

CLINICAL HISTORY
A 27‐year‐old woman in the last trimester of her second pregnancy presented with a 5‐month history of difficulty in swallowing, hoarseness and increasing headaches. She reported that the same symptoms had occurred during the first pregnancy but had fully resolved after delivery. On neurological examination, a facial paresis and deafness were present on the right side as well as significant dysfunction of the right IX, X and XI cranial nerves.
RADIOLOGY
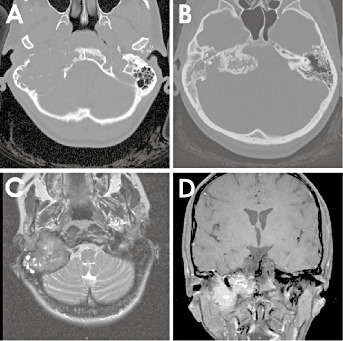
Axial CT showed the destruction of the mastoid portion of the petrous bone by the lower parts of the tumor (Figure 1A) while the upper parts revealed massive calcifications with compression of the brainstem (Figure 1B). MRI demonstrated a lobulated temporal bone mass with invasion of the right jugular foramen. T2‐weighted, axial MRI of the mastoid part of the tumor exhibited an intermediate signal indicating solid tumor (Figure 1C) and showed hypointense signal corresponding to calcification at the level of the brainstem compression. Postcontrast MRI demonstrated enhancement of the non‐calcified parts of the tumor (Figure 1D). (For additional MRI images go to: http://path.upmc.edu/divisions/neurpath/bpath/cases/case130.html).
Figure 1.
PATHOLOGY
Grossly the tumor was brown and dark red and had a firm consistency with varying calcified portions. Microscopic examination disclosed two well‐demarcated components (Figure 2A). Predominating were highly cellular areas consisting of ovoid to spindled cells and numerous osteoclast‐like multinucleated giant cells that frequently contained cytoplasmic vacuoles (Figure 2B).
Figure 2.
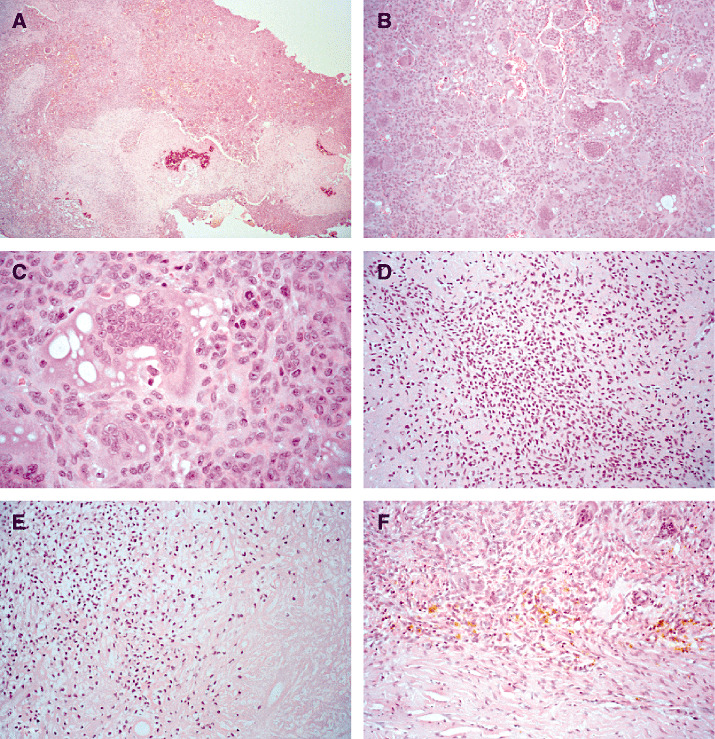
In these areas a few typical mitoses occurred (Figure 2C). The minor lobulated areas consisted of round or polygonal‐shaped cells with ovoid, grooved, hyperchromatic nuclei, surrounded by eosiphilic chondroid matrix (Figure 2D). Here, most cells lacked well‐defined cellular margins and occasionally showed perinuclear empty spaces (Figure 2E). Calcifications were predominantly found in the minor lobulated areas of the tumor and displayed a patchy or geographic pattern (Figure 2A). Intercellular calcifications, so‐called chicken‐wire‐like calcifications, were not observed. Very rarely small foci of necrosis without signs of calcification were found, most likely because of prior tumor embolization (Figure 2E, bottom right side). No marked nuclear atypia was seen. Within the tumor and at the tumor margins occasionally some siderophages appeared (Figure 2F).
