

Abstract
The description of neuroglia by Virchow in 1848 may be considered the starting point of our understanding of primary brain tumors. At the beginning of the 20th century, surgical removal of primary brain tumors became possible, and therefore, tissue for microscopic analysis and clinical data on survival became available. During this time, research on gliomas beyond improving surgical procedures focused on their classification. The classification schemes developed emphasized parameters for sorting tumors with regard to (i) cytological aspects; (ii) presumed tumor cell origin; (iii) histological appearance of the tissue; or (iv) clinical outcome. Over the years, experimental studies have greatly improved our knowledge on gliomas. Gliomas induced by viruses, chemicals, radiation, transgenes and knock‐out technology contributed to the understanding of their pathogenesis and still serve as preclinical models for the testing of novel therapies. Recent advances in developmental neurobiology and the identification of stem cells provided new insights into the origin of brain tumors and the molecular mechanisms of tumor formation. This review briefly compiles the evolution of our concepts on gliomas, focusing on the latest developments.
Keywords: glioma, history, review
PREREQUISITES FOR UNDERSTANDING BRAIN TUMOURS
Systematic analysis of gliomas became possible following the availability of tumor tissue. While the first reports on gliomas relied on autopsy studies, it was the pioneering work of Bennett and Godlee and their followers that made surgical material available. Their seminal report (9) initiated the development of modern glioma surgery. However, it was not only surgical skills that needed to be developed. Anesthesia caused by inhalation of nitrous oxide entered dentistry in the middle of the 19th century, and in 1846, Morton and Warren made their famous appearance in the Massachusetts General Hospital when they removed a mandibular tumor from a patient rendered unconscious by ether inhalation. Further advancement in anesthesia allowed intracranial surgery. Another essential precondition for glioma surgery was the development of techniques allowing preoperative tumor localization. Localization efforts in the early years of glioma surgery relied on meticulous analysis of neurologic deficits. The development and adaptation of pneumencephalography by Dandy in 1918 and arteriography by Moniz in 1927 allowed for the first time the preoperative visualization of brain tumors, and the introduction of computed tomography by Cormack and Hounsfield and magnetic resonance imaging by Mansfield and Lauterbur resulted in high resolution images indispensable in today's neurosurgery. Thus, the direct access to tissue was an invaluable factor for developing concepts on glioma. This access and the interest in the postoperative clinical course of the patients placed neurosurgeons in key positions for generating glioma classification schemes.
CLASSIFICATION AND GRADING OF BRAIN TUMORS FROM THE BEGINNING TO THE PRESENT DAY
Early tumor classification had already relied on comparing tumor features with those of normal tissue. This concept was formulated by J. Müller in 1838 (63). This principle was extended to include the developmental stage of the tumor cells and was systematically applied to brain tumors by Bailey and Cushing. Their book, A Classification of the Tumours of the Glioma Group on a Histogenetic Basis with a Correlated Study of Prognosis, was published in 1926 and introduced the concept still at the basis of tumor classification currently in use (5). Their classification relied on histological features observed in glial tumors and comparison with mature glial cells. Brain tumors with cells resembling astrocytes were termed astrocytomas; the tumor cells of ependymoma shared an appearance with ependymal cells; and pinealoma was composed of tumor cells resembling those of the pineal gland. A scheme of this early classification is shown in Figure 1. The names of several tumor entities relate to cell types no longer recognized today. Examples are spongioblastoma to the spongioblast (or by other authors glioblastoma referring to the glioblast) and medulloblastoma to the medulloblast. It is noteworthy that this first comprehensive classification system already outlined the concept of tumors arising from immature precursor cells or, employing a very timely term, from neuroectodermal stem cells.
Figure 1.
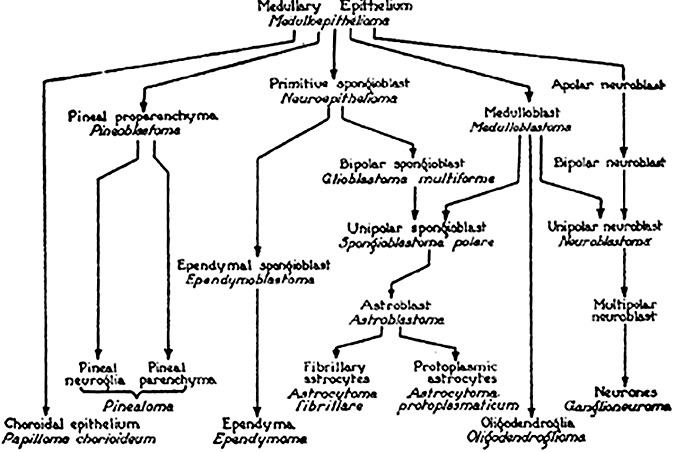
Classification scheme of primary brain tumors by Bailey and Cushing. Tumors were expected to arise from cells with corresponding differentiation. For example, medulloepithelioma would originate from the medullary epithelium or oligodendroglioma would originate from oligodendroglia (adapted from Bailey P (1948) Intracranial Tumors, 2nd ed. Thomas: Springfield, IL).
Several other investigators proposed classification schemes limited to gliomas: Roussy and Oberling in 1931, Penfield in 1932, Bergstrand in 1932, and Rio Hortega beginning in 1932 and extending into the following years. However, none of these classification systems became as widely accepted as that of Bailey and Cushing. The interested reader is referred to K. Zülch giving an extensive overview on the historical development of classification (103).
In 1949, Kernohan et al introduced a simplified classification that recognized four tumor grades for gliomas (45). His approach focused on cytological criteria and gained wide recognition among pathologists. A subsequent proposal by Ringertz in 1950 defining three tumor grades received less attention (76). Currently, the most widely used classification and grading system for brain tumors is that of the World Health Organization (WHO). The WHO classification was first presented in 1979 and is the result of compromises agreed on by a panel of expert neuropathologists led by Zülch (104). The involvement of large numbers of specialists, first by Zülch and later under the tender guidance of Kleihues, may be one of the keys to the success of the WHO grading. The most recent update has just been made available (53). Another approach to the classification of astrocytic brain tumors was proposed by Daumas‐Duport et al in 1988 (19). This approach relies on only four criteria for tumor grading, thus greatly reducing the complexity of the task. More complex is an attempt based on analysis of histological and clinical parameters and computer‐assisted analysis forwarded (TESTAST 268) by Schmitt in 1992 (78). Interestingly, a study on 102 supratentorial astrocytomas reported very similar data on survival regardless whether WHO, Daumas‐Duport, Kernohan or TESTAST 268 classification was used for diagnosis (43). Future development will definitely include molecular parameters. Although the most recent WHO release has not emphasized the importance of molecular markers, it is not unrealistic to expect molecular analysis to become central to classification and grading.
