

Abstract
Intergroup Radiation Therapy Oncology Group Trial 9402 study, a phase III trial of chemotherapy plus radiotherapy (PCV‐plus‐RT) vs. radiotherapy alone for pure and mixed anaplastic oligodendroglioma confirmed the prognostic significance of 1p 19q deletion and showed that only progression‐free survival (PFS) was prolonged in PCV‐plus‐RT‐treated patients and only in association with 1p 19q deletion. We reviewed tumor histopathology, separating 115 tumors deemed to be classic for oligodendroglioma (CFO) from 132 lacking classic features of oligodendroglioma (NCFO) and evaluated the relationship of histopathology and 1p 19q status to treatment and outcome. The study disclosed: (i) overall survival (OS) of patients with CFO was significantly longer than for patients with NCFO (P < 0.0001) and was not affected by necrosis. Median OS for CFO patients with and without necrosis was 6.6 and 6.3 years (OS log‐rank P = not significant), respectively, in contrast to NCFO showing 1.9 and 3.3 years respectively (OS log‐rank P = 0.014). (ii) Classic oligodendroglial morphology was highly associated with 1p 19q deletion, present in 80% of CFO and only in 13% of NCFO. (iii) On multivariate analysis, both classic oligodendroglial morphology and 1p 19q deletion remained significantly associated with PFS and OS. (iv) Patients with CFO treated with PCV‐plus‐RT showed a trend toward increased survival compared with CFO treated with RT (P = 0.08). Median OS was not reached in the PCV‐plus‐RT group and was 6.3 years in RT group. These findings suggest that classic oligodendroglial morphology combined with 1p 19q deletion may in the future be predictive of chemotherapeutic response and survival.
Keywords: anaplastic oligodendroglioma, anaplastic oligoastrocytoma, 1p 19q deletion
INTRODUCTION
Concurrent deletion of chromosomal arms 1p and 19q, a distinctive feature of oligodendroglioma, has been reported in up to 80% of these tumors 11, 19. In most cases, deletions involve one entire copy of 1p and 19q chromosome arms, and this loss appears to be mediated by an unbalanced t(1;19)(q10;p10) translocation 6, 10, 20. This combined deletion of chromosomal arms 1p and 19q is a relevant prognostic marker in patients with anaplastic oligodendroglial tumors and likely predicts responsiveness to combined radiotherapy and chemotherapy 2, 3, 7, 8, 21, 22.
Although there is a strong association between combined 1p 19q deletion and classic oligodendroglial morphology, this association is not absolute. As we observed in a recent study, in which the tumor histopathology was reviewed independently by five neuropathologists with experience in diagnosis of brain tumors, 14% of tumors showing classic oligodendroglial morphology, according to consensus, lacked 1p 19q deletion, while 18% of tumors classified as not having classic oligodendroglial features demonstrated combined 1p 19q deletion (14). Our results further suggested that, in addition to 1p/19q status, histologic features contributed information to the prediction of outcome in patients with anaplastic oligodendrogliomas and that the prognostic significance may be limited to patients with tumors showing classic histopathology.
Histologic classification of high grade gliomas can be challenging, because many high grade gliomas do not show classic defined histological features, but rather a non‐classic, ambiguous morphology resulting in considerable inter‐observer variability and limited diagnostic reproducibility (4). This is particularly true for oligodendroglial tumors, including pure oligodendrogliomas and especially mixed oligoastrocytomas, this last one a category of tumors in which objective diagnostic criteria are controversial and difficult to apply. As recently described, the clinical recognition of the more favorable prognosis of anaplastic oligodendrogliomas and of their frequent responsiveness to treatment, coupled with the pathologists desire to assure that “no patient is deprived of a potentially effective chemotherapy”, has further loosened diagnostic criteria and blurred the histopathological line separating glioblastoma and oligodendroglioma 1, 17. Currently, up to 25% of malignant gliomas are diagnosed as malignant oligodendrogliomas and oligoastrocytomas 4, 5, 18.
Two separate phase III clinical trials comparing outcome in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas treated with chemotherapy and radiotherapy compared with radiotherapy alone included patients diagnosed with anaplastic oligodendrogliomas or anaplastic mixed oligoastrocytomas with at least 25% oligodendroglial elements either by the local pathologists (22) or by central review before random assignment to treatment group (3). Both studies confirmed the prognostic value of combined 1p 19q deletion in identifying “less aggressive” tumors, although neither study showed significant difference in overall survival (OS) between treatment arms, either as a whole, or within genetic groups defined by 1p/19q status. No details were provided regarding the correlation of morphology (oligodendroglial and oligoastrocytic) and 1p 19q status. A follow‐up paper on European Organization for Research and Treatment of Cancer (EORTC) trial 26951, addressing this aspect from the point of view of concordance between the morphologic diagnosis made by the local pathologist and used for patient enrollment and that made by a neuropathologist consensus panel found a very low concordance especially for the diagnosis of anaplastic oligoastrocytoma (8%). This resulted in inclusion of cases, which were controversial with respect to the presence of oligodendroglioma, a finding to which the authors attributed the low frequency of 1p 19q deletion in their series (25%), greatly limiting subsequent correlative analysis (12).
In the present study, we hypothesized that tumor histopathology is a factor associated with outcome in anaplastic oligodendroglial tumors. In particular, we asked whether tumors with classic oligodendroglial morphology were less aggressive compared with those which lacked classic features for oligodendroglioma. We critically reviewed the tumor histopathological features of the patients enrolled in Intergroup Radiation Therapy Oncology Group Trial (RTOG) 9402 and evaluated the relationship of histopathology and combined 1p 19q deletion to treatment and outcome.
MATERIALS AND METHODS
Histopathology review
RTOG trial 9402 was a phase III trial of chemotherapy plus radiotherapy (PCV‐plus‐RT) compared with radiotherapy alone. Patients with newly diagnosed supratentorial anaplastic gliomas of pure oligodendroglial or mixed oligoastrocytic morphology according to mandatory pre‐enrollment central pathology review were eligible (3). To be an oligoastrocytoma a 25% or greater oligodendroglial morphology was required. The definition of anaplasia was based on evaluation of five microscopic features: tumor cellularity, nuclear pleomorphism, mitotic activity, endothelial vascular proliferation and necrosis. For a tumor to be anaplastic, it had to contain at least two anaplastic features, one of which was frequent mitoses (>5 mitoses per 10 high‐power fields) or endothelial vascular proliferation. Two neuropathologists (KA and CG) undertook independently a retrospective histological review of the entire study set, which included hematoxylin and eosin (H&E) slides from 247 of the original 289 patients enrolled in the study (85%). Each neuropathologist was given the slide set and was asked to score each slide based on whether it had or did not have the features of classic oligodendroglioma. Features typically considered classic for oligodendroglioma (CFO), including cellular monomorphism, round/regular nuclei, presence of nodules, microcalcifications, microcysts, chicken wire vasculature were also recorded. Cases, in which the reviewers differed, were reviewed by a third neuropathologist (PCB). Consensus was defined as either agreement between the two original reviewers or at least among two (of three) neuropathologists in the remaining cases.
