

Abstract
Cytologic pleomorphism has been described in a limited number of benign pineal tumors, namely pineocytoma (PC) and pineal parenchymal tumors (PPTs) of intermediate differentiation (PPTID). We examined the clinicopathologic features in a retrospective series of 14 cases (seven females and seven males aged from 10 to 65 years) of pleomorphic PPT. Seven cases were PC, with no mitoses and with areas of tumoral cells forming large pineocytomatous rosettes and other areas with giant cells containing hyperchromatic nuclei. The other seven were PPTID, presenting few mitoses (≤2), a Ki67 proliferation index between 3% and 7%, and predominantly composed of small neoplastic cells and scattered giant cells, sometimes multinucleated. In the 14 tumors, the proportion of pleomorphic areas was variable. Most tumoral cells showed extensive neuronal differentiation with strong expression of neuron‐specific enolase, synaptophysin and neurofilaments. Some of the neoplastic cells expressed S100 protein. The follow‐up period ranged from 1.2 to 13 years and only one PC and one PPTID progressed after stereotactic biopsy or incomplete resection. The lack of invasiveness and the low proliferation index of these tumors suggest a benign clinical course despite the marked pleomorphism, the latter of which can lead to upgrading.
Keywords: pineal parenchymal tumors, pineocytoma, pleomorphic, neuronal differentiation
INTRODUCTION
Pineal parenchymal tumors (PPTs) represent about 30% of pineal region neoplasms and are rare tumors accounting for less than 0.1% of all brain tumors. They are subdivided into pineocytoma (PC), pineoblastoma and PTT of intermediate differentiation (PPTID) 9, 15, 16. In PC, cytologic pleomorphism accompanied by ganglion cells intensely expressing neuronal markers has been described 9, 11. Moreover, cytologic pleomorphism has been recently reported in one case of PPTID (20). However, because of the rarity of PPT, these are the only tumors with cytologic pleomorphism reported in the literature. The presence of pleomorphic cells may lead to upgrading of tumors. In this study, we report 14 cases of PPT presenting cytologic pleomorphism from our cooperative series of more than 100 patients presenting with PPT.
MATERIALS AND METHODS
Patients
One hundred and ten out of 433 tumor samples of the pineal region obtained between 1979 and 2006 in the different centers were retained as PPT after histopathological review. Fourteen of these showed pleomorphism and were studied using light microscopy and immunohistochemistry. Five of these cases have been included in a previous study analyzing the correlation of histological features with prognosis in 66 cases of PPT (9).
Light microscopy
Surgically removed tissues were fixed in 4% paraformaldehyde/15% picric acid, formol or formol‐zinc and embedded in paraffin, then cut into 4 µm sections. Routine staining was performed using hemalin phloxin saffron, hemalin eosin saffron or hemalin eosin. Mitoses were counted in 10 randomly selected fields at high magnification (×400) and expressed as the number of mitoses in 10 high‐power fields (HPFs). The number of vessels was scored as 0, + or ++ when, respectively, absent, moderate or high. For a subset of tumors, cytological imprints were prepared and stained with toluidine blue.
Immunohistochemistry
With the exception of those intended for testing for the presence of S100 protein, the sections were heated for 2 × 5 minutes in citrate buffer in a microwave oven, then, stained with the specific antiserum. Immunohistochemical staining was performed using the avidin‐biotin technique and either polyclonal antiserum against S100 protein or monoclonal antibodies against neuron‐specific enolase (NSE), synaptophysin (SYN), neurofilament (NF, 70 and 200 kDa forms), chromogranin A (ChgA), NeuN, vimentin (VIM), glial fibrillary acidic protein (GFAP), or CD34 (see Table 2 for sources). The intensity of labeling was scored as 0 when absent or +/− or + when, respectively, moderate or high. Immunohistochemistry with anti‐Ki67 antibody (MIB1 clone, Dako) was performed on samples fixed in formol or formol‐zinc in order to estimate cell proliferation.
Table 2.
