

Abstract
To evaluate the usefulness of neuronal intranuclear inclusions and neuropil inclusions for the pathological assessment of Huntington’s disease (HD), their presence in neocortex was assessed by ubiquitin and N‐terminal huntingtin immunohistochemistry in a consecutive series of 195 autopsy brains of individuals with a positive or tentative clinical diagnosis of, or at risk for, HD. The findings were correlated with striatal pathology (n = 190), CAG repeat length (n = 85) and original pathological diagnosis (n = 186). The antibodies detected both these inclusions in 181 patients with HD pathology ≥ Vonsattel et al’s grade I, five patients lacking striatal tissue for review, and two at‐risk individuals with grade 0 and grade I HD pathology, respectively. One patient with HD‐like pathology and two patients and four at‐risk individuals without HD pathology lacked HD inclusions. In the genetically analyzed cases, the inclusions were exclusively and consistently observed in association with repeat expansion [(CAG)n ≥ 39, n = 81]. Thirteen inclusion‐positive cases, including the grade 0 at‐risk individual, had a false negative original pathological diagnosis of HD and four had an unjustly questionable diagnosis. A false positive diagnosis was made in the inclusion‐negative case with HD‐like pathology. These results indicate that immunohistochemical analysis for HD inclusions facilitates the pathological evaluation of HD and enhances its accuracy.
INTRODUCTION
Huntington’s disease (HD) is an autosomal dominant progressive neurodegenerative disorder characterized by involuntary movements, cognitive decline and behavioral disturbances.
CAG repeat analysis is the gold standard of the diagnosis of HD. If genetic analysis is not feasible, the diagnosis needs to be based on clinical features, family history and pathological confirmation (41, 46).
Brain samples of a consecutive series of 203 subjects autopsied for pathological assessment of HD are available in the Leiden HD pathology archives. DNA analysis could be performed in 85 of these cases and revealed discrepant genetic and pathological diagnoses in four. Such diagnostic discrepancies have also been reported by others (35, 36, 51). Evidently, the pathological evaluation of HD is not invariably straightforward.
Recently, neuronal intranuclear inclusions (NII) and inclusions in neuronal processes (neuropil inclusions) (NI) have been observed in the brains of HD transgenic (Tg) mice and in human HD brain. These inclusions are detectable by immunohistochemistry using antibodies to the N‐terminus of huntingtin, that is, the HD gene product, or ubiquitin (9, 10, 13, 21, 38). HD inclusions involve the neocortex of the human HD brain and are also present in other regions, such as the caudate, putamen and allocortex (3, 13, 26). Neocortical NII density correlates with CAG repeat length (3, 13).
The aim of this study was to evaluate the usefulness of HD inclusions for the pathological assessment of HD. Therefore, the presence of NII and NI was assessed in the neocortex of 195 cases of our archives using ubiquitin and N‐terminal huntingtin immunohistochemistry. The findings were correlated with striatal pathology graded according to the scheme of Vonsattel et al (33, 49) in 190 cases. CAG repeat lengths were available in 85 cases, and an original pathological diagnosis was on record in 186 cases.
MATERIAL AND METHODS
Between 1954 and April 2004, brain samples of 203 individuals either with a positive or tentative clinical diagnosis of, or at risk for, HD were collected in the Leiden HD pathology archives. This number comprises the majority of the subjects autopsied for pathological assessment of HD in the Netherlands during that time. Informed consent to brain autopsy and use of the tissues for research purposes was obtained from the patients and/or their relatives. Clinical data and data on the family history were recorded from the medical histories made available to the Leiden HD pathology archives. A family history was considered positive when at least one other pathologically or genetically established patient with HD was known in the family. A family history was considered negative when no other siblings had any signs or symptoms of HD, and both parents were alive and healthy or lived without neurological or psychiatric disorders over the age of 65 years. A family history was considered suspect when one parent had an ambiguous history or had died before the age of 65 years, or when the family history disclosed a neurological (parkinsonism) or psychiatric disorder, or suicide. If no information could not be obtained, the family history was considered unknown (41).
