

Abstract
Meningioma tumor growth involves the subarachnoid space that contains the cerebrospinal fluid. Modeling tumor growth in this microenvironment has been associated with widespread leptomeningeal dissemination, which is uncharacteristic of human meningiomas. Consequently, survival times and tumor properties are varied, limiting their utility in testing experimental therapies. We report the development and characterization of a reproducible orthotopic skull‐base meningioma model in athymic mice using the IOMM‐Lee cell line. Localized tumor growth was obtained by using optimal cell densities and matrigel as the implantation medium. Survival times were within a narrow range of 17–21 days. The xenografts grew locally compressing surrounding brain tissue. These tumors had histopathologic characteristics of anaplastic meningiomas including high cellularity, nuclear pleomorphism, cellular pattern loss, necrosis and conspicuous mitosis. Similar to human meningiomas, considerable invasion of the dura and skull and some invasion of adjacent brain along perivascular tracts were observed. The pattern of hypoxia was also similar to human malignant meningiomas. We use bioluminescent imaging to non‐invasively monitor the growth of the xenografts and determine the survival benefit from temozolomide treatment. Thus, we describe a malignant meningioma model system that will be useful for investigating the biology of meningiomas and for preclinical assessment of therapeutic agents.
Keywords: bioluminescent imaging, IOMM‐Lee, meningioma, orthotopic, skull base, xenografts
INTRODUCTION
Meningiomas are common tumors of the central nervous system that originate from the meningeal covering (22), and therefore can occur in any location along the entire neural axis. They are a source of considerable morbidity and mortality because of their location and the existence of aggressive variants (18). Nevertheless, meningiomas remain a poorly understood cancer. A major obstacle to achieving an improved understanding of the molecular basis of meningioma tumorigenesis, and to evaluating experimental therapies for meningioma treatment, has been the scarcity of in vitro and in vivo model systems. Recently, considerable progress has been made in successfully growing meningioma cell lines in vitro 1, 20. These cell lines represent promising new tools for investigating the biology of meningiomas. However, in vivo meningioma model systems still have problems that restrict their utility 11, 16, 27.
Attempts at propagating available meningioma cell lines as orthotopic xenografts have been unable to recapitulate the human tumor growth pattern, as implantation of tumor cells results in widespread leptomeningeal dissemination throughout the subdural and intraventricular space 16, 27. Consequently, quantification of tumor growth as well as response to therapeutic agents is difficult compromising the utility of the model systems (16). The purpose of this study was to locally constrain meningioma tumor growth and develop a clinically relevant meningioma model system in athymic mice. Because skull base meningiomas are challenging to remove surgically and patients with these meningiomas have a worse prognosis 15, 17, we chose to develop a skull base model using the malignant meningioma cell line, IOMM‐Lee, which is tumorigenic in vivo and grows at a rapid rate (13). By modifying these cells with a luciferase reporter, we have been able to additionally use bioluminescent imaging (BLI) to monitor in vivo growth of these cells, as well as their response to temozolomide therapy.
MATERIALS AND METHODS
Generation of enhanced green fluorescent protein and firefly luciferase expressing IOMM‐Lee cells
The intraosseous malignant meningioma derived cell line, IOMM‐Lee, was used in all the experiments described in this report 13, 14. IOMM‐Lee cells were electroporated (Gene Pulser X Cell, Biorad, Hercules, CA, USA) with the enhanced green fluorescent protein (EGFP)‐N3 plasmid (BD Biosciences, San Jose, CA, USA) and single clones expressing EGFP were selected in 600 µg/mL G418. IOMM‐Lee cells were tagged with firefly luciferase (fluc) under the control of the spleen focus forming virus promoter using lentiviral‐mediated gene transfer (9). Lentiviruses were generated by cotransfection of 293T cells with plasmids for gag‐pol, env and fluc. The 48 h post‐transfection filtered supernatant was used to infect IOMM‐Lee cells.
Intracranial IOMM‐Lee transplantations
All animal experiments were conducted following protocols approved by the University of California, San Francisco, Institutional Animal Care and Use Committee. Five‐ to six‐week‐old female athymic mice were anesthetized with Ketamine/Xylazine and fixed in a Model 940 stereotactic frame (David Kopf Instruments, Tujunga, CA, USA). The skull base region was reached by using the following injection coordinates: 2 mm to the right of the bregma, 2 mm posterior to the bregma and 5.8 mm below the skull surface. The indicated cell numbers and volumes of IOMM‐Lee suspended in phosphate buffered saline (PBS) or matrigel were implanted using a Model 5000 Microinjection Unit (David Kopf Instruments, Tujunga, CA, USA) loaded with a 5 µL Hamilton 7105 syringe. For injection volumes of 0.5 µL, cells were steadily implanted over a period of 70 s and the needle was left in place for 1 minute before it was withdrawn slowly. The skull burr‐hole was sealed with bone‐wax and the skin incision was closed with 7 mm staples. Control mice were implanted with matrigel alone. The mice were monitored closely and were euthanized if they exhibited any neurological symptoms or had >15% weight loss or at predefined times post‐implantation. The estimated survival times were the times from cell implantation to euthanasia.