List and sources of antibodies used and immunohistochemical results obtained. Abbreviations: NSE = neuron‐specific enolase; SYN = synaptophysin; NF = neurofilament; ChgA = chromogranin A; VIM = vimentin; GFAP = glial fibrillary acidic protein.
| Antigen | Antibody source | Number of tumors analyzed | − (%) | +/− (%) | + (%) |
|---|---|---|---|---|---|
| NSE | H14 clone, Dako (Trappes, France) | 8 | 0 | 0 | 100 |
| SYN | SY38 clone, Dako (Trappes, France) | 14 | 0 | 0 | 100 |
| NF | 2F11 clone, Dako (Trappes, France) | 14 | 0 | 0 | 100 |
| ChgA | Dak A3 clone, Dako (Trappes, France) | 13 | 23 | 8 | 69 |
| NeuN | Clone A60, Chemicon, Euromedex (Mundolsheim, France) | 5 | 100 | 0 | 0 |
| CD34 | Clone QBEND10, Immunotech (Marseille, France) | 2 | 50 | 0 | 50 |
| S100‐P | Biomeda 23/11 (Paris) or Dako (Trappes, France) | 12 | 8 | 17 | 75 |
| VIM | V9 clone, Dako (Trappes, France) | 11 | 36 | 36 | 27 |
| GFAP | 6F2 clone, Dako (Trappes, France) | 13 | 100 | 0 | 0 |
RESULTS
Clinical data
The main clinical data are summarized in Table 1. Seven cases, with a male predominance (six males and one female), were pleomorphic PC without mitoses and considered as low grade tumors (grade I). The other seven cases, with a female predominance (one male and six females), were tumors with ≤2 mitoses and a Ki67 proliferation index between 3% and 7% and were considered as PPTID grade II. The patients' ages ranged from 10 to 65 years for the PCs (mean ± SEM = 47.4 ± 7.1), and from 24 to 60 years for the PPTIDs (mean ± SEM = 42.0 ± 6.1).
Table 1.
Clinical and morphological findings in pleomorphic PPT. Abbreviations: HPF = high‐power field; NA = not available; ND = not done; 0 = no or very low; + = moderate number; ++ = high number of vessels; RT = radiotherapy.
| Case | Age (years)/sex | Tumor size (mm) | Operation | Histological type | Grade | Complementary adjuvant treatment | Follow‐up | Mitoses/10 HPFs | Vessels | Ki67 (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 10/M | 20 | Complete resection | PC | I | No | No recurrence, alive at 13 years | 0 | 0 | ND |
| 2 | 42/M | 30 | Complete resection | PC | I | No | Lost to follow‐up | 0 | + | ND |
| 3 | 44/M | 30 | Stereotactic biopsy | PC | I | Focal RT | Progression at 8 years, death | 0 | + | ND |
| 4 | 53/M | NA | Ventriculoscopy Stereotactic biopsy Complete resection | PC | I | Focal RT | Alive at 2 years | 0 | + | 0 |
| 5 | 54/M | 30 | Complete resection | PC | I | No | Alive at 4 years | 0 | + | ND |
| 6 | 64/F | 20 | Complete resection | PC | I | No | No recurrence, alive at 13 years | 0 | + | ND |
| 7 | 65/M | 20 | Complete resection | PC | I | Focal RT | No recurrence, death at 7 years | 0 | + | ND |
| 8 | 24/F | NA | Ventriculoscopy and biopsy Complete resection | PPTID | II | RT | Alive at 6 years | 1 | + | 3 |
| 9 | 24/F | 40 | Biopsy Resection | PPTID | II | No | No recurrence, alive at 3.5 years | 0 | + | 4 |
| 10 | 33/F | 50 | Complete resection | PPTID | II | No | No recurrence, alive at 1.2 years | 0 | + | 6 |
| 11 | 37/F | 30 | Endoscopic biopsy Incomplete resection | PPTID | II | No | Progression, alive at 1.4 years | 0 | 0 | 4 |
| 12 | 57/F | NA | Stereostatic biopsy Incomplete resection | PPTID | II | RT | Alive at 6.6 years | 2 | ++ | 4 |
| 13 | 59/M | 29 | Stereostatic biopsy Endoscopic biopsy Incomplete resection | PPTID | II | No | Death (pneumopathy) at 1.9 years | 0 | ++ | 5 |
| 14 | 60/F | 26 | Ventriculocisternostomy Complete resection | PPTID | II | No | Alive at 1.7 years | 2 | + | 7 |
Cases 1, 2, 3, 6 and 7 have been included in a previous study (9).
