

Abstract
Tau protein is involved in microtubule assembly and stabilization. Filamentous deposits made of tau constitute a defining characteristic of several neurodegenerative diseases. The relevance of tau dysfunction for neurodegeneration has been clarified through the identification of mutations in the Tau gene in cases with frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP‐17). Although the mechanisms by which these mutations lead to nerve cell death are only incompletely understood, it is clear that they cause the formation of tau filaments with distinct morphologies and isoform compositions. The range of tau pathology identified in FTDP‐17 recapitulates that in sporadic tauopathies, indicating a major role for tau dysfunction in these diseases.
FRONTOTEMPORAL DEMENTIA
Frontotemporal dementia (FTD) has a prevalence of 5–15 cases per 100 000 in the age group of 45–65 years 7, 91, 100). Clinically, it is characterized by severe behavioral changes and language difficulties, with cognitive dysfunction appearing later (75). Most cases of FTD are sporadic, with 20%–30% being familial 87, 100). The major pathological hallmark of FTD is an atrophy of the frontotemporal cortex, with neuronal loss, gliosis and spongiosis of the superficial layers. Cases of FTD are pathologically heterogeneous and can be divided into three subtypes based on the presence of protein inclusions in the brain: those with tau‐positive inclusions, those with ubiquitin‐positive, tau‐negative inclusions and those lacking distinctive histopathology (66). The term Pick’s disease is now reserved for cases of FTD with intraneuronal argyrophilic inclusions, so‐called Pick bodies, which consist of abnormal tau protein. Pick bodies are present in 10%–30% of sporadic FTD cases 42, 67, 100).
Around 30% of cases of familial FTD are caused by Tau mutations and characterized by tau pathology 72, 87, 100). A substantial proportion (20%–40%) of hereditary FTD does not have Tau mutations 54, 89, 98). Some of these families are characterized by ubiquitin‐positive, tau‐negative inclusions and show linkage to chromosome 17q21‐22. Recently, it has been shown that they have mutations in the Progranulin gene 4, 13). Ubiquitin inclusions are also present in hereditary FTD linked to loci on chromosome 9 45, 121). Mutations in the Valosin‐containing protein gene on chromosome 9p21‐p12 have been identified and shown to lead to hereditary FTD with inclusion body myopathy and Paget’s disease of the bone (124). Furthermore, FTD in a Danish family has been linked to chromosome 3 (3). The disease in this family and some cases with FTD and amyotrophic lateral sclerosis is caused by mutations in the CHMP2B (charged multivesicular body protein 2B) gene 80, 103). CHMP2B mutations are associated with ubiquitin inclusions 49, 80) and occasional tau pathology 36, 127). Ubiquitin inclusions in familial cases of FTD have been shown to contain TDP‐43 (TAR DNA‐binding protein‐43) 1, 49, 78).
TAU PROTEIN AND NEURODEGENERATION
Filamentous tau inclusions are the pathological hallmark of a number of neurodegenerative disorders, of which Alzheimer’s disease (AD) is the most common. It is characterized by the presence of intraneuronal neurofibrillary tangles (NFTs) and extracellular amyloid‐β plaques. NFTs consist predominantly of paired helical filaments (PHFs) that contain tau protein in a hyperphosphorylated state 12, 22, 30, 31, 62). Tauopathies, such as progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Pick’s disease and argyrophilic grain disease (AGD) show tau pathology in the absence of amyloid‐β plaques and are distinguished by different types and distributions of tau‐positive inclusions (62).
Tau is involved in microtubule (MT) assembly and stabilization. In adult human brain there are six tau isoforms that are produced from a single gene by alternative mRNA splicing of exons 2, 3 and 10 (Figure 1) (28). They differ by the presence of three or four repeats, which constitute the MT‐binding domains of tau 53, 61). Three isoforms contain three repeats each, encoded by exons 9, 11 and 12, whereas alternatively spliced exon 10 encodes the additional repeat that distinguishes three‐ from four‐repeat forms (29). In adult human brain, the ratio between three‐ and four‐repeat tau isoforms is close to 1 (27). Tau is a phosphoprotein and in the human diseases it becomes hyperphosphorylated and assembles into filaments (62). The etiological role of tau protein in neurodegeneration has been convincingly demonstrated through the identification of mutations in Tau in familial disorders with dementia and/or parkinsonism 48, 86, 109).
Figure 1.
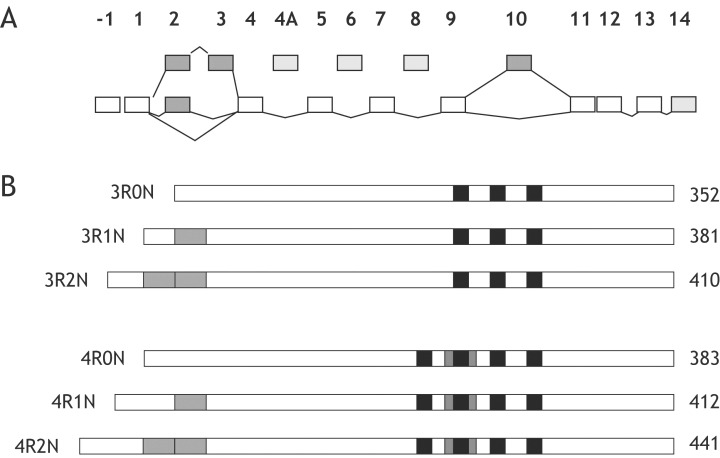
Schematic representation of the Tau gene and the six tau isoforms expressed in adult human brain. A. The human Tau gene contains 16 exons, of which exons 2, 3 and 10 (dark gray boxes) are alternatively spliced. Exons 4A, 6 and 8 (light gray boxes) are not transcribed in human brain. B. Six tau isoforms are generated by alternative mRNA splicing of exons 2, 3 and 10 (dark gray boxes). They range from 352 to 441 amino acids in length. The black boxes represent the microtubule‐binding repeats of tau.
MUTATIONS IN TAU
Frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP‐17) is an autosomal‐dominantly inherited disease that consistently shows severe frontotemporal atrophy with neuronal loss, gliosis and spongiform changes in layer 2. Involvement of medial temporal lobe structures, for example, entorhinal cortex, hippocampus and amygdala, is more variable. Degeneration of substantia nigra and basal ganglia is common 23, 107). Despite substantial neuropathological heterogeneity, the formation of abundant tau inclusions in nerve cells or in both nerve cells and glia is an invariant feature of FTDP‐17. In 1998, exonic and intronic mutations in Tau were identified in cases of FTDP‐17, demonstrating that genetic defects in Tau can cause neurodegeneration 48, 86, 109).
Molecular mechanisms. Thirty‐nine mutations in Tau have been identified to date (Figure 2). They can be grouped according to their positions in the gene, which in turn defines their effects on tau mRNA and protein, as well as the type of resultant pathology. The majority of mutations occurs in the coding region of Tau and includes missense, deletion and silent mutations. Intronic mutations located close to the splice‐donor site of the intron following exon 10 are also common. Most coding region mutations are located in the MT‐binding region (exons 9–12) or close to it (exon 13). Two mutations in exon 1 of Tau have also been reported 39, 88). Mutations in exons 1, 9, 12, and 13 affect all six tau isoforms. In contrast, mutations in exon 10 affect only four‐repeat tau isoforms or their expression 25, 35, 40, 44, 77, 82, 119). The primary effect of the intronic mutations and some mutations that affect splicing regulatory elements is exerted at the level of mRNA splicing and leads to an altered expression of tau isoforms 11, 48, 109, 117). Tau mutations can be classified as those that affect tau‐microtubule interactions and/or fibril formation, and those that affect exon 10 splicing. This distinction is not absolute, as some mutations have multiple effects.
Figure 2.
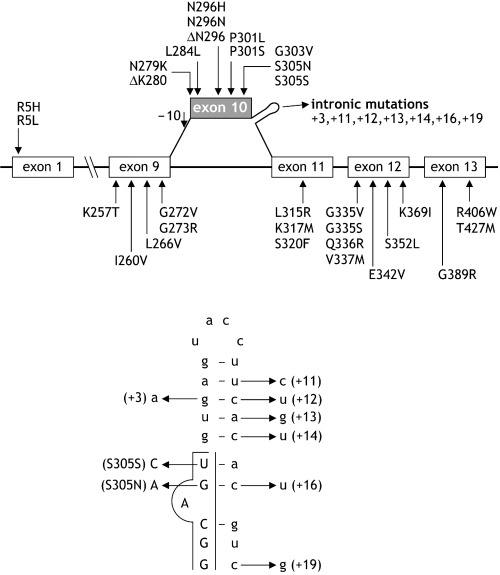
Overview of Tau mutations identified in frontotemporal dementia and parkinsonism linked to chromosome 17. All known coding region mutations are located in exons 1, 9, 10, 11, 12 and 13. The alternatively spliced exon 10 is depicted in gray. Mutations are numbered according to the longest tau isoform (441 amino acids).
Mutations altering tau‐MT interactions and fibril formation. In accordance with their location in the MT‐binding region of tau, most missense mutations reduce the affinity of tau for MTs, as indicated by a reduction in the ability of mutant tau to promote MT assembly 14, 37, 44). This mechanism applies to coding region mutations in exons 9, 11, 12 and 13. The P301L and P301S mutations in exon 10 also share this mechanism, whereas the Q336R mutation in exon 12 slightly increases the ability of tau to promote MT assembly (84). A reduction in MT binding has also been reported for mutations located outside the repeat region, that is, exon 1 mutations 39, 57). This effect may be mediated through a conformational change in the amino‐terminal projection domain, leading to alterations in tau trafficking and/or compartmentalization, affecting tau interactions with MTs and possibly altering the regulation of MT dynamics. Additional functional changes could be caused through the introduction or removal of potential phosphorylation sites by some mutations, such as K257T, P301S and S320F 37, 94, 99)
Several mutations affecting tau‐MT interactions also have pro‐fibrillogenic effects (32). They have a direct stimulatory effect on heparin‐induced tau filament assembly in vitro, with some mutations leading to the aggregation of specific tau isoforms. For example, the K257T mutation stimulates heparin‐induced assembly of three‐repeat tau into filaments in vitro, but has no effect on the fibrillogenesis of four‐repeat tau (94). This may mirror the pathogenic events occurring in vivo, since filaments extracted from brains of patients with this mutation are predominantly made of three‐repeat tau (94).
