

Abstract
Controversy surrounds the recent 2007 WHO Classification of Tumours of the Nervous System. A number of nosologic issues remain to be resolved, some a reflection of conceptual disagreement, others the result of inadequate data to permit their definitive resolution. Among these and discussed herein are (i) the nosologic place of highly anaplastic oligoastrocytic tumors, (ii) the forms and significance of microvascular changes in high‐grade gliomas, (iii) the makeup of the glioneuronal tumors category, (iv) the subclassification of pineal parenchymal tumors of intermediate type, and (v) the classification of principle forms of mesenchymal neoplasms, specifically hemangiopericytoma and solitary fibrous tumor. These issues and others are the substance of this and an upcoming companion article.
Keywords: brain tumors, classification, controversial issues, pathology, World Health Organization
INTRODUCTION
The last four decades have seen the formulation of four editions of the World Health Organization Classification of Tumours of the Central Nervous System 24, 25, 33, 76, three in rapid succession over the past 14 years alone. Each brought with it revisions reflecting changes of concept, some fundamental, as well as minor or subtle alterations. Not all changes met with overall, unanimous approval by members of the working group. Although each edition represented a marked improvement over prior efforts, knotty problems remained. This and a companion article will address controversies surrounding the 2007 WHO classification, leaving readers to draw their own conclusions.
GRADE IV OLIGOASTROCYTOMA VS. “GLIOBLASTOMA WITH OLIGODENDROGLIAL COMPONENT”—CLARITY OR CONFUSION
This important issue of dissension has its basis in persistence of the antiquated term “glioblastoma” in an era in which all of diagnostic surgical pathology has focused upon cellular differentiation rather than morphologic epiphenomena. In gliomas, the latter include “microvascular proliferation” (see below) and necrosis. Neither contributes to a cell‐based diagnosis, serving only to establish tumor grade. Harkening back to Cushing and the concept of the “glioblast” also serves no purpose in present day tumor nosology. Instead, it is a reminder of the days when it was considered appropriate to combine very high‐grade astrocytomas, oligodendrogliomas and ependymomas featuring vascular proliferation and necrosis into a “glioblastoma” category (76). A relevant quote from the 1979 WHO “blue book” states: “Some typical glioblastomas show no evidence of a more differentiated tumor, whereas others are predominantly glioblastomas with focal areas of recognizable astrocytoma, less commonly oligodendroglioma or, exceptionally, ependymoma. Any of these gliomas may in fact terminate as a glioblastoma.” That concept was abandoned years ago when the 1993 version of the WHO blue book clearly stated that glioblastomas are poorly differentiated astrocytic tumors corresponding to grade IV (25). This served to remove some of the misconception and confusion surrounding glioblastoma. It could only have been bettered by introducing the alternative term “grade IV astrocytoma,” thus bringing the classification into line with the practice of establishing cell‐based diagnoses. Indeed, only tumor classifications predicated upon cellular differentiation should carry the day.
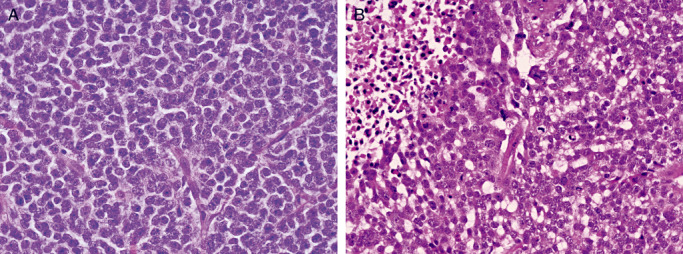
Thus, it is not surprisingly that the most contentious issues to arise at the 2007 Heidelberg conference was a nosologic shift to include tumors featuring both oligodendroglial and astrocytic components in addition to necrosis (Figure 1), in substance grade IV oligoastrocytoma, into the glioblastoma category under the pattern designation “glioblastoma with oligodendroglial component”. High‐grade oligoastrocytic tumors with a variable, often minor oligodendroglial component (oligoastrocytomas) have long been recognized, and are familiar to pathologists and neurosurgeons/oncologists alike 3, 40. In contrast, the term “glioblastoma with oligodendroglial component” has a much shorter track record. The designation has been used in the research setting 15, 31, 45. In clinical studies it has been applied to occasional cases of “glioblastoma with long term survival”7, 35, in one large series of “glioblastomas” in which an oligodendroglial element was seen in 20% of cases (20), and in another wherein 4% of tumors consisted in part (up to 30%) of oligodendrocytes (72). The term “oligodendroglial component” has even been appended to WHO grade III or anaplastic astrocytoma (8), somehow totally circumventing use of the time‐honored oligoastrocytoma category. 31, 72. Thus, it is not surprising that some members of the 2007 working group considered formal adoption of the term “glioblastoma with oligodendroglial component” to be not only a conceptual error, but an invitation to diagnostic and therapeutic confusion, particularly as 15–20% of such tumors show chromosome 1p and 19q co‐deletion, a feature of oligodendroglial neoplasia (41). Introduction of this cumbersome designation is hard to understand, as even its proponents consider it synonymous with grade IV oligoastrocytoma.
Figure 1.
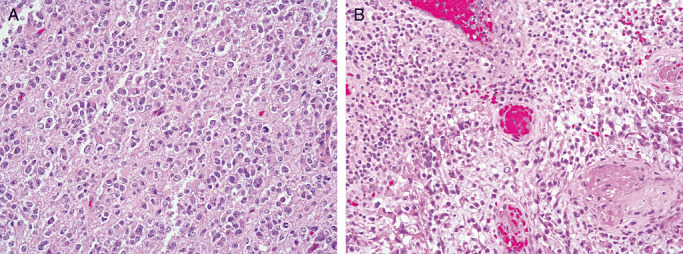
WHO grade IV “Glioblastoma with oligodendroglial features” (A,B) differs from WHO grade III oligoastrocytoma simply by the presence of necrosis in the latter.
Interestingly, justification for this terminologic shift is sought in a recent study showing that high oligoastrocytoma featuring anaplasia and necrosis, a tumor already referred to by some as grade IV oligoastrocytoma, to have a less favorable prognosis than either WHO grade III astrocytoma or oligoastrocytoma, but one more favorable than WHO grade IV astrocytoma (glioblastoma) 3, 41. What better case could be made for endorsing the term “WHO grade IV oligoastrocytoma?”
