

Abstract
Work done over the past decade has led to a molecular understanding of frontotemporal lobar degeneration (FTLD), a deadly disease that afflicts patients in mid‐life. It is a common cause of dementia, second only to Alzheimer’s disease in the population below 65 years of age. Neuroanatomical and neurobiological substrates have been identified for the three major subtypes of FTLD and these discoveries have broadened the FTLD spectrum to include amyotrophic lateral sclerosis (ALS). Mutations in MAPT were found to cause frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP‐17), a familial disorder with filamentous tau inclusions in nerve cells and glial cells. FTDP‐17 can result in clinical syndromes that closely resemble progressive supranuclear palsy, corticobasal degeneration and Pick’s disease. More recently, mutations in three genes (VCP, CHMP2B and PGRN) have been found to cause FTLD with ubiquitin‐positive, tau‐negative neuronal inclusions (FTLD‐U). They explain a large proportion of inherited FTLD‐U. It remains to be seen whether dementia lacking distinctive histopathology (DLDH) constitutes a third disease category, as many of these cases are now being reclassified as FTLD‐U. Recently, TAR DNA‐binding protein‐43 (TDP‐43) has been identified as a key protein of the ubiquitin inclusions of FTLD‐U and ALS. Thus, for familial forms of FTLD and related disorders, we now know the primary etiologies and accumulating proteins. These findings are pivotal for dissecting the pathways by which different etiologies lead to the varied clinicopathological presentations of FTLD.
INTRODUCTION
Frontotemporal lobar degeneration (FTLD) is the second most common form of cortical dementia in the population below the age of 65 years. Clinically, it is characterized by changes in personality/behavior and/or language dysfunction (aphasia), and results in at least three distinct clinical syndromes: frontotemporal dementia, semantic dementia and primary progressive aphasia (PPA). Extrapyramidal features can also be present and are an important component of corticobasal degeneration (CBD) and progressive supranuclear palsy (PSP).
Over the past decade, much effort has gone into the histopathological characterization of FTLD and, based on the presence or absence of the microtubule‐associated protein tau (MAPT) in neuronal inclusions, FTLD is now classified as either a tauopathy or a non‐tauopathy disorder. Tauopathy disorders (FTLD‐τ) include Pick’s disease, PSP, CBD, and frontotemporal dementia and parkinsonism linked to chromosome‐17 (FTDP‐17). The non‐tauopathy disorders include FTLD with ubiquitin‐positive neuronal inclusions (FTLD‐U) and dementia lacking distinctive histopathology (DLDH). However, considerable heterogeneity is observed both clinically and neuropathologically and many of the neurobehavioral syndromes and pathologies overlap (Figure 1). This partly matches with the high genetic complexity observed in FTLD.
Figure 1.
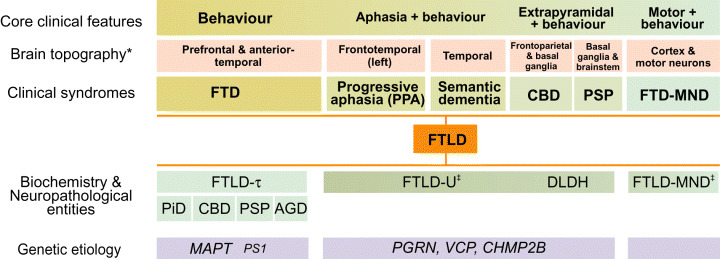
Genotypes, proteotypes and phenotypes of FTLD. Clinical and pathological syndromes of FTD/FTLD constitute a clinicopathologic spectrum. The spectrum continues on the left with Alzheimer’s dementia‐frontal variant and on the right with ALS (*evident on both neuropathology and neuroimaging; ‡the inclusion proteopathy could be TDP‐43). AGD = argyrophilic grain disease; ALS = amyotrophic lateral sclerosis; CBD = corticobasal degeneration; FTD = frontotemporal dementia; FTLD = frontotemporal lobar degeneration; MND = motor neuron disease; PiD = Pick’s disease; PSP = progressive supranuclear palsy.
Despite this heterogeneity, the past few years have witnessed enormous progress, both in terms of understanding the pathological complexity and in identifying the genetic etiologies of FTLD. If a rapidly changing classification and a new terminology are a measure of increasing knowledge, then progress in FTLD has been rapid indeed. As a result, CBD and PSP are now grouped under the umbrella of FTLD‐τ (62). Over the past 3 years, the first FTLD‐U‐causing genes have been identified. They are the valosin‐containing protein gene or VCP on chromosome 9p21‐p12 (109), the charged multivesicular body protein 2B gene or CHMP2B on chromosome 3p13 (97) and the recently identified progranulin gene or PGRN on chromosome 17q21‐22 (6, 16). Identification of PGRN was particularly exciting, as it provided an explanation for the amazing coincidence of the presence of two important genes linked with the same disease phenotype on chromosome 17q21‐22. Moreover, TAR DNA‐binding protein (TDP‐43) has recently been identified as a key protein in the inclusions of FTLD‐U and related disorders (75). It remains to be seen whether mutations in the TDP‐43 gene can also cause FTLD‐U. The central theme of this review is to identify commonalities and overlaps between the different clinicopathological entities, and to support the viewpoint that FTLD is part of a spectrum of diseases that includes amyotrophic lateral sclerosis (ALS).
CLINICAL SYNDROMES COMPRISING FTLD
FTLD usually occurs between the ages of 35 and 75 years and is the second most common form of cortical dementia in the presenium, after Alzheimer’s disease (AD). The personality changes and aphasia observed in patients allow one to distinguish between the three prototypical clinical syndromes of frontotemporal dementia, semantic dementia and PPA. Recently, highly specific (57, 74) and very sensitive (62) diagnostic criteria have been established for these disorders.
Frontotemporal dementia (FTD) is the most common clinical manifestation of FTLD, accounting for 5%–10% of all dementia patients, 10%–20% of which are younger than 65 years (31, 74, 89). In these patients, progressive deterioration in personality occurs, initially with a relative preservation of language and memory. Social and personal conduct is profoundly altered, accompanied by inertia and loss of volition. Repetitive, compulsive and stereotypic behavior is common. Although linguistically correct, speech output is reduced. Mutism may ensue later in the course of the disease. The absence of early neurological signs and findings of focal abnormalities in the frontotemporal lobes on neuroimaging contribute to the clinical diagnosis. However, in some patients, only behavioral changes are observed and these patients are referred to as having FTD‐behavioral variant. FTD can also be accompanied by signs of parkinsonism, as in CBD, a clinical syndrome where progressive asymmetrical rigidity and apraxia, often accompanied by aphasia, are the most common symptoms. Similarly, progressive aphasia and behavioral features can also accompany PSP, which is the second most common cause of parkinsonism, after Parkinson’s disease. PSP is characterized by progressive axial rigidity, bradykinesia, vertical gaze palsy and dysarthria. Rarely, FTD is accompanied by motor neuron disease (FTD‐MND), where patients have features of both ALS and FTD. Other complex forms of FTD associated with additional phenotypes have been described, especially in familial forms. Thus, families linked to chromosome 3p13 (CHMP2B gene) or to chromosome 9q21 have parkinsonism, as is often observed in FTDP‐17; chromosome 20p‐linked families have motor disturbances; and chromosome 9p21‐p12‐linked families (VCP) present with the unusual triad of inclusion body myopathy (IBM), Paget’s disease of the bone (PDB) and FTD (IBM‐PDB‐FTD) [reviewed in (82)].
Semantic dementia and PPA are alternative presentations of FTLD, where progressive changes in language function are an early and predominant feature that precedes behavioral symptoms. In semantic dementia, loss of verbal and nonverbal skills is the core feature, as evidenced by impairment in naming and word comprehension in the context of a fluent, effortless and grammatically correct speech output. An inability to recognize the meaning of visual stimuli is a striking feature; however, visuospatial skills and day‐to‐day memory are well preserved. Late in the course of the disease, patients may become mute. By contrast, PPA is a disorder of expressive language that is characterized by a progressive reduction in speech production, with phonological and grammatical errors and word retrieval difficulties, with preservation of daily life activities and evidence of relatively normal nonverbal abilities on neuropsychological testing. The understanding of word meaning is reasonably well preserved, while difficulties in reading and writing may occur. With the progression of disease, patients may become mute. These overlapping neurobehavioral syndromes have overlapping and heterogeneous pathological correlates.
NEUROPATHOLOGY OF FTLD
The current consensus on the pathological classification of FTLD is based on (i) histopathological presence or absence of neuronal inclusions; (ii) immunohistochemical identification and biochemical characterization of proteins accumulating in the neuronal/glial inclusions; (iii) anatomical distribution of the underlying histopathology.
