

Abstract
Multiple sclerosis (MS) is an inflammatory demyelinating disease of the central nervous system (CNS). Adhesion molecules play important roles in cell–cell and cell–extracellular matrix (ECM) interactions in inflammation. Blocking the interaction between inflammatory cells and vascular endothelia can prevent cell entry into tissues and harmful inflammatory responses, that is, autoimmunity, but could also limit immunosurveillance by anti‐viral T cells in sites of infection or latency. Development of progressive multifocal leukoencephalopathy in patients treated with antibody against very late antigen (VLA)‐4 prompted us to explore an alternative therapeutic approach. We used an antibody against the integrin α2, VLA‐2, that interacts with ECM, not vascular endothelium. SJL/J mice were sensitized with myelin proteolipid protein (PLP)139–151 peptide to induce experimental autoimmune encephalomyelitis (EAE), an animal model for MS. Treatment of mice with VLA‐2 antibody suppressed clinical signs and CNS inflammation of EAE, when antibody was given immediately after disease onset. In contrast, VLA‐4 or VLA‐2 antibody treatment of mice during the priming or remission phase of EAE had minor effects on the disease’s clinical course. No differences were found in lymphoproliferative responses to PLP139–151 among treatment groups. Data suggest that blocking cell–ECM interactions can be an alternative therapy for MS.
INTRODUCTION
Adhesion molecules play important roles in many aspects of the immune response, including lymphocyte‐endothelial cell adhesion, providing a co‐stimulatory signal for antigen specific T‐cell proliferation and fostering interactions between the antigen presenting cells and T cells (7, 13, 22, 23, 62). The integrin family of adhesion receptors is composed of at least eight different β subunits and 18 different α subunits that dimerize to form at least 24 different heterodimers (24). Integrins mediate cell–cell interactions as well as cell interaction with the extracellular matrix (ECM). They can also activate intracellular signaling cascades, including tyrosine, serine/threonine and mitogen‐activated protein kinase activities associated with neurotrophic factors and cytokines. Integrins exert potent affects over a diverse array of cellular functions, including proliferation, differentiation, process outgrowth, gene expression, survival, cell migration and inflammation. Thus integrins serve a major role as environmental sensors for the cell. In the central nervous system (CNS), Pinkstaff et al (55) showed mRNA expression of α1, α3, α4, α5, α6, α7, α8, αV, β1, β4 and β5, but not of α2, β2 and β3, integrin subunits in adult rat brain.
Multiple sclerosis (MS) is a serious neurological disorder in which demyelination and inflammation occur in the white matter of the CNS. MS has been hypothesized to have a viral component linked to an autoimmune component (19). The adhesion of leukocytes to brain endothelium and their subsequent migration into the CNS is facilitated by the interaction between various cell surface adhesion molecules and their endothelial cell ligands. In animal models of MS, including experimental autoimmune encephalomyelitis (EAE) and Theiler’s murine encephalomyelitis virus infection, adhesion molecules are highly expressed on T cells and vascular endothelium in the CNS (4, 27, 67). In vivo administration of monoclonal antibodies (4, 31, 32, 33, 61, 83) or antisense oligonucleotide (52) specific to α4 integrin or its ligand can suppress EAE induced by either active sensitization or adoptive transfer of myelin specific T cells. These therapies are thought to disrupt T cell–endothelial cell interactions and to block T‐cell infiltration from the circulation into the CNS (62).
Natalizumab (Tysabri®/Antegren®) is a humanized monoclonal antibody raised against human α4 integrin which blocks the engagement of α4β1 integrin [very late antigen (VLA)‐4, CD49d/CD29] with vascular cell adhesion molecule (VCAM)‐1 (CD106) on the endothelium of various organs, including the CNS, as well as the engagement of α4β7 integrin with endothelial mucosal addressin cell adhesion molecule‐1 in the gut (40). In November 2004, the US Food and Drug Administration approved natalizumab for the treatment of MS. However, natalizumab was withdrawn from the market, because two patients with MS (35, 39) and one patient with Crohn’s disease (73) who were treated with natalizumab developed progressive multifocal leukoencephalopathy, a demyelinating disease caused by JC virus (14, 38). As VLA‐4 is upregulated in memory T cells, treatment with VLA‐4 antibody could block the interaction between JC virus‐specific CD8+ T cells and endothelium in the CNS (11, 38), leading to reactivation of JC virus. Thus, the potential for these immunomodulatory antibody therapies to reactivate latent virus represents a major emerging issue in the development of new, more targeted therapies for MS.
The collagen binding integrins, such as α1 and α2, are distinct from the other ECM binding integrins, such as α4. These contain a collagen binding domain, specifically the “inserted” domain or I domain, comprising about 180 amino acids in the α chain. The integrin α2β1 (also designated VLA‐2, GPIaIIa, CD49b/CD29) recognizes fibrillar collagens, such as type I collagen, with higher affinity than the non‐fibrillar collagens, such as type IV, whereas the α1β1 (VLA‐1, CD49a/CD29) integrin exhibits higher binding affinity to the non‐fibrillar collagens (21, 34).
de Fougerolles et al (10) have demonstrated that blocking VLA‐1 and VLA‐2 could modulate CD4+ T cell‐mediated delayed type hypersensitivity responses (20) as well as CD8+ T cell‐mediated contact hypersensitivity responses (81). Treatment of mice with VLA‐1 or VLA‐2 antibody led to a marked decrease in cellular infiltration and edema, whereas a nonspecific inflammation provoked by croton oil was not affected. In addition, Andreasen et al (1) showed upregulation of VLA‐1 and VLA‐2 on T cells in lymphocytic choriomeningitis virus infection in mice, and that treatment with VLA‐1 or VLA‐2 antibody suppressed lymphocytic choriomeningitis virus induced CD8+ T cell‐mediated inflammation in the footpad, which requires migration of cells into the ECM to induce measurable swelling. As various cell types express VLA‐2, such as activated T cells (50, 57) [reviewed in (23)], monocytes, neutrophils (78), mast cells (12), platelets (29) and endothelial cells (79), this treatment most likely leads to alterations in these cells’ ability to interact with one another that would normally result in a specific inflammatory response.
To explore alternative therapies targeting adhesion molecules, we administered VLA‐2 (α2) antibody to mice with EAE induced using myelin proteolipid protein (PLP)139–151 peptide. Treatment with VLA‐2 antibody suppressed clinical signs of EAE when the antibody was given immediately after the onset of disease, but not during the priming or remission phases of EAE. Treatment with VLA‐2 antibody also decreased the disease incidence during the chronic stage. In contrast, treatment with VLA‐4 (α4) antibody after disease onset had a mild effect on the clinical course. No difference in lymphoproliferative responses to PLP was observed between the different antibody regimes. Blocking the interaction between inflammatory cells and the ECM could potentially provide an alternative therapy for certain organ‐specific autoimmune diseases.
MATERIALS AND METHODS
EAE induction and analysis.
Female SJL/J mice (Jackson Laboratory, Bar Harbor, ME) were injected subcutaneously at the base of the tail with 100 nmol of modified PLP139–151 peptide (HSLGKWLGHPDKF) (Core Facility, University of Utah Huntsman Cancer Institute, Salt Lake City, UT) (3, 68) in Imject® Freund’s Incomplete Adjuvant (Pierce Biotechnology, Rockford, IL) containing Mycobacterium tuberculosis H37 Ra (Difco Laboratories, Detroit, MI) (CFA). The final concentration of M. tuberculosis in the PLP/CFA solution was 2 mg/mL. Mice were weighed and observed for clinical signs for 2 months. EAE signs were assessed according to the following criteria (68): 0 = no clinical disease; 1 = loss of tail tonicity; 2 = mild hind leg paresis; 3 = moderate hind leg paralysis; 4 = complete paraplegia; and 5 = quadriplegia, moribund state or death. Animal handling and protocols were approved by the IACUC at the University of Utah. A cumulative disease score was calculated by adding the daily scores of an individual mouse over the observation period, using Origin version 7.5 (OriginLab Corporation, Northampton, MA).
VLA‐2 or VLA‐4 antibody treatment.