Tissue processing and immunohistochemistry
EGFP fluorescence in tumor cells was analyzed in formalin fixed mouse heads after removal of the skull with a Leica MZ Fluo III stereomicroscope equipped with a Leica GFP‐plus filter set. For histopathologic examination, mouse heads with the skull intact were fixed in 10% neutral buffered formalin for 48 h, decalcified in Decal Rapid Bone Decalcifier (American Histology, Lodi, CA, USA) for 24 h, and embedded in paraffin. Serial 10 µm thick sections were cut, numbered and processed for either hematoxylin and eosin (H&E) staining or immunohistochemistry. Immunohistochemistry was performed for vimentin using the clone Vim 3B4 antibody (1:100; Dako Corporation, Carpinteria, CA, USA), for carbonic anhydrase 9 (CA9) using the NB 100‐417 antibody (1:1000; Novus Biologicals, Littleton, CO, USA) and for the Ki67 antigen using the clone MIB‐1 antibody (1:100; Dako Corporation, Carpinteria, CA, USA) as described earlier (28). For calculating the MIB‐labeling index, a total of 1000 nuclei in three hot‐spots were counted.
BLI of luciferase––IOMM‐Lee xenografts
BLI of intracranial xenografts was performed using the IVIS Lumina System (Xenogen Corp., Alameda, CA, USA) coupled to the data‐acquisition LivingImage software (Xenogen Corp.). Before imaging, the mice were anesthetized with Ketamine/Xylazine. Thirty mg/mL of luciferin (potassium salt; Gold Biotechnology, St Louis, MO, USA) in PBS was injected intraperitoneally at a dose of 150 mg/kg body weight. Images were acquired between 10 and 20 minutes post‐luciferin administration and peak luminescent signal was recorded. Signal intensity was quantitated as the sum of all detected photon counts within a region of interest using the LivingImage software package (6).
Temozolomide treatment and statistical analysis
For assaying in vitro sensitivity to temozolomide (TMZ), 100 000 IOMM‐Lee and IOMM‐Lee‐Luc cells were plated in six well plates and the indicated concentration of TMZ was added at 24, 48 and 72 h. At 144 h, the cells were trypsinized, resuspended in 1 mL media and processed using the MTT Cell Proliferation Assay (ATCC, Manassas, VA, USA) following manufacturer's direction. Absorbance was read at 590 nm. The luminescence of IOMM‐Lee‐Luc cells treated with TMZ as described above was also read at 144 h in six well plates using the IVIS Lumina System in the presence of 30 µL of 30 mg/mL luciferin (potassium salt; Gold Biotechnology, St Louis, MO, USA). All in vitro experiments were performed in triplicate. For assaying in vivo sensitivity to TMZ, five mice bearing IOMM‐Lee xenografts were orally administered with 120 mg/kg of TMZ for four consecutive days starting on day 10. Five mice bearing IOMM‐Lee xenografts constituted the control group and received no treatment. The Kaplan‐Meier estimator was used to generate the survival curves (12). Differences between survival curves were compared using a log‐rank test (19).
Methylation‐specific polymerase chain reaction (MSP)
Genomic DNA from IOMM‐Lee cells was isolated using the DNeasy kit (Qiagen, Valencia, CA, USA). The methylation status of the O6‐methylguanine‐DNA methyltransferase (MGMT) gene promoter was determined by MSP as described earlier 4, 7. Bisulfite treatment of isolated DNA was performed using the EZ DNA Methylation Gold kit (Zymo Research, Orange, CA, USA), followed by polymerase chain reaction (PCR) amplification to distinguish methylated and unmethylated DNA using PCR conditions and primers described earlier (7).
RESULTS
Fluorescent and bioluminescent tagging of IOMM‐Lee cells
In order to accurately assess the extent of leptomeningeal dissemination, IOMM‐Lee cells were fluorescently labeled with EGFP and a single high‐level EGFP expressing clone, designated IOMM‐Lee‐EGFP2, was selected for further analysis. For BLI, IOMM‐Lee cells were transduced with lentivirus encoding firefly luciferase, and transduced cell pools (IOMM‐Lee‐Luc) were injected in mice. IOMM‐Lee‐EGFP2, IOMM‐Lee‐Luc and parental IOMM‐Lee had similar growth curves in vitro (data not shown). Also, IOMM‐Lee‐EGFP2 and parental IOMM‐Lee had similar growth curves as subcutaneous tumors in athymic mice (data not shown). Thus, there was no indication of fluorescent and bioluminescent labeling altering the growth properties of IOMM‐Lee parental cells.
Estimated survival times
We injected varying amounts and concentrations of IOMM‐Lee‐EGFP2 cells in PBS or matrigel into the skull base region of athymic mice and calculated estimated survival times based on the appearance of neurological symptoms or weight loss. All the mice injected with IOMM‐Lee‐EGFP2 cells developed tumors. Thus, the tumor take rate with this cell line is 100%. Considerable variability in survival times and widespread leptomeningeal dissemination throughout the skull base and subarachnoid space was observed when PBS was used as an implantation medium (data not shown). When matrigel was used as an implantation medium, both intra‐ and inter‐group variability in survival times were observed, with the extent of the former depending on both cell number and cell volume injected (Figure 1). For example, mice injected with 500 000 cells/5.0 µL died as early as 8 days or as late as 17 days (Figure 1), and this variability was caused by differences in tumor cell dissemination between mice receiving this amount and concentration of cells (data not shown). More consistent survival times with mice dying within 5 days of each other was observed for injections of 3 million cells/3.0 µL and 50 000 cells/0.5 µL (Figure 1). Of these two conditions, mice injected using the latter condition had slightly longer estimated survival times of 17–21 days and were chosen for use in subsequent investigations (see below).