Mutations altering tau mRNA splicing. All intronic mutations and some coding region mutations affect the splicing of exon 10 and alter the ratio of tau isoforms. The vast majority of intronic mutations are located in the intron following exon 10 (I10; Figure 2). Exon trapping has shown that most intronic mutations increase the splicing‐in of exon 10. This leads to the increased expression of four‐repeat tau isoforms, which assemble into filaments in the brains of mutation carriers (62). The +19 and +29 mutations have been reported to reduce exon 10 splicing, leading to an increase in three‐repeat tau (113). The +29 change was reported as a rare polymorphism (18), before being considered a mutation (113). The family has now been found to have a Progranulin mutation (85), establishing that it is a benign polymorphism. A few exonic mutations also alter the ratio of tau isoforms by increasing the splicing‐in of exon 10. In particular, missense mutations N279K, G303V and S305N, and silent mutations L284L, N296N and S305S increase exon 10 splicing 11, 18, 38, 97, 110, 113), though through different mechanisms, as detailed below. Furthermore, the magnitude of the increase in splicing of exon 10 differs between mutations, depending on their position 32, 38, 71, 109).
Tau mutations alter splicing by disrupting the secondary structure of the mRNA splice site and by modifying regulatory sequences. Secondary structure analysis has predicted the presence of a stable, folded RNA stem‐loop at the boundary of exon 10 and the downstream intron (Figure 2) 48, 109, 122). All intronic mutations analyzed to date affect the thermodynamic stability of this stem‐loop and disrupt its structure (122). The S305N mutation changes the last amino acid of exon 10, thus reducing the thermodynamic stability of the stem‐loop 50, 122) and, similar to the +3 intronic mutation, increases the binding of U1 snRNP to the 5′ splice site. This leads to increased splicing of exon 10 109, 122). Instead, the silent S305S mutation disrupts the stem‐loop structure without altering U1 snRNP binding (112).
An alternative mechanism is based on the disruption of splicing regulatory sequences. Several Tau mutations in the coding region (N279K, L284L, N296N, S305N) disrupt splicing regulatory sequences, leading to splicing‐in of exon 10 and excessive production of 4R tau (17). An array of local splicing‐enhancer and splicing‐inhibitor elements that modulate the 5′ and 3′ splice sites of exon 10 has also been identified. Mutations N279K and L284L strengthen an exon splicing‐enhancer element located in the 5′ region of exon 10, resulting in increased levels of exon 10‐containing mRNA and soluble 4R tau 17, 18). The same mechanism has recently been found for a mutation located at position –10 of the intron that precedes exon 10 (68). The silent mutation N296N leads to increased levels of exon 10‐containing transcripts by disrupting an exon splicing‐silencer 17, 110) or by creating an exon splicing‐enhancer sequence (34). Conversely, it has been reported that the +19 mutation increases the splicing‐out of exon 10 by altering a splicing silencer sequence 17, 113).
Intronic mutations and some coding region mutations thus affect tau mRNA splicing and lead to an altered ratio of three‐ to four‐repeat isoforms. In particular, most mutations increase the expression of four‐repeat tau and result in the formation of tau deposits consisting mainly or exclusively of four‐repeat tau, indicating the importance of a normal balance between three‐ and four‐repeat tau for neuronal function (26).
Mutations altering tau‐MT interactions, fibril formation and tau mRNA splicing. Several missense mutations located in exon 10 potentially exert their effects at both protein and RNA levels. Mutations ΔK280, ΔN296 and N296H greatly reduce the ability of tau to promote MT assembly in vitro 34, 51, 95, 130). Instead, mutations S305N and Q336R slightly stimulate the ability of tau to promote MT assembly 38, 84). Divergent findings have been reported for the effects of these mutations on heparin‐induced tau filament assembly, ranging from a stimulatory 5, 34) to no 32, 130) effect.
Effects at the mRNA level have also been demonstrated for these mutations. The N296H and S305N mutations increase the splicing‐in of exon 10 34, 38). Accordingly, accumulation of sarkosyl‐insoluble four‐repeat tau has been observed in the brain of an individual with the N296H mutation (51). It has been reported that the ΔN296 mutation leads either to increased (130) or unaltered (34) exon 10 splicing. The ΔK280 mutation represents an exception, since it reduces the splicing‐in of exon 10, possibly leading to an overproduction of 3R tau (18). This effect has recently been confirmed by quantitative analysis of 3R and 4R mRNA levels in brain tissue from a patient with this mutation (117). Experimentally, the ΔK280 mutation also reduces the ability of tau to promote microtubule assembly (37). The significantly greater amount of 3R tau in the sarkosyl‐insoluble fraction from the brain of the proband with the ΔK280 mutation has established that the primary effect of this mutation is at the RNA level (117).
A mutation in exon 12 (E342V) also affects tau at both RNA and protein levels. It reduces the ability of tau to promote MT assembly in vitro and appears to affect the splicing‐in of exon 10. Indeed, it has been reported that exon 10‐containing tau mRNA is increased in a patient with the E342V mutation (63). This was accompanied by an increase in four‐repeat tau without N‐terminal inserts and a decrease in four‐repeat tau with N‐terminal inserts (63). These findings suggest that the E342V mutation may cause FTDP‐17 through unprecedented mechanisms that alter splicing of exons 2, 3, and 10, to preferentially increase four‐repeat tau without N‐terminal inserts, and to promote tau filament assembly.
In summary, mutations in Tau exert their pathogenic effects through three primary mechanisms: (i) alteration of MT assembly; (i) promotion of tau filament formation; (iii) alteration of tau mRNA splicing and isoform expression. A few mutations only act through one of these mechanisms, but most share several modes of action. The primary mechanism appears to determine the type of pathology that will develop in the brain, at least with regard to the isoform composition of tau deposits.
GENETIC EPIDEMIOLOGY
An etiological role for Tau was first suspected, when a family with the so‐called dementia‐disinhibition‐parkinsonism‐amyotrophy complex showed significant linkage to chromosome 17q21‐22. 65, 125). Subsequently, this linkage was also found in several familial disorders described as different nosological entities in the older literature, including hereditary Pick’s disease, familial subcortical gliosis and autosomal‐dominant dementia with widespread NFTs 23, 41, 59, 105, 106, 115). The descriptive term FTDP‐17 now denotes these entities (23). Pathological tau changes are present in neuronal and glial cells of the brain in most of these disorders. In 1998, three research groups identified mutations in Tau in eight families with FTDP‐17 48, 86, 109) and a total of 39 different Tau mutations has been identified to date in families from Europe, the USA, Japan and Australia (Figure 2). Interestingly, Pick’s disease‐like pathology has been described in a patient with a novel Presenilin‐1 mutation who lacked amyloid‐β deposits in brain (16).
FTDP‐17 usually shows an autosomal‐dominant mode of inheritance, but recessive forms have also been described 79, 89). The mode of inheritance is less obvious in some families, due to a lack of information of family history and the possibility of non‐paternity (73). Incomplete penetrance was convincingly demonstrated for the L315R mutation (40). Some families with mutations N279K and P301L are large, with more than 20 affected members over several generations 116, 126). Other families consist of only a few affected members, and sometimes a mutation has only been identified in the proband (43). This may reflect the fact that affected relatives from earlier generations had clinical diagnoses other than FTD during life, including multiple sclerosis and Parkinson’s disease 43, 116).
A few Tau mutations have been identified worldwide, 2, 7, 15, 48, 57, 95, 118, 128), whereas others have only been described in single families 40, 43, 63, 94, 99, 131). Intronic mutations have been found almost exclusively outside Continental Europe 48, 83, 129). P301L appears to be the most prevalent mutation, with more than affected 20 families in the USA, France, the Netherlands, Japan, Italy and Poland 19, 48, 57, 58, 70, 87, 95, 132). It is not clear whether this high frequency can be explained by a common ancestor. Haplotype analysis has shown that the N279K and R406W mutations have occurred as independent events in several families 90, 92, 118). In contrast, all families with the +16 intronic mutation share a common haplotype, probably derived from a common ancestor in Wales, although recombinational events have resulted in a reduced size of shared alleles in some cases (82).
The frequency of Tau mutations in FTD varies between different studies 6, 20, 46, 87, 95, 100, 132). The high prevalence of individuals with a clinical diagnosis of FTD in the Netherlands (14%) and North West Britain (10%) contrasts with the much lower prevalence in the USA (6%) and elsewhere (0%) 20, 114, 132). The frequency is obviously higher in familial cases of FTD, but even here it varies considerably between the USA (8%) and Europe (20%–30%). It is unclear whether this geographical variation reflects true differences in prevalence between specific populations or is the result of different methods of case ascertainment. A selection bias towards familial cases is unlikely, since the percentage of familial cases was similar in these studies 87, 100). The frequency of Tau mutations is higher in cases of familial FTD with tau pathology 72, 87). All studies agree that the absence of tau pathology in familial FTD cases excludes the presence of Tau mutations 72, 87).
AGE AT ONSET
The clinical presentation correlates to some extent with the type or location of mutations within Tau. However, the inter‐ and intrafamilial variation in age at onset may be considerable for some mutations. For others, the average age at onset in families from different continents is remarkably similar, despite different genetic backgrounds and environments 2, 7, 15, 57).
The age at onset for P301L and several other mutations is usually between 45 and 65 years. Dementia may occasionally present between 65 and 70 years of age, but never after the age of 70 years 7, 116). Intronic mutations show the same distribution of age at onset, although a few cases have an earlier onset (around the age of 40) 83, 106). Clinical symptoms may develop even earlier, between 20 and 30 years, in patients with mutations P301S, L315R, G335S and G335V 10, 40, 79, 104, 111), or between 30 and 44 years in patients with mutations L266V and N279K 43, 56, 126). Late‐onset dementia after the age of 70 years may also occur, as observed for cases with the R5H and I260V mutations 35, 39). The healthy status of an 82‐year‐old carrier with the L315R mutation suggests that genetic or environmental factors can play a role in determining the age of onset (40). The duration of illness is on the average between 8 and 10 years for patients with FTDP‐17. An exception is mutation R406W, which is characterized by a slow rate of disease progression lasting up to 25 years 92, 116). Patients with an early age at onset often show a more aggressive disease progression, leading to death within 5 years 10, 40, 102, 104).