Despite strong objection, both during and after the Heidelberg Meeting, the term “glioblastoma with oligodendroglial component” persists 26, 71. Its imprecision will be the nidus of confusion, not only to pathologists but to surgeons and oncologists alike. The prognostic difference between oligoastrocytoma of grades III and IV based upon the absence or presence of necrosis, is well known (3). It is precisely for that reason that oligodendroglial tumor treatment protocols relying upon these distinctions, particularly those predicated upon a clear distinction between very high‐grade oligoastrocytoma and glioblastoma, as we know it, cannot accommodate this designation. From the therapeutic viewpoint, this is the crux of the matter. As a result, “glioblastoma with oligodendroglial component” may not be universally adopted. Indeed, what may occur is informal use of the term “grade IV oligoastrocytoma” in an effort to maintain nosologic continuity with the grade III lesion. Lastly, employing the term “glioblastoma with oligodendroglial component” may have a negative effect upon patients and treating physicians alike, inducing despair and therapeutic nihilism. Indeed, it may discourage vigorous pursuit of ancillary studies such as FISH for 1p/19q co‐deletion, now a common practice in dealing with oligoastrocytomas. Even experienced clinicians may be negatively affected by the designation “glioblastoma”, no matter how favorably qualified.
The present departure from a strict definition of glioblastoma as an astrocytic neoplasm and the inherent difficulty in confidently identifying a minor oligodendroglial component may play havoc with the evolving definition of the tumor and our emerging knowledge of so‐called primary and secondary types. The latter, originating by progressive anaplasia from astrocytomas of WHO grades II or III, may well be viewed as truly astrocytic in nature, whereas primary glioblastoma with their small, undifferentiated‐appearing cells may correspond to tumor originating in or composed in part of neuroepithelial stem cells capable of glioneuronal differentiation. Such stem cells have been demonstrated in human brain (“neuronal stem cells”) 21, 56 and in brain tumors (“brain tumor stem cells”) 12, 56. This would explain the occurrence of glioblastomas with true epithelial differentiation (62) as well as examples with PNET components (51), an issue further discussed below.
THE PROGNOSTIC SIGNIFICANCE OF MICROVASCULAR PATTERNS IN GLIOMAS
Whether subtle or conspicuous, increased vascularity is a well‐known feature of gliomas. It varies, not only in degree, but in type. As a reflection of the metabolic state of a tumor and its production of a particular combination of factors that regulate vasculo‐ and angiogenesis, it may simply consist of increased prominence of otherwise normal‐appearing vessels, or a conspicuous increase in vascular density. Examples of the former include the accentuated geometric cortical vasculature that characterizes oligodendroglioma (58), and subtle increase in white matter vasculature that accompanies low‐ to intermediate‐grade astrocytomas of the diffuse or infiltrative type. In addition to such “physiologic hypervascularity”, high‐grade gliomas often feature conspicuous vascular proliferation, either in the form of glomeruloid vessels or as so‐called “endothelial proliferation” (Figure 2). Such neovascularity exhibits MIB‐1 labeling indices five times higher (10% vs. 2%) than that of the above‐noted, less conspicuous vasculature (73).
Figure 2.
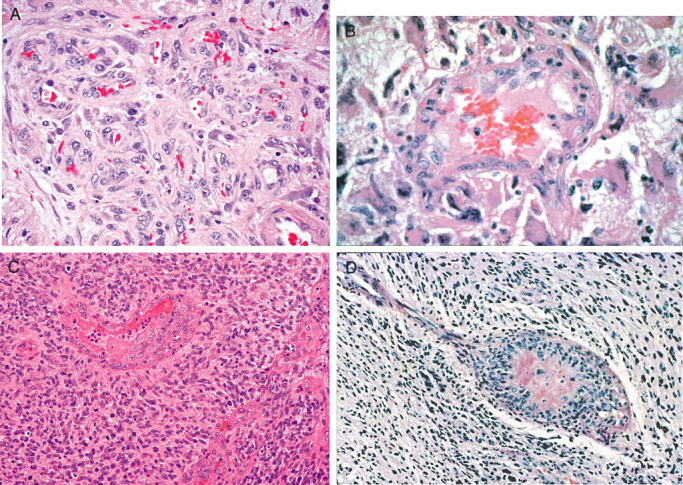
Vascular alterations in gliomas include both complex capillary tangles termed “glomeruloid vasculature” (A), and larger, often single lumen, multi‐layered vessels termed “endothelial proliferation” (B–D). Both vascular patterns feature proliferation of primarily pericytic or smooth muscle cells.
First introduced by Daumas‐Duport et al in 1988, the term “endothelial proliferation” denotes apparent multi‐layering of endothelium, typically in single‐lumened vessels of small to intermediate dimension (5) (Figure 2). In this sizable, 287 case, systematically studied series of ordinarily infiltrative astrocytic tumors, this vascular change was largely limited to glioblastomas (WHO grade IV astrocytoma). Multivariate analysis found both it and necrosis to be strongly associated with survival, the P‐value being less than 0.0001 for both. When subsequent immunohistochemical studies, particularly of glomeruloid vessels, showed that pericytes and smooth muscle cells rather than endothelial cells contribute most to the complex vascularity of gliomas 14, 44, 74 (Figure 2), an alternative, all‐encompassing designation was introduced. The term “microvascular proliferation” was coined to de‐emphasize the contribution of endothelium (74). Unfortunately, its inherent imprecision as an umbrella term obscured the distinction between the innocuous alteration (glomeruloid vascular proliferation) and the one of demonstrated prognostic importance (“endothelial proliferation”). The 2007 WHO classification does nothing to clarity the issue. The 2007 WHO chapter, Glioblastoma, mentions both in the same breath (26). The issue of what comprises “microvascular proliferation” is similarly obscured in the chapters Anaplastic Astrocytoma (28) and Anaplastic Oligoastrocytoma (71).