At the end of his or her review, the neuropathologist was asked what specific diagnosis according to WHO criteria he or she would render.
Study design and treatment
Clinical trial design and treatment as well as surveillance and follow‐up have been previously described elsewhere (3). Briefly, patients age ≥18 years with newly diagnosed anaplastic gliomas of pure oligodendroglial or mixed oligoastrocytic morphology were eligible (see above). Eligible patients also had a Karnofsky performance score (KPS) of ≥60 and adequate hematologic, pulmonary, renal and hepatic function [absolute neutrophil count ≥ 1.5 × 109/L, platelets ≥ 150 × 109/L, oxygen diffusing capacity ≥ 60%, serum creatinine ≤ 1.5 × the upper limit of normal (ULN), total bilirubin ≤ 1.5 × ULN, and liver function tests < 2 × ULN]. All study centers had institutional review board approval, and all patients consented in writing. Patients were randomly assigned at RTOG headquarters within 8 weeks of surgical diagnosis to either PCV followed by focal RT (the experimental arm) or to RT alone (the control arm). Patients were stratified by age <50 years vs. 50 years or older, KPS of 60–70 vs. ≥80 years, and moderately anaplastic (2–3 anaplastic features) vs. highly anaplastic tumors (4–5 anaplastic features). Patients began the assigned treatment within 1 week of random assignment. In the experimental arm, up to four cycles of PCV were to be administered every 6 weeks before RT as follows: lomustine 130 mg/m2 orally on day 1; procarbazine 75 mg/m2 orally daily on days 8–21; and vincristine 1.4 mg/m2 intravenously on days 8 and 29. Doses were calculated using actual body weight, reduced for toxicity and not escalated. There was no upper limit on the dose of vincristine. Compliance was assessed by case reviews.
1p 19q Fluorescent in situ hybridization
Fluorescent in situ hybridization (FISH) studies on tumor tissue sections to detect chromosomes 1p and 19q deletion were performed as previously described (9).
Briefly, for each case, a paraffin‐embedded tumor block was selected based on tumor content, including the highest grade component and representation of the predominant morphology of the individual case. Tumor sections immediately adjacent to the first H&E stained slide were deparaffinized, dehydrated, microwave treated in citrate buffer (pH 6.0) for 10 minutes, digested in pepsin solution (4 mg/mL in 0.9% NaCl, pH 1.5) for 15 minutes at 37°C, rinsed in two times standard saline citrate (SSC) at room temperature for 5 minutes and air dried. Dual‐probe hybridization was performed using a digoxigenin‐labeled locus‐specific 1p or 19q probe and a SpectrumGreenlabeled probe (Vysis, Downers Grove, IL, USA) mapping to 1q and 19p, respectively. Probes and target DNA were denatured simultaneously in an 80°C oven for 5 minutes, followed by overnight incubation at 37°C. Slides were then washed in 1.5 mol urea/0.1 times SSC at 45°C for 10 minutes (three times) and in two times SSC at room temperature for 2 minutes. After washing, the digoxigenin‐labeled probes were detected using a rhodamine detection kit (Oncor, Gaithersburg, MD, USA). Nuclei were counterstained with 4,6‐diamidino‐2‐phenylindole and the antifade compound p‐phenylenediamine. A Zeiss (Thornwood, NY, USA) Axioplan microscope equipped with a triple‐pass filter (DAPI/Green/Orange; Vysis, Downers Grove, IL) was used to assess the number of FISH signals for each locus‐specific FISH probe. Approximately 300 non‐overlapping nuclei were enumerated per hybridization. Ranges for normal FISH probe copy number were established by hybridizing and enumerating gliosis specimens (data not shown) and through extensive comparisons of FISH, LOH and CGH data.
Statistical analysis
Inter‐reviewer agreement was assessed using the kappa statistic. Chi‐squared statistics were used to test for associations between categorical variables. Overall and progression‐free survival (PFS) were estimated by the Kaplan–Meier method with testing carried out with the log‐rank statistic. The Cox proportional hazards model was fitted to adjust for stratification factors and other confounding clinical and genetic variables.
RESULTS
Histopathology review
Based on histological review, 115 (of 247, 47%) cases were assigned to the CFO group and the remaining 132 (of 247, 53%) cases to the group in which the classic features of oligodendroglioma were not present (not classic for oligodendroglioma—NCFO). Consensus was reached between the first two reviewers (A and B) in 192 (of 247) cases (78%), 70% for CFO and 84% for NCFO, respectively, with a corresponding overall kappa statistic of 0.55 (95% CI 0.44, 0.65) indicating a moderate amount of inter‐reviewer agreement. On review of the 55 cases in which there was disagreement among the first two reviewers, reviewer C appeared to agree more frequently with reviewer A than B (69% and 31% of times, respectively). Consensus among reviewers (A, B, C) was summarized in Table 1.
Table 1.
Consensus among the three neuropathology reviewers (A, B, C). Abbreviations: CFO = tumor with features classic for oligodendroglioma; NCFO = tumor with features not classic for oligodendroglioma.
| A&B | A&C | B&C | Total | |
|---|---|---|---|---|
| CFO | 81 | 25 | 9 | 115 |
| NCFO | 111 | 13 | 8 | 132 |
| Total | 192 | 38 | 17 | 247 |
Figure 1 illustrates one example of a tumor showing classic features of oligodendroglioma (CFO) (panels A, B) and one example of a tumor in which the CFO were not present (NCFO) (panels C, D).
Figure 1.