CLINICAL PRESENTATION
Two major clinical subtypes can be distinguished among FTDP‐17 patients: dementia‐predominant and parkinsonism‐predominant (128). Both subtypes can occur in patients with the same mutation and even within the same family. The dementia phenotype is usually associated with mutations G272V and P301L 7, 74, 116). Personality changes are characteristic, with disinhibition, jocularity and asocial behavior as salient features. Apathy and loss of initiative are also prominent in the initial presentation. Obsessive‐compulsive behavior in some patients or the occurrence of paranoid delusions and hallucinations in others, may initially suggest a psychiatric disorder 102, 111, 114, 116). Memory problems may dominate the clinical presentation to such an extent that a clinical diagnosis of AD is considered during life 92, 99). Patients may have trouble with planning, suffer from a loss of concentration and develop judgment impairment and loss of insight sufficient to disrupt their social and professional lives. Emotional bluntness is often embarrassing for family members. Other common clinical features are hyperorality and roaming behavior.
Patients develop early language difficulties consisting of word finding problems and stereotyped words and phrases are frequently used 10, 15, 43, 64, 116, 126). Semantic paraphasias and impaired language comprehension are sometimes observed (83), but a true semantic dementia has not been reported. Paucity of speech results in mutism within 5 years in all patients, except for a long preservation of language abilities in some patients with the R406W mutation (116). Partial or generalized epileptic seizures are a specific feature of some cases with the P301S mutation, whereas mental retardation has been reported in a patient with the +11 intronic mutation 71, 104).
While parkinsonism is the dominant clinical presentation in some, but not all, patients with mutation N279K or the intronic mutations 51, 71, 83, 126, 128), it may also occur in other cases (123). Characteristic features include gait impairment, rigidity, bradykinesia, postural instability and resting tremor, with no or only a transient effect of levodopa treatment. Corticospinal tract signs are occasionally present in the parkinsonism‐predominant type (2). Vertical gaze palsy, saccadic eye movements and axial rigidity are early symptoms in patients with a few mutations (ΔN296 and S305N) and are consistent with a clinical diagnosis of PSP 81, 112). Patients with parkinsonism may develop these symptoms later in the disease 106, 126). Unilateral rigidity, dystonia and contractures, in combination with impaired eye movements, occur in cases with some mutations and suggest a clinical diagnosis of CBD 10, 15, 112). These observations indicate that there is a clinical overlap between FTDP‐17, PSP and CBD (10). At present, the question whether to consider these conditions as clinical phenotypes of a single disease or as distinct clinical entities is irrelevant. The main issue is to determine whether FTDP‐17, as well as sporadic and familial PSP and CBD, all have the same underlying pathophysiology.
NEUROPSYCHOLOGY
Patients with FTDP‐17 differ in cognitive function from those with AD. They have a relatively intact episodic memory and do not lose their way. Orientation and visuoconstructive functions are usually intact. Intelligence scores are normally low. Verbal fluency, abstract thinking and executive functions, including planning and mental set‐shifting (Wisconsin Card Sorting Test), are impaired, reflecting frontal dysfunction. Attention and concentration are decreased, often contributing to low test results 7, 116). Poor performance on formal memory tests may be present, but immediate and delayed recall in verbal and nonverbal memory tests is often relatively preserved (104). Language dysfunction may consist of inefficient word retrieval, anomia and sometimes mildly impaired comprehension, although it is never consistent with the cognitive profile of semantic dementia. The pattern of cognitive dysfunction is similar for all individuals with Tau mutations. Cognitive function may be impaired decades before the presentation of dementia, since asymptomatic mutation carriers have already reduced verbal fluency, attention and motor speed and set shifting in their twenties and early thirties 7, 21, 24).
INVESTIGATIONS
A clinical diagnosis of FTDP‐17 can be supported by neuroimaging. Frontotemporal atrophy is the most common neuroradiological feature 7, 116, 129). Patients with some mutations show a predominantly temporal pattern of atrophy (Figure 3), often asymmetric 10, 39, 112), and occasionally also have hippocampal atrophy. Diffuse cerebral atrophy is a common finding in other patients, especially those with intronic mutations 15, 33, 71, 81, 92, 106). Single‐Photon Emission Computed Tomography (SPECT) shows hypoperfusion of the anterior part of the brain early in the disease, even in patients with normal brain morphology (74). Glucose metabolism is reduced in frontal and temporal lobes of the brain on Positron Emission Tomography (73). In patients with parkinsonism, FluoroDopa metabolism in the globus pallidus is significantly impaired (126). SPECT with the radiologand 123I‐N‐omega‐fluoropropyl‐2beta‐carbomethoxy‐3beta‐(4‐iodophenyl)nortropane (FP‐CIT) shows a severe symmetrical decrease of presynaptic dopamine transporter binding in the striatum in individuals with the P301S mutation (104). Electroencephalography tends to be normal (7), except for the interictal epileptic discharges described in some patients with the P301S mutation (104). Denervation potentials and fasciculations on electromyography reflecting anterior horn cell disease have been found in one series (74), but understanding their significance awaits further investigation. Normal levels of total and hyperphosphorylated tau are found in cerebrospinal fluid in FTD with tau pathology (101). This finding has yet to be explained in the context of increased levels of cerebrospinal fluid (CSF) tau in AD and PSP 52, 120).
Figure 3.
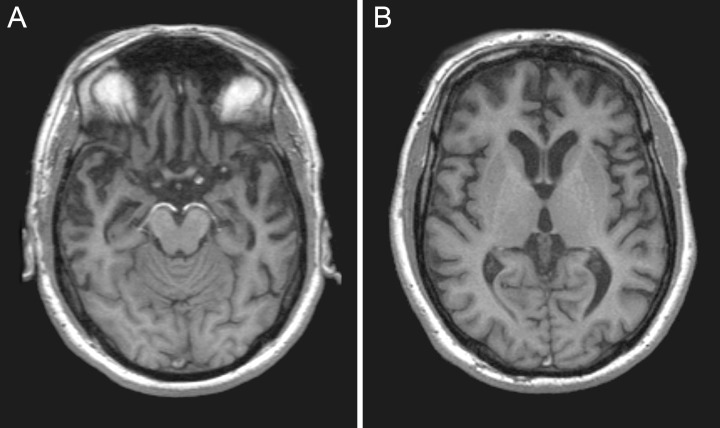
Magnetic resonance imaging. Moderate atrophy of the anterior temporal lobes in a patient with the P301L mutation in exon 10 of Tau.
PATHOLOGY
Macroscopic features. FTDP‐17 is characterized by nerve cell loss and gliosis in cerebral cortex, subcortical nuclei, white matter and brainstem. For the most common mutations (N279K, P301L, intronic), the pathological variation is known, whereas for others, observations have been confined to single patients. The brain weight at autopsy is often reduced, being frequently less than 1000 g. The lateral ventricles can be grossly enlarged (83). The brain shows frontal and temporal atrophy in patients with most mutations, often in a knife‐edge pattern. Temporal atrophy is most prominent in the anterior part, with relative preservation of the posterior part of the superior temporal gyrus 7, 93). The atrophy occasionally extends into the parietal lobe 43, 104). Frontotemporal atrophy may be mild in patients with some mutations 47, 88, 93, 106). Atrophy of the hippocampus and amygdala often accompanies temporal lobe atrophy 7, 43, 83), although these structures can also be normal 70, 115). Subcortical nuclei range from normal 84, 88, 92) to severely atrophic 43, 60, 70, 71, 93). Substantia nigra and locus coeruleus show depigmentation in some patients 52, 60, 74, 92, 93), but may be normal in others 94, 99, 115). The brainstem is sometimes atrophic, whereas the dentate nucleus of the cerebellum usually has a normal appearance 8, 73, 74).
Microscopy. Severe neuronal cell loss and gliosis are present in the frontal and/or temporal cortex in patients with most, but not all, Tau mutations 47, 88, 93). Ballooned cells can often be found in deep cortical layers 52, 93). The hippocampus usually shows loss of pyramidal cells in cornu ammonis and subiculum, although there is no or only focal neuronal loss in some cases 60, 93, 110, 115). The amygdala is affected in most cases. Subcortical nuclei show neuronal loss 94, 129) or are normal 33, 47, 50, 51, 56, 60, 76, 84, 102, 109, 110). Gliosis and/or loss of myelin of the subcortical white matter may be present 73, 104), with occasional degeneration of the corticospinal tract (51). Substantia nigra and locus coeruleus often exhibit severe neuronal loss 83, 111), but may be normal in patients with mutations in exons 12 and 13 64, 115, 116). Neuronal loss may also be present in other brainstem nuclei, the dentate nucleus of the cerebellum and the spinal cord 71, 88, 93, 106, 111). Diffuse and neuritic amyloid‐β plaques have been described in cortical regions in several cases 18, 39, 60, 70, 116). It appears likely that their presence is a secondary and coincident pathological feature in older patients (69). Silver staining (Bodian, Bielschowsky, methenamine silver and Gallyas) has been used to visualize neuronal and glial inclusions. These techniques have given positive staining of NFTs, neuropil threads, dystrophic neurites, coiled bodies and astrocytic processes 10, 35, 39, 43, 56, 64, 73, 88, 93, 109, 116). Pick or Pick‐like bodies have also been identified in this manner (51).