EVOLUTION OF TECHNIQUES FOR GLIOMA CLASSIFICATION AND GRADING
Microscopic evaluation of tumors followed the identification of cells and the subsequent development of tissue‐staining methods. A major breakthrough was the development and application of staining and impregnation techniques by Golgi, Ramon y Cajal, Del Rio‐Hortega and others allowing identification of neural cells in the central nervous system (CNS). An early report already distinguished neuroglia of the CNS from fibers of connective tissue and described the proliferation of neuroglia in pathologic processes (95). Histology became the dominating source for diagnosing and classifying brain tumors and still is the basis of daily routine in neuropathology.
Histochemistry has been applied to research on brain tumors, in particular in the middle of the last century, and generated a large body of data; however, the impact of histochemical analysis on the diagnosis and classification of brain tumors can be neglected. The advent of electron microscopy added a novel dimension to the analysis of gliomas. This technique allowed the investigation of individual cellular compartments and organelles and the identification of specific structures of cell lineage such as synaptic vesicles in neuronal cells or cilia in ependymal cells. Ultrastructural findings are still helpful in today's diagnosis of neoplastic intracranial lesions, for example as with the demonstration of Birbeck bodies in histiocytosis. However, electron microscopy as a tool for providing diagnostic information beyond that obtained by standard histology has been nearly completely replaced by immunohistochemistry. The advent of immunohistochemistry marked a novel era in tumor characterization. Rubinstein was among the first neuropathologists to recognize the potential of antibodies to cell lineage‐specific antigens for tumor classification. This novel concept had to be accompanied by development of cell culture systems in order to obtain the appropriate antigens. Lineage‐specific antibodies led to the reclassification of several tumor types and currently belong to the most powerful tools for glioma classification and for characterization of rare glioma entities. For example, the glial nature of pleomorphic xanthoastrocytoma has been proven by detection of glial fibrillary acidic protein (GFAP) in the tumor cells. Previously, this tumor entity had been assumed to be of meningothelial origin (44). Another tumor entity, recently identified as a glioma by its expression of GFAP, is the chordoid glioma of third ventricle (13). The identification of several newly characterized tumor entities relied strongly on immunohistochemistry, and their inclusion into the newest update of the WHO classification (53) demonstrates the overwhelming impact of this technique for current and future diagnosis of brain tumors. The rosette‐forming glioneuronal tumor of the fourth ventricle with its typical expression of neuronal antigens shown in Figure 2 may serve as an example for tumors of previously uncertain nature only recently included in the WHO classification system. Today, a large panel of antibodies against glial, neuronal, epithelial and mesenchymal antigens represents an essential tool for the classification of brain tumors. The introduction of antibodies targeting epitopes such as Ki67 in proliferating cells established the role of immunohistochemistry in tumor grading.
Figure 2.
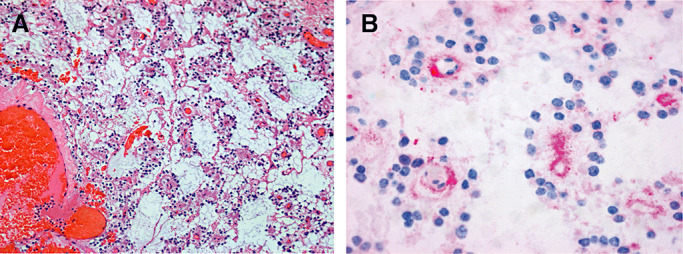
A. Neurocytic rosettes and perivascular pseudorosettes in rosette‐forming glioneuronal tumor of the fourth ventricle; HE × 100. B. Binding of antibodies to synaptophysin (SYN) in the neuropil lining delicate vessels in the perivascular pseudorosettes; SYN × 400.
The development of tools for the analysis of chromosomes and DNA allowed systematic examination of brain tumors for cytogenetic and molecular genetic alterations. Among the first consistent cytogenetic changes in solid tumors, loss of a small chromosome belonging to the G group, later to be identified as chromosome 22, was discovered by Zang and Zinger in meningiomas (98). Analysis of cytogenetic alterations in gliomas has been the topic of many studies and has led to the identification of some consistent patterns such as losses of chromosome 10, the short arm of chromosome 9 and trisomy of chromosome 7 in glioblastoma; however, individual gliomas turned out to be very heterogeneous in their chromosomal configuration and even a fraction of pilocytic astrocytoma WHO grade I already demonstrated unexpectedly altered karyotypes. A very consistent pattern of deletions characterizes the majority of oligodendroglial tumors. A combined loss of one entire copy each of chromosomal arms 1p and 19q is seen in approximately 70% of these tumors. Two recent reports were able to provide an explanation for the combined deletion. It could be shown that a translocation is responsible for this trait so typical of oligodendroglioma (32, 41).
With the advent of PCR technology, it became possible to screen large series of tumors for alterations of their associated genes. Although oncogenes turned out to be affected only in small fractions of brain tumors, several tumor suppressor genes were reported to have high mutational frequencies. Prominent examples are point mutations in the TP53 gene, observed in approximately 50% of diffuse astrocytomas WHO grade II and anaplastic astrocytomas WHO grade III (93), while the PTEN gene is mutated in roughly 25% of glioblastomas WHO grade IV (23). These analyses allowed the identification of molecular subgroups of brain tumors that cannot otherwise be distinguished on morphological grounds alone. In some instances, such molecular examination is of clinical relevance. Patients with oligodendrogliomas exhibiting combined deletions of chromosomal arms 1p and 19q, have a significantly better outcome than patients with oligodendrogliomas not showing this feature (15). Moreover, patients with glioblastoma live longer and respond better to alkylating chemotherapy if their tumors have low O‐6‐methylguanine‐DNA methyltransferase (MGMT) activity caused by the silencing of this gene by promoter methylation (36). So far, the clinical impact of these analyses has been modest, because due to lack of effective alternative therapy, glioblastoma or oligodendroglioma patients without MGMT inactivation also receive chemotherapy with alkylating agents. However, molecular analyses already assist in the diagnosis of tumors of indeterminate differentiation and gliomas with only small samples available.