Monoclonal antibody against rat α2 (clone Ha1/29) was raised in Armenian hamsters (47). The antibody cross reacts with mouse α2 (10, 29). Adhesion studies showed that the antibody had an EC50 for inhibiting rat platelet binding to type I collagen of 0.36 nM and of mouse platelets of 2 nM (data not shown). Anti‐mouse CD49d/VLA‐4 (clone PS/2) which recognizes the α4 chain of the integrin, was obtained from SouthernBiotech (Birmingham, AL) (49, 65). Mice were injected intraperitoneally with 75 µg (5 mg/kg) of antibody against VLA‐2 or VLA‐4. Control mice were given hamster antibody against trinitrophenol (TNP)‐keyhole limpet hemocyanin (clone A19‐3) (BD Biosciences, San Diego, CA). Mice were randomly separated into several groups. Mice were administered antibody during the induction phase of EAE (days 0, 2, 4, 7, 9), during the effector phase (days 10, 11, 12, 14, 16, 18, 20) or during the relapse and remission period (days 18, 21, 23, 25, 27, 29, 32, 34, 36).
Histology.
Mice were euthanized using isoflurane when moribund, or after the 2‐month observation period. Mice were perfused with phosphate‐buffered saline followed by a 4% paraformaldehyde solution in phosphate‐buffered saline. CNS tissues were removed, and the brains were divided into five coronal slabs and the spinal cords into 10 to 12 transverse slabs, and the tissues were embedded in paraffin. Four‐micrometer thick tissue sections were stained with Luxol fast blue for myelin visualization. Histological scoring was performed as described previously (68, 69). For scoring spinal cord sections, each spinal cord section was divided into quadrants: the anterior funiculus, the posterior funiculus and each lateral funiculus. Any quadrant containing meningitis or demyelination was given a score of 1 in that pathologic class. The total number of positive quadrants for each pathologic class was determined, then divided by the total number of quadrants present on the slide and multiplied by 100 to give the percent involvement for each pathologic class. Brain sections were scored for meningitis (0 = no meningitis, 1 = mild cellular infiltrates, 2 = moderate cellular infiltrates, 3 = severe cellular infiltrates), perivascular cuffing (0 = no cuffing, 1 = 1 to 10 lesions, 2 = 11 to 20 lesions, 3 = 21 to 30 lesions, 4 = 31 to 40 lesions, 5 = over 40 lesions) and demyelination (0 = no demyelination, 1 = mild demyelination, 2 = moderate demyelination, 3 = severe demyelination). Each score from the brain was combined for a maximum score of 11 per mouse (70).
Lymphoproliferative assay.
Prior to paraformaldehyde perfusion, inguinal lymph nodes and spleens were removed and pooled. Mononuclear cells (MNCs) were isolated with Histopaque®‐1083 (Sigma, St. Louis, MO). A volume of 200 µL containing 2 × 105 cells in RPMI‐1640 supplemented with 1% glutamine, 1% antibiotics, 50 µM 2‐mercaptoethanol, and 10% fetal bovine serum was added to each well of 96 well plates. PLP139–151, PLP178–191 (NTWTTCQSIAFPSK) (68), myelin basic protein (MBP)84–104 (VHFFKNIVTPRTPPPSQGKGR) (37, 59, 75) or myelin oligodendrocyte glycoprotein (MOG)92–106 (DEGGYTCFFRDHSYQ) (69, 70, 71) peptide was added such that the final concentration of peptide was 50 µg/mL. The cells were cultured at 37°C in 5% CO2 for 4 days, pulsed with 1 µCi of tritiated thymidine per well, and then cultured for another 18 to 24 h. Cultures were harvested onto filters using a multi‐well cell harvester and 3H uptake was measured using an LS 6500 Multi‐Purpose Scintillation Counter (Beckman Coulter, Inc, Fullerton, CA). Results were expressed as mean counts per minute of triplicate wells or stimulation index (experimental counts per minute/counts per minute in wells without antigen stimulation).
Serum antibody against hamster IgG.
Blood was collected from EAE mice when sacrificed (approximately 2 months after sensitization). We used an enzyme‐linked immunosorbent assay to measure serum levels of anti‐hamster immunoglobulin (Ig)G (70, 71). We coated 96 well flat‐bottom Immmuno Plate, MaxiSorp (Nalge Nunc International, Rochester, NY) with hamster IgG overnight. After blocking, serial dilutions of sera were added to the plates and incubated for 90 minutes. After washing, a peroxidase conjugated anti‐mouse IgG (H + L) (minimum cross‐reactivity to bovine, chicken, goat, guinea pig, hamster, horse, human, rabbit, rat and sheep serum protein, Rockland, Gilbertsville, PA) was added for 90 minutes. The plates were colorized using o‐phenylenediamine dihydrochloride (Sigma) and were read at 492 nm using a Titertek Multiskan Plus MK II spectrophotometer (Flow Laboratories, McLean, VA). Optical densities (ODs) were calculated by subtracting ODs of the wells incubated with no sera.
RESULTS
VLA‐2 antibody administered during the effector phase suppresses EAE.
By day 10, half of control PLP139–151 sensitized mice had obvious weight loss and 6% of the mice developed mild paresis of the tail. The disease progressed until day 14, when mice had hind limb paralysis (effector phase, first attack). By day 20, most mice regained weight and recovered (remission). Around 3 weeks after sensitization, mice developed a second attack of EAE (relapse). Mice developed additional relapses with incomplete recovery during the 2‐month observation period.
Mice were treated with VLA‐2 antibody on days 10, 11, 12, 14, 16, 18 and 20 post sensitization, during the entire period of the first attack (defined as the effector phase); antibody treatment was initiated after half of the mice had weight loss and treatment was suspended during the remission (Figure 1A, VLA‐2 early). Mice given VLA‐2 antibody had a lower incidence of paralysis and a statistically significant diminution of the mean maximum clinical scores compared with that of control mice (treated with control antibody) during the first attack of EAE [mean maximum clinical score ± standard error of the mean (SEM): VLA‐2 treatment, 1.5 ± 0.3; control, 2.5 ± 0.2, P < 0.01, t‐test] (Table 1 and Figure 1A).
Figure 1.
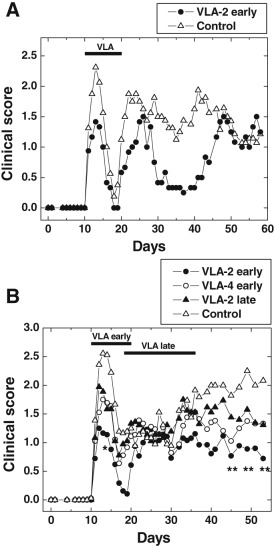
Suppression of experimental autoimmune encephalomyelitis (EAE) by very late antigen (VLA)‐2 antibody during the effector phase. Mice were sensitized with myelin proteolipid protein (PLP)139–151 on day 0. The figure shows mean clinical scores on a given day. A. Groups of mice received VLA‐2 antibody or control antibody on days 10, 11, 12, 14, 16, 18 and 20. VLA‐2 antibody administration suppressed EAE. Each experimental group consisted of six to nine mice. B. VLA‐2 vs. VLA‐4 antibody treatment in EAE. Groups of mice received VLA‐2 antibody or VLA‐4 antibody on days 10, 11, 12, 14, 16, 18 and 20 (effector phase) or VLA‐2 antibody or control antibody on days 18, 21, 23, 25, 27, 29, 32, 34 and 36 (remission and relapse). VLA‐2 antibody suppressed both acute and chronic diseases compared with control mice if it was given during effector phase (* day 14, P < 0.05; ** days 45, 49 and 53, P < 0.01). No significant suppression of disease was observed in mice treated with VLA‐4 antibody during the effector phase or mice treated with VLA‐2 antibody during the remission and relapse phase. Each experimental group consisted of 18 to 20 mice.
Table 1.
Incidence of experimental autoimmune encephalomyelitis (EAE) in mice treated with VLA‐2 or VLA‐4 antibody.*
| Treatment | Cumulative clinical scores† | Mice with clinical sings/total number of mice (%)‡ | |||
|---|---|---|---|---|---|
| Acute stage | Chronic stage | Entire period | Acute disease§ | Chronic disease¶ | |
| Control | 13.1 ± 1.7 | 55.3 ± 7.6 | 70.1 ± 9.2 | 23/28 (82) | 24/26 (92) |
| VLA‐2 early | 5.2 ± 1.1** | 30.0 ± 7.2 | 35.4 ± 8.1 | 16/24 (67) | 15/19 (79) |
| VLA‐2 late | 10.3 ± 1.6 | 44.9 ± 6.5 | 55.2 ± 7.7 | 23/27 (85) | 26/27 (96) |
| VLA‐4 early | 9.9 ± 1.2 | 40.3 ± 9.0 | 50.6 ± 10.4 | 17/18 (94) | 16/18 (89) |
EAE was induced in mice using myelin proteolipid protein (PLP)139–151 in complete Freund’s adjuvant (CFA) on day 0. Groups of mice were treated with antibody against very late antigen (VLA)‐2 or VLA‐4 during the effector phase of EAE (early) or during the remission and relapse of EAE (late). Control mice received isotype control antibody.