Figure 1.
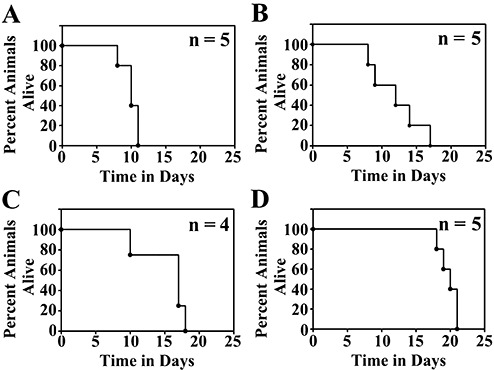
Estimated survival curves of athymic mice implanted with IOMM‐Lee‐EGFP2 cells in the skull base region. Athymic mice implanted with 3 000 000 cells/3 µL (A), 500 000 cells/5 µL (B), 50 000 cells/1 µL (C) or 50 000 cells/0.5 µL (D) were euthanized when they exhibited neurological symptoms or weight loss. Kaplan Meier plots for each implantation condition and the numbers of mice (n) in each group are shown.
Localized meningioma tumor growth
Macroscopic analysis of IOMM‐Lee‐EGFP2 xenograft growth revealed that the tumor mass did not invade the surface of the brain, and typically adhered to periosteal membranes when the skull and brain were separated (Figure 2). Examination of EGFP fluorescence revealed that tumor growth was confined to the site of implantation with minimal tumor cell dissemination to surrounding locations (Figure 2). The brain was visibly compressed at the location of the tumor.
Figure 2.
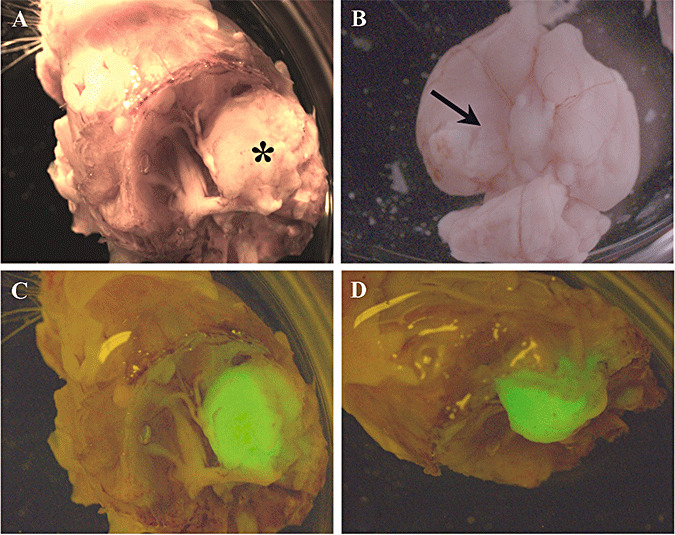
Macroscopic view of IOMM‐Lee skull base meningioma xenografts. IOMM‐Lee‐EGFP2 cells were implanted in the skull base region using matrigel as the implantation medium to obtain localized tumor growth. The tumor mass (asterisk in A) was observed between the brain and the skull, and adhered to the skull when the skull (A) and brain (B) were separated. Compression of the brain was observed (arrow in B). Minimal leptomeningeal dissemination was observed as assessed by the distribution of the fluorescent EGFP label (C,D).
Histopathology of IOMM‐Lee xenografts
Histopathologic analyses were performed on tumors that were fixed, processed and sectioned in situ with the skull and brain intact. Xenografts displayed histopathologic features that were reminiscent of human anaplastic meningiomas, and included high cellularity, prominent nuclear pleomorphism marked by a high nuclear to cytoplasmic ratio and prominent nucleoli. Cells were typically arranged in syncytial‐like, highly cellular sheets with variable amounts of micro and geographic necrosis. Mitotic figures were conspicuous and the MIB‐1 labeling indices were typically 30%.
Growth characteristics of IOMM‐Lee xenografts
To evaluate xenograft growth patterns, brains with intact skulls were resected from mice injected with IOMM‐Lee‐Luc cells (50 000/0.5 µL) and sacrificed at days 3, 6, 9, 12, 16 post‐implantation or when they exhibited weight loss and/or neurological symptoms. Fixed and embedded brain and skull tissue was serially sectioned, and examined by conventional H&E analysis or after staining for human vimentin. Progressive tumor growth was evident from day 3 onward, and by day 6, the tumor appeared to erode into the adjacent skull while primarily compressing the brain with early multifocal invasion into perivascular spaces (Figure 3). As the tumor mass enlarged, the boundary with the brain remained well‐demarcated except for regions of microinvasion (3, 4). By day 12 the xenografts had conspicuous necrotic zones, and the tumors had breached the pia and were invading the brain along perivascular and cranial nerve tracts (Figure 4). Immunohistochemical staining for human vimentin gave no indication of tumor dissemination at locations distant from the site of tumor implantation, and therefore meningioma tumor growth was localized to the site of tumor implantation.
