

Abstract
Alzheimer's disease is the commonest dementia. One major characteristic of its pathology is accumulation of amyloid‐β (Aβ) as insoluble deposits in brain parenchyma and in blood vessel walls [cerebral amyloid angiopathy (CAA)]. The distribution of Aβ deposits in the basement membranes of cerebral capillaries and arteries corresponds to the perivascular drainage pathways by which interstitial fluid (ISF) and solutes are eliminated from the brain—effectively the lymphatic drainage of the brain. Theoretical models suggest that vessel pulsations supply the motive force for perivascular drainage of ISF and solutes. As arteries stiffen with age, the amplitude of pulsations is reduced and insoluble Aβ is deposited in ISF drainage pathways as CAA, thus, further impeding the drainage of soluble Aβ. Failure of perivascular drainage of Aβ and deposition of Aβ in the walls of arteries has two major consequences: (i) intracerebral hemorrhage associated with rupture of Aβ‐laden arteries in CAA; and (ii) Alzheimer's disease in which failure of elimination of ISF, Aβ and other soluble metabolites from the brain alters homeostasis and the neuronal environment resulting in cognitive decline and dementia. Therapeutic strategies that improve elimination of Aβ and other soluble metabolites from the brain may prevent cognitive decline in Alzheimer's disease.
Keywords: Amyloid‐beta, cerebral Amyloid Angiopathy, perivascular drainage, Alzheimer's disease, intracerebral hemorrhage, cerebrovascular disease
INTRODUCTION
Dementia is a major problem in the elderly population with the prevalence doubling every 5 years after the age of 65 years (39). Some 2.3% of those over the age of 65 years and up to 25% over 85 years in the UK show clinical evidence of moderate to severe cognitive impairment (24). Alzheimer's disease is the commonest type of dementia, but it often overlaps with vascular dementia because of cerebral infarction (83). Imaging studies suggest that 40% of patients with Alzheimer's disease also have some degree of vascular dementia (103) and this is supported by pathological studies (25).
ALZHEIMER'S DISEASE
Alzheimer's disease is characterized pathologically by the accumulation of ubiquitinated hyperphosphorylated tau in neurofibrillary tangles within neurons and by the deposition of amyloid‐β (Aβ) in brain tissue and in the walls of cerebral blood vessels as cerebral amyloid angiopathy (CAA) (26). A diagnosis of Alzheimer's disease is made in post‐mortem brains by the presence of insoluble plaques of Aβ and by the number and distribution of neurofibrillary changes that include neuritic amyloid plaques, neurofibrillary tangles and neuropil threads 14, 55. Clinicopathological studies, however, suggest that three features in particular correlate with clinical dementia. They are (i) the number and distribution of neurofibrillary changes 13, 14; (ii) a raised level of soluble Aβ in the brain 49, 53; and (iii) the severity of CAA (17).
Several major pieces of evidence suggest a key role for Aβ in the pathogenesis of Alzheimer's disease (92). There is the association of Down's syndrome with the pathological changes of Alzheimer's disease (50) and its link with chromosome 21 that contains the gene for amyloid precursor protein (APP) (92). There is a well‐established link between mutations in the APP and presenilin genes and familial Alzheimer's disease with the overproduction of Aβ, especially Aβ1–42 (31). In late onset sporadic Alzheimer's disease, the major genetic link is with the apolipoprotein E (APOE) locus (20); the protein APOE co‐localizes with Aβ in the brain in Alzheimer's disease (58). In transgenic mice expressing human APP and human tau transgenes, Aβ deposition develops prior to the accumulation of tau in neurons, suggesting a causal relationship (63). Finally, oligomers of Aβ appear to be toxic to neurons, and injection of Aβ into mice with mutations of the tau gene accelerates hyperphosphorylation of tau and the formation of neurofibrillary tangles (92).
In this review we focus upon the reasons why amyloid accumulates in the elderly and Alzheimer brain and examine how this may influence therapeutic strategies for Alzheimer's disease.
Accumulation of Aβ in the elderly and Alzheimer brain
Amyloid in the brains of elderly individuals and in Alzheimer's disease is mainly composed of Aβ peptides, 40 or 42 amino acids in length, derived from the trans‐membrane protein APP by the action of two aspartyl proteases, β‐ and γ‐secretase (93). Although Aβ is a soluble peptide, it is converted into an intermediate insoluble pre‐fibrillar form and into 8–10 nm amyloid fibrils with a β‐pleated sheet structure under the influence of concentration of Aβ, pH and a variety of tissue factors (69). Soluble and insoluble forms of Aβ are both present in the brain in Alzheimer's disease. The diffuse and focal cored or neuritic plaques of Aβ are a prominent histological feature of Alzheimer's disease in sections stained by immunohistochemistry with antibodies directed against Aβ(26). However, it is the high levels of soluble Aβ in the brain, measured biochemically, that correlate with cognitive decline and dementia rather than the number of plaques of insoluble Aβ49, 53.
The 40‐amino‐acid‐long Aβ (Aβ1–40) is more soluble than the longer Aβ1–42 and they differ in their distribution in brain and vessel walls. Aβ1–40 tends to be the major form in the amyloid in artery walls in CAA; whereas, Aβ1–42 is more prominent in the plaques in brain tissue 38, 90. Aβ in plaques and in artery walls is co‐distributed with APOE (58), suggesting that APOE may have a role as a chaperone for Aβ(69).
Although synaptic activity regulates the level of Aβ in interstitial fluid (ISF) (19), little is known about the normal physiological function of Aβ(33). But as we will seek to demonstrate here, Aβ in its insoluble forms has a major pathological role in blocking the drainage of ISF and solutes from the brain.
Increased production of Aβ or failure of elimination in Alzheimer's disease?
Mutations in the APP and presenilin genes are associated with an overproduction of Aβ in familial forms of Alzheimer's disease, particularly of Aβ1–42 78, 79. It was the discovery of such mutations in the 1990s that resulted in the formulation of the amyloid cascade hypothesis. This states that Alzheimer's disease is caused by increased production of Aβ, a cleavage product of APP 31, 32. Increased production of Aβ may occur in the 5% of cases of Alzheimer's disease that are familial. However, there is no firm evidence for overproduction of Aβ in the much more common sporadic late onset Alzheimer's disease in which the major susceptibility gene is the APOE locus (20). From the evidence available today, it seems likely that the decreased elimination of Aβ that occurs with advancing age is a major cause of Alzheimer's disease. Even in familial forms of Alzheimer's disease, the onset of dementia is often delayed until the fifth or sixth decades (26), suggesting that failure of elimination of Aβ with age also plays a part in the pathogenesis of familial types of the disease. In these cases, the overproduction of aberrant forms of Aβ may overwhelm the mechanisms for elimination of Aβ at an earlier age than in sporadic Alzheimer's disease.
Mechanisms for the elimination of Aβ from the brain
Several mechanisms for elimination of Aβ have been identified, and they fall into three main categories:
-
1
(i) Enzymic degradation of Aβ in the brain parenchyma by proteases, such as, neprilysin 28, 54 and insulin‐degrading enzyme (IDE) (47).
-
2
(ii) Direct absorption of Aβ into the blood via low‐density lipoprotein receptor‐related protein (LRP)‐1 at a rate estimated for Aβ1–40 at 0.21 pmol/minute/g ISF and at a slower rate for Aβ1–42 (8). P‐glycoprotein expressed on the luminal aspect of endothelial cells also contributes to the removal of Aβ from the brain into the blood (18).
-
3
(iii) Perivascular drainage of Aβ with other solutes and ISF along capillary and artery walls 67, 100. This is some sixfold slower than absorption into the blood via LRP (8) but perivascular drainage appears to compensate when the LRP mechanism is blocked or fails (80) and when neprilysin levels in the brain are reduced (54).
In this review, we concentrate mainly on the failure of perivascular drainage of Aβ that occurs in the aging brain. We relate this failure to CAA, to Alzheimer's disease and to the spectrum of arterial disease that may also result from failure of elimination of proteins along perivascular pathways. This condition can be defined as “protein‐elimination failure arteriopathy” (PEFA) which is seen throughout the body but is most commonly associated with the central and peripheral nervous systems (see below). In order to put PEFA into context, we will first review how fluid and solutes are eliminated from the central nervous system (CNS).
EXTRACELLULAR FLUIDS AND THE CENTRAL NERVOUS SYSTEM
Cerebrospinal fluid (CSF) and ISF are the two extracellular fluids associated with the brain and spinal cord (95). Failure of CSF drainage is associated with hydrocephalus (95), and failure of ISF drainage is associated with CAA, intracerebral hemorrhage (ICH), dementia and the uncommon condition of giant tumefactive perivascular spaces (74).