†Cumulative disease scores were calculated by adding the daily scores of an individual mouse during the acute stage (from day 0 to 18) or the chronic stage (from day 18 to 53) or over the entire observation period (day 0 to 53). The data are mean ± SEM from two independent experiments. The mice receiving VLA‐2 antibody during the effector phase (VLA‐2 early) had significantly lower cumulative scores during the acute stage, compared with control mice (**P < 0.01, ANOVA).
Number of mice with any clinical signs/total number of mice from two independent experiments. Although the VLA‐2 early treatment group had a statistically lower acute disease incidence in one of two experiments (P < 0.05, chi‐square test), no statistical difference was seen in acute or chronic disease incidence between the groups when the data from two experiments were combined.
Mice developed clinical signs, including tail and/or hindlimb paralysis with or without incontinence, around 10 to 14 days after sensitization with PLP (acute disease). The acute disease subsided completely or incompletely on day 18 (remission).
From day 20, mice showed the second episode of paralysis of tail and/or hind limb, and a few relapses of disease were observed during the 2‐month observation period (chronic disease).
Mice treated with VLA‐2 antibody developed less paralytic disease than control mice in the first attack (VLA‐2 treatment, 67%; control, 82%) and subsequent relapse (VLA‐2 treatment, 79%; control, 92%) (Table 1). Although VLA‐2 antibody was administered until day 20, suppression of clinical signs of EAE was seen to extend to days 30 to 45 in the first experiment (Figure 1A) and to days 40 to 50 in the second experiment (Figure 1B). VLA‐2 antibody treatment resulted in significantly lower cumulative clinical scores during the first attack, compared with control (P < 0.01, ANOVA, Table 1), while during the chronic stage, mice receiving VLA‐2 antibody had a tendency to have lower cumulative scores than control mice (P = 0.06, ANOVA).
Treatment with VLA‐4 antibody during the effector phase.
Antibody against VLA‐4 has been shown to suppress several types of EAE (4, 6, 33, 65, 74, 83). We tested whether administration of VLA‐4 antibody during the effector phase could suppress EAE induced actively with PLP139–151 peptide. Unlike VLA‐2 antibody, we did not see statistically significant suppression of EAE compared with control mice (Figure 1B), although VLA‐4 antibody treatment tended to reduce the disease severity during the chronic stage (Figure 1B). Disease incidence during the first attack (VLA‐4 treatment, 94%; control, 82%) and relapse (VLA‐4 treatment, 89%; control, 92%) were similar between the VLA‐4 antibody‐treated groups vs. control (Table 1). However, while the mean clinical score appeared to be reduced, there was no statistical difference, compared with the control group (Table 1).
Treatment with VLA‐2 antibody during the remission and relapse phase.
We also tested whether we could modulate EAE in mice by giving VLA‐2 antibody during the remission phase, that is, when mice recovered from acute disease but had not shown the signs of the second attack (relapse). Day 18 was chosen for initiating treatment, because mice demonstrated a clear recovery from the acute disease at this time. Although mice were treated with VLA‐2 antibody 3 times a week for 3 weeks (9 injections total), a significant suppression of EAE during or after treatment with VLA‐2 antibody was not observed (Figure 1B, VLA‐2 late). The incidence of mice with a relapse during the chronic phase was also similar between control mice vs. mice treated with VLA‐2 antibody during the remission phase (control, 92%; VLA‐2 treatment, 96%) (Table 1). However, the mean clinical score during the chronic stage was reduced to a similar degree as seen with the VLA‐4 antibody group, although there was no statistical difference, compared with the control group (Figure 1B, Table 1).
VLA‐2 antibody suppresses inflammatory demyelination in the spinal cord.
In all groups, inflammatory demyelinating lesions were more pronounced in the spinal cord than in the brain. Thus, we compared the severity of spinal cord pathology between mice receiving VLA‐2 antibody during the effector phase or relapse and remission, mice receiving VLA‐4 antibody during the effector phase, vs. mice receiving control antibody. MNC infiltration in the meninges and demyelination was observed most frequently in the gracile fasciculus of the posterior funiculus and the ventral root exit zone in the anterolateral funiculus. Subpial demyelination was occasionally seen. The cuneatus fasciculus of the posterior funiculus and dorsal nerve roots were preserved. Perivascular cuffing in the white matter was mild. Mice treated with VLA‐2 antibody at the disease onset (VLA‐2 early, Figure 2A) developed mild inflammatory demyelinating lesions, while some mice in the same group had no lesions in the CNS. Mice treated with VLA‐4 antibody during the effector phase (VLA‐4 early, Figure 2B) or mice treated with VLA‐2 antibody during the relapse and remission (VLA‐2 late, Figure 2C) tended to have less neuropathology, compared with mice treated with control antibody (Control, Figure 2D).
Figure 2.
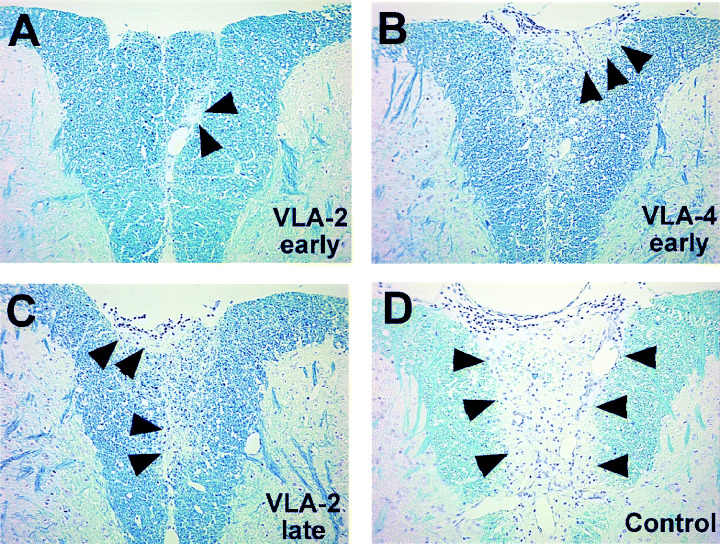
Representative spinal cord pathology of experimental autoimmune encephalomyelitis (EAE) mice treated with very late antigen (VLA)‐2, VLA‐4 or control antibody. A. Mild demyelination (arrowheads) was seen in the posterior funiculus of the spinal cord of mice treated with VLA‐2 antibody during the effector phase (VLA‐2 early). B. VLA‐4 antibody treatment during the effector phase (VLA‐4 early) or (C) VLA‐2 antibody treatment during relapse and remission phase (VLA‐2 late) resulted in moderate demyelinating lesions (arrowhead) in the gracile fasciculus of the posterior funiculus with meningitis. D. In control antibody treatment (control), EAE mice developed mononuclear cell meningitis with totally demyelinated gracile fasciculus of the posterior funiculus (arrowheads). Luxol fast blue stain. Magnification × 83.
Comparing spinal cord pathology scores, we found that, on day 47, mice receiving either VLA‐2 (VLA‐2 early) or VLA‐4 (VLA‐4 early) antibody during the effector phase had reduced meningitis, demyelination and overall spinal cord pathology scores than those mice receiving VLA‐2 (VLA‐2 late) or control (Control) antibody during the remission and relapse phase (Figure 3A). Two months after PLP sensitization (Figure 3B), mice receiving VLA‐2 antibody during the effector phase (VLA‐2 early) had the lowest spinal cord pathology scores in all categories. Mice receiving VLA‐4 antibody during the effector phase (VLA‐4 early) or VLA‐2 during the relapse and remission phase (VLA‐2 late) tended to have lower spinal cord pathology scores, compared with those in control mice (Control, Figure 3B). Thus, the mean pathology scores in the spinal cord correlated with the mean clinical scores for each treatment. Statistically, however, there was no significant difference among groups in the spinal cord. This is most likely caused by a large range of pathology scores among individual mice even within the same groups; each group contained a few mice with no neuropathology.
Figure 3.