On computer tomography (CT), the tumors were non invasive, well‐circumscribed masses that were isodense to hypodense on non‐contrast CT. Homogeneous enhancement was seen with the use of contrast. On magnetic resonance imaging, the tumors exhibited low isointensity or mixed signal intensity on T1‐weighted images ( Figure 1A). Homogeneous to slightly heterogeneous contrast enhancement was seen on the contrast enhanced images (Figure 1B).
Figure 1.
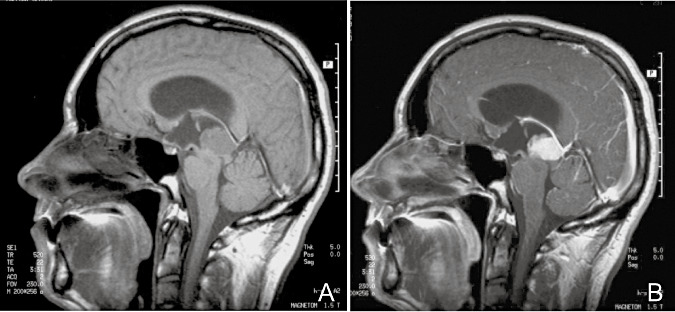
A. Sagittal T1‐weighted magnetic resonance imaging (MRI) scan in a patient with pleomorphic pineal parenchymal tumor (case 3). B. Sagittal T1‐weighted MRI scan in the same patient after gadolinium injection, showing homogeneous enhancement.
Tumor size (maximal diameter as assessed in preoperative imaging studies) ranged from 20 to 50 mm and was not different between the PCs (25.0 ± 2.2) and PPTIDs (35.0 ± 4.4). Complete resection of the tumor was achieved in nine patients; in the other five cases, only incomplete resection of the tumor was feasible or surgery remained restricted to diagnostic biopsy. Nine patients had no treatment after surgery, while five received radiotherapy, focally in three cases.
Detailed follow‐up information was available for 13 patients. The follow‐up period ranged from 1.2 to 13 years. Progression was only seen in two patients, one 8 years after PC was diagnosed by stereotactic biopsy and one PPTID patient 1.4 years after incomplete resection. Of the seven patients so far followed up for 5 years or known to have died in this period, four out of four with PC survived (one dying at 7 years and one at 8 years) and two out of three with PPTID survived, one dying of pneumopathy at 1.9 years.
HISTOPATHOLOGICAL FEATURES
The pleomorphic PCs presented areas with tumoral cells forming large pineocytomatous rosettes of variable number and size and areas with giant cells with hyperchromatic nuclei ( Figure 2A). The cytological imprint showed pinealocytes with small round nuclei and other larger cells with pleomorphic nuclei (Figure 2A inset). The pleomorphic PPTIDs were predominantly composed of small neoplastic cells with a high nuclear to cytoplasmic ratios, dense chromatin and irregularly shaped nuclei along with scattered giant cells, which were sometimes multinucleated (Figure 2B). The proportion of pleomorphic areas varied between tumors and could represent more than half of the tumor. Only three tumors presented any mitotic figures and these were few (1–2 per 10 HPFs). Despite the marked pleomorphism, neither endothelial proliferation nor necrosis was found. In all tumors, numerous vessels were seen.
Figure 2.