Cerebrospinal fluid
CSF has received more attention than ISF in the past, probably because it is readily accessible in vivo due to its relatively large pool of 30 mL CSF in the ventricles and 110 mL in the subarachnoid spaces (9). Produced by the choroid plexus at a rate of 350 µL/minute in humans (22), CSF passes through the ventricular system into the subarachnoid spaces. In humans, CSF appears to act as a buoyancy fluid and movement of CSF is related to pulsations in the vascular system (4). In humans, most of the CSF drains into the blood via arachnoid villi and granulations in the major venous sinuses 95, 96. In non‐primates, such as rat, rabbit and sheep, arachnoid villi are less well developed than in humans and in these species some 50% of the CSF drains via the cribriform plate and nasal lymphatics to regional lymph nodes in the neck 12, 21, 41.
A layer of ependyma separates CSF in the ventricles from the periventricular brain tissue, but ependymal cells lack tight junctions and fluid appears to drain from periventricular white matter into the ventricles (1). The ependymal lining around the ventricles is often incomplete in adult brains. In hydrocephalus, in which drainage of CSF from the ventricles is impeded, CSF passes into periventricular white matter, often through breaks in the ependyma 95, 99. There may be communication between CSF in the subarachnoid space and the brain and spinal cord in rodents (1); but in the human brain, 94, 96, a layer of pia mater, subpial collagen and the glia limitans separate the CSF in the subarachnoid space from nervous system tissue in the brain and spinal cord 3, 59. Pharmacological agents injected into the spinal CSF have little or no functional effect on the spinal cord (2), suggesting that there is a functional barrier between CSF in the subarachnoid space and the CNS.
Interstitial fluid
ISF in the brain and spinal cord is derived partly from the blood and partly from tissue metabolism (1), and amounts to 280 mL in the human brain (9). CSF may contribute to ISF, but the amount is uncertain (1). There are no lymphatic vessels in the mammalian brain that are comparable in structure to those in the rest of the body. However, it is estimated that the rate of drainage of ISF from the brain is 0.11–0.29 µL/minute/g of brain 1, 86 and that this is comparable to the average lymphatic drainage in the rest of the body (86).
If there are no lymphatic vessels, how do ISF and solutes drain from the brain?
Recent studies using fluorescent tracers and laser confocal microscopy have shown that ISF and solutes drain from the mouse brain along basement membranes of capillaries and arteries (15). By combining these results with earlier studies it is possible to identify a continuous route for the drainage of ISF from the brain to cervical lymph nodes. This perivascular lymphatic drainage pathway is blocked by Aβ in CAA and Alzheimer's disease.
Perivascular drainage of ISF and solutes from the brain
The details of the drainage pathway by which fluorescent tracers are eliminated from the mouse brain (15) are highly relevant to CAA. Formalin‐fixable fluorescent 3 kDa dextran (about the same molecular weight as Aβ) and fluorescent 40 kDa ovalbumin were injected (volume 0.5 µL) into the caudate putamen of mice, and the distribution of the tracers was monitored at 5 minutes to 24 h. Fluorescent dextran and ovalbumin tracers initially spread diffusely within the extracellular spaces of gray matter. By 5 minutes after injection, however, both tracers were detected within the basement membranes of capillaries and in basement membranes surrounding smooth muscle cells of the tunica media of artery walls. By 3 h, the 0.5 µL of tracer had been removed from brain parenchyma and from the basement membranes of capillaries and artery walls. Although no tracer could be detected in the basement membranes within the walls of leptomeningeal arteries, tracers were present in perivascular macrophages on the outer aspect of the leptomeningeal artery walls. This suggests that (i) tracers had reached the leptomeningeal vessels on their passage out of the brain; and (ii) a proportion of tracer had moved centrifugally through the walls of arteries during its passage out of the brain.
Figure 1 summarizes the perivascular drainage pathway for ISF and solutes out of the brain (15). Tracers in the extracellular spaces of the brain enter the capillary basement membranes between the endothelial layer and the surrounding astrocytes and then pass into the basement membranes between the smooth muscle cells in the tunica media of arteries. No tracer was detected in basement membranes of arterial endothelium or in basement membranes between the artery wall and the surrounding glia limitans (15).
Figure 1.
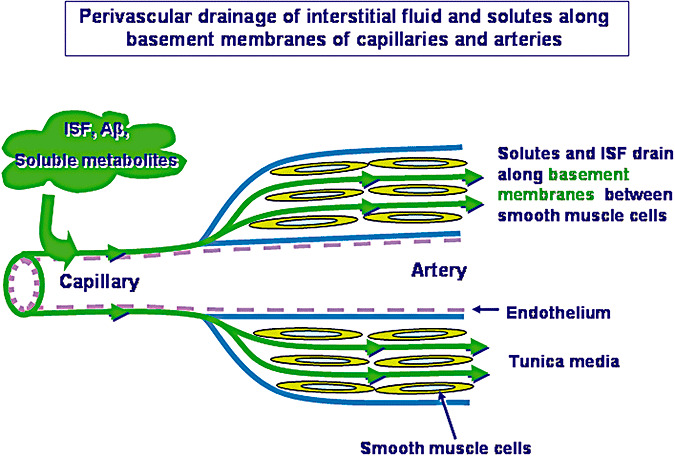
Perivascular pathways for the drainage of interstitial fluid (ISF), solutes, including amyloid‐β (Aβ), from the brain. Injection studies show that tracers diffuse through the extracellular spaces of the brain and enter basement membranes of capillaries to drain out of the brain along basement membranes in the tunica media of arteries. The drainage pathway is depicted here by the green lines and arrows. Basement membranes of the arterial endothelium and on the outer aspect of the arterial wall are devoid of tracer and are coloured blue. Endothelium is pink. From the results of the study by Carare et al. (15).
Lymphatic drainage of ISF and solutes
No fluorescent tracer was detected in cervical lymph nodes in the study reported above (15), possibly because of the very small amount of tracer injected. However, previous studies have shown that ISF and solutes drain from the brain to cervical lymph nodes 21, 86. Evans blue albumin, horseradish peroxidase and radioiodine‐labeled serum albumin injected as tracers into rodent or rabbit brains have been located in the adventitia of arteries in the circle of Willis at the base of the brain (86), but not in the walls of the carotid arteries in the neck. The presence of tracer in the adventitia of carotid arteries ceased abruptly at the skull base, suggesting that tracers drain from artery walls to cervical lymphatics (86).
The immunological significance of lymphatic drainage of ISF and solutes is emphasized in experiments showing that removal of the cervical lymph nodes in the rat interferes with B and T lymphocyte‐mediated immune reactions in the brain 34, 35, 45, 66. Despite the role played by the drainage of ISF and solutes in immunological reactions in the brain, there is little evidence that lymphocytes or macrophages drain from the brain to regional lymph nodes with ISF. The perivascular drainage pathways along vascular basement membranes do not seem to have the capacity for the drainage of cells (15). The absence of direct drainage of inflammatory cells from brain to lymph nodes may be a factor in the relative immunological privilege in the brain 30, 66
Motive force for the perivascular drainage of solutes from the brain
Movement of CSF within the ventricles, aqueduct and at the foramen magnum is driven by the pulsations of intracranial arteries (4). Less is known about the motive force for the perivascular transport of ISF and solutes from the brain. However, theoretical studies suggest that the contrary (or reflection) wave that follows each pulse wave is the force driving ISF and solutes along artery walls in the reverse direction to the flow of blood (75). Such a model would require some form of attachment or valve‐like mechanism to prevent back flow during the pulse wave. It is possible that changes in the conformation of basement membranes during expansion and recoil of the artery wall may play such a valve‐like role, but direct evidence for this is not yet available.
CEREBRAL AMYLOID ANGIOPATHY
Deposition of Aβ in blood vessel walls as CAA is a feature of elderly humans, Alzheimer's disease and a number of hereditary disorders (69). Leptomeningeal and cortical arteries are most frequently affected, and capillaries in the human brain are the least often involved. CAA is also a feature of transgenic mice bearing mutant human genes for APP; such mice produce increased amounts of Aβ(38). ICH is a major and often fatal complication of CAA, but CAA is also associated with Alzheimer's disease (5).
Aβ is deposited in perivascular drainage pathways in CAA (100). In the early stages of CAA, Aβ has almost exactly the same distribution as soluble tracers that are draining from the brain along basement membranes in the walls of capillaries and arteries15, 67, 100. Capillary basement membranes can be visualized by staining for collagen IV (Figure 2A). Aβ is deposited in the basement membranes of capillaries (Figure 2B) 67, 100, often forming compact nodules (Drusen) 67, 76, 100 (Figure 2C). Collagen IV in basement membranes surrounding smooth muscle cells in the tunica media of arteries is also well demonstrated by immunohistochemistry (Figure 3A). Staining for Aβ shows that it is deposited in basement membranes between smooth muscle cells in artery walls in CAA 67, 100 (Figure 3B).
Figure 2.
