

Abstract
Improving clinical tests are allowing us to more precisely classify autism spectrum disorders and diagnose them at earlier ages. This raises the possibility of earlier and potentially more effective therapeutic interventions. To fully capitalize on this opportunity, however, will require better understanding of the neurobiological changes underlying this devastating group of developmental disorders. It is becoming clear that the normal trajectory of neurodevelopment is altered in autism, with aberrations in brain growth, neuronal patterning and cortical connectivity. Changes to the structure and function of synapses and dendrites have also been strongly implicated in the pathology of autism by morphological, genetic and animal modeling studies. Finally, environmental factors are likely to interact with the underlying genetic profile, and foster the clinical heterogeneity seen in autism spectrum disorders. In this review we attempt to link the molecular pathways altered in autism to the neurodevelopmental and clinical changes that characterize the disease. We focus on signaling molecules such as neurotrophin, Reelin, PTEN and hepatocyte growth factor, neurotransmitters such as serotonin and glutamate, and synaptic proteins such as neurexin, SHANK and neuroligin. We also discuss evidence implicating oxidative stress, neuroglial activation and neuroimmunity in autism.
INTRODUCTION
Autism spectrum disorders (ASD) are the most devastating conditions in the broad range of developmental abnormalities known as “pervasive developmental disorders”(175). ASD comprise a complex and heterogeneous group of conditions that include autism, Rett and Asperger syndromes, and pervasive developmental disorder‐otherwise nonspecified (2). The main clinical features of ASD are stereotypic behaviors and marked impairment in communication, social skills and cognition 129, 174. Clinical signs of ASD are frequently present at 3 years of age and recent prospective studies in toddlers indicate that abnormalities in social, communication and play behavior that may represent early indicators of autism can be detected as early as 14 months of age (124). Abnormalities in language development, mental retardation and epilepsy are frequent problems in the clinical profiles of patients with autism, and some patients may exhibit features of clinical regression, in which neurodevelopmental milestones are lost and/or other clinical signs worsen (174). ASD are clinically heterogeneous and can be associated in up to 10% of patients with well‐described neurological and genetic disorders, such as tuberous sclerosis, fragile X, Rett's and Down syndromes, although in most patients the causes are still unknown 159, 176 (see review by London). The heterogeneity and clinical variability of autism has prompted some researchers to use the term autisms instead of autism (81).
The stereotypic behaviors and marked delay or disruption of communication and social behavior trajectories that characterize ASD indicate that crucial neuroanatomic structures and neurodevelopmental pathways may be affected during intra‐uterine and/or early postnatal brain development. Several lines of research indicate that ASD are associated with disarrangement of neuronal organization, cortical connectivity and neurotransmitter pathways. While the causes of these abnormalities are still being identified, it is generally believed that genetic as well as environmental factors are involved in the pathogenesis of ASD 98, 147, 164. This review focuses on the current knowledge of molecular and cellular factors that may contribute to pathogenic mechanisms in ASD, and examines how they might affect the development and functioning of the central nervous system (CNS).
THE NEUROANATOMICAL AND NEURODEVELOPMENTAL BASIS OF ASD
Different approaches, including clinical assessment, neuroimaging and neuropathological studies have been used to assess the structural and morphological brain abnormalities in ASD. One consistent finding in ASD is altered brain growth, which has been extensively documented by Courchesne et al (54). The clinical onset of autism appears to be preceded by two phases of brain growth abnormalities: a reduced head size at birth, then a sudden and excessive increase between 1–2 months and 6–14 months of age 54, 57. Furthermore, these reports and other recent neuroimaging studies have shown that an abnormal pattern of brain overgrowth also occurs in areas of the frontal lobe, cerebellum and limbic structures between 2 and 4 years of age, a pattern that is followed by abnormal slowness in brain growth 54, 55, 57, 192. These brain regions are intimately involved in the development of social, communication and motor abilities that are impaired in ASD. For example, social orienting deficits in ASD were linked to abnormalities in frontal brain mechanisms involved in associating rewards with goal‐directed activity 62, 201. A recent clinical study found that a head circumference >75th percentile is associated with more impaired adaptive behaviors and with less impairment in IQ measures and motor and verbal language development (182). Neuroimaging studies have also demonstrated an overall enlargement of brain volume associated with increased subcortical white matter in the frontal lobe, and abnormal patterns of growth in the cerebral cortex, amygdala and hippoccampal formations (see review by Herbert (95)). A detailed parcellation study of the cerebral white matter showed increased volume of the subcortical or outer radiate white matter in all lobes, but most remarkable in the frontal lobe, supporting the view that an overgrowth of intrahemispheric and cortico‐cortical connections rather than interhemispheric connections occur in patient with autism and language‐associated developmental disorders 96, 97. Other studies of cortical and cerebral white matter volumes are indicative of inter‐regional disconnectivity 95, 96, 97, potentially resulting in poor integration within and across neurobehavioral developmental domains 56, 117.
Other novel neuroimaging approaches such as diffusion tensor imaging (DTI) and functional magnetic resonance imaging (fMRI) have also demonstrated disruption of white matter tracts and disconnection between brain regions in patients with autism. DTI of the brain reveals reduced fractional anisotropy values in white matter adjacent to the ventromedial prefrontal cortices, anterior cingulated gyrus and superior temporal regions, suggesting disruption of white matter tracts in brain regions involved in social functioning (9). Interestingly, fMRI of the brain has also shown abnormal patterns of activation and synchronization across different cortical and subcortical regions. This includes reduction in the functional connectivity and decreased correlation of the time series involved in higher order tasks that include language, working memory, problem solving and social cognition (reviewed by Minshew (147)).
Post‐mortem neuropathological studies also show disturbances in neuronal and cortical organization (reviewed in this issue by Casanova). Indeed, cytoarchitectural organizational abnormalities of the cerebral cortex, cerebellum, and other subcortical structures appear to be the most prominent neuropathological changes in autism 7, 112. An unusual laminar cytoarchitecture with packed small neurons has been described in the classical neuropathological studies by Kemper and Bauman, but no abnormalities in the external configuration of the cerebral cortex were noted (112). Cerebellar and brainstem pathology was also prominent, with loss and atrophy of Purkinje cells, predominantly in the posterolateral neocerebellar cortex. Kemper and Bauman 11, 112 have delineated at least three different types of pathological abnormalities in autism: (i) a curtailment of the normal development of neurons in the forebrain limbic system, (ii) an apparent decrease in the cerebellar Purkinje cell population, and (iii) age‐related changes in neuronal size and number in the nucleus of the diagonal band of Broca, the cerebellar nuclei and the inferior olive. Most recently, studies of the amygdala showed an abnormal pattern of growth with an overall decrease number of neurons 190, 191. These observations suggest that delays and disarrangements in neuronal maturation are important in the pathogenesis of autism (55), although the possibility that Purkinje cells or other neurons were initially present and subsequently degenerated must also be considered. In addition to these cytoarchitectural abnormalities, the structure and number of cortical mini‐columns, narrow chains of neurons that extend vertically across layers 2–6 (151) to form anatomical and functional units, appear to be abnormal in ASD. Mini‐columns in brain from patients with ASD are more numerous, smaller, and less compact in their cellular configuration in the frontal and temporal regions, as compared with controls ((34) and review by Casanova in this issue).
Taken together, clinical, neuroimaging and neuropathological studies support the hypothesis that autisms are disorders of neuronal‐cortical organization that cause alterations of information processing at different levels of the nervous system, from synaptic and dendritic organization to pathway connectivity and brain structure 81, 147. These neurobiological alterations likely affect the developmental trajectory of social behavior and communication during early stages of childhood (124) and appear to be influenced by both genetic and environmental factors (Figure 1). Some of the morphological abnormalities (eg, minicolumnar disorganization) suggest the events involved in the pathogenesis of ASD occur early during neurodevelopment, perhaps during first and second trimester of gestation. However, there is still uncertainty about the precise timing of the neuronal and cortical changes in ASD. For example, there is lack of clear gyral or cortical lamination abnormalities (103), a common feature of neurodevelopmental disorders originating at early stages such as those that occur during the first or second trimester.
Figure 1.
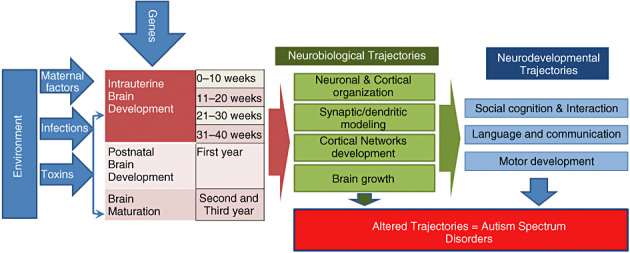
Genetic and environmental factors that influence intrauterine and early postnatal brain development likely alter neurobiological and neurodevelopmental trajectories that determine the clinical core of ASD.
GENETICS AND NEUROBIOLOGY OF ASD
The major role of genetics in autism is clear, as a concordance rate of 60% to 92% is seen in monozygotic twins. Recent studies have further documented the genetic complexity of ASD, and highlight the polygenic nature of the disorder 160, 187, 194, 205, 220. From these and other analyses, it is clear that molecular pathways with the potential to disrupt neurodevelopmental trajectories in utero or after birth are involved in the pathogenesis of ASD. Such pathways may be associated with many different developmental processes, from neuronal migration and cortical organization to synaptic and dendritic conformation. Environmental factors (159), including both maternal/intrauterine and postnatal events, may well modify the underlying genetic substrate and lead to greater abnormalities in neuronal organization and cortical network development. In the sections below, we further discuss the range of neurobiological changes in ASD, and associate them when possible with potential genetic etiologies. We have attempted to use a neuroanatomical framework in organizing this part of the review (Figure 2), while recognizing that many of the molecular pathways implicated in autism have effects on multiple CNS processes.
Figure 2.
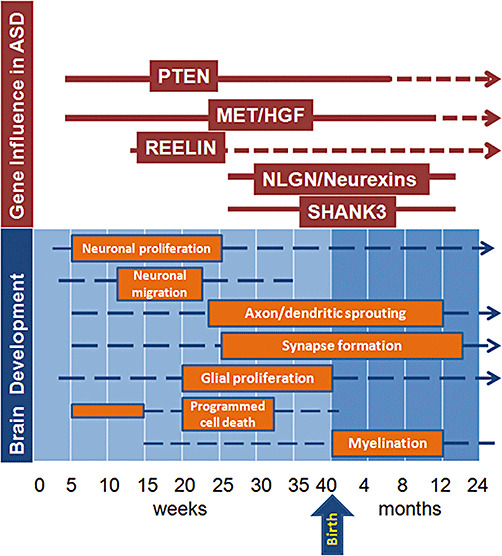
Multiple genes associated with autism spectrum disorders (ASD) appear to influence neurodevelopment at different stages of prenatal and postnatal life. These genes have specific periods of influence (red solid line) during defined stages of brain development (orange boxes), but their influence may extend to later stages of development including adult life (red broken lines). (Brain development graphic concept based on review by de Graaf‐Peters and Hadders‐Algra. (63))
Neuronal and cortical organization. Molecular pathways critical for normal neuronal and cortical organization that have been implicated in patients with ASD include those directed by growth factors such as hepatocyte growth factor (HGF) and its receptor MET, neurotrophic factors such as brain‐derived neurotrophic factor (BDNF), serotonin and other neurotransmitters, and signaling proteins such as Reelin.
MET and the HGF pathway. Both genetic and protein expression studies have associated the receptor MET and its ligand HGF with ASD. A recent case–control study demonstrated a strong association of a single nucleotide polymorphism (G‐to‐C) in a common 5′ promoter of the MET gene with ASD. The relative risk of ASD diagnosis was 2.27 in subjects with the C/C as compared with the G/G genotype (32). This study is especially relevant because the MET gene is located at 7q31 in one of the regions most commonly associated by genetic linkage studies with ASD 104, 220. MET is a transmembrane receptor that possesses tyrosine kinase activity 14, 24 and is activated by binding to HGF, also termed scatter factor or hepatopoietin A. HGF and MET, are present in both developing and adult mammalian brains, suggesting important functions across a broad range of neurodevelopment (115). HGF acts as a neurotrophic factor for motor, sensory and parasympathetic neurons (203), and influences neuronal migration 169, 170 and dendritic development (91). The HGF/MET pathway also plays a role in regulating dendritic morphology in the developing cerebral cortex and promoting neurite outgrowth (170). Decreased levels of MET itself and altered levels of mRNA of proteins associated with the HGF/MET pathway have been documented in brain tissues from patients with ASD (33). In addition to these genetic observations and brain tissue findings, we have documented increased levels of HGF in cerebrospinal fluid (CSF) of patients with autism (211), suggesting a potential compensatory feedback mechanism.