The brain samples were fixed in 10% buffered formalin and/or frozen. Fixation duration varied from hours to years. Fixed brain samples were cut according to protocol and tissue blocks were embedded in paraffin. Hematoxylin and eosin and Luxol fast blue staining were performed routinely. For immunohistochemistry, 5‐µm‐thick tissue sections of neocortex were incubated overnight with the primary antibodies, including a monoclonal antibody to ubiquitin (Chemicon, Temecula, CA) (1:2000) and antiserum H7 (26) (1:1000) and monoclonal antibody 5374 (Chemicon) (1:50), both directed to the N‐terminus of huntingtin, followed by either biotinylated rabbit antimouse or swine antirabbit immunoglobulin (Dako, Glostrup, Denmark), Vectastain ABC Elite kit (Vector, Burlingame, CA) and 3,3′‐diaminobenzidine/H2O2 solution. For immunostaining with the N‐terminal huntingtin antibodies, sections were pretreated by boiling in citrate buffer (pH 6.0) for 20 minutes. If tissue specimens were not available, immunohistochemistry was performed on archival hematoxylin and eosin‐stained sections decolorized in 4% HCL and 96% alcohol. In two selected cases, sections of the striatum, including the tail of the caudate, were also processed for ubiquitin and N‐terminal huntingtin immunohistochemistry.
If relevant, the pathological assessment also involved brain areas other than the neocortex and striatum and included as well modified Bielschowsky staining and/or immunohistochemistry as described above using monoclonal antibody AT8 against phosphorylated Ser‐202 and Threonin‐205 of tau protein (Innogenetics, Ghent, Belgium) (1:50), monoclonal antibody TAU‐2 (Sigma, St Louis, MO) (1:1000), monoclonal antibody 6F/3D against amyloid β protein (DAKO) (1:10) after pretreatment with trypsin for 30 minutes followed by formic acid 85% for 60 minutes and/or a monoclonal antibody against α‐synuclein (Zymed, San Francisco, CA) (1:100) after pretreatment as described above for the N‐terminal huntingtin antibodies.
Assessment of the presence of NII and NI and neuropathological examination, including grading of disease severity according to the scheme of Vonsattel et al (49), revised in 1994 to include at‐risk cases in addition to symptomatic cases (33), were undertaken by one of us (MM‐S). The cases were assessed as grades 0–IV or as ungraded, the latter allowing for a confident pathological diagnosis of HD, but with macroscopic descriptions and/or tissue sampling insufficient to reliably grade disease severity. In all cases, tissue assessments were carried out blind to the clinical and family history, genetic status and original neuropathological diagnosis. Grading and assessment of the presence of neocortical NII and NI were performed independently of each other.
For CAG repeat analysis, DNA was isolated from stored antemortal peripheral blood samples using the PUREGENETM nucleic acid purification chemistries and the AUTOPURETM Instrument (Gentra Systems, Minneapolis, MN) or from frozen or formalin‐fixed (one case) brain tissue using the QIAamp® DNA Mini Kit (QIAGEN, Venlo, the Netherlands) according to the manufacturer’s instructions. Formalin‐fixed brain tissue (25 mg samples) was prewashed with phosphate buffer solution. Polymerase chain reaction (PCR) amplification of the CAG repeat was performed according to standard procedures. The following primers were used: HD1‐FAM: 5′‐ATGAAGGCCTTCGAG TCCCTCAAGTCCTTC‐3′; HD2: 5′‐AA ACTCACGG TCGGT GCAGCGGCTC CTCAG‐3′; HD3: 5′‐GGCGGTGGC GGCTGTTG CTGCTGC‐3′. HD1 and HD2 were used to amplify the CAG repeat including the CCG repeat, and HD1 and HD3 to amplify the CAG repeat alone. PCR products were electrophoresed on the ABI PRISMTM 310 Genetic Analyzer (Applied Biosystems, Foster City, CA) in the GeneScan Analysis mode. Data were processed with GeneScan AnalysisTM software (Applied Biosystems) and downstream analysis was performed in Genotyper Fragment AnalysisTM software (Applied Biosystems). The repeat contains 6–26 CAGs in normal, stable alleles; 27–35 CAGs in large normal, but expandable alleles; 36–39 CAGs in expanded alleles with reduced penetrance; and ≥40 CAGs in fully penetrant alleles (45).