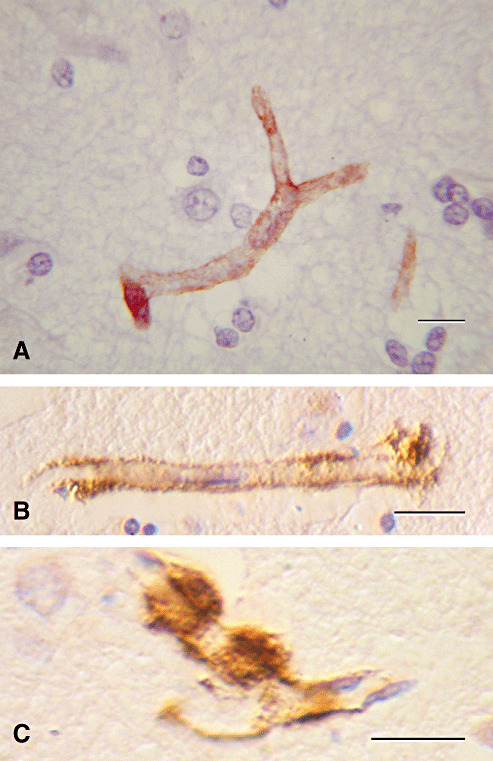
Amyloid angiopathy involving capillaries in Alzheimer's disease. Amyloid‐β (Aβ) is deposited in the basement membranes of cerebral capillaries. A. Normal capillary showing collagen IV (brown) in the basement membrane. B. Aβ (brown) in the basement membrane of a cerebral capillary. C. Small hemispherical “Drusen” of Aβ on the outer aspect of a capillary basement membrane laden with Aβ (brown). Immunohistochemistry for (A) collagen IV (Novocastra monoclonal antibody) (B) and (C) Aβ. Reproduced with permission from Preston et al (67). Bars = 10 µm.
Figure 3.
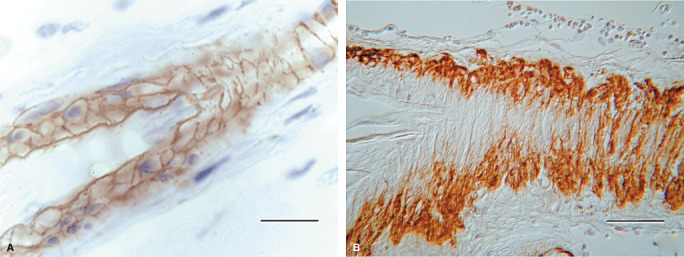
Cerebral amyloid angiopathy with deposition of amyloid‐β (Aβ) in basement membranes of arteries. A. A normal leptomeningeal artery showing collagen IV (brown) in the basement membranes between the smooth muscle cells in the tunica media. Nuclei of the smooth muscle cells are stained blue. B. Tangential section through a leptomeningeal artery showing Aβ in the basement membranes of the tunica media. Immunohistochemistry for (A) collagen IV (Novocastra monoclonal antibody) (B) Aβ (Dako pan‐Aβ monoclonal antibody) Bars = 20 µm.
Smear preparations of fresh brain tissue give a slightly different view of the relationships between Aβ and vessel walls in CAA (67). Figure 4A shows a capillary loop with amyloid in the basement membrane and a feathery outcrop of amyloid in the surrounding brain tissue. The brittle nature of the amyloid deposits in CAA is apparent in Figure 4B in which plates of Aβ in an artery wall appear to have shattered.
Figure 4.
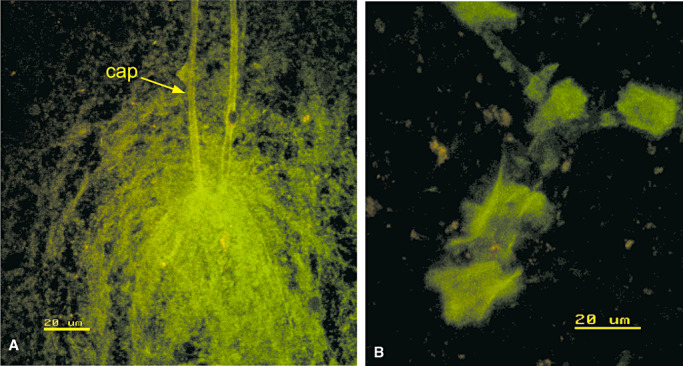
Cerebral amyloid angiopathy in smear preparations. A. A capillary loop (cap) with amyloid in its basement membrane (green) and a sheath of amyloid fibers in the surrounding brain. B. An artery with plates of amyloid (green) in the wall that appear to have been shattered during the preparation of the smear. A and B stained by thioflavin S. Confocal images. Reproduced with permission from Preston et al (67).
Ultrastructural studies show that fibrils of Aβ are initially deposited in the lamina densa of the basement membranes in the media between smooth muscle cells (102), and this increases the thickness of the basement membrane. Eventually smooth muscle cells die and the artery wall becomes almost completely replaced by Aβ(69).
In many ways, Aβ acts as a natural tracer for the perivascular drainage of solutes and ISF; they both drain along the same perivascular basement membrane pathway out of the brain (Figure 1). Deposits of Aβ are usually only seen in the walls of cortical vessels and the smaller leptomeningeal arteries by immunohistochemistry. Biochemical studies, however, have shown that Aβ is present in the leptomeningeal arteries in individuals aged 20–90 years (20 years was the youngest age tested) (82). Aβ was present in the walls of middle cerebral arteries and basilar arteries at the base of the brain, but no Aβ was detected in the walls of internal carotid arteries in the neck (82). This supports the observations in experimental animals that solutes draining from the brain along perivascular pathways leave the artery walls at the base of the skull probably to drain to regional lymph nodes in the neck.
Effects of Aβ deposition on vessel walls in CAA
Basement membranes
The initial effect of deposits of Aβ on the walls of capillaries and arteries is a change in the protein composition of basement membranes. Capillary basement membranes contain collagen IV (Figure 2A), laminin and fibronectin. In normal arteries, collagen IV is present in basement membranes throughout the width of the vessel wall (Figure 3A); whereas, laminin, fibronectin and perlecan are predominantly located in basement membranes at the periphery of the artery and in the endothelial basement membrane (84).
The heparan sulphate proteoglycan, perlecan, is associated with deposits of Aβ in the brain and accelerates Aβ fibril formation in vitro (36); whereas, laminin binds to Aβ in basement membranes and is a potent inhibitor of Aβ amyloid fibril formation (16). There appears to be a role for the heparan sulphate proteoglycan, agrin, in the accumulation of Aβ in blood vessel walls in the Dutch type of familial CAA but not in Alzheimer's disease (91).
Major changes occur in the basement membranes when Aβ is deposited in the walls of arteries and capillaries in CAA. There is a decrease in collagen IV (Figure 5) (105), laminin (Figure 6) and perlecan in artery walls whereas fibronectin remains relatively unaffected. Loss of collagen IV and laminin from capillary basement membranes occurs at sites of Aβ deposition (5, 6). However, the amount of fibronectin associated with the walls of capillaries is increased at sites of Aβ deposition (Figure 7) (84).
Figure 5.
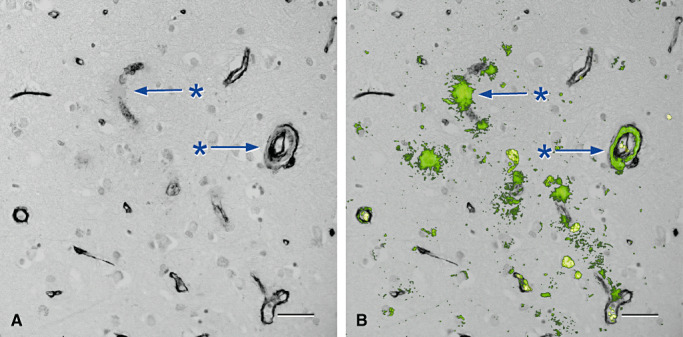
Loss of collagen IV in association with deposition of amyloid in the walls of arteries and capillaries in cerebral amyloid angiopathy (84) . A and B are from the same microscope field. A. Cerebral cortex stained only for collagen IV (black) showing focal loss of collagen IV from capillary (*, upper arrow) and artery (*, lower arrow) walls. B. The same vessels (* and arrows) show amyloid (green) replacing collagen IV in capillary and artery walls. The endothelial and outer basement membranes in the artery wall are selectively preserved and spared from amyloid deposits. Erythrocytes within vessel lumina are yellow. Immunohistochemistry for collagen IV (Novocastra monoclonal antibody), counterstained with Congo red for amyloid that appears green in this confocal hybrid image. Bars = 40 µm in both illustrations.
Figure 6.