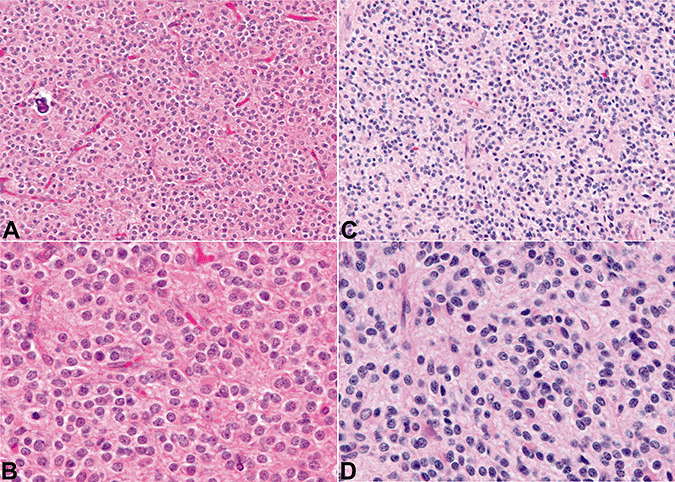
The figure illustrates one example of a tumor showing classic features of oligodendroglioma (A,B) and one example in which the classic features of oligodendroglioma were not present (C,D).
When reviewers A and B were asked what specific WHO diagnosis they would render “today” for the tumors, it was apparent that two different approaches were used, one accepting the concept/existence of mixed tumors, the other skeptical about it. For this second reviewer, all the CFO should be classified as anaplastic oligodendrogliomas (WHO, III) while most of the NCFO tumors would be divided in anaplastic astrocytomas (WHO, III) and glioblastoma (WHO, IV). The other reviewer would diagnose most of the CFO as anaplastic oligodendrogliomas (WHO, III), while among the NCFO tumors the term anaplastic oligoastrocytoma (WHO, III or IV‐ glioblastoma with oligodendroglial features in the new WHO classification) would be used liberally and only a minority would be classified as pure astrocytomas either anaplastic (WHO, III) or glioblastoma (WHO, IV). Some of the tumors among the NCFO tumors were considered anaplastic astrocytomas (WHO, III) and glioblastomas (WHO, IV) by one or more of the pathologists (A, B, C) in this study. This lack of concordance in diagnostic styles limited any meaningful analysis to the dichotomous CFO vs. NCFO variable.
Histopathology, necrosis and survival analysis
Data concerning necrosis were derived from original trial database: necrosis was reported as present in 100 tumors (including 46 CFO and 54 NCFO), absent in 128 tumors (including 57 CFO and 71 NCFO), while the form was left blank in 19 cases. At the time of this review, only one or two representative slides from the original tumor were available for review, while at the time of central review “all available slides” were reviewed. Since necrosis may be a focal finding, the data derived from the original database were thought to be the most appropriate for evaluation of this variable.
Patients with CFO with and without necrosis had similar OS (P = 0.99). Their OS was significantly longer than OS of patients with NCFO irrespective of the presence of necrosis (Figure 2). Patients with NCFO without necrosis in addition had a significantly longer OS than patients with NCFO with necrosis. Median OS for patients with CFO with and without necrosis was 6.6 and 6.3 years (OS log‐rank P = ns), respectively, in contrast to NCFO showing 1.9 and 3.3 years (OS log‐rank P = 0.014).
Figure 2.
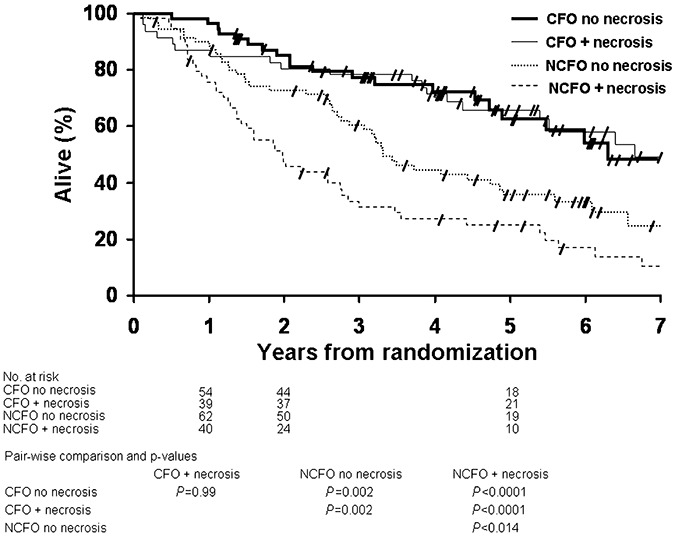
Kaplan–Meier estimates of overall survival by histopathology (CFO and NCFO), with and without presence of necrosis. Pair‐wise comparisons and P‐values are tabulated below the graphic. CFO = tumor with features classic for oligodendroglioma; NCFO = tumor with features not classic for oligodendroglioma.
PFS of patients with CFO without necrosis was significantly longer than PFS of patients with CFO with necrosis (P = 0.005) and of patients with NCFO with (P < 0.0001) and without (P = 0.0009) necrosis (Figure 3).
Figure 3.
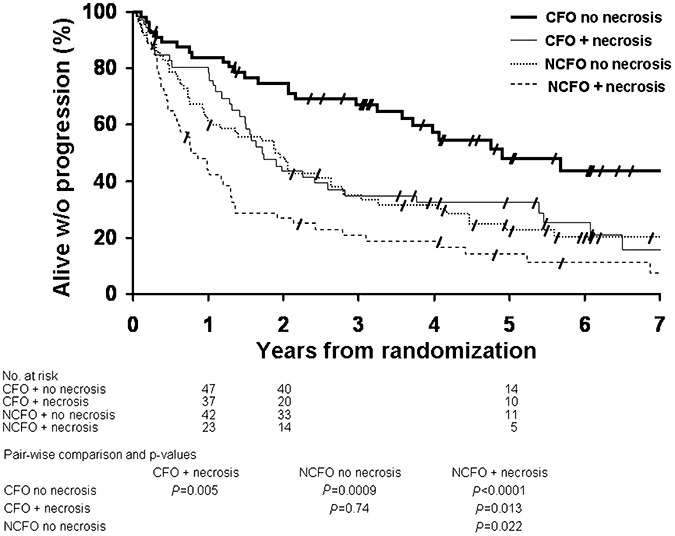
Kaplan–Meier estimates of progression‐free survival by histopathology (CFO and NCFO), with and without presence of necrosis. Pair‐wise comparisons and P‐values are tabulated below the graphic. CFO = tumor with features classic for oligodendroglioma; NCFO = tumor with features not classic for oligodendroglioma.