No doubt, the term “microvascular proliferation” will persist. It should not, however, be employed as an unqualified umbrella term for hypervascularity of all types. If used to denote either glomeruloid vasculature or what Daumas‐Duport et al once termed “endothelial proliferation”(5), admittedly a misnomer, it should be stated as such.
Lastly, the significance of vascular thrombosis in gliomas varies, as do the types of necrosis observed. On occasion, degenerative vascular changes, as seen in pilocytic astrocytoma (65) and ependymoma (37), are accompanied by infarction, that is, broad zones of necrosis unaccompanied by peri‐necrotic palisading of tumor cells. In that setting, infarct‐like necrosis, unlike palisading necrosis, is of no prognostic significance. In contrast, any form of necrosis accompanying anaplastic features in diffuse or infiltrative astrocytic tumors indicate a very negative prognosis (5). One recent study has shown that vascular thrombosis in diffuse astrocytic tumors is largely limited to high‐grade tumors and, for practical purposes, equates with WHO grade IV or “glioblastoma”(67). Therefore, the prognostic relevance of both necrosis and vascular thrombosis depends entirely upon a tumor's histologic type. They do not represent universally ominous findings.
GLIONEURONAL TUMORS—A RUNAWAY TUMOR CATEGORY?
Before embarking upon a discussion of specific glioneuronal tumors, a few words regarding the category and the diagnostic criteria of glioneuronal differentiation are in order. The term “divergent differentiation” denotes a substantial shift in differentiation of a tumor toward one or more other distinct cell types. The process, key to normal development and elegantly expressed in the central nervous system (CNS), has its most obvious neoplastic manifestation in teratomas.
The increasing recognition of divergent, particularly neuronal differentiation in common gliomas has its basis in the routine application of immunohistochemistry. Although the method enjoys great popularity, when push comes to shove, electron microscopy can play as much if not more of a role in exploring differentiation by providing precise morphologic data. This, of course, begs the question, “What is the ‘gold standard’ in the identification of neuronal differentiation?” In many instances, routine histochemistry is inadequate, disclosing only obvious, mature neurons. Immunohistochemistry is also plagued by vagaries. Glaring examples include neuron‐specific enolase and S‐100 protein, both of which are known to be unreliable markers of neuronal and glial differentiation, respectively. It is precisely for this reason that batteries of markers are employed to maximize diagnostic specificity. In contrast, ultrastructural parameters are relatively free of the nonspecificities so much a part of routine immunohistochemistry. Despite waning expertise in the general and neuropathology communities, electron microscopy and its variant technologies, such as immunoelectron microscopy, remain powerful tools. Although perhaps not applicable in routine practice, its role in determining and confirming tumor differentiation and thus shaping nosologic concepts remains essential.
Discomforting as it may be to see time‐honored classification schemes threatened, the notion that parenchymal tumors of the CNS are either glial or neuronal in nature has long been abandoned. The finding of divergent glioneuronal differentiation in some CNS tumors meets with little surprise. For example, in embryonal tumors of the CNS, such as medulloepithelioma, medulloblastoma and primitive neuroectodermal tumor, the occurrence of divergent glial and/or neuronal differentiation is almost “physiologic”. Such tumors may also feature mesenchymal components, elements even more deserving of the designation “divergent”(36). In tumors featuring mature cellular elements, the two lines of differentiation may be histologically obvious (ganglioglioma and the more recently described extraventricular neurocytoma, papillary glioneuronal tumor, and rosette‐forming glioneuronal tumor) 1, 2, 29, 30. Divergent glioneuronal differentiation in tumors associated with phakomatoses, diseases known to be associated with such dysgenetic lesions as subependymal giant cell astrocytoma of the tuberous sclerosis complex, comes as little surprise (19). The same is true of “quasi‐hamartomatous” lesions such as ganglioglioma (55) and dysembryoplastic neuroepithelial tumors (6), both of which are often associated with cortical dysplasia.
Unlike the finding of mature neurons in conventional ganglion cell tumors such as ganglioglioma, the cytologic features of neuronal cells in newcomers to the glioneuronal category are quite varied. For example, less obviously neuronal cells termed “ganglioid cells” are a key feature of “infantile desmoplastic ganglioglioma”, a rare pediatric variant of ganglion cell tumor (69). Whereas the spectrum of neuronal cells in papillary glioneuronal tumor is remarkably broad, ranging from neurocytes through ganglioid to mature ganglion cells (29), in rosette‐forming glioneuronal tumor the diagnostic Homer Wright‐like rosettes consist entirely of neurocytes (29). Lastly, the oligodendrocyte‐like cells of dysembryoplastic neuroepithelial tumor only reluctantly express neuronal features (18).
Also of note in the context of this section are phenotypically astrocytic tumors in which neuronal differentiation tends to be solely an immunophenotypic and/or ultrastructural feature. For example, recognizable ganglion cells are infrequent in subependymal giant cell astrocytoma 19, 22, 32. The same is true in conventional pleomorphic xanthoastrocytoma 13, 17, 53 wherein ganglion cells are rarely a significant component 10, 17, 50, 75.
More surprising is the immunohistochemical finding of divergent neuronal differentiation in tumors long considered patently glial in nature. Relatively recent reports document its occurrence in the full spectrum of gliomas, including anaplastic astrocytoma 23, 54, 68, oligodendroglioma 34, 52 and ependymoma (61). Such differentiation often takes the form of neurocyte islands (Figure 3). The emergence of mesenchymal elements, fibrous, chondroid, osseous and even myogenic, in gliosarcomas (27), occurs more frequently in high‐grade astrocytomas (63) than in oligodendroglioma (60) or ependymoma 59, 61. Differentiation toward true epithelium of squamous, glandular or neuroendocrine type is very rare and occurs most often in WHO grade IV astrocytoma (glioblastoma) 9, 43.
Figure 3.
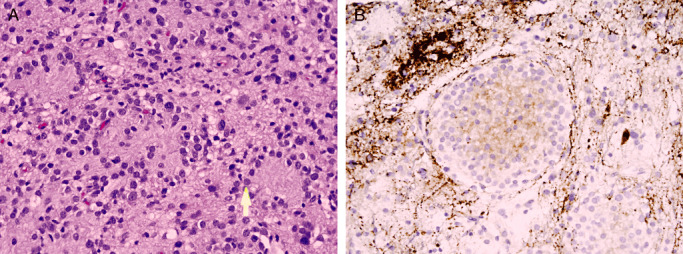
Neuronal differentiation on a WHO grade II astrocytoma is here seen as neurocyte islands (A). The latter are synaptophysin positive (B).