Interestingly, the multifunctional roles of the HGF/MET pathway also involve the immune system, as studies have demonstrated expression of MET in dendritic cells (161) and during activation of monocytes (12). HGF‐stimulated monocytes increased the expression of chemoattractant factors including MCP‐1, MIP‐2β, MIP‐1α and IL‐8 (13). HGF also exhibited immunosuppressive effects without up‐regulation of IL‐10 or TGF‐β(161), findings that suggest HGF/MET signaling is involved in regulation of the inflammatory responses. Because some of the non‐neurological manifestations of ASD include immune and gastrointestinal problems, the dysregulation of HGF/MET may provide a link between dysfunction of the CNS and other organs.
Reelin. RELN, which encodes the protein Reelin is another gene playing a critical role in cortical patterning that may be involved in autism. Reelin is a secreted extracellular matrix protein that controls neuronal migration, cortical layering and other aspects of brain development via interactions with lipoprotein receptors (reviewed by Forster (77)). It was initially implicated in ASD based on associations between a polymorphic GCG repeat immediately 5′ of the RELN gene and autism in both case‐control and family‐based studies in an Italian population (166). The fact that RELN is located on the distal long arm of chromosome 7 at a locus (7q22) associated with autism susceptibility added further support to the concept that Reelin function might be important, as did the reduced levels of Reelin found in post‐mortem studies of autistic brains (73). Attempts to confirm these intriguing preliminary findings have yielded varied results. Some reports have supported an association between genetic changes in the RELN locus and autism 196, 199, 224, while others have not 22, 66, 118.Transgenic mouse studies are also suggestive, but not definitive, with some social changes and defects in cortical layering observed in mice mutant in RELN alleles (186).
Neurotrophins. Neurotrophic growth factors, or neurotrophins, are good candidates for involvement in ASD because of their fundamental roles in guiding CNS development and cortical organization, and their abnormal expression patterns in autistic individuals. The core functions of neurotrophins during neurodevelopment include regulation of cell proliferation, migration and survival, and extend to include the modulation of axonal and dendritic outgrowth, synapse formation and other neuroplastic processes (5). The neurotrophin family consists of at least four proteins, including nerve growth factor, BDNF, neurotrophin‐3 and neurotrophin‐4 (92). Their potential role in pathogenic ASD pathways has been examined in several studies involving a heterogeneous groups of neurodevelopmental disorders 146, 155, 179.
Neurotrophins and their receptors are expressed in the neocortex and hippocampus (102) and these patterns of neurotrophin expression are activity‐dependent and regulated by sensory inputs, electrical activity and stimulation (102), (138). BDNF and its receptor, trkB, are densely expressed on cortical and hippocampal neurons, and influence both axonal and dendritic growth in a highly neuron‐specific and age‐dependent manner (139). In rodents, the expression of the trkB receptor peaks in the first 2 weeks postnatally, but BDNF action on cortical plasticity continues into adulthood 119, 139. With maturation, trkB becomes enriched at the site of glutamatergic synapses and therefore uniquely able to modulate experience‐dependent plasticity (85).
Interestingly, abnormalities in neurotrophins, especially BDNF, have been implicated in the etiology of several brain disorders that show altered cortical maturation and plasticity, such as schizophrenia and depression 158, 197. Genetic studies and expression of BDNF in serum of patients with ASD have pointed out potential links to the pathogenesis of autism. Nelson et al found elevated levels of BDNF and NT4/5 by assessment of archived neonatal blood samples of ASD patients (155). Elevation of BDNF was also reported in a study of 18 Japanese children with ASD as compared with controls (148), and the authors suggested hyperactivity of this growth factor may be involved in neurobiological abnormalities in autism. Similar findings were reported in a study of American children with ASD, where elevation of BDNF was demonstrated along with the presence of auto‐antibodies against BDNF 47, 153, 206.
It is still unknown how these observations fit into the neurodevelopmental pathogenesis of ASD, and it is unclear whether the increase in BDNF is a primary pathogenic mechanism or a secondary reaction to cortical abnormalities in ASD. However, one report suggesting that genetic changes in autistic individuals account for altered neurotrophin levels supports the notion that BDNF dysregulation could be a primary factor in the development of autism. CADPS2 is a gene found in the AUTS1 susceptibility locus for autism on 7q31 (42). Sadakata et al have recently shown that CADPS2 is aberrantly spliced in some autistic patients, and that Cadps2 knockout mice have autistic‐like phenotypes. CADPS2 regulates the exocytosis of dense‐core vesicles, including BDNF‐containing vesicles. In addition, the cellular distribution of BDNF in the brain largely overlaps with that of CADPS2 183, 184.
Neurotransmitters. Several lines of research suggest that abnormalities in serotoninergic, GABAergic and glutamatergic pathways occur in autism (reviewed by Zimmerman (225)). Neurotransmitter function in the CNS is linked not only to synaptic neuronal interactions, but also to other roles including brain maturation and cortical organization. Neurotransmitters and their receptors may act as paracrine signaling molecules in the immature brain and help control mechanisms that govern neuronal migration and positioning (134). It is well known that activation of specific GABA and glutamate receptors (GluRs) occurs during cell migration, and is involved in regulating radial and tangential migration (134). Because of these diverse functions, neurotransmitters and their receptors are clearly capable of playing central roles in the wide variety of neurobiological alterations associated with ASD.
The role of serotonin in autism has been explored using biomarker, neuroimaging and genetic approaches (193). The most relevant brain imaging studies used positron emission tomography to show that young children with autism lacked the developmental peak in brain 5‐HT synthesis capacity seen in typically developing infants (36), (41). Reduced synthesis of 5‐HT was observed in dentatothalamocortical pathways, with simultaneous increases in the contralateral dentate cerebellar nucleus (41). More recently, SPECT studies demonstrated significant reductions in 5‐HT2A binding in the cerebral cortex (152). Elevated levels of serotonin in the platelets of patients with autism has also been observed by a number of groups 29, 48, 123. In contrast, studies that assess changes in 5‐HT receptors in platelets or whole blood of individuals with autism show decreased 5‐HT2 receptor binding 51, 140.
Genetic studies have also identified abnormalities in serotonin‐related genes. Tryptophan hydroxylase‐2 (TPH2) is the rate‐limiting enzyme in 5‐HT synthesis in the CNS, and one group found a particular variant of TPH2 to be associated with autism (53). A second study, however, was not able to confirm this (181). Polymorphisms in the promoter region of the serotonin transporter gene SLC6A4 have also been reported to be associated with autism and cortical gray matter volume 39, 52, 67, 204, 213, 215. Finally, the gene ITGB3 has been proposed as a regulator of serotonin levels in autism based on genetic association studies 214, 215. Synergistic interaction between the SLC6A4 and ITGB3 loci has also been suggested (58).
Another line of research supporting serotonin as a neurobiological factor in ASD comes from pharmacological interventions. Drugs acting on the 5‐HT2 receptor 28, 143 alter the serotonin system and have caused behavioral improvements in autistic patients 94, 101, 114, 150, 168. Specifically, the selective serotonin reuptake inhibitor fluoxetine causes improvements in social behavior while decreasing aggressive and stereotyped behaviors in children with autism 6, 27, 50, 64, 72, 82. Interestingly, approaches that decrease CNS serotonin such as tryptophan depletion exacerbated symptoms in patients with ASD 49, 142.
A wide range of studies suggest that changes in serotonin and other neurotransmitters can result in aberrant cortical development. 5‐HT afferents from the brainstem raphe nuclei innervate cerebral cortex during a critical time in cortical morphogenesis. Similar to the peak in serotonin synthesis at 2 years of age in humans, rodents show a transient peak in serotonin levels in the first few days after birth 46, 100. At this time, layer IV of the sensory areas of cortex exhibits dense patches of staining for serotonin and 5‐HTTs, particularly in the “barrel field” in primary somatosensory cortex 18, 60, 78, 178. In vivo, it appears that too little or too much serotonin is detrimental to cortical development. Experimental approaches in rodents with neonatal systemic 5‐HT depletion reveal delayed development of several cortical layers (162), the aberrant appearance of thalamocortical afferent patterning in the barrel field (18) and an ultimate decrease in the size of the barrel field 156, 165. Altered dendritic and synaptic development appears to be at the root of serotonin's effects 137, 219, as barrel formation is restored in MAOA and 5‐HTT single and double knockouts by the blockade of serotonin synthesis, or the additional knockout of 5‐HT1B receptors, which normally inhibit glutamate release (185).
The interaction of serotonin pathways with neurotrophins such as BDNF suggests a potential interplay between these factors in ASD pathogenesis. BDNF and serotonin show co‐regulation in response to environmental factors 25, 136. During brain development, factors such as perinatal stress or environmental enrichment lead to long‐term alterations in BDNF expression in brain and blood plasma 25, 79. In rodent models, maternal infection can cause long‐term increases in BDNF within the cerebral cortex and other brain areas that eventually affect the development of serotoninergic pathways (83). Another example of this interaction comes from mice heterozygous for BDNF (BDNF+/–) that display premature, age‐associated loss in forebrain serotonergic innervation (130). Similarly, 5‐HTT function is impaired in the brains of BDNF+/– mice (61). Localized increases in BDNF expression promote 5‐HT fiber sprouting after injury 88, 133. In turn, 5‐HT depletion via inhibition of synthesis is accompanied by decreases in BDNF levels in the mature hippocampus (223). Such decreases in BDNF expression may be mediated by serotonergic mechanisms in that 5‐HT2A receptor antagonists have been shown to block stress induced decreases in BDNF expression in the hippocampus and cortex (210).
Excitatory neurotransmitter signaling via glutamate receptors (GluRs) also likely plays a role in cortical development (134), and has the potential for involvement in the pathogenesis of ASD. Candidate genes‐screening and association analyses showed that the kainate receptor GluR6 105, 198, 202, metabotropic GluR8 (GRM8) (195) and one of four N‐methyl‐D‐aspartate (NMDA) receptor 2 subunits, GRIN2A (8), appear to be associated with ASD. Interestingly, cDNA micro‐array techniques along with other mRNA and protein studies of brain tissues from patients with autism identified significant increases in expression of several genes associated with glutamatergic pathways, including excitatory amino acid transporter 1 and glutamate receptor AMPA 1 (173). Such disturbances of the glutamatergic system may well affect cortical development and plasticity, as experimental evidence suggests that GluRs play roles in the activity‐dependent refinement of synaptic connectivity (65). GluRs are classified broadly into two groups, ionotropic sites, linked to ion channels and metabotropic sites, linked to second messengers (144). The ionotropic sites include those activated by the exogenous agonists, NMDA, amino‐3‐hydroxy‐5‐methyl‐4‐isoxazole propionic acid (AMPA) and kainate (KA). NMDA receptors influence both the retraction of incorrectly placed axon arbors and synapses and the elaboration of correctly positioned terminals. NMDA receptors also have well‐documented roles in cortical development and activity‐dependent plasticity 89, 134.
GABAergic pathways also play important roles during brain development, and the interplay of glutamatergic and GABAergic systems facilitates modeling of the cerebral cortex by positioning of principal, pyramidal and interneurons (134). The establishment of the GABAergic system and the migration of GABAergic interneurons are crucial for the development of an inhibitory cortical system that regulates the excitatory processes mediated by glutamatergic pathways (127). A balance between excitation and inhibition is crucial for normal development, and its disruption may produce profound consequences for CNS function and homeostasis (126). GABAergic interneurons are also important for processing of information across cortical domains and are part of the structure of mini‐columns, an essential module involved in the physiopathology of cortical dysfunction in autism (35). The potential involvement of the GABAergic system in the pathogenesis of ASD has been suggested by clinical, neuropathological and genetic studies. Elevated levels of GABA in platelets (180) and reduction in the GABAergic receptor system has been documented by studies of brain tissues from patients with autism 16, 17. The location of three genes for subunits of the GABAA receptor, GABRB3, GABRA5 and GABRG3 on the proximal 15q arm (189) prompted genetic studies in ASD that yielded inconsistent results (reviewed by Schmitz (188)). One study that evaluated fourteen GABA receptor subunit genes found an association between GABRA4 and a potential increase in the risk of autism through interaction with GABRB1 (131).