Following the identification of mutations in individual genes, it became evident that distinct signaling pathways could be activated or inactivated in different positions. A prominent example in glioma is the disruption of cell cycle control by alterations either in RB or upstream in CDKN2A (90). Such observations led to an interest in altered gene expression as an alternative to dysfunction caused by mutations. With the advent of SAGE (serial analysis of gene expression) and microarray technology, it became possible to generate expression maps of brain tumors. A pioneering study highlighted typical expression profiles distinguishing medulloblastomas from other primitive neuroectodermal tumors and from atypical teratoid rhabdoid tumors (69). Distinct glioma types also exhibit characteristic expression profiles, and recent research focuses on identification of qualifiers of diagnostic, prognostic or therapeutic value. For example, class distinctions based on the expression profile in a series of malignant non‐classic glioma proved superior to histological evaluation in predicting clinical course (64). Gene expression profiles also can separate genetic subsets of gliomas indistinguishable on morphological grounds, such as primary and secondary glioblastoma (54, 88). The combination of expression arrays with screening technology allowing determination of the genomic status such as matrix‐/array‐CGH, or with screening technology allowing assessment of methylation status of promoters will provide more insight into mechanisms underlying expression. Much emphasis will be given to the fact that tumors of comparable histology may exhibit entirely different genomic and signaling pathway alterations.
BRAIN TUMOR MODELS
Cell lines developed from sporadic mouse glioma such as the spontaneous murine astrocytoma (VM/Dk background) and GL261 (C57/BL6 background) cell lines (33, 79) have been very helpful for the understanding of glioma. However, sporadic brain tumors in laboratory animals remain a rare event and additional animal models have to be devised for the induction of brain tumors, mostly in rats or mice.
Chemical carcinogenesis
Experimental brain tumors became widely available with the systematic administration of alkylating agents to laboratory animals. One of the most successful systems relied on a transplacental model. Female rats were inoculated with a single dose of ethylnitrosourea (ENU) at day 15 of pregnancy. Although this treatment had little effect on the adult animals, all offspring exhibited teratogenic or carcinogenic effects of ENU. Aside from congenital malformations, a significant number of offspring developed gliomas or nerve sheath tumors, the latter most frequently involving the trigeminal nerve (22). In subsequent studies of ENU‐induced gliomas, the predominant mutations in the c‐neu gene resulted in a substitution from valine to glutamic acid in position 664 contained in the transmembrane region (7).
Viral carcinogenesis
In parallel with the development of chemically induced rodent tumors, the induction of brain tumors by virus proved successful in several models. JC virus induced tumors resembling medulloblastoma in Syrian hamsters (102) and adenovirus 12 did so in mice; Simian virus 40 (SV40)‐induced ependymomas in hamsters (30); and avian sarcoma virus B‐77 caused astrocytic tumors in rats (17). Many studies have detected viruses in human glioma. Measles virus (26), SV40 (84) or JC virus (75) have been suggested as potential glioma‐inducing agents. However, in contrast to other tumor entities, such as cervical cancer or nasopharyngeal carcinoma, the issue that viruses cause human glioma remains controversial, with many conflicting reports being published. It should be kept in mind that early polio vaccines were contaminated with SV40, and a Swedish study estimates that at least 700 000 patients in Sweden were inoculated with these preparations. However, long‐term follow‐up did not find increased glioma incidence in patients exposed to SV40‐contaminated preparations (65).
Transplantation models with genetically engineered vectors
Several methods have been used to induce experimental brain tumors by transplantation of cells containing genetically engineered vectors. The transfection and expression of diverse activated oncogenes in fetal brain tissue transplanted into adult rats resulted in endothelial hemangiomas with polyoma middle T or astrocytic and mesenchymal tumors with v‐src (1). One of the lessons learned from these experiments was the synergistic effect of activated oncogenes in these tumors. Although injection of fetal rat cells transfected with either Ha‐ras or v‐myc into syngeneic adult recipients yielded tumors in at best half of the transplanted animals, the vast majority of animals receiving both oncogenes developed tumors (96). However, morphological features of these xenografts are not always similar to those of human tumors, and the genetic alterations inducing these experimental tumors are often not the same as those in the human counterparts.
Therefore, much attention has been paid to generating tumor mouse models by engineering neural cells with those mutated genes or oncogenes shown to be of importance in human glioma. TP53 is among the most frequently mutated genes in diffuse human astrocytoma (93), and neurofibromatosis type 1 (NF1) patients frequently develop astrocytic tumors; most frequently, they develop pilocytic astrocytoma WHO grade I rather than anaplastic astrocytoma or glioblastoma. Mice with germline mutations in TP53 and NF1 were shown to consistently develop malignant astrocytoma within 6 months of age (74). In another example, platelet‐derived growth factor (PDGF) overexpression induced oligodendroglioma upon targeting nestin‐expressing cells, and induced oligodendrogliomas or oligoastrocytomas if GFAP‐expressing cells were targeted (18). Additional inactivation of InK4a‐Arf in these mice resulted in more aggressive gliomas and earlier time of onset. In contrast to the early transplantation models, these refined models recapitulate molecular steps of human glioma formation, and therefore, can serve as models for experimental therapy.
Transplantation of gliomas in human recipients has been studied by several authors. Subcutaneous injection of autologous glioblastoma yielded viable grafts in approximately half of the patients. One lesson learned from these quite problematic experiments was that transplants tended to change morphology, with sarcomatous portions becoming the predominant tumor component (57). Of more current importance is the risk of glioblastoma being transmitted by transplantation of organs. Although metastasis of malignant glioma to visceral organs is exceedingly rare, recipients of organs from donors with malignant gliomas have developed this malignancy in the transplant (42).
CONCEPTS ON THE ORIGIN OF GLIOMA
The current predominant hypothesis for the origin of cancer is the somatic mutation theory. This concept is based on the assumption that cancer arises from a single cell that has acquired cell cycle‐relevant disturbances mostly in the form of somatic mutations. This theory dates back to Boveri who already had the vision of chromosomes promoting proliferation and mediating inhibition in tumor cells (12). Contrasting with this is another concept termed the field theory (97) that emphasizes the interaction of neighboring cells. This theory is based on the assumption that tumors not only grow by cellular proliferation but also by neoplastic conversion of preformed fields. The field theory has been applied to gliomas (91) without becoming a central dogma to explain the origin of these tumors.
The overwhelming body of data on carcinogenesis was gathered following the concept of individual cells being the founders of gliomas. The experimental designs of studies following this theory had the conceptual advantage of reducing the complexity of the question by addressing single cells, single chromosomes and single genes. However, even this reductionistic model is ever gaining complexity because of the identification of novel tumor‐relevant mechanisms such as epigenetic regulation of gene products by promoter methylation status, by histone acetylation status or by regulatory micro RNAs.