Immunohistochemistry. Tau staining is more widespread than silver staining. A large number of phosphorylation‐dependent anti‐tau antibodies have been used, with antibodies AT8 and PHF1 giving the strongest staining. Different types of neuronal tau inclusions are associated with distinct Tau mutations; they include diffuse or punctate staining, Pick bodies, NFTs and pretangles (Figure 4). Pick bodies usually do not stain with antibody 12E8 (9), which is specific for tau phosphorylated at S262 and/or S356. Cases with the G389R mutation are an exception (73). Tau‐positive inclusions are most often found in frontotemporal cortex and subcortical nuclei, but can also occur in midbrain, brainstem, cerebellum and spinal cord. Tau‐positive inclusions often mirror the severity of neuronal loss, but can also be prominent in regions with less severe nerve cell loss 40, 93). Depending on the type of mutation, tau pathology may be confined to nerve cells (94) or may be most severe in astrocytes and oligodendroglia 10, 35, 56, 93). NFTs are often most numerous in frontotemporal cortex, but may be more widespread in subcortical nuclei and brainstem 51, 93, 110, 111). Flame‐shaped or globose NFTs are associated with intronic mutations and some mutations in exons 12 and 13 60, 93, 105, 111, 115). Extracellular tangles are only occasionally present (84). Granule cells of the dentate gyrus of the hippocampus frequently contain Pick‐like bodies and pyramidal cells of the cornu ammonis sector and subiculum often show NFTs 60, 108); however, occasionally, the hippocampus is free of tau pathology (115).
Figure 4.
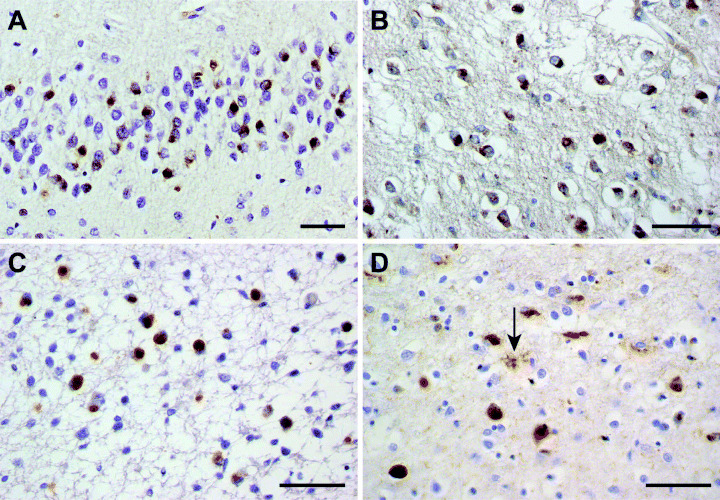
Tau pathology in cases with the G272V and ΔK280 Tau mutations. Tau‐immunoreactive Pick bodies in the dentate gyrus of the hippocampus (A,C), the caudate nucleus (B) and the frontal cortex (D). Note the tau‐positive astrocyte (arrowed) in (D). (A,B), case with mutation G272V in exon 9 of Tau. (C,D), case with mutation ΔK280 in exon 10 of Tau.
Substantia nigra and locus coeruleus often show NFTs, neuropil threads and glial inclusions 60, 93, 111), but lack tau pathology altogether in some patients with mutations in exons 12 and 13 64, 92, 99, 115). Cerebellar nuclei, brainstem and spinal cord may show a few NFTs, neuropil threads and glial inclusions. Neuropil threads are often present in variable severity and distribution, whereas thick axonal swellings are occasionally seen (73).
Pre‐tangles are characteristic of cases with the P301L mutation 108, 116), but also occur in patients with other mutations. The P301L mutation gives rise to characteristic ring‐like tau staining around the nucleus 70, 108, 116). Several mutations are associated with Pick bodies, which vary in number in cortex, hippocampus, amygdala and subcortical nuclei 40, 43, 76, 94, 108, 117). They usually stain with three‐repeat specific anti‐tau antibodies and are negative with four‐repeat specific antibodies.
Glial tau pathology can be more severe than neuronal pathology. Glial tangles and coiled bodies are more abundant in white matter than in cortex in cases with inclusions consisting predominantly of four‐repeat tau isoforms 10, 51, 60, 71, 110). Tufted astrocytes characteristic of PSP are found in the cortex and subcortical nuclei in patients with several exonic and intronic mutations 43, 55, 60, 88, 129). Their presence in brains from patients with mutations affecting all six tau isoforms (R5H, L266V, S305N, L315R) contrasts with the observation of abundant glial tau pathology associated with mutations that only affect four‐repeat tau or increase the relative amount of four‐repeat tau. Another interesting observation is that tufted astrocytes associated with these mutations appear to be more abundant in cortical areas with severe neuronal loss than in areas that are less severely involved 40, 55). This suggests that astrocytic inclusions may develop later than neuronal deposits or may be longer‐lived. Phagocytosis of neuronal deposits by astrocytes provides a possible explanation, but the connection between a given Tau mutation and astrocytic tau pathology awaits clarification.
Biochemistry. Immunoblotting of sarkosyl‐insoluble tau shows distinct profiles according to the location of Tau mutations. For mutations in exon 10 and at the splice‐donor site of the intron following exon 10, four‐repeat tau isoforms predominate. Two different mechanisms underlie the deposition of four‐repeat tau. First, the intronic mutations and some mutations in exon 10 (at positions 279, 296 and 305) produce a twofold to tenfold increase in exon 10+ transcripts over exon 10– transcripts 38, 44, 109). The resulting overexpression of soluble four‐repeat tau leads to its deposition. Second, mutations in exons 9–13 result in reduced binding to microtubules 37, 40, 99). For the P301L mutation in exon 10, this change affects only four‐repeat tau and results in its aggregation (96). Neuronal and glial tau pathology is usually associated with the deposition of four‐repeat tau isoforms, while neuronal tau pathology is associated with all six isoforms. In cases with mutations V337M and R406W, NFTs consist of all six tau isoforms, as expected 105, 111, 116). Pick bodies contain exclusively or predominantly three‐repeat tau in cases with mutations K257T, L266V and G272V in exon 9 and mutation ΔK280 in exon 10 9, 43, 55, 94, 117), whereas for other mutations, Pick bodies contain both three‐ and four‐repeat tau isoforms, as judged by western blotting 40, 63, 73, 76, 99). Although associated glial tau pathology may be responsible for the presence of four‐repeat tau in these cases, staining with specific anti‐tau antibodies has also shown a mixture of three‐ and four‐repeat tau in Pick bodies in a case with the Q336R mutation (84).
The association between mutation location and insoluble tau isoform profiles is less clear for other mutations 40, 64, 99). Unlike the observation of a predominance of three‐repeat tau in some exon 9 mutations, only four‐repeat tau was found in the case of mutation I260V in exon 9 (35). The neuronal and glial tau pathology in the R5L mutation in exon 1 consists mainly of four‐repeat tau, despite the fact all six isoforms carry this mutation (88).
Electron microscopy. The inclusions of FTDP‐17 consist of abnormal filaments made of hyperphosphorylated tau protein 62, 106). These filaments can be studied in sarkosyl‐insoluble brain extracts or in fixed brain tissue. Tau filaments show specific morphologies, which vary with the different mutations. PHFs with a diameter of 8–20 nm and a periodicity of 80 nm are found in AD, but also in FTDP‐17 with mutations in exons 12 and 13 64, 102, 105, 108, 111). Slender twisted filaments or ribbons with an irregular periodicity of 90–130 nm are characteristic of intronic and some exonic mutations with a predominance of four‐repeat tau 2, 47, 93, 106). Narrow, irregularly twisted filaments or ribbons of 15 nm and a periodicity of 130 nm or greater are found in some other cases, including those with mutation P301L 70, 94, 108). Several mutations produce predominantly straight filaments 10, 43, 84). Phosphorylation‐dependent and phosphorylation‐independent anti‐tau antibodies decorate these filaments.
CONCLUSION
The recognition of the etiological role of Tau in some disorders with dementia and parkinsonism has implications for clinical practice, that is, genetic counseling. It has shown that dysfunction or dysregulation of tau protein can cause neurodegenerative changes accompanied by the presence of hyperphosphorylated tau protein deposits in neurons and glial cells. In addition, observations from clinical and pathological studies in other tauopathies, such as PSP, CBD, Pick’s disease and AGD, support the hypothesis that tau plays an important role in the pathogenesis of these disorders (26). Some functional consequences of Tau mutations have been clarified, such as their effects on microtubule assembly and the change in the ratio of three‐ vs. four‐repeat tau. However, it is unknown how this change in isoform composition leads to neuronal and glial tau pathology. The correlation between the location of mutations in Tau and tau pathology or tau isoform profiles is weaker than believed earlier. It is not known how missense mutations outside exons 9–12 lead to a reduction in microtubule binding. Furthermore, the significance of glial tau pathology in both FTDP‐17 and related disorders awaits further explanation. The factors contributing to the variation in clinical phenotype, including incomplete penetrance, remain to be determined. Elucidation of the genetic factors that are necessary to cause PSP, CBD, Pick’s disease and AGD will be a major step forward in the understanding of the mechanisms modulating the tauopathy phenotypes.
ACKNOWLEDGEMENTS
We would like to thank Mrs. J.A.C. Romeijn for assistance in preparing the manuscript, and T. de Vries Lentsch for the photography and artwork. MGS would like to acknowledge the support of the UK Medical Research Council, the Alzheimer’s Research Trust and the EU FP6 Integrated Project “APOPIS”.