Synaptic and dendritic changes. An early review focused on the neurobiology of autism and Rett syndrome helped introduce the concept that experience‐dependent synaptic plasticity might be disrupted in such developmental disorders (227). Dendritic abnormalities can also be observed in ASD. Indeed, decreased dendritic branching in CA1 and CA4 was reported in one of the earliest analyses of pathological changes in autism (177). Several leads from genetic studies have also implicated synaptic changes in autism. These include alterations in the genes encoding Neuroligins 3 and 4, their binding partners Neurexins 1 and 3, SHANK and contactin‐associated‐protein‐like 2 (CNTNAP2). The neuroligins, a family of five postsynaptic cell adhesion molecules, were the first of these to be associated with autism. In 2003, Jamain et al reported that Neuroligins 3 and 4 were mutated in ASD patients (106). They initially examined the locus because it is located at Xp22.3, a chromosomal region deleted in several autistic females. When they screened 36 pairs of affected siblings and 122 trios with autism, they found one Swedish family harboring a frameshift mutation leading to a premature stop codon in NLGN4, and another Swedish family with a mutation affecting a highly conserved residue in NLGN3. The NLGN4 mutation is predicted to represent a genetic null allele, while the changes in NLGN3 result in a protein that does not efficiently traffic to the cell surface and appears to have altered binding abilities 40, 44.
Subsequent attempts to confirm the role of neuroligin mutations in patients with ASD have yielded mixed results. Laumonnier et al reported a frameshift mutation in the NLGN4 gene in a large French family with mental retardation, some of whom also had autism (125). A mixed cohort of 148 autistic patients from the USA and Portugal contained about 3% with missense mutations in conserved regions of NLGN4, but no changes in NLGN3 (218). A functional analysis found that the R704C mutation described by Yan et al weakened the binding of neuroligin to syntrophin, suggesting they could be biologically significant (217). A Finnish study of 100 families with autism yielded a modest association of the disease symptoms with the NLGN1, three and four loci, but no functional mutations were identified in the 30 cases sequenced (221). It has also been suggested that the splicing pattern of the neuroligins is altered in autistic individuals (207). In contrast, however, studies of 96 autistic patients in Quebec (80), 196 in Toronto (212) and 124 from an international molecular genetic study of autism (15) did not identify any genetic alterations interpreted as being causally linked to autism. Furthermore, in at least one family deletion of NLGN4 was not associated with autistic symptoms (132).
Given these somewhat conflicting findings, the recent discovery that neurexin, a major protein partner of the neuroligin family, is altered in some autistic individuals provides key support for the concept that this synaptogenic pathway is involved in ASD development. Feng et al screened three beta‐neurexin genes in 203 patients with autism, as well as in 535 controls (74). They found two putative missense mutations predicted to cause structural changes in four autistic cases, but in none of the controls. Neurexin are presynaptic proteins, and represent the binding partners for postsynaptic neuroligins. This interaction is thought to trigger postsynaptic differentiation and control the balance of inhibitory GABAergic and stimulatory glutamatergic inputs 87, 171.
SHANK3, another synaptic protein which can bind neuroligins, was also recently implicated in autism. It was initially investigated because of its location on chromosome 22 in a region lost or rearranged in patients with ASD. This microdeletion syndrome involving 22q13.3 is characterized by multiple developmental delays, dysmorphic features and autistic behavior (135). SHANK3, also known as ProSAP2, is one of three genes located in the minimal involved region. It encodes a type of protein found in excitatory synapses that serves as a scaffold and can bind to neuroligins (145). Shank proteins have been proposed as master organizers of postsynaptic density because of their ability to nucleate multimeric protein complexes in dendritic spines. Durand et al recently sequenced all SHANK3 exons in 227 individuals with ASD and in 190 controls (69). They identified alterations in a small percentage of patients, and showed that mutations in a single copy could be associated with language and/or social communication disorders.
Abnormalities in brain growth. Head circumference was found to be abnormally large in a subset of autistic patients by Kanner in 1943, and approximately 20% of children with autism have macrocephaly 76, 122. As described above, a wide range of imaging studies have more precisely delineated abnormalities in the growth of the brain as a whole, and of specific regions and structures. Potential molecular causes of these size changes are beginning to be discovered. For example, a polymorphism in the HOXA1 homeobox gene has been associated with increased head circumference in patients with autism (45). The cellular basis of brain overgrowth is not yet clear, but several theories have been advanced. One hypothesis is a reduction in the pruning and consolidation of synapses during development, leading to an increased number of neurites. Increased numbers of neurons or glia in the brain, either through initial overproduction or reduction of cell death, are additional possibilities. These and other theories are discussed in more detail in a recent review of brain growth in autism (141). Finally, it is possible that hypertrophy of individual cells may cause the brain size increase. An intriguing candidate potentially involved in the regulation of brain size in autism via this final mechanism is the gene PTEN (phosphatase and tensin homolog on chromosome 10).
PTEN was initially evaluated in ASD patients because it is mutated in Cowden syndrome, a rare autosomal dominant condition characterized by numerous hamartomas and an increased risk of cancer (167). Inherited PTEN mutations are also found in patients with Bannayan–Riley–Ruvalcavba (BRRS) and Proteus syndromes. Macrocephaly is a feature of Cowden syndrome patients, and some of these individuals were reported to be autistic 84, 167. Macrocephaly and autistic behavior has also been reported in a patient with BRRS (228). Given these commonalities between inherited PTEN syndromes and autism, Butler et al sequenced the PTEN gene in 18 autistic patients with macrocephaly, and found three with heterozygous germline mutations (30). A more recent screen of 88 patients with ASD and macrocephaly identified one with a misssense mutation in PTEN, but no partial or whole gene deletions (31). Several additional cases of autistic individuals with PTEN mutations have also been reported recently, leading to the recommendation that such testing be routinely performed 19, 99. It is not yet clear if PTEN mutations in autistic individuals are always associated with increased head size, or if normocephalic autistic patients might also have disruptions in PTEN function. It will also be interesting to determine if other members of the signaling cascades regulated by PTEN are altered in autism.
PTEN is a phosphatase that regulates signaling through the phosphoinositol 3‐kinase (PI3K) pathway. It has multiple downstream effects, and regulates cellular proliferation, differentiation and migration. In neoplasms, PTEN acts as a tumor suppressor, with loss of function mutations and deletions causing increased proliferation and decreased cell death. In postmitotic neurons, however, loss of PTEN function leads to the hypertrophic growth without proliferation, resulting in formation of aberrant ganglion cells and a phenotype highly similar to that seen in Lhermitte–Duclos disease, which is associated with Cowden syndrome (120).
PTEN has subsequently been deleted from postmitotic neurons of the cerebral cortex and dentate gyrus in transgenic mice, leading to some very interesting behavioral and neuropathological changes (121). These animals showed progressive macrocephaly, but also were impoverished in their social interactions. For example, while wild‐type animals will preferentially interact with a mouse they have not previously encountered, PTEN deficient animals did not. Indeed, the transgenic animals were as likely to interact with an inanimate object as a social target animal. These behavioral changes may be caused by multifaceted neuropathological changes, as in addition to increased neuronal size the authors found alterations in axons, dendrites and synapses in the transgenic animals. Specifically, in mutant animals they documented enlargement of mossy fiber tracts, ectopic granule axons, dendritic hypertrophy and a dramatic increase in the number of presynaptic vesicle. These changes are consistent with a previous report implicating the AKT/mTOR pathway, which functions downstream of PTEN, in dendritic arborization (109).
Tuberous sclerosis (TS) is another genetically defined neurodevelopmental disorder caused by alterations in genetic signaling pathways that converge with those controlled by PTEN. TS patients are frequently also diagnosed with autism, with estimated rates of ASD ranging from 17% to 68% (200). Some investigators have found that the numbers or location of cortical tubers in TS is correlated with autistic behaviors, suggesting these discrete structural lesions might cause the association 20, 71. Others, however, did not find that the number or site of cortical tubers correlated with autistic behaviors 3, 21. In order to examine this pathway, we performed a preliminary immunohistochemical investigation of S6 ribosomal protein phosphorylation in post‐mortem brains from five autistic children and an equal number of matched controls, but did not identify any major changes (70).
NEUROIMMUNITY, ENVIRONMENT AND NON‐GENETIC FACTORS IN ASD
It is clear that genetics alone do not determine the entire ASD phenotype, and that other non‐genetic factors must play roles as modifiers of processes determined by genetic susceptibility. Environment and epigenetic factors both have the ability to influence pathogenic mechanisms of cortical and neuronal function. Among environmental factors, maternal influences and exposure to neurotoxins and potential environmental pollutants have been the focus of attention in recent investigations. These may interact with the neuroimmune system and disrupt neurodevelopmental pathways resulting in alterations of neurobehavioral trajectories such as those that occur in ASD 124, 129. A recent study, for example, found that patients with autism and larger head sizes show a significant association with a history of allergic/immune disorders both in the patient and in first‐degree relatives (182).
Neuroglia and neuroimmunity in ASD. Neuroglial cells such as astrocytes and microglia, along with perivascular macrophages and endothelial cells, play important roles in neuronal function and homeostasis 1, 10, 68, 157, 216. Both microglia and astroglia are fundamentally involved in cortical organization, neuroaxonal guidance and synaptic plasticity 75, 209. Neuroglial cells contribute in a number of ways to the regulation of immune responses in the CNS. Astrocytes, for example, play an important role in the detoxification of excess excitatory amino acids (154), maintenance of the integrity of the blood–brain barrier (172), production of neurotrophic factors (10) and the metabolism of glutamate (154). In normal homeostatic conditions, astrocytes facilitate neuronal survival by producing growth factors and mediating uptake/removal of excitotoxic neurotransmitters, such as glutamate, from the synaptic microenvironment (154). However, during astroglial activation secondary to injury or in response to neuronal dysfunction, astrocytes can produce several factors that may modulate inflammatory responses. For example, they secrete pro‐inflammatory cytokines, chemokines and metalloproteinases that can magnify immune reactions within the CNS (10). Microglial and astroglial activation is an important factor in the neuroglial responses to injury or dysfunction. Microglia are involved in synaptic stripping, cortical plasticity and immune surveillance 1, 86. Changes in astroglia and microglia can therefore produce marked neuronal dysfunction that is likely to be associated with mechanisms of neuronal dysfunction observed in autism. These neuroglial changes are mediated by the production of oxidative species, cytokines, chemokines and other neuroactive substances (10).
There has been growing interest in the role of immunity and immunological dysfunction in the pathogenesis of ASD (reviewed by Pardo (163) and Ashwood (4)). Several reports link the presence of immunological dysfunction with autism, and some studies suggest that up to 60% of patients with ASD have various types of systemic immune dysfunction, either as part of cellular or humoral immune responses 116, 128, 208. A few earlier case reports found pathological evidence of immunological reactions within the CNS, such as lymphocyte infiltration and microglial nodules 7, 90. Several reports using different methodologies and small patient populations have shown increases in pro‐inflammatory cytokines in peripheral blood samples in ASD (see review by Ashwood (4)). Most recently, Molloy et al found an increased pattern of production of Th2‐associated cytokines in leukocytes from autistic subjects (149).
Neuropathological studies of post‐mortem brain tissues from autistic patients demonstrate an active and ongoing neuroinflammatory process in the cerebral cortex and white matter characterized by astroglial and neuroglial activation. These findings support a role for neuroimmune responses in the pathogenesis of ASD (211). As both astroglia and microglia are involved in pathogenic inflammatory mechanisms common to many different disorders of the CNS, it is possible that different factors (eg, genetic susceptibility, maternal factors, prenatal environmental exposures) may trigger the development of these neuroglial reactions. Furthermore, protein array techniques used to establish the profiles of immune mediators demonstrated that cytokines/chemokines such as MCP‐1, IL‐6 and TGFβ1, which are mainly derived from activated neuroglia, are the most prevalent cytokines in brain tissues (211). Similar findings were seen in CSF from autistic patients. Preliminary studies also show that serum concentrations of subsets of cytokines and chemokines, such as MCP‐1 and IL‐6, parallel the CSF levels, suggesting that serum levels may be useful as surrogate markers of neuroinflammatory activity in autistic subjects. These findings strongly suggest that neuroimmune reactions are part of the neuropathological processes in ASD, and that immune responses are among the factors that may contribute to CNS dysfunction. However, the significance of the neuroinflammatory response to the specific neuropathologies and behavioral disruptions in ASD, and its position in the etiology of ASD, requires further exploration.
Oxidative stress. Oxidative stress is another possible cause of Purkinje cell loss and other neuroanatomical changes described in autistic brains (reviewed in 37, 113). Oxidative stress occurs when the levels of reactive oxygen species exceed the antioxidant capacities of a cell, often leading to cell death. Because of its very high oxygen demands and limited antioxidant capacity, the brain is thought to be relatively vulnerable to oxidative stress (111). Several studies have shown decreased levels of antioxidants such as superoxide dismutase, transferrin and ceruloplasmin in the blood or serum of patients with ASD 38, 108, 222. Significant elevations in biomarker profiles indicating increased oxidative stress, such as increased lipid peroxidation, have also been documented in autism 38, 107, 229. Interestingly, in one report the alterations in antioxidant proteins were linked specifically to regressive autism, suggesting a postnatal environmental effect (38). Polymorphisms in metabolic pathway genes may contribute to the increased oxidative stress in autism (108). Advanced glycation end products have also been reported to be elevated in both the brain tissue and serum of autistic patients, a change which can also lead to increased oxidative damage 23, 110.