RESULTS
HD NII/NI (n = 195).
Tissue specimens and/or archival paraffin sections of the neocortex were available from 195/203 subjects. The antibody against ubiquitin and both N‐terminal huntingtin antibodies detected NII as well as NI in 188 of these 195 cases (Figure 1). HD inclusions were tau‐ and α‐synuclein‐negative. Neither HD NII nor NI were observed on the remaining seven brains (Table 1).
Figure 1.
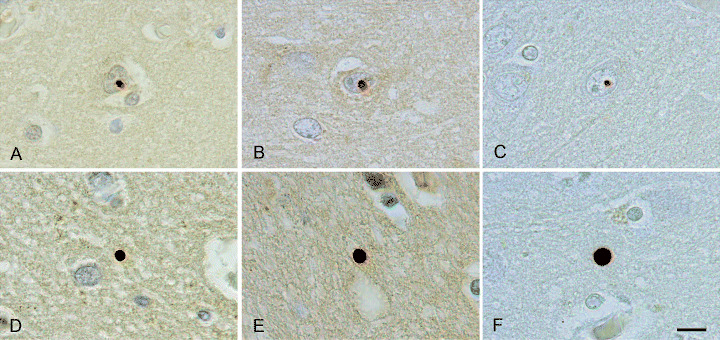
Neuronal intranuclear inclusions (A–C) and neuropil inclusions (D–F) in the neocortex of the brains of two at‐risk individuals with grade I (A,B,D,E) and grade 0 (C,F) HD pathology, respectively. Immunostaining was performed with an antibody against ubiquitin (A,D) and antiserum H7 (B,E) and antibody 5374 (C,F) both directed to the N‐terminus of huntingtin. Scale bar (A–F) 10 µm.
Table 1.
Clinical and genetic data of 195 cases with immunohistochemical analysis for Huntington’s disease (HD) inclusions.
| HD inclusions† | Clinical diagnosis | Family history | (CAG)n | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HD | HD? | At‐risk | +/S | − | U/NR | ≥39 | ≤26 | ND | ||
| Positive | 188‡ | 175 | 11 | 2 | 166 | 7§ | 15 | 81 | 0 | 107 |
| Negative | 7¶ | 0 | 3 | 4 | 6 | 0 | 1 | 0 | 4 | 3 |
ubiquitinated N‐terminal huntingtin‐positive neuronal intranuclear and neuropil inclusions; + = positive; − = negative; S = suspect; U = unknown; NR = not reported; ND = not determined.
102 males and 86 females, age at death 11–88 years (mean 57.4 years).
Years after autopsy, DNA was obtained from the parents of one of these cases (case 9, Table 3). The father appeared to have an intermediate CAG repeat length.
Two males and five females, age at death 55–90 years (mean 67.8 years).
HD inclusions and clinical diagnosis (n = 195).
A positive or tentative clinical diagnosis of HD was made in 186/188 inclusion‐positive cases, supported in the vast majority of cases by a positive or suspect family history. The remaining two cases with HD inclusions were at risk. Of the seven inclusion‐negative cases, three had a tentative clinical diagnosis of HD and four were at risk of inheriting the disease (Table 1).
HD inclusions and retrospective HD pathology assessment (n = 190/195).
In 190 cases, including 183 with and seven without neocortical HD inclusions, striatal tissue was available for review. Pathological changes of HD ≥ grade I were observed in 182/183 inclusion‐positive cases. The remaining case did not show HD pathology (grade 0). Of the seven inclusion‐negative cases, six were free of HD pathology, whereas one case showed HD‐like pathology (Table 2).
