosci 28:464–471. [DOI] [PubMed] [Google Scholar]
- 36. Nakao N, Brundin P, Funa K, Lindvall O, Odin P (1995) Trophic and protective actions of brain‐derived neurotrophic factor on striatal DARPP‐32‐containing neurons in vitro. Brain Res Dev Brain Res 90:92–101. [DOI] [PubMed] [Google Scholar]
- 37. Ozbas‐Gerceker F, Gorter JA, Redeker S, Ramkema M, Van Der Valk P, Baayen JC et al (2004) Neurotrophin receptor immunoreactivity in the hippocampus of patients with mesial temporal lobe epilepsy. Neuropathol Appl Neurobiol 30:651–664. [DOI] [PubMed] [Google Scholar]
- 38. Perez‐Navarro E, Alberch J, Neveu I, Arenas E (1999) Brain‐derived neurotrophic factor, neurotrophin‐3 and neurotrophin‐4/5 differentially regulate the phenotype and prevent degenerative changes in striatal projection neurons after excitotoxicity in vivo. Neuroscience 91:1257–1264. [DOI] [PubMed] [Google Scholar]
- 39. Perez‐Navarro E, Canudas AM, Akerund P, Alberch J, Arenas E (2000) Brain‐derived neurotrophic factor, neurotrophin‐3, and neurotrophin‐4/5 prevent the death of striatal projection neurons in a rodent model of Huntington's disease. J Neurochem 75:2190–2199. [DOI] [PubMed] [Google Scholar]
- 40. Pruunsild P, Kazantseva A, Aid T, Palm K, Timmusk T (2007) Dissecting the human BDNF locus: bidirectional transcription, complex splicing, and multiple promoters. Genomics 90:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reichardt LF (2006) Neurotrophin‐regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci 361:1545–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reiner A, Albin RL, Anderson KD, D’Amato CJ, Penney JB, Young AB (1988) Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA 85:5733–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rigamonti D, Bolognini D, Mutti C, Zuccato C, Tartari M, Sola F et al (2007) Loss of huntingtin function complemented by small molecules acting as RE1/NRSE silencer modulators. J Biol Chem 282:24554–24562. [DOI] [PubMed] [Google Scholar]
- 44. Rosas HD, Koroshetz WJ, Chen YI, Skeuse C, Vangel M, Cudkowicz ME et al (2003) Evidence for more widespread cerebral pathology in early HD: an MRI‐based morphometric analysis. Neurology 60:1615–1620. [DOI] [PubMed] [Google Scholar]
- 45. Rosas HD, Hevelone ND, Zaleta AK, Greve DN, Salat DH, Fischl B (2005) Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology 65:745–747. [DOI] [PubMed] [Google Scholar]
- 46. Spires TL, Grote HE, Varshney NK, Cordery PM, Van Dellen A, Blakemore C, Hannan AJ (2004) Environmental enrichment rescues protein deficits in a mouse model of Huntington's disease, indicating a possible disease mechanism. J Neurosci 24:2270–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stoilov P, Castren E, Stamm S (2002) Analysis of the human TrkB gene genomic organization reveals novel TrkB isoforms, unusual gene length, and splicing mechanism. Biochem Biophys Res Commun 290:1054–1065. [DOI] [PubMed] [Google Scholar]
- 48. Tan J, Shepherd RK (2006) Aminoglycoside‐induced degeneration of adult spiral ganglion neurons involves differential modulation of tyrosine kinase B and p75 neurotrophin receptor signaling. Am J Pathol 169:528–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME (1998) Ca2+ influx regulates BDNF transcription by a CREB family transcription factor‐dependent mechanism. Neuron 20:709–726. [DOI] [PubMed] [Google Scholar]
- 50. Teng HK, Teng KK, Lee R, Wright S, Tevar S, Almeida RD et al (2005) ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci 25:5455–5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. The Huntington's Disease Collaborative Research Group (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72:971–983. [DOI] [PubMed] [Google Scholar]
- 52. Trottier Y, Devys D, Imbert G, Saudou F, An I, Lutz Y et al (1995) Cellular localization of the Huntington's disease protein and discrimination of the normal and mutated form. Nat Genet 10:104–110. [DOI] [PubMed] [Google Scholar]
- 53. Ventimiglia R, Mather PE, Jones BE, Lindsay RM (1995) The neurotrophins BDNF, NT‐3 and NT‐4/5 promote survival and morphological and biochemical differentiation of striatal neurons in vitro. Eur J Neurosci 7:213–222. [DOI] [PubMed] [Google Scholar]
- 54. Widmer HR, Hefti F (1994) Neurotrophin‐4/5 promotes survival and differentiation of rat striatal neurons developing in culture. Eur J Neurosci 6:1669–1679. [DOI] [PubMed] [Google Scholar]
- 55. Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P et al (2002) Increased sensitivity to N‐methyl‐D‐aspartate receptor‐mediated excitotoxicity in a mouse model of Huntington's disease. Neuron 33:849–860. [DOI] [PubMed] [Google Scholar]
- 56. Zuccato C, Cattaneo E (2007) Role of brain‐derived neurotrophic factor in Huntington's disease. Prog Neurobiol 81:294–330. [DOI] [PubMed] [Google Scholar]
- 57. Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L et al (2001) Loss of Huntingtin‐Mediated BDNF Gene Transcription in Huntington's disease. Science 293:493–498. [DOI] [PubMed] [Google Scholar]
- 58. Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L et al (2003) Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE‐controlled neuronal genes. Nat Genet 35:76–83. [DOI] [PubMed] [Google Scholar]
- 59. Zuccato C, Liber D, Ramos C, Tarditi A, Rigamonti D, Tartari M et al (2005) Progressive loss of BDNF in a mouse model of Huntington's disease and rescue by BDNF delivery. Pharmacol Res 52:133–139. [DOI] [PubMed] [Google Scholar]
- 60. Zuccato C, Tartari M, Goffredo D, Cattaneo E, Rigamonti D (2005) From target identification to drug screening assays for neurodegenerative diseases. Pharmacol Res 52:245–251. [DOI] [PubMed] [Google Scholar]
- 61. Zuccato C, Belyaev N, Conforti P, Ooi L, Tartari M, Papadimou E et al (2007) Widespread disruption of REST/NRSF occupancy of its target genes in Huntington's disease. J Neurosci 27:6972–6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Correlation between post‐mortem interval (PMI) and (A) brain‐derived neurotrophic factor (BDNF) protein and (B) mRNA level in control and Huntington’s disease patients. No correlation between BDNF level and PMI was observed, according to Spearman test.
Figure S2. A selection of RNA used in this study.
Supporting info item
Supporting info item