Histopathology and combined 1p 19q deletion
Data on 1p 19q status were available in 196 (of 247) cases from which H&E slides were available for review (79%). Ninety‐one of the 196 cases (46%) showed combined 1p 19q deletion (46%) and the remaining 105 (54%) did not. Of these 105 cases there were 60 cases with intact 1p 19q (31%); 30 cases with 19q deletion only (15%); and 15 cases with 1p deletion only (8%). Among CFO tumors, data on 1p 19q status were available in 97 (of 115) cases, from which H&E slides were available for review (84%). These included 78 cases showing a combined 1p 19q deletion (80%) and 19 cases (20%) without combined deletion (five cases with intact 1p 19q (5%); eight cases with 19q deletion only (8%); and six cases with 1p deletion only (6%)). Among NCFO tumors, data on 1p 19q status were available in 99 (of 132) cases from which H&E slides were available for review (75%). These included 13 cases showing a combined 1p 19q deletion (13%) and 86 cases (87%) without combined deletion (55 cases with intact 1p 19q (56%); 22 cases with 19q deletion only (22%); and nine cases with 1p deletion only (9%)). Data regarding 1p 19q in CFO and NCFO are summarized in Table 2. Figure 4 illustrates an example of a CFO not showing 1p 19q deletion (panel A) and one example of a tumor NCFO, with 1p 19q deletion (panel B).
Table 2.
1p 19q status in CFO and NCFO. Abbreviations: CFO = tumor with features classic for oligodendroglioma; NCFO = tumor with features not classic for oligodendroglioma.
| 1p 19q deletion | 1p 19q intact | 19q loss | 1p loss | Total (n) | |
|---|---|---|---|---|---|
| CFO | 78 (80%) | 5 (5%) | 8 (8%) | 6 (6%) | 97 |
| NCFO | 13 (13%) | 55 (56%) | 22 (22%) | 9 (9%) | 99 |
| Total (n) | 91 | 60 | 30 | 15 | 196 |
Figure 4.
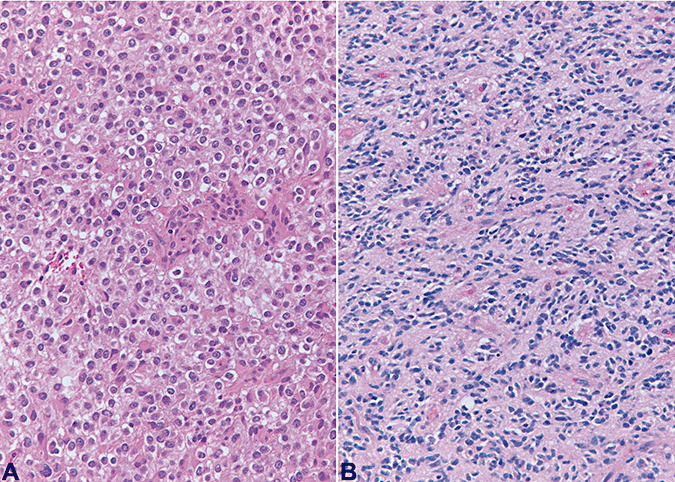
The figure illustrates an example of a CFO tumor which did not show 1p 19q deletion (panel A) and one example of a NCFO tumor with 1p 19q deletion (panel B). CFO = tumor with features classic for oligodendroglioma; NCFO = tumor with features not classic for oligodendroglioma.
Histopathology, combined 1p 19q deletion and survival analysis
Data on OS of patients with CFO and NCFO tumors, with or without combined 1p 19q deletion are summarized in Figure 5. OS of patients with CFO with 1p 19q deletion was significantly longer than OS of patients with CFO, but without 1p 19q deletion (P = 0.015) and of patients with NCFO without 1p 19q deletion (P < 0.0001). OS of patients with NCFO with 1p 19q deletion was significantly longer than OS of patients with NCFO without 1p 19q deletion (P = 0.04). Although the OS curves of patients with CFO and NCFO with 1p 19q deletion were distinctly separate, the difference was not statistically significant (P = 0.14). Median OS had not been reached in patients with CFO with 1p 19q deletion, while it was 5.5 years in patients with CFO without 1p 19 q deletion, 6.1 years in patients with NCFO with 1p 19q deletion and 2.6 years in patients with NCFO without 1p 19q deletion, respectively.
Figure 5.
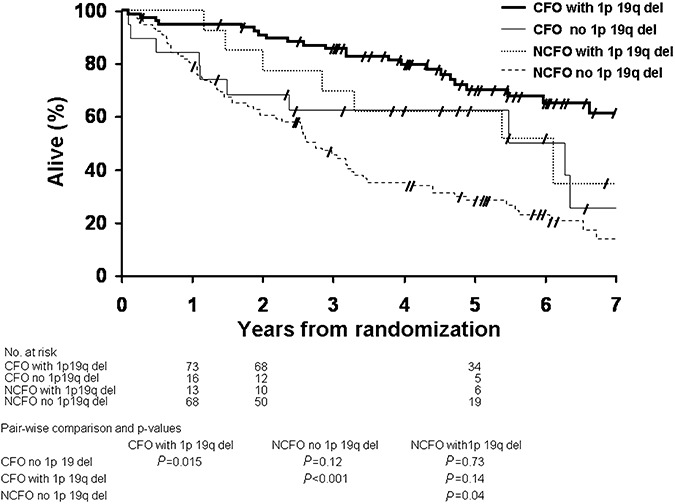
Kaplan‐Meier estimates of overall survival by histopathology (CFO and NCFO) and genotype (1p 19q deletion status). Pair‐wise comparisons and P‐values are tabulated below the graphic. CFO = tumor with features classic for oligodendroglioma; NCFO = tumor with features not classic for oligodendroglioma.
PFS of patients with CFO tumors with 1p 19q deletion was significantly longer than OS of patients with CFO tumors without 1p 19q deletion (P = 0.016) and of patients with NCFO tumors without 1p 19q deletion (P < 0.0001). PFS of patients with NCFO tumors with 1p 19q deletion was not significantly longer than PFS of patients with CFO tumors with 1p 19q deletion (P = 0.53), patients with CFO tumors without 1p 19q deletion (P = 0.26) and patients with NCFO tumors without 1p 19q deletion (P = 0.07). Median PFS was 4.5 years in patients with CFO tumors with 1p 19q deletion, 1.1 years in patients with CFO tumors without 1p 19q deletion, 1.2 years in patients with NCFO tumors with 1p 19 q deletion and 1.3 years in patients with NCFO tumors without 1p 19 q deletion, respectively.