A first distinction is made based on the presence of tau inclusions (Figure 2). Tau is the MAPT gene product and adult human brain expresses six tau isoforms that are derived from a single gene by alternative mRNA splicing. Three isoforms contain three microtubule‐binding repeats each (3R‐τ); the other three isoforms have an additional repeat encoded by exon 10 of Tau (4R‐τ). Thus, based on the presence or absence of filamentous tau inclusions, FTLD is differentiated into tauopathy and non‐tauopathy. The tauopathy group is further divided into 3R‐τ and 4R‐τ subgroups, that is, 3R‐τ in Pick’s disease, 4R‐τ in CBD and PSP and all six isoforms in AD. However, this is only a rough guide, because overlaps frequently occur, especially in Pick’s disease.
Figure 2.
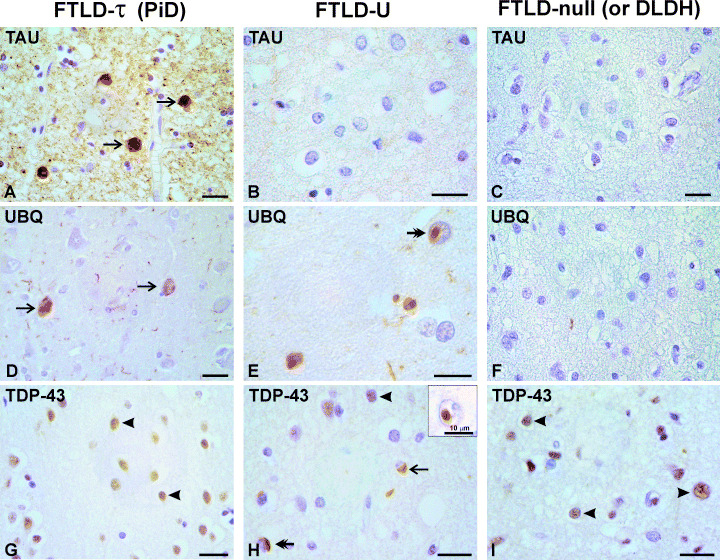
Pathological features of FTLD‐τ, FTLD‐U and DLDH. Tau (A–C), ubiquitin (D–F) and TDP‐43 (G,H,I) immunostaining is shown on serial sections from superior frontal cortex of Pick’s disease (left panel), FTLD‐U with a PGRN IVS0 + 5G > C mutation (middle panel), and DLDH (right panel). Tau‐reactive cytoplasmic inclusions (arrows) are present in Pick’s disease (A), but absent in FTLD‐U (B) and DLDH (C). Ubiquitin‐reactive inclusions are present in Pick’s disease (D) and FTLD‐U (E), but not in DLDH (F). TDP‐43 commonly stains neuronal nuclei (arrowheads in G,H,I) and also both cytoplasmic and intranuclear inclusions in FTLD‐U (arrow and double‐headed arrows in H). In neurons with inclusions, the normal nuclear staining of TDP‐43 appears reduced. Tau, ubiquitin, and TDP‐43 were stained with mAb AT8 (Innogenetics), rabbit serum against polyubiquitin (Dako, Glostrup, Denmark) and TDP‐43 (R&D systems, Abingdon, UK), respectively, using ABC‐HRP/DAB immunochemical system. Scale bar represents 20 µm. DLDH = dementia lacking distinctive histopathology; FTLD = frontotemporal lobar degeneration.
Among the forms of FTLD without tau deposits, two overlapping pathologies are observed. The first type is characterized by the presence of ubiquitin‐positive neuronal inclusions, also called FTD without motor neuron disease (MND), but with MND‐type inclusions or, more commonly, FTLD‐U. The second type is that without ubiquitin‐positive inclusions and is referred to as DLDH.
Tauopathy FTLD
Pick’s disease. The first form of FTLD was described by Arnold Pick in 1892 in patients with behavioral and aphasic clinical presentations and a severe circumscribed atrophy of the frontotemporal lobes at autopsy (79). Subsequently, many investigators, including Alzheimer, described the histopathological features of argyrophilic inclusions (later referred to as Pick bodies) and swollen achromatic cells (later referred to as Pick cells), and the eponym Pick’s disease was suggested. This term is now restricted to cases of FTLD with Pick bodies. Pick’s disease is often considered to be the prototypical FTLD; hence the concern that this might lead to the erroneous view that all cases of FTLD are tauopathies (46, 106).
The classical atrophy observed in patients with Pick’s disease is a “knife‐edge” atrophy of the frontal and temporal lobes of the cerebral cortex. The posterior part of the superior temporal lobe is typically spared. Affected brain regions show severe neuronal loss and astrogliosis, but the chief histological abnormality is the presence of Pick bodies in the dentate gyrus, pyramidal cells of the CA1 sector and subiculum of the hippocampus, the neocortex and several subcortical nuclei. In the neocortex, Pick bodies are frequently located in layers II and VI, in contrast to the predominance of neurofibrillary tangles in layers III and V in AD. By electron microscopy, Pick bodies contain intermediate filaments, 15 nm straight filaments and some paired helical filaments (21). Biochemical characterization shows that the insoluble tau in Pick bodies consists of 3R isoforms (11, 53, 105). However, recent studies have demonstrated much greater biochemical heterogeneity and up to 50% of patients with Pick’s disease had either at least as much 4R‐τ as 3R‐τ, or even a predominance of 4R‐τ (71, 117). Familial forms of Pick’s disease occur, in particular in the context of FTDP‐17 (53, 106).
Frontotemporal dementia and Parkinsonism linked to chromosome 17 (FTDP‐17). An autosomal‐dominantly inherited form of frontotemporal dementia with parkinsonism was linked to chromosome 17q21‐22 in 1994 (111). In the following years, 13 additional families with FTD and parkinsonism with linkage to 17q21‐22 were identified. In 1997, a consensus conference introduced the term FTDP‐17 to describe these patients (27) and the following year mutations in MAPT were reported in the majority of these families (38, 84, 99). At present, 39 mutations in MAPT have been identified in 115 families. Interestingly, MAPT mutations have also been identified in cases with CBD and with PSP (53, 87, 91). For a complete update, visit (http://www.molgen.ua.ac.be/FTDMutations).
Clinically, MAPT mutation carriers present with disinhibition, loss of initiative, obsessive‐compulsive behavior and/or psychosis, followed by cognitive decline. In most patients, extrapyramidal symptoms occur only late in the clinical course, but considerable heterogeneity is observed both between mutations and within families with the same mutation (27). Age at onset is also highly variable and ranges from the early 20s to late 70s, with a mean age around 50 years (27, 70, 92). Neuropathologically, FTDP‐17 presents with atrophy of the frontal and temporal lobes with neuronal loss, astrocytic gliosis and spongiosis in the superficial cortical layers. Basal ganglia are also affected (27). Filamentous tau inclusions in neuronal and glial cells are a characteristic finding (98). Biochemically and neuropathologically, many cases of FTDP‐17 resemble sporadic Pick’s disease.
Corticobasal degeneration, progressive supranuclear palsy and argyrophilic grain disease. CBD and PSP are two clinical manifestations of FTLD‐τ. CBD has diverse clinical presentations, such as progressive asymmetrical rigidity and apraxia, progressive aphasia and frontal lobe dementia. Because of this diversity, a diagnosis of CBD is not dependent on a specific clinical presentation, but rather on a defined neuropathology. The minimal pathologic features for CBD are cortical and striatal tau‐positive neuronal and glial lesions, especially astrocytic plaques and thread‐like lesions in both white and gray matter, alongside neuronal loss in focal cortical regions and substantia nigra (22).
PSP has early behavioral manifestations, rendering differentiation from FTD difficult. Pathologically, it is characterized by atrophy of brainstem and basal ganglia, with corresponding neuronal loss, gliosis, and a high density of tangle‐like tau pathology, neuropil threads and glial fibrillary tangles in both astrocytes (tufted astrocytes) and oligodendrocytes (coiled bodies) (32). However, atypical or variant PSP, where the severity and distribution of abnormalities deviate from the above, is also common (32). A link between CBD and PSP is that they are both 4R‐tauopathies (11, 71, 105).
A relatively new entity in the 4R‐τ group is argyrophilic grain disease (AGD). Because the specific disease manifestations of AGD are currently unclear, it has so far remained a neuropathological entity (9, 103, 104). Argyrophilic grains are present in the hippocampal region and the amygdala, and are accompanied by coiled bodies in the underlying white matter and ballooned neurons in the limbic region. The frequency of AGD is estimated to be ∼5% at routine autopsy (65, 103).