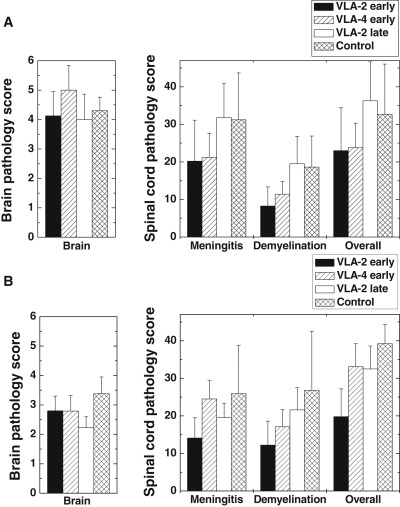
Neuropathology score of experimental autoimmune encephalomyelitis mice treated with very late antigen (VLA)‐2 or VLA‐4 antibody. Mice were sensitized with myelin proteolipid protein (PLP)139–151 on day 0. Groups of mice received VLA‐2 antibody or VLA‐4 antibody during the effector phase or VLA‐2 or control antibody during the remission and relapse phase. A. On day 47, VLA‐2 early and VLA‐4 early groups had lower meningitis, demyelination and overall spinal cord pathology scores than VLA‐2 late and control groups. B. Two months after PLP sensitization, VLA‐2 early group had the lowest meningitis, demyelination and overall spinal cord pathology score between groups. VLA‐4 early and VLA‐2 late groups had slightly lower spinal cord pathology scores than the control group. No difference was seen in brain pathology scores 47 days or 2 months post sensitization. Each treatment group consisted of four or five mice on day 47, and nine to 13 mice on 2 months post sensitization. Values are the means ± SEM.
In addition to spinal cord pathology, we examined brain pathology in treated EAE mice. In all groups, mild MNC meningitis was detected near the hippocampus and cerebral peduncle. Low levels of perivascular cuffing composed of MNCs were observed mainly in the cerebellar white matter and brainstem. Demyelination was seen in the brainstem in only a few mice. There were no differences in the brain pathology scores among the groups at 47 days or 2 months post sensitization.
Meningitis and demyelination correlate with clinical signs of EAE.
We next examined which pathologic features in the spinal cord were associated with clinical signs. Overall neuropathology score, which includes meningitis, perivascular cuffing and demyelination, most strongly correlated with clinical scores (r = 0.9, P < 0.01, Figure 4D). Meningitis (r = 0.84, P < 0.01, Figure 4A) and demyelination (r = 0.79, P < 0.01, Figure 4C) also strongly correlated with clinical signs, while perivascular cuffing showed some association with clinical signs (r = 0.54, P < 0.01, Figure 4B). In contrast, there was no correlation between brain pathology and clinical signs (r = 0.14, P = 0.51, data not shown).
Figure 4.
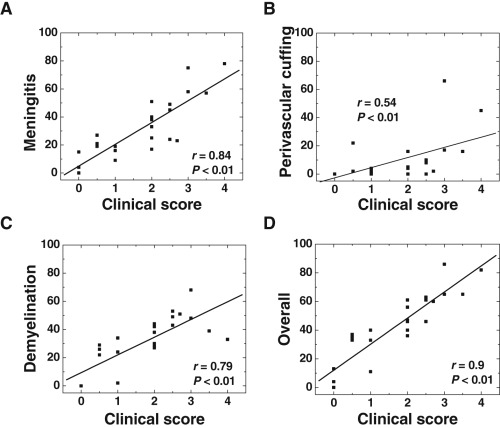
Correlation of spinal cord pathology with clinical scores of experimental autoimmune encephalomyelitis (EAE). We compared spinal cord pathology with clinical scores of EAE, 2 months after proteolipid protein sensitization. We scored severities of meningitis (A), perivascular cuffing (B), demyelination (C) as well as overall neuropathology (D) that included meningitis, perivascular cuffing and/or demyelination. Meningitis, demyelination and overall scores were strongly correlated with clinical signs. The data were from pooled spinal cord samples of 24 mice treated either with very late antigen (VLA)‐2, VLA‐4, or control antibody.
VLA‐2 antibody does not suppress EAE during the induction phase.
Adhesion molecules have been shown to play a role in not only homing but also priming of T cells (52). To determine the role of VLA‐2 at the time of priming with PLP139–151 peptide, we administered VLA‐2 antibody to mice during the induction phase but prior to mice having any clinical signs (days 0, 2, 4, 7 and 9). Mice treated with VLA‐2 antibody had a similar disease onset, clinical scores and weight loss compared with mice treated with control antibody (Figure 5). Although clinical EAE scores, based on the extent of paralysis, and weight changes were identical between the two groups during the first attack, mice treated with VLA‐2 antibody showed less movement, had ruffled fur and a hunched back, which are not seen in EAE, and required hand‐feeding and watering for survival. During the chronic stage, mice treated with VLA‐2 antibody tended to have higher clinical scores than control mice (mean maximum score: VLA‐2 treatment, 3.1 ± 0.5; control group, 2.6 ± 0.6), however, this difference did not achieve statistical significance (P > 0.05, t‐test). Incidences of acute and chronic diseases were identical between the two groups: 86% of mice developed acute attack and relapse in both groups.
Figure 5.
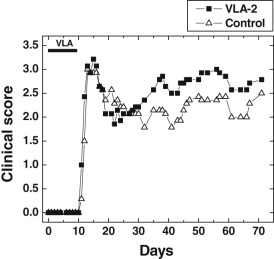
No suppression of experimental autoimmune encephalomyelitis by very late antigen (VLA)‐2 antibody during the induction phase. Mice were sensitized with myelin proteolipid protein (PLP)139–151 on day 0. Groups of mice received VLA‐2 antibody or control antibody on days 0, 2, 4, 7 and 9. No significant difference was seen in mean clinical scores between the two groups. Each experimental group consisted of seven mice.
We also compared neuropathology between control and VLA‐2 treatment groups, when mice were euthanized. No differences in the severity or distribution of the lesions in the CNS were observed. Similarly, there was no difference in neuropathology score in the spinal cord (P > 0.05, mean pathology score ± SEM: meningitis, control 36.1 ± 8.0 vs. VLA‐2 treatment 26.6 ± 7.4; perivascular cuffing, control 8.3 ± 3.3 vs. VLA‐2 treatment 7.3 ± 2.6; demyelination, control 45.1 ± 8.7 vs. VLA‐2 treatment 50.7 ± 9.6; overall, control 58.7 ± 8.6 vs. VLA‐2 treatment 60.4 ± 8.8). In the brain, there were no differences in the severity or distribution of lesions between the two groups (P > 0.05, mean brain pathology score ± SEM: control, 5 ± 0.6; VLA‐2 treatment, 5.4 ± 0.7).
Lymphoproliferative responses to PLP or other CNS antigens did not correlate with treatment or clinical signs.
We tested whether PLP specific T‐cell immune responses correlated with severity of clinical signs or treatment with VLA‐2 or VLA‐4 antibody. MNCs were isolated from spleens and lymph nodes, and examined for lymphoproliferative responses to myelin antigens using 3H thymidine incorporation. In all groups sensitized with PLP139–151, we found high anti‐PLP139–151 specific T‐cell proliferation in mice treated with VLA‐2 antibody during the induction phase (Figure 6A), during the effector phase (VLA‐2 early, Figure 6B) or during remission (VLA‐2 late, Figure 6B), similar to levels seen in control groups. A similarly high degree of T‐cell proliferation to PLP139–151 in asymptomatic mice compared with mice having severe paralysis was also seen (data not shown). Others have reported that relapses correlate with epitope spreading to PLP178–191, MOG92–106 or MBP84–104 in PLP139–151 induced EAE in SJL mice (65, 75). However, we did not see significant epitope spreading in any groups. MNCs from mice treated with VLA‐2 antibody during either effector phase or remission, tended to have a high spontaneous proliferation (autoproliferation) in wells without antigen stimuli, compared with EAE mice receiving control antibody, which resulted in lower stimulation indices in groups treated with VLA‐2 antibody (Figure 6C). Mice treated with VLA‐4 antibody during the effector phase had similar lymphoproliferative responses as T cells from control mice (data not shown).
Figure 6.
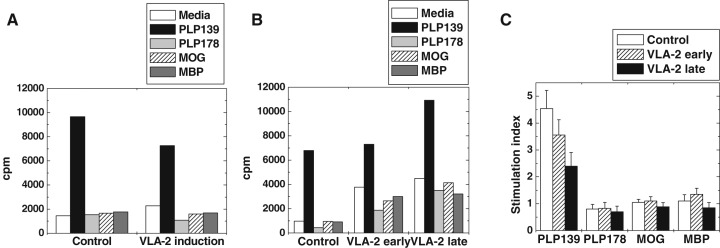
Lymphoproliferative responses to myelin antigens in experimental autoimmune encephalomyelitis (EAE) mice treated with very late antigen (VLA)‐2 antibody. Mononuclear cells (MNCs) were isolated from spleens or lymph nodes and incubated with myelin proteolipid protein (PLP)139–151 (PLP139), PLP178–191 (PLP178), MOG92–106 (MOG) or MBP84–104 (MBP) for 5 days. A. Comparably high lymphoproliferative responses to PLP139–151 were detected both in EAE mice which received control antibody (control) or VLA‐2 antibody during the induction phase of EAE. B. EAE mice treated with control antibody (control), or with VLA‐2 antibody during the effector phase (VLA‐2 early) or during remission (VLA‐2 late) had high lymphoproliferation to PLP139–151. C. Significant lymphoproliferation was detected only to PLP139–151, but not other myelin antigens. Because of high spontaneous proliferation, mice treated with VLA‐2 antibody during the effector phase (VLA‐2 early) or remission (VLA‐2 late) tended to have lower stimulation indices in PLP139–151 stimulation, compared with mice injected with control antibody (control). We did not detect significant epitope spreading to any other myelin antigens. Results are means of 4 (A), 6 (B) or 11 (C) lymphoproliferative assays using MNCs from spleens or lymph nodes pooled from two to three mice.