One question of intense current research addresses the nature of the cells giving rise to gliomas. It has been a matter of debate for a long time whether gliomas arise from differentiated glial cells or originate from less differentiated or immature and possibly multipotent precursor or stem cells. Experimental murine models were interpreted quite differently, ranging from the notion that these experimental tumors could be traced to mature cells based on histologic resemblance of tumor with differentiated cells in the CNS, to the idea that they arise preferentially from immature cells supported by the enhanced efficiency of transplacental induction of malignant gliomas in the fetal stage (22). The latter model clearly shows the vulnerability of immature fetal cells for neoplastic transformation. The potential of fetal cells to generate tumors was later shown in transfection and transplantation experiments. Viral vectors carrying oncogenes were used to transform cells from fetal rat brain in vitro, followed by transplantation into syngeneic adult animals. Depending on the constructs used, tumors of different morphology and differentiation have been induced (96). Recently, the stem cell concept has gained much attention. Intracerebral xenografts of as few as 100 CD133‐positive glioma (stem) cells into NOD‐SCID mice successfully initiated tumor growth, while as many as up to 105 CD133‐negative glioma cells failed to form a tumor (83).
The tumor stem cell concept
The recognition that within the tumor there is a cell population that shares common features with their normal stem cell counterparts has opened a new window to look at neoplasia. This cancer stem cell‐like population, also known as tumor‐initiating cells, seems to represent the most therapy‐resistant cell population within a tumor and has therefore become a major focus in devising future therapeutic strategies.
The term “stem cell” was first applied by the evolutionary biologist Ernst Haeckel (35) to unicellular organisms, as putative phylogenetic origins of multicellular organisms. Nowadays, the term “stem cell” indicates the unlimited capacity to self‐renew and differentiate. These two features were first detected in cells of the hematopoietic system in the late 19th century, establishing them as the prototypical stem cell (71). Meanwhile, stem cells have been detected in many adult tissues including the intestine, the skin and also the CNS. The detection of cells capable of generating neurons contradicts the dogma of the postmitotic stage of adult brain dating back to Ramon y Cajal, who stated that “in adult centres the nerve paths are something fixed, ended immutable. Everything may die, nothing may be regenerated” (72). It was Joseph Altman who in 1965 demonstrated the existence of ongoing cell division in the hippocampus and olfactory bulb of the rat (2). Since then, multiple studies have repeatedly shown the existence of continuing neurogenesis in the subventricular zone (SVZ) and the dentate gyrus of the hippocampus (DG). Stem cells in these regions can self‐renew, proliferate and differentiate into neuronal or glial lineage. Thus, they undergo asymmetric divisions that generate other stem cells or more committed neural progenitor cells.
Stem cells with the potential to self‐renew are currently identified by the expression of CD133 or nestin, while more committed progenitor cells express differentiation markers such as NeuN for the neuronal or GFAP for the astrocytic lineage. However, GFAP is also expressed by slowly proliferating stem cells, so‐called type B cells. These cells are radial glia acting as precursor intermediates between immature neuroepithelial cells and differentiating neuronal progeny in the SVZ (3, 61). Adult neural stem cells (NSCs) in the DG also retain radial glial properties (reviewed in 10, 21, 27, 56, 68, 87).
Another important aspect of stem cell biology is the so‐called stem cell niche. This niche is a specialized microenvironment harboring heterologous cell types that tightly regulate stem cell renewal, proliferation and differentiation, thus preventing depletion or over‐proliferation of the stem cell pool. In the neurogenic niche, endothelial cells have been identified as regulators of proliferation and neurogenesis (81). Soluble mediators of cellular responses mainly arising from neighboring astrocytes such as wingless‐related proteins (WNTs), their antagonists, soluble Notch modulators, FGFs and Hedgehog (HH) also influence the ability of stem cells to proliferate and differentiate and may even affect the architecture of the niche (77). In the SVZ, the extracellular matrix protein tenascin C increases stem cell propensity to generate glial progenitors (29). The cytoarchitecture of the niche influences the production of metabolites, such as reactive oxygen species (ROS). ROS seems to be a means to intricately regulate cellular functions, linking cell density to proliferation status (51). On the other hand, NSCs are highly sensitive when it comes to elevated levels of ROS accompanying irradiation injury—both in vivo and in vitro studies have shown that irradiation depletes NSCs by inducing elevated ROS levels in these cells (50). It has been extensively demonstrated that injury to the CNS which involves increased production of ROS, expansion of endothelial cells, and recruitment and activation of astrocytes and microglia to the lesion site induces the expansion of NSCs (24, 60, 99).
Cancer neural stem cells: a cell type and a concept
The first evidence for the existence of cancer stem cells was found in acute myeloid leukemia (AML). Bonnet and Dick identified a subset of cells capable of giving rise to a phenocopy of the original AML in immunodeficient mice (11). The ability to reproduce the original tumor pathology by xenotransplantation followed by serial transplantation is widely accepted as the criterion to define cancer stem cells. In CNS tumors, neural cancer stem cells (NCSCs) have been found to be enriched in the population expressing the marker CD133 (89), or in the population able to efflux the Hoechst 33342 dye, the “side population” (SP) (46). The latter property is based on the overactivation of ATP‐binding cassette (ABC) transporter proteins (31, 100). By this means, NCSCs have been identified in glioblastoma multiforme GBM, medulloblastoma and ependymoma (37, 40, 82, 86). Later, NCSCs were also isolated from GBM by applying the same conditions used to isolate normal NSCs at a frequency comparable to that of cultures of initially sorted CD133‐positive cells from the tumor (28).
NCSCs share common signaling pathways for self‐renewal, proliferation and differentiation with normal NSCs. Human CD133 GBM cells express bone morphogenetic proteins (BMPs) and BMP‐receptors, which normally induce astrocytic differentiation of NSCs (67). Also, Notch signaling pathway induces proliferation of both NSCs and NCSCs (25, 39, 70, 81). The polycomb transcription factor Bmi‐1, acting as a regulator of chromatin remodeling, regulate self renewal of NSCs and tumor cells (14, 58). Bmi‐1 induces self‐renewal by repressing the tumor suppressors INK4a and ARF. Expression of Bmi‐1 is highly increased in medulloblastomas (48, 59). Sonic hedgehog is required for proliferation of granule cerebellar precursors, and activating mutations in this pathway lead to development of cerebellar medulloblastomas (reviewed in 66).