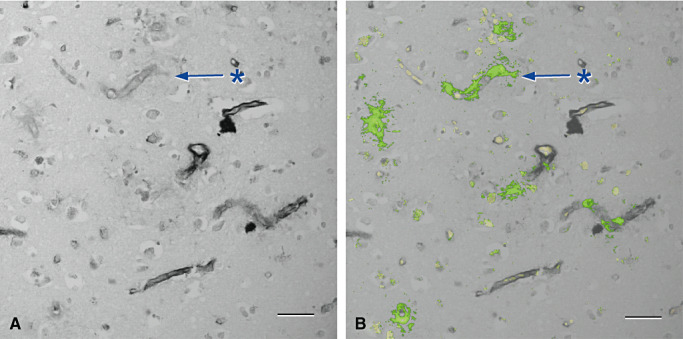
Laminin is lost from regions of amyloid deposition in the walls of capillaries in cerebral amyloid angiopathy (84) . A and B are from the same microscope field. A. cerebral cortex stained only for laminin (black) showing focal loss of laminin from capillary walls (*). B. The same vessels show amyloid (green) replacing laminin in the capillary basement membranes (*). Erythrocytes within capillaries are yellow. Immunohistochemistry for laminin, (Novocastra monoclonal antibody) counterstained with Congo red for amyloid that appears green in this confocal hybrid image. Bars = 40 µm in both illustrations.
Figure 7.
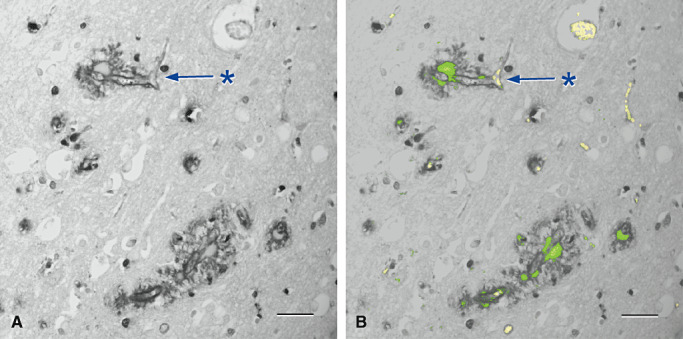
Fibronectin is increased adjacent to regions of amyloid deposition in the walls of capillaries in cerebral amyloid angiopathy (84) . A and B are from the same microscope field. A. Cerebral cortex showing an increase in fibronectin (black) in the brain parenchyma around capillaries (*). B. The same vessels show amyloid (green) in association with the increase in fibronectin (*). Erythrocytes within vessel lumina are yellow. Immunohistochemistry for fibronectin (Novocastra monoclonal antibody), counterstained with Congo red for amyloid that appears green in this confocal hybrid image. Bars = 40 µm in both illustrations.
There is selective preservation of collagen IV in basement membranes of the arterial endothelium and in the outermost basement membranes of the tunica media in cortical arteries in CAA (Figure 5). These two basement membrane layers are also devoid of experimental tracers when they drain along perivascular pathways of intracerebral arteries (Figure 1) (15). Lack of involvement of the endothelial basement membranes in arteries affected by CAA may mean that the endothelium is selectively preserved in these vessels (105) and may account for the low incidence of arterial thrombosis associated with CAA. Preservation of the outer basement membrane suggests that Aβ drains longitudinally into artery walls from capillary basement membranes and not radially from the brain, even though Aβ often accumulates in the periarterial glia limitans (67).
Destructive changes in artery walls in CAA
With increasing amounts of Aβ deposited in the walls of arteries in CAA, there is loss of smooth muscle cells from the tunica media (67), focal aneurysm formation, fibrinoid necrosis and separation of the outer part of the vessel from the inner layers—so called double barreling of arteries 51, 69. Intracortical arteries are not usually affected as severely as leptomeningeal arteries by destructive changes, but may show extensive replacement of smooth muscle cells by Aβ.
Inflammatory changes associated with CAA
An uncommon but distinctive feature of CAA is an angiodestructive inflammation. It is often granulomatous and affects mainly leptomeningeal arteries laden with Aβ(77). Patients may present with alteration in mental state, headaches and seizures and with hyperintense lesions in the white matter on MRI 42, 77. The APOE ε4 genotype is strongly related to CAA‐related inflammation (42).
Pathogenesis of CAA
When Scholz described CAA in 1938 (76), he suggested that amyloid in blood vessel walls was derived from the blood. Later it was proposed that the Aβ in CAA was mainly derived from smooth muscle cells within the artery walls (102). In common with most cells in the body, smooth muscle cells produce APP and produce measurable amounts of Aβ in cell culture (29). It was then proposed that Aβ is entrapped in perivascular ISF drainage pathways in CAA in the human brain (100). Support for this proposal came from transgenic mice that produce mutant human Aβ only in the brain, and develop prominent CAA (38). However, Aβ produced by smooth muscle cells artery walls may contribute to or even initiate deposition of Aβ in the perivascular drainage pathways 29, 62.
Almost all cases of CAA occur in middle aged or elderly individuals, so one major question is why does old age predispose to CAA? Several factors have been identified that may help to answer this question.
Factors impeding perivascular elimination of Aβ and other amyloids in CAA
Nature of the amyloid
Apart from Aβ, a number of other chemically distinct amyloids are associated with CAA; these include cystatin C, transthyretin, gelsolin, prion protein, ABri and ADan (69). Furthermore, the structure of Aβ and the proportions of Aβ1–40 and 1–42 may vary in familial CAA and familial Alzheimer's disease because of mutations in the APP or presenilin genes (69). Such variations may affect the capacity of the amyloidogenic proteins to drain along perivascular pathways in aging arteries.
Failure of degradation of Aβ or absorption into the blood
A reduction in the degradation of Aβ by neprilysin and other proteases in elderly human brains (54) and in transgenic mice (28) is associated with increased severity of CAA. Furthermore, the Aβ molecules that are produced by the Dutch, Flemish, Italian and Arctic mutant APP genes are resistant to degradation by neprilysin (89). Diversion of Aβ into perivascular drainage pathways may be the major reason for the increased severity of the CAA in these disorders (37). Aβ is also diverted into perivascular drainage pathways when absorption of Aβ into the blood via the LRP mechanism is reduced (80).
Failure of transport of soluble amyloid along aging arteries
Clinical disease and severe deposition of Aβ in the brain and in CAA do not usually occur until adulthood or middle age even in cases of familial Alzheimer's disease and familial CAA 69, 78. In the sporadic forms of CAA, significant deposition of Aβ is unusual before the seventh decade (26). Theoretical models suggest that perivascular drainage of ISF, and Aβ is driven by vessel pulsations (75). The character of pulsations in arteries changes with age as vessels become stiffer with arteriosclerosis (57). This may be a major factor in slowing the drainage of Aβ and allowing it to form insoluble amyloid fibrils within vascular basement membranes. Aβ CAA is a prominent feature of many transgenic mouse models in which there is production of mutant human APP (38). CAA presents at about 12 months of age, but changes in cerebral artery similar to the aging of human arteries occur before the Aβ is deposited (81). Changes in vessel tone resulting from cholinergic deafferentation in the rabbit also result in CAA (7). From these observations it seems that age changes and stiffening of artery walls and cholinergic deafferentation impede the perivascular drainage of Aβ, and that this induces CAA and, ultimately, the deposition of Aβ in the brain in Alzheimer's disease 6, 98.
Production of Aβ by smooth muscle cells
Insoluble Aβ1–42 appears to be deposited first in artery walls and this is followed by the much more abundant deposition of more soluble Aβ1–40 in CAA (90). One explanation for this observation may be that elimination of Aβ1–42, produced by smooth muscle cells in the walls of aging arteries, fails and that this provides a nidus for the accumulation of the more soluble Aβ1–40 draining from the brain in perivascular pathways 29, 62.
Inherent defects or age changes in vascular basement membranes
Aβ relates to various protein constituents in vascular basement membranes especially heparan sulphate proteoglycan that promotes amyloid fibril formation 43, 44 and laminin that inhibits amyloid fibril formation (16). Ultrastructural changes occur in vascular basement membranes with age; they become thicker and accumulate collagen (27). Such changes may interfere with perivascular drainage of Aβ. Transgenic mice that express transforming growth factor β1 at low levels in astrocytes, show thickening of capillary basement membranes and deposition of Aβ in the walls of cerebral blood vessels at the age of 6 months (104). It is not yet known whether genetic variations in constituents of basement membranes results in increased deposition of Aβ in aging vessel walls.
Influence of branching pattern of the vessels
Accumulation of Aβ in artery walls in CAA is often discontinuous along the length of the vessel wall and may show focal accumulation at the sites of vessel branching (100), suggesting some impedance of drainage at vessel branches. Viewing the cerebral vascular tree as a whole, there are some differences between mice and men in the pattern of development of CAA. Tracing the development of CAA in 10‐ to 26‐month‐old transgenic mice showed a progression of CAA starting in the arteries at the base of the brain and then involving the smaller branches of the cerebral arteries on the superior surfaces of the brain (23). At a microscopical level, initial deposition of Aβ had a banding pattern determined by the organization of the vascular smooth muscle cells. In humans, Aβ deposition that is detectable by histological techniques is usually confined to the smaller branches of the leptomeningeal arteries and arteries in the cortex (100).