REFERENCES
- 1. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T (2006) TDP‐43 is a component of ubiquitin‐positive tau‐negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611. [DOI] [PubMed] [Google Scholar]
- 2. Arima K, Kowalska A, Hasegawa M, Mukoyama M, Watanabe R, Kawai M, Takahashi K, Iwatsubo T, Tabira T, Sunohara N (2000) Two brothers with frontotemporal dementia and parkinsonism with an N279K mutation of the tau gene. Neurology 54:1787–1795. [DOI] [PubMed] [Google Scholar]
- 3. Ashworth A, Lloyd S, Brown J, Gydesen S, Sorensen SA, Brun A, Englund E, Humphreys C, Housman D, Badura M, Stanton V Jr, Taylor K, Cameron J, Munroe D, Johansson J, Rossor M, Fisher EM, Collinge J (1999) Molecular genetic characterisation of frontotemporal dementia on chromosome 3. Dement Geriatr Cogn Disord 10(Suppl. 1): 93–101. [DOI] [PubMed] [Google Scholar]
- 4. Baker M, Mackenzie IR, Pickering‐Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M (2006) Mutations in progranulin cause tau‐negative frontotemporal dementia linked to chromosome 17. Nature 442:916–919. [DOI] [PubMed] [Google Scholar]
- 5. Von Bergen M, Barghorn S, Li L, Marx A, Biernat J, Mandelkow EM, Mandelkow E (2001) Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta‐structure. J Biol Chem 276:48165–48174. [DOI] [PubMed] [Google Scholar]
- 6. Binetti G, Nicosia F, Benussi L, Ghidoni R, Feudatari E, Barbiero L, Signorini S, Villa A, Mattioli F, Zanetti O, Alberici A (2003) Prevalence of TAU mutations in an Italian clinical series of familial frontotemporal patients. Neurosci Lett 338:85–87. [DOI] [PubMed] [Google Scholar]
- 7. Bird TD, Nochlin D, Poorkaj P, Cherrier M, Kaye J, Payami H, Peskind E, Lampe TH, Nemens E, Boyer PJ, Schellenberg GD (1999) A clinical pathological comparison of three families with frontotemporal dementia and identical mutations in the tau gene (P301L). Brain 122:741–756. [DOI] [PubMed] [Google Scholar]
- 8. Bird T, Knopman D, Van Swieten J, Rosso S, Feldman H, Tanabe H, Graff‐Raford N, Geschwind D, Verpillat P, Hutton M (2003) Epidemiology and genetics of frontotemporal dementia/Pick’s disease. Ann Neurol 54(Suppl. 5):S29–S31. [DOI] [PubMed] [Google Scholar]
- 9. Bronner IF, Ter Meulen BC, Azmani A, Severijnen LA, Willemsen R, Kamphorst W, Ravid R, Heutink P, Van Swieten JC (2005) Hereditary Pick’s disease with the G272V tau mutation shows predominant three‐repeat tau pathology. Brain 128:2645–2653. [DOI] [PubMed] [Google Scholar]
- 10. Bugiani O, Murrell JR, Giaccone G, Hasegawa M, Ghigo G, Tabaton M, Morbin M, Primavera A, Carella F, Solaro C, Grisoli M, Savoiardo M, Spillantini MG, Tagliavini F, Goedert M, Ghetti B (1999) Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J Neuropathol Exp Neurol 58:667–677. [DOI] [PubMed] [Google Scholar]
- 11. Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZS, Miller B, Li D, Payami H, Awert F, Markopoulou K, Andreadis A, D’Souza I, Lee VM, Reed L, Trojanowski JQ, Zhukareva V, Bird T, Schellenberg G, Wilhelmsen KC (1998) Pathogenic implications of mutations in the tau gene in pallido‐ponto‐nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci USA 95:13103–13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Crowther RA (1991) Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc Natl Acad Sci USA 88:2288–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cruts M, Gijselinck I, Van Der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, Van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van Den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar‐Singh S, Van Broeckhoven C (2006) Null mutations in progranulin cause ubiquitin‐positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–924. [DOI] [PubMed] [Google Scholar]
- 14. Dayanandan R, Van Slegtenhorst M, Mack TG, Ko L, Yen SH, Leroy K, Brion JP, Anderton BH, Hutton M, Lovestone S (1999) Mutations in tau reduce its microtubule binding properties in intact cells and affect its phosphorylation. FEBS Lett 446:228–232. [DOI] [PubMed] [Google Scholar]
- 15. Delisle MB, Murrell JR, Richardson R, Trofatter JA, Rascol O, Soulages X, Mohr M, Calvas P, Ghetti B (1999) A mutation at codon 279 (N279K) in exon 10 of the Tau gene causes a tauopathy with dementia and supranuclear palsy. Acta Neuropathol (Berl) 98:62–77. [DOI] [PubMed] [Google Scholar]
- 16. Dermaut B, Kumar‐Singh S, Engelborghs S, Theuns J, Rademakers R, Saerens J, Pickut BA, Peeters K, Van Den Broeck M, Vennekens K, Claes S, Cruts M, Cras P, Martin JJ, Van Broeckhoven C, De Deyn PP (2004) A novel presenilin 1 mutation associated with Pick’s disease but not beta‐amyloid plaques. Ann Neurol 55:617–626. [DOI] [PubMed] [Google Scholar]
- 17. D’Souza I, Schellenberg GD (2002) Tau Exon 10 expression involves a bipartite intron 10 regulatory sequence and weak 5′ and 3′ splice sites. J Biol Chem 277:26587–26599. [DOI] [PubMed] [Google Scholar]
- 18. D’Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD, Schellenberg GD (1999) Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism‐chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA 96:5598–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dumanchin C, Camuzat A, Campion D, Verpillat P, Hannequin D, Dubois B, Saugier‐Veber P, Martin C, Penet C, Charbonnier F, Agid Y, Frebourg T, Brice A (1998) Segregation of a missense mutation in the microtubule‐associated protein tau gene with familial frontotemporal dementia and parkinsonism. Hum Mol Genet 7:1825–1829. [DOI] [PubMed] [Google Scholar]
- 20. Fabre SF, Forsell C, Viitanen M, Sjogren M, Wallin A, Blennow K, Blomberg M, Andersen C, Wahlund LO, Lannfelt L (2001) Clinic‐based cases with frontotemporal dementia show increased cerebrospinal fluid tau and high apolipoprotein E epsilon4 frequency, but no tau gene mutations. Exp Neurol 168:413–418. [DOI] [PubMed] [Google Scholar]
- 21. Ferman TJ, McRae CA, Arvanitakis Z, Tsuboi Y, Vo A, Wszolek ZK (2003) Early and pre‐symptomatic neuropsychological dysfunction in the PPND family with the N279K tau mutation. Parkinsonism Relat Disord 9:265–270. [DOI] [PubMed] [Google Scholar]
- 22. Flament S, Delacourte A, Hemon B, Défossez A (1989) Characterization of two pathological tau protein, variants in Alzheimer brain cortices. J Neurol Sci 92:133–141. [DOI] [PubMed] [Google Scholar]
- 23. Foster NL, Wilhelmsen K, Sima AA, Jones MZ, D’Amato CJ, Gilman S (1997) Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Conference participants. Ann Neurol 41:706–715. [DOI] [PubMed] [Google Scholar]
- 24. Geschwind DH, Robidoux J, Alarcon M, Miller BL, Wilhelmsen KC, Cummings JL, Nasreddine ZS (2001) Dementia and neurodevelopmental predisposition: cognitive dysfunction in presymptomatic subjects precedes dementia by decades in frontotemporal dementia. Ann Neurol 50:741–746. [DOI] [PubMed] [Google Scholar]
- 25. Giaccone G, Rossi G, Farina L, Marcon G, Di Fede G, Catania M, Morbin M, Sacco L, Bugiani O, Tagliavini F (2005) Familial frontotemporal dementia associated with the novel MAPT mutation T427M. J Neurol 252:1543–1545. [DOI] [PubMed] [Google Scholar]
- 26. Goedert M (2004) Tau protein and neurodegeneration. Semin Cell Dev Biol 15:45–49. [DOI] [PubMed] [Google Scholar]
- 27. Goedert M, Jakes R (1990) Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J 9:4225–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA (1989) Multiple isoforms of human microtubule‐associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3:519–526. [DOI] [PubMed] [Google Scholar]
- 29. Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA (1989) Cloning and sequencing of the cDNA encoding an isoform of microtubule‐associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J 8:393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goedert M, Spillantini MG, Cairns NJ, Crowther RA (1992) Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron 8:159–168. [DOI] [PubMed] [Google Scholar]
- 31. Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA (1996) Assembly of microtubule‐associated protein tau into Alzheimer‐like filaments induced by sulphated glycosaminoglycans. Nature 383:550–553. [DOI] [PubMed] [Google Scholar]
- 32. Goedert M, Jakes R, Crowther RA (1999) Effects of frontotemporal dementia FTDP‐17 mutations on heparin‐induced assembly of tau filaments. FEBS Lett 450:306–311. [DOI] [PubMed] [Google Scholar]
- 33. Goedert M, Spillantini MG, Crowther RA, Chen SG, Parchi P, Tabaton M, Lanska DJ, Markesbery WR, Wilhelmsen KC, Dickson DW, Petersen RB, Gambetti P (1999) Tau gene mutation in familial progressive subcortical gliosis. Nat Med 5:454–457. [DOI] [PubMed] [Google Scholar]
- 34. Grover A, DeTure M, Yen SH, Hutton M (2002) Effects on splicing and protein function of three mutations in codon N296 of tau in vitro. Neurosci Lett 323:33–36. [DOI] [PubMed] [Google Scholar]