Of particular interest in this vein is a recent publication reporting diffuse astrocytomas, especially high‐grade, giant cell‐containing examples in which phenotypically astrocytic cells immunolabel for one or more neuronal markers (70) (Figure 4). Dubbed “malignant glioneuronal tumor”, the suggestion was made that it differs in behavior from its conventional counterparts, featuring a greater tendency to craniospinal and systemic metastasis but, nonetheless, a more favorable prognosis. The study as well as the concept has its problems. The spectrum of glioneuronal neoplasms, although considerably expanded in recent years, consists almost entirely of benign lesions, their designation as specific entities being predicated more upon distinctive morphology than upon differences in biologic behavior. Whereas the addition of glioneuronal tumor with neuropil islands (68) to the glioneuronal tumor category, a move initially given consideration at the 2007 Heidelberg meeting, would have represented a significant departure, inclusion of the “malignant glioneuronal tumors” described by Varlet et al (70) would change the landscape of the category entirely. One further criticism with respect to these tumors is that the study relied upon neurofilament protein immunoreactivity as the baseline diagnostic criterion (70). Accordingly, otherwise classic glioblastomas with neurofilament positivity would be reclassified as “malignant glioneuronal tumor.” Although confocal microscopy did show dual GFAP and neurofilament protein double labeling in a small number of cases, no ultrastructural correlation was provided. Relying solely upon immunochemistry, particularly focusing upon a single stain that yields highly variable results from one laboratory to another, is an insufficient basis upon which to establish a nosologic entity. Thus, for good reason, “malignant glioneuronal tumor” is not, at present, an entity in the WHO Classification of Tumours of the Nervous System. Only systematic translational studies predicated upon immunobatteries and ultrastructural correlation on the one hand and therapeutic/outcome data on the other could establish a clinically meaningful nosologic place for such tumors.
Figure 4.
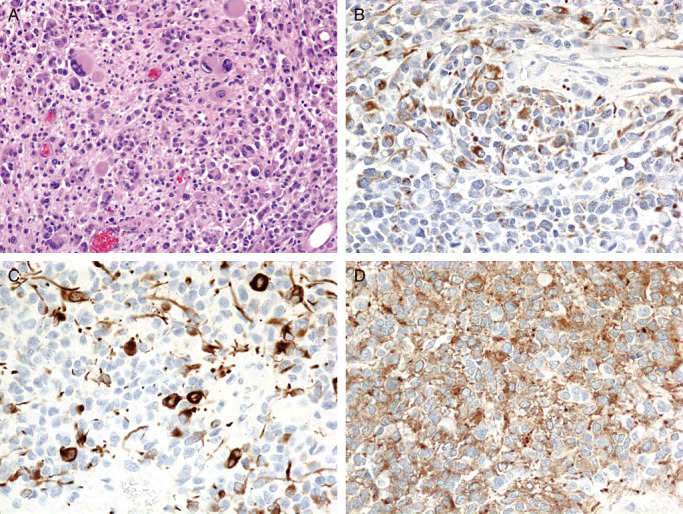
So‐called “malignant glioneuronal tumor” in which pleomorphic cells in an otherwise typical glioblastoma (A) show GFAP (B), neurofilament protein (C), and synaptophysin immunoreactivity (D).
It could be argued that the category of glioneuronal tumors is not the rightful place for malignant, phenotypically glial tumors with neuronal differentiation, be it gangliocytic or neurocytic. This would preserve the inherently favorable prognosis presently associated this category.
PINEAL PARENCHYMAL TUMOR OF INTERMEDIATE DIFFERENTIATION—THE NEED FOR PRECISE CRITERIA
The term “pineal parenchymal tumor of intermediate differentiation” was adopted by the 2000 WHO (46) pursuant to a study (66), suggesting that such lesions are associated with a prognosis intermediate between that of pineocytoma and pineoblastoma. Diffuse in histologic pattern (Figure 5), featuring occasional Homer Wright type rather than pineocytomatous rosettes and lacking the primitive, small cell appearance and necrosis of pineoblastoma, this lesion with its variable, but often low‐level proliferative activity, is associated with a lesser likelihood of craniospinal metastasis than the latter. Nonetheless, its morphologic spectrum is considerable, varying somewhat in cellularity, nuclear cytoplasmic ratio, cytologic atypia and mitotic activity (Figure 5). Thus, the designation “intermediate differentiation” begged for refinement. As experience had increased, it became obvious that the term included both prognostically favorable and unfavorable lesions. The question—“Where to draw the line?” It was clear that a large, multi‐institutional effort was required in order to establish criteria for low and higher grade variants of differing prognoses. The large cooperative study of Fevre‐Montague et al (11) confirmed their existence, but it was felt by the 2007 WHO working group that the criteria proffered fell short of arriving at a statistically meaningful morphologic breakpoint. This was not surprising, as proliferative activity may vary, even in pineocytomas. Furthermore, degree of cellular differentiation, a criterion focused upon by prior histologic (66) and ultrastructural (42) studies, also reckons into classification and prognostication. The approach at stratification employed by Jouvet et al relied mainly upon two criteria, proliferative activity (mitoses) and immunoreactivity for neurofilament protein, the latter being sparse in higher grade examples (Figure 5). The working group decided that even larger studies based upon additional parameters and more complete, extended follow‐up were needed. Nonetheless, the strength of Jouvet's study is that it confirmed the existence of an intermediate differentiation category comprising a sufficiently broad pathobiologic spectrum as to require its division into at least two prognostic subcategories.
Figure 5.
Pineal parenchymal tumor of intermediate differentiation variation illustrated in low‐ (A) and high‐grade (B) form. Note differences in nuclear atypia and proliferative activity. Note necrosis in the high‐grade variant (B).