Association of CAA with apolipoprotein E
APOE co‐localizes with Aβ in plaques in brain parenchyma and with Aβ in vessel walls in CAA (58). The ε4 allele of APOE is a risk factor for the development of CAA and APOE ε2 allele is associated with fibrinoid necrosis in CAA vessels and with ICH (51). APOE ε4 allele is also a risk factor for Alzheimer's disease (20) and for capillary CAA (87). As yet, the exact reasons for the association of CAA with APOE polymorphisms are not clear, but it is possible that APOE is associated with fibrillogenesis of Aβ within brain tissue and in perivascular drainage pathways (46).
PROTEIN‐ELIMINATION FAILURE ARTERIOPATHIES (PEFA)
CAA involving the deposition of Aβ in the walls of cerebral arteries can be considered as a PEFA and part of a general pathological phenomenon that involves other proteins and arteries in organs other than the brain.
Perivascular drainage of ISF and soluble proteins is highly developed in the CNS but it may not be unique to the CNS as it appears to occur in other organs. A variety of amyloid proteins, other than the different types of Aβ, is deposited in the walls of cerebral arteries; they include cystatin C, gelsolin and transthyretin, which may be associated with familial CAA and ICH or dementia (69). However, cystatin C and transthyretin are also deposited in arteries in other organs.
Arteries in peripheral nerves are particularly affected in familial transthyretin amyloid peripheral neuropathy (68). Like the brain, nerves do not posses conventional lymphatics and in transthyretin amyloid neuropathy, deposits of amyloid are seen within the endoneurium and in artery walls (68). In cystatin C amyloidosis, amyloid is deposited not only in the walls of cerebral arteries but also in arteries in other organs and in nonvascular basement membranes (65).
Elimination of proteins that are endogenous to artery walls appears to fail in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) (72). This disorder is caused by single missense mutations or small deletions in the Notch3 gene encoding a transmembrane receptor Notch3 40, 72. Notch signaling is essential during development when it regulates cellular differentiation but in adults, Notch3 is expressed only in vascular smooth muscle cells (40). In CADASIL, electron‐dense granular osmiophilic material (GOM) and Notch3 protein accumulate in arterial walls throughout the body because of impaired clearance and result in destruction of vascular smooth muscle cells (40). The major effects in humans are seen in the cerebral arteries (40) but in transgenic mice expressing mutant Notch3, GOM deposits and Notch3 accumulation is seen within both the cerebral and peripheral arteries. Arteries in the tail are severely affected in the transgenic mice with degeneration of vascular smooth muscle cells (72); this may be due to the length of the artery, and thus be analogous to the situation in the brain.
These examples of PEFA suggest that drainage of ISF and solutes occurs along arteries in organs other than the brain and, when drainage fails, they are liable to develop PEFA.
COMPLICATIONS ARISING FROM THE FAILURE OF PERIVASCULAR DRAINAGE OF AMYLOID FROM THE BRAIN
Failure of elimination of Aβ from the brain along perivascular drainage pathways is associated with two major disorders, (i) CAA‐related ICH and (ii) Alzheimer's disease. The two disorders appear to be largely distinct in their clinical occurrence and manifestations 97, 106.
Intracerebral haemorrhage associated with CAA
ICH related to CAA is associated with a number of hereditary disorders but most cases are sporadic. Most of the familial cases of ICH related to CAA are due to missense mutations in the part of the APP gene that codes for Aβ(106). No genetic abnormality in the presenilin genes has been identified, at least in the Dutch type of ICH (11). Hereditary cerebral hemorrhage with amyloidosis‐Dutch type is the most fully documented of the familial cases. It presents clinically with strokes, mostly hemorrhagic, at a mean age of 50 years; cognitive decline is generally only present after the first stroke 10, 106. Pathologically, patients show severe amyloid angiopathy particularly involving occipital and cerebellar leptomeningeal arteries and cortical arteries (106). Many of the vessels with CAA have secondary degenerative changes including aneurysm formation.
There are a number of other types of familial CAA associated with mutations in the APP gene including the Italian, Iowa, Flemish and Piedmont types that have similar presentations to the Dutch type (106). Cystatin C CAA is associated with Icelandic type of familial ICH 48, 65
Sporadic Aβ CAA is associated with 20–30% of spontaneous ICH (106). The hemorrhages are distributed preferentially in the temporal and occipital lobes (71) similar to hemorrhages in the Dutch type of familial CAA (106). This region of the brain is supplied largely by the posterior cerebral artery circulation which may indicate a preference for this part of the cerebral circulation to develop CAA (97). Although there is no association between genetic abnormalities in the APP gene in sporadic CAA, APOE ε4 is associated with increased deposition of Aβ in artery walls (64) and APOE ε2 is associated with hemorrhage and the vasculopathic features of CAA, particularly fibrinoid necrosis (52). There are other factors involved in the pathogenesis of sporadic CAA, in particular, the failure of degradation of Aβ by neprilysin in the elderly population (54).
Alzheimer's disease
CAA is associated with dementia in Alzheimer's disease 5, 17 and in the types of familial CAA that are not primarily associated with ICH, for example the Arctic and the ABri and ADan types of CAA 69, 106.
Dementia in Alzheimer's disease is the most important complication resulting from the failure of perivascular elimination of Aβ from the brain. Deposition of insoluble Aβ in artery walls impedes the elimination of soluble Aβ, resulting in the accumulation of insoluble plaques of Aβ and eventually a rise in the level of soluble Aβ and other metabolites in the brain. It may be the alteration in brain homeostasis and deterioration of the neuronal environment that are the major causes of cognitive decline in Alzheimer's disease.
Figure 8 suggests a possible sequence of events that leads to failure of homeostasis in the extracellular environment of neurons in the brain in Alzheimer's disease, largely because of blocking of perivascular drainage of soluble metabolites by Aβ in CAA.
Figure 8.
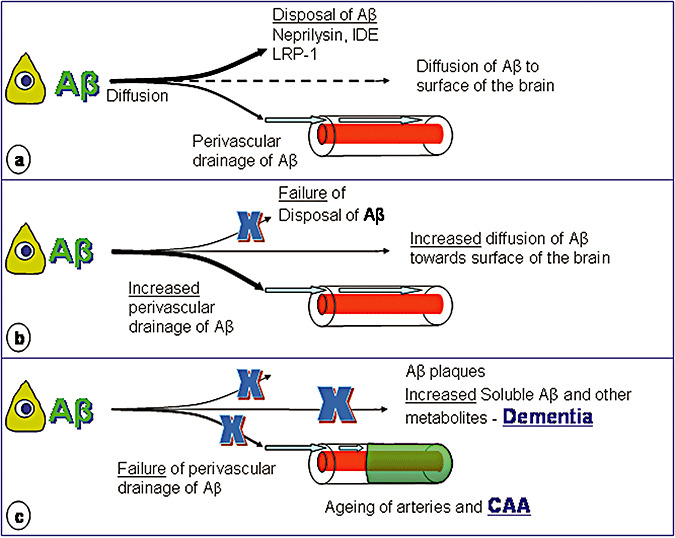
Failure of Elimination of amyloid‐β (Aβ) from the brain in Alzheimer's disease. A. In the normal young brain, Aβ is produced by neurons and other cells, diffuses through the extracellular spaces and is either degraded by neprilysin and insulin‐degrading enzyme (IDE) or absorbed into the blood via lipoprotein receptor‐related protein (LRP)‐1 mediated mechanisms. Some Aβ also drains with interstitial fluid along perivascular pathways in the walls of capillaries and arteries and a small proportion may diffuse toward the surface of the brain. B. With age disposal of Aβ by neprilysin, IDE and LRP‐1 mediated mechanisms fails and more Aβ is diverted to the perivascular drainage pathways. C. As arteries stiffen with age, perivascular drainage of Aβ becomes less efficient and ultimately fails because of blockage of the pathways by deposits of amyloid fibrils—cerebral amyloid angiopathy (CAA). Insoluble Aβ is deposited as plaques in the brain parenchyma and this interferes with diffusion of Aβ and other solutes through the extracellular spaces. Eventually, perivascular drainage fails and levels of soluble Aβ and other soluble metabolites in the brain rise and disturb homeostasis of the neuronal environment, resulting in neuronal malfunction, cognitive decline and dementia.
Aβ is produced throughout life by cells in the brain and diffuses through the extracellular spaces to be degraded by neprilysin and IDE or absorbed into the blood via LRP mediated mechanisms (Figure 8A). Aβ also drains along perivascular pathways.