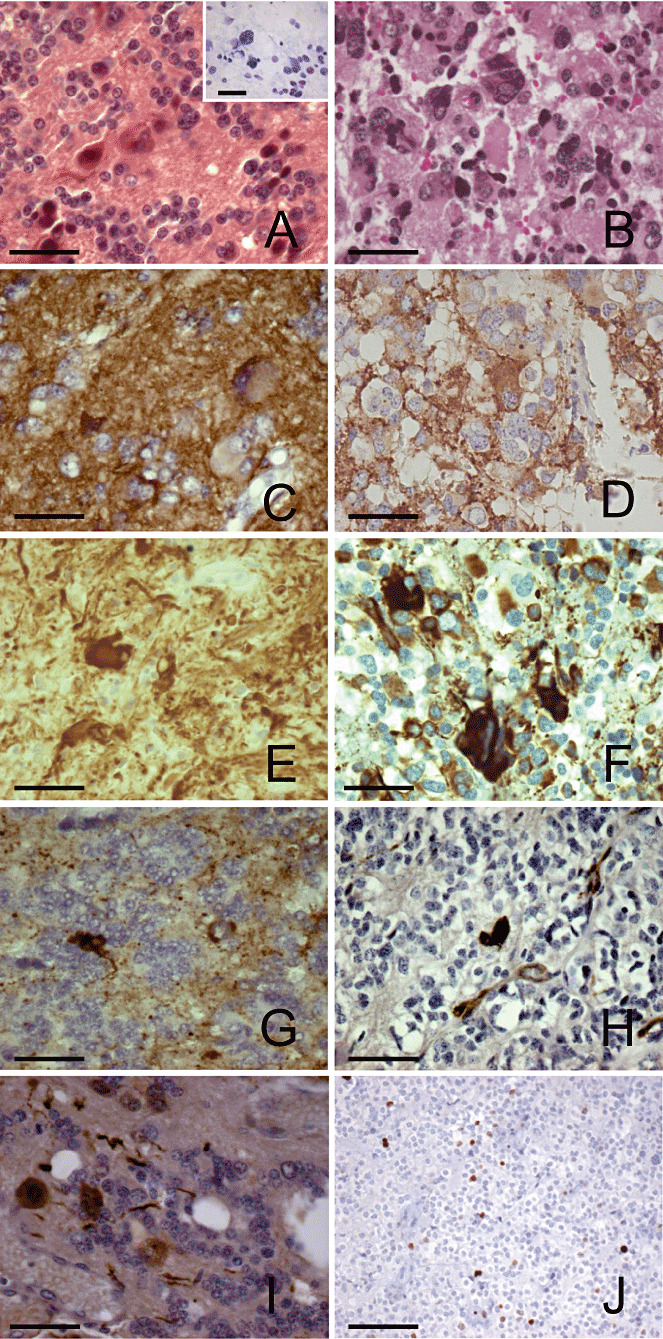
Light microscopic and immunohistochemical features of pleomorphic pineal parenchymal tumors (PPTs). A. Pleomorphic pineocytoma (PC) with pineocytomatous rosettes and giant cells with hyperchromatic nuclei (hemalin phloxine saffron staining). Inset: cytologic imprint of a pleomorphic PC showing tumoral cells with small round nuclei and larger cells with pleomorphic nuclei (toluidine blue staining). B. Pleomorphic PPT of intermediate differentiation (PPTID) with marked cytologic pleomorphism (hemalin eosin staining). Synaptophysin immuno‐labeling in a pleomorphic PC (C) and a pleomorphic PPTID (D). Expression of neurofilament in one pleomorphic PC (E) and in one pleomorphic PPTID (F), with intense labeling of giant cells. G. Chromogranin A labeling in a pleomorphic PC. H. CD34 labeling in a pleomorphic PC showing some positive giant cells. I. Nuclear and cytoplasmic S100 protein staining in giant cells of a pleomorphic PC. J. Areas of a pleomorphic PPTID showing nuclei stained for Ki67. Scale bars = 50 µm in A–I, 100 µm in J.