HEMANGIOPERICYTOMA AND SOLITARY FIBROUS TUMOR—THE NEUROPATHOLOGIC APPROACH
Once termed “angioblastic meningioma” in the mistaken belief it was meningothelial in nature, what is now hemangiopericytoma is an easily recognized, prognostically important mesenchymal neoplasm. The newer designation was adopted when ultrastructural (64), immunohistochemical 49, 57 and genetic studies (16) showed no link to conventional meningioma, benign or malignant. Unfortunately, clearcut pericytic features were also lacking. Unlike the trend in soft tissue pathology, to abandon hemangiopericytoma as a diagnostic entity and to reassign it piecemeal to other tumor categories based on morphologic, immunohistochemical and genetic grounds, the neuropathology community has opted to retain the designation. This is justified. Unlike systemic hemangiopericytoma, most of which are benign, those affecting the CNS are considered malignant by definition, existing in low‐ and high‐grade form (38) with predictably aggressive clinical behavior, including high rates of recurrence and late metastases.
In the majority of instances, the distinction of hemangiopericytoma from solitary fibrous tumor, a lesion described as occurring in the CNS (4) and rather recently added to the WHO Classification of Tumours of the Nervous System (48), poses no problem (Figure 6). As a rule, the latter are benign, their behavior resembling that of WHO grade I meningioma. Histologic malignancy in solitary fibrous tumors, an infrequent event, is more often focal than widespread 39, 47. Nonetheless, the resemblance of malignant solitary fibrous tumor to hemangiopericytoma may be striking (Figure 6). Their uniform, strong immunoreactivity for C34 and total lack of EMA staining, as well as ultrastructural features which more closely resemble those of fibroblasts, aid in the distinction from both hemangiopericytoma and meningioma. Further justifying retention of hemangiopericytoma in the WHO classification is the sheer volume of old and modern therapeutic literature regarding “angioblastic meningioma” and hemangiopericytoma. Experience with solitary fibrous tumor of the CNS is also accumulating. Arbitrarily abandoning the term hemangiopericytoma or combining the lesions would engender more than nosologic confusion. All things considered, there are both diagnostic and therapeutic reasons for retaining the distinction. Both the 2000 and 2007 WHO committees opted to do so.
Figure 6.
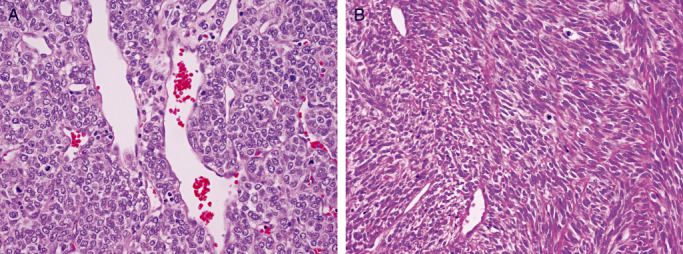
Malignant hemangiopericytoma (A) as well as solitary fibrous tumor (B) may show considerable morphologic overlap. Note transition from typical cytology of solitary fibrous tumor to more cellular, hemangiopericytoma‐like tissue (B).
CONCLUSION
The 2007 WHO Classification of Tumours of the CNS is, rightfully, the international standard of tumor classification. It masterfully summarizes our knowledge of the subject. Necessarily, as what has become an illustrated, “state of the art” book, it deals little with divergence of opinion on a number of important issues. Hopefully this and an upcoming companion commentary on what remains knotty issues will both prompt consideration and focus efforts upon their resolution.
A philosophical comment is in order. The human need to classify or impose conceptual order exemplified by Linnaeus (1707–1778) continues in all spheres of knowledge. In pathology, the process has very human implications. In the spirit of the WHO, it must maintain practicality and, above all, worldwide utility, regardless of available methodology. This underscores the need to maintain a balance between those basic necessities on the one hand and concept as well as technologic advances on the other.
REFERENCES
- 1. Becker AJ, Wiestler OD, Figarella‐Branger D, Blumcke I (2007) Ganglioglioma and gangliocytoma. In WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 103–105. International Agency for Research on Cancer (IARC): Lyon. [Google Scholar]
- 2. Brat DJ, Scheithauer BW, Eberhart CG, Burger PC (2001) Extraventricular neurocytomas: pathologic features and clinical outcome. Am J Surg Pathol 25:1252–1260. [DOI] [PubMed] [Google Scholar]
- 3. Buckner JC, O'Fallon JR, Dinapoli RP, Schomberg PJ, Farr G, Schaefer P et al (2007) Prognosis in patients with anaplastic oligoastrocytoma is associated with histologic grade. J Neurooncol 84:279–286. [DOI] [PubMed] [Google Scholar]
- 4. Carneiro SS, Scheithauer BW, Nascimento AG, Hirose T, Davis DH (1996) Solitary fibrous tumor of the meninges: a lesion distinct from fibrous meningioma. A clinicopathologic and immunohistochemical study. Am J Clin Pathol 106:217–224. [DOI] [PubMed] [Google Scholar]
- 5. Daumas‐Duport C, Scheithauer B, O'Fallon J, Kelly P (1988) Grading of astrocytomas. A simple and reproducible method. Cancer 62:2152–2165. [DOI] [PubMed] [Google Scholar]
- 6. Daumas‐Duport C, Scheithauer BW, Chodkiewicz JP, Laws ER, Jr ., Vedrenne C (1988) Dysembryoplastic neuroepithelial tumor: a surgically curable tumor of young patients with intractable partial seizures. Report of thirty‐nine cases. Neurosurgery 23: 545–556. [DOI] [PubMed] [Google Scholar]
- 7. Deb P, Sharma MC, Mahapatra AK, Agarwal D, Sarkar C (2005) Glioblastoma multiforme with long term survival. Neurol India 53:329–332. [PubMed] [Google Scholar]