Non‐tauopathy FTLD
FTLD with tau‐negative, ubiquitin‐positive inclusions (FTLD‐U). Recent estimates have suggested that patients lacking tau pathology account for more than 50% of autopsy‐confirmed FTLD (25, 41, 43, 45, 56). This proportion will grow even larger as many patients previously thought to be having DLDH show FTLD‐U pathology on re‐examination (see later). FTLD‐U occurs as both familial and sporadic forms. To date, 20 families have been described with non‐tauopathy FTLD linked to defined genetic loci and newly identified genes on various chromosomes, including FTLD‐U families linked to chromosome 17q21‐22.
FTLD‐U is characterized by the presence of ubiquitin‐positive neuronal cytoplasmic inclusions (NCIs) and lentiform intranuclear inclusions (NIIs) in layer II of frontal and temporal cortex, in striatum and in the dentate fascia of the hippocampus (56, 58, 63, 102). Because these inclusions were first described in patients with MND (76), they are sometimes referred to as FTLD with MND‐type inclusions but without MND (40). Besides inclusions, superficial laminar spongiosis and chronic degenerative changes of the frontotemporal and, sometimes, parietal regions are the most consistent features. Ultrastructural analysis of the inclusions has shown the presence of tubofilamentous structures with a diameter of 10–18 nm (82). Patients with sporadic FTLD‐U have a similar pathology, except that NIIs are only rarely present (7). Moreover, FTLD‐MND is characterized by only a few or no inclusions in caudate nucleus and frontotemporal cortex, but with ubiquitin‐positive granular inclusions in the dentate fascia and lower motor neurons (66, 110). The latter features are reminiscent of ALS.
For a long time, the nature of the protein components of tau‐negative, ubiquitin‐positive inclusions remained unknown; however, recent studies have shed important light on this issue. In a type of FTLD‐U called neuronal intermediate filament inclusion dementia (NIFID), neurofilaments have been identified as the major component of the inclusions (42). NIFID patients present relatively early with FTD and may also suffer from parkinsonism and MND. Atrophy of frontotemporal cortex and caudate nucleus is a common feature (13, 42). For other cases of familial FTLD‐U, a number of minor proteins has been recognized. For instance, p62 was reported to be present in the inclusions (82), similar to AD, Pick’s disease and Lewy body dementia (2, 50). HSP70 has also been found in some FTLD‐U inclusions (82).
Recently, TDP‐43, a nuclear protein that functions in the regulation of transcription and alternative splicing, has been identified as a major inclusion protein in FTLD‐U and ALS (3, 75). In the inclusions, TDP‐43 is hyperphosphorylated and ubiquitinated. It constitutes a common pathologic substrate that links sporadic and familial FTLD‐U with ALS.
Dementia lacking distinctive histopathology. The third subtype of FTLD, for which sporadic and familial forms have been described, is characterized by neuronal cell loss and gliosis in the absence of protein inclusions (47). DLDH is less common than previously thought, as many patients are now being reclassified as having FTLD‐U (44, 61). Thus, until recently, hereditary dysphasic disinhibition dementia 2 (HDDD2) was considered to be a classical form of inherited DLDH. However, recent work (72) has shown that it is caused by a PGRN mutation and that ubiquitin‐positive inclusions are present in brain. Earlier, a selective loss of all six tau isoforms, with no corresponding change in tau mRNA levels, was reported in DLDH and HDDD2 brains, but the significance of these findings remains uncertain (116).
GENETICS AND MOLECULAR PATHOLOGY OF FTLD
Tauopathy FTLD. Following the identification of mutations in MAPT in FTDP‐17, considerable progress has been made in genotype‐phenotype analysis and many types of tau pathology have been identified. It is now clear that MAPT mutations give rise to FTLD‐τ pathology. Similarly, sporadic forms of PSP and CBD are also linked to MAPT in some populations, where inheritance of the H1 haplotype of MAPT is a risk factor (5, 20, 36, 69, 88).
Only rarely have MAPT variants been reported to be associated with “non‐tauopathy” (80, 100). Recently, these patients have been shown to harbor mutations in PGRN, indicating that the reported MAPT variants were benign polymorphisms (81). Similarly, a family with the presenilin 1 (PSEN1) insArg352 change (1, 8, 94) was shown to have a PGRN mutation (81). In contrast, the PSEN1 Gly183Val mutation associated with “Pick’s disease tauopathy”, which we reported earlier (19), does not carry a mutation in PGRN. This mutation affects a splice donor signal and is predicted to produce, apart from the full‐length missense transcript, at least two other aberrantly spliced transcripts that, when not degraded, would lead to short C‐truncated proteins (19). Although the precise disease mechanism remains unidentified, presenilin 1 protein is reduced by ∼20% in brain of a Gly183Val carrier. There is also an experimental amyloid precursor protein‐independent reduction in γ‐secretase activity in presenilin null mouse fibroblasts. The involvement of a splice donor signal and the reduced protein suggest that the Gly183Val mutation is a more complicated mutation than initially believed (Tolia et al, unpub. data). Interestingly, another PSEN1 mutation associated with FTD, Leu113Pro, is also affecting a splice donor site (90). Association of these splice‐site mutations with FTD is interesting in the light of the presence of alternative PSEN1 transcripts in FTD brain (24), the loss of presenilin function associated with AD‐causing mutations (49) and the finding that PSEN conditional knock‐out mice show tau‐like neurodegeneration (95). Future studies will address whether and how presenilin loss contributes to neurodegeneration involving tau.
Non‐tauopathy FTLD. The study of FTLD‐U is presently at a particularly exciting stage. The remainder of this review will therefore focus on the recent advances. As mentioned above, approximately 20 families with non‐tauopathy FTLD have been linked genetically to loci on chromosomes 3p13, 9q21, 9p21, 17q21‐22 and 20p (6, 16, 97, 109). To date, pathogenic mutations in three genes have been identified. Mutations in a fourth gene, dynactin (DCTN1), have been identified mainly in ALS patients, but with some overlap with FTD (85) (Table 1).
Table 1.
Genes involved in FTLD.
VCP missense mutations cause IBM‐PDB‐FTD. IBM‐PDB‐FTD is a rare autosomal‐dominant degenerative disorder of the brain (mean age at onset of 54 years for FTD), muscle (adult‐onset distal and proximal muscle weakness), and bone (early‐onset PDB) (48). It has recently been shown to be caused by mutations in the VCP gene (109). VCP is an abundantly expressed 97‐kDa valosin‐containing protein and a prominent member of the AAA‐ATPase gene superfamily (ATPase associated with a variety of activities) (83). It acts as a molecular chaperone in a variety of cellular activities, including cell cycle regulation, programmed cell death, stress response, transcriptional regulation, cytosolic protein degradation and endoplasmic reticulum‐associated degradation of misfolded proteins. Almost all these activities are directly or indirectly regulated by the ubiquitin‐proteasome (Ub‐Pr) system. VCP forms a stable barrel‐like homo‐hexameric structure with a two‐tier ring made up of the two conserved AAA domains. It binds to the Ub‐Pr machinery through its N‐terminal domain. Both N‐ and C‐terminal regions are connected to the AAA domains via linker sequences (18). The hexameric structure predicts a dominant‐negative effect for the known missense mutations. To date, eight mutations have been reported in 17 IBM‐PDB‐FTD families; they involve mostly the Ub‐binding N‐terminal domain, but can also be found in the AAA domains and the linker sequences (114).
The neuropathology associated with VCP mutations is not unique, although lenticular NIIs appear to be particularly abundant and the dentate gyrus has been reported to lack significant pathology (26, 82, 96). Family DR7, reported earlier as FTLD without additional features (82), has now been shown to carry a novel VCP mutation Arg159His. FTLD without additional features has also been described in two families segregating with mutations Arg93Cys and Arg155Cys in VCP (30). Interestingly, VCP‐like immunoreactivity is observed only rarely in the ubiquitin inclusions in these patients (26, 82).
CHMP2B mutations cause a small subset of FTLD‐U. In an autosomal‐dominant form of FTD in a large family of Danish descent and linked to a locus on chromosome 3p13 (10), a complex C‐truncating mutation in CHMP2B was identified (97). In addition, a Gly442Thr missense mutation was also identified in one individual with semantic dementia from a large European FTD series (97). A large screen of patients with familial FTD from the US, the UK and the Netherlands failed to reveal additional mutations, suggesting that CHMP2B mutations are not a common cause of FTLD (14, 93).
Neuropathologically, the Danish family was initially reported to have DLDH, but subsequent studies showed the presence of ubiquitin‐positive NCIs (35). Besides giving rise to FTD, CHMP2B mutations may also cause additional phenotypes. For instance, mutations have been identified in two ALS patients, one of whom had also FTD (78). However, the sequence change in the latter was also present in a control individual, rendering its significance uncertain.