Table 2.
Retrospective and original pathological diagnosis of Huntington’s disease (HD) in 195 cases with immunohistochemical analysis for HD inclusions.
CAG repeat length and HD inclusions (n = 85/195)/retrospective HD pathology assessment (n = 84/190).
In 81 inclusion‐positive and four inclusion‐negative cases, CAG repeat analysis could be performed. All of the 81 inclusion‐positive cases had expanded repeats [five (CAG)n 39; 65 (CAG)n 40–49; nine (CAG)n 50–59; two (CAG)n 80–89][range (CAG)n 39–86]. HD pathology assessment was possible in 80 of these cases and revealed pathological changes of the disease ≥ grade II. The repeats of the four inclusion‐negative cases were within the normal range. These cases did not show HD pathology (Table 2).
Retrospective and original pathological diagnosis (n = 184/190).
A record of the original pathological assessment was available in 184/190 cases retrospectively evaluated for HD pathology. Of these, 177 cases were inclusion‐positive and seven were inclusion‐negative.
Discrepant retrospective and original pathological diagnosis (n = 18/184)
Inclusion‐positive cases.
Thirteen of 177 inclusion‐positive cases, including one of the two at‐risk individuals with HD inclusions, had received an original pathological diagnosis negative for HD (Table 3). The latter individual was the only inclusion‐positive case of our series not showing HD pathology on review. She had a genetically confirmed family history of HD and died suddenly from pulmonary embolism at age 39. NI were readily detectable in the neocortex of her brain, but NII were scarce. The caudate, including the tail, and the putamen showed NII and occasional NI (1, 2). Attempts to extract DNA for CAG repeat analysis from formalin‐fixed brain tissue of this case failed repeatedly both in our hands and elsewhere (D. Geschwind, pers. comm.). In another four inclusion‐positive cases, the original diagnosis was questionable of HD (Table 3).
Table 3.
Pathological, genetic, and clinical data of 18 cases with discrepant retrospective and original pathological diagnoses of Huntington’s disease (HD).
| Number/age/sex | Brain weight (g) | Pathological assessment | (CAG)n | Clinical diagnosis HD/chorea | Family history (41) | |||
|---|---|---|---|---|---|---|---|---|
| Original | Revised | |||||||
| HD | NII/NI | HD/Grade | Additional/other diagnoses | |||||
| 1/39/F | ND | − | +/+ | +/0† | ND | ND | at‐risk | + |
| 2/47/M | ND | − | +/+ | +/I | ND | ND | +?/− | S |
| 3/71/M | 1270 | − | +/+ | +/I | Lac infarcts | ND | +?/+? | − |
| 4/64/F | 1070 | − | +/+ | +/II | Str ascl | ND | + | S |
| 5/66/F | 1340 | − | +/+ | +/II | Men‐enc | 17–43 | + | S |
| 6/70/F | 1210 | − | +/+ | +/II | ND | ND | + | U |
| 7/72/F | 1285 | − | +/+ | +/II | Str ascl | ND | + | S |
| 8/74/F | ND | − | +/+ | +/II | Str ascl | ND | + | S |
| 9/50/M | 1260 | − | +/+ | +/III | ND | 27–45 | + | −‡ |
| 10/51/F | 1100 | − | +/+ | +/III | ND | 15–43 | + | S |
| 11/73/F | 1030 | − | +/+ | +/III | AD§ | 18–42 | + | S |
| 12/83/F | ND | − | +/+ | +/III | AD¶, Str ascl | ND | + | U |
| 13/80/F | ND | − | +/+ | +/ungraded | ND | ND | +? | S |
| 14/36/M | ND | +? | +/+ | +/II | ND | ND | + | + |
| 15/70/M | ND | +? | +/+ | +/II | Str ascl | ND | +?/+ | S |
| 16/57/M | ND | +? | +/+ | +/III | Caudate infarct†† | ND | +?/+ | S |
| 17/61/M | 980 | +? | +/+ | +/IV† | Old contusion | ND | +?/+ | S |
| 18/45/F | ND | + | −/− | −/HD‐like‡‡ | ChAc | ND | +?/+ | S |
Reviewed by J‐P Vonsattel (NY, USA).