Maternal factors. A final interesting area of research has focused on the potential role of maternal factors in the pathogenesis of autism. A study by Comi and Zimmerman (43) showed that the mean number of autoimmune disorders was greater in families with autism, and that 46% of ASD patient's families had two or more members with autoimmune disorders. As the number of family members with autoimmune disorders increased from one to three, the risk of autism was greater, with an odds ratio that increased from 1.9 to 5.5. The most common autoimmune disorders observed were type 1 diabetes, adult rheumatoid arthritis, hypothyroidism and systemic lupus erythematosus. However, a large population‐based case–control study found no significant differences in the proportion of case and control mothers with a diagnosis of autoimmune disease in the 4‐year period surrounding pregnancy (59). Tissue‐based studies have also suggested a role for maternal autoimmune factors in the pathogenesis of autism. The presence of maternal auto‐antibodies that cross‐react with brain epitopes was demonstrated by two studies 26, 226. In one study by Zimmerman et al, serum from 11 mothers and their children with autism was compared with serum from controls in its ability to bind adult rat brain proteins using immunoblot techniques. In another study by Van de Water et al (26), of 61 mothers of patients with autism, seven of the plasma samples (11.5%) contained maternal antibody cross‐reactive with human fetal brain proteins 73 kDa and 37 kDa in size. This was not observed in the control group of mothers with typical developing or non‐autistic developmentally delayed children. The presence of such antibodies in the plasma of some mothers suggests the transfer of maternal antibodies during early development could interact with fetal CNS proteins, affecting neurodevelopmental pathways and increasing the risk of ASD.
SUMMARY
In this review, we have attempted to briefly summarize some of what is currently known about the neurobiological causes of autism. Autism and related developmental disorders are clinically heterogeneous, and are likely caused by a range of factors. This heterogeneity has made it difficult to tease out the individual causal elements of this devastating disease. Slowly, however, genetic and environmental alterations are being defined, including the molecular and genetic changes affecting brain growth and development described above. This improving understanding may ultimately lead to new strategies for the prevention or cure of ASD. An encouraging recent report provides an example of such therapeutic progress, as Hayashi et al have shown that the symptoms of fragile X syndrome in mice can be reversed by inhibition of a specific kinase (93). Similar studies targeting a broad range of molecular factors involved in autism will hopefully eventually allow us to treat the growing number of patients afflicted with ASD.
ACKNOWLEDGMENTS
Dr Pardo is supported by the Peter Emch Fund for Autism Research, The Bart A McLean Fund for Neuroimmunology Research, Cure Autism Now and NIH‐NIDA (K08‐DA16160). Dr Eberhart receives support from 1R01NS055089‐01A2.
REFERENCES
- 1. Aloisi F (2001) Immune function of microglia. Glia 36:165–179. [DOI] [PubMed] [Google Scholar]
- 2. American Psychiatric Association (1994) Diagnostic and Statistical Manual of Mental Disorders, IV. American Psychiatric Association, Washington, DC. [Google Scholar]
- 3. Asano E, Chugani DC, Muzik O, Behen M, Janisse J, Rothermel R, Mangner TJ, Chakraborty PK, Chugani HT (2001) Autism in tuberous sclerosis complex is related to both cortical and subcortical dysfunction. Neurology 57:1269–1277. [DOI] [PubMed] [Google Scholar]
- 4. Ashwood P, Wills S, Van de WJ (2006) The immune response in autism: a new frontier for autism research. J Leukoc Biol 80:1–15. [DOI] [PubMed] [Google Scholar]
- 5. Avital A, Goshen I, Kamsler A, Segal M, Iverfeldt K, Richter‐Levin G, Yirmiya R (2003) Impaired interleukin‐1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus 13:826–834. [DOI] [PubMed] [Google Scholar]
- 6. Baghdadli A, Gonnier V, Aussilloux C (2002) [Review of psychopharmacological treatments in adolescents and adults with autistic disorders. Encephale 28:248–254. [PubMed] [Google Scholar]
- 7. Bailey A, Luthert P, Dean A, Harding B, Janota I, Montgomery M, Yirmiya R (1998) A clinicopathological study of autism. Brain 121:889–905. [DOI] [PubMed] [Google Scholar]
- 8. Barnby G, Abbott A, Sykes N, Morris A, Weeks DE, Mott R, Lamb J, Bailey AJ, Monaco AP (2005) Candidate‐gene screening and association analysis at the autism‐susceptibility locus on chromosome 16p: evidence of association at GRIN2A and ABAT. Am J Hum Genet 76:950–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barnea‐Goraly N, Kwon H, Menon V, Eliez S, Lotspeich L, Reiss AL (2004) White matter structure in autism: preliminary evidence from diffusion tensor imaging. Biol Psychiatry 55:323–326. [DOI] [PubMed] [Google Scholar]
- 10. Bauer J, Rauschka H, Lassmann H (2001) Inflammation in the nervous system: the human perspective. Glia 36:235–243. [DOI] [PubMed] [Google Scholar]
- 11. Bauman ML, Kemper TL (2005) Structural Brain Anatomy in Autism: what is the evidence? In: The Neurobiology of Autism, 2nd edn. Bauman ML, Kemper TL (eds), pp. 136–149. The Johns Hopkins University Press: Baltimore. [Google Scholar]
- 12. Beilmann M, Odenthal M, Jung W, Vande Woude GF, Dienes HP, Schirmacher P (1997) Neoexpression of the c‐met/hepatocyte growth factor‐scatter factor receptor gene in activated monocytes. Blood 90:4450–4458. [PubMed] [Google Scholar]
- 13. Beilmann M, Vande Woude GF, Dienes HP, Schirmacher P (2000) Hepatocyte growth factor‐stimulated invasiveness of monocytes. Blood 95:3964–3969. [PubMed] [Google Scholar]
- 14. Birchmeier C, Gherardi E (1998) Developmental roles of HGF/SF and its receptor, the c‐Met tyrosine kinase. Trends Cell Biol 8:404–410. [DOI] [PubMed] [Google Scholar]
- 15. Blasi F, Bacchelli E, Pesaresi G, Carone S, Bailey AJ, Maestrini E (2006) Absence of coding mutations in the X‐linked genes neuroligin 3 and neuroligin 4 in individuals with autism from the IMGSAC collection. Am J Med Genet B Neuropsychiatr Genet 141:220–221. [DOI] [PubMed] [Google Scholar]
- 16. Blatt GJ (2005) GABAergic cerebellar system in autism: a neuropathological and developmental perspective. Int Rev Neurobiol 71:167–178. [DOI] [PubMed] [Google Scholar]
- 17. Blatt GJ, Fitzgerald CM, Guptill JT, Booker AB, Kemper TL, Bauman ML (2001) Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J Autism Dev Disord 31:537–543. [DOI] [PubMed] [Google Scholar]
- 18. Blue ME, Erzurumlu RS, Jhaveri S (1991) A comparison of pattern formation by thalamocortical and serotonergic afferents in the rat barrel field cortex. Cereb Cortex 1:380–389. [DOI] [PubMed] [Google Scholar]
- 19. Boccone L, Dessi V, Zappu A, Piga S, Piludu MB, Rais M, Massidda C, De VS, Cao A, Loudianos G (2006) Bannayan‐Riley‐Ruvalcaba syndrome with reactive nodular lymphoid hyperplasia and autism and a PTEN mutation. Am J Med Genet A 140:1965–1969. [DOI] [PubMed] [Google Scholar]
- 20. Bolton PF, Griffiths PD (1997) Association of tuberous sclerosis of temporal lobes with autism and atypical autism. Lancet 349:392–395. [DOI] [PubMed] [Google Scholar]
- 21. Bolton PF, Park RJ, Higgins JN, Griffiths PD, Pickles A (2002) Neuro‐epileptic determinants of autism spectrum disorders in tuberous sclerosis complex. Brain 125:1247–1255. [DOI] [PubMed] [Google Scholar]
- 22. Bonora E, Beyer KS, Lamb JA, Parr JR, Klauck SM, Benner A, Paolucci M, Abbott A, Ragoussis I, Poustka A, Bailey AJ, Monaco AP (2003) Analysis of reelin as a candidate gene for autism. Mol Psychiatry 8:885–892. [DOI] [PubMed] [Google Scholar]
- 23. Boso M, Emanuele E, Minoretti P, Arra M, Politi P, Ucelli di NS, Barale F (2006) Alterations of circulating endogenous secretory RAGE and S100A9 levels indicating dysfunction of the AGE‐RAGE axis in autism. Neurosci Lett 410:169–173. [DOI] [PubMed] [Google Scholar]
- 24. Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, Aaronson SA (1991) Identification of the hepatocyte growth factor receptor as the c‐met proto‐oncogene product. Science 251:802–804. [DOI] [PubMed] [Google Scholar]
- 25. Branchi I, Francia N, Alleva E (2004) Epigenetic control of neurobehavioural plasticity: the role of neurotrophins. Behav Pharmacol 15:353–362. [DOI] [PubMed] [Google Scholar]
- 26. Braunschweig D, Krakowiak P, Ashwood P, Hertz‐Picciotto I, Hansen L, Croen L, Pessah IN, Van de Water J. (2007) Maternal Plasma Antibodies to Human Fetal Brain in Autism. 6th International Meeting for Autism Research, Seattle, WA.