As neprilysin, IDE and LRP disposal mechanisms fail with age, more Aβ is diverted into perivascular drainage pathways in the walls of aging arteries (Figure 8B). There may also be an increase in the diffusion of Aβ toward the surface of the brain and deposition of insoluble Aβ in the extracellular spaces of gray matter to form plaques.
Perivascular elimination of Aβ fails as age changes and stiffening in artery walls reduce the motive force for the drainage of Aβ. Drainage is further reduced by the deposition of fibrillar Aβ in the drainage pathways themselves (Figure 8C). Deposition of Aβ as CAA has the effects of blocking the drainage of soluble metabolites, including Aβ, and loss of homeostasis of the extracellular environment of neurons in the brain. Loss of homeostasis and deterioration in the extracellular environment of neurons may play a major role in neuronal dysfunction and cell death in Alzheimer's disease. Furthermore, soluble oligomers of Aβ may also be one of the toxic metabolites (92)
Impaired drainage of ISF may also partly account for the increase in fluid in the white matter (leukoaraiosois) in Alzheimer's disease (70), and reduced perfusion reserve in the white matter (88).
Therapeutic strategies for Alzheimer's disease
Preventing the accumulation of Aβ in perivascular drainage pathways seems to be a valid therapeutic target in Alzheimer's disease. Immunotherapy removes established plaques of insoluble Aβ from the brain 60, 61, 101 and relieves the restricted diffusion of solutes through the extracellular spaces of the brain 56, 85. However, immunotherapy does not appear to reduce the burden of Aβ in CAA 60, 61, 101. Age changes with stiffening of artery walls appear to be related to the failure of perivascular elimination of Aβ and the development of CAA in both humans and mice 81, 98. Therapies to reduce cerebrovascular disease may retard the development of CAA. However, reducing the amount of Aβ entering the perivascular drainage pathways, by increasing the level of neprilysin in the brain, or by improving LRP‐related clearance of Aβ into the blood, (73) may also be sustainable therapeutic strategies.
ACKNOWLEDGMENT
This work was supported by the Alzheimer's Research Trust.
REFERENCES
- 1. Abbott NJ (2004) Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem Int 45:545–552. [DOI] [PubMed] [Google Scholar]
- 2. Aird RB (1986) A study of intrathecal, cerebrospinal fluid‐to‐brain exchange. Exp Neurol 86:342–358. [DOI] [PubMed] [Google Scholar]
- 3. Alcolado R, Weller RO, Parrish EP, Garrod D (1988) The cranial arachnoid and pia mater in man: anatomical and ultrastructural observations. Neuropathol Appl Neurobiol 14:1–17. [DOI] [PubMed] [Google Scholar]
- 4. Alperin NJ, Lee SH, Loth F, Raskin PB, Lichtor T (2000) MR‐intracranial pressure (ICP): a method to measure intracranial elastance and pressure noninvasively by means of MR Imaging: baboon and human study. Radiology 217:877–885. [DOI] [PubMed] [Google Scholar]
- 5. Attems J, Quass M, Jellinger KA, Lintner F (2007) Topographical distribution of cerebral amyloid angiopathy and its effect on cognitive decline are influenced by Alzheimer's disease pathology. J Neurol Sci 257:49–55. [DOI] [PubMed] [Google Scholar]
- 6. Beach TG, Kuo YM, Spiegel K, Emmerling MR, Sue LI, Kokjohn K, Roher AE (2000) The cholinergic deficit coincides with Abeta deposition at the earliest histopathologic stages of Alzheimer's disease. J Neuropathol Exp Neurol 59:308–313. [DOI] [PubMed] [Google Scholar]
- 7. Beach TG, Potter PE, Kuo YM, Emmerling MR, Durham RA, Webster SD et al (2000) Cholinergic deafferentation of the rabbit cortex: a new animal model of Abeta deposition. Neurosci Lett 283:9–12. [DOI] [PubMed] [Google Scholar]
- 8. Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV (2007) Transport pathways for clearance of human Alzheimer's amyloid beta‐peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab 27:909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bergsneider M (2001) Evolving concepts of Cerebrospinal Fluid. Neurosurg Clin N Am 36:631–638. [PubMed] [Google Scholar]
- 10. Bornebroek M, Haan J, Maat‐Schieman ML, Van Duinen SG, Roos RA (1996) Hereditary cerebral hemorrhage with amyloidosis‐Dutch type (HCHWA‐D): I—A review of clinical, radiologic and genetic aspects. Brain Pathol 6:111–114. [DOI] [PubMed] [Google Scholar]
- 11. Bornebroek M, Haan J, Backhovens H, Deutz P, Van Buchem MA et al (1997) Presenilin‐1 polymorphism and hereditary cerebral hemorrhage with amyloidosis, Dutch type. Ann Neurol 42:108–110. [DOI] [PubMed] [Google Scholar]
- 12. Boulton M, Flessner M, Armstrong D, Mohamed R, Hay J, Johnston M (1999) Contribution of extracranial lymphatics and arachnoid villi to the clearance of a CSF tracer in the rat. Am J Physiol 276:R818–R823. [DOI] [PubMed] [Google Scholar]
- 13. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol (Berl) 82:239–259. [DOI] [PubMed] [Google Scholar]
- 14. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol (Berl) 112:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carare RO, Bernardes‐Silva M, Newman TA, Page AM, Nicoll JAR, Perry VH, Weller RO (2008) Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries. Significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol Jan 16 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 16. Castillo GM, Lukito W, Peskind E, Raskind M, Kirschner DA, Yee AG, Snow AD (2000) Laminin inhibition of beta‐amyloid protein (Abeta) fibrillogenesis and identification of an Abeta binding site localized to the globular domain repeats on the laminin a chain. J Neurosci Res 62:451–462. [DOI] [PubMed] [Google Scholar]
- 17. CFAS NGoM (2001) Pathological correlates of late‐onset dementia in a multicentre, community‐based population in England and Wales. Lancet 357:169–175. [DOI] [PubMed] [Google Scholar]
- 18. Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB et al (2005) P‐glycoprotein deficiency at the blood‐brain barrier increases amyloid‐beta deposition in an Alzheimer's disease mouse model. J Clin Invest 115:3285–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC et al (2005) Synaptic activity regulates interstitial fluid amyloid‐beta levels in vivo. Neuron 48:913–922. [DOI] [PubMed] [Google Scholar]
- 20. Coon KD, Myers AJ, Craig DW, Webster JA, Pearson JV, Lince DH et al (2007) A high‐density whole‐genome association study reveals that APOE is the major susceptibility gene for sporadic late‐onset Alzheimer's disease. J Clin Psychiatry 68:611–612. [DOI] [PubMed] [Google Scholar]
- 21. Cserr HF, Knopf PM (1992) Cervical lymphatics, the blood‐brain barrier and the immunoreactivity of the brain: a new view. Immunol Today 13:507–512. [DOI] [PubMed] [Google Scholar]
- 22. Davson H, Welch K, Segal MB (1987) Physiology and Pathophysiology of the Cerebrospinal Fluid. Churchill Livingstone: Edinburgh. [Google Scholar]
- 23. Domnitz SB, Robbins EM, Hoang AW, Garcia‐Alloza M, Hyman BT, Rebeck GW et al (2005) Progression of cerebral amyloid angiopathy in transgenic mouse models of Alzheimer's disease. J Neuropathol Exp Neurol 64:588–594. [DOI] [PubMed] [Google Scholar]
- 24. Ely M, Melzer D, Opit I, Brayne C (1996) Estimating the numbers and characteristics of elderly people with cognitive disability in local populations. Res Policy Plan 14:13–18. [Google Scholar]
- 25. Esiri MM, Nagy Z, Smith MZ, Barnetson L, Smith AD (1999) Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer's disease. Lancet 354:919–920. [DOI] [PubMed] [Google Scholar]
- 26. Esiri MM, Lee VMY, Trojanowski JQ (eds) (2004) The Neuropathology of Dementia, 2nd edn. Cambridge University Press: Cambridge. [Google Scholar]
- 27. Farkas E, De Jong GI, De Vos RA, Jansen Steur EN, Luiten PG (2000) Pathological features of cerebral cortical capillaries are doubled in Alzheimer's disease and Parkinson's disease. Acta Neuropathol (Berl) 100:395–402. [DOI] [PubMed] [Google Scholar]