Years after this patient’s death, his father was shown to have an intermediate CAG repeat length.
CERAD infrequent, Braak I/II (47).
CERAD infrequent, Braak III/IV (47).
Considered related to an arteriovenous malformation.
Creutzfeldt‐Jakob disease excluded by G Janssen (Utrecht, The Netherlands).
Abbreviations and symbols: NII/NI = ubiquitinated N‐terminal huntingtin‐positive neuronal intranuclear inclusions/neuropil inclusions; Grade = grade according to Vonsattel et al (33, 49); Chorea = choreiform movements (indicated if relevant); F = female; − = negative; + = positive; M = male; +? = questionable; Lac = lacunar; Str ascl = striatal arteriosclerosis; Men‐enc = old meningo‐encephalitis; S = suspect; U = unknown; AD = Alzheimer’s disease; ChAc = chorea‐acanthocytosis; CERAD = Consortium to Establish a Registry for AD; ND = not determined.
Figure 2.
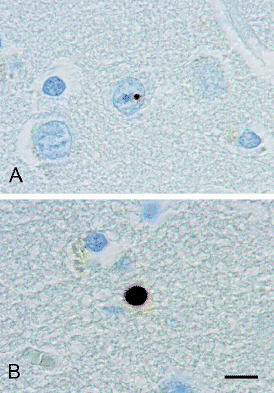
Neuronal intranuclear inclusion (A) and neuropil inclusion (B) in the tail of the caudate of the brain of the at‐risk individual with grade 0 HD pathology. Immunostaining was performed with antibody 5374 directed to the N‐terminus of huntingtin. Scale bar (A,B) 10 µm.
Inclusion‐negative cases.
One of the seven cases without HD inclusions had an original pathological diagnosis positive for HD. On review, a diagnosis of autosomal recessive chorea‐acanthocytosis (ChAc) was made. This subject was the product of a consanguineous marriage and developed involuntary smacking movements of the tongue, difficulties in swallowing and a tendency to fall because of stretch spasms of one leg at age 38. In addition, the clinical history recorded choreiform movements, dysarthria, behavioral disturbances, intellectual decline and elevated serum creatine kinase levels. No mention was made of acanthocytes in the peripheral blood smear. She died at age 45. Her brain showed HD‐like pathology with almost complete loss of small neurons and fierce astrocytic gliosis in the neostriatum. These changes were relatively less severe rostroventrally and in the dorsolateral part of the caudal putamen and the caudate tail. Oligodendrocytes were more numerous than may be usual in high‐grade HD neostriatum (49). Immunohistochemical analysis for prion disease was negative. Recently, one of her children was found to have a heterozygous CHAC gene mutation, very likely causing aberrant protein production. The pathological, genetic and clinical data of the inclusion‐negative cases are summarized in Table 4.
Table 4.
Pathological, genetic and clinical data of seven cases without Huntington’s disease (HD) neuronal intranuclear and neuropil inclusions.
| Number/age/sex | Brain weight (g) | Pathological assessment | (CAG)n | Clinical diagnosis HD/chorea | Family history (41) | ||
|---|---|---|---|---|---|---|---|
| Original | Revised | ||||||
| HD | HD/grade | Additional/other diagnoses | |||||
| 1/55/F | — | − | −/0 | Pontine hem | 20–26 | at risk | S |
| 2/62/M | 1250 | − | −/0 | Infarct right caudate | — | at risk | S |
| 3/64/M | 1360 | − | −/0 | Cong malformation† | 15–18 | at risk | + |
| 4/90/F | — | − | −/0 | AD‡ | 19–24 | at risk | + |
| 5/58/F | 1000 | − | −/0 | AD§, brain calcinosis | — | +?¶/− | S |
| 6/74/F | — | − | −/0 | PSP symbol | 16–17 | +?††/+ | U |
| 7/45/F | — | + | −/HD‐like‡‡ | ChAc | — | +?/+ | S |
Partial agenesis of the corpus callosum [reviewed by H Vinters (LA, USA)].