Like VCP, CHMP2B is widely expressed in brain and although the exact function of CHMP2B is unknown, its yeast ortholog Vsp2 is part of the ESCRTIII complex (endosomal secretory complex required for transport). This process enables the sorting of transmembrane proteins and trafficking along late endosomes to multivesicular bodies (MVBs) and lysosomes. Dysfunction of these components results in the inability of the MVB to internalize membrane‐bound cargo and results in poor protein turnover (4). Accordingly, epitope‐tagged mutant isoforms of CHMP2B cause aberrant late endosomal trafficking in PC12 cells (97), although it is not known whether this is through a gain of function or (dominant) loss‐of‐function mechanism. The Danish CHMP2B mutation causes altered splicing, resulting in two aberrant transcripts that lead to a protein lacking the 36 C‐terminal amino acids of CHMP2B. In one of these transcripts, there is addition of a non‐physiological C‐terminal 29 amino acid sequence, which may cause an aberrant gain of toxic function (67, 97).
PGRN loss‐of‐function mutations cause FTLDU‐17. The identification of mutations in PGRN was an exciting event in the study of FTLD. Previously, FTLD‐U had been shown to be linked to the chromosome 17q21—MAPT region in a number of families, including ours (54, 60, 86, 115). However, extensive analysis of the 140 kb MAPT genomic region, including intronic and regulatory sequences, failed to detect any pathogenic mutation (15). At the same time, biochemical analysis of tau from frozen brain did not reveal any abnormalities in the distribution of tau isoforms at either protein or mRNA level (60). An extensive mutational analysis of genes near MAPT, which was carried out in parallel to the above work, led to the identification of PGRN as the FTLDU‐17 gene (6, 16). Subsequent studies in other FTLDU‐17 families and in the HDDD2 family confirmed this finding (6, 16, 28, 37, 52, 72). PGRN encodes a 68.5 kDa secreted precursor glycoprotein composed of a signal peptide followed by 7.5 tandem repeats of a 12‐cysteinyl granulin motifs that can be proteolytically cleaved to form a family of 6 kDa granulin peptides (33). PGRN is a widely expressed multifunctional growth factor and both PGRN and granulins have important roles in development, cell cycle progression, cell motility, wound repair and inflammation (17, 33, 34). PGRN is expressed in neurons of cerebral cortex, hippocampus and cerebellum. A high expression of PGRN is associated with a variety of tumors, including glioblastoma (17, 33, 55).
So far, seven unique PGRN null mutations have been identified in 67 FTD patients (AD&FTD Mutation database: http://www.molgen.ua.ac.be/ADmutations). Their frequency is estimated to be 5%–11% in the sporadic, and 13%–25% in the familial FTD population from Belgium, the USA and France, indicating that (null) mutations in PGRN are a major cause of FTD (16, 28, 52). Four classes of PGRN mutations have been identified. The first class includes mutations where the transcripts do not leave the nucleus, that is, the splice‐donor site mutation of the PGRN exon 0 (IVS0 + 5G > C), where nuclear retention signals remain in the unspliced transcript and prevent it from leaving the nucleus, where it is destroyed (16). Although this is so far the only mutation in this class, it provides one of the most convincing pieces of evidence in support of the notion that PGRN mutations are loss‐of‐function mutations. The second class of mutations is where the transcript reaches the cytoplasm, but the protein is not produced efficiently. It includes additional splice site, frameshift and nonsense mutations that result in nonsense‐mediated decay of the mutant transcripts (6, 16, 64). This class also includes mutations in the Met start codon, which disrupt the Kozak sequence and result in a substantial reduction in PGRN expression (6, 16, 28). A third class is where the protein is mislocalized; this includes the missense mutation in the signal peptide identified in the HDDD2 family (72). It abolishes recognition of the signal peptide by the signal recognition particle and hampers its translocation into the endoplasmic reticulum. The final class includes coding variants, the significance of which is currently not understood (28).
Neuropathologically, cases with PGRN mutations show numerous lentiform NIIs in neocortex and striatum, as well as less well‐formed NCIs, especially in the hippocampus (59) (Kumar‐Singh et al, unpub. data). Thus, both PGRN and VCP mutation carriers have abundant NIIs, which is an infrequent finding in patients with sporadic FTLD‐U. PGRN staining showed that, although immunoreactivity was localized to a subset of cortical neurons and up‐regulated in activated microglia, ubiquitin inclusions were not stained (6, 16). By contrast, anti‐TDP‐43 antibodies stained a substantial number of inclusions in PGRN mutation carriers (75) (Kumar‐Singh et al, unpub. data). The mechanisms by which PGRN loss‐of‐function causes FTLD‐U are unclear, but parallels might be drawn from cell culture work, where PGRN has been shown to abrogate the requirement for insulin‐like growth factor 1 receptor for growth (112), perhaps by promoting the activation of phosphatidylinositol 3′‐kinase and mitogen‐activated protein kinase pathways with sustained expression of cyclin B (113).
TDP‐43 may define the underlying proteopathy. TDP‐43 is a major component of ubiquitin‐positive NIIs and NCIs of sporadic and familial FTLD‐U (3, 75). TDP‐43–positive deposits were also identified in ALS, suggesting that their formation is a common downstream denominator of FTLD‐U/ALS ubiquitinopathies. TDP‐43 was first identified as a ubiquitously expressed 43‐kD nuclear protein that binds to the TAR DNA in the human immunodeficiency virus 1 long terminal repeat, where it functions as a transcriptional repressor (77). A second function as an activator of exon skipping was identified later (12). The primary transcript of the human TARDBP gene undergoes alternative splicing to generate 8 distinct mRNAs (108) and the protein undergoes phosphorylation. The levels of nuclear TDP‐43 are reduced in neurons in FTLD, especially those with inclusions (75). It is presently not known whether a loss of TDP‐43 function contributes to the FTLD‐U phenotype.
CONCLUSION
From the large body of clinical, neuropathological, biochemical and genetic data discussed here, it is clear that FTLD is heterogeneous, as indicated by the existence of FTLD‐τ, FTLD‐U, DLDH, FTD‐MND and ALS. Not only is heterogeneity observed between disease forms, but it is also seen between families with the same mutation and even within families. Thus, these diseases most likely represent a clinicopathological spectrum. DLDH is becoming less common and a number of findings suggest that FTLD‐U and ALS are at two ends of the same spectrum. Thus, they have overlapping clinical features, that is, ALS patients can develop dementia and, conversely, patients with FTLD frequently develop ALS. FTLD and ALS also have an overlapping spectrum of pathology. Furthermore, they share common etiologies, that is, ALS is associated with mutations in CHMP2B (78) and PGRN (60) and a mutation in DCTN1—an ALS gene—may cause FTD in some family members (73). Interestingly, two recent reports have identified an FTLD‐ALS locus on chromosome 9p13.2‐21.3 (68, 107). Even where common etiologies of ALS and FTLD are not directly evident, common disease pathways may be involved, that is, PGRN stimulates the expression of vascular endothelial growth factor (VEGF) (101) and mutations in angiogenin or polymorphisms in VEGF are associated with ALS (29, 51). Another example is that VCP can bind to Dorfin (an E3 ligase) and contributes to its ability to ubiquitinate superoxide dismutase 1, which, when mutant, causes ALS (39). And lastly, TDP‐43 is a component of the ubiquitin‐positive inclusions of both FTLD‐U and ALS.
The cellular pathways that the currently known FTLD‐U gene products are involved in are protein turnover—that is, the endosomal‐lysosomal system (CHMP2B), the unfolded protein response and the Ub‐Pr system (VCP) and cell signaling (PGRN). It is presently unclear how disruption of these pathways can lead to FTLD‐U and the accumulation of TDP‐43. The latter contains two RNA‐binding domains and it is well documented that such domains can also be involved in protein–protein interactions (23). Could this mean that other proteins are also sequestered in the inclusions and that the resultant loss of their function is an even more proximate event? Judging by the recent progress, it appears likely that in times to come we will learn how different triggering factors can lead to FTLD and related disorders.
ACKNOWLEDGMENTS
The authors thank the personnel of the BioBank of the Born‐Bunge Institute for providing pathological specimens and the Genetic Service Facility (http://www.vibgeneticservicefacility.be) for the genetic analyses. The research of the authors is supported by the Special Research Fund of the University of Antwerp, the Fund for Scientific Research Flanders (FWO‐F), the Interuniversity Attraction Poles (IUAP) program P5/19 of the Belgian Science Policy Office (BELSPO), the Stichting Alzheimer Onderzoek, Belgium, EU contract LSHM‐CT‐2003‐503330 (APOPIS) and a Zenith Award from the US Alzheimer Association.