DIAGNOSIS
Chondroblastoma of the petrous portion of the temporal bone.
DISCUSSION
Chondroblastoma is an unusual benign tumor that occurs preferentially in long bones such as the tibia, humerus and femur (11, 16). Its origin may also be from pelvis, spine, ribs and scapula, and less commonly from calcaneus, patella and tarsal bones. It corresponds to less than 1% of all primary bone tumors (3, 4, 16) and it is found predominantly in the first two decades of life (1, 10). So far the exact basic cellular phenotype remains unclear. While chondroblastomas have long been considered to derive from chondroblasts, these tumors might be reclassified as osteoid‐forming neoplasms (1).
Cranial chondroblastomas are extremely rare and the first case was originally described by Denko and Krauel in 1955 (5). Chondroblastomas of the skull tend to occur in older patients. The average age for patients with temporal bone chondroblastomas is 53.6 years old (8). The most frequent location of the chondroblastomas involving the cranial bones is the squamous portion of the temporal bone and 30% to 50% of the lesions may have calcific foci (7). The extremely uncommon tumor location in the petrosal bone without squamosal involvement in the present case may be related to persistent cartilaginous centers of the chondrocranium (12). Swelling in the temporal region, plugged sensation in the ear and hearing loss are the most frequently reported complaints in cases of cranial chondroblastomas (15). In the present case, the deficits of the lower cranial nerves, that presented as a transitory dysfunction during the first pregnancy and then evolved into a permanent deficit during the second gestation, is an atypical clinical manifestation and may be related to the increase of the venous pressure during pregnancy.
Misinterpretation of the histology of chondroblastomas may occur because—as in the present case—the multinucleated giant cell containing stroma may predominate over tumor components revealing the typical chondroid differentiation with chondroblasts and a chondroid matrix (2). Thus, its differential diagnosis includes, in particular, giant cell tumor. In contrast to chondroblastomas, giant cell tumors rarely show calcifications and do not contain areas with a chondroid matrix (6). In doubtful cases in which—different to the present case—unequivocal cartilaginous (chondroblastic) areas are not obvious, differential diagnosis of a giant cell tumor may be ruled out by S‐100 immunoreactivity. Tumor cells in giant cell tumors do not express S‐100, according to their histiocytic origin (9). In contrast, S‐100 immunoreactivity is of no help in distinguishing chondroblastoma and cases of clear cell chondrosarcoma, a very rare subtype of chondrosarcoma (1, 13, 17). In contrast to chondroblastoma, clear cell chondrosarcoma affects mainly adults in the third to fifth decade of life, lacks eosinophilic cytoplasm of tumor cells and reveals less well defined cell borders (1).
Additionally, in clear cell chondrosarcoma only minor amounts of a chondroid matrix are intermingled (6). Furthermore, some cases of chondroblastoma are associated with a secondary aneurysmal bone cyst (6). Thus, tumor regions revealing typical aspects of an aneurysmal bone cyst do not exclude chondroblastoma as the underlying primary disease.
Wide resection should be attempted in treatment of chondroblastomas and radiation therapy should be reserved for recurrence or incomplete tumor removal, as sarcomatous degeneration has been reported after radiotherapy (14).
ACKNOWLEDGMENTS
The authors thank Professor G. Delling (Division of Osteopathology, Institute of Pathology, University Hospital Eppendorf, Hamburg, Germany) for his reference diagnosis on the present case.