Histopathology, combined 1p 19q deletion and tumor location
Data regarding tumor location were available in all 247 cases from which H&E slides were reviewed. These included 151 cases, in which the tumor involved only one lobe (61%), in order of decreasing frequency frontal (n = 105, 70%), temporal (n = 23, 15%), parietal (n = 22, 15%), occipital (n = 1, <1%). Ninety‐six tumors involved more than one lobe and/or the corpus callosum (39%). Among CFO tumors, 72 (of 115) involved only one lobe (63%), in order of decreasing frequency frontal (n = 56, 78%), parietal (n = 8, 11%), temporal (n = 7, 10%), occipital (n = 1, 1%). Loss of 1p 19q was identified in 38 (of 46) (83 %) CFO of frontal, four (of four) (100%) of temporal and six (of eight) (75%) of parietal lobes. Among NCFO, 79 (of 132) involved only one lobe (60%), in order of decreasing frequency frontal (n = 49, 62%), temporal (n = 16, 20%), parietal (n = 14, 18%). Loss of 1p 19q was identified in eight (of 39) (21%) NCFO of frontal, one (of 13) (8%) of temporal and 0 (of five) of parietal lobes. There was no association between 1p 19q status and tumor location.
Histopathology, treatment and survival analysis
Patients with CFO tumors had a significantly longer OS compared with NCFO patients, and this difference was seen in both treatment arms (Figure 6). Median OS of CFO patients was 6.3 years when treated with RT and had not been reached in patients treated with PCV‐plus‐RT. Median survival in NCFO patients was 3.1 years when treated with RT and 2.7 in patients treated with PCV‐plus‐RT. The OS curve of the CFO patients treated with PCV‐plus‐RT was distinct from the one of patients treated with RT. The P‐value of 0.08, in light of the separation of the two curves, may suggest a statistical trend toward improved OS as compared with the RT‐only treatment arm (P = 0.08).
Figure 6.
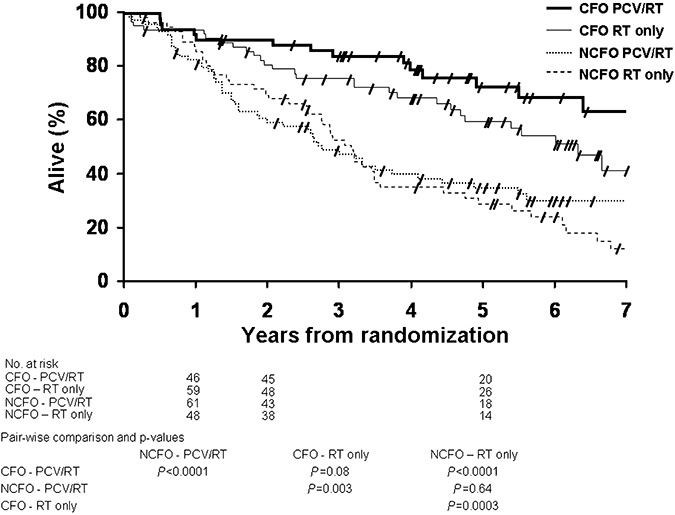
Kaplan‐Meier estimates of overall survival by histopathology (CFO and NCFO) and treatment (PCV‐RT and RT only). Pair‐wise comparisons and p‐values are tabulated below the graphic. CFO = tumor with features classic for oligodendroglioma; NCFO = tumor with features not classic for oligodendroglioma; PCV = procarbazine, lomustine and vincristine; RT = radiotherapy.
PFS was significantly longer in CFO patients treated with PCV‐plus‐RT as compared with patients treated with RT (P = 0.0005) and to NCFO patients treated either with PCV plus RT (P = 0.0004) or with RT (P < 0.0001), (Figure 7). Median PFS of CFO patients was 6.5 years when treated with PCV‐plus‐RT as compared with 2.2 years when treated with RT. Median PFS of NCFO patients was 1.2 years when treated with RT and 1.4 years in patients treated with PCV‐plus‐RT.
Figure 7.
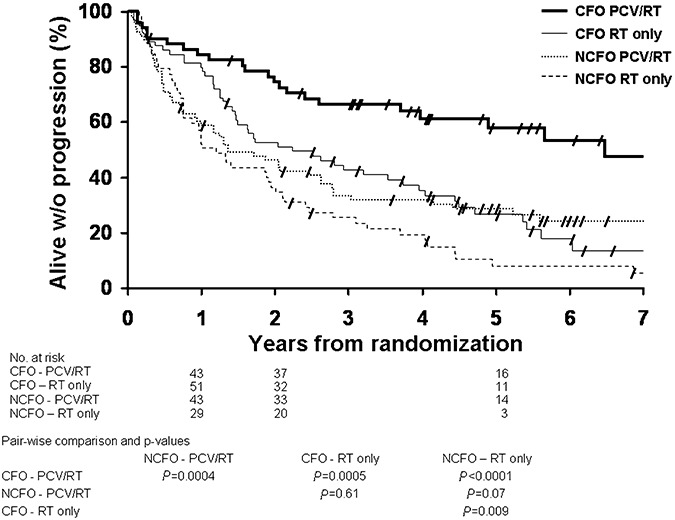
Kaplan‐Meier estimates of progression‐free survival by histopathology (CFO and NCFO) and treatment (PCV‐RT and RT only). Pair‐wise comparisons and p‐values are tabulated below the graphic. CFO = tumor with features classic for oligodendroglioma; NCFO = tumor with features not classic for oligodendroglioma; PCV = procarbazine, lomustine and vincristine; RT = radiotherapy.
Cox multivariate regression analysis
The influence of histopathological, genetic and clinical variables on OS was analyzed in the Cox proportional hazards models with assigned treatment, age, KPS and number of anaplastic features as fixed covariates (3, 4). Co‐deletion of 1p and 19q chromosomes, classic oligodendroglial histopathology, younger age, normal neurological function, absence of endothelial proliferation, unifocality of disease on baseline imaging and PCV therapy were independently associated with longer OS time, whereas sex, race and necrosis were not. Co‐deletion of 1p and 19q chromosomes, unifocality of disease on baseline imaging, PCV therapy, younger age and absence of endothelial proliferation and predicted longer PFS time.
Table 3.