REFERENCES
- 1. Amtul Z, Lewis PA, Piper S, Crook R, Baker M, Findlay K, Singleton A, Hogg M, Younkin L, Younkin SG, Hardy J, Hutton M, Boeve BF, Tang‐Wai D, Golde TE (2002) A presenilin 1 mutation associated with familial frontotemporal dementia inhibits gamma‐secretase cleavage of APP and notch. Neurobiol Dis 9:269–273. [DOI] [PubMed] [Google Scholar]
- 2. Arai T, Nonaka T, Hasegawa M, Akiyama H, Yoshida M, Hashizume Y, Tsuchiya K, Oda T, Ikeda K (2003) Neuronal and glial inclusions in frontotemporal dementia with or without motor neuron disease are immunopositive for p62. Neurosci Lett 342:41–44. [DOI] [PubMed] [Google Scholar]
- 3. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T (2006) TDP‐43 is a component of ubiquitin‐positive tau‐negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611. [DOI] [PubMed] [Google Scholar]
- 4. Babst M, Katzmann DJ, Estepa‐Sabal EJ, Meerloo T, Emr SD (2002) Escrt‐III: an endosome‐associated heterooligomeric protein complex required for mvb sorting. Dev Cell 3:271–282. [DOI] [PubMed] [Google Scholar]
- 5. Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez‐tur J, Hardy J, Lynch T, Bigio E, Hutton M (1999) Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet 8:711–715. [DOI] [PubMed] [Google Scholar]
- 6. Baker M, Mackenzie IR, Pickering‐Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M (2006) Mutations in progranulin cause tau‐negative frontotemporal dementia linked to chromosome 17. Nature 442: 916–919. [DOI] [PubMed] [Google Scholar]
- 7. Bigio EH, Johnson NA, Rademaker AW, Fung BB, Mesulam MM, Siddique N, Dellefave L, Caliendo J, Freeman S, Siddique T (2004) Neuronal ubiquitinated intranuclear inclusions in familial and non‐familial frontotemporal dementia of the motor neuron disease type associated with amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 63:801–811. [DOI] [PubMed] [Google Scholar]
- 8. Boeve BF, Baker M, Dickson DW, Parisi JE, Giannini C, Josephs KA, Hutton M, Pickering‐Brown SM, Rademakers R, Tang‐Wai D, Jack CR Jr, Kantarci K, Shiung MM, Golde T, Smith GE, Geda YE, Knopman DS, Petersen RC (2006) Frontotemporal dementia and parkinsonism associated with the IVS1 + 1G−> A mutation in progranulin: a clinicopathologic study. Brain 129:3103–3114. [DOI] [PubMed] [Google Scholar]
- 9. Braak H, Braak E (1989) Cortical and subcortical argyrophilic grains characterize a disease associated with adult onset dementia. Neuropathol Appl Neurobiol 15:13–26. [DOI] [PubMed] [Google Scholar]
- 10. Brown J, Ashworth A, Gydesen S, Sorensen A, Rossor M, Hardy J, Collinge J (1995) Familial non‐specific dementia maps to chromosome 3. Hum Mol Genet 4:1625–1628. [DOI] [PubMed] [Google Scholar]
- 11. Buée L, Delacourte A (1999) Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP‐17 and Pick’s disease. Brain Pathol 9:681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE (2001) Nuclear factor TDP‐43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J 20:1774–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cairns NJ, Grossman M, Arnold SE, Burn DJ, Jaros E, Perry RH, Duyckaerts C, Stankoff B, Pillon B, Skullerud K, Cruz‐Sanchez FF, Bigio EH, Mackenzie IR, Gearing M, Juncos JL, Glass JD, Yokoo H, Nakazato Y, Mosaheb S, Thorpe JR, Uryu K, Lee VM, Trojanowski JQ (2004) Clinical and neuropathologic variation in neuronal intermediate filament inclusion disease. Neurology 63:1376–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cannon A, Baker M, Boeve B, Josephs K, Knopman D, Petersen R, Parisi J, Dickison D, Adamson J, Snowden J, Neary D, Mann D, Hutton M, Pickering‐Brown SM (2006) CHMP2B mutations are not a common cause of frontotemporal lobar degeneration. Neurosci Lett 398:83–84. [DOI] [PubMed] [Google Scholar]
- 15. Cruts M, Rademakers R, Gijselinck I, Van Der ZJ, Dermaut B, De Pooter T, De Rijk P, Del Favero J, Van Broeckhoven C (2005) Genomic architecture of human 17q21 linked to frontotemporal dementia uncovers a highly homologous family of low‐copy repeats in the tau region. Hum Mol Genet 14:1753–1762. [DOI] [PubMed] [Google Scholar]
- 16. Cruts M, Gijselinck I, Van Der ZJ, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, Van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den BM, Cuijt I, Vennekens K, De Deyn PP, Kumar‐Singh S, Van Broeckhoven C (2006) Null mutations in progranulin cause ubiquitin‐positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–924. [DOI] [PubMed] [Google Scholar]
- 17. Daniel R, He Z, Carmichael KP, Halper J, Bateman A (2000) Cellular localization of gene expression for progranulin. J Histochem Cytochem 48:999–1009. [DOI] [PubMed] [Google Scholar]
- 18. DeLaBarre B, Brunger AT (2003) Complete structure of p97/valosin‐containing protein reveals communication between nucleotide domains. Nat Struct Biol 10:856–863. [DOI] [PubMed] [Google Scholar]
- 19. Dermaut B, Kumar‐Singh S, Engelborghs S, Theuns J, Rademakers R, Sacrens J, Pickut BA, Peeters K, Van den Broeck M, Vennekens K, Claes S, Cruts M, Cras P, Martin JJ, Van Broeckhoven C, De Deyn PP (2004) A novel presenilin 1 mutation associated with Pick’s disease but not beta‐amyloid plaques. Ann Neurol 55:617–626. [DOI] [PubMed] [Google Scholar]
- 20. Di Maria E, Tabaton M, Vigo T, Abbruzzese G, Bellone E, Donati C, Frasson E, Marchese R, Montagna P, Munoz DG, Pramstaller PP, Zanusso G, Ajmar F, Mandich P (2000) Corticobasal degeneration shares a common genetic background with progressive supranuclear palsy. Ann Neurol 47: 374–377. [DOI] [PubMed] [Google Scholar]
- 21. Dickson DW (1998) Pick’s disease: a modern approach. Brain Pathol 8:339–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, Jellinger K, Lantos PL, Lippa CF, Mirra SS, Tabaton M, Vonsattel JP, Wakabayashi K, Litvan I (2002) Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 61:935–946. [DOI] [PubMed] [Google Scholar]
- 23. Dreyfuss G, Matunis MJ, Pinol‐Roma S, Burd CG (1993) hnRNP proteins and the biogenesis of mRNA. Ann Rev Biochem 62:289–321. [DOI] [PubMed] [Google Scholar]
- 24. Evin G, Smith MJ, Tziotis A, McLean C, Canterford L, Sharples RA, Cappai R, Weidemann A, Beyreuther K, Cotton RG, Masters CL, Culvenor JG (2002) Alternative transcripts of presenilin‐1 associated with frontotemporal dementia. Neuroreport 13:719–723. [DOI] [PubMed] [Google Scholar]
- 25. Forman MS, Farmer J, Johnson JK, Clark CM, Arnold SE, Coslett HB, Chatterjee A, Hurtig HI, Karlawish JH, Rosen HJ, Van Deerlin V, Lee VM, Miller BL, Trojanowski JQ, Grossman M (2006) Frontotemporal dementia: clinicopathological correlations. Ann Neurol 59:952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA, Jhaveri BS, Karlawish JH, Pestronk A, Smith TW, Tu PH, Watts GD, Markesbery WR, Smith CD, Kimonis VE (2006) Novel ubiquitin neuropathology in frontotemporal dementia with valosin‐containing protein gene mutations. J Neuropathol Exp Neurol 65:571–581. [DOI] [PubMed] [Google Scholar]
- 27. Foster NL, Wilhelmsen K, Sima AA, Jones MZ, D’Amato CJ, Gilman S (1997) Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Ann Neurol 41:706–715. [DOI] [PubMed] [Google Scholar]
- 28. Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, Crook R, Melquist S, Kuntz K, Petersen R, Josephs K, Pickering‐Brown SM, Graff‐Radford N, Uitti R, Dickson D, Wszolek Z, Gonzalez J, Beach TG, Bigio E, Johnson N, Weintraub S, Mesulam M, White CL III, Woodruff B, Caselli R, Hsiung GY, Feldman H, Knopman D, Hutton M, Rademakers R (2006) Mutations in progranulin are a major cause of ubiquitin‐positive frontotemporal lobar degeneration. Hum Mol Genet 15:2988–3001. [DOI] [PubMed] [Google Scholar]
- 29. Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, Patterson V, Swingler R, Kieran D, Prehn J, Morrison KE, Green A, Acharya KR, Brown RH Jr, Hardiman O (2006) ANG mutations segregate with familial and “sporadic” amyotrophic lateral sclerosis. Nat Genet 38:411–413. [DOI] [PubMed] [Google Scholar]
- 30. Guyant‐Marechal L, Laquerriere A, Duyckaerts C, Dumanchin C, Bou J, Dugny F, Le BI, Frebourg T, Hannequin D, Campion D (2006) Valosin‐containing protein gene mutations: clinical and neuropathologic features. Neurology 67:644–651. [DOI] [PubMed] [Google Scholar]
- 31. Harvey RJ, Skelton‐Robinson M, Rossor MN (2003) The prevalence and causes of dementia in people under the age of 65 years. J Neurol Neurosurg Psychiatry 74:1206–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, McKee A, Tabaton M, Litvan I (1994) Preliminary NINDS neuropathologic criteria for Steele‐Richardson‐Olszewski syndrome (progressive supranuclear palsy). Neurology 44:2015–2019. [DOI] [PubMed] [Google Scholar]
- 33. He Z, Bateman A (2003) Progranulin (granulin‐epithelin precursor, PC‐cell‐derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med 81:600–612. [DOI] [PubMed] [Google Scholar]
- 34. He Z, Ong CH, Halper J, Bateman A (2003) Progranulin is a mediator of the wound response. Nat Med 9:225–229. [DOI] [PubMed] [Google Scholar]
- 35. Holm I, Englund E, the FReJA Consortium (2006) Ubiquitin‐positive inclusions in frontotemporal dementia linked to chromosome 3 (FTD‐3). 16th International Congress on Neuropathology, San Francisco, USA, September 9–16 [Abstract.