Figure 3.
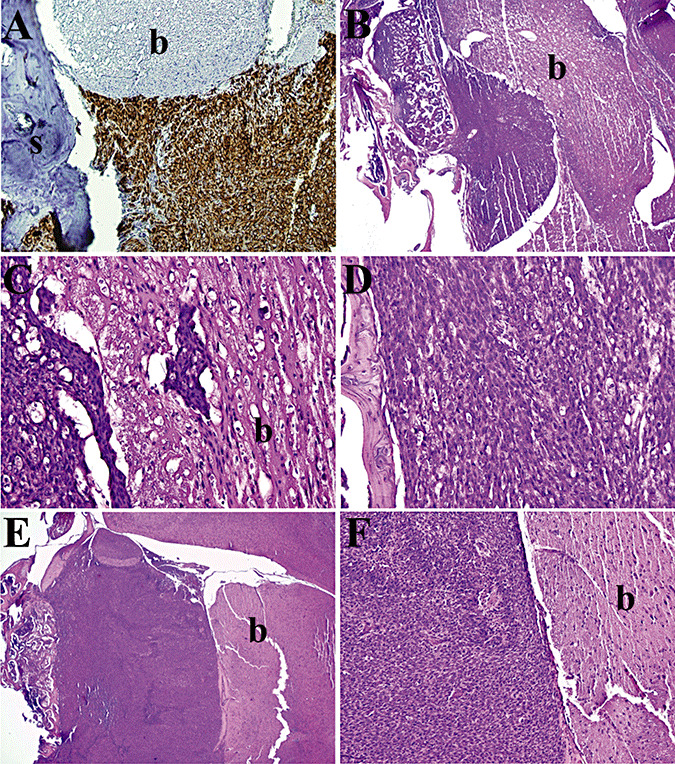
Growth Characteristics of IOMM‐Lee skull base xenografts. Tissue sections of IOMM‐Lee xenografts on day 6 (A), day 9 (B–D) and day 12 (E,F) were stained with human vimentin (A) or hematoxylin and eosin (B–F) and examined to evaluate the pattern of tumor growth. IOMM‐Lee tumor growth maintained a well‐demarcated boundary with the brain, except for microinvasion of the brain by small clumps of tumor cells (C). s, skull; b, brain.
Figure 4.
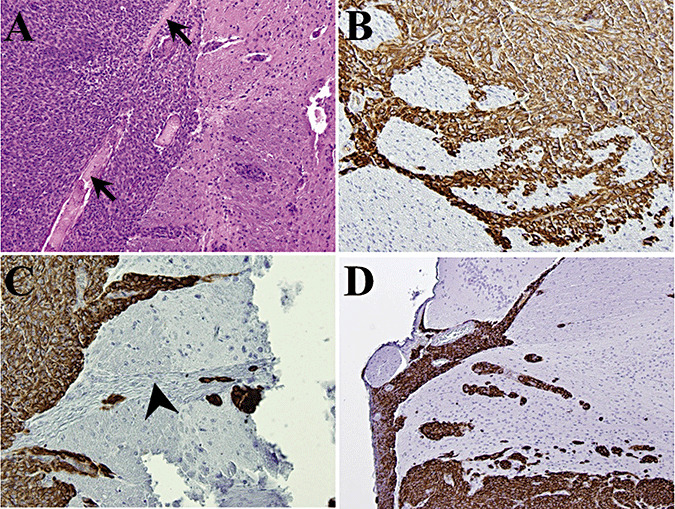
Pattern of brain invasion by IOMM‐Lee xenografts. Tissue sections of IOMM‐Lee xenografts on day 12 (A–C) or day 16 (D) were stained with human vimentin (B–D) or hematoxylin and eosin (A) and examined to evaluate the pattern of invasion of brain. By day 12, the tumor had breached the pia (A) and was invading the brain along perivascular (B) and cranial nerve (C) tracts. A similar pattern of invasion was observed in day 16 tumors (D). Arrow, pia; arrowhead, cranial nerve.
Tumor hypoxia is an endogenous characteristic of malignant meningiomas, is associated with higher‐grade histology as well as aggressive clinical behavior (28). With respect to this animal model of malignant meningioma, we assessed the appearance and prevalence of hypoxia by CA9 immunohistochemistry (28). Regions of hypoxia were first visible in day 9 xenografts (Figure 5), prior to the appearance of necrosis. Extensive hypoxic cell numbers were observed at the brain interface suggesting that this edge of the tumor was slower at recruiting vessels from the neuropil as opposed to non‐brain interface tissue. Necrosis was first observed in day 12 tumors (Figure 5), and similar to human meningiomas, CA9 staining was zonal, found in viable cells surrounding regions of necrosis and also in regions not associated with any visible necrosis.
Figure 5.