Cox proportional hazards model for overall survival. Abbreviations: PCV = procarbazine, lomustine and vincristine; RT = radiotherapy; KPS = Karnofsky performance score; CFO = tumor with features classic for oligodendroglioma; NCFO = tumor with features not classic for oligodendroglioma.
| Variable (Bolded value has favorable outcome) | P‐value | Hazard ratio (95% CI) |
|---|---|---|
| Assigned treatment (PCV+RT vs. RT alone) | 0.18 | 0.76 (0.50, 1.14) |
| Age (<50 vs. ≥50) | <0.0001 | 0.35 (0.22, 0.56) |
| KPS (80–100 vs. 60–70) | 0.018 | 0.49 (0.27, 0.89) |
| Necrosis (No vs. Yes) | 0.66 | 0.90 (0.55, 1.46) |
| Endothelial proliferation (No vs. Yes) | 0.045 | 0.60 (0.36, 0.99) |
| Multifocal disease (No vs. Yes) | <0.0001 | 0.21 (0.10, 0.43) |
| Histopathology review (CFO vs. NCFO) | 0.0007 | 0.40 (0.24, 0.68) |
| 1p/19q (deleted vs. not deleted) | 0.0015 | 0.43 (0.26, 0.72) |
Model derived from stepwise selection of all variables found significant in model with fixed covariates of assigned treatment and stratification variables of age, KPS, necrosis, endothelial proliferation (reference variables).
Variable(s) not included in final model: pretreatment steroid use, surgery, neurologic function.
Variable(s) dropped from modeling as not significant with reference variables: gender, race.
Hazard ratios expressed are HRfavorable/HRunfavorable.
Table 4.
Cox proportional hazards model for progression‐free survival. Abbreviations: PCV = procarbazine, lomustine and vincristine; RT = radiotherapy; KPS = Karnofsky performance score; CFO = tumor with features classic for oligodendroglioma; NCFO = tumor with features not classic for oligodendroglioma.
| Variable (Bolded value has favorable outcome) | P‐value | Hazard ratio (95% CI) |
|---|---|---|
| Assigned treatment (PCV+RT vs. RT alone) | 0.0012 | 0.55 (0.38, 0.79) |
| Age (<50 vs. ≥50) | 0.03 | 0.65 (0.44, 0.97) |
| KPS (80–100 vs. 60–70) | 0.007 | 0.46 (0.26, 0.81) |
| Necrosis (No vs. Yes) | 0.07 | 0.66 (0.43, 1.03) |
| Endothelial proliferation (No vs. Yes) | 0.03 | 0.61 (0.38, 0.96) |
| Multifocal disease (No vs. Yes) | 0.0012 | 0.35 (0.18, 0.66) |
| Histopathology review (CFO vs. NCFO) | 0.20 | 0.73 (0.45, 1.18) |
| 1p/19q (deleted vs. not deleted) | <0.0001 | 0.36 (0.25, 0.53) |
Model derived from stepwise selection of all variables found significant in model with fixed covariates of assigned treatment and stratification variables of age, KPS, necrosis, endothelial proliferation (reference variables).
Variable(s) not included in final model: pretreatment steroid use, surgery, neurologic function.
Variable(s) dropped from modeling as not significant with reference variables: gender, race.
Hazard ratios expressed are HRfavorable/HRunfavorable.
DISCUSSION
To further explore the relationship between histopathology and 1p/19q status in oligodendroglial tumors, following our recent experience (14), we reviewed the histological features of the tumors from patients enrolled in RTOG 9402 (3). This study, as well as the EORTC study 26951, conceived in the early 1990s, tested the hypothesis that combined chemotherapy and radiotherapy would prolong the survival of patients with newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas compared with radiotherapy alone. Both studies enrolled patients diagnosed with anaplastic oligodendrogliomas or anaplastic mixed oligoastrocytomas with at least 25% oligodendroglial elements either by the local pathologists (22) or by central review on all patients before random assignment to treatment group (3). The cohorts of patients in these two clinical trials, because of their histological enrollment criteria included patients with classic oligodendrogliomas as well as patients with oligoastrocytomas, a much more controversial entity, for which diagnostic criteria are poorly defined and have only limited reproducibility (4).
Our review of RTOG 9402 gave us an opportunity to explore to some extent the issue of morphological classification and grading of high grade diffuse oligodendroglial tumors, a controversial issue recently addressed by Miller et al (15). RTOG 9402 included anaplastic oligodendroglial tumors, which we divided in two groups: tumors with classic oligodendroglial features (CFO) and tumor in which classic oligodendroglial features were not present (NCFO). Our study confirmed that classic oligodendroglial morphology is a strong predictor of OS and PFS (1, 2). As suggested by previous studies 5, 15 as well as by the 2007 WHO classification, our review confirmed that the use of necrosis to stratify classic anaplastic oligodendrogliomas into grade III and grade IV would not result in a stratification into distinct prognostic groups. Among these CFO tumors, number of anaplastic features (data not shown) and in particular presence of necrosis do not appear to be of prognostic significance (Figure 2). However, PFS was significantly longer in patients with CFO without necrosis compared with those with necrosis (Figure 3). In contrast, patients whose tumors were classified as not having classic oligodendroglial features (NCFO) and showed necrosis demonstrated both significantly decreased OS and PFS compared with patients with NCFO without necrosis (2, 3). It is important to emphasize, however, that among the consensus NCFO tumors there was little concordance among reviewers to a specific diagnosis. It is probable that these NCFO tumors, which likely were classified originally as mixed oligoastrocytomas at the time of patient enrollment, today might be classified differently by different neuropathologists with a spectrum of diagnoses ranging from anaplastic oligoastrocytomas to anaplastic astrocytoma and glioblastoma. Some of these may be classified as “oligoastrocytoma, grade IV”(15) or glioblastoma with oligodendroglial features (WHO 2007) (13). When we look at the median OS of patients with NCFO tumors without necrosis (39.6 months) and those with necrosis (21.8 months) in RTOG 9402, the lengths of OS time are comparable or longer than the ones recently reported for anaplastic astrocytomas (40.1 months), anaplastic oligoastrocytomas with necrosis (22.8 months) and glioblastoma (9.8 months), respectively(15). These tumors are the ones, which most likely will benefit from adjunct objective ancillary studies, such as molecular genetics given the lack of concordance based on morphological grounds, perhaps because of their “ambiguous morphology”4, 17. The high inter‐observer variation in the morphological diagnosis of these tumors was emphasized by the recent review of EORTC trial 26951, in which central review by a panel of neuropathologists confirmed the original diagnosis in 52% of anaplastic oligodendroglioma, but only in 8% of anaplastic oligoastrocytoma, with alternative consensus and/or majority diagnoses including anaplastic oligodendroglioma, low grade glioma and glioblastoma (12).