Some of the pathways important for NSC biology are deregulated in GBM. The tumor suppressor PTEN controls proliferation and migration of NSCs and is often inactivated in glioblastomas (23, 34). WNT signals that regulate neurogenesis of NSCs are involved in the pathogenesis of medulloblastoma (49, 55). Also, the machinery involved in the control of asymmetric divisions in normal NSCs is frequently found to be deregulated in cancer. Accordingly, Numb, a regulator of asymmetric divisions and Notch signaling acting as an anti‐proliferative and pro‐differentiation signal, is down‐regulated in cerebellar progenitors and their malignant derivative, medulloblastoma (16, 20, 92, 94). Also, expression of atypical protein kinase C dramatically increases the number of divisions of neuroblasts in Drosophila and has been identified as an oncogene in lung cancer (47, 73). In addition, symmetric divisions provide the perfect setting for the appearance of aneuploidy and other secondary mutations by relaxing the control on mitotic spindles (62). Thus, asymmetric divisions of NSCs might be a mechanism to prevent tumourigenic degeneration. Injury to the CNS, such as stroke, increases the number of symmetric division of NSCs (99). Thus, though not yet documented, injury to the CNS might increase the risk of tumor formation in the CNS of genetically predisposed individuals.
Neural stem cells as the tumour's “cell of origin”
One of the central questions in cancer stem cell biology is whether CNS tumors arise by aberrant proliferation of normal NSCs. This notion is supported by the fact that the location within the CNS of ependymoma and pilocytic astrocytomas determines their transcriptional signatures, which in turn share a high similarity with the radial glia signature from the respective location (80, 86). Along this line, the cell of origin of tumors with inactivated p53 and activated Ras locates to the stem cell niche of the SVZ. Mut1 mice are engineered to lack p53 in the germline and neurofibromin in GFAP‐expressing cells. All Mut1 mice developed astrocytomas ranging from low‐grade to GBM, and the early presymptomatic lesions were detected within the SVZ (101). This suggests that GFAP‐positive cells within the SVZ are more prone to develop astrocytomas induced by deficiency of p53/neurofibromin or more predisposed towards tumorigenesis by the microenvironment. Likewise, PDGF in nestin‐positive neural progenitors or GFAP‐positive cells were predisposed to glioma formation, with the highest incidence shown by targeting nestin‐positive cells (18). However, other glioma‐associated genetic lesions such as deregulation of EGFR signaling and INK4a/ARF deficiency could induce GBM formation either from adult astrocytes or NSCs (4). Also, microarray and animal models suggest that the cell of origin of most medulloblastoma is the external granule cell, a self‐renewing but not multipotent cell type (69). Thus, several studies on astrocytomas initiation still support the model of cellular dedifferentiation and transformation (28, 83). Altogether, available data suggest that NSCs can be the source of many CNS tumors, and that most malignant tumors, independent of their source of origin, do contain putative NCSCs.
Clinical implications
Actual treatment strategies of brain tumors encompass surgical resection followed by adjuvant chemo‐ and radiotherapy. New imaging techniques and better protocols for adjuvant therapy have slightly increased patient's life expectancy. However, resistance to apoptosis and the highly invasive behavior of those tumor cells that escape surgery lead to the formation of secondary tumors. NCSCs are highly resistant to the effects of chemotherapeutic drugs (38, 52). This resistance can be explained in part by a higher expression of the multidrug resistance gene BCRP1, DNA repair genes such as MGMT and/or genes that inhibit apoptosis. Treatment with the chemotherapeutic drug temozolomide prolongs the life of patients with GBM; however, this treatment can even increase the number of CD133‐positive cells in primary GBM (85), thereby promoting the expansion of cells resistant to treatment in the long run. NCSCs are also the most radiation‐resistant cell population within GBM. These cells can be rendered less resistant to radiation by inhibiting the Chk1 and Chk2 kinases that control DNA repair mechanisms (6). Thus, it may be worthwhile considering modifications of current therapy protocols for brain tumors in order to control the undesirable effects on NCSCs.
The appreciation that these therapy‐resistant cells might be isolated from the tumor mass and used as targets to develop new therapies might open new and unexpected therapeutic possibilities. To this end, treatment with bone morphogenetic proteins of CD133‐positive cells may induce their differentiation and loss of tumor‐forming capacity (67). Pharmacologic inhibitors of Notch specifically depleted the NCSC population in medulloblastomas and prevented tumor formation in xenotransplantation experiments (25). However, NCSCs do also exist within a niche, often coined as the neo stem cell‐niche. A deregulated niche might contribute to the formation of brain tumors. Therefore, the idea of targeting the neo‐niche might prove to be advantageous to patients. A further potential application of stem cell features for therapy was reported by Benedetti and colleagues. Transplanted neural stem cells have been shown to migrate towards the brain tumor, and thus, may serve as carriers of factors that promote the tumor's regression. In this regard, NSCs genetically engineered to express interleukin‐4 reduced the tumor's growth and increased the survival of tumor‐bearing mice (8).
CLOSING REMARKS
The focus of this review on the most recent insights into glioma biology reflects the excitement about novel concepts that hopefully will allow us to further understand the origin of these tumors. However, a retrospective view on brain tumor research spanning an entire century teaches that most of our initial interpretations need to be put into perspective. Diagnostic uncertainties were thought to be solved with the advent of electron microscopy and again with the introduction of immunohistochemistry. The success in induction of experimental tumors by carcinogens was expected to identify environmental hazards of relevance for the genesis of brain tumors. Radiation and chemotherapy was seen as the tools to conquer cancer. Current enthusiasm is focusing on molecular methods expected to revolutionize our concepts of cancer and diagnostic and therapeutic approaches. We are detecting chromosomal representation and gene transcription in individual gliomas, and next, we will be able to compare the proteome of these tumors. Therapies aimed at attacking the tumor cells or at modifying the immune response by transfer of recombinant are currently being tested. Indeed, most of the novel concepts developed and most of the advances in laboratory techniques have enriched our knowledge on gliomas. However, the still gloomy fate of patients diagnosed with malignant glioma today reminds us that we are far away from having satisfactory solutions. The recognition of multiple and very different parameters affecting tumor biology is quite likely to result in adaptation of molecular screening procedures and incorporation in diagnostic routine and in therapies tailored to individual patients. We will work further to understand the genesis of brain tumors and to refine the tools for diagnosing and treating gliomas.
ACKNOWLEDGMENTS
We thank Dr. F. Scaravilli for helpful suggestions and for critically reading the manuscript.