- 8. Donahue B, Scott CB, Nelson JS, Rotman M, Murray KJ, Nelson DF et al (1997) Influence of an oligodendroglial component on the survival of patients with anaplastic astrocytomas: a report of Radiation Therapy Oncology Group 83‐02. Int J Radiat Oncol 38:911–914. [DOI] [PubMed] [Google Scholar]
- 9. Du Plessis DG, Rutherfoord GS, Joyce KA, Walker C (2004) Phenotypic and genotypic characterization of glioblastoma multiforme with epithelial differentiation and adenoid formations. Clin Neuropathol 23:141–148. [PubMed] [Google Scholar]
- 10. Evans AJ, Fayaz I, Cusimano MD, Laperriere N, Bilbao JM (2000) Combined pleomorphic xanthoastrocytoma‐ganglioglioma of the cerebellum. Arch Pathol Lab Med 124:1707–1709. [DOI] [PubMed] [Google Scholar]
- 11. Fevre‐Montange M, Hasselblatt M, Figarella‐Branger D, Chauveinc L, Champier J, Saint‐Pierre G et al (2006) Prognosis and histopathologic features in papillary tumors of the pineal region: a retrospective multicenter study of 31 cases. J Neuropath Exp Neur 65:1004–1011. [DOI] [PubMed] [Google Scholar]
- 12. Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S et al (2004) Isolation and characterization of tumorigenic, stem‐like neural precursors from human glioblastoma. Cancer Res 64: 7011–7021. [DOI] [PubMed] [Google Scholar]
- 13. Giannini C, Scheithauer BW, Lopes MB, Hirose T, Kros JM, VandenBerg SR (2002) Immunophenotype of pleomorphic xanthoastrocytoma. Am J Surg Pathol 26:479–485. [DOI] [PubMed] [Google Scholar]
- 14. Haddad SF, Moore SA, Schelper RL, Goeken JA (1992) Vascular smooth muscle hyperplasia underlies the formation of glomeruloid vascular structures of glioblastoma multiforme. J Neuropath Exp Neur 51:488–492. [DOI] [PubMed] [Google Scholar]
- 15. He J, Mokhtari K, Sanson M, Marie Y, Kujas M, Huguet S et al (2001) Glioblastomas with an oligodendroglial component: a pathological and molecular study. J Neuropath Exp Neur 60:863–871. [DOI] [PubMed] [Google Scholar]
- 16. Herath SE, Stalboerger PG, Dahl RJ, Parisi JE, Jenkins RB (1994) Cytogenetic studies of four hemangiopericytomas. Cancer Genet Cytogen 72:137–140. [DOI] [PubMed] [Google Scholar]
- 17. Hirose T, Giannini C, Scheithauer BW (2001) Ultrastructural features of pleomorphic xanthoastrocytoma: a comparative study with glioblastoma multiforme. Ultrastruct Pathol 25:469–478. [DOI] [PubMed] [Google Scholar]
- 18. Hirose T, Scheithauer BW, Lopes MB, VandenBerg SR (1994) Dysembryoplastic neuroeptihelial tumor (DNT): an immunohistochemical and ultrastructural study. J Neuropath Exp Neur 53:184–195. [DOI] [PubMed] [Google Scholar]
- 19. Hirose T, Scheithauer BW, Lopes MB, Gerber HA, Altermatt HJ, Hukee MJ et al (1995) Tuber and subependymal giant cell astrocytoma associated with tuberous sclerosis: an immunohistochemical, ultrastructural, and immunoelectron and microscopic study. Acta Neuropathol 90:387–399. [DOI] [PubMed] [Google Scholar]
- 20. Homma T, Fukushima T, Vaccarella S, Yonekawa Y, Di Patre PL, Franceschi S, Ohgaki H (2006) Correlation among pathology, genotype, and patient outcomes in glioblastoma. J Neuropath Exp Neur 65:846–854. [DOI] [PubMed] [Google Scholar]
- 21. Jin K, Galvan V (2007) Endogenous neural stem cells in the adult brain. J Neuroimmune Pharmacol 2:236–242. [DOI] [PubMed] [Google Scholar]
- 22. Katsetos CD, Del Valle L, Geddes JF, Assimakopoulou M, Legido A, Boyd JC et al (2001) Aberrant localization of the neuronal class III beta‐tubulin in astrocytomas. Arch Pathol Lab Med 125:613–624. [DOI] [PubMed] [Google Scholar]
- 23. Keyvani K, Rickert CH, Von Wild K, Paulus W (2001) Rosetted glioneuronal tumor: a case with proliferating neuronal nodules. Acta Neuropathol 101:525–528. [DOI] [PubMed] [Google Scholar]
- 24. Kleihues P, Cavenee WK (2000) World Health Organization Classification of Tumours—Pathology and Genetics. Tumours of the Nervous System. IARC Press: Lyon. [Google Scholar]
- 25. Kleihues P, Burger PC, Scheithauer BW (1993) Histological Typing of Tumours of the Central Nervous System, 2nd edn. World Health Organization. Springer‐Verlag: Berlin. [Google Scholar]
- 26. Kleihues P, Burger PC, Aldape KD, Brat DJ, Biernat W, Bigner DD et al (2007) Glioblastoma. In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 33–46. International Agency for Research on Cancer (IARC): Lyon. [Google Scholar]
- 27. Kleihues P, Burger PC, Aldape KD, Brat DJ, Biernat W, Bigner DD et al (2007) Gliosarcoma. In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 48–49. International Agency for Research on Cancer (IARC): Lyon. [Google Scholar]
- 28. Kleihues P, Burger PC, Rosenblum MK, Paulus W, Scheithauer BW (2007) Anaplastic astrocytoma. In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 30–32. International Agency for Research on Cancer (IARC): Lyon. [Google Scholar]
- 29. Komori T, Scheithauer BW, Anthony DC, Rosenblum MK, McLendon RE, Scott RM et al (1998) Papillary glioneuronal tumor: a new variant of mixed neuronal‐glial neoplasm. Am J Surg Pathol 22:1171–1183. [DOI] [PubMed] [Google Scholar]
- 30. Komori T, Scheithauer BW, Hirose T (2002) A rosette‐forming glioneuronal tumor of the fourth ventricle: infratentorial form of dysembryoplastic neuroepithelial tumor? Am J Surg Pathol 26:582–591. [DOI] [PubMed] [Google Scholar]