- 35. Grover A, England E, Baker M, Sahara N, Adamson J, Granger B, Houlden H, Passant U, Yen SH, DeTure M, Hutton M (2003) A novel tau mutation in exon 9 (1260V) causes a four‐repeat tauopathy. Exp Neurol 184:131–140. [DOI] [PubMed] [Google Scholar]
- 36. Gydesen S, Brown JM, Brun A, Chakrabarti L, Gade A, Johannsen P, Rossor M, Thusgaard T, Grove A, Yancopoulou D, Spillantini MG, Fisher EM, Collinge J, Sorensen SA (2002) Chromosome 3 linked frontotemporal dementia (FTD‐3). Neurology 59:1585–1594. [DOI] [PubMed] [Google Scholar]
- 37. Hasegawa M, Smith MJ, Goedert M (1998) Tau proteins with FTDP‐17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett 437:207–210. [DOI] [PubMed] [Google Scholar]
- 38. Hasegawa M, Smith MJ, Iijima M, Tabira T, Goedert M (1999) FTDP‐17 mutations N279K and S305N in tau produce increased splicing of exon 10. FEBS Lett 443:93–96. [DOI] [PubMed] [Google Scholar]
- 39. Hayashi S, Toyoshima Y, Hasegawa M, Umeda Y, Wakabayashi K, Tokiguchi S, Iwatsubo T, Takahashi H (2002) Late‐onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann Neurol 51:525–530. [DOI] [PubMed] [Google Scholar]
- 40. Van Herpen E, Rosso SM, Serverijnen LA, Yoshida H, Breedveld G, Van de Graaf R, Kamphorst W, Ravid R, Willemsen R, Dooijes D, Majoor‐Krakauer D, Kros JM, Crowther RA, Goedert M, Heutink P, Van Swieten JC (2003) Variable phenotypic expression and extensive tau pathology in two families with the novel tau mutation L315R. Ann Neurol 54:573–581. [DOI] [PubMed] [Google Scholar]
- 41. Heutink P (2000) Untangling tau‐related dementia. Hum Mol Genet 9:979–986. [DOI] [PubMed] [Google Scholar]
- 42. Hodges JR, Davies RR, Xuereb JH, Casey B, Broe M, Bak TH, Kril JJ, Halliday GM (2004) Clinicopathological correlates in frontotemporal dementia. Ann Neurol 56:399–406. [DOI] [PubMed] [Google Scholar]
- 43. Hogg M, Grujic ZM, Baker M, Demirci S, Guillozet AL, Sweet AP, Herzog LL, Weintraub S, Mesulam MM, LaPointe NE, Gamblin TC, Berry RW, BinDer LI, De Silva R, Lees A, Espinoza M, Davies P, Grover A, Sahara N, Ishizawa T, Dickson D, Yen SH, Hutton M, Bigio EH (2003) The L266V tau mutation is associated with frontotemporal dementia and Pick‐like 3R and 4R tauopathy. Acta Neuropathol (Berl) 106:323–336. [DOI] [PubMed] [Google Scholar]
- 44. Hong M, Zhukareva V, Vogelsberg‐Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM (1998) Mutation‐specific functional impairments in distinct tau isoforms of hereditary FTDP‐17. Science 282:1914–1917. [DOI] [PubMed] [Google Scholar]
- 45. Hosler BA, Siddique T, Sapp PC, Sailor W, Huang MC, Hossain A, Daube JR, Nance M, Fan C, Kaplan J, Hung WY, McKenna‐Yasek D, Haines JL, Pericak‐Vance MA, Horvitz HR, Brown RH Jr (2000) Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21‐q22. JAMA 284:1664–1669. [DOI] [PubMed] [Google Scholar]
- 46. Houlden H, Baker M, Adamson J, Grover A, Waring S, Dickson D, Lynch T, Boeve B, Petersen RC, Pickering‐Brown S, Owen F, Neary D, Craufurd D, Snowden J, Mann D, Hutton M (1999) Frequency of tau mutations in three series of non‐Alzheimer’s degenerative dementia. Ann Neurol 46:243–248. [DOI] [PubMed] [Google Scholar]
- 47. Hulette CM, Pericak‐Vance MA, Roses AD, Schmechel DE, Yamaoka LH, Gaskell PC, Welsh‐Bohmer KA, Crowther RA, Spillantini MG (1999) Neuropathological features of frontotemporal dementia and parkinsonism linked to chromosome 17q21‐22 (FTDP‐17): duke Family 1684. J Neuropathol Exp Neurol 58:859–866. [DOI] [PubMed] [Google Scholar]
- 48. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering‐Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, De Graaff E, Wauters E, Van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, Van Swieten J, Mann D, Lynch T, Heutink P (1998) Association of missense and 5′‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature 393:702–705. [DOI] [PubMed] [Google Scholar]
- 49. Igniatovic T, Xuereb J, Hodges J, Spillantini MG (2007) TDP‐43 in ubiquitin inclusions in familial and sporadic semantic dementia. Neuropathol Appl Neurobiol (Suppl.):in press. [Google Scholar]
- 50. Iijima M, Tabira T, Poorkaj P, Schellenberg GD, Trojanowski JQ, Lee VM, Schmidt ML, Takahashi K, Nabika T, Matsumoto T, Yamashita Y, Yoshioka S, Ishino H (1999) A distinct familial presenile dementia with a novel missense mutation in the tau gene. Neuroreport 10:497–501. [DOI] [PubMed] [Google Scholar]
- 51. Iseki E, Matsumura T, Marui W, Hino H, Odawara T, Sugiyama N, Suzuki K, Sawada H, Arai T, Kosaka K (2001) Familial frontotemporal dementia and parkinsonism with a novel N296H mutation in exon 10 of the tau gene and a widespread tau accumulation in the glial cells. Acta Neuropathol (Berl) 102:285–292. [DOI] [PubMed] [Google Scholar]
- 52. Itoh N, Arai H, Urakami K, Ishiguro K, Ohno H, Hampel H, Buerger K, Wiltfang J, Otto M, Kretzschmar H, Moeller HJ, Imagawa M, Kohno H, Nakashima K, Kuzuhara S, Sasaki H, Imahori K (2001) Large‐scale, multicenter study of cerebrospinal fluid tau protein phosphorylated at serine 199 for the antemortem diagnosis of Alzheimer’s disease. Ann Neurol 50:150–156. [DOI] [PubMed] [Google Scholar]
- 53. Kar S, Fan J, Smith MJ, Goedert M, Amos LA (2003) Repeat motifs of tau bind to the insides of microtubules in the absence of taxol. EMBO J 22:70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kertesz A, Kawarai T, Rogaeva E, St George‐Hyslop PH, Poorkaj P, Bird TD, Munoz DG (2000) Familial frontotemporal dementia with ubiquitin‐positive, tau‐negative inclusions. Neurology 54: 818–827. [DOI] [PubMed] [Google Scholar]
- 55. Kobayashi T, Ota S, Tanaka K, Ito Y, Hasegawa M, Umeda Y, Motoi Y, Takanashi M, Yasuhara M, Anno M, Mizuno Y, Mori H (2003) A novel L266V mutation of the tau gene causes frontotemporal dementia with a unique tau pathology. Ann Neurol 53:133–137. [DOI] [PubMed] [Google Scholar]
- 56. Kobayashi K, Hayashi M, Kidani T, Ujike H, Iijima M, Ishihara T, Nakano H, Sugimori K, Shimazaki M, Kuroda S, Koshino Y (2004) Pick’s disease pathology of a missense mutation of S305N of frontotemporal dementia and parkinsonism linked to chromosome 17: another phenotype of S305N. Dement Geriatr Cogn Disord 17:293–297. [DOI] [PubMed] [Google Scholar]
- 57. Kodama K, Okada S, Iseki E, Kowalska A, Tabira T, Hosoi N, Yamanouchi N, Noda S, Komatsu N, Nakazato M, Kumakiri C, Yazaki M, Sato T (2000) Familial frontotemporal dementia with a P301L tau mutation in Japan. J Neurol Sci 176:57–64. [DOI] [PubMed] [Google Scholar]
- 58. Kowalska A, Asada T, Arima K, Kumakiri C, Kozubski W, Takahashi K, Tabira T (2001) Genetic analysis in patients with familial and sporadic frontotemporal dementia: two tau mutations in only familial cases and no association with apolipoprotein epsilon4. Dement Geriatr Cogn Disord 12:387–392. [DOI] [PubMed] [Google Scholar]
- 59. Lanska DJ, Currier RD, Cohen M, Gambetti P, Smith EE, Bebin J, Jackson JF, Whitehouse PJ, Markesbery WR (1994) Familial progressive subcortical gliosis. Neurology 44:1633–1643. [DOI] [PubMed] [Google Scholar]
- 60. Lantos PL, Cairns NJ, Khan MN, King A, Revesz T, Janssen JC, Morris H, Rossor MN (2002) Neuropathologic variation in frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology 58:1169–1175. [DOI] [PubMed] [Google Scholar]
- 61. Lee G, Neve RL, Kosik KS (1989) The microtubule binding domain of tau protein. Neuron 2:1615–1624. [DOI] [PubMed] [Google Scholar]
- 62. Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159. [DOI] [PubMed] [Google Scholar]
- 63. Lippa CF, Zhukareva V, Kawarai T, Uryu K, Shafiq M, Nee LE, Grafman J, Liang Y, St George‐Hyslop PH, Trojanowski JQ, Lee VM (2000) Frontotemporal dementia with novel tau pathology and a Glu342Val tau mutation. Ann Neurol 48:850–858. [PubMed] [Google Scholar]
- 64. Lippa CF, Zhukareva V, Kawarai T, Uryu K, Shafiq M, Nee LE, Grafman J, Liang Y, St George‐Hyslop PH, Trojanowski JQ, Lee VM (2000) Frontotemporal dementia with novel tau pathology and a Glu342Val tau mutation. Ann Neurol 48:850–858. [PubMed] [Google Scholar]
- 65. Lynch T, Sano M, Marder KS, Bell KL, Foster NL, Defendini RF, Sima AA, Keohane C, Nygaard TG, Fahn S (1994) Clinical characteristics of a family with chromosome 17‐linked disinhibition‐dementia‐parkinsonism‐amyotrophy complex. Neurology 44:1878–1884. [DOI] [PubMed] [Google Scholar]
- 66. Mackenzie IRA, Shi J, Shaw CL, DuPlessis D, Neary D, Snowden JS, Mann DMA (2006) Dementia lacking distinctive histology (DLDH) revisited. Acta Neuropathol 112:551–559. [DOI] [PubMed] [Google Scholar]
- 67. McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ (2001) Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol 58: 1803–1809. [DOI] [PubMed] [Google Scholar]