REFERENCES
- 1. Aguzzi A, Kleihues P, Heckl K, Wiestler OD (1991) Cell type‐specific tumour induction in neural transplants by retrovirus‐mediated oncogene transfer. Oncogene 6:113–118. [PubMed] [Google Scholar]
- 2. Altman J, Das GD (1965) Post‐natal origin of microneurones in the rat brain. Nature 207:953–956. [DOI] [PubMed] [Google Scholar]
- 3. Alvarez‐Buylla A, Garcia‐Verdugo JM, Tramontin AD (2001) A unified hypothesis on the lineage of neural stem cells. Nat Rev 2:287–293. [DOI] [PubMed] [Google Scholar]
- 4. Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ et al (2002) Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 1:269–277. [DOI] [PubMed] [Google Scholar]
- 5. Bailey P, Cushing H (1926) A Classification of The Tumors of The Glioma Group on a Histogenetic Basis with a Correlated Study of Prognosis. Lippincot JB: Philadelphia. [Google Scholar]
- 6. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB et al (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444:756–760. [DOI] [PubMed] [Google Scholar]
- 7. Bargmann CI, Hung MC, Weinberg RA (1986) Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell 45:649–657. [DOI] [PubMed] [Google Scholar]
- 8. Benedetti S, Pirola B, Pollo B, Magrassi L, Bruzzone MG, Rigamonti D et al (2000) Gene therapy of experimental brain tumors using neural progenitor cells. Nat Med 6:447–450. [DOI] [PubMed] [Google Scholar]
- 9. Bennett H, Godlee R (1884) Excision of a tumour from the brain. Lancet 2:1090–1091. [Google Scholar]
- 10. Bonfanti L, Peretto P (2007) Radial glial origin of the adult neural stem cells in the subventricular zone. Prog Neurobiol 83:24–36. [DOI] [PubMed] [Google Scholar]
- 11. Bonnet D, Dick JE (1997) Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3:730–737. [DOI] [PubMed] [Google Scholar]
- 12. Boveri T (1914) Zur Frage der Entstehung maligner Tumoren. Gustav Fisher: Jena. [Google Scholar]
- 13. Brat DJ, Scheithauer BW, Staugaitis SM, Cortez SC, Brecher K, Burger PC (1998) Third ventricular chordoid glioma: a distinct clinicopathologic entity. J Neuropathol Exp Neurol 57:283–290. [DOI] [PubMed] [Google Scholar]
- 14. Bruggeman SW, Valk‐Lingbeek ME, Van Der Stoop PP, Jacobs JJ, Kieboom K, Tanger E et al (2005) Ink4a and Arf differentially affect cell proliferation and neural stem cell self‐renewal in Bmi1‐deficient mice. Genes Dev 19:1438–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR et al (1998) Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 90:1473–1479. [DOI] [PubMed] [Google Scholar]
- 16. Cayouette M, Raff M (2002) Asymmetric segregation of Numb: a mechanism for neural specification from Drosophila to mammals. Nat Neurosci 5:1265–1269. [DOI] [PubMed] [Google Scholar]
- 17. Copeland DD, Talley FA, Bigner DD (1976) The fine structure of intracranial neoplasms induced by the inoculation of avian sarcoma virus in neonatal and adult rats. Am J Pathol 83:149–176. [PMC free article] [PubMed] [Google Scholar]
- 18. Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC (2001) PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev 15:1913–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Daumas‐Duport C, Scheithauer B, O'Fallon J, Kelly P (1988) Grading of astrocytomas. A simple and reproducible method. Cancer 62:2152–2165. [DOI] [PubMed] [Google Scholar]
- 20. Di Marcotullio L, Ferretti E, Greco A, De Smaele E, Po A, Sico MA et al (2006) Numb is a suppressor of Hedgehog signalling and targets Gli1 for Itch‐dependent ubiquitination. Nat Cell Biol 8:1415–1423. [DOI] [PubMed] [Google Scholar]
- 21. Doetsch F, Caille I, Lim DA, Garcia‐Verdugo JM, Alvarez‐Buylla A (1999) Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97:703–716. [DOI] [PubMed] [Google Scholar]
- 22. Druckrey H, Ivankovic S, Preussmann R (1966) Teratogenic and carcinogenic effects in the offspring after single injection of ethylnitrosourea to pregnant rats. Nature 210:1378–1379. [DOI] [PubMed] [Google Scholar]
- 23. Duerr EM, Rollbrocker B, Hayashi Y, Peters N, Meyer‐Puttlitz B, Louis DN et al (1998) PTEN mutations in gliomas and glioneuronal tumors. Oncogene 16:2259–2264. [DOI] [PubMed] [Google Scholar]
- 24. Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O (2003) Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci USA 100:13632–13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li YM, Eberhart CG (2006) Notch pathway inhibition depletes stem‐like cells and blocks engraftment in embryonal brain tumors. Cancer Res 66:7445–7452. [DOI] [PubMed] [Google Scholar]
- 26. Fleury H, Pasquier PD (1977) Replication of measles virus in a cell culture from a glioblastoma of human origin. J Neuropathol Exp Neurol 36:842–845. [DOI] [PubMed] [Google Scholar]
- 27. Gage FH (2002) Neurogenesis in the adult brain. J Neurosci 22:612–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S et al (2004) Isolation and characterization of tumorigenic, stem‐like neural precursors from human glioblastoma. Cancer Res 64:7011–7021. [DOI] [PubMed] [Google Scholar]
- 29. Garcion E, Halilagic A, Faissner A, Ffrench‐Constant C (2004) Generation of an environmental niche for neural stem cell development by the extracellular matrix molecule tenascin C. Development 131:3423–3432. [DOI] [PubMed] [Google Scholar]
- 30. Gerber P, Kirschstein RL (1962) SV40‐induced ependymomas in newborn hamsters. I. Virus‐tumour relationships. Virology 18:582–588. [DOI] [PubMed] [Google Scholar]
- 31. Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC (1996) Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med 183:1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Griffin CA, Burger P, Morsberger L, Yonescu R, Swierczynski S, Weingart JD, Murphy KM (2006) Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J Neuropathol Exp Neurol 65:988–994. [DOI] [PubMed] [Google Scholar]
- 33. Grosser BI (1966) 11‐beta‐Hydroxysteroid metabolism by mouse brain and glioma 261. J Neurochem 13:475–478. [DOI] [PubMed] [Google Scholar]
- 34. Groszer M, Erickson R, Scripture‐Adams DD, Lesche R, Trumpp A, Zack JA et al (2001) Negative regulation of neural stem/progenitor cell proliferation by the Pten tumour suppressor gene in vivo. Science 294:2186–2189. [DOI] [PubMed] [Google Scholar]
- 35. Haeckel E (1874) Anthropogenie, 1st edn. Engelmann Wilhelm: Leipzig. [Google Scholar]
- 36. Hegi ME, Diserens AC, Gorlia T, Hamou MF, De Tribolet N, Weller M et al (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003. [DOI] [PubMed] [Google Scholar]