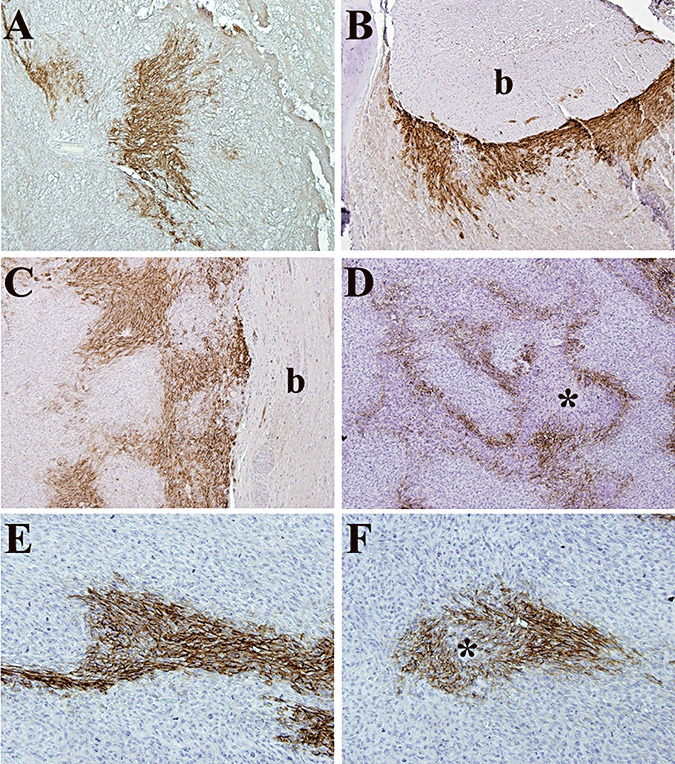
Pattern of hypoxia in IOMM‐Lee skull‐base xenografts. Immunohistochemistry with a polyclonal antibody against carbonic anhydrase 9 (CA9) was performed to evaluate the prevalence and distribution of hypoxia in tissue sections of IOMM‐Lee xenografts on day 9 (A), day 12 (B), day 16 (C) and day 21 (D–F) post‐implantation. Similar to human meningiomas, CA9 expression was zonal and sometimes not associated with any visible necrosis (E). The brain interface had considerable hypoxia (B,C) compared with the skull interface. b, brain; *, necrosis.
BLI of meningioma tumor burden
BLI was used to quantitate intracranial meningioma tumor growth rates in advance of testing tumor response to alkylator therapy. Luminescence readings were detectable as early as day 3, and growth curves displayed consistent patterns of exponential increase between mice. Mice exhibited weight loss and neurological symptoms when photon counts of 3–4 × 107 were reached. In total, the BLI results showed reproducible growth of skull base xenografts, and provided pilot data for use in timing the administration of therapy in the subsequent experiment.
Xenograft response to TMZ therapy
To assess the efficacy of alkylator therapy, we treated IOMM‐Lee and IOMM‐Lee‐Luc cells in culture with TMZ (Figure 6A,B). IOMM‐Lee was sensitive to TMZ in vitro and the bioluminescent labeling did not alter the sensitivity to TMZ. Sensitivity to TMZ has been associated with MGMT promoter hypermethylation, and the MGMT promoter was methylated in IOMM‐Lee cells (Figure 6C). To assess the in vivo sensitivity of IOMM‐Lee xenografts to TMZ, tumor growth in control and TMZ‐treated mice were followed using BLI. TMZ treatment resulted in a significant survival benefit to the mice (Figure 6, P = 0.003). While control group mice died by 17–21 days, one TMZ‐treated mouse died on day 38 and the remaining four were still alive on day 43. To plot BLI tumor growth curves, each mouse's luminescence measurements were normalized against their own day 10 luminescence reading, allowing each mouse to serve as its own control. TMZ treatment arrested the rapid, exponential growth of the xenografts and resulted in reduced normalized luminescence readings (Figure 7). Recovery of exponential tumor growth was observed in the TMZ‐treated mouse that died on day 38, while the remaining four mice had stable luminescent measurements till day 43 (Figure 7).
Figure 6.
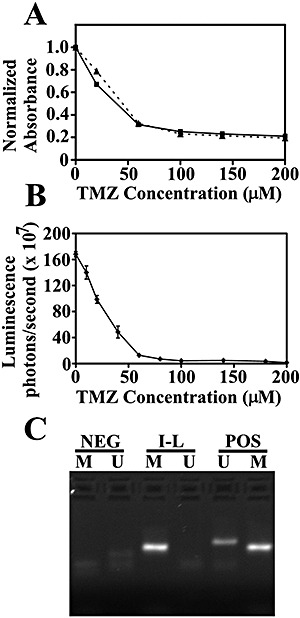
Efficacy of temozolomide (TMZ) in cultured IOMM‐Lee cells and methylation status of the O6‐methylguanine‐DNA methyltransferase (MGMT) promoter. A. IOMM‐Lee (squares and solid line) or IOMM‐Lee‐Luc cells (triangles and dashed line) were treated with the indicated concentration of TMZ and the number of viable cells was calculated using the MTT Cell Proliferation assay. Absorbance values were normalized to the no treatment control and plotted against TMZ concentration. The number of viable cells at different TMZ concentrations was similar for IOMM‐Lee and IOMM‐Lee‐Luc cells. B. IOMM‐Lee‐Luc cells in culture were treated with the indicated concentration of TMZ and luminescence was measured at 144 h. Luminescence readings are plotted against TMZ concentration. C. Methylation‐specific polymerase chain reaction was performed to determine the methylation status of the MGMT gene. The presence of a visible polymerase chain reaction product in the lane U indicates the presence of unmethylated MGMT gene and the presence of product in the lane M indicates the presence of methylated MGMT gene. MGMT is methylated in IOMM‐Lee. Abbreviations: NEG = negative controls; I‐L = IOMM‐Lee; POS = positive controls.