Although the nomenclature differed (CFO vs. NCFO compared with anaplastic oligodendroglioma (AO) and anaplastic oligoastrocytoma (AOA), similar to the findings of our previous study (14), the presence of classic oligodendroglial features predicted 1p/19q loss, with 80% of CFO tumors showing combined 1p 19q loss. However, although classic oligodendroglial morphology was highly associated with combined 1p 19q loss, 20% of CFO tumors had intact 1p and/or 19q chromosomes and 13% of NCFO tumors demonstrated combined 1p 19q deletion. Similar frequencies of 1p 19q deletion were found in the study by Miller et al (15), (89% for AO and 19% for AOA). Classic oligodendroglial tumors, which did not show combined 1p 19q deletion (n = 19) had significantly worse OS compared with CFO with combined 1p 19q deletion, while NCFO with combined 1p 19q deletion had significantly longer OS compared with NCFO without 1p 19q deletion. Similar results were found in the review of EORTC cases, which show a significant difference in OS between anaplastic oligodendrogliomas with and without deletion (12). These findings, supported by the multivariate analysis results, confirm that both histopathological features and 1p 19q deletion status findings contribute independent information to the prediction of outcome in patients with anaplastic oligodendrogliomas, as suggested by our previous study (14) and other studies 15, 22. It appears that CFO with 1p 19q deletion have the most favorable prognosis and NCFO tumors without 1p 19q deletion the least favorable prognosis. Classic oligodendroglial tumors without 1p 19q deletion appear to have an intermediate OS, significantly different from both the above mentioned groups. We did not detect significant differences in OS between patients with NCFO with combined 1p 19q deletion and patients with CFO with combined 1p 19q deletion, but the number of NCFO showing combined 1p 19q deletion (n = 13) is too small to derive robust statistical conclusions. Similar data were presented in the paper by Miller et al (15) (supplemental figure A5, JCO 24:5419), in which the relative infrequency of 1p/19q retention in AO (12%) and 1p/19q co‐deletion in AOA (19%) as well as the fact that the genetic data were predominantly acquired in more recently diagnosed patients (52% and 56% of AOA and AO patients diagnosed from 2003 to 2005, respectively) may have influenced results.
It would be of interest to know why 20% of CFOs in our study, a number comparable with the ones obtained in other studies, do not show combined 1p 19q deletion. It is unclear if some of these CFO not showing 1p 19q deletion are “true” negative or might be considered “false” negative. A “false” negative could result either from tumor heterogeneity of the 1p 19q deletion status and failure to sample 1p 19q deleted regions or it might represent a failure to detect the presence of deletion in a sufficient number of cells when tumor cell density is too low compared with non‐tumoral cells to detect the presence of the deletion. The fact that the OS of patients with CFO, which do not show 1p 19q deletion is significantly shorter than for patients with CFO with 1p 19q deletion makes the possibility that these cases are simply “false negative” unlikely. Testing of these samples for the presence or absence of the newly described 1q/19p translocation may shed further light on this finding, especially in light of the findings that fusion may be present in tumors, which do not show 1p 19q deletion by FISH (9).
It has been suggested that the frequency of chromosomal losses of 1p and 19q in oligodendrogliomas may be related to tumor location, with a low rate of allelic loss in tumors of the temporal and a high rate in tumors of the frontal, parietal and occipital lobes16, 23. In this series, both CFO and NCFO tumors occurred most frequently in the frontal lobes compared with other lobes (78% and 62%, respectively) and occurrence of 1p 19q deletion appears to correlate closely with histopathology, but be independent of tumor location. Among CFO, frequency of 1p 19q deletion seemed to be independent of location occurring in 83% (38 of 46) of frontal CFO, 100% (4 of 4) of temporal and 75% (6 of 8) of parietal tumors. Among NCFO, the maximum frequency of 1p 19q deletion was in tumors occurring in the frontal lobe (21%), but the numbers are small.
In RTOG 9402, as well as in the EORTC study, PCV chemotherapy appeared to improve PFS but not OS, although in RTOG 9402 the increase in PFS was significant only in patients with 1p/19q loss and not in patients with 1p 19q intact 3, 22. Interestingly, when treatment outcome is examined after a careful review of tumor morphology and distinction of CFO from NCFO, among patients with classic oligodendroglial tumors a trend toward increased OS was seen in the PCV‐plus‐RT compared with the “RT‐only” arm. Inspection of the two curves shows a distinct separation, and the median survival had not been reached in the CFO, PCV‐plus‐RT treated arm. Since a similar finding was not seen with 1p 19q status (3), this suggests that the combination of classic histopathology with 1p 19q deletion may in the future be predictive for chemotherapeutic response in anaplastic oligodendrogliomas. Analysis of survival data at a later time point, after median survival has been reached in both groups, will be important in confirming or refuting this hypothesis. PFS was significantly longer in CFO treated with PCV‐plus‐RT compared with those treated with RT only and to NCFO irrespective of treatment. The independent significance of classic morphology as a prognostic and likely a predictive factor remained true in multivariate analysis.
From our review, it appears important to consider both classic oligodendroglial morphology and 1p 19q deletion in predicting outcome. In the original report of the RTOG 9402 trial (3) as well as of the EORTC trial 26951 (22), the authors simply emphasized the importance of the combined 1p 19q loss in predicting longer OS and possibly the increased responsiveness to treatment. Both studies in their conclusions underscored the need to design specific future clinical trials separating patients with 1p 19q deletion. Our findings suggest that strictly defined classic histopathology may be an important additional consideration in study design, although it has the limitation to some extent of being assessed subjectively. In the EORTC report, the authors lamented the considerable inter‐observer variation in the histologic diagnosis of oligodendroglial tumors, to which they attribute the low frequency (25%) of samples carrying the combined 1p 19q deletion, the “chemotherapy‐sensitive oligodendroglial tumor subgroup” that their trial was originally aiming to study (22). Interestingly, the authors proceeded to suggest that, provided that oligodendroglial tumors showing combined 1p 19q deletion were isolated by genotyping and addressed separately, “oligodendroglial tumors having no 1p 19q loss had similar prognosis to anaplastic astrocytomas, and until new molecular markers were identified, there was no reason to keep them apart based on rather subjective histologic criteria”. Our results contradict these conclusions and suggest that the 1p 19q intact cases are a heterogeneous group of tumors, likely not uniform clinically or genetically. For example, in our study, CFO without 1p 19q deletion had a more favorable prognosis than NCFO without 1p 19q deletion. The clinical and molecular spectrum of oligodendroglial tumors with intact 1p 19q alleles will be better understood in the coming years than they are today. The EORTC suggestion that the spectrum of “anaplastic gliomas”, at least for the purpose of clinical trials, should now be divided into “anaplastic 1p 19q co‐deleted oligodendrogliomas” and “all anaplastic non‐1p 19q deleted gliomas” may simply represent a pragmatic compromise in 2007. Lastly, it is our impression that, although objective inter‐observer variation in the histologic diagnosis of oligodendroglial tumors may in part have contributed to the “tumor mix” in EORTC trial 26951, it is likely that central pathology review prior to patient enrollment by a neuropathologist, as performed in the RTOG 9402 study, rather than relying on the local hospital diagnosis, would have significantly changed the proportion of enrolled patients. The collective experience of both the RTOG 9402 and EORTC 26951 anaplastic oligodendroglioma trials indicate the importance of central review as one component of patient eligibility to establish uniformity of entry criteria in future trials involving these tumors.