- 37. Hemmati HD, Nakano I, Lazareff JA, Masterman‐Smith M, Geschwind DH, Bronner‐Fraser M, Kornblum HI (2003) Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA 100:15178–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hirschmann‐Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U et al (2004) A distinct “side population” of cells with high drug efflux capacity in human tumour cells. Proc Natl Acad Sci USA 101:14228–14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hitoshi S, Alexson T, Tropepe V, Donoviel D, Elia AJ, Nye JS et al (2002) Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev 16:846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA (2002) Human cortical glial tumors contain neural stem‐like cells expressing astroglial and neuronal markers in vitro. Glia 39:193–206. [DOI] [PubMed] [Google Scholar]
- 41. Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M et al (2006) A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 66:9852–9861. [DOI] [PubMed] [Google Scholar]
- 42. Jonas S, Bechstein WO, Lemmens HP, Neuhaus R, Thalmann U, Neuhaus P (1996) Liver graft‐transmitted glioblastoma multiforme. A case report and experience with 13 multiorgan donors suffering from primary cerebral neoplasia. Transpl Int 9:426–429. [DOI] [PubMed] [Google Scholar]
- 43. Karak AK, Singh R, Tandon PN, Sarkar C (2000) A comparative survival evaluation and assessment of interclassification concordance in adult supratentorial astrocytic tumors. Pathol Oncol Res 6:46–52. [DOI] [PubMed] [Google Scholar]
- 44. Kepes JJ, Rubinstein LJ, Eng LF (1979) Pleomorphic xanthoastrocytoma: a distinctive meningocerebral glioma of young subjects with relatively favorable prognosis. A study of 12 cases. Cancer 44:1839–1852. [DOI] [PubMed] [Google Scholar]
- 45. Kernohan JW, Mabon RF, Svien HJ, Adson AW (1949) A simplified classification of gliomas. Proc Staff Meet Mayo Clin 24:71–75. [PubMed] [Google Scholar]
- 46. Kim M, Morshead CM (2003) Distinct populations of forebrain neural stem and progenitor cells can be isolated using side‐population analysis. J Neurosci 23:10703–10709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee CY, Robinson KJ, Doe CQ (2006) Lgl, Pins and aPKC regulate neuroblast self‐renewal versus differentiation. Nature 439:594–598. [DOI] [PubMed] [Google Scholar]
- 48. Leung C, Lingbeek M, Shakhova O, Liu J, Tanger E, Saremaslani P et al (2004) Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature 428:337–341. [DOI] [PubMed] [Google Scholar]
- 49. Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio A et al (2005) Wnt signalling regulates adult hippocampal neurogenesis. Nature 437:1370–1375. [DOI] [PubMed] [Google Scholar]
- 50. Limoli CL, Giedzinski E, Rola R, Otsuka S, Palmer TD, Fike JR (2004) Radiation response of neural precursor cells: linking cellular sensitivity to cell cycle checkpoints, apoptosis and oxidative stress. Radiat Res 161:17–27. [DOI] [PubMed] [Google Scholar]
- 51. Limoli CL, Rola R, Giedzinski E, Mantha S, Huang TT, Fike JR (2004) Cell‐density‐dependent regulation of neural precursor cell function. Proc Natl Acad Sci USA 101:16052–16057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR et al (2006) Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer 5:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Louis D, Ohgaki H, Wiestler O, Cavenee W (2007) World health organization classification of tumours of the central nervous system. In: World Health Organization Classification of Tumours. Bosman F, Jaffe E, Lakhani S, Ohgaki H (eds). IARC: Lyon. [Google Scholar]
- 54. Maher EA, Brennan C, Wen PY, Durso L, Ligon KL, Richardson A et al (2006) Marked genomic differences characterize primary and secondary glioblastoma subtypes and identify two distinct molecular and clinical secondary glioblastoma entities. Cancer Res 66:11502–11513. [DOI] [PubMed] [Google Scholar]
- 55. Marino S (2005) Medulloblastoma: developmental mechanisms out of control. Trends Mol Med 11: 17–22. [DOI] [PubMed] [Google Scholar]
- 56. Merkle FT, Tramontin AD, Garcia‐Verdugo JM, Alvarez‐Buylla A (2004) Radial glia give rise to adult neural stem cells in the subventricular zone. Proc Natl Acad Sci USA 101:17528–17532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mitts MG, Walker AE (1964) Autotransplantation of gliomas. J Neuropathol Exp Neurol 23:324–333. [DOI] [PubMed] [Google Scholar]
- 58. Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ (2003) Bmi‐1 dependence distinguishes neural stem cell self‐renewal from progenitor proliferation. Nature 425:962–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J et al (2006) Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443:448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Monje ML, Mizumatsu S, Fike JR, Palmer TD (2002) Irradiation induces neural precursor‐cell dysfunction. Nat Med 8:955–962. [DOI] [PubMed] [Google Scholar]
- 61. Mori T, Buffo A, Gotz M (2005) The novel roles of glial cells revisited: the contribution of radial glia and astrocytes to neurogenesis. Curr Top Dev Biol 69:67–99. [DOI] [PubMed] [Google Scholar]
- 62. Morrison SJ, Kimble J (2006) Asymmetric and symmetric stem‐cell divisions in development and cancer. Nature 441:1068–1074. [DOI] [PubMed] [Google Scholar]
- 63. Müller J (1838) Über den feineren Bau und die Formen der Krankhaften Geschwülste. Reimer: Berlin. [Google Scholar]
- 64. Nutt CL, Mani D, Betensky R, Tamayo P, Cairncross J, Ladd C et al (2003) Gene expression‐based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res 63:1602–1607. [PubMed] [Google Scholar]
- 65. Olin P, Giesecke J (1998) Potential exposure to SV40 in polio vaccines used in Sweden during 1957: no impact on cancer incidence rates 1960 to 1993. Dev Biol Stand 94:227–233. [PubMed] [Google Scholar]
- 66. Oliver TG, Wechsler‐Reya RJ (2004) Getting at the root and stem of brain tumors. Neuron 42:885–888. [DOI] [PubMed] [Google Scholar]
- 67. Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G et al (2006) Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour‐initiating cells. Nature 444:761–765. [DOI] [PubMed] [Google Scholar]
- 68. Pinto L, Gotz M (2007) Radial glial cell heterogeneity—the source of diverse progeny in the CNS. Prog Neurobiol 83:2–23. [DOI] [PubMed] [Google Scholar]
- 69. Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME et al (2002) Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 415:436–442. [DOI] [PubMed] [Google Scholar]