Antibody responses against VLA‐2 antibody do not correlate with clinical signs of EAE.
Injections of antibody against adhesion molecules could induce antibody responses to the therapeutic antibody produced in a different species; such antibody responses potentially neutralize inoculated antibody (40, 76). Antibody responses against even humanized antibody have been reported (18). We titrated antibody levels against hamster IgG in sera from mice which were administered the VLA‐2 antibody (made in hamster). As controls, we used sera from PLP139–151 induced EAE mice that received no antibody. Mice were bled when euthanized about 2 months after sensitization. As VLA‐2 antibody is Armenian hamster IgG, the levels of serum antibody against hamster IgG were measured using an enzyme‐linked immunosorbent assay as described previously (68, 69). We found serum anti‐hamster IgG in all mice that received VLA‐2 antibody. All mice developed similar titers of anti‐hamster IgG regardless of their clinical signs of EAE. In contrast, we did not detect serum anti‐hamster IgG in controls (mice not receiving hamster IgG) (mean OD ± SEM at 1:210 dilution: VLA‐2 antibody group, 0.38 ± 0.01; control, 0), while control EAE mice developed high antibody responses against PLP139–151 (data not shown).
DISCUSSION
The interaction of T cells with the surrounding ECM environment can have a considerable impact on a variety of T‐cell functions. ECM has been shown to provide a costimulus for T‐cell proliferation (57) and cytokine production when T cells are cultured with CD3 antibody (50). VLA‐2 antibody can inhibit collagen induced T‐cell proliferation as well as tumor necrosis factor α secretion from activated T cells. Interaction of integrins and ECM may either positively contribute to migration or, alternatively, favor anchoring and immobilization of lymphocytes at inflammatory sites. VLA‐2 blockade has been shown to attenuate T‐cell locomotion, altering locomotion and orientation in a three‐dimensional collagen matrix (15). In addition, retention of inflammatory cells can lead to activation of CNS cells such as microglia/macrophages and astrocytes producing enhanced pathology.
In our studies, VLA‐2 antibody treatment was most effective when antibody was given immediately after the onset of the disease. Although we do not know the precise mechanism(s) by which VLA‐2 antibody suppressed EAE, blockade of interaction between VLA‐2 and ECM could alter encephalitogenic T‐cell functions in the CNS, such as cytokine production and locomotion, leading to suppression of meningitis and demyelination. In contrast, VLA‐2 antibody did not suppress EAE when antibody was given during the induction phase. In addition, treatment of mice with VLA‐2 antibody did not alter lymphoproliferative responses to PLP or any other myelin antigens, regardless of the timing of antibody administration. Thus, VLA‐2 seems not to play a role in priming of T cells to the initial inciting self‐antigen or development of epitope spreading to other self‐peptides.
VLA‐2 can interact with laminin and collagen. In the CNS, immunoreactivity of laminin has been demonstrated in some particular areas, such as the hippocampus (82), and collagen is encountered primarily in association with blood vessels and is virtually absent from the parenchyma. Expression of laminin has been shown to be altered under pathological conditions (80). Thus, future investigation of VLA‐2 expression and its ligands in inflammatory sites might further our understanding of the role of VLA‐2 in disease.
Activation‐induced cell death (AICD) has been suggested to play an important role in the regulation of immune response, including deletion of autoreactive T cells in EAE (8). Gendron et al (17) showed that engagement of VLA‐2 with collagen type I, but not with fibronectin or laminin, reduced both AICD and Fas‐induced apoptosis of human T cells in vitro. They also found that the inhibition of apoptosis of T cells was abrogated in the presence of blocking VLA‐2 antibodies, suggesting the potential importance that interactions between VLA‐2 and collagen may have on the control of T lymphocyte homeostasis and their persistence in chronic inflammatory diseases. This is in contrast to the effect of VLA‐4 on apoptosis of a human antigen specific T‐cell clone, where AICD was enhanced by the co‐ligation of the T‐cell receptor (TCR) and VLA‐4 or leukocyte function‐associated antigen‐1 (CD11a/CD18) in vitro (9). Depending on the antibody, a VLA‐4 antibody, 9C10, can induce apoptosis in mouse T cells in vitro, while the other adhesion blocking VLA‐4 antibodies, PS/2 and R1/2, do not induce apoptosis (64). In vivo administration of VLA‐4 or VCAM‐1 antibody resulted in an increase in T‐cell apoptosis in rats with experimental autoimmune neuritis (41). It is not yet clear whether the antibodies used in this study were blocking or stimulating antibody, as the name of the VLA‐4 antibody producing cell clone was not provided. In our study, we used adhesion blocking VLA‐2 (Ha1/29) and VLA‐4 (PS/2) antibodies. Thus, our VLA‐2 antibody could abrogate inhibition of AICD of T cells, leading to an increase in apoptosis of PLP specific autoreactive T cells. In contrast, VLA‐4 antibody could block enhancement of AICD of T cells, resulting in a decrease in apoptosis of encephalitogenic T cells.
DX5, monoclonal VLA‐2 antibody, has been used as a marker for murine natural killer (NK) and NKT cells in mice (2). Although the function of VLA‐2 on NK and NKT cells is largely unknown, both NK and NKT cells have been suggested to play an important role in some, but not all, EAE models and MS patients (25, 26, 46, 51). Fritz and Zhao (16) demonstrated that DX5+ NK1.1+ TCR αβ+ T cells regulated recovery from acute EAE in an adoptive transfer model using TCR β‐chain knockout C57BL/6 mice. As SJL/J mice, which we used in our current experiments, contain few or no NK/NKT cells (30), modulation of regulatory NK/NKT cells by VLA‐2 antibody would likely play a minor role, if any, in the suppression of EAE by VLA‐2 antibody treatment. In contrast to mice, VLA‐2 is not a consistent marker on human NK cells, and its expression changes depending on the conditions. Nevertheless, NK and NKT cell function should be monitored in future studies in humans.
Neutrophil locomotion and recruitment to extravascular tissue has been shown to be dependent on VLA‐2, but not on VLA‐4 (78). The inflammatory infiltrate in MOG induced primary and secondary progressive EAE is dominated by neutrophils (69, 70, 71). In this model, few T cells exist in demyelinating lesions during the progressive stage of EAE, although EAE seems to be initiated by T cells. Demyelinating pathology with neutrophilic infiltration has been demonstrated in other forms of EAE and MS (28, 42, 43, 44, 45, 54, 60). Thus, treatment with VLA‐2 antibody, but not with VLA‐4 antibody, may be effective in other clinical types of MS and EAE.
Infiltrating macrophages and activated microglia are involved in the inflammatory response in the CNS initiated by autoreactive T cells (5, 56). The macrophages and microglia produce cytokines such as tumor necrosis factor and IL‐1 that can intensify disease. Limiting the influx of macrophages should also limit the extent of pathology in the CNS.
In the current experiment, treatment with VLA‐4 antibody during the effector phase did not have a marked influence on EAE. While the effect of antibody against α4 integrin on development of new brain lesions in MS has been studied in three published, randomized, controlled phase II trials (58), careful evaluation is required in all studies. The first clinical study using natalizumab resulted in a rebound increase in the relapse rate after stopping treatment (72). This post‐treatment rebound is similar to what has been observed in EAE studies, using anti‐VLA‐4 treatment (65, 66).