The immunohistochemical results are reported in Table 2 and illustrated in Figure 2. The tumor cells were positive for the neuronal markers SYN (Figure 2C,D), NSE, and NF (Figure 2E,F). Some giant cells were intensively labeled, especially with anti‐NF antibodies (Figure 2E,F). In some cases, the neoplastic cells were reactive for ChgA (Figure 2G). In a subset of tumors analyzed (one PC and four PPTIDs), no NeuN immunoreactivity was seen. Of the two tumors analyzed, a pleomorphic PC expressed CD34 only in vascular endothelial cells, while a PPTID also expressed this glycoprotein in giant cells (Figure 2H). In some tumors, vimentin expression was restricted to vessels, while, in others, it was also seen in tumor cells. Only interstitial and glial cells were labeled with anti‐GFAP antibody. Giant cells in some tumors expressed S100 protein (Figure 2I). The Ki67 labeling index varied from 3% to 7% (4.7 ± 0.5) in the maximally stained areas in the PPTIDs (Figure 2J).
DISCUSSION
Pleomorphic variants of PPT were first described in PC, with the presence of giant cells with irregular, hyperchromatic nuclei, sometimes multinucleated, in tumors also presenting characteristic pineocytomatous rosettes 9, 11. Cytologic pleomorphism has also been reported in one case of PPTID with moderate proliferative activity (20). An accurate histological diagnosis is essential for these rare tumors because optimal therapeutic management depends on the tumor grading. Our large cooperative series of PPTs made it possible for us to collect 14 clinically followed cases of pleomorphic PPT presenting features of either PC or PPTID. Except for one young boy, the PC patients were older than 40 years. More than half of the patients with PPTID were younger than 40 years and this tumor seems to be observed in younger patients, in accordance with the previously reported case (20). The male predominance in PC and the female predominance in PPTID remain to be verified with a higher number of cases, as no sex predilection has been reported for PPT 15, 16. Tumor recurrence was only observed in two cases. Moreover, the disease progression in the patients presenting with PC could be due to incomplete tumor resection. Based on our previous experience, completeness of excision seems to be the most important factor for avoiding disease progression.
Seven cases were diagnosed as PC because of the presence of underlying typical areas with an isomorphic cytology and large fibrillary pineocytomatous rosettes and immunoreactivity for a variety of neuronal markers 8, 9, which make it possible to distinguish these tumors from glioneuronal neoplasms 4, 18, 22, which generally express glial markers. The large number of cells labeled for neuronal markers, including many pleomorphic cells, confirmed previous findings that a neuronal immunophenotype is common in PC 5, 9, 11. Moreover, we previously showed by microarray analysis that two variants of PC, one typical and one pleomorphic, had very similar gene profiles and the two tumors were clearly classified together by cluster analysis based on the entire gene set, notwithstanding the presence of pleomorphic cells (6). The other seven cases, diagnosed as PPTID in our series, were rather similar to the case presented by Sasaki et al (20) and to case 2 reported by Kuchelmeister et al (11). They had moderate proliferative activity, as indicated by only a few mitotic figures and slightly increased Ki67 index, and can be considered as grade II tumors, as reported in the recent PPT classification of the World Health Organisation (16), with a prognosis not as favorable as that of PC. Only one of these cases received radiation therapy and had a 6‐year survival, but, as the follow‐up of the other PPTID cases was quite short, the benefit of such treatment in preventing local tumor regrowth is still uncertain.