Figure 7.
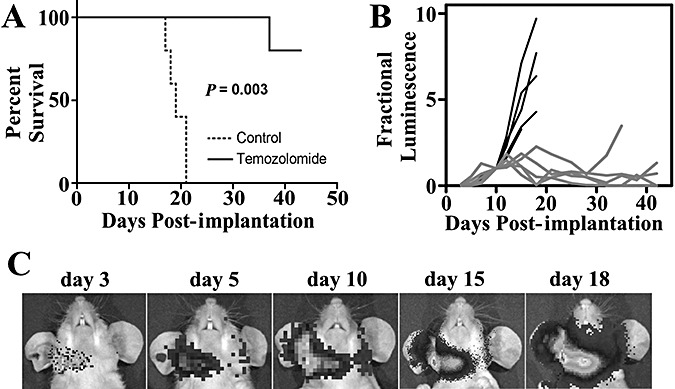
Bioluminescent imaging of IOMM‐Lee xenografts, and their response to temozolomide (TMZ). Ten mice received injections of IOMM‐Lee‐Luc cells and were randomized into two groups that received either no treatment or treatment with 120 mg/kg TMZ for four consecutive days. A. Survival curves of athymic mice from control (dotted line) and TMZ‐treatment (solid line) groups are plotted. A significant survival benefit (P = 0.003) for mice treated with TMZ was observed. B. Luminescence readings for each mouse were normalized against its own day 10 luminescence reading. Normalized BLI plots associated with monitoring of intracranial tumor growth for control (black line) and TMZ‐treatment (gray line) groups are shown. C. Representative images from one control group mouse at the indicated time points post‐implantation of IOMM‐Lee‐Luc cells are shown.
DISCUSSION
Animal models are essential preclinical tools in the study of the molecular mechanisms of cancer and for evaluating anti‐tumor therapies. A small number of meningioma animal models exist but every one of these systems has issues that limit their utility and/or clinical relevance. For example, in the genetic model of meningiomas, which is based on the knockout of the NF2 gene specifically in arachnoidal cells, only 20% of the mice develop tumors after 11 to 14 months (11). Other model systems have utilized non‐orthotopic locations that do not take micro‐environmental influences of tumor growth into consideration. Rodent studies on the chemotherapeutic agent, hydroxyurea, were performed using meningioma cells grown in the galea (25), and tests on celecoxib were performed in subcutaneous tumors (21). Orthotopic meningioma model systems utilizing primary and established meningioma cell lines have been described 16, 27. However, primary meningioma cell lines senesce after a few passages and therefore these model systems are not reproducible 16, 27. Orthotopic models with established meningioma cell lines have been associated with widespread leptomeningeal dissemination limiting the ability to quantitate the extent of tumor growth in these systems.
In this study, we present a well‐characterized, reproducible, clinically relevant skull base malignant meningioma xenograft model system in which we have overcome the problem of leptomeningeal dissemination. We show that localized meningioma tumor growth is sensitive to particular cell densities and is dependent on using matrigel as the implantation agent. In addition, we show that mice bearing localized xenografts had reproducible survival times within a range of 5 days. IOMM‐Lee has been extensively used in meningioma research 23, 26 and has previously been implanted into the skull base region of mice 16, 27. However, localized tumor growth using this cell line has not previously been attained. Prior studies utilized PBS as the implantation agent, and it is likely that the use of matrigel would have reduced observed tumor cell dissemination. Previous reports of survival times with mice bearing IOMM‐Lee orthotopic xenografts were similar to the survival times in this study 16, 24.
Preclinical cancer models should ideally resemble the human disease and mimic human tumor‐host interactions. Human meningiomas are generally well‐demarcated lesions that remain spherical or globular even after they attain considerable size (3). Consistent with the growth pattern of human meningiomas, IOMM‐Lee xenografts grew as well‐demarcated lesions between the skull and brain and remained globular even when the tumor mass was large. Malignant meningiomas commonly penetrate dura and invade bone. IOMM‐Lee xenografts adhere to the skull and exhibit considerable invasion of the skull. Considerable compression of the brain was also observed. Human meningiomas grow as separate entities adjacent to the brain but often insinuate themselves into the subjacent cortex as small tongues or pegs of neoplastic tissue that follow the course of superficial blood vessels (3). IOMM‐Lee xenografts followed a similar pattern of brain invasion, with small cell clumps invading the surrounding brain along perivascular and cranial nerve tracts.