- 28. Farris W, Schütz SG, Cirrito JR, Shankar GM, Sun X, George A et al (2007) Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am J Pathol 171:241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frackowiak J, Potempska A, LeVine H, Haske T, Dickson D, Mazur‐Kolecka B (2005) Extracellular deposits of a beta produced in cultures of Alzheimer's disease brain vascular smooth muscle cells. J Neuropathol Exp Neurol 64:82–90. [DOI] [PubMed] [Google Scholar]
- 30. Galea I, Bechmann I, Perry VH (2007) What is immune privilege (not)? Trends Immunol 28:12–18. [DOI] [PubMed] [Google Scholar]
- 31. Hardy J (2006) Alzheimer's disease: the amyloid cascade hypothesis: an update and reappraisal. J Alzheimers Dis 9:151–153. [DOI] [PubMed] [Google Scholar]
- 32. Hardy J (2006) Amyloid double trouble. Nat Genet 38:11–12. [DOI] [PubMed] [Google Scholar]
- 33. Hardy J, Cullen K (2006) Amyloid at the blood vessel wall. Nat Med 12:756–757. [DOI] [PubMed] [Google Scholar]
- 34. Harling‐Berg CJ, Park TJ, Knopf PM (1999) Role of the cervical lymphatics in the Th2‐type hierarchy of CNS immune regulation. J Neuroimmunol 101:111–127. [DOI] [PubMed] [Google Scholar]
- 35. Harling Berg C, Knopf PM, Merriam J, Cserr HF (1989) Role of cervical lymph nodes in the systemic humoral immune response to human serum albumin microinfused into rat cerebrospinal fluid. J Neuroimmunol 25:2–3. [DOI] [PubMed] [Google Scholar]
- 36. Hart M, Li L, Tokunaga T, Lindsey JR, Hassell JR, Snow AD, Fukuchi K (2001) Overproduction of perlecan core protein in cultured cells and transgenic mice. J Pathol 194:262–269. [DOI] [PubMed] [Google Scholar]
- 37. Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD et al (2004) Aβ is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci 7:954–960. [DOI] [PubMed] [Google Scholar]
- 38. Herzig MC, Van Nostrand WE, Jucker M (2006) Mechanism of cerebral beta‐amyloid angiopathy: murine and cellular models. Brain Pathol 16:40–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jorm AF, Korten AE, Henderson AS (1987) The prevalence of dementia: a quantitative integration of the literature. Acta Psychiatr Scand 76:465–479. [DOI] [PubMed] [Google Scholar]
- 40. Kalimo H, Ruchoux M‐M, Vitanen M, Kalaria RN (2002) CADASIL: a common form of hereditary arteriopathy causing brain infarcts and dementia. Brain Pathol 12:371–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kida S, Pantazis A, Weller RO (1993) CSF drains directly from the subarachnoid space into nasal lymphatics in the rat. Anatomy, histology and immunological significance. Neuropathol Appl Neurobiol 19:480–488. [DOI] [PubMed] [Google Scholar]
- 42. Kinnecom C, Lev MH, Wendell L, Smith EE, Rosand J, Frosch MP, Greenberg SM (2007) Course of cerebral amyloid angiopathy‐related inflammation. Neurology 68:1411–1416. [DOI] [PubMed] [Google Scholar]
- 43. Kisilevsky R, Szarek WA, Ancsin J, Vohra R, Li Z, Marone S (2004) Novel glycosaminoglycan precursors as antiamyloid agents: part IV. J Mol Neurosci 24:167–172. [DOI] [PubMed] [Google Scholar]
- 44. Kisilevsky R, Ancsin JB, Szarek WA, Petanceska S (2007) Heparan sulfate as a therapeutic target in amyloidogenesis: prospects and possible complications. Amyloid 14:21–32. [DOI] [PubMed] [Google Scholar]
- 45. Lake J, Weller RO, Phillips MJ, Needham M (1999) Lymphocyte targeting of the brain in adoptive transfer cryolesion‐EAE. J Pathol 187:259–265. [DOI] [PubMed] [Google Scholar]
- 46. Lashley T, Holton JL, Verbeek MM, Rostagno A, Bojsen‐Møller M, David G et al (2006) Molecular chaperons, amyloid and preamyloid lesions in the BRI2 gene‐related dementias: a morphological study. Neuropathol Appl Neurobiol 32:492–504. [DOI] [PubMed] [Google Scholar]
- 47. Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X et al (2003) Enhanced proteolysis of beta‐amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 40:1087–1093. [DOI] [PubMed] [Google Scholar]
- 48. Levy E, Jaskolski M, Grubb A (2006) The role of cystatin C in cerebral amyloid angiopathy and stroke: cell biology and animal models. Brain Pathol 16:60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L et al (1999) Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol 155:853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mann DM, Yates PO, Marcyniuk B (1984) Alzheimer's presenile dementia, senile dementia of Alzheimer type and Down's syndrome in middle age form an age related continuum of pathological changes. Neuropathol Appl Neurobiol 10:185–207. [DOI] [PubMed] [Google Scholar]
- 51. McCarron MO, Nicoll JA, Stewart J, Ironside JW, Mann DM, Love S et al (1999) The apolipoprotein E epsilon2 allele and the pathological features in cerebral amyloid angiopathy‐related hemorrhage. J Neuropathol Exp Neurol 58:711–718. [DOI] [PubMed] [Google Scholar]
- 52. McCarron MO, Nicoll JAR, Stewart J, Ironside JW, Mass DMA, Love S et al (1999) The apolipoprotein E epsilon 2 allele and the pathological features in cerebral amyloid angiopathy‐related hemorrhage. J Neuropathol Exp Neurol 58:711–718. [DOI] [PubMed] [Google Scholar]
- 53. McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K et al (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46:860–866. [DOI] [PubMed] [Google Scholar]
- 54. Miners JS, Van Helmond Z, Chalmers K, Wilcock G, Love S, Kehoe PG (2006) Decreased expression and activity of neprilysin in Alzheimer's disease are associated with cerebral amyloid angiopathy. J Neuropathol Exp Neurol 65:1012–1021. [DOI] [PubMed] [Google Scholar]
- 55. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization for the neuropathologic assessment of Alzheimer's disease. Neurology 41:479–486. [DOI] [PubMed] [Google Scholar]
- 56. Mueggler T, Rausch M, Meyer‐Luehmann M, Staufenbiel M, Jucker M, Rudin M (2004) Restricted diffusion in the brain of transgenic mice with cerebral amyloidosis. Eur J Neurosci 20:811–817. [DOI] [PubMed] [Google Scholar]
- 57. Nagasawa S, Handa H, Okumura A, Naruo Y, Moritake K, Hayashi K (1979) Mechanical properties of human cerebral arteries. Part 1: effects of age and vascular smooth muscle activation. Surg Neurol 12:297–304. [PubMed] [Google Scholar]
- 58. Navarro A, Del Valle E, Astudillo A, González del Rey C, Tolivia J (2003) Immunohistochemical study of distribution of apolipoproteins E and D in human cerebral beta amyloid deposits. Exp Neurol 184:697–704. [DOI] [PubMed] [Google Scholar]
- 59. Nicholas DS, Weller RO (1988) The fine anatomy of the human spinal meninges. A light and scanning electron microscopy study. J Neurosurg 69:276–282. [DOI] [PubMed] [Google Scholar]
- 60. Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO (2003) Neuropathology of human Alzheimer disease after immunization with amyloid‐beta peptide: a case report. Nat Med 9:448–452. [DOI] [PubMed] [Google Scholar]
- 61. Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P et al (2006) Abeta species removal after Abeta42 immunization. J Neuropathol Exp Neurol 65:1040–1048. [DOI] [PubMed] [Google Scholar]
- 62. Nicoll JAR, Yamada M, Frackowiak J, Mazur Kolecka B, Weller RO (2004) Cerebral amyloid angiopathy plays a direct role in the pathogenesis of Alzheimer's disease. Pro‐CAA position statement. Neurobiol Aging 25:589–597. [DOI] [PubMed] [Google Scholar]
- 63. Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM (2003) Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging 24:1063–1070. [DOI] [PubMed] [Google Scholar]
- 64. Olichney JM, Hansen LA, Galasko D, Saitoh T, Hofstetter CR, Katzman R, Thal LJ (1996) The apolipoprotein E epsilon 4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in Alzheimer's disease and Lewy body variant. Neurology 47:190–196. [DOI] [PubMed] [Google Scholar]
- 65. Palsdottir A, Snorradottir AO, Thorsteinsson L (2006) Hereditary cystatin C amyloid angiopathy: genetic, clinical, and pathological aspects. Brain Pathol 16:55–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Phillips MJ, Needham M, Weller RO (1997) Role of cervical lymph nodes in autoimmune encephalomyelitis in the Lewis rat. J Pathol 182:457–464. [DOI] [PubMed] [Google Scholar]
- 67. Preston SD, Steart PV, Wilkinson A, Nicoll JAR, Weller RO (2003) Capillary and arterial amyloid angiopathy in Alzheimer's disease: defining the perivascular route for the elimination of amyloid beta from the human brain. Neuropathol Appl Neurobiol 29:106–117. [DOI] [PubMed] [Google Scholar]