- 36. Houlden H, Baker M, Morris HR, MacDonald N, Pickering‐Brown S, Adamson J, Lees AJ, Rossor MN, Quinn NP, Kertesz A, Khan MN, Hardy J, Lantos PL, George‐Hyslop P, Munoz DG, Mann D, Lang AE, Bergeron C, Bigio EH, Litvan I, Bhatia KP, Dickson D, Wood NW, Hutton M (2001) Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology 56: 1702–1706. [DOI] [PubMed] [Google Scholar]
- 37. Huey ED, Grafman J, Wassermann EM, Pietrini P, Tierney MC, Ghetti B, Spina S, Baker M, Hutton M, Elder JW, Berger SL, Heflin KA, Hardy J, Momeni P (2006) Characteristics of frontotemporal dementia patients with a Progranulin mutation. Ann Neurol 60:374–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering‐Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, De Graaff E, Wauters E, Van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, Van Swieten J, Mann D, Lynch T, Heutink P (1998) Association of missense and 5′‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature 393:702–705. [DOI] [PubMed] [Google Scholar]
- 39. Ishigaki S, Hishikawa N, Niwa J, Iemura S, Natsume T, Hori S, Kakizuka A, Tanaka K, Sobue G (2004) Physical and functional interaction between Dorfin and Valosin‐containing protein that are colocalized in ubiquitylated inclusions in neurodegenerative disorders. J Biol Chem 279:51376–51385. [DOI] [PubMed] [Google Scholar]
- 40. Jackson M, Lennox G, Lowe J (1996) Motor neurone disease‐inclusion dementia. Neurodegeneration 5:339–350. [DOI] [PubMed] [Google Scholar]
- 41. Johnson JK, Diehl J, Mendez MF, Neuhaus J, Shapira JS, Forman M, Chute DJ, Roberson ED, Pace‐Savitsky C, Neumann M, Chow TW, Rosen HJ, Forstl H, Kurz A, Miller BL (2005) Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol 62:925–930. [DOI] [PubMed] [Google Scholar]
- 42. Josephs KA, Holton JL, Rossor MN, Braendgaard H, Ozawa T, Fox NC, Petersen RC, Pearl GS, Ganguly M, Rosa P, Laursen H, Parisi JE, Waldemar G, Quinn NP, Dickson DW, Revesz T (2003) Neurofilament inclusion body disease: a new proteinopathy? Brain 126:2291–2303. [DOI] [PubMed] [Google Scholar]
- 43. Josephs KA, Holton JL, Rossor MN, Godbolt AK, Ozawa T, Strand K, Khan N, Al Sarraj S, Revesz T (2004) Frontotemporal lobar degeneration and ubiquitin immunohistochemistry. Neuropathol Appl Neurobiol 30:369–373. [DOI] [PubMed] [Google Scholar]
- 44. Josephs KA, Jones AG, Dickson DW (2004) Hippocampal sclerosis and ubiquitin‐positive inclusions in dementia lacking distinctive histopathology. Dement Geriatr Cogn Disord 17:342–345. [DOI] [PubMed] [Google Scholar]
- 45. Katsuse O, Dickson DW (2005) Ubiquitin immunohistochemistry of frontotemporal lobar degeneration differentiates cases with and without motor neuron disease. Alzheimer Dis Assoc Disord 19(Suppl.1):S37–S43. [DOI] [PubMed] [Google Scholar]
- 46. Kertesz A, Munoz DG, Hillis A (2003) Preferred terminology. Ann Neurol 54(Suppl.5):S3–S6. [DOI] [PubMed] [Google Scholar]
- 47. Knopman DS, Mastri AR, Frey WH, Sung JH, Rustan T (1990) Dementia lacking distinctive histologic features: a common non‐Alzheimer degenerative dementia. Neurology 40:251–256. [DOI] [PubMed] [Google Scholar]
- 48. Kovach MJ, Waggoner B, Leal SM, Gelber D, Khardori R, Levenstien MA, Shanks CA, Gregg G, Al Lozi MT, Miller T, Rakowicz W, Lopate G, Florence J, Glosser G, Simmons Z, Morris JC, Whyte MP, Pestronk A, Kimonis VE (2001) Clinical delineation and localization to chromosome 9p13.3‐p12 of a unique dominant disorder in four families: hereditary inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Mol Genet Metab 74:458–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kumar‐Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens K, Corsmit E, Cruts M, Dermaut B, Wang R, Van Broeckhoven C (2006) Mean age‐of‐onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum Mutat 27:686–695. [DOI] [PubMed] [Google Scholar]
- 50. Kuusisto E, Suuronen T, Salminen A (2001) Ubiquitin‐binding protein p62 expression is induced during apoptosis and proteasomal inhibition in neuronal cells. Biochem Biophys Res Commun 280:223–228. [DOI] [PubMed] [Google Scholar]
- 51. Lambrechts D, Storkebaum E, Morimoto M, Del Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, Andersson J, Van MI, Al Chalabi A, Bornes S, Musson R, Hansen V, Beckman L, Adolfsson R, Pall HS, Prats H, Vermeire S, Rutgeerts P, Katayama S, Awata T, Leigh N, Lang‐Lazdunski L, Dewerchin M, Shaw C, Moons L, Vlietinck R, Morrison KE, Robberecht W, Van Broeckhoven C, Collen D, Andersen PM, Carmeliet P (2003) VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet 34:383–394. [DOI] [PubMed] [Google Scholar]
- 52. Le Ber I, Guedj E, Gabelle A, Verpillat P, Volteau M, Thomas‐Anterion C, Decousus M, Hannequin D, Vera P, Lacomblez L, Camuzat A, Didic M, Puel M, Lotterie JA, Golfier V, Bernard AM, Vercelletto M, Magne C, Sellal F, Namer I, Michel BF, Pasquier J, Salachas F, Bochet J, Brice A, Habert MO, Dubois B (2006) Demographic, neurological and behavioural characteristics and brain perfusion SPECT in frontal variant of frontotemporal dementia. Brain 129:3051–3065. [DOI] [PubMed] [Google Scholar]
- 53. Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159. [DOI] [PubMed] [Google Scholar]
- 54. Lendon CL, Lynch T, Norton J, McKeel DW Jr, Busfield F, Craddock N, Chakraverty S, Gopalakrishnan G, Shears SD, Grimmett W, Wilhelmsen KC, Hansen L, Morris JC, Goate AM (1998) Hereditary dysphasic disinhibition dementia: a frontotemporal dementia linked to 17q21‐22. Neurology 50:1546–1555. [DOI] [PubMed] [Google Scholar]
- 55. Liau LM, Lallone RL, Seitz RS, Buznikov A, Gregg JP, Kornblum HI, Nelson SF, Bronstein JM (2000) Identification of a human glioma‐associated growth factor gene, granulin, using differential immuno‐absorption. Cancer Res 60:1353–1360. [PubMed] [Google Scholar]
- 56. Lipton AM, White CL III, Bigio EH (2004) Frontotemporal lobar degeneration with motor neuron disease‐type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol (Berl) 108:379–385. [DOI] [PubMed] [Google Scholar]
- 57. Lund and Manchester Criteria (1994) Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups. J Neurol Neurosurg Psychiatry 57:416–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mackenzie IR, Feldman HH (2005) Ubiquitin immunohistochemistry suggests classic motor neuron disease, motor neuron disease with dementia, and frontotemporal dementia of the motor neuron disease type represent a clinicopathologic spectrum. J Neuropathol Exp Neurol 64:730–739. [DOI] [PubMed] [Google Scholar]
- 59. Mackenzie IR, Baker M, Pickering‐Brown S, Hsiung GY, Lindholm C, Dwosh E, Gass J, Cannon A, Rademakers R, Hutton M, Feldman HH (2006) The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain 129:3081–3090. [DOI] [PubMed] [Google Scholar]