CERAD moderate, Braak III/IV (47).
CERAD moderate, Braak V/VI (47).
Differential diagnosis of Parkinson’s disease symbol (25).
Differential diagnosis of PSP.
Creutzfeldt‐Jakob disease excluded by G Janssen (Utrecht, The Netherlands).
Abbreviations and symbols: Chorea = choreiform movements (indicated if relevant); F = female; M = male; − = negative; Hem = hemorrhage; S = suspect; Cong = congenital; + = positive; AD = Alzheimer’s disease; +? = questionable; PSP = progressive supranuclear palsy; U = unknown; ChAc = chorea‐acanthocytosis; CERAD = Consortium to Establish a Registry for AD.
No original pathological diagnosis on record and/or no striatal tissue for HD pathology review (n = 11).
A record of the initial evaluation was not available in 6/182 inclusion‐positive cases showing histopathological changes of HD on review. One of these was the other of the two at‐risk individuals with HD inclusions. The family history of this individual was suspect at the time of autopsy and confirmed both pathologically and genetically afterward. He did not show involuntary movements until the time of his death from pneumonia at age 49, but he did have “the psychic disturbances” of the disease according to his family. His brain showed grade I HD pathology. NII and NI were readily detectable in the neocortex (Figure 1) and neostriatum, including the tail of the caudate.
Striatal tissue for HD pathology review was not available in 5/195 inclusion‐positive cases. Two of these were patients with an original pathological diagnosis of HD and three were patients without a record of the initial assessment. DNA analysis could be performed in one of the latter and revealed an HD CAG repeat of 50.
Other findings.
Arteriosclerosis‐related ischemic changes were observed in the striatum in 5/17 cases with a false negative or unjustly questionable original pathological diagnosis of HD. Two cases, including one of the latter, displayed pathological changes of Alzheimer’s disease. Evidence of an old meningo‐encephalitis was observed in another case. Two brains harbored cerebral infarctions, also affecting the caudate in one case, and one brain had remnants of a cerebral contusion encroaching on the neostriatum (Table 3).
DISCUSSION
This study shows the sensitivity of HD inclusions in the confirmation of the diagnosis of HD. Both NII and NI were observed in the neocortex of the brains of 81/81 symptomatic subjects with expanded repeats. Some studies have reported a lack of either NI or NII or both in the neocortex of genetically established patients with HD (3, 7). However, our analysis concerns a large number of patients and the finding of a consistent association of HD inclusions and repeat expansion in these cases is in line with the results of several other reports (13, 17, 19, 40). None of the above studies included patients with grade 0 HD pathology.
HD inclusions have also been found in the brains of asymptomatic HD gene carriers (13, 16, 19, 37). Two such individuals were reported to harbor NII as well as NI in the neocortex. They had 42 and 48 repeats, respectively, and were 32 years old. Both had grade I HD pathology (13, 19, 37). The caudate tail was examined in another three cases and contained NII but not NI. One of these cases was a 44‐year‐old individual with 37 repeats, who died more than 30 years before the expected clinical onset (16). Both NII and NI were observed in the neocortex as well as the caudate tail of two at‐risk cases of the present series. The brain of one of these individuals did not show histopathological changes of HD, but the presence of HD inclusions identified her as a possible HD gene carrier.
In HD Tg mice, the appearance of HD inclusions precedes both the onset of disease symptoms and the development of neurodegeneration (9, 10, 48). This sequence of events seems to be the case in human HD brain as well (13, 16, 19, 37, this study). However, when or where HD NII and NI, respectively, first appear in a particular brain remains a question. In addition, although clinical disease has not been reported in individuals with less than 36 CAG repeats, the repeat threshold for immunohistochemically detectable inclusion formation is not known. So far, the smallest repeat size reported to be associated with HD inclusions was found in an asymptomatic subject and amounted to 37 repeats (16). All other findings have concerned patients or asymptomatic individuals with CAG repeat sizes of 39 or over (3, 13, 16, 17, 19, 40, this study). Thus, the relation of HD inclusions and CAG repeat length needs further elucidation.