- 31. Kraus JA, Lamszus K, Glesmann N, Beck M, Wolter M, Sabel M et al (2001) Molecular genetic alterations in glioblastomas with oligodendroglial component. Acta Neuropathol 101:311–320. [DOI] [PubMed] [Google Scholar]
- 32. Lopes MB, Altermatt HJ, Scheithauer BW, Shepherd CW, VandenBerg SR (1996) Immunohistochemical characterization of subependymal giant cell astrocytomas. Acta Neuropathol 91:368–375. [DOI] [PubMed] [Google Scholar]
- 33. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A et al (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Makuria AT, Henderson FC, Rushing EJ, Hartmann DP, Azumi N, Ozdemirli M (2007) Oligodendroglioma with neurocytic differentiation versus atypical extraventricular neurocytoma: a case report of unusual pathologic findings of a spinal cord tumor. J Neurooncol 82:199–205. [DOI] [PubMed] [Google Scholar]
- 35. McLendon RE, Halperin EC (2003) Is the long‐term survival of patients with intracranial glioblastoma multiforme overstated? Cancer 98:1745–1748. [DOI] [PubMed] [Google Scholar]
- 36. McLendon RE, Judkins AR, Eberhart CG, Fuller GN, Sarkar C, Ng H‐K (2007) Central nervous system primitive neuroectodermal tumours (PNETs). In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 141–143. International Agency for Research on Cancer (IARC): Lyon. [Google Scholar]
- 37. McLendon RE, Wiestler OD, Kros JM, Korshunov A, Ng H‐K (2007) Ependymoma. In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 74–78. International Agency for Research on Cancer (IARC): Lyon. [Google Scholar]
- 38. Mena H, Ribas JL, Pezeshkpour GH, Cowan DN, Parisi JE (1991) Hemangiopericytoma of the central nervous system: a review of 94 cases. Hum Pathol 22:84–91. [DOI] [PubMed] [Google Scholar]
- 39. Metellus P, Bouvier C, Guyotat J, Fuentes S, Jouvet A, Vasiljevic A et al (2007) Solitary fibrous tumors of the central nervous system: clinicopathological and therapeutic considerations of 18 cases. Neurosurgery 60:715–722. Discussion 22. [DOI] [PubMed] [Google Scholar]
- 40. Miller CR, Perry A (2007) Glioblastoma. Arch Pathol Lab Med 131:397–406. [DOI] [PubMed] [Google Scholar]
- 41. Miller CR, Dunham CP, Scheithauer BW, Perry A (2006) Significance of necrosis in grading of oligodendroglial neoplasms: a clinicopathologic and genetic study of newly diagnosed high‐grade gliomas. J Clin Oncol 24:5419–5426. [DOI] [PubMed] [Google Scholar]
- 42. Min KW, Scheithauer BW, Bauserman SC (1994) Pineal parenchymal tumors: an ultrastructural study with prognostic implications. Ultrastruct Pathol 18:69–85. [DOI] [PubMed] [Google Scholar]
- 43. Mueller W, Lass U, Herms J, Kuchelmeister K, Bergmann M, Von Deimling A (2001) Clonal analysis in glioblastoma with epithelial differentiation. Brain pathol (Zurich, Switzerland) 11:39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nagashima T, Hoshino T, Cho KG (1987) Proliferative potential of vascular components in human glioblastoma multiforme. Acta Neuropathol 73:301–305. [DOI] [PubMed] [Google Scholar]
- 45. Nagasaka T, Gunji M, Hosokai N, Hayashi K, Ikeda H, Ito M, Inao S (2007) FISH 1p/19q deletion/imbalance for molecular subclassification of glioblastoma. Brain Tumor Path 24:1–5. [DOI] [PubMed] [Google Scholar]
- 46. Nakazato Y, Jouvet A, Scheithauer BW (2007) Pineal parenchymal tumour of intermediate differentiation. In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 124–125. International Agency for Research on Cancer (IARC): Lyon. [Google Scholar]
- 47. Ogawa K, Tada T, Takahashi S, Sugiyama N, Inaguma S, Takahashi SS, Shirai T (2004) Malignant solitary fibrous tumor of the meninges. Virchows Arch 444:459–464. [DOI] [PubMed] [Google Scholar]
- 48. Paulus W, Scheithauer BW (2000) Mesenchymal, non‐meningothelial tumors. In: Pathology and Genetics of Tumours of the Nervous System. Kleihues P, Cavanee WK (eds), pp. 142–148. International Agency For Research on Cancer: Lyon. [Google Scholar]
- 49. Perry A, Scheithauer BW, Nascimento AG (1997) The immunophenotypic spectrum of meningeal hemangiopericytoma: a comparison with fibrous meningioma and solitary fibrous tumor of meninges. Am J Surg Pathol 21:1354–1360. [DOI] [PubMed] [Google Scholar]
- 50. Perry A, Giannini C, Scheithauer BW, Rojiani AM, Yachnis AT, Seo IS et al (1997) Composite pleomorphic xanthoastrocytoma and ganglioglioma: report of four cases and review of the literature. Am J Surg Pathol 21:763–771. [DOI] [PubMed] [Google Scholar]
- 51. Perry A, Miller CR, Gujrati M, Scheithauer BW, Jost SC, Raghavan R et al (2008) Malignant Gliomas with Primitive Neuroectodermal Tumor‐like Components: A clinicopathologic and genetic study of 52 Cases. Brain Pathol in press. (doi:10.1111/j.1750‐3639.2008.00167.x.) [DOI] [PMC free article] [PubMed]
- 52. Perry A, Scheithauer BW, Macaulay RJ, Raffel C, Roth KA, Kros JM (2002) Oligodendrogliomas with neurocytic differentiation. A report of 4 cases with diagnostic and histogenetic implications. J Neuropath Exp Neur 61:947–955. [DOI] [PubMed] [Google Scholar]
- 53. Powell SZ, Yachnis AT, Rorke LB, Rojiani AM, Eskin TA (1996) Divergent differentiation in pleomorphic xanthoastrocytoma. Evidence for a neuronal element and possible relationship to ganglion cell tumors. Am J Surg Pathol 20:80–85. [DOI] [PubMed] [Google Scholar]
- 54. Prayson RA, Abramovich CM (2000) Glioneuronal tumor with neuropil‐like islands. Hum Pathol 31:1435–1438. [PubMed] [Google Scholar]