- 60. Mackenzie IR, Baker M, West G, Woulfe J, Qadi N, Gass J, Cannon A, Adamson J, Feldman H, Lindholm C, Melquist S, Pettman R, Sadovnick AD, Dwosh E, Whiteheart SW, Hutton M, Pickering‐Brown SM (2006) A family with tau‐negative frontotemporal dementia and neuronal intranuclear inclusions linked to chromosome 17. Brain 129: 853–867. [DOI] [PubMed] [Google Scholar]
- 61. Mackenzie IR, Shi J, Shaw CL, Duplessis D, Neary D, Snowden JS, Mann DM (2006) Dementia lacking distinctive histology (DLDH) revisited. Acta Neuropathol (Berl) 112:551–559. [DOI] [PubMed] [Google Scholar]
- 62. McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ (2001) Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol 58: 1803–1809. [DOI] [PubMed] [Google Scholar]
- 63. Mann DM, McDonagh AM, Snowden J, Neary D, Pickering‐Brown SM (2000) Molecular classification of the dementias. Lancet 355:626. [DOI] [PubMed] [Google Scholar]
- 64. Maquat LE (2004) Nonsense‐mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 5:89–99. [DOI] [PubMed] [Google Scholar]
- 65. Martinez‐Lage P, Munoz DG (1997) Prevalence and disease associations of argyrophilic grains of Braak. J Neuropathol Exp Neurol 56:157–164. [DOI] [PubMed] [Google Scholar]
- 66. Mitsuyama Y, Takamiya S (1979) Presenile dementia with motor neuron disease in Japan. A new entity? Arch Neurol 36:592–593. [DOI] [PubMed] [Google Scholar]
- 67. Momeni P, Bell J, Duckworth J, Hutton M, Mann D, Brown SP, Hardy J (2006) Sequence analysis of all identified open reading frames on the frontal temporal dementia haplotype on chromosome 3 fails to identify unique coding variants except in CHMP2B. Neurosci Lett 410:77–79. [DOI] [PubMed] [Google Scholar]
- 68. Morita M, Al Chalabi A, Andersen PM, Hosler B, Sapp P, Englund E, Mitchell JE, Habgood JJ, De Belleroche J, Xi J, Jongjaroenprasert W, Horvitz HR, Gunnarsson LG, Brown RH Jr (2006) A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology 66:839–844. [DOI] [PubMed] [Google Scholar]
- 69. Morris HR, Janssen JC, Bandmann O, Daniel SE, Rossor MN, Lees AJ, Wood NW (1999) The tau gene A0 polymorphism in progressive supranuclear palsy and related neurodegenerative diseases. J Neurol Neurosurg Psychiatry 66:665–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Morris HR, Khan MN, Janssen JC, Brown JM, Perez‐Tur J, Baker M, Ozansoy M, Hardy J, Hutton M, Wood NW, Lees AJ, Revesz T, Lantos P, Rossor MN (2001) The genetic and pathological classification of familial frontotemporal dementia. Arch Neurol 58:1813–1816. [DOI] [PubMed] [Google Scholar]
- 71. Mott RT, Dickson DW, Trojanowski JQ, Zhukareva V, Lee VM, Forman M, Van Deerlin V, Ervin JF, Wang DS, Schmechel DE, Hulette CM (2005) Neuropathologic, biochemical, and molecular characterization of the frontotemporal dementias. J Neuropathol Exp Neurol 64:420–428. [DOI] [PubMed] [Google Scholar]
- 72. Mukherjee O, Pastor P, Cairns NJ, Chakraverty S, Kauwe JS, Shears S, Behrens MI, Budde J, Hinrichs AL, Norton J, Levitch D, Taylor‐Reinwald L, Gitcho M, Tu PH, Tenenholz GL, Liscic RM, Armendariz J, Morris JC, Goate AM (2006) HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin‐positive, tau‐negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol 60:314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Munch C, Rosenbohm A, Sperfeld AD, Uttner I, Reske S, Krause BJ, Sedlmeier R, Meyer T, Hanemann CO, Stumm G, Ludolph AC (2005) Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann Neurol 58:777–780. [DOI] [PubMed] [Google Scholar]
- 74. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51:1546–1554. [DOI] [PubMed] [Google Scholar]
- 75. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VMY (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. [DOI] [PubMed] [Google Scholar]
- 76. Okamoto K, Hirai S, Yamazaki T, Sun XY, Nakazato Y (1991) New ubiquitin‐positive intraneuronal inclusions in the extra‐motor cortices in patients with amyotrophic lateral sclerosis. Neurosci Lett 129:233–236. [DOI] [PubMed] [Google Scholar]
- 77. Ou SH, Wu F, Harrich D, Garcia‐Martinez LF, Gaynor RB (1995) Cloning and characterization of a novel cellular protein, TDP‐43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 69:3584–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Parkinson N, Ince PG, Smith MO, Highley R, Skibinski G, Andersen PM, Morrison KE, Pall HS, Hardiman O, Collinge J, Shaw PJ, Fisher EM (2006) ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 267:1074–1077. [DOI] [PubMed] [Google Scholar]
- 79. Pick A (1892) Über die Beziehungen der senilen Hirnatrophie zur Aphasie. Prager Med Wochenschr 17:165–167. [Google Scholar]
- 80. Pickering‐Brown SM, Richardson AMT, Snowden JS, McDonagh AM, Burns A, Braude W, Baker M, Liu WK, Yen SH, Hardy J, Hutton M, Davies Y, Allsop D, Craufurd D, Neary D, Mann DMA (2002) Inherited frontotemporal dementia in nine British families associated with intronic mutations in the tau gene. Brain 125:732–751. [DOI] [PubMed] [Google Scholar]
- 81. Pickering‐Brown SM, Baker M, Gass J, Boeve BF, Loy CT, Brooks WS, Mackenzie IRA, Martins RN, Kwok JBJ, Halliday GM, Kril J, Schofield PR, Mann DMA, Hutton M (2006) Mutations in progranulin explain atypical phenotypes with variants in MAPT. Brain 129:3124–3126. [DOI] [PubMed] [Google Scholar]
- 82. Pirici D, Van Der Zee J, Vandenberghe R, Rademakers R, Dermaut B, Cruts M, Vennekens K, Cuijt I, Lübke U, Ceuterick C, Martin JJ, Van Broeckhoven C, Kumar‐Singh S (2006) Characterization of ubiquitinated intraneuronal inclusions in a novel Belgian frontotemporal lobar degeneration family. J Neuropathol Exp Neurol 65:289–301. [DOI] [PubMed] [Google Scholar]
- 83. Pleasure IT, Black MM, Keen JH (1993) Valosin‐containing protein, VCP, is a ubiquitous clathrin‐binding protein. Nature 365:459–462. [DOI] [PubMed] [Google Scholar]
- 84. Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43:815–825. [DOI] [PubMed] [Google Scholar]
- 85. Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH Jr, Ludlow CL, Fischbeck KH (2003) Mutant dynactin in motor neuron disease. Nat Genet 33:455–456. [DOI] [PubMed] [Google Scholar]
- 86. Rademakers R, Cruts M, Dermaut B, Sleegers K, Rosso SM, Van Den Broeck M, Backhovens H, Van Swieten J, Van Duijn CM, Van Broeckhoven C (2002) Tau negative frontal lobe dementia at 17q21: significant finemapping of the candidate region to a 4.8 cM interval. Mol Psychiatry 7:1064–1074. [DOI] [PubMed] [Google Scholar]
- 87. Rademakers R, Cruts M, Van Broeckhoven C (2004) The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat 24:277–295. [DOI] [PubMed] [Google Scholar]
- 88. Rademakers R, Melquist S, Cruts M, Theuns J, Del Favero J, Poorkaj P, Baker M, Sleegers K, Crook R, De Pooter T, Bel KS, Adamson J, Van den BD, Van den BM, Gass J, Corsmit E, De Rijk P, Thomas N, Engelborghs S, Heckman M, Litvan I, Crook J, De Deyn PP, Dickson D, Schellenberg GD, Van Broeckhoven C, Hutton ML (2005) High‐density SNP haplotyping suggests altered regulation of tau gene expression in progressive supranuclear palsy. Hum Mol Genet 14:3281–3292. [DOI] [PubMed] [Google Scholar]