Seventeen inclusion‐positive cases of this series had received an original pathological diagnosis either negative for, or questionable of, HD (Table 3). One of these cases was at risk of inheriting HD. The absence of HD pathology in this individual’s brain precluded a correct diagnosis on conventional examination. Each one of the remaining 16 cases appeared to show pathological changes of the disease on review. However, factors liable to affect the accuracy of the histopathological evaluation of HD, that is, mild pathology, coincidental other neuropathology and atypical clinical features (51), were frequent among these cases (Table 3) and may have influenced the initial assessments. The usefulness of HD inclusion detection in such instances is obvious.
A growing number of neurodegenerative diseases, among others the triplet repeat disorders Huntington’s disease‐like 2 (HDL2), spinocerebellar ataxia (SCA) types 1–3, 7, and 17, and dentatorubral‐pallidoluysian atrophy (DRPLA), is associated with ubiquitinated NII (14, 18, 22, 24, 27, 28, 39, 44). HDL2 is nearly identical to HD both clinically and pathologically, including the presence of ubiquitin‐positive NII in the neocortex. The disease is generally rare but almost as common as HD in black South Africans (28). The SCAs and DRPLA may overlap clinically with HD. Of these, particularly SCA17 and DRPLA may be associated with neostriatal neuronal loss and/or gliosis and also show ubiquitinated NII in the neocortex (1, 2, 3, 5, 15, 20, 42). N‐terminal huntingtin immunohistochemistry may serve to distinguish HD NII from NII associated with other disorders. This also holds for the differentiation of neocortical HD NI from ubiquitin‐positive, tau‐negative neurites accumulating in this region in other conditions (4, 6, 11, 12, 27, 29, 32, 34). Charles et al (8) have demonstrated α‐synuclein immunoreactivity of NII and NI in HD patients and Tg mice. Observations by others in a cell culture system support this finding (50). However, HD inclusions did not immunostain for α‐synuclein in the present study.
One case in the present study had an original pathological diagnosis positive for HD but no HD inclusions. On review, the diagnosis was amended to recessive ChAc. This condition shares clinical and pathological features with HD, including neostriatal neuronal loss and gliosis of the striatum (30, 36, 43). The absence of ubiquitinated NII and NI also distinguishes an HD phenocopy with HD‐like pathology, proposed as HDL4, from HD (35). This disorder does not have a specific gene or mutation identified, nor does another HD phenocopy, HDL3. Bilateral caudate atrophy is seen on brain imaging in the latter, but there are no reports on the neuropathology of HDL3 (23). HD‐like pathology is also a feature of the phenocopy HDL1. The association of HDL1 with an insertional mutation in the prion protein gene further illustrates the genetic heterogeneity of clinically diagnosed HD (31).
In summary, immunohistochemistry for the detection of HD inclusions may serve to support a pathological diagnosis of HD irrespective of the family history, clinical symptoms or the presence of histopathological changes of the disease. The absence of HD inclusions should caution the pathologist to make a diagnosis of HD. The pathological evaluation of HD should include immunohistochemical analysis for HD inclusions, which is of particular importance in the case DNA analysis is not feasible.