- 27. Buchsbaum MS, Hollander E, Haznedar MM, Tang C, Spiegel‐Cohen J, Wei TC, Solimando A, Buchsbaum BR, Robins D, Bienstock C, Cartwright C, Mosovich S (2001) Effect of fluoxetine on regional cerebral metabolism in autistic spectrum disorders: a pilot study. Int J Neuropsychopharmacol 4:119–125. [DOI] [PubMed] [Google Scholar]
- 28. Buitelaar JK, Willemsen‐Swinkels SH (2000) Medication treatment in subjects with autistic spectrum disorders. Eur Child Adolesc Psychiatry 9(Suppl.1):I85–I97. [DOI] [PubMed] [Google Scholar]
- 29. Burgess NK, Sweeten TL, McMahon WM, Fujinami RS (2006) Hyperserotoninemia and altered immunity in autism. J Autism Dev Disord 36:697–704. [DOI] [PubMed] [Google Scholar]
- 30. Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C (2005) Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 42:318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Buxbaum JD, Cai G, Chaste P, Nygren G, Goldsmith J, Reichert J, Anckarsater H, Rastam M, Smith CJ, Silverman JM, Hollander E, Leboyer M, Gillberg C, Verloes A, Betancur C (2007) Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet 144:484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Campbell DB, Sutcliffe JS, Ebert PJ, Militerni R, Bravaccio C, Trillo S, Elia M, Schneider C, Melmed R, Sacco R, Persico AM, Levitt P (2006) A genetic variant that disrupts MET transcription is associated with autism. Proc Natl Acad Sci USA 103:16834–16839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Campbell DB, D'Oronzio R, Garbett K, Ebert PJ, Mirnics K, Levitt P, Persico AM (2007) Disruption of cerebral cortex MET signaling in autism spectrum disorder. Ann Neurol (in press). [DOI] [PubMed] [Google Scholar]
- 34. Casanova MF, Buxhoeveden DP, Switala AE, Roy E (2002) Minicolumnar pathology in autism. Neurology 58:428–432. [DOI] [PubMed] [Google Scholar]
- 35. Casanova MF, Buxhoeveden D, Gomez J (2003) Disruption in the inhibitory architecture of the cell minicolumn: implications for autisim. Neuroscientist 9:496–507. [DOI] [PubMed] [Google Scholar]
- 36. Chandana SR, Behen ME, Juhasz C, Muzik O, Rothermel RD, Mangner TJ, Chakraborty PK, Chugani HT, Chugani DC (2005) Significance of abnormalities in developmental trajectory and asymmetry of cortical serotonin synthesis in autism. Int J Dev Neurosci 23:171–182. [DOI] [PubMed] [Google Scholar]
- 37. Chauhan A, Chauhan V (2006) Oxidative stress in autism. Pathophysiology 13:171–181. [DOI] [PubMed] [Google Scholar]
- 38. Chauhan A, Chauhan V, Brown WT, Cohen I (2004) Oxidative stress in autism: increased lipid peroxidation and reduced serum levels of ceruloplasmin and transferrin—the antioxidant proteins. Life Sci 75:2539–2549. [DOI] [PubMed] [Google Scholar]
- 39. Cho IH, Yoo HJ, Park M, Lee YS, Kim SA (2007): Family‐based association study of 5‐HTTLPR and the 5‐HT2A receptor gene polymorphisms with autism spectrum disorder in Korean trios. Brain Res 1139:34–41. [DOI] [PubMed] [Google Scholar]
- 40. Chubykin AA, Liu X, Comoletti D, Tsigelny I, Taylor P, Sudhof TC (2005) Dissection of synapse induction by neuroligins: effect of a neuroligin mutation associated with autism. J Biol Chem 280:22365–22374. [DOI] [PubMed] [Google Scholar]
- 41. Chugani DC, Muzik O, Behen M, Rothermel R, Janisse JJ, Lee J, Chugani HT (1999) Developmental changes in brain serotonin synthesis capacity in autistic and nonautistic children. Ann Neurol 45:287–295. [DOI] [PubMed] [Google Scholar]
- 42. Cisternas FA, Vincent JB, Scherer SW, Ray PN (2003) Cloning and characterization of human CADPS and CADPS2, new members of the Ca2 +‐dependent activator for secretion protein family. Genomics 81:279–291. [DOI] [PubMed] [Google Scholar]
- 43. Comi AM, Zimmerman AW, Frye VH, Law PA, Peeden JN (1999) Familial clustering of autoimmune disorders and evaluation of medical risk factors in autism. J Child Neurol 14:388–394. [DOI] [PubMed] [Google Scholar]
- 44. Comoletti D, De JA, Jennings LL, Flynn RE, Gaietta G, Tsigelny I, Ellisman MH, Taylor P (2004) The Arg451Cys‐neuroligin‐3 mutation associated with autism reveals a defect in protein processing. J Neurosci 24:4889–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Conciatori M, Stodgell CJ, Hyman SL, O'Bara M, Militerni R, Bravaccio C, Trillo S, Montecchi F, Schneider C, Melmed R, Elia M, Crawford L, Spence SJ, Muscarella L, Guarnieri V, D'Agruma L, Quattrone A, Zelante L, Rabinowitz D, Pascucci T, Puglisi‐Allegra S, Reichelt KL, Rodier PM, Persico AM (2004) Association between the HOXA1 A218G polymorphism and increased head circumference in patients with autism. Biol Psychiatry 55:413–419. [DOI] [PubMed] [Google Scholar]
- 46. Connell S, Karikari C, Hohmann CF (2004) Sex‐specific development of cortical monoamine levels in mouse. Brain Res 151:187–191. [DOI] [PubMed] [Google Scholar]
- 47. Connolly AM, Chez M, Streif EM, Keeling RM, Golumbek PT, Kwon JM, Riviello JJ, Robinson RG, Neuman RJ, Deuel RM (2006) Brain‐derived neurotrophic factor and autoantibodies to neural antigens in sera of children with autistic spectrum disorders, Landau‐Kleffner syndrome, and epilepsy. Biol Psychiatry 59:354–363. [DOI] [PubMed] [Google Scholar]
- 48. Cook EH (1990) Autism: review of neurochemical investigation. Synapse 6:292–308. [DOI] [PubMed] [Google Scholar]
- 49. Cook EH, Leventhal BL (1996) The serotonin system in autism. Curr Opin Pediatr 8:348–354. [DOI] [PubMed] [Google Scholar]
- 50. Cook EH Jr, Rowlett R, Jaselskis C, Leventhal BL (1992) Fluoxetine treatment of children and adults with autistic disorder and mental retardation. J Am Acad Child Adolesc Psychiatry 31:739–745. [DOI] [PubMed] [Google Scholar]
- 51. Cook EH Jr, Arora RC, Anderson GM, Berry‐Kravis EM, Yan SY, Yeoh HC, Sklena PJ, Charak DA, Leventhal BL (1993) Platelet serotonin studies in hyperserotonemic relatives of children with autistic disorder. Life Sci 52:2005–2015. [DOI] [PubMed] [Google Scholar]
- 52. Cook EH Jr, Courchesne R, Lord C, Cox NJ, Yan S, Lincoln A, Haas R, Courchesne E, Leventhal BL (1997) Evidence of linkage between the serotonin transporter and autistic disorder. Mol Psychiatry 2:247–250. [DOI] [PubMed] [Google Scholar]
- 53. Coon H, Dunn D, Lainhart J, Miller J, Hamil C, Battaglia A, Tancredi R, Leppert MF, Weiss R, McMahon W (2005) Possible association between autism and variants in the brain‐expressed tryptophan hydroxylase gene (TPH2). Am J Med Genet B Neuropsychiatr Genet 135:42–46. [DOI] [PubMed] [Google Scholar]
- 54. Courchesne E (2004) Brain development in autism: early overgrowth followed by premature arrest of growth. Ment Retard Dev Disabil Res Rev 10:106–111. [DOI] [PubMed] [Google Scholar]
- 55. Courchesne E, Pierce K (2005) Brain overgrowth in autism during a critical time in development: implications for frontal pyramidal neuron and interneuron development and connectivity. Int J Dev Neurosci 23:153–170. [DOI] [PubMed] [Google Scholar]
- 56. Courchesne E, Pierce K (2005) Why the frontal cortex in autism might be talking only to itself: local over‐connectivity but long‐distance disconnection. Curr Opin Neurobiol 15:225–230. [DOI] [PubMed] [Google Scholar]
- 57. Courchesne E, Redcay E, Kennedy DP (2004) The autistic brain: birth through adulthood. Curr Opin Neurol 17:489–496. [DOI] [PubMed] [Google Scholar]
- 58. Coutinho AM, Sousa I, Martins M, Correia C, Morgadinho T, Bento C, Marques C, Ataide A, Miguel TS, Moore JH, Oliveira G, Vicente AM (2007) Evidence for epistasis between SLC6A4 and ITGB3 in autism etiology and in the determination of platelet serotonin levels. Hum Genet 121:243–256. [DOI] [PubMed] [Google Scholar]
- 59. Croen LA, Grether JK, Yoshida CK, Odouli R, Van de WJ (2005) Maternal autoimmune diseases, asthma and allergies, and childhood autism spectrum disorders: a case‐control study. Arch Pediatr Adolesc Med 159:151–157. [DOI] [PubMed] [Google Scholar]
- 60. D'Amato RJ, Blue ME, Largent BL, Lynch DR, Ledbetter DJ, Molliver ME, Snyder SH (1987) Ontogeny of the serotonergic projection to rat neocortex: transient expression of a dense innervation to primary sensory areas. Proc Natl Acad Sci USA 84:4322–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Daws LC, Munn JL, Valdez MF, Frosto‐Burke T, Hensler JG (2007) Serotonin transporter function, but not expression, is dependent on brain‐derived neurotrophic factor (BDNF): in vivo studies in BDNF‐deficient mice. J Neurochem 101:641–651. [DOI] [PubMed] [Google Scholar]
- 62. Dawson G, Munson J, Estes A, Osterling J, McPartland J, Toth K, Carver L, Abbott R (2002) Neurocognitive function and joint attention ability in young children with autism spectrum disorder versus developmental delay. Child Dev 73:345–358. [DOI] [PubMed] [Google Scholar]
- 63. De Graaf‐Peters VB, Hadders‐Algra M (2006) Ontogeny of the human central nervous system: what is happening when? Early Hum Dev 82:257–266. [DOI] [PubMed] [Google Scholar]
- 64. DeLong GR, Teague LA, Swain Kamran M (1998) Effects of fluoxetine treatment in young children with idiopathic autism. Dev Med Child Neurol 40:551–562. [DOI] [PubMed] [Google Scholar]
- 65. Derkach VA, Oh MC, Guire ES, Soderling TR (2007) Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev 8:101–113. [DOI] [PubMed] [Google Scholar]
- 66. Devlin B, Bennett P, Dawson G, Figlewicz DA, Grigorenko EL, McMahon W, Minshew N, Pauls D, Smith M, Spence MA, Rodier PM, Stodgell C, Schellenberg GD (2004) Alleles of a reelin CGG repeat do not convey liability to autism in a sample from the CPEA network. Am J Med Genet B Neuropsychiatr Genet 126:46–50. [DOI] [PubMed] [Google Scholar]
- 67. Devlin B, Cook EH Jr, Coon H, Dawson G, Grigorenko EL, McMahon W et al (2005) Autism and the serotonin transporter: the long and short of it. Mol Psychiatry 10:1110–1116. [DOI] [PubMed] [Google Scholar]
- 68. Dong Y, Benveniste EN (2001) Immune function of astrocytes. Glia 36:180–190. [DOI] [PubMed] [Google Scholar]
- 69. Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsater H, Sponheim E, Goubran‐Botros H, Delorme R, Chabane N, Mouren‐Simeoni MC, De MP, Bieth E, Roge B, Heron D, Burglen L, Gillberg C, Leboyer M, Bourgeron T (2007) Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet 39:25–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Eberhart CG, Copeland J, Abel TW (2006) Brief report: s6 ribosomal protein phosphorylation in autistic frontal cortex and cerebellum: a tissue array analysis. J Autism Dev Disord 36:1131–1135. [DOI] [PubMed] [Google Scholar]
- 71. Eluvathingal TJ, Behen ME, Chugani HT, Janisse J, Bernardi B, Chakraborty P, Juhasz C, Muzik O, Chugani DC (2006) Cerebellar lesions in tuberous sclerosis complex: neurobehavioral and neuroimaging correlates. J Child Neurol 21:846–851. [DOI] [PubMed] [Google Scholar]
- 72. Fatemi SH, Realmuto GM, Khan L, Thuras P (1998) Fluoxetine in treatment of adolescent patients with autism: a longitudinal open trial. J Autism Dev Disord 28:303–307. [DOI] [PubMed] [Google Scholar]
- 73. Fatemi SH, Snow AV, Stary JM, Raghi‐Niknam M, Reutiman TJ, Lee S, Brooks AI, Pearce DA (2005) Reelin signaling is impaired in autism. Biol Psychiatry 57:777–787. [DOI] [PubMed] [Google Scholar]
- 74. Feng J, Schroer R, Yan J, Song W, Yang C, Bockholt A, Cook EH, Jr. , Skinner C, Schwartz CE, Sommer SS (2006) High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci Lett 409:10–13. [DOI] [PubMed] [Google Scholar]
- 75. Fields RD, Stevens‐Graham B (2002) New insights into neuron‐glia communication. Science 298:556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fombonne E, Roge B, Claverie J, Courty S, Fremolle J (1999) Microcephaly and macrocephaly in autism. J Autism Dev Disord 29:113–119. [DOI] [PubMed] [Google Scholar]