In the second study, treatment with natalizumab reduced the mean number of relapses and new lesions, compared with controls during the 6‐month treatment (new lesion: control, 9.6 per patient; natalizumab, 0.7 per patient) (48). However, a 6‐month post‐treatment follow‐up study showed no difference in the numbers of relapses or new lesions between control and treated groups (new lesions: control, 2.7 per patient; natalizumab, 2.8 per patient). The above results can be explained by exacerbation of disease by placebo, rather than by suppression of disease by natalizumab, during the treatment, as: (i) the number of new lesions in the control group decreased from 9.6 to 2.7, while those in the treated group increased from 0.7 to 2.8, during the 6‐month follow‐up period; and (ii) during the follow‐up period, the volume of enhancing lesions decreased in the control group (from 1169 mm3 to 442 mm3), but increased in the natalizumab‐treated group (from 156 mm3 to 306 mm3). There was a trend toward an increased rate of infections in the natalizumab‐treated patients. In the third study, a significant decrease in lesion volume was observed at 1 and 3 weeks, but not 14 weeks, after a single injection of natalizumab (53). Steroids were required to treat relapses in 3% of the placebo group, 12% of the 1‐mg/kg natalizumab group and 8% of the 3‐mg group during the study.
Recently, treatment with VLA‐4 antibody has been shown to influence not only memory and effector T cells but also humoral immunity and mucosal immunity (77). In addition, VLA‐4 and other adhesion molecules have been shown to be expressed on CD25+CD4+ regulatory T cells (36, 63). Thus, treatment with VLA‐4 antibody could suppress not only memory and effector T cells, but also other immune cells. The balance of such humoral vs. cellular immune responses and/or regulatory vs. effector T cells positive for certain adhesion molecules may change depending on the disease course, for example, between remission vs. relapses in MS and its animal models. In this context, timing of treatment with anti‐adhesion molecules can result in discordant effects as in our current experiments or experiments reported by others (65, 66).
In conclusion, the above findings have important implications for the treatment of demyelinating diseases of the CNS, indicating that VLA‐2 plays a contributory role in clinical and pathological disease in EAE and is a potential target for early therapeutic intervention in the treatment of MS.
ACKNOWLEDGMENTS
We thank Nikki Jo Kirkman BS and Jane E. Libbey MS for many helpful discussions, and Sarah E. Doyle BS, Faris Hasanovic, Lincoln A. Hunt BS, J. Wes Peterson, Steven R. Wheelwright, Reina Yamaji, Nathan J. Young and Aminatu R. Yusuf for excellent technical assistance. We are grateful to Ms Kathleen Borick for preparation of the manuscript. This work was supported by NIH grant 5 R01 NS40350 and funding from Targeted Molecules Corporation. Target Molecules Corporation was acquired by Chromos Molecular Systems February 2, 2006.
REFERENCES
- 1. Andreasen SØ, Thomsen AR, Koteliansky VE, Novobrantseva TI, Sprague AG, De Fougerolles AR, Christensen JP (2003) Expression and functional importance of collagen‐binding integrins, α1β1 and α2β1, on virus‐activated T cells. J Immunol 171:2804–2811. [DOI] [PubMed] [Google Scholar]
- 2. Arase H, Saito T, Phillips JH, Lanier LL (2001) Cutting edge: The mouse NK cell‐associated antigen recognized by DX5 monoclonal antibody is CD49b (α2 integrin, very late antigen‐2). J Immunol 167:1141–1144. [DOI] [PubMed] [Google Scholar]
- 3. Barnett LA, Whitton JL, Wada Y, Fujinami RS (1993) Enhancement of autoimmune disease using recombinant vaccinia virus encoding myelin proteolipid protein [published erratum appears in J Neuroimmunol 48:120, 1993] J Neuroimmunol 44:15–25. [DOI] [PubMed] [Google Scholar]
- 4. Baron JL, Madri JA, Ruddle NH, Hashim G, Janeway CA Jr (1993) Surface expression of α4 integrin by CD4 T cells is required for their entry into brain parenchyma. J Exp Med 177:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benveniste EN (1997) Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med 75:165–173. [DOI] [PubMed] [Google Scholar]
- 6. Brocke S, Piercy C, Steinman L, Weissman IL, Veromaa T (1999) Antibodies to CD44 and integrin α4, but not L‐selectin, prevent central nervous system inflammation and experimental encephalomyelitis by blocking secondary leukocyte recruitment. Proc Natl Acad Sci USA 96:6896–6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carlos TM, Harlan JM (1994) Leukocyte‐endothelial adhesion molecules. Blood 84:2068–2101. [PubMed] [Google Scholar]
- 8. Critchfield JM, Racke MK, Zúñiga‐Pflücker JC, Cannella B, Raine CS, Goverman J, Lenardo MJ (1994) T cell deletion in high antigen dose therapy of autoimmune encephalomyelitis. Science 263:1139–1143. [DOI] [PubMed] [Google Scholar]
- 9. Damle NK, Klussman K, Leytze G, Aruffo A, Linsley PS, Ledbetter JA (1993) Costimulation with integrin ligands intercellular adhesion molecule‐1 or vascular cell adhesion molecule‐1 augments activation‐induced death of antigen‐specific CD4+ T lymphocytes. J Immunol 151:2368–2379. [PubMed] [Google Scholar]
- 10. De Fougerolles AR, Sprague AG, Nickerson‐Nutter CL, Chi‐Rosso G, Rennert PD, Gardner H, Gotwals PJ, Lobb RR, Koteliansky VE (2000) Regulation of inflammation by collagen‐binding integrins α1β1 and α2β1 in models of hypersensitivity and arthritis. J Clin Invest 105:721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Du Pasquier RA, Schmitz JE, Jean‐Jacques J, Zheng Y, Gordon J, Khalili K, Letvin NL, Koralnik IJ (2004) Detection of JC virus‐specific cytotoxic T lymphocytes in healthy individuals. J Virol 78:10206–10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Edelson BT, Li Z, Pappan LK, Zutter MM (2004) Mast cell‐mediated inflammatory responses require the α2β1 integrin. Blood 103:2214–2220. [DOI] [PubMed] [Google Scholar]
- 13. Engelhardt B (1998) The role of α4‐integrin in T lymphocyte migration into the inflamed and noninflamed central nervous system. Curr Top Microbiol Immunol 231:51–64. [DOI] [PubMed] [Google Scholar]
- 14. Finkelstein SD (1997) Polyomaviruses and progressive multifocal leukoencephalopathy. In: Pathology of Infectious Diseases. Connor DH, Chandler FW, Schwartz DA, Manz HJ, Lack EE (eds), pp. 265–272. Appleton & Lange: Stamford. [Google Scholar]
- 15. Friedl P, Noble PB, Zänker KS (1995) T lymphocyte locomotion in a three‐dimensional collagen matrix. Expression and function of cell adhesion molecules. J Immunol 154:4973–4985. [PubMed] [Google Scholar]
- 16. Fritz RB, Zhao M‐L (2001) Regulation of experimental autoimmune encephalomyelitis in the C57BL/6J mouse by NK1.1+, DX5+, αβ+ T cells. J Immunol 166:4209–4215. [DOI] [PubMed] [Google Scholar]
- 17. Gendron S, Couture J, Aoudjit F (2003) Integrin α2β1 inhibits Fas‐mediated apoptosis in T lymphocytes by protein phosphatase 2A‐dependent activation of the MAPK/ERK pathway. J Biol Chem 278:48633–48643. [DOI] [PubMed] [Google Scholar]
- 18. Ghosh S, Goldin E, Gordon FH, Malchow HA, Rask‐Madsen J, Rutgeerts P, Vyhnálek P, Zádorová Z, Chir B, Palmer T, Donoghue S (2003) Natalizumab for active Crohn’s disease. (the Natalizumab Pan‐European Study Group). N Engl J Med 348:24–32. [DOI] [PubMed] [Google Scholar]
- 19. Gilden DH (2005) Infectious causes of multiple sclerosis. Lancet Neurol 4:195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gocinski BL, Tigelaar RE (1990) Roles of CD4+ and CD8+ T cells in murine contact sensitivity revealed by in vivo monoclonal antibody depletion. J Immunol 144:4121–4128. [PubMed] [Google Scholar]