Tumor hypoxia is significantly associated with higher histopathologic grade in meningiomas and is indicative of an aggressive meningioma phenotype (28). Similar to human meningiomas, the pattern of hypoxia in IOMM‐Lee xenografts was found in the classic perinecrotic pattern in larger tumors with visible necrosis. Additionally, as in human meningiomas, hypoxia was found in histologically viable regions of tumors not associated with any visible necrosis. Interestingly, the amount of hypoxia at the brain interface was considerably greater than that at the skull interface, suggesting that the skull interface had easier access to a vascular supply. While the contribution of tumor hypoxia to meningioma growth is unknown, the current model system will allow such investigations and also allow testing of therapeutic strategies that target both normoxic and hypoxic cells.
BLI is an extremely accurate predictor of intracranial tumor burden and it has recently been shown using intracranial glioblastoma xenografts that there is an excellent correlation between intracranial tumor photon emission and tumor volume (6). BLI tumor growth curves for the IOMM‐Lee xenografts were consistent between mice and BLI could accurately predict survival. Thus, BLI is ideal for following intracranial meningioma tumor growth and for monitoring therapeutic response. Meningioma tumor growth in animals has previously been monitored using contrast enhanced magnetic resonance imaging (MRI) (27). While MRI accurately follows tumor growth, it is a less cost–effective technique and relatively few researchers have access to such a facility. Immortalized benign meningioma cell lines have recently been developed in several laboratories 5, 20. These cell lines grow very slowly in mice and require long in vivo observation periods of several months. It is anticipated that BLI will be especially useful in following the tumor growth and monitoring the therapeutic response of these slower growing but more common meningiomas.
TMZ, a DNA methylating agent, has schedule‐dependent anti‐tumor activity against a variety of malignancies, including gliomas and melanomas (8). Currently, it is routinely used in the clinic to treat malignant gliomas. The efficacy of TMZ against malignant meningiomas has not been evaluated. In the current study, we show that TMZ was an effective therapy against rodent IOMM‐Lee xenografts and resulted in a considerable survival benefit. Methylation of the promoter of the DNA‐repair gene, MGMT, has been associated with sensitivity to TMZ in glioblastoma patients (10). The MGMT promoter was methylated in IOMM‐Lee cells. Sixteen percent of all meningiomas have aberrant methylation of the MGMT promoter (2) and patients bearing these tumors could possibly respond to TMZ therapy. However, more detailed preclinical studies using this chemotherapeutic agent are necessary before conclusions on the effectiveness of TMZ as a therapeutic agent for meningiomas are made.
In summary, we have developed a rodent preclinical meningioma model system and show that it can be used to evaluate therapeutic regimens. This system has several features that mimic the human meningioma growth pattern and will enable us to dissect the biology of meningioma tumorigenesis, evaluate tumor‐host interactions unique to meningiomas and test the toxicity and efficacy of novel therapeutic approaches.
ACKNOWLEDGMENTS
We thank Cynthia Cowdrey for technical assistance with tissue processing, Lily Hu for technical assistance with generation of the luciferase lentivirus, Jennifer Ayers‐Ringler for technical assistance with the MGMT promoter methylation assay and Drs Katharine Striedinger and Jeanette Hyer for useful discussions and review of the manuscript. This study was supported by NIH/NINDS grant 1R03NS054829‐01 to AL and SPORE grant P50CA097257 to CDJ.
REFERENCES
- 1. Baia GS, Slocum AL, Hyer JD, Misra A, Sehati N, Vandenberg SR et al (2006) A genetic strategy to overcome the senescence of primary meningioma cell cultures. J Neurooncol 78:113–121. [DOI] [PubMed] [Google Scholar]
- 2. Bello MJ, Aminoso C, Lopez‐Marin I, Arjona D, Gonzalez‐Gomez P, Alonso ME et al (2004) DNA methylation of multiple promoter‐associated CpG islands in meningiomas: relationship with the allelic status at 1p and 22q. Acta Neuropathol (Berl) 108:413–421. [DOI] [PubMed] [Google Scholar]
- 3. Burger PC, Scheithauer BW, Vogel FS (2002) Surgical Pathology of the Nervous System and Its Coverings, 4th edn. Churchill Livingstone: New York. [Google Scholar]
- 4. Cankovic M, Mikkelsen T, Rosenblum ML, Zarbo RJ (2007) A simplified laboratory validated assay for MGMT promoter hypermethylation analysis of glioma specimens from formalin‐fixed paraffin‐embedded tissue. Lab Invest 87:392–397. [DOI] [PubMed] [Google Scholar]