- 77. Forster E, Jossin Y, Zhao S, Chai X, Frotscher M, Goffinet AM (2006) Recent progress in understanding the role of Reelin in radial neuronal migration, with specific emphasis on the dentate gyrus. Eur J Neurosci 23:901–909. [DOI] [PubMed] [Google Scholar]
- 78. Fujimiya M, Hosoda S, Kitahama K, Kimura H, Maeda T (1986) Early development of serotonin neuron in the rat brain as studied by immunohistochemistry combined with tryptophan administration. Brain Dev 8:335–342. [DOI] [PubMed] [Google Scholar]
- 79. Garoflos E, Panagiotaropoulos T, Pondiki S, Stamatakis A, Philippidis E, Stylianopoulou F (2005) Cellular mechanisms underlying the effects of an early experience on cognitive abilities and affective states. Ann Gen Psychiatry 4:8 [electronic resource. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gauthier J, Bonnel A, St‐Onge J, Karemera L, Laurent S, Mottron L, Fombonne E, Joober R, Rouleau GA (2005) NLGN3/NLGN4 gene mutations are not responsible for autism in the Quebec population. Am J Med Genet B Neuropsychiatr Genet 132:74–75. [DOI] [PubMed] [Google Scholar]
- 81. Geschwind DH, Levitt P (2007) Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol 17:103–111. [DOI] [PubMed] [Google Scholar]
- 82. Ghaziuddin M, Tsai L, Ghaziuddin N (1991) Fluoxetine in autism with depression. J Am Acad Child Adolesc Psychiatry 30:508–509. [DOI] [PubMed] [Google Scholar]
- 83. Gilmore JH, Jarskog LF, Vadlamudi S (2003) Maternal infection regulates BDNF and NGF expression in fetal and neonatal brain and maternal‐fetal unit of the rat. J Neuroimmunol 138:49–55. [DOI] [PubMed] [Google Scholar]
- 84. Goffin A, Hoefsloot LH, Bosgoed E, Swillen A, Fryns JP (2001) PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet 105:521–524. [DOI] [PubMed] [Google Scholar]
- 85. Gomes RA, Hampton C, El‐Sabeawy F, Sabo SL, McAllister AK (2006) The dynamic distribution of TrkB receptors before, during, and after synapse formation between cortical neurons. J Neurosci 26:11487–11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Graeber MB, Bise K, Mehraein P (1993) Synaptic stripping in the human facial nucleus. Acta Neuropathol (Berl) 86:179–181. [DOI] [PubMed] [Google Scholar]
- 87. Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM (2004) Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119:1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Grider MH, Mamounas LA, Le W, Shine HD (2005) In situ expression of brain‐derived neurotrophic factor or neurotrophin‐3 promotes sprouting of cortical serotonergic axons following a neurotoxic lesion. J Neurosci Res 82:404–412. [DOI] [PubMed] [Google Scholar]
- 89. Groc L, Gustafsson B, Hanse E (2006) AMPA signalling in nascent glutamatergic synapses: there and not there! Trends Neurosci 29:132–139. [DOI] [PubMed] [Google Scholar]
- 90. Guerin P, Lyon G, Barthelemy C, Sostak E, Chevrollier V, Garreau B, Lelord G (1996) Neuropathological study of a case of autistic syndrome with severe mental retardation. Dev Med Child Neurol 38:203–211. [DOI] [PubMed] [Google Scholar]
- 91. Gutierrez H, Dolcet X, Tolcos M, Davies A (2004) HGF regulates the development of cortical pyramidal dendrites. Development 131:3717–3726. [DOI] [PubMed] [Google Scholar]
- 92. Hallbook F (1999) Evolution of the vertebrate neurotrophin and Trk receptor gene families. Curr Opin Neurobiol 9:616–621. [DOI] [PubMed] [Google Scholar]
- 93. Hayashi ML, Rao BS, Seo JS, Choi HS, Dolan BM, Choi SY, Chattarji S, Tonegawa S (2007) Inhibition of p21‐activated kinase rescues symptoms of fragile X syndrome in mice. Proc Natl Acad Sci USA 104:11489–11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hazell P (2007) Drug therapy for attention‐deficit/hyperactivity disorder‐like symptoms in autistic disorder. J Paediatr Child Health 43:19–24. [DOI] [PubMed] [Google Scholar]
- 95. Herbert MR (2005) Large brains in autism: the challenge of pervasive abnormality. Neuroscientist 11:417–440. [DOI] [PubMed] [Google Scholar]
- 96. Herbert MR, Ziegler DA, Deutsch CK, O'Brien LM, Lange N, Bakardjiev A, Hodgson J, Adrien KT, Steele S, Makris N, Kennedy D, Harris GJ, Caviness VS, Jr (2003) Dissociations of cerebral cortex, subcortical and cerebral white matter volumes in autistic boys. Brain 126:1182–1192. [DOI] [PubMed] [Google Scholar]
- 97. Herbert MR, Ziegler DA, Makris N, Filipek PA, Kemper TL, Normandin JJ, Sanders HA, Kennedy DN, Caviness VS, Jr (2004) Localization of white matter volume increase in autism and developmental language disorder. Ann Neurol 55:530–540. [DOI] [PubMed] [Google Scholar]
- 98. Herbert MR, Russo JP, Yang S, Roohi J, Blaxill M, Kahler SG et al (2006) Autism and environmental genomics. Neurotoxicology 27:671–684. [DOI] [PubMed] [Google Scholar]
- 99. Herman GE, Butter E, Enrile B, Pastore M, Prior TW, Sommer A (2007) Increasing knowledge of PTEN germline mutations: two additional patients with autism and macrocephaly. Am J Med Genet A 143:589–593. [DOI] [PubMed] [Google Scholar]
- 100. Hohmann CF, Hamon R, Batshaw ML, Coyle JT (1988) Transient postnatal elevation of serotonin levels in mouse neocortex. Brain Res 471:163–166. [DOI] [PubMed] [Google Scholar]
- 101. Hollander E, Phillips A, Chaplin W, Zagursky K, Novotny S, Wasserman S, Iyengar R (2005) A placebo controlled crossover trial of liquid fluoxetine on repetitive behaviors in childhood and adolescent autism. Neuropsychopharmacology 30:582–589. [DOI] [PubMed] [Google Scholar]
- 102. Huang EJ, Reichardt LF (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24:677–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Hutsler JJ, Love T, Zhang H (2007) Histological and magnetic resonance imaging assessment of cortical layering and thickness in autism spectrum disorders. Biol Psychiatry 61:449–457. [DOI] [PubMed] [Google Scholar]
- 104. International Molecular Genetic Study of Autism Consortium (2001) Further characterization of the autism susceptibility locus AUTS1 on chromosome 7q. Hum Mol Genet 10:973–982. [DOI] [PubMed] [Google Scholar]
- 105. Jamain S, Betancur C, Quach H, Philippe A, Fellous M, Giros B, Gillberg C, Leboyer M, Bourgeron T (2002) Linkage and association of the glutamate receptor 6 gene with autism. Mol Psychiatry 7:302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T (2003) Mutations of the X‐linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet 34:27–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, Gaylor DW, Neubrander JA (2004) Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr 80:1611–1617. [DOI] [PubMed] [Google Scholar]
- 108. James SJ, Melnyk S, Jernigan S, Cleves MA, Halsted CH, Wong DH, Cutler P, Bock K, Boris M, Bradstreet JJ, Baker SM, Gaylor DW (2006) Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am J Med Genet B Neuropsychiatr Genet 141:947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M (2005) Control of dendritic arborization by the phosphoinositide‐3′‐kinase‐Akt‐mammalian target of rapamycin pathway. J Neurosci 25:11300–11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Junaid MA, Pullarkat RK (2001) Proteomic approach for the elucidation of biological defects in autism. J Autism Dev Disord 31:557–560. [DOI] [PubMed] [Google Scholar]
- 111. Juurlink BH, Paterson PG (1998) Review of oxidative stress in brain and spinal cord injury: suggestions for pharmacological and nutritional management strategies. J Spinal Cord Med 21:309–334. [DOI] [PubMed] [Google Scholar]
- 112. Kemper TL, Bauman M (1998) Neuropathology of infantile autism. J Neuropathol Exp Neurol 57:645–652. [DOI] [PubMed] [Google Scholar]
- 113. Kern JK, Jones AM (2006) Evidence of toxicity, oxidative stress, and neuronal insult in autism. J Toxicol Environ Health B Crit Rev 9:485–499. [DOI] [PubMed] [Google Scholar]
- 114. Kolevzon A, Mathewson KA, Hollander E (2006) Selective serotonin reuptake inhibitors in autism: a review of efficacy and tolerability. J Clin Psychiatry 67:407–414. [DOI] [PubMed] [Google Scholar]
- 115. Korhonen L, Sjoholm U, Takei N, Kern MA, Schirmacher P, Castren E, Lindholm D (2000) Expression of c‐Met in developing rat hippocampus: evidence for HGF as a neurotrophic factor for calbindin D‐expressing neurons. Eur J Neurosci 12:3453–3461. [DOI] [PubMed] [Google Scholar]
- 116. Korvatska E, Van de WJ, Anders TF, Gershwin ME (2002) Genetic and immunologic considerations in autism. Neurobiol Dis 9:107–125. [DOI] [PubMed] [Google Scholar]
- 117. Koshino H, Carpenter PA, Minshew NJ, Cherkassky VL, Keller TA, Just MA (2005) Functional connectivity in an fMRI working memory task in high‐functioning autism. Neuroimage 24:810–821. [DOI] [PubMed] [Google Scholar]
- 118. Krebs MO, Betancur C, Leroy S, Bourdel MC, Gillberg C, Leboyer M (2002) Absence of association between a polymorphic GGC repeat in the 5′ untranslated region of the reelin gene and autism. Mol Psychiatry 7:801–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kuipers SD, Bramham CR (2006) Brain‐derived neurotrophic factor mechanisms and function in adult synaptic plasticity: new insights and implications for therapy. Curr Opin Drug Discov Devel 9:580–586. [PubMed] [Google Scholar]
- 120. Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ (2001) Pten regulates neuronal soma size: a mouse model of Lhermitte‐Duclos disease. Nat Genet 29:404–411. [DOI] [PubMed] [Google Scholar]
- 121. Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF (2006) Pten regulates neuronal arborization and social interaction in mice. Neuron 50:377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Lainhart JE, Piven J, Wzorek M, Landa R, Santangelo SL, Coon H, Folstein SE (1997) Macrocephaly in children and adults with autism. J Am Acad Child Adolesc Psychiatry 36:282–290. [DOI] [PubMed] [Google Scholar]
- 123. Lam KS, Aman MG, Arnold LE (2006) Neurochemical correlates of autistic disorder: a review of the literature. Res Dev Disabil 27:254–289. [DOI] [PubMed] [Google Scholar]
- 124. Landa RJ, Holman KC, Garrett‐Mayer E (2007) Social and communication development in toddlers with early and later diagnosis of autism spectrum disorders. Arch Gen Psychiatry 64:853–864. [DOI] [PubMed] [Google Scholar]
- 125. Laumonnier F, Bonnet‐Brilhault F, Gomot M, Blanc R, David A, Moizard MP, Raynaud M, Ronce N, Lemonnier E, Calvas P, Laudier B, Chelly J, Fryns JP, Ropers HH, Hamel BC, Andres C, Barthelemy C, Moraine C, Briault S (2004) X‐linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet 74:552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Levitt P (2005) Disruption of interneuron development. Epilepsia 46(Suppl.7):22–28. [DOI] [PubMed] [Google Scholar]
- 127. Levitt P, Eagleson KL, Powell EM (2004) Regulation of neocortical interneuron development and the implications for neurodevelopmental disorders. Trends Neurosci 27:400–406. [DOI] [PubMed] [Google Scholar]
- 128. Licinio J, Alvarado I, Wong ML (2002) Autoimmunity in autism. Mol Psychiatry 7:329. [DOI] [PubMed] [Google Scholar]
- 129. Lord C, Cook EH, Leventhal BL, Amaral DG (2000) Autism spectrum disorders. Neuron 28:355–363. [DOI] [PubMed] [Google Scholar]
- 130. Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, Tessarollo L (1999)Brain‐derived neurotrophic factor‐deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci USA 96:15239–15244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Ma DQ, Whitehead PL, Menold MM, Martin ER, Shley‐Koch AE, Mei H, Ritchie MD, DeLong GR, Abramson RK, Wright HH, Cuccaro ML, Hussman JP, Gilbert JR, Pericak‐Vance MA (2005) Identification of significant association and gene‐gene interaction of GABA receptor subunit genes in autism. Am J Hum Genet 77:377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Macarov M, Zeigler M, Newman JP, Strich D, Sury V, Tennenbaum A, Meiner V (2007) Deletions of VCX‐A and NLGN4: a variable phenotype including normal intellect. J Intellect Disabil Res 51:329–333. [DOI] [PubMed] [Google Scholar]