- 21. Gullberg D, Gehlsen KR, Turner DC, Ahlen K, Zijenah LS, Barnes MJ, Rubin K (1992) Analysis of alpha 1 beta 1, alpha 2 beta 1 and alpha 3 beta 1 integrins in cell—collagen interactions: identification of conformation dependent alpha 1 beta 1 binding sites in collagen type I. EMBO J 11:3865–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heino J (2000) The collagen receptor integrins have distinct ligand recognition and signaling functions. Matrix Biol 19:319–323. [DOI] [PubMed] [Google Scholar]
- 23. Hemler ME (1990) VLA proteins in the integrin family: structures, functions, and their role on leukocytes. Annu Rev Immunol 8:365–400. [DOI] [PubMed] [Google Scholar]
- 24. Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110:673–687. [DOI] [PubMed] [Google Scholar]
- 25. Illés Z, Kondo T, Newcombe J, Oka N, Tabira T, Yamamura T (2000) Differential expression of NK T cell Vα24JαQ invariant TCR chain in the lesions of multiple sclerosis and chronic inflammatory demyelinating polyneuropathy. J Immunol 164:4375–4381. [DOI] [PubMed] [Google Scholar]
- 26. Illés Z, Shimamura M, Newcombe J, Oka N, Yamamura T (2004) Accumulation of Vα7.2‐Jα33 invariant T cells in human autoimmune inflammatory lesions in the nervous system. Int Immunol 16:223–230. [DOI] [PubMed] [Google Scholar]
- 27. Inoue A, Koh C‐S, Yamazaki M, Ichikawa M, Isobe M, Ishihara Y, Yagita H, Kim BS (1997) Anti‐adhesion molecule therapy in Theiler’s murine encephalomyelitis virus‐induced demyelinating disease. Int Immunol 9:1837–1847. [DOI] [PubMed] [Google Scholar]
- 28. Ishizu T, Osoegawa M, Mei F‐J, Kikuchi H, Tanaka M, Takakura Y, Minohara M, Murai H, Mihara F, Taniwaki T, Kira J (2005) Intrathecal activation of the IL‐17/IL‐8 axis in opticospinal multiple sclerosis. Brain 128:988–1002. [DOI] [PubMed] [Google Scholar]
- 29. Jarvis GE, Best D, Watson SP (2004) Glycoprotein VI/Fc receptor γ chain‐independent tyrosine phosphorylation and activation of murine platelets by collagen. Biochem J 383:581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kaminsky SG, Nakamura I, Cudkowicz G (1983) Selective defect of natural killer and killer cell activity against lymphomas in SJL mice: low responsiveness to interferon inducers. J Immunol 130:1980–1984. [PubMed] [Google Scholar]
- 31. Kanwar JR, Harrison JEB, Wang D, Leung E, Mueller W, Wagner N, Krissansen GW (2000) β7 integrins contribute to demyelinating disease of the central nervous system. J Neuroimmunol 103:146–152. [DOI] [PubMed] [Google Scholar]
- 32. Kanwar JR, Kanwar RK, Wang D, Krissansen GW (2000) Prevention of a chronic progressive form of experimental autoimmune encephalomyelitis by an antibody against mucosal addressin cell adhesion molecule‐1, given early in the course of disease progression. Immunol Cell Biol 78:641–645. [DOI] [PubMed] [Google Scholar]
- 33. Kent SJ, Karlik SJ, Cannon C, Hines DK, Yednock TA, Fritz LC, Horner HC (1995) A monoclonal antibody to α4 integrin suppresses and reverses active experimental allergic encephalomyelitis. J Neuroimmunol 58:1–10. [DOI] [PubMed] [Google Scholar]
- 34. Kern A, Eble J, Golbik R, Kühn K (1993) Interaction of type IV collagen with the isolated integrins α1β1 and α2β1. Eur J Biochem 215:151–159. [DOI] [PubMed] [Google Scholar]
- 35. Kleinschmidt‐DeMasters BK, Tyler KL (2005) Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta‐1a for multiple sclerosis. N Engl J Med 353:369–374. [DOI] [PubMed] [Google Scholar]
- 36. Kohm AP, Carpentier PA, Anger HA, Miller SD (2002) Cutting edge: CD4+CD25+ regulatory T cells suppress antigen‐specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol 169:4712–4716. [DOI] [PubMed] [Google Scholar]
- 37. Kono DH, Urban JL, Horvath SJ, Ando DG, Saavedra RA, Hood L (1988) Two minor determinants of myelin basic protein induce experimental allergic encephalomyelitis in SJL/J mice. J Exp Med 168:213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Koralnik IJ (2004) New insights into progressive multifocal leukoencephalopathy. Curr Opin Neurol 17:365–370. [DOI] [PubMed] [Google Scholar]
- 39. Langer‐Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D (2005) Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med 353:375–381. [DOI] [PubMed] [Google Scholar]
- 40. Léger OJP, Yednock TA, Tanner L, Horner HC, Hines DK, Keen S, Saldanha J, Jones ST, Fritz LC, Bendig MM (1997) Humanization of a mouse antibody against human alpha‐4 integrin: A potential therapeutic for the treatment of multiple sclerosis. Hum Antibodies 8:3–16. [PubMed] [Google Scholar]
- 41. Leussink VI, Zettl UK, Jander S, Pepinsky RB, Lobb RR, Stoll G, Toyka KV, Gold R (2002) Blockade of signaling via the very late antigen (VLA‐4) and its counterligand vascular cell adhesion molecule‐1 (VCAM‐1) causes increased T cell apoptosis in experimental autoimmune neuritis. Acta Neuropathol (Berl) 103:131–136. [DOI] [PubMed] [Google Scholar]
- 42. Levine S (1974) Hyperacute, neutrophilic, and localized forms of experimental allergic encephalomyelitis: a review. Acta Neuropathol (Berl) 28:179–189. [DOI] [PubMed] [Google Scholar]
- 43. Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM, Trebst C, Weinshenker B, Wingerchuk D, Parisi JE, Lassmann H (2002) A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain 125:1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Määttä JA, Erälinna J‐P, Röyttä M, Salmi AA, Hinkkanen AE (1996) Physical state of the neuroantigen in adjuvant emulsions determines encephalitogenic status in the BALB/c mouse. J Immunol Methods 190:133–141. [DOI] [PubMed] [Google Scholar]
- 45. Määttä JA, Sjoholm UR, Nygardas PT, Salmi AA, Hinkkanen AE (1998) Neutrophils secreting tumor necrosis factor alpha infiltrate the central nervous system of BALB/c mice with experimental autoimmune encephalomyelitis. J Neuroimmunol 90:162–175. [DOI] [PubMed] [Google Scholar]
- 46. Matsumoto Y, Kohyama K, Aikawa Y, Shin T, Kawazoe Y, Suzuki Y, Tanuma N (1998) Role of natural killer cells and TCRγδ T cells in acute autoimmune encephalomyelitis. Eur J Immunol 28:1681–1688. [DOI] [PubMed] [Google Scholar]
- 47. Mendrick DL, Kelly DM (1993) Temporal expression of VLA‐2 and modulation of its ligand specificity by rat glomerular epithelial cells in vitro . Lab Invest 69:690–702. [PubMed] [Google Scholar]
- 48. Miller DH, Khan OA, Sheremata WA, Blumhardt LD, Rice GPA, Libonati MA, Willmer‐Hulme AJ, Dalton CM, Miszkiel KA, O’Connor PW (2003) A controlled trial of Natalizumab for relapsing multiple sclerosis. (the International Natalizumab Multiple Sclerosis Trial Group). N Engl J Med 348:15–23. [DOI] [PubMed] [Google Scholar]
- 49. Miyake K, Weissman IL, Greenberger JS, Kincade PW (1991) Evidence for a role of the integrin VLA‐4 in lympho‐hemopoiesis. J Exp Med 173:599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Miyake S, Sakurai T, Okumura K, Yagita H (1994) Identification of collagen and laminin receptor integrins on murine T lymphocytes. Eur J Immunol 24:2000–2005. [DOI] [PubMed] [Google Scholar]
- 51. Miyamoto K, Miyake S, Yamamura T (2001) A synthetic glycolipid prevents autoimmune encephalomyelitis by inducing TH2 bias of natural killer T cells. Nature 413:531–534. [DOI] [PubMed] [Google Scholar]
- 52. Myers KJ, Witchell DR, Graham MJ, Koo S, Butler M, Condon TP (2005) Antisense oligonucleotide blockade of alpha 4 integrin prevents and reverses clinical symptoms in murine experimental autoimmune encephalomyelitis. J Neuroimmunol 160:12–24. [DOI] [PubMed] [Google Scholar]
- 53. O’Connor PW, Goodman A, Willmer‐Hulme AJ, Libonati MA, Metz L, Murray RS, Sheremata WA, Vollmer TL, Stone LA (2004) Randomized multicenter trial of natalizumab in acute MS relapses: Clinical and MRI effects (the Natalizumab Multiple Sclerosis Trial Group). Neurology 62:2038–2043. [DOI] [PubMed] [Google Scholar]