- 55. Prayson RA, Khajavi K, Comair YG (1995) Cortical architectural abnormalities and MIB1 immunoreactivity in gangliogliomas: a study of 60 patients with intracranial tumors. J Neuropath Exp Neur 54:513–520. [DOI] [PubMed] [Google Scholar]
- 56. Quinones‐Hinojosa A, Chaichana K (2007) The human subventricular zone: a source of new cells and a potential source of brain tumors. Exp Neurol 205:313–324. [DOI] [PubMed] [Google Scholar]
- 57. Rajaram V, Brat DJ, Perry A (2004) Anaplastic meningioma versus meningeal hemangiopericytoma: immunohistochemical and genetic markers. Hum Pathol 35:1413–1418. [DOI] [PubMed] [Google Scholar]
- 58. Reifenberger G, Kros JM, Louis DN, Collins VP (2007) Oligodendroglioma. In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 54–59. International Agency for Research on Cancer (IARC): Lyon. [Google Scholar]
- 59. Rodriguez FJ, Scheithauer BW, Fourney DR, Robinson CA (2008) Ependymoma and intraparenchymal calcifying pseudoneoplasm of the neural axis: incidental collision or unique reactive phenomenon? Acta Neuropathol 115:363–366. [DOI] [PubMed] [Google Scholar]
- 60. Rodriguez FJ, Scheithauer BW, Jenkins R, Burger PC, Rudzinskiy P, Vlodavsky E et al (2007) Gliosarcoma arising in oligodendroglial tumors (“oligosarcoma”): a clinicopathologic study. Am J Surg Pathol 31:351–362. [DOI] [PubMed] [Google Scholar]
- 61. Rodriguez FJ, Scheithauer BW, Robbins PD, Burger PC, Hessler RB, Perry A et al (2007) Ependymomas with neuronal differentiation: a morphologic and immunohistochemical spectrum. Acta Neuropathol 113:313–324. [DOI] [PubMed] [Google Scholar]
- 62. Rodriguez FJ, Scheithauer BW, Giannini C, Bryant S, Jenkins RB (2008) Epithelial and pseudoepithelial morphology in glioblastoma and gliosarcoma: comparative pathologic and molecular study of different subtypes. Mod Pathol 21:323A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Salvati M, Caroli E, Raco A, Giangaspero F, Delfini R, Ferrante L (2005) Gliosarcomas: analysis of 11 cases do two subtypes exist? J Neurooncol 74:59–63. [DOI] [PubMed] [Google Scholar]
- 64. Scheithauer BW, Bruner JM (1987) Central nervous system tumors. Clin Lab Med 7:157–179. [PubMed] [Google Scholar]
- 65. Scheithauer BW, Hawkins C, Tihan T, VandenBerg SR, Burger PC (2007) Pilocytic astrocytoma. In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 14–20. International Agency for Research on Cancer (IARC): Lyon. [Google Scholar]
- 66. Schild SE, Scheithauer BW, Schomberg PJ, Hook CC, Kelly PJ, Frick L et al (1993) Pineal parenchymal tumors. Clinical, pathologic, and therapeutic aspects. Cancer 72:870–880. [DOI] [PubMed] [Google Scholar]
- 67. Tehrani N, Friedman TM, Olson JJ, Brat DJ (2007) Intravascular thrombosis is more frequent in glioblastoma than other central nervous system malignancies (abstract 47.4). Proceedings of Experimental Biology, Washington DC.
- 68. Teo JG, Gultekin SH, Bilsky M, Gutin P, Rosenblum MK (1999) A distinctive glioneuronal tumor of the adult cerebrum with neuropil‐like (including “rosetted”) islands: report of 4 cases. Am J Surg Pathol 23:502–510. [DOI] [PubMed] [Google Scholar]
- 69. VandenBerg SR (1993) Desmoplastic infantile ganglioglioma and desmoplastic cerebral astrocytoma of infancy. Brain Pathol (Zurich, Switzerland) 3:275–281. [DOI] [PubMed] [Google Scholar]
- 70. Varlet P, Soni D, Miquel C, Roux FX, Meder JF, Chneiweiss H, Daumas‐Duport C (2004) New variants of malignant glioneuronal tumors: a clinicopathological study of 40 cases. Neurosurgery 55:1377–1391. Discussion 91‐2. [DOI] [PubMed] [Google Scholar]
- 71. Von Deimling A, Reifenberger G, Kros JM, Louis DN, Collins VP (2007) Anaplastic oligoastrocytoma. In: WHO Classification of Tumours of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 66–67. International Agency for Research on Cancer (IARC): Lyon. [Google Scholar]
- 72. Vordermark D, Ruprecht K, Rieckmann P, Roggendorf W, Vince GH, Warmuth‐Metz M et al (2006) Glioblastoma multiforme with oligodendroglial component (GBMO): favorable outcome after post‐operative radiotherapy and chemotherapy with nimustine (ACNU) and teniposide (VM26). BMC Cancer 6:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Watanabe K, Ogala N, Von Ammon K, Yonekawa Y, Nagai M, Ohgaki H, Kleihues P (1996) Immunohistochemical assessments of p53 protein accumulation and tumor growth fraction during the progression of astrocytomas. In: Brain Tumour Research Therapy. Nagai M (ed.), pp. 255–262. Springer‐Verlag: Tokyo. [Google Scholar]
- 74. Wesseling P, Schlingemann RO, Rietveld FJ, Link M, Burger PC, Ruiter DJ (1995) Early and extensive contribution of pericytes/vascular smooth muscle cells to microvascular proliferation in glioblastoma multiforme: an immuno‐light and immuno‐electron microscopic study. J Neuropath Exp Neur 54:304–310. [DOI] [PubMed] [Google Scholar]
- 75. Yeh DJ, Hessler RB, Stevens EA, Lee MR (2003) Composite pleomorphic xanthoastrocytoma‐ganglioglioma presenting as a suprasellar mass: case report. Neurosurgery 52:1465–1468. Discussion 8–9. [DOI] [PubMed] [Google Scholar]
- 76. Zulch KJ (1979) Histological Typing of Tumours of the Central Nervous System (Number 21). International Histological Classification of Tumours World Health Organization: Geneva. [Google Scholar]