- 68. Malkani R, D’Souza I, Gwinn‐Hardy K, Schellenberg GD, Hardy J, Momeni P (2006) A MAPT mutation in a regulatory element upstream of exon 10 causes frontotemporal dementia. Neurobiol Dis 22:401–403. [DOI] [PubMed] [Google Scholar]
- 69. Mann DM, McDonagh AM, Pickering‐Brown SM, Kowa H, Iwatsubo T (2001) Amyloid beta protein deposition in patients with frontotemporal lobar degeneration: relationship to age and apolipoprotein E genotype. Neurosci Lett 304:161–164. [DOI] [PubMed] [Google Scholar]
- 70. Mirra SS, Murrell JR, Gearing M, Spillantini MG, Goedert M, Crowther RA, Levey AI, Jones R, Green J, Shoffner JM, Wainer BH, Schmidt ML, Trojanowski JQ, Ghetti B (1999) Tau pathology in a family with dementia and a P301L mutation in tau. J Neuropathol Exp Neurol 58:335–345. [DOI] [PubMed] [Google Scholar]
- 71. Miyamoto K, Kowalska A, Hasegawa M, Tabira T, Takahashi K, Araki W, Akiguchi I, Ikemoto A (2001) Familial frontotemporal dementia and parkinsonism with a novel mutation at an intron 10+11‐splice site in the tau gene. Ann Neurol 50:117–120. [DOI] [PubMed] [Google Scholar]
- 72. Morris HR, Khan MN, Janssen JC, Brown JM, Perez‐Tur J, Baker M, Ozansoy M, Hardy J, Hutton M, Wood NW, Lees AJ, Revesz T, Lantos P, Rossor MN (2001) The genetic and pathological classification of familial frontotemporal dementia. Arch Neurol 58:1813–1816. [DOI] [PubMed] [Google Scholar]
- 73. Murrell JR, Spillantini MG, Zolo P, Guazzelli M, Smith MJ, Hasegawa M, Redi F, Crowther RA, Pietrini P, Ghetti B, Goedert M (1999) Tau gene mutation G389R causes a tauopathy with abundant Pick body‐like inclusions and axonal deposits. J Neuropathol Exp Neurol 58:1207–1226. [DOI] [PubMed] [Google Scholar]
- 74. Nasreddine ZS, Loginov M, Clark LN, Lamarche J, Miller BL, Lamontagne A, Zhukareva V, Lee VM, Wilhelmsen KC, Geschwind DH (1999) From genotype to phenotype: a clinical pathological, and biochemical investigation of frontotemporal dementia and parkinsonism (FTDP‐17) caused by the P301L tau mutation. Ann Neurol 45:704–715. [DOI] [PubMed] [Google Scholar]
- 75. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51:1546–1554. [DOI] [PubMed] [Google Scholar]
- 76. Neumann M, Schulz‐Schaeffer W, Crowther RA, Smith MJ, Spillantini MG, Goedert M, Kretzschmar HA (2001) Pick’s disease associated with the novel Tau gene mutation K369I. Ann Neurol 50:503–513. [DOI] [PubMed] [Google Scholar]
- 77. Neumann M, Diekmann S, Bertsch U, Vanmassenhove B, Bogerts B, Kretzschmar HA (2005) Novel G335V mutation in the tau gene associated with early onset familial frontotemporal dementia. Neurogenetics 6:91–95. [DOI] [PubMed] [Google Scholar]
- 78. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. [DOI] [PubMed] [Google Scholar]
- 79. Nicholl DJ, Greenstone MA, Clarke CE, Rizzu P, Crooks D, Crowe A, Trojanowski JQ, Lee VM, Heutink P (2003) An English kindred with a novel recessive tauopathy and respiratory failure. Ann Neurol 54:682–686. [DOI] [PubMed] [Google Scholar]
- 80. Parkinson N, Ince PG, Smith MO, Highley R, Skibinski G, Andersen PM, Morrison KE, Pall HS, Hardiman O, Collinge J, Shaw PJ, Fisher EM (2006) ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67:1074–1077. [DOI] [PubMed] [Google Scholar]
- 81. Pastor P, Pastor E, Carnero C, Vela R, Garcia T, Amer G, Tolosa E, Oliva R (2001) Familial atypical progressive supranuclear palsy associated with homozigosity for the delN296 mutation in the tau gene. Ann Neurol 49:263–267. [DOI] [PubMed] [Google Scholar]
- 82. Pickering‐Brown S, Baker M, Bird T, Trojanowski J, Lee V, Morris H, Rossor M, Janssen JC, Neary D, Craufurd D, Richardson A, Snowden J, Hardy J, Mann D, Hutton M (2004) Evidence of a founder effect in families with frontotemporal dementia that harbor the tau +16 splice mutation. Am J Med Genet 125B:79–82. [DOI] [PubMed] [Google Scholar]
- 83. Pickering‐Brown SM, Richardson AM, Snowden JS, McDonagh AM, Burns A, Braude W, Baker M, Liu WK, Yen SH, Hardy J, Hutton M, Davies Y, Allsop D, Craufurd D, Neary D, Mann DM (2002) Inherited frontotemporal dementia in nine British families associated with intronic mutations in the tau gene. Brain 125:732–751. [DOI] [PubMed] [Google Scholar]
- 84. Pickering‐Brown SM, Baker M, Nonaka T, Ikeda K, Sharma S, Mackenzie J, Simpson SA, Moore JW, Snowden JS, De Silva R, Revesz T, Hasegawa M, Hutton M, Mann DM (2004) Frontotemporal dementia with Pick‐type histology associated with Q336R mutation in the tau gene. Brain 127:1415–1426. [DOI] [PubMed] [Google Scholar]
- 85. Pickering‐Brown SM, Baker M, Gass J, Boeve BF, Loy CT, Brooks WS, Mackenzie IR, Martins RN, Kwok JB, Halliday GM, Kril J, Schofield PR, Mann DM, Hutton M (2006) Mutations in progranulin explain atypical phenotypes with variants in MAPT. Brain 129:3124–3126. [DOI] [PubMed] [Google Scholar]
- 86. Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43:815–825. [DOI] [PubMed] [Google Scholar]
- 87. Poorkaj P, Grossman M, Steinbart E, Payami H, Sadovnick A, Nochlin D, Tabira T, Trojanowski JQ, Borson S, Galasko D, Reich S, Quinn B, Schellenberg G, Bird TD (2001) Frequency of tau gene mutations in familial and sporadic cases of non‐Alzheimer dementia. Arch Neurol 58:383–387. [DOI] [PubMed] [Google Scholar]
- 88. Poorkaj P, Muma NA, Zhukareva V, Cochran EJ, Shannon KM, Hurtig H, Koller WC, Bird TD, Trojanowski JQ, Lee VM, Schellenberg GD (2002) An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol 52: 511–516. [DOI] [PubMed] [Google Scholar]
- 89. Rademakers R, Cruts M, Dermaut B, Sleegers K, Rosso SM, Van Den Broeck M, Backhovens H, Van Swieten J, Van Duijn CM, Van Broeckhoven C (2002) Tau negative frontal lobe dementia at 17q21: significant finemapping of the candidate region to a 4.8 cM interval. Mol Psychiatry 7:1064–1074. [DOI] [PubMed] [Google Scholar]
- 90. Rademakers R, Dermaut B, Peeters K, Cruts M, Heutink P, Goate A, Van Broeckhoven C (2003) Tau (MAPT) mutation Arg406Trp presenting clinically with Alzheimer disease does not share a common founder in Western Europe. Hum Mutat 22:409–411. [DOI] [PubMed] [Google Scholar]
- 91. Ratnavalli E, Brayne C, Dawson L, Hodges JR (2002) The prevalence of frontotemporal dementia. Neurology 58:1615–1621. [DOI] [PubMed] [Google Scholar]
- 92. Reed LA, Grabowski TJ, Schmidt ML, Morris JC, Goate A, Solodkin A, Van HG, Schelper RL, Talbot CJ, Wragg MA, Trojanowski JQ (1997) Autosomal dominant dementia with widespread neurofibrillary tangles. Ann Neurol 42:564–572. [DOI] [PubMed] [Google Scholar]
- 93. Reed LA, Schmidt ML, Wszolek ZK, Balin BJ, Soontornniyomkij V, Lee VM, Trojanowski JQ, Schelper RL (1998) The neuropathology of a chromosome 17‐linked autosomal dominant parkinsonism and dementia (“pallido‐ponto‐nigral degeneration”). J Neuropathol Exp Neurol 57:588–601. [DOI] [PubMed] [Google Scholar]
- 94. Rizzini C, Goedert M, Hodges JR, Smith MJ, Jakes R, Hills R, Xuereb JH, Crowther RA, Spillantini MG (2000) Tau gene mutation K257T causes a tauopathy similar to Pick’s disease. J Neuropathol Exp Neurol 59:990–1001. [DOI] [PubMed] [Google Scholar]
- 95. Rizzu P, Van Swieten J, Joosse M, Hasegawa M, Stevens M, Tibben A, Niermeijer MF, Hillebrand M, Ravid R, Oostra BA, Goedert M, Van Duijn C, Heutink P (1999) High prevalence of mutations in the microtubule‐associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet 64:414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Rizzu P, Joosse M, Ravid R, Hoogeveen A, Kamphorst W, Van Swieten JC, Willemsen R, Heutink P (2000) Mutation‐dependent aggregation of tau protein and its selective depletion from the soluble fraction in brain of P301L FTDP‐17 patients. Hum Mol Genet 9:3075–3082. [DOI] [PubMed] [Google Scholar]
- 97. Ros R, Thobois S, Streichenberger N, Kopp N, Sanchez MP, Perez M, Hoenicka J, Avila J, Honnorat J, De Yebenes JG (2005) A new mutation of the tau gene, G303V, in early‐onset familial progressive supranuclear palsy. Arch Neurol 62:1444–1450. [DOI] [PubMed] [Google Scholar]
- 98. Rosso SM, Kamphorst W, De Graaf B, Willemsen R, Ravid R, Niermeijer MF, Spillantini MG, Heutink P, Van Swieten JC (2001) Familial frontotemporal dementia with ubiquitin‐positive inclusions is linked to chromosome 17q21‐22. Brain 124:1948–1957. [DOI] [PubMed] [Google Scholar]
- 99. Rosso SM, Van Herpen E, Deelen W, Kamphorst W, Severijnen LA, Willemsen R, Ravid R, Niermeijer MF, Dooijes D, Smith MJ, Goedert M, Heutink P, Van Swieten JC (2002) A novel tau mutation, S320F, causes a tauopathy with inclusions similar to those in Pick’s disease. Ann Neurol 51:373–376. [DOI] [PubMed] [Google Scholar]