- 54. Piddlesden SJ, Storch MK, Hibbs M, Freeman AM, Lassmann H, Morgan BP (1994) Soluble recombinant complement receptor 1 inhibits inflammation and demyelination in antibody‐mediated demyelinating experimental allergic encephalomyelitis. J Immunol 152:5477–5484. [PubMed] [Google Scholar]
- 55. Pinkstaff JK, Detterich J, Lynch G, Gall C (1999) Integrin subunit gene expression is regionally differentiated in adult brain. J Neurosci 19:1541–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Raivich G, Banati R (2004) Brain microglia and blood‐derived macrophages: molecular profiles and functional roles in multiple sclerosis and animal models of autoimmune demyelinating disease. Brain Res Brain Res Rev 46:261–281. [DOI] [PubMed] [Google Scholar]
- 57. Rao WH, Hales JM, Camp RDR (2000) Potent costimulation of effector T lymphocytes by human collagen type I. J Immunol 165:4935–4940. [DOI] [PubMed] [Google Scholar]
- 58. Rice GPA, Hartung H‐P, Calabresi PA (2005) Anti‐α4 integrin therapy for multiple sclerosis: Mechanisms and rationale. Neurology 64:1336–1342. [DOI] [PubMed] [Google Scholar]
- 59. Sakai K, Zamvil SS, Mitchell DJ, Lim M, Rothbard JB, Steinman L (1988) Characterization of a major encephalitogenic T cell epitope in SJL/J mice with synthetic oligopeptides of myelin basic protein. J Neuroimmunol 19:21–32. [DOI] [PubMed] [Google Scholar]
- 60. Sakuma H, Kohyama K, Park IK, Miyakoshi A, Tanuma N, Matsumoto Y (2004) Clinicopathological study of a myelin oligodendrocyte glycoprotein‐induced demyelinating disease in LEW.1AV1 rats. Brain 127:2201–2213. [DOI] [PubMed] [Google Scholar]
- 61. Soilu‐Hänninen M, Röyttä M, Salmi A, Salonen R (1997) Therapy with antibody against leukocyte integrin VLA‐4 (CD49d) is effective and safe in virus‐facilitated experimental allergic encephalomyelitis. J Neuroimmunol 72:95–105. [DOI] [PubMed] [Google Scholar]
- 62. Springer TA (1995) Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu Rev Physiol 57:827–872. [DOI] [PubMed] [Google Scholar]
- 63. Stassen M, Fondel S, Bopp T, Richter C, Müller C, Kubach J, Becker C, Knop J, Enk AH, Schmitt S, Schmitt E, Jonuleit H (2004) Human CD25+ regulatory T cells: two subsets defined by the integrins α4β7 or α4β1 confer distinct suppressive properties upon CD4+ T helper cells. Eur J Immunol 34:1303–1311. [DOI] [PubMed] [Google Scholar]
- 64. Tchilian EZ, Owen JJT , Jenkinson EJ (1997) Anti‐α4 integrin antibody induces apoptosis in murine thymocytes and staphylococcal enterotoxin B‐activated lymph node T cells. Immunology 92:321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Theien BE, Vanderlugt CL, Eagar TN, Nickerson‐Nutter C, Nazareno R, Kuchroo VK, Miller SD (2001) Discordant effects of anti‐VLA‐4 treatment before and after onset of relapsing experimental autoimmune encephalomyelitis. J Clin Invest 107:995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Theien BE, Vanderlugt CL, Nickerson‐Nutter C, Cornebise M, Scott DM, Perper SJ, Whalley ET, Miller SD (2003) Differential effects of treatment with a small‐molecule VLA‐4 antagonist before and after onset of relapsing EAE. Blood 102:4464–4471. [DOI] [PubMed] [Google Scholar]
- 67. Tsunoda I, Fujinami RS (1996) Two models for multiple sclerosis: Experimental allergic encephalomyelitis and Theiler’s murine encephalomyelitis virus. J Neuropathol Exp Neurol 55:673–686. [DOI] [PubMed] [Google Scholar]
- 68. Tsunoda I, Kuang L‐Q, Tolley ND, Whitton JL, Fujinami RS (1998) Enhancement of experimental allergic encephalomyelitis (EAE) by DNA immunization with myelin proteolipid protein (PLP) plasmid DNA. J Neuropathol Exp Neurol 57:758–767. [DOI] [PubMed] [Google Scholar]
- 69. Tsunoda I, Kuang L‐Q, Theil DJ, Fujinami RS (2000) Antibody association with a novel model for primary progressive multiple sclerosis: Induction of relapsing‐remitting and progressive forms of EAE in H2S mouse strains. Brain Pathol 10:402–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tsunoda I, Kuang L‐Q, Igenge IZM, Fujinami RS (2005) Converting relapsing remitting to secondary progressive experimental allergic encephalomyelitis (EAE) by ultraviolet B irradiation. J Neuroimmunol 160:122–134. [DOI] [PubMed] [Google Scholar]
- 71. Tsunoda I, Libbey JE, Kuang L‐Q, Terry EJ, Fujinami RS (2005) Massive apoptosis in lymphoid organs in animal models for primary and secondary progressive multiple sclerosis. Am J Pathol 167:1631–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tubridy N, Behan PO, Capildeo R, Chaudhuri A, Forbes R, Hawkins CP, Hughes RAC, Palace J, Sharrack B, Swingler R, Young C, Moseley IF, MacManus DG, Donoghue S, Miller DH (1999) The effect of anti‐α4 integrin antibody on brain lesion activity in MS. The UK Antegren Study Group. Neurology 53:466–472. [DOI] [PubMed] [Google Scholar]
- 73. Van Assche G, Van Ranst M, Sciot R, Dubois B, Vermeire S, Noman M, Verbeeck J, Geboes K, Robberecht W, Rutgeerts P (2005) Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N Engl J Med 353:362–368. [DOI] [PubMed] [Google Scholar]
- 74. Van Der Laan LJW, Van Der Goes A, Wauben MHM, Ruuls SR, Döpp EA, De Groot CJA, Kuijpers TW, Elices MJ, Dijkstra CD (2002) Beneficial effect of modified peptide inhibitor of α4 integrins on experimental allergic encephalomyelitis in Lewis rats. J Neurosci Res 67:191–199. [DOI] [PubMed] [Google Scholar]
- 75. Vanderlugt CL, Neville KL, Nikcevich KM, Eagar TN, Bluestone JA, Miller SD (2000) Pathologic role and temporal appearance of newly emerging autoepitopes in relapsing experimental autoimmune encephalomyelitis. J Immunol 164:670–678. [DOI] [PubMed] [Google Scholar]
- 76. Verdun E, Isoardo G, Oggero A, Ferrero B, Ghezzi A, Montanari E, Zaffaroni M, Durelli L (2002) Autoantibodies in multiple sclerosis patients before and during IFN‐β1b treatment: Are they correlated with the occurrence of autoimmune diseases? J Interferon Cytokine Res 22:245–255. [DOI] [PubMed] [Google Scholar]
- 77. Von Andrian UH, Engelhardt B (2003) α4 integrins as therapeutic targets in autoimmune disease. N Engl J Med 348:68–72. [DOI] [PubMed] [Google Scholar]
- 78. Werr J, Johansson J, Eriksson EE, Hedqvist P, Ruoslahti E, Lindbom L (2000) Integrin α2β1 (VLA‐2) is a principal receptor used by neutrophils for locomotion in extravascular tissue. Blood 95: 1804–1809. [PubMed] [Google Scholar]
- 79. Wu JE, Santoro SA (1994) Complex patterns of expression suggest extensive roles for the α2β1 integrin in murine development. Dev Dyn 199:292–314. [DOI] [PubMed] [Google Scholar]
- 80. Wyss‐Coray T, Feng L, Masliah E, Ruppe MD, Lee HS, Toggas SM, Rockenstein EM, Mucke L (1995) Increased central nervous system production of extracellular matrix components and development of hydrocephalus in transgenic mice overexpressing transforming growth factor‐β1. Am J Pathol 147:53–67. [PMC free article] [PubMed] [Google Scholar]
- 81. Xu H, Banerjee A, Dilulio NA, Fairchild RL (1997) Development of effector CD8+ T cells in contact hypersensitivity occurs independently of CD4+ T cells. J Immunol 158:4721–4728. [PubMed] [Google Scholar]
- 82. Yamamoto T, Iwasaki Y, Yamamoto H, Konno H, Isemura M (1988) Intraneuronal laminin‐like molecule in the central nervous system: demonstration of its unique differential distribution. J Neurol Sci 84:1–13. [DOI] [PubMed] [Google Scholar]
- 83. Yednock TA, Cannon C, Fritz LC, Sanchez‐Madrid F, Steinman L, Karin N (1992) Prevention of experimental autoimmune encephalomyelitis by antibodies against α4β1 integrin. Nature 356:63–66. [DOI] [PubMed] [Google Scholar]