The sometimes multinucleated giant cells are thought to be neoplastic and may derive from fusion of mononuclear tumor cells (20). Two types of cells have been described in the neonatal pineal gland (13) and we can hypothesize that both are capable of neoplastic transformation. The above authors showed that the two types of cells differentially express NSE and S100 protein and that the proportion of each type varies throughout postnatal evolution. At birth, the predominant cell type exhibits rosette formation and expresses S100 protein, but not NSE. The number of the second type of cell, which expresses NSE, but not S100 protein, increases gradually with age, and these cells are probably the origin of pineal parenchymal cells. As oncogenesis may recapitulate ontogeny of the human pineal gland (12), both types of cell might be seen in the tumor. Moreover, the presence of another type of cell, which is slightly larger than a pinealocyte and has non‐homogeneously condensed nuclear chromatin, has been reported in the fetal pineal gland (14). The pleomorphism in PPT may derive from these different subtypes of cells found in the human pineal gland during development. The expression of S100 protein in some giant cells may be related to the presence of this protein in cells which may represent immature pinealocytes, as seen in the developing pineal gland (13). CD34, expressed in vascular endothelial cells, was also found in giant cells in one PPTID. This expression might correspond to an undifferentiated level of these tumoral cells. Indeed, this marker has been found in neoplastic and malformative lesions, most notably in gangliogliomas 2, 3 and pleomorphic xanthoastrocytomas (19), and seems to be associated with a displastic or non‐differentiated cell type.
The pleomorphism encountered in PPT has been described in other low‐grade tumors, such as astrocytoma in the pineal gland (17) and hypothalamus (1), pituicytoma (21) and pleomorphic xanthoastrocytoma 7, 10. This marked pleomorphism may lead to upgrading of these tumors and incorrect therapy.
In conclusion, PC and PPTID are rare tumors which can present with marked pleomorphism, potentially leading to the misclassification and upgrading of these tumors. The short postoperative period for some tumors did not allow assessment of the biological behavior of these tumors. However, the lack of invasiveness and the low number of proliferating cells suggest a benign clinical course for these pleomorphic neoplasms.
ACKNOWLEDGMENTS
We thank the neurosurgeons B. Bataille (Poitiers), P. Cornu (Paris), P. Decq (Créteil), A. Goutelle (Lyon), S. Gaillard (Paris), F. Grisoli (Marseille), C. Lapras (Lyon), P. Mertens (Lyon), K. Mourier (Paris), C. Mottolese (Lyon), G. Robert (Paris), F.E. Roux (Toulouse), J.P. Sichez (Paris) and R. Van Effentere (Paris) for their contribution to the study.
We also thank E. Gros and M. Soutrenon for technical assistance, S. Badet for secretarial assistance, and T. Barkas for linguistic help.
This work was supported by INSERM, the Association for Cancer Research (ARC 3277) and the ADERTU.
REFERENCES
- 1. Abe T, Inoue R, Isono M, Ishii K, Fujiki M, Kamida T et al (2006) Benign pleomorphic astrocytoma in the hypothalamus‐case report. Neurol Med Chir (Tokyo) 46:101–103. [DOI] [PubMed] [Google Scholar]
- 2. Blumcke I, Wiestler OD (2002) Gangliogliomas: an intriguing tumor entity associated with focal epilepsies. J Neuropathol Exp Neurol 61:575–584. [DOI] [PubMed] [Google Scholar]
- 3. Blumcke I, Giencke K, Wardelmann F, Beyenburg S, Kral T, Sarioglu N et al (1999) The CD34 epitope is expressed in neoplastic and malformative lesions associated with chronic, focal epilepsies. Acta Neuropathol 97:481–490. [DOI] [PubMed] [Google Scholar]
- 4. Faillot T, Sichez JP, Capelle L, Kujas M, Fohanno D (1998) Ganglioglioma of the pineal region: case report and review of the literature. Surg Neurol 49:104–107. [DOI] [PubMed] [Google Scholar]