- 70. Purow BW, Haque RM, Noel MW, Su Q, Burdick MJ, Lee J et al (2005) Expression of Notch‐1 and its ligands, Delta‐like‐1 and Jagged‐1, is critical for glioma cell survival and proliferation. Cancer Res 65:2353–2363. [DOI] [PubMed] [Google Scholar]
- 71. Ramalho‐Santos M, Willenbring H (2007) On the origin of the term “Stem Cell. Cell Stem Cell 1:35–38. [DOI] [PubMed] [Google Scholar]
- 72. Ramon y Cajal S (1928) Degeneration & Regeneration of the Nervous System. University Press: Oxford. [Google Scholar]
- 73. Regala RP, Weems C, Jamieson L, Khoor A, Edell ES, Lohse CM, Fields AP (2005) Atypical protein kinase C iota is an oncogene in human non‐small cell lung cancer. Cancer Res 65:8905–8911. [DOI] [PubMed] [Google Scholar]
- 74. Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T (2000) Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain‐specific effects. Nat Genet 26:109–113. [DOI] [PubMed] [Google Scholar]
- 75. Rencic A, Gordon J, Otte J, Curtis M, Kovatich A, Zoltick P et al (1996) Detection of JC virus DNA sequence and expression of the viral oncoprotein, tumour antigen, in brain of immunocompetent patient with oligoastrocytoma. Proc Natl Acad Sci USA 93:7352–7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ringertz N (1950) Grading of gliomas. Acta Pathol Microbiol Scand 27:51–64. [PubMed] [Google Scholar]
- 77. Scadden DT (2006) The stem‐cell niche as an entity of action. Nature 441:1075–1079. [DOI] [PubMed] [Google Scholar]
- 78. Schmitt HP, Oberwittler C (1992) Computer‐aided classification of malignancy in astrocytomas. II. The value of categorically evaluated histologic and non‐histologic features for a numerical classifier. Anal Cell Pathol 4:409–419. [PubMed] [Google Scholar]
- 79. Serano RD, Pegram CN, Bigner DD (1980) Tumorigenic cell culture lines from a spontaneous VM/Dk murine astrocytoma (SMA). Acta Neuropathol (Berl) 51:53–64. [DOI] [PubMed] [Google Scholar]
- 80. Sharma MK, Mansur DB, Reifenberger G, Perry A, Leonard JR, Aldape KD et al (2007) Distinct genetic signatures among pilocytic astrocytomas relate to their brain region origin. Cancer Res 67:890–900. [DOI] [PubMed] [Google Scholar]
- 81. Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N et al (2004) Endothelial cells stimulate self‐renewal and expand neurogenesis of neural stem cells. Science 304:1338–1340. [DOI] [PubMed] [Google Scholar]
- 82. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB (2003) Identification of a cancer stem cell in human brain tumors. Cancer Res 63:5821–5828. [PubMed] [Google Scholar]
- 83. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T et al (2004) Identification of human brain tumour initiating cells. Nature 432:396–401. [DOI] [PubMed] [Google Scholar]
- 84. Tabuchi K, Kirsch WM, Van Buskirk JJ (1978) Immunocytochemical evidence of SV 40‐related T antigen in two human brain tumours of ependymal origin. Acta Neurochir (Wien) 43:239–249. [DOI] [PubMed] [Google Scholar]
- 85. Tang C, Chua CL, Ang BT (2007) Insights into the cancer stem cell model of glioma tumorigenesis. Ann Acad Med Singapore 36:352–356. [PubMed] [Google Scholar]
- 86. Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P et al (2005) Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 8:323–335. [DOI] [PubMed] [Google Scholar]
- 87. Tramontin AD, Garcia‐Verdugo JM, Lim DA, Alvarez‐Buylla A (2003) Postnatal development of radial glia and the ventricular zone (VZ): a continuum of the neural stem cell compartment. Cereb Cortex 13:580–587. [DOI] [PubMed] [Google Scholar]
- 88. Tso CL, Freije WA, Day A, Chen Z, Merriman B, Perlina A et al (2006) Distinct transcription profiles of primary and secondary glioblastoma subgroups. Cancer Res 66:159–167. [DOI] [PubMed] [Google Scholar]
- 89. Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV et al (2000) Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci USA 97:14720–14725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ueki K, Ono Y, Henson JW, Von Deimling A, Louis DN (1996) CDKN2/p16 or RB alterations occur in the majority of glioblastomas and are inversely correlated. Cancer Res 56:150–153. [PubMed] [Google Scholar]
- 91. Van Der Horst L (1955) Application of the field theory to gliomata. J Neuropathol Exp Neurol 14:369–375. [PubMed] [Google Scholar]
- 92. Verdi JM, Bashirullah A, Goldhawk DE, Kubu CJ, Jamali M, Meakin SO, Lipshitz HD (1999) Distinct human NUMB isoforms regulate differentiation vs. proliferation in the neuronal lineage. Proc Natl Acad Sci USA 96:10472–10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Von Deimling A, Eibl RH, Ohgaki H, Louis DN, Von Ammon K, Petersen I et al (1992) p53 mutations are associated with 17p allelic loss in grade II and grade III astrocytoma. Cancer Res 52:2987–2990. [PubMed] [Google Scholar]
- 94. Wakamatsu Y, Maynard TM, Jones SU, Weston JA (1999) NUMB localizes in the basal cortex of mitotic avian neuroepithelial cells and modulates neuronal differentiation by binding to NOTCH‐1. Neuron 23:71–81. [DOI] [PubMed] [Google Scholar]
- 95. Weigert C (1895) Beiträge zur Kenntnis der normalen meschlichen Neuroglia. Diesterweg: Frankfurt. [Google Scholar]
- 96. Wiestler OD, Aguzzi A, Schneemann M, Eibl R, Von Deimling A, Kleihues P (1992) Oncogene complementation in fetal brain transplants. Cancer Res 52:3760–3767. [PubMed] [Google Scholar]
- 97. Willis R (1953) Pathology of Tumours. Butterworth and Company: London. [Google Scholar]
- 98. Zang KD, Singer H (1967) Chromosomal constitution of meningiomas. Nature 216:84–85. [DOI] [PubMed] [Google Scholar]
- 99. Zhang R, Zhang Z, Zhang C, Zhang L, Robin A, Wang Y et al (2004) Stroke transiently increases subventricular zone cell division from asymmetric to symmetric and increases neuronal differentiation in the adult rat. J Neurosci 24:5810–5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ et al (2001) The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side‐population phenotype. Nat Med 7:1028–1034. [DOI] [PubMed] [Google Scholar]
- 101. Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP et al (2005) Early inactivation of p53 tumour suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell 8:119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zu Rhein GM (1983) Studies of JC virus‐induced nervous system tumors in the Syrian hamster: a review. Prog Clin Biol Res 105:205–221. [PubMed] [Google Scholar]
- 103. Zulch K (1986) Brain Tumors. Their Biology and Pathology, 3rd edn. Springer Verlag: Berlin. [Google Scholar]
- 104. Zülch KJ (1979) Histological Typing of Tumours of the Central Nervous System. World Health Organization: Geneva. [Google Scholar]