REFERENCES
- 1. Aigner T (2002) Towards a new understanding and classification of chondrogenic neoplasias of the skeleton—biochemistry and cell biology of chondrosarcoma and its variants. Virchows Arch 441:219–230. [DOI] [PubMed] [Google Scholar]
- 2. Ben Salem D, Allaoui M, Dumousset E, Ponnelle T, Justrabo E, Martin D, Sautreaux JL (2002) Chondroblastoma of the temporal bone associated with a persistent hypoglossal artery. Acta Neurochir 144:1315–1318. [DOI] [PubMed] [Google Scholar]
- 3. Dahlin DC, Ivins JC (1972) Benign chondroblastoma: a study of 125 cases. Cancer 30:401–413. [DOI] [PubMed] [Google Scholar]
- 4. Dahlin DC, Unni KK (1986) Bone Tumors: general Aspects and Data on 8542 Cases, 4th edn. Thomas: Springfield, IL. [Google Scholar]
- 5. Denko JV, Krauel LH (1955) Benign chondroblastoma of bone: an unusual localization in temporal bone. Arch Pathol 59:710–711. [PubMed] [Google Scholar]
- 6. Fechner RE, Mills SE (1992) Tumors of the Bones and Joints, 3rd edn. Armed Forces Institute of Pathology: Washington, DC. [Google Scholar]
- 7. Flowers CH, Rodriguez J, Naseem M, Reyes MM, Verano AS (1995) MR of benign chondroblastoma of the temporal bone. AJNR Am J Neuroradiol 16:414–416. [PMC free article] [PubMed] [Google Scholar]
- 8. Hong SM, Park YK, Ro JY (1999) Chondroblastoma of the temporal bone: a clinicopathologic study of five cases. J Korean Med Sci 14:559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Karabela‐Bouropoulou V, Markaki S, Prevedorou D, Vidali N (1989) A combined immunohistochemical and histochemical approach on the differential diagnosis of giant cell epiphyseal neoplasms. Pathol Res Pract 184:184–187. [DOI] [PubMed] [Google Scholar]
- 10. Kobayashi Y, Murakami R, Toba M, Ichikawa T, Kanazawa R, Sanno N, Shimura T, Sawada N, Hosone M, Kumazaki T (2001) Chondroblastoma of the temporal bone. Skeletal Radiol 30:714–718. [DOI] [PubMed] [Google Scholar]
- 11. Kurt A‐M, Unni KK, Sim FH, McLeod RA (1989) Chondroblastoma of bone. Hum Pathol 20:965–976. [DOI] [PubMed] [Google Scholar]
- 12. Mann SS, Naidich TP, Towbin RB, Doundoulakis SH (2000) Imaging of postnatal maturation of the skull base. Neuroimag Clin North Am 10:1–21. [PubMed] [Google Scholar]
- 13. Monda L, Wick MR (1985) S‐100 immunostaining in the differential diagnosis of chondroblastoma. Hum Pathol 16:287–293. [DOI] [PubMed] [Google Scholar]
- 14. Steiner GC (1965) Postradiation sarcoma of bone. Cancer 18:603–612. [DOI] [PubMed] [Google Scholar]
- 15. Tanohata K, Noda M, Katoh H, Okazaki A, Sugiyama S, Onishi S, Tanida T, Maehara T (1986) Chondroblastoma of the temporal bone. Neuroradiology 28:367–370. [DOI] [PubMed] [Google Scholar]
- 16. Turcotte RE, Kurt A‐M, Sim FH, Unni KK, McLeod RA (1993) Chondroblastoma. Hum Pathol 24:944–949. [DOI] [PubMed] [Google Scholar]
- 17. Weiss AP, Dorfmann HD (1986) S‐100 protein in human cartilage lesions. J Bone Joint Surg Am 68:521–526. [PubMed] [Google Scholar]