- 68. Reilly MM, Staunton H (1996) Peripheral nerve amyloidosis. Brain Pathol 6:163–177. [DOI] [PubMed] [Google Scholar]
- 69. Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL (2003) Cerebral Amyloid Angiopathies: a Pathologic, Biochemical, and Genetic View. J Neuropathol Exp Neurol 62:885–898. [DOI] [PubMed] [Google Scholar]
- 70. Roher AE, Kuo Y‐M, Esh C, Knebel C, Weiss N, Kalback W et al (2003) Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer's disease. Mol Med 9:112–122. [PMC free article] [PubMed] [Google Scholar]
- 71. Rosand J, Muzikansky A, Kumar A, Wisco JJ, Smith EE, Betensky RA, Greenberg SM (2005) Spatial clustering of hemorrhages in probable cerebral amyloid angiopathy. Ann Neurol 58:459–462. [DOI] [PubMed] [Google Scholar]
- 72. Ruchoux MM, Domenga V, Brulin P, Maciazek J, Limol S, Tournier‐Lasserve E, Joutel A (2003) Transgenic mice expressing mutant Notch3 develop vascular alterations characteristic of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Am J Pathol 162:329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R et al (2007) Clearance of amyloid‐beta by circulating lipoprotein receptors. Nat Med 13:1029–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Salzman KL, Osborn AG, House P, Jinkins JR, Ditchfield A, Cooper JA, Weller RO (2005) Giant Tumefactive Perivascular Spaces. Am J Neuroradiol 26:298–305. [PMC free article] [PubMed] [Google Scholar]
- 75. Schley D, Carare‐Nnadi R, Please CP, Perry VH, Weller RO (2006) Mechanisms to explain the reverse perivascular transport of solutes out of the brain. J Theor Biol 238:962–974. [DOI] [PubMed] [Google Scholar]
- 76. Scholz W (1938) Studien zur Pathologie der Hirngefässe II. Die drusige Entartung der Hirnarterien und ‐capillaren. (Ein Form seniler Gefässerkrankung). Zeitshrift für die gesamte Neurologie und Psychiatrie 162:694–715. [Google Scholar]
- 77. Scolding NJ, Joseph F, Kirby PA, Mazanti I, Gray F, Mikol J et al (2005) Abeta‐related angiitis: primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain 128:500–515. [DOI] [PubMed] [Google Scholar]
- 78. Selkoe DJ (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81:741–766. [DOI] [PubMed] [Google Scholar]
- 79. Selkoe DJ (2006) Amyloid beta‐peptide is produced by cultured cells during normal metabolism: a reprise. J Alzheimers Dis 9:163–168. [DOI] [PubMed] [Google Scholar]
- 80. Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B et al (2000) Clearance of Alzheimer's amyloid‐beta(1–40) peptide from brain by LDL receptor‐related protein‐1 at the blood‐brain barrier. J Clin Invest 106:1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shin HK, Jones PB, Garcia‐Alloza M, Borrelli L, Greenberg SM, Bacskai BJ et al (2007) Age‐dependent cerebrovascular dysfunction in a transgenic mouse model of cerebral amyloid angiopathy. Brain 130:2310–2319. [DOI] [PubMed] [Google Scholar]
- 82. Shinkai Y, Yoshimura M, Ito Y, Odaka A, Suzuki N, Yanagisawa K, Ihara Y (1995) Amyloid beta‐proteins 1–40 and 1–42(43) in the soluble fraction of extra‐ and intracranial blood vessels. Ann Neurol 38:421–428. [DOI] [PubMed] [Google Scholar]
- 83. Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR (1997) Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 277:813–817. [PubMed] [Google Scholar]
- 84. Subash M, Weller RO (2003) Cerebral amyloid angiopathy: differential preservation of endothelial cell basement membranes. Neuropath Appl Neurobiol 29:184. [Google Scholar]
- 85. Sykova E, Vorisek I, Antonova T, Mazel T, Meyer‐Luehmann M et al (2005) Changes in extracellular space size and geometry in APP23 transgenic mice: a model of Alzheimer's disease. Proc Natl Acad Sci USA 102:479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Szentistvanyi I, Patlak CS, Ellis RA, Cserr HF (1984) Drainage of interstitial fluid from different regions of rat brain. Am J Physiol 246:F835–F844. [DOI] [PubMed] [Google Scholar]
- 87. Thal DR, Ghebremedhin E, Rub U, Yamaguchi H, Tredici KD, Braak H (2002) Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol 61:282–293. [DOI] [PubMed] [Google Scholar]
- 88. Tomura N, Sasaki K, Kidani H, Nishii T, Yasuda K, Ishiyama K et al (2007) Reduced perfusion reserve in leukoaraiosis demonstrated using acetazolamide challenge 123I‐IMP SPECT. J Comput Assist Tomogr 31:884–887. [DOI] [PubMed] [Google Scholar]
- 89. Tsubuki S, Takaki Y, Saido TC (2003) Dutch, Flemish, Italian, and Arctic mutations of APP and resistance of Abeta to physiologically relevant proteolytic degradation. Lancet 361:1957–1958. [DOI] [PubMed] [Google Scholar]
- 90. Van Dorpe J, Smeijers L, Dewachter I, Nuyens D, Spittaels K, Van Den Haute C et al (2000) Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the london mutant of human APP in neurons. Am J Pathol 157:1283–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Van Horssen J, Otte‐Höller I, David G, Maat‐Schieman ML, Van Den Heuvel LP, Wesseling P et al (2001) Heparan sulfate proteoglycan expression in cerebrovascular amyloid beta deposits in Alzheimer's disease and hereditary cerebral hemorrhage with amyloidosis (Dutch) brains. Acta Neuropathol (Berl) 102:604–614. [DOI] [PubMed] [Google Scholar]
- 92. Walsh DM, Selkoe DJ (2007) Abeta oligomers—a decade of discovery. J Neurochem 101:1172–1184. [DOI] [PubMed] [Google Scholar]
- 93. Walsh DM, Minogue AM, Sala Frigerio C, Fadeeva JV, Wasco W, Selkoe DJ (2007) The APP family of proteins: similarities and differences. Biochem Soc Trans 35:416–420. [DOI] [PubMed] [Google Scholar]
- 94. Weller RO (1995) Fluid compartments and fluid balance in the central nervous system. In: Gray's Anatomy, 38th edn. Williams PL (eds), pp. 1202–1224. Churchill Livingstone: Edinburgh. [Google Scholar]
- 95. Weller RO (1998) Pathology of cerebrospinal fluid and interstitial fluid of the CNS: significance for Alzheimer disease, prion disorders and multiple sclerosis. J Neuropathol Exp Neurol 57:885–894. [DOI] [PubMed] [Google Scholar]
- 96. Weller RO (2005) Microscopic morphology and histology of the human meninges. Morphologie 89:22–34. [DOI] [PubMed] [Google Scholar]
- 97. Weller RO, Nicoll JA (2005) Cerebral amyloid angiopathy: both viper and maggot in the brain. Ann Neurol 58:348–350. [DOI] [PubMed] [Google Scholar]
- 98. Weller RO, Nicoll JAR (2003) Cerebral Amyloid Angiopathy: pathogenesis and effects on the ageing and Alzheimer brain. Neurol Res 25:611–616. [DOI] [PubMed] [Google Scholar]
- 99. Weller RO, Wisniewski H (1969) Histological and ultrastructural changes with experimental hydrocephalus in adult rabbits. Brain 92:819–828. [DOI] [PubMed] [Google Scholar]
- 100. Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE (1998) Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer's disease. Am J Pathol 153:725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D (2004) Passive immunotherapy against Abeta in aged APP‐transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation 1:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wisniewski HM, Wegiel J (1994) Beta‐amyloid formation by myocytes of leptomeningeal vessels. Acta Neuropathol (Berl) 87:233–241. [DOI] [PubMed] [Google Scholar]
- 103. Wu CC, Mungas D, Eberling JL, Reed BR, Jagust WJ (2002) Imaging interactions between Alzheimer's disease and cerebrovascular disease. Ann NY Acad Sci 977:403–410. [DOI] [PubMed] [Google Scholar]
- 104. Wyss Coray T, Lin C, Sanan DA, Mucke L, Masliah E (2000) Chronic overproduction of transforming growth factor‐beta1 by astrocytes promotes Alzheimer's disease‐like microvascular degeneration in transgenic mice. Am J Pathol 156:139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zhang WW, Lempessi H, Olsson Y (1998) Amyloid angiopathy of the human brain: immunohistochemical studies using markers for components of extracellular matrix, smooth muscle actin and endothelial cells. Acta Neuropathol (Berl) 96:558–563. [DOI] [PubMed] [Google Scholar]
- 106. Zhang‐Nunes SX, Maat‐Schieman ML, Van Duinen SG, Roos RA, Frosch MP, Greenberg SM (2006) The cerebral beta‐amyloid angiopathies: hereditary and sporadic. Brain Pathol 16:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]