- 133. Mamounas LA, Altar CA, Blue ME, Kaplan DR, Tessarollo L, Lyons WE (2000) BDNF promotes the regenerative sprouting, but not survival, of injured serotonergic axons in the adult rat brain. J Neurosci 20:771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Manent JB, Represa A (2007) Neurotransmitters and brain maturation: early paracrine actions of GABA and glutamate modulate neuronal migration. Neuroscientist 13:268–279. [DOI] [PubMed] [Google Scholar]
- 135. Manning MA, Cassidy SB, Clericuzio C, Cherry AM, Schwartz S, Hudgins L, Enns GM, Hoyme HE (2004) Terminal 22q deletion syndrome: a newly recognized cause of speech and language disability in the autism spectrum. Pediatrics 114:451–457. [DOI] [PubMed] [Google Scholar]
- 136. Mattson MP, Maudsley S, Martin B (2004) BDNF and 5‐HT: a dynamic duo in age‐related neuronal plasticity and neurodegenerative disorders. Trends Neurosci 27:589–594. [DOI] [PubMed] [Google Scholar]
- 137. Mazer C, Muneyyirci J, Taheny K, Raio N, Borella A, Whitaker‐Azmitia P (1997) Serotonin depletion during synaptogenesis leads to decreased synaptic density and learning deficits in the adult rat: a possible model of neurodevelopmental disorders with cognitive deficits. Brain Res 760:68–73. [DOI] [PubMed] [Google Scholar]
- 138. McAllister AK (2001) Neurotrophins and neuronal differentiation in the central nervous system. Cell Mol Life Sci 58:1054–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. McAllister AK (2002) Neurotrophins and cortical development. Results Probl Cell Differ 39:89–112. [DOI] [PubMed] [Google Scholar]
- 140. McBride PA, Anderson GM, Hertzig ME, Snow ME, Thompson SM, Khait VD et al (1998) Effects of diagnosis, race, and puberty on platelet serotonin levels in autism and mental retardation. J Am Acad Child Adolesc Psychiatry 37:767–776. [DOI] [PubMed] [Google Scholar]
- 141. McCaffery P, Deutsch CK (2005) Macrocephaly and the control of brain growth in autistic disorders. Prog Neurobiol 77:38–56. [DOI] [PubMed] [Google Scholar]
- 142. McDougle CJ, Naylor ST, Cohen DJ, Aghajanian GK, Heninger GR, Price LH (1996) Effects of tryptophan depletion in drug‐free adults with autistic disorder. Arch Gen Psychiatry 53:993–1000. [DOI] [PubMed] [Google Scholar]
- 143. McDougle CJ, Scahill L, McCracken JT, Aman MG, Tierney E, Arnold LE, Freeman BJ, Martin A, McGough JJ, Cronin P, Posey DJ, Riddle MA, Ritz L, Swiezy NB, Vitiello B, Volkmar FR, Votolato NA, Walson P (2000) Research Units on Pediatric Psychopharmacology (RUPP) Autism Network. Background and rationale for an initial controlled study of risperidone. Child Adolesc Psychiatr Clin N Am 9:201–224. [PubMed] [Google Scholar]
- 144. Meldrum BS (2000) Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr 130:1007. [DOI] [PubMed] [Google Scholar]
- 145. Meyer G, Varoqueaux F, Neeb A, Oschlies M, Brose N (2004) The complexity of PDZ domain‐mediated interactions at glutamatergic synapses: a case study on neuroligin. Neuropharmacology 47:724–733. [DOI] [PubMed] [Google Scholar]
- 146. Minshew N (2001) What are neuropeptides and neurotrophins and why are they important in autism? J Autism Dev Disord 31:517. [DOI] [PubMed] [Google Scholar]
- 147. Minshew NJ, Williams DL (2007) The new neurobiology of autism: cortex, connectivity, and neuronal organization. Arch Neurol 64:945–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Miyazaki K, Narita N, Sakuta R, Miyahara T, Naruse H, Okado N, Narita M (2004) Serum neurotrophin concentrations in autism and mental retardation: a pilot study. Brain Dev 26:292–295. [DOI] [PubMed] [Google Scholar]
- 149. Molloy CA, Morrow AL, Meinzen‐Derr J, Schleifer K, Dienger K, Manning‐Court P, Altaye M, Wills‐Karp M (2006) Elevated cytokine levels in children with autism spectrum disorder. J Neuroimmunol 172:198–205. [DOI] [PubMed] [Google Scholar]
- 150. Moore ML, Eichner SF, Jones JR (2004) Treating functional impairment of autism with selective serotonin‐reuptake inhibitors. Ann Pharmacother 38:1515–1519. [DOI] [PubMed] [Google Scholar]
- 151. Mountcastle VB (1997) The columnar organization of the neocortex. Brain 120:701–722. [DOI] [PubMed] [Google Scholar]
- 152. Murphy DG, Daly E, Schmitz N, Toal F, Murphy K, Curran S et al (2006) Cortical serotonin 5‐HT2A receptor binding and social communication in adults with Asperger's syndrome: an in vivo SPECT study. Am J Psychiatry 163:934–936. [DOI] [PubMed] [Google Scholar]
- 153. Nassenstein C, Kutschker J, Tumes D, Braun A (2006) Neuro‐immune interaction in allergic asthma: role of neurotrophins. Biochem Soc Trans 34:591–593. [DOI] [PubMed] [Google Scholar]
- 154. Nedergaard M, Takano T, Hansen AJ (2002) Beyond the role of glutamate as a neurotransmitter. Nat Rev Neurosci 3:748–755. [DOI] [PubMed] [Google Scholar]
- 155. Nelson KB, Grether JK, Croen LA, Dambrosia JM, Dickens BF, Jelliffe LL, Hansen RL, Phillips TM (2001) Neuropeptides and neurotrophins in neonatal blood of children with autism or mental retardation. Ann Neurol 49:597–606. [PubMed] [Google Scholar]
- 156. Nett‐Clarke CA, Leslie MJ, Lane RD, Rhoades RW (1994) Effect of serotonin depletion on vibrissa‐related patterns of thalamic afferents in the rat's somatosensory cortex. J Neurosci 14:7594–7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Neumann H (2001) Control of glial immune function by neurons. Glia 36:191–199. [DOI] [PubMed] [Google Scholar]
- 158. Neumeister A, Yuan P, Young TA, Bonne O, Luckenbaugh DA, Charney DS, Manji H (2005) Effects of tryptophan depletion on serum levels of brain‐derived neurotrophic factor in unmedicated patients with remitted depression and healthy subjects. Am J Psychiatry 162:805–807. [DOI] [PubMed] [Google Scholar]
- 159. Newschaffer CJ, Croen LA, Daniels J, Giarelli E, Grether JK, Levy SE, Mandell DS, Miller LA, Pinto‐Martin J, Reaven J, Reynolds AM, Rice CE, Schendel D, Windham GC (2007) The epidemiology of autism spectrum disorders. Annu Rev Public Health 28:235–258. [DOI] [PubMed] [Google Scholar]
- 160. Nishimura Y, Martin CL, Vazquez‐Lopez A, Spence SJ, Varez‐Retuerto AI, Sigman M, Steindler C, Pellegrini S, Schanen NC, Warren ST, Geschwind DH (2007) Genome‐wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum Mol Genet 16:1682–1698. [DOI] [PubMed] [Google Scholar]
- 161. Okunishi K, Dohi M, Nakagome K, Tanaka R, Mizuno S, Matsumoto K, Miyazaki Ji, Nakamura T, Yamamoto K (2005) A novel role of hepatocyte growth factor as an immune regulator through suppressing dendritic cell function. J Immunol 175:4745–4753. [DOI] [PubMed] [Google Scholar]
- 162. Osterheld‐Haas MC, Hornung JP (1996) Laminar development of the mouse barrel cortex: effects of neurotoxins against monoamines. Exp Brain Res 110:183–195. [DOI] [PubMed] [Google Scholar]
- 163. Pardo CA, Vargas DL, Zimmerman AW (2005) Immunity, neuroglia and neuroinflammation in autism. Int Rev Psychiatry 17:485–495. [DOI] [PubMed] [Google Scholar]
- 164. Persico AM, Bourgeron T (2006) Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci 29:349–358. [DOI] [PubMed] [Google Scholar]
- 165. Persico AM, Altamura C, Calia E, Puglisi‐Allegra S, Ventura R, Lucchese F, Keller F (2000) Serotonin depletion and barrel cortex development: impact of growth impairment vs. serotonin effects on thalamocortical endings. Cereb Cortex 10:181–191. [DOI] [PubMed] [Google Scholar]
- 166. Persico AM, D'Agruma L, Maiorano N, Totaro A, Militerni R, Bravaccio C, Wassink TH, Schneider C, Melmed R, Trillo S, Montecchi F, Palermo M, Pascucci T, Puglisi‐Allegra S, Reichelt KL, Conciatori M, Marino R, Quattrocchi CC, Baldi A, Zelante L, Gasparini P, Keller F (2001) Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol Psychiatry 6:150–159. [DOI] [PubMed] [Google Scholar]
- 167. Pilarski R, Eng C (2004) Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet 41:323–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Posey DJ, Erickson CA, Stigler KA, McDougle CJ (2006) The use of selective serotonin reuptake inhibitors in autism and related disorders. J Child Adolesc Psychopharmacol 16:181–186. [DOI] [PubMed] [Google Scholar]
- 169. Powell EM, Mars WM, Levitt P (2001) Hepatocyte growth factor/scatter factor is a motogen for interneurons migrating from the ventral to dorsal telencephalon. Neuron 30:79–89. [DOI] [PubMed] [Google Scholar]
- 170. Powell EM, Muhlfriedel S, Bolz J, Levitt P (2003) Differential regulation of thalamic and cortical axonal growth by hepatocyte growth factor/scatter factor. Dev Neurosci 25:197–206. [DOI] [PubMed] [Google Scholar]
- 171. Prange O, Wong TP, Gerrow K, Wang YT, El‐Husseini A (2004) A balance between excitatory and inhibitory synapses is controlled by PSD‐95 and neuroligin. Proc Natl Acad Sci USA 101:13915–13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Prat A, Biernacki K, Wosik K, Antel JP (2001) Glial cell influence on the human blood‐brain barrier. Glia 36:145–155. [DOI] [PubMed] [Google Scholar]
- 173. Purcell AE, Jeon OH, Zimmerman AW, Blue ME, Pevsner J (2001) Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 57:1618–1628. [DOI] [PubMed] [Google Scholar]
- 174. Rapin I (1997) Autism. N Engl J Med 337:97–104. [DOI] [PubMed] [Google Scholar]
- 175. Rapin I (2002) The autistic‐spectrum disorders. N Engl J Med 347:302–303. [DOI] [PubMed] [Google Scholar]
- 176. Rapin I, Katzman R (1998) Neurobiology of autism. Ann Neurol 43:7–14. [DOI] [PubMed] [Google Scholar]
- 177. Raymond GV, Bauman ML, Kemper TL (1996) Hippocampus in autism: a Golgi analysis. Acta Neuropathol (Berl) 91:117–119. [DOI] [PubMed] [Google Scholar]
- 178. Rhoades RW, Nett‐Clarke CA, Chiaia NL, White FA, MacDonald GJ, Haring JH, Jacquin MF (1990) Development and lesion induced reorganization of the cortical representation of the rat's body surface as revealed by immunocytochemistry for serotonin. J Comp Neurol 293:190–207. [DOI] [PubMed] [Google Scholar]
- 179. Riikonen R (2003) Neurotrophic factors in the pathogenesis of Rett syndrome. J Child Neurol 18:693–697. [DOI] [PubMed] [Google Scholar]
- 180. Rolf LH, Haarmann FY, Grotemeyer KH, Kehrer H (1993) Serotonin and amino acid content in platelets of autistic children. Acta Psychiatr Scand 87:312–316. [DOI] [PubMed] [Google Scholar]
- 181. Sacco R, Papaleo V, Hager J, Rousseau F, Moessner R, Militerni R, Bravaccio C, Trillo S, Schneider C, Melmed R, Elia M, Curatolo P, Manzi B, Pascucci T, Puglisi‐Allegra S, Reichelt KL, Persico AM (2007) Case‐control and family‐based association studies of candidate genes in autistic disorder and its endophenotypes: tPH2 and GLO1. BMC Med Genet 8:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Sacco R, Militerni R, Frolli A, Bravaccio C, Gritti A, Elia M et al (2007) Clinical, morphological, and biochemical correlates of head circumference in autism. Biol Psychiatry i. [DOI] [PubMed] [Google Scholar]
- 183. Sadakata T, Kakegawa W, Mizoguchi A, Washida M, Katoh‐Semba R, Shutoh F, Okamoto T, Nakashima H, Kimura K, Tanaka M, Sekine Y, Itohara S, Yuzaki M, Nagao S, Furuichi T (2007) Impaired cerebellar development and function in mice lacking CAPS2, a protein involved in neurotrophin release. J Neurosci 27:2472–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Sadakata T, Washida M, Furuichi T (2007) Alternative splicing variations in mouse CAPS2: differential expression and functional properties of splicing variants. BMC Neurosci 8:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Salichon N, Gaspar P, Upton AL, Picaud S, Hanoun N, Hamon M, De Maeyer E, Murphy DL, Mossner R, Lesch KP, Hen R, Seif I (2001) Excessive activation of serotonin (5‐HT) 1B receptors disrupts the formation of sensory maps in monoamine oxidase a and 5‐ht transporter knock‐out mice. J Neurosci 21:884–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Salinger WL, Ladrow P, Wheeler C (2003) Behavioral phenotype of the reeler mutant mouse: effects of RELN gene dosage and social isolation. Behav Neurosci 117:1257–1275. [DOI] [PubMed] [Google Scholar]