REFERENCES
- 1. Burger PC (2002) What is an oligodendroglioma? Brain Pathol 12:257–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DH, Hammond RR et al (1998) Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 90:1473–1479. [DOI] [PubMed] [Google Scholar]
- 3. Cairncross G, Berkey B, Shaw E, Jenkins R, Scheithauer B, Brachman D et al (2006) Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol 24:2707–2714. [DOI] [PubMed] [Google Scholar]
- 4. Coons SW, Johnson PC, Scheithauer BW, Yates AJ, Pearl DK (1997) Improving diagnostic accuracy and interobserver concordance in the classification and grading of primary gliomas. Cancer 79:1381–1393. [DOI] [PubMed] [Google Scholar]
- 5. Giannini C, Scheithauer BW, Weaver AL, Burger PC, Kros JM, Mork S et al (2001) Oligodendrogliomas: reproducibility and prognostic value of histologic diagnosis and grading. J Neuropathol Exp Neurol 60:248–262. [DOI] [PubMed] [Google Scholar]
- 6. Griffin CA, Burger P, Morsberger L, Yonescu R, Swierczynski S, Weingart JD et al (2006) Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J Neuropathol Exp Neurol 65:988–994. [DOI] [PubMed] [Google Scholar]
- 7. Hashimoto N, Murakami M, Takahashi Y, Fujimoto M, Inazawa J, Mineura K (2003) Correlation between genetic alteration and long‐term clinical outcome of patients with oligodendroglial tumors, with identification of a consistent region of deletion on chromosome arm 1p. Cancer 97:2254–2261. [DOI] [PubMed] [Google Scholar]
- 8. Ino Y, Betensky RA, Zlatescu MC, Sasaki H, MacDonald BR, Stemmer‐Rachamimor AO et al (2001) Molecular subtypes of anaplastic oligodendroglioma: implications for patient management at diagnosis. Clin Cancer Res 7:839–845. [PubMed] [Google Scholar]
- 9. Jenkins RB, Curran W, Scfot CB, Cairncross G (2001) Pilot evaluation of 1p and 19q deletions in anaplastic oligodendrogliomas collected by a national cooperative cancer treatment group. Am J Clin Oncol 24:506–508. [DOI] [PubMed] [Google Scholar]
- 10. Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M et al (2006) A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 66:9852–9861. [DOI] [PubMed] [Google Scholar]
- 11. Jeuken JW, Von Deimling A, Wesseling P (2004) Molecular pathogenesis of oligodendroglial tumors. J Neurooncol 70:161–181. [DOI] [PubMed] [Google Scholar]
- 12. Kros JM, Gorlia T, Kouwenhoven MC, Zheng P, Collins VP, Figarella‐Branger D et al (2007) Panel review of anaplastic oligodendroglioma from EORTC trial 26951: assessment of consensus in diagnosis, influence of 1p/19q loss and correlations with outcome. J Neuropathol Exp Neurol 66:545–551. [DOI] [PubMed] [Google Scholar]
- 13. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (2007) WHO Classification of Tumours of The Central Nervous System, 4th edn. International Agency for Cancer Research: Lyon. [Google Scholar]
- 14. McDonald JM, See SJ, Tremont IW, Colman H, Gilbert MR, Groves M et al (2005) Links the prognostic impact of histology and 1p/19q status in anaplastic oligodendroglial tumors. Cancer 104:1468–1477. [DOI] [PubMed] [Google Scholar]
- 15. Miller CR, Dunham CP, Scheithauer BW, Perry A (2006) Significance of necrosis in grading of oligodendroglial neoplasms: a clinicopathologic and genetic study of newly diagnosed high‐grade gliomas. J Clin Oncol 24:5419–5426. [DOI] [PubMed] [Google Scholar]
- 16. Mueller W, Hartmann C, Hoffmann A, Lanksch W, Kiwi J, Tonn J et al (2002) Genetic signature of oligoastrocytomas correlates with tumor location and denotes distinct molecular subsets. Am J Pathol 161:313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nutt CL, Mani DR, Betensky RA, Tamayo P, Cairncross JG, Ladd C et al (2003) Gene expression‐based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res 63:1602–1607. [PubMed] [Google Scholar]
- 18. Puduvalli VK, Hashmi M, McAllister LD, Levin VA, Hess KR, Prados M et al (2003) Anaplastic oligodendrogliomas: prognostic factors for tumor recurrence and survival. Oncology 65:259–266. [DOI] [PubMed] [Google Scholar]
- 19. Reifenberger G, Louis DN (2003) Oligodendroglioma: toward molecular definitions in diagnostic neuro‐oncology. J Neuropathol Exp Neurol 62:111–126. [DOI] [PubMed] [Google Scholar]
- 20. Smith JS, Tachibana I, Lee HK, Qian J, Pohl U, Mohrenweiser HW et al (2000) Mapping of the chromosome 19 q‐arm glioma tumor suppressor gene using fluorescence in situ hybridization and novel microsatellite markers. Genes Chromosomes Cancer 29:16–25. [DOI] [PubMed] [Google Scholar]
- 21. Thiessen B, Maguire JA, McNeil K, Huntsman D, Martin MA, Horsman D (2003) Loss of heterozygosity for loci on chromosome arms 1p and 10q in oligodendroglial tumors: relationship to outcome and chemosensitivity. J Neurooncol 64:271–278. [DOI] [PubMed] [Google Scholar]
- 22. Van den Bent MJ, Carpentier AF, Brandes AA, Sanson M, Taphoorn MJ, Bernsen HJ et al (2006) Adjuvant procarbazine, lomustine, and vincristine improves progression‐free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol 24:2715–2722. [DOI] [PubMed] [Google Scholar]
- 23. Zlatescu MC, TehraniYazdi A, Sasaki H, Megyesi JF, Betensky RA, Louis DN, Cairncross JG (2001) Tumor location and growth pattern correlate with genetic signature in oligodendroglial neoplasms. Cancer Res 61:6713–6715. [PubMed] [Google Scholar]