- 5. Fèvre‐Montange M, Jouvet A, Privat K, Korf HW, Champier J, Reboul A et al (1998) Immunohistochemical, ultrastructural, biochemical and in vitro studies of a pineocytoma. Acta Neuropathol (Berl) 95:532–539. [DOI] [PubMed] [Google Scholar]
- 6. Fèvre‐Montange M, Champier J, Szathmari A, Wierinckx A, Mottolese C, Guyotat J et al (2006) Microarray analysis reveals differential gene expression patterns in tumors of the pineal region. J Neuropathol Exp Neurol 65:675–684. [DOI] [PubMed] [Google Scholar]
- 7. Giannini C, Paulus W, Louis DN, Liberski P (2007) Pleomorphic xanthoastrocyma. In: World Health Organization Classification of Tumours of the Central Nervous System, Chapter 1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 22–24. IARC: Lyon. [Google Scholar]
- 8. Jouvet A, Fèvre‐Montange M, Besançon R, Derrington E, Saint‐Pierre G, Belin MF et al (1994) Structural and ultrastructural characteristics of human pineal gland, and pineal parenchymal tumors. Acta Neuropathol (Berl) 88:334–348. [DOI] [PubMed] [Google Scholar]
- 9. Jouvet A, Saint‐Pierre G, Fauchon F, Privat K, Bouffet E, Ruchoux MM et al (2000) Pineal parenchymal tumors: a correlation of histological features with prognosis in 66 cases. Brain Pathol 10:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kepes JJ, Rubinstein LJ, Eng LF (1979) Pleomorphic xanthoastrocytoma: a distinctive meningocerebral glioma of young subjects with relatively favorable prognosis. A study of 12 cases. Cancer 44:1839–1852. [DOI] [PubMed] [Google Scholar]
- 11. Kuchelmeister K, Von Borcke IM, Klein H, Bergmann M, Gullotta F (1994) Pleomorphic pineocytoma with extensive neuronal differentiation: report of two cases. Acta Neuropathol (Berl) 88:448–453. [DOI] [PubMed] [Google Scholar]
- 12. Min KW (1995) Pineal parenchymal tumors: “oncogeny” recapitulates ontogeny. J Child Neurol 10:490. [DOI] [PubMed] [Google Scholar]
- 13. Min KW, Seo IS, Song J (1987) Postnatal evolution of the human pineal gland. An immunohistochemical study. Lab Invest 57:724–728. [PubMed] [Google Scholar]
- 14. Moller M (1974) The ultrastructure of the human fetal pineal gland. I. Cell types and blood vessels. Cell Tissue Res 152:13–30. [DOI] [PubMed] [Google Scholar]
- 15. Nakazato Y, Jouvet A, Scheithauer BW (2007) Pineocytoma. In: World Health Organization Classification of Tumours of the Central Nervous System, Chapter 7. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 122–123. IARC: Lyon. [Google Scholar]
- 16. Nakazato Y, Jouvet A, Scheithauer BW (2007) Pineal parenchymal tumour of intermediate differentiation. In: World Health Organization Classification of Tumours of the Central Nervous System, Chapter 7. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 124–125. IARC: Lyon. [Google Scholar]
- 17. Nitta J, Tada T, Kyoshima K, Goto T, Ishii K, Hongo K, Kobayashi S (2001) Atypical pleomorphic astrocytoma in the pineal gland: case report. Neurosurgery 49:1458–1460. [DOI] [PubMed] [Google Scholar]
- 18. Pages A, Pages M (1980) Ganglioglioma of the pineal body. General review apropos of a case. Ann Anat Pathol (Paris) 25:273–279. [PubMed] [Google Scholar]
- 19. Reifenberger G, Kailich K, Wiestler OD, Blumcke I (2003) Expression of the CD34 antigen in pleomorphic xanthoastrocytomas. Acta Neuropathol 105:358–364. [DOI] [PubMed] [Google Scholar]
- 20. Sasaki A, Horiguchi K, Nakazato Y (2006) Pineal parenchymal tumor of intermediate differentiation with cytologic pleomorphism. Neuropathology 26:212–217. [DOI] [PubMed] [Google Scholar]
- 21. Takei H, Goodman JC, Tanaka S, Bhattacharjee MB, Bahrami A, Powell SZ (2005) Pituicytoma incidentally found at autopsy. Pathol Int 55:745–749. [DOI] [PubMed] [Google Scholar]
- 22. Tender GC, Smith RD (2004) Pineal ganglioglioma in a young girl: a case report and review of the literature. J La State Med Soc 156:316–318. [PubMed] [Google Scholar]