- 89. Ratnavalli E, Brayne C, Dawson K, Hodges JR (2002) The prevalence of frontotemporal dementia. Neurology 58:1615–1621. [DOI] [PubMed] [Google Scholar]
- 90. Raux G, Gantier R, Thomas‐Anterion C, Boulliat J, Verpillat P, Hannequin D, Brice A, Frebourg T, Campion D (2000) Dementia with prominent frontotemporal features associated with L113P presenilin 1 mutation. Neurology 55:1577–1578. [DOI] [PubMed] [Google Scholar]
- 91. Reed LA, Wszolek ZK, Hutton M (2001) Phenotypic correlations in FTDP‐17. Neurobiol Aging 22:89–107. [DOI] [PubMed] [Google Scholar]
- 92. Rizzu P, Van Swieten JC, Joosse M, Hasegawa M, Stevens M, Tibben A, Niermeijer MF, Hillebrand M, Ravid R, Oostra BA, Goedert M, Van Duijn CM, Heutink P (1999) High prevalence of mutations in the microtubule‐associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet 64:414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rizzu P, Van Mil SE, Anar B, Rosso SM, Kaat LD, Heutink P, Van Swieten JC (2006) CHMP2B mutations are not a cause of dementia in Dutch patients with familial and sporadic frontotemporal dementia. Am J Med Genet B Neuropsychiatr Genet 141:944–946. [DOI] [PubMed] [Google Scholar]
- 94. Rogaeva E, Fafel K, Song Y, Medeiros B, Sato C, Liang Y, Richard E, Rogaev E, Frommelt P, Sadovnick AD, Meschino W, Rockwood K, Boss M, Mayeux R, St George‐Hyslop P (2001) Screening for PS1 mutations in a referral‐based series of AD cases: 21 novel mutations. Neurology 57:621–625. [DOI] [PubMed] [Google Scholar]
- 95. Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ III, Kandel ER, Duff K, Kirkwood A, Shen J (2004) Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age‐dependent neurodegeneration. Neuron 42:23–36. [DOI] [PubMed] [Google Scholar]
- 96. Schroder R, Watts GD, Mehta SG, Evert BO, Broich P, Fliessbach K, Pauls K, Hans VH, Kimonis V, Thal DR (2005) Mutant valosin‐containing protein causes a novel type of frontotemporal dementia. Ann Neurol 57:457–461. [DOI] [PubMed] [Google Scholar]
- 97. Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, Brandner S, Brun A, Rossor MN, Gade A, Johannsen P, Sorensen SA, Gydesen S, Fisher EM, Collinge J (2005) Mutations in the endosomal ESCRTIII‐complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37:806–808. [DOI] [PubMed] [Google Scholar]
- 98. Spillantini MG, Bird TD, Ghetti B (1998) Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol 8:387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 95: 7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Stanford PM, Shepherd CE, Halliday GM, Brooks WS, Schofield PW, Brodaty H, Martins RN, Kwok JBJ, Schofield PR (2003) Mutations in the tau gene that cause an increase in three repeat tau and frontotemporal dementia. Brain 126:814–826. [DOI] [PubMed] [Google Scholar]
- 101. Tangkeangsirisin W, Serrero G (2004) PC cell‐derived growth factor (PCDGF/GP88, progranulin) stimulates migration, invasiveness and VEGF expression in breast cancer cells. Carcinogenesis 25:1587–1592. [DOI] [PubMed] [Google Scholar]
- 102. Taniguchi S, McDonagh AM, Pickering‐Brown SM, Umeda Y, Iwatsubo T, Hasegawa M, Mann DM (2004) The neuropathology of frontotemporal lobar degeneration with respect to the cytological and biochemical characteristics of tau protein. Neuropathol Appl Neurobiol 30:1–18. [DOI] [PubMed] [Google Scholar]
- 103. Togo T, Cookson N, Dickson DW (2002) Argyrophilic grain disease: neuropathology, frequency in a dementia brain bank and lack of relationship with apolipoprotein E. Brain Pathol 12:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Togo T, Sahara N, Yen SH, Cookson N, Ishizawa T, Hutton M, De Silva R, Lees A, Dickson DW (2002) Argyrophilic grain disease is a sporadic 4‐repeat tauopathy. J Neuropathol Exp Neurol 61:547–556. [DOI] [PubMed] [Google Scholar]
- 105. Tolnay M, Probst A (1999) Tau protein pathology in Alzheimer’s disease and related disorders. Neuropathol Appl Neurobiol 25:171–187. [DOI] [PubMed] [Google Scholar]
- 106. Tolnay M, Probst A (2002) Frontotemporal lobar degeneration—tau as a pied piper? Neurogenetics 4:63–75. [DOI] [PubMed] [Google Scholar]
- 107. Vance C, Al Chalabi A, Ruddy D, Smith BN, Hu X, Sreedharan J, Siddique T, Schelhaas HJ, Kusters B, Troost D, Baas F, De Jong V, Shaw CE (2006) Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2‐21.3. Brain 129:868–876. [DOI] [PubMed] [Google Scholar]
- 108. Wang HY, Wang IF, Bose J, Shen CK (2004) Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics 83:130–139. [DOI] [PubMed] [Google Scholar]
- 109. Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin‐containing protein. Nat Genet 36:377–381. [DOI] [PubMed] [Google Scholar]
- 110. Wightman G, Anderson VE, Martin J, Swash M, Anderton BH, Neary D, Mann D, Luthert P, Leigh PN (1992) Hippocampal and neocortical ubiquitin‐immunoreactive inclusions in amyotrophic lateral sclerosis with dementia. Neurosci Lett 139:269–274. [DOI] [PubMed] [Google Scholar]
- 111. Wilhelmsen KC, Lynch T, Pavlou E, Higgins M, Nygaard TG (1994) Localization of disinhibition‐dementia‐parkinsonism‐amyotrophy complex to 17q21‐22. Am J Hum Genet 55:1159–1165. [PMC free article] [PubMed] [Google Scholar]
- 112. Xu SQ, Tang D, Chamberlain S, Pronk G, Masiarz FR, Kaur S, Prisco M, Zanocco‐Marani T, Baserga R (1998) The granulin/epithelin precursor abrogates the requirement for the insulin‐like growth factor 1 receptor for growth in vitro. J Biol Chem 273:20078–20083. [DOI] [PubMed] [Google Scholar]
- 113. Zanocco‐Marani T, Bateman A, Romano G, Valentinis B, He ZH, Baserga R (1999) Biological activities and signaling pathways of the granulin/epithelin precursor. Cancer Res 59:5331–5340. [PubMed] [Google Scholar]
- 114. Van Der Zee J, Gijselinck I, Pirici D, Kumar‐Singh S, Cruts M, Van Broeckhoven C (2007) Frontotemporal dementia with ubiquitin‐positive inclusions: a molecular genetic update. Neurodegener Dis (in press). [DOI] [PubMed] [Google Scholar]
- 115. Van Der Zee J, Rademakers R, Engelborghs S, Gijselinck I, Bogaerts V, Vandenberghe R, Santens P, Caekebeke J, De Pooter T, Peeters K, Lubke U, Van den BM, Martin JJ, Cruts M, De Deyn PP, Van Broeckhoven C, Dermaut B (2006) A Belgian ancestral haplotype harbours a highly prevalent mutation for 17q21‐linked tau‐negative FTLD. Brain 129:841–852. [DOI] [PubMed] [Google Scholar]
- 116. Zhukareva V, Vogelsberg‐Ragaglia V, Van Deerlin VM, Bruce J, Shuck T, Grossman M, Clark CM, Arnold SE, Masliah E, Galasko D, Trojanowski JQ, Lee VM (2001) Loss of brain tau defines novel sporadic and familial tauopathies with frontotemporal dementia. Ann Neurol 49:165–175. [DOI] [PubMed] [Google Scholar]
- 117. Zhukareva V, Mann D, Pickering‐Brown S, Uryu K, Shuck T, Shah K, Grossman M, Miller BL, Hulette CM, Feinstein SC, Trojanowski JQ, Lee VM (2002) Sporadic Pick’s disease: a tauopathy characterized by a spectrum of pathological tau isoforms in gray and white matter. Ann Neurol 51:730–739. [DOI] [PubMed] [Google Scholar]