- 100. Rosso SM, Donker Kaat L, Baks T, Joosse M, De Koning I, Pijnenburg Y, De Jong D, Dooijes D, Kamphorst W, Ravid R, Niermeijer MF, Verheij F, Kremer HP, Scheltens P, Van Duijn CM, Heutink P, Van Swieten JC (2003) Frontotemporal dementia in the Netherlands: patient characteristics and prevalence estimates from a population‐based study. Brain 126:2016–2022. [DOI] [PubMed] [Google Scholar]
- 101. Rosso SM, Van Herpen E, Pijnenburg YA, Schoonenboom NS, Scheltens P, Heutink P, Van Swieten JC (2003) Total tau and phosphorylated tau 181 levels in the cerebrospinal fluid of patients with frontotemporal dementia due to P301L and G272V tau mutations. Arch Neurol 60:1209–1213. [DOI] [PubMed] [Google Scholar]
- 102. Saito Y, Geyer A, Sasaki R, Kuzuhara S, Nanba E, Miyasaka T, Suzuki K, Murayama S (2002) Early‐onset, rapidly progressive familial tauopathy with R406W mutation. Neurology 58:811–813. [DOI] [PubMed] [Google Scholar]
- 103. Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, Brandner S, Brun A, Rossor MN, Gade A, Johannsen P, Sorensen SA, Gydesen S, Fisher EM, Collinge J (2005) Mutations in the endosomal ESCRTIII‐complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37:806–808. [DOI] [PubMed] [Google Scholar]
- 104. Sperfeld AD, Collatz MB, Baier H, Palmbach M, Storch A, Schwarz J, Tatsch K, Reske S, Joosse M, Heutink P, Ludolph AC (1999) FTDP‐17: an early‐onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann Neurol 46:708–715. [DOI] [PubMed] [Google Scholar]
- 105. Spillantini MG, Crowther RA, Goedert M (1996) Comparison of the neurofibrillary pathology in Alzheimer’s disease and familial presenile dementia with tangles. Acta Neuropathol (Berl) 92:42–48. [DOI] [PubMed] [Google Scholar]
- 106. Spillantini MG, Goedert M, Crowther RA, Murrell JR, Farlow MR, Ghetti B (1997) Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci USA 94:4113–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Spillantini MG, Bird TD, Ghetti B (1998) Frontotemporal dementia and parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol 8:387–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Spillantini MG, Crowther RA, Kamphorst W, Heutink P, Van Swieten J (1998) Tau pathology in two Dutch families with mutations in the microtubule‐binding region of tau. Am J Pathol 153:1359–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 95:7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Spillantini MG, Yoshida H, Rizzini C, Lantos PL, Khan N, Rossor MN, Goedert M, Brown J (2000) A novel tau mutation (N296N) in familial dementia with swollen achromatic neurons and corticobasal inclusion bodies. Ann Neurol 48:939–943. [DOI] [PubMed] [Google Scholar]
- 111. Spina S, Murrell JR, Yoshida H, Ghetti B, Bermingham N, Sweeney B, Dlouhy SR, Crowther RA, Goedert M, Keohane C (2006) The novel Tau mutation G335S: clinical, neuropathological and molecular characterization. Acta Neuropathol (Berl):2006 Dec 22 [e‐pub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 112. Stanford PM, Halliday GM, Brooks WS, Kwok JB, Storey CE, Creasey H, Morris JG, Fulham MJ, Schofield PR (2000) Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: expansion of the disease phenotype caused by tau gene mutations. Brain 123:880–893. [DOI] [PubMed] [Google Scholar]
- 113. Stanford PM, Shepherd CE, Halliday GM, Brooks WS, Schofield PW, Brodaty H, Martins RN, Kwok JB, Schofield PR (2003) Mutations in the tau gene that cause an increase in three repeat tau and frontotemporal dementia. Brain 126:814–826. [DOI] [PubMed] [Google Scholar]
- 114. Stanford PM, Brooks WS, Teber ET, Hallupp M, McLean C, Halliday GM, Martins RN, Kwok JB, Schofield PR (2004) Frequency of tau mutations in familial and sporadic frontotemporal dementia and other tauopathies. J Neurol 251:1098–1104. [DOI] [PubMed] [Google Scholar]
- 115. Sumi SM, Bird TD, Nochlin D, Raskind MA (1992) Familial presenile dementia with psychosis associated with cortical neurofibrillary tangles and degeneration of the amygdala. Neurology 42:120–127. [DOI] [PubMed] [Google Scholar]
- 116. Van Swieten JC, Stevens M, Rosso SM, Rizzu P, Joosse M, De Koning I, Kamphorst W, Ravid R, Spillantini MG, Niermeijer MF, Heutink P (1999) Phenotypic variation in hereditary frontotemporal dementia with tau mutations. Ann Neurol 46:617–626. [DOI] [PubMed] [Google Scholar]
- 117. Van Swieten JC, Bronner IF, Azmani A, Severijnen LA, Kamphorst W, Ravid R, Rizzu P, Willemsen R, Heutink P (2007) The ΔK280 mutation in MAP tau favours exon 10 skipping in vivo. J Neuropathol Exp Neurol 66:17–25. [DOI] [PubMed] [Google Scholar]
- 118. Tanaka R, Kobayashi T, Motoi Y, Anno M, Mizuno Y, Mori H (2000) A case of frontotemporal dementia with tau P301L mutation in the Far East. J Neurol 247:705–707. [DOI] [PubMed] [Google Scholar]
- 119. Tsuboi Y, Baker M, Hutton ML, Uitti RJ, Rascol O, Delisle MB, Soulages X, Murrell JR, Ghetti B, Yasuda M, Komure O, Kuno S, Arima K, Sunohara N, Kobayashi T, Mizuno Y, Wszolek ZK (2002) Clinical and genetic studies of families with the tau N279K mutation (FTDP‐17). Neurology 59:1791–1793. [DOI] [PubMed] [Google Scholar]
- 120. Urakami K, Wada K, Arai H, Sasaki H, Kanai M, Shoji M, Ishizu H, Kashihara K, Yamamoto M, Tsuchiya‐Ikemoto K, Morimatsu M, Takashima H, Nakagawa M, Kurokawa K, Maruyama H, Kaseda Y, Nakamura S, Hasegawa K, Oono H, Hikasa C, Ikeda K, Yamagata K, Wakutani Y, Takeshima T, Nakashima K (2001) Diagnostic significance of tau protein in cerebrospinal fluid from patients with corticobasal degeneration or progressive supranuclear palsy. J Neurol Sci 183:95–98. [DOI] [PubMed] [Google Scholar]
- 121. Vance C, Al‐Chalabi A, Ruddy D, Smith BN, Hu X, Sreedharan J, Siddique T, Schelhaas HJ, Kusters B, Troost D, Baas F, De Jong V, Shaw CE (2006) Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2‐21.3. Brain 129:868–876. [DOI] [PubMed] [Google Scholar]
- 122. Varani L, Hasegawa M, Spillantini MG, Smith MJ, Murrell JR, Ghetti B, Klug A, Goedert M, Varani G (1999) Structure of tau exon 10 splicing regulatory element RNA and destabilization by mutations of frontotemporal dementia and parkinsonism linked to chromosome 17. Proc Natl Acad Sci USA 96:8229–8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Walker RH, Friedman J, Wiener J, Hobler R, Gwinn‐Hardy K, Adam A, DeWolfe J, Gibbs R, Baker M, Farrer M, Hutton M, Hardy J (2002) A family with a tau P301L mutation presenting with parkinsonism. Parkinsonism Relat Disord 9:121–123. [DOI] [PubMed] [Google Scholar]
- 124. Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin‐containing protein. Nat Genet 36:377–381. [DOI] [PubMed] [Google Scholar]
- 125. Wilhelmsen KC, Lynch T, Pavlou E, Higgins M, Nygaard TG (1994) Localization of disinhibition‐dementia‐parkinsonism‐amyotrophy complex to 17q21‐22. Am J Hum Genet 55:1159–1165. [PMC free article] [PubMed] [Google Scholar]
- 126. Wszolek ZK, Pfeiffer RF, Bhatt MH, Schelper RL, Cordes M, Snow BJ, Rodnitzky RL, Wolters EC, Arwert F, Calne DB (1992) Rapidly progressive autosomal dominant parkinsonism and dementia with pallido‐ponto‐nigral degeneration. Ann Neurol 32:312–320. [DOI] [PubMed] [Google Scholar]
- 127. Yancopoulou D, Crowther RA, Chakrabarti L, Gydesen S, Brown JM, Spillantini MG (2003) Tau protein in frontotemporal dementia linked to chromosome 3 (FTD‐3). J Neuropathol Exp Neurol 62:878–882. [DOI] [PubMed] [Google Scholar]
- 128. Yasuda M, Kawamata T, Komure O, Kuno S, D’Souza I, Poorkaj P, Kawai J, Tanimukai S, Yamamoto Y, Hasegawa H, Sasahara M, Hazama F, Schellenberg GD, Tanaka C (1999) A mutation in the microtubule‐associated protein tau in pallido‐nigro‐luysian degeneration. Neurology 53:864–868. [DOI] [PubMed] [Google Scholar]
- 129. Yasuda M, Takamatsu J, D’Souza I, Crowther RA, Kawamata T, Hasegawa M, Hasegawa H, Spillantini MG, Tanimukai S, Poorkaj P, Varani L, Varani G, Iwatsubo T, Goedert M, Schellenberg DG, Tanaka C (2000) A novel mutation at posititon +12 in the intron following exon 10 of the tau gene in familial frontotemporal dementia (FTD‐Kumamoto). Ann Neurol 47:422–429. [PubMed] [Google Scholar]
- 130. Yoshida H, Crowther RA, Goedert M (2002) Functional effects of tau gene mutations deltaN296 and N296H. J Neurochem 80:548–551. [DOI] [PubMed] [Google Scholar]
- 131. Van Der Zee J, Rademakers R, Engelborghs S, Gijselinck I, Bogaerts V, Vandenberghe R, Santens P, Caekebeke J, De Pooter T, Peeters K, Lubke U, Van Den Broeck M, Martin JJ, Cruts M, De Deyn PP, Van Broeckhoven C, Dermaut B (2006) A Belgian ancestral haplotype harbours a highly prevalent mutation for 17q21‐linked tau‐negative FTLD. Brain 129:841–852. [DOI] [PubMed] [Google Scholar]
- 132. Zekanowski C, Peplonska B, Styczynska M, Gustaw K, Kuznicki J, Barcikowska M (2003) Mutation screening of the MAPT and STH genes in Polish patients with clinically diagnosed frontotemporal dementia. Dement Geriatr Cogn Disord 16:126–131. [DOI] [PubMed] [Google Scholar]