- 187. Schellenberg GD, Dawson G, Sung YJ, Estes A, Munson J, Rosenthal E, Rothstein J, Flodman P, Smith M, Coon H, Leong L, Yu CE, Stodgell C, Rodier PM, Spence MA, Minshew N, McMahon WM, Wijsman EM (2006) Evidence for multiple loci from a genome scan of autism kindreds. Mol Psychiatry 11:1049–1060. [DOI] [PubMed] [Google Scholar]
- 188. Schmitz C, Van Kooten IA, Hof PR, Van Engeland H, Patterson PH, Steinbusch HW (2005) Autism: neuropathology, alterations of the GABAergic system, and animal models. Int Rev Neurobiol 71:1–26. [DOI] [PubMed] [Google Scholar]
- 189. Schroer RJ, Phelan MC, Michaelis RC, Crawford EC, Skinner SA, Cuccaro M, Simensen RJ, Bishop J, Skinner C, Fender D, Stevenson RE (1998) Autism and maternally derived aberrations of chromosome 15q. Am J Med Genet 76:327–336. [DOI] [PubMed] [Google Scholar]
- 190. Schumann CM, Amaral DG (2005) Stereological estimation of the number of neurons in the human amygdaloid complex. J Comp Neurol 491:320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Schumann CM, Amaral DG (2006) Stereological analysis of amygdala neuron number in autism. J Neurosci 26:7674–7679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Schumann CM, Hamstra J, Goodlin‐Jones BL, Lotspeich LJ, Kwon H, Buonocore MH, Lammers CR, Reiss AL, Amaral DG (2004) The amygdala is enlarged in children but not adolescents with autism; the hippocampus is enlarged at all ages. J Neurosci 24:6392–6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Scott MM, Deneris ES (2005) Making and breaking serotonin neurons and autism. Int J Dev Neurosci 23:277–285. [DOI] [PubMed] [Google Scholar]
- 194. Sebat J, Lakshmi B, Malhotra D, Troge J, Lese‐Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee YH, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimaki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King MC, Skuse D, Geschwind DH, Gilliam TC, Ye K, Wigler M (2007) Strong association of de novo copy number mutations with autism. Science 316:445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Serajee FJ, Zhong H, Nabi R, Huq AH (2003) The metabotropic glutamate receptor 8 gene at 7q31: partial duplication and possible association with autism. J Med Genet 40:e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Serajee FJ, Zhong H, Mahbubul Huq AH (2006) Association of Reelin gene polymorphisms with autism. Genomics 87:75–83. [DOI] [PubMed] [Google Scholar]
- 197. Shoval G, Weizman A (2005) The possible role of neurotrophins in the pathogenesis and therapy of schizophrenia. Eur Neuropsychopharmacol 15:319–329. [DOI] [PubMed] [Google Scholar]
- 198. Shuang M, Liu J, Jia MX, Yang JZ, Wu SP, Gong XH, Ling YS, Ruan Y, Yang XL, Zhang D (2004) Family‐based association study between autism and glutamate receptor 6 gene in Chinese Han trios. Am J Med Genet B Neuropsychiatr Genet 131:48–50. [DOI] [PubMed] [Google Scholar]
- 199. Skaar DA, Shao Y, Haines JL, Stenger JE, Jaworski J, Martin ER, DeLong GR, Moore JH, McCauley JL, Sutcliffe JS, Shley‐Koch AE, Cuccaro ML, Folstein SE, Gilbert JR, Pericak‐Vance MA (2005) Analysis of the RELN gene as a genetic risk factor for autism. Mol Psychiatry 10:563–571. [DOI] [PubMed] [Google Scholar]
- 200. Smalley SL (1998) Autism and tuberous sclerosis. J Autism Dev Disord 28:407–414. [DOI] [PubMed] [Google Scholar]
- 201. Smith IM, Nichols SL, Issekutz K, Blake K (2005) Behavioral profiles and symptoms of autism in CHARGE syndrome: preliminary Canadian epidemiological data. Am J Med Genet A 133:248–256. [DOI] [PubMed] [Google Scholar]
- 202. Strutz‐Seebohm N, Korniychuk G, Schwarz R, Baltaev R, Ureche ON, Mack AF, DeLong GR, Moore JH, McCauley JL, Sutcliffe JS, Shley‐Koch AE, Cuccaro ML, Folstein SE, Gilbert JR, Pericak‐Vance MA (2006) Functional significance of the kainate receptor GluR6(M836I) mutation that is linked to autism. Cell Physiol Biochem 18:287–294. [DOI] [PubMed] [Google Scholar]
- 203. Sun W, Funakoshi H, Nakamura T (2002) Localization and functional role of hepatocyte growth factor (HGF) and its receptor c‐met in the rat developing cerebral cortex. Brain Res Mol Brain Res 103:36–48. [DOI] [PubMed] [Google Scholar]
- 204. Sutcliffe JS, Delahanty RJ, Prasad HC, McCauley JL, Han Q, Jiang L, Li C, Folstein SE, Blakely RD (2005) Allelic heterogeneity at the serotonin transporter locus (SLC6A4) confers susceptibility to autism and rigid‐compulsive behaviors. Am J Hum Genet 77:265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, Vincent JB, Skaug JL, Thompson AP, Senman L, Feuk L, Qian C, Bryson SE, Jones MB, Marshall CR, Scherer SW, Vieland VJ, Bartlett C, Mangin LV, Goedken R, Segre A, Pericak‐Vance MA, Cuccaro ML, Gilbert JR, Wright HH, Abramson RK, Betancur C, Bourgeron T, Gillberg C, Leboyer M, Buxbaum JD, Davis KL, Hollander E, Silverman JM, Hallmayer J, Lotspeich L, Sutcliffe JS, Haines JL, Folstein SE, Piven J, Wassink TH, Sheffield V, Geschwind DH, Bucan M, Brown WT, Cantor RM, Constantino JN, Gilliam TC, Herbert M, Lajonchere C, Ledbetter DH, Lese‐Martin C, Miller J, Nelson S, Samango‐Sprouse CA, Spence S, State M, Tanzi RE, Coon H, Dawson G, Devlin B, Estes A, Flodman P, Klei L, McMahon WM, Minshew N, Munson J, Korvatska E, Rodier PM, Schellenberg GD, Smith M, Spence MA, Stodgell C, Tepper PG, Wijsman EM, Yu CE, Roge B, Mantoulan C, Wittemeyer K, Poustka A, Felder B, Klauck SM, Schuster C, Poustka F, Bolte S, Feineis‐Matthews S, Herbrecht E, Schmotzer G, Tsiantis J, Papanikolaou K, Maestrini E, Bacchelli E, Blasi F, Carone S, Toma C, Van EH, De JM, Kemner C, Koop F, Langemeijer M, Hijimans C, Staal WG, Baird G, Bolton PF, Rutter ML, Weisblatt E, Green J, Aldred C, Wilkinson JA, Pickles A, Le CA, Berney T, McConachie H, Bailey AJ, Francis K, Honeyman G, Hutchinson A, Parr JR, Wallace S, Monaco AP, Barnby G, Kobayashi K, Lamb JA, Sousa I, Sykes N, Cook EH, Guter SJ, Leventhal BL, Salt J, Lord C, Corsello C, Hus V, Weeks DE, Volkmar F, Tauber M, Fombonne E, Shih A (2007) Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet 39:319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Tabakman R, Lecht S, Sephanova S, Rien‐Zakay H, Lazarovici P (2004) Interactions between the cells of the immune and nervous system: neurotrophins as neuroprotection mediators in CNS injury. Prog Brain Res 146:387–401. [DOI] [PubMed] [Google Scholar]
- 207. Talebizadeh Z, Lam DY, Theodoro MF, Bittel DC, Lushington GH, Butler MG (2006) Novel splice isoforms for NLGN3 and NLGN4 with possible implications in autism. J Med Genet 43:e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Torrente F, Ashwood P, Day R, Machado N, Furlano RI, Anthony A, Davies SE, Wakefield AJ, Thomson MA, Walker‐Smith JA, Murch SH (2002) Small intestinal enteropathy with epithelial IgG and complement deposition in children with regressive autism. Mol Psychiatry 7:375–382, 334. [DOI] [PubMed] [Google Scholar]
- 209. Ullian EM, Christopherson KS, Barres BA (2004) Role for glia in synaptogenesis. Glia 47:209–216. [DOI] [PubMed] [Google Scholar]
- 210. Vaidya VA, Terwilliger RM, Duman RS (1999) Role of 5‐HT2A receptors in the stress‐induced down‐regulation of brain‐derived neurotrophic factor expression in rat hippocampus. Neurosci Lett 262:1–4. [DOI] [PubMed] [Google Scholar]
- 211. Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA (2005) Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol 57:67–81. [DOI] [PubMed] [Google Scholar]
- 212. Vincent JB, Kolozsvari D, Roberts WS, Bolton PF, Gurling HM, Scherer SW (2004) Mutation screening of X‐chromosomal neuroligin genes: no mutations in 196 autism probands. Am J Med Genet B Neuropsychiatr Genet 129:82–84. [DOI] [PubMed] [Google Scholar]
- 213. Wassink TH, Hazlett HC, Epping EA, Arndt S, Dager SR, Schellenberg GD, Dawson G, Piven J (2007) Cerebral cortical gray matter overgrowth and functional variation of the serotonin transporter gene in autism. Arch Gen Psychiatry 64:709–717. [DOI] [PubMed] [Google Scholar]
- 214. Weiss LA, Kosova G, Delahanty RJ, Jiang L, Cook EH, Ober C, Sutcliffe JS (2006) Variation in ITGB3 is associated with whole‐blood serotonin level and autism susceptibility. Eur J Hum Genet 14:923–931. [DOI] [PubMed] [Google Scholar]
- 215. Weiss LA, Ober C, Cook EH Jr (2006) ITGB3 shows genetic and expression interaction with SLC6A4. Hum Genet 120:93–100. [DOI] [PubMed] [Google Scholar]
- 216. Williams KC, Hickey WF (2002) Central nervous system damage, monocytes and macrophages, and neurological disorders in AIDS. Annu Rev Neurosci 25:537–562. [DOI] [PubMed] [Google Scholar]
- 217. Yamakawa H, Oyama S, Mitsuhashi H, Sasagawa N, Uchino S, Kohsaka S, Ishiura S (2007)Neuroligins 3 and 4X interact with syntrophin‐gamma2, and the interactions are affected by autism‐related mutations. Biochem Biophys Res Commun 355:41–46. [DOI] [PubMed] [Google Scholar]
- 218. Yan J, Oliveira G, Coutinho A, Yang C, Feng J, Katz C, Sram J, Bockholt A, Jones IR, Craddock N, Cook EH, Jr. , Vicente A, Sommer SS (2005) Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol Psychiatry 10:329–332. [DOI] [PubMed] [Google Scholar]
- 219. Yan W, Wilson CC, Haring JH (1997) 5‐HT1a receptors mediate the neurotrophic effect of serotonin on developing dentate granule cells. Brain Res 98:185–190. [DOI] [PubMed] [Google Scholar]
- 220. Yang MS, Gill M (2007) A review of gene linkage, association and expression studies in autism and an assessment of convergent evidence. Int J Dev Neurosci 25:69–85. [DOI] [PubMed] [Google Scholar]
- 221. Ylisaukko‐Oja T, Rehnstrom K, Auranen M, Vanhala R, Alen R, Kempas E, Ellonen P, Turunen JA, Makkonen I, Riikonen R, Nieminen‐von WT, Von WL, Peltonen L, Jarvela I (2005) Analysis of four neuroligin genes as candidates for autism. Eur J Hum Genet 13:1285–1292. [DOI] [PubMed] [Google Scholar]
- 222. Yorbik O, Sayal A, Akay C, Akbiyik DI, Sohmen T (2002) Investigation of antioxidant enzymes in children with autistic disorder. Prostaglandins Leukot Essent Fatty Acids 67:341–343. [DOI] [PubMed] [Google Scholar]
- 223. Zetterstrom TS, Pei Q, Madhav TR, Coppell AL, Lewis L, Grahame‐Smith DG (1999) Manipulations of brain 5‐HT levels affect gene expression for BDNF in rat brain. Neuropharmacology 38:1063–1073. [DOI] [PubMed] [Google Scholar]
- 224. Zhang H, Liu X, Zhang C, Mundo E, Macciardi F, Grayson DR, Guidotti AR, Holden JJ (2002) Reelin gene alleles and susceptibility to autism spectrum disorders. Mol Psychiatry 7:1012–1017. [DOI] [PubMed] [Google Scholar]
- 225. Zimmerman AW, Connors SL, Pardo CA (2006) Neuroimmunology and neurotransmitters in autism. In: Autism: A Neurological Disorder of Early Brain Development. Tuchman R, Rapin I (eds), pp. 141–159. International Child Neurology Association: London, Mac Keith Press. [Google Scholar]
- 226. Zimmerman AW, Connors SL, Matteson KJ, Lee LC, Singer HS, Castaneda JA, Pearce DA (2007) Maternal antibrain antibodies in autism. Brain Behav Immun 21:351–357. [DOI] [PubMed] [Google Scholar]
- 227. Zoghbi HY (2003) Postnatal neurodevelopmental disorders: meeting at the synapse? Science 302:826–830. [DOI] [PubMed] [Google Scholar]
- 228. Zori RT, Marsh DJ, Graham GE, Marliss EB, Eng C (1998) Germline PTEN mutation in a family with Cowden syndrome and Bannayan‐Riley‐Ruvalcaba syndrome. Am J Med Genet 80:399–402. [PubMed] [Google Scholar]
- 229. Zoroglu SS, Armutcu F, Ozen S, Gurel A, Sivasli E, Yetkin O, Meram I (2004) Increased oxidative stress and altered activities of erythrocyte free radical scavenging enzymes in autism. Eur Arch Psychiatry Clin Neurosci 254:143–147. [DOI] [PubMed] [Google Scholar]