- 5. Cargioli TG, Ugur HC, Ramakrishna N, Chan J, Black PM, Carroll RS (2007) Establishment of an in vivo meningioma model with human telomerase reverse transcriptase. Neurosurgery 60:750–759; discussion 759–760. [DOI] [PubMed] [Google Scholar]
- 6. Dinca EB, Sarkaria JN, Schroeder MA, Carlson BL, Voicu R, Berger MS, James CD (2007) Bioluminescence monitoring of intracranial gliolastoma xenograft response to primary and salvage temozolomide therapy. J Neurosurg 107:610–616. [DOI] [PubMed] [Google Scholar]
- 7. Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG (1999) Inactivation of the DNA repair gene O6‐methylguanine‐DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 59:793–797. [PubMed] [Google Scholar]
- 8. Friedman HS, Kerby T, Calvert H (2000) Temozolomide and treatment of malignant glioma. Clin Cancer Res 6:2585–2597. [PubMed] [Google Scholar]
- 9. Hasegawa K, Pham L, O'Connor MK, Federspiel MJ, Russell SJ, Peng KW (2006) Dual therapy of ovarian cancer using measles viruses expressing carcinoembryonic antigen and sodium iodide symporter. Clin Cancer Res 12:1868–1875. [DOI] [PubMed] [Google Scholar]
- 10. Hegi ME, Diserens AC, Gorlia T, Hamou MF, De Tribolet N, Weller M et al (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003. [DOI] [PubMed] [Google Scholar]
- 11. Kalamarides M, Niwa‐Kawakita M, Leblois H, Abramowski V, Perricaudet M, Janin A et al (2002) Nf2 gene inactivation in arachnoidal cells is rate‐limiting for meningioma development in the mouse. Genes Dev 16:1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kaplan EL, Meier P (1958) Non‐parametric estimation from incomplete observations. J Am Stat Assoc 53:457–481. [Google Scholar]
- 13. Lee WH (1990) Characterization of a newly established malignant meningioma cell line of the human brain: IOMM‐Lee. Neurosurgery 27:389–395. [PubMed] [Google Scholar]
- 14. Lee WH, Tu YC, Liu MY (1988) Primary intraosseous malignant meningioma of the skull: case report. Neurosurgery 23:505–508. [DOI] [PubMed] [Google Scholar]
- 15. Mathiesen T, Lindquist C, Kihlstrom L, Karlsson B (1996) Recurrence of cranial base meningiomas. Neurosurgery 39:2–7. [DOI] [PubMed] [Google Scholar]
- 16. McCutcheon IE, Friend KE, Gerdes TM, Zhang BM, Wildrick DM, Fuller GN (2000) Intracranial injection of human meningioma cells in athymic mice: an orthotopic model for meningioma growth. J Neurosurg 92:306–314. [DOI] [PubMed] [Google Scholar]
- 17. Mendenhall WM, Morris CG, Amdur RJ, Foote KD, Friedman WA (2003) Radiotherapy alone or after subtotal resection for benign skull base meningiomas. Cancer 98:1473–1482. [DOI] [PubMed] [Google Scholar]
- 18. Modha A, Gutin PH (2005) Diagnosis and treatment of atypical and anaplastic meningiomas: a review. Neurosurgery 57:538–550. [DOI] [PubMed] [Google Scholar]
- 19. Peto R, Peto J (1972) Asymptotically efficient rank invariant procedures. J R Stat Soc Ser A Stat Soc 135:185–207. [Google Scholar]
- 20. Puttmann S, Senner V, Braune S, Hillmann B, Exeler R, Rickert CH, Paulus W (2005) Establishment of a benign meningioma cell line by hTERT‐mediated immortalization. Lab Invest 85:1163–1171. [DOI] [PubMed] [Google Scholar]
- 21. Ragel BT, Jensen RL, Gillespie DL, Prescott SM, Couldwell WT (2006) Celecoxib inhibits meningioma tumor growth in a mouse xenograft model. Cancer 109:588–597. [DOI] [PubMed] [Google Scholar]
- 22. Riemenschneider MJ, Perry A, Reifenberger G (2006) Histological classification and molecular genetics of meningiomas. Lancet Neurol 5:1045–1054. [DOI] [PubMed] [Google Scholar]
- 23. Robb VA, Li W, Gutmann DH (2004) Disruption of 14‐3‐3 binding does not impair Protein 4.1B growth suppression. Oncogene 23:3589–3596. [DOI] [PubMed] [Google Scholar]
- 24. Salhia B, Rutka JT, Lingwood C, Nutikka A, Van Furth WR (2002) The treatment of malignant meningioma with verotoxin. Neoplasia 4:304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schrell UM, Rittig MG, Anders M, Kiesewetter F, Marschalek R, Koch UH, Fahlbusch R (1997) Hydroxyurea for treatment of unresectable and recurrent meningiomas. I. Inhibition of primary human meningioma cells in culture and in meningioma transplants by induction of the apoptotic pathway. J Neurosurg 86:845–852. [DOI] [PubMed] [Google Scholar]
- 26. Surace EI, Lusis E, Haipek CA, Gutmann DH (2004) Functional significance of S6K overexpression in meningioma progression. Ann Neurol 56:295–298. [DOI] [PubMed] [Google Scholar]
- 27. Van Furth WR, Laughlin S, Taylor MD, Salhia B, Mainprize T, Henkelman M et al (2003) Imaging of murine brain tumors using a 1.5 Tesla clinical MRI system. Can J Neurol Sci 30:326–332. [DOI] [PubMed] [Google Scholar]
- 28. Yoo H, Baia GS, Smith JS, McDermott MW, Bollen AW, Vandenberg SR et al (2007) Expression of the hypoxia marker carbonic anhydrase 9 is associated with anaplastic phenotypes in meningiomas. Clin Cancer Res 13:68–75. [DOI] [PubMed] [Google Scholar]