ACKNOWLEDGMENTS
We are greatly indebted to: the patients and their families for their consent to brain autopsy; Maria Vegter‐van der Vlis for providing genealogical data and Frans Prins for preparing the photographs; the referring physicians and pathologists, particularly Dirk Troost (Amsterdam, The Netherlands), Rob de Vos (Enschede, The Netherlands) and Pieter Wesseling (Nijmegen, The Netherlands), who provided tissue specimens for pathology review; Jean‐Paul Vonsattel (NY, USA) for reviewing two cases of the present series; Harry Vinters (LA, USA) for reviewing the neuropathology in the at‐risk individual with congenital brain malformation; Gerard Janssen (Utrecht, The Netherlands) for performing screening for Creutzfeldt‐Jakob disease in the ChAc case; Anthony Monaco and Carol Dobson‐Stone (Oxford, UK) for CHAC mutational screening in a child of the latter case; and Dan Geschwind (LA, USA) for his attempts to isolate DNA from formalin‐fixed brain tissue for CAG repeat analysis. Elly Ippel (Utrecht, The Netherlands) was involved in the counseling of the family of the ChAc patient. We thank Axel Wintzen for his critical comments on the article.
REFERENCES
- 1. Bauer P, Laccone F, Rolfs A, Wüllner U, Bösch S, Peters H, Liebscher S, Scheible M, Epplen JT, Weber BHF, Holinski‐Feder E, Weinrich‐Schwaiger H, Morris‐Rosendahl DJ, Andrich J, Riess O (2004) Trinucleotide repeat expansion in SCA17/TBP in white patients with Huntington’s disease‐like phenotype. J Med Genet 41:230–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Becher MW, Rubinsztein DC, Leggo J, Wagster MV, Stine OC, Ranen NG, Franz ML, Abbott MH, Sherr M, MacMillan JC, Barron L, Porteous M, Harper PS, Ross CA (1997) Dentatorubral and pallioluysian atrophy (DRPLA). Mov Disord 12:519–530. [DOI] [PubMed] [Google Scholar]
- 3. Becher MW, Kotzuk JA, Sharp AH, Davies SW, Bates GP, Price DL, Ross CA (1998) Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis 4:387–397. [DOI] [PubMed] [Google Scholar]
- 4. Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson’s disease‐related pathology. Cell Tissue Res 318:121–134. [DOI] [PubMed] [Google Scholar]
- 5. Bruni AC, Takahashi Fujigasaki J, Maltecca F, Foncin JF, Servadio A, Casari G, D’Adamo P, Maletta R, Curcio SAM, De Michele G, Filla A, El Hachimi KH, Duyckaerts C (2004) Behavioral disorder, dementia, ataxia, and rigidity in a large family with TATA box‐binding protein mutation. Arch Neurol 61:1314–1320. [DOI] [PubMed] [Google Scholar]
- 6. Cammarata S, Tabaton M (1992) Ubiquitin‐reactive axons have a widespread distribution and are unrelated to prion protein plaques in Creutzfeldt‐Jakob disease. J Neurol Sci 110:32–36. [DOI] [PubMed] [Google Scholar]
- 7. Caramins M, Halliday G, McCusker E, Trent RJ (2003) Genetically confirmed clinical Huntington’s disease with no observable cell loss. J Neurol Neurosurg Psychiatry 74:968–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Charles V, Mezey E, Reddy PH, Dehejia A, Young TA, Polymeropoulos MH, Brownstein MJ, Tagle DA (2000) Alpha‐synuclein immunoreactivity of Huntington polyglutamine aggregates in striatum and cortex of Huntington’s disease patients and transgenic mouse models. Neurosci Lett 289:29–32. [DOI] [PubMed] [Google Scholar]
- 9. Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wankner EE, Mangiarini L, Bates GP (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90:537–548. [DOI] [PubMed] [Google Scholar]
- 10. Davies SW, Turmaine M, Cozens BA, Raza AS, Mahal A, Mangiarini L, Bates GP (1999) From neuronal inclusions to neurodegeneration: neuropathological investigation of a transgenic mouse model of Huntington’s disease. Philos Trans R Soc Lond B Biol Sci 354:971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dickson DW, Wertkin A, Kress Y, Ksiezak‐Reding H, Yen SH (1990) Ubiquitin immunoreactive structures in normal human brains. Distribution and developmental aspects. Lab Invest 63:87–99. [PubMed] [Google Scholar]
- 12. Dickson TC, King CE, McCormack GH, Vickers JC (1999) Neurochemical diversity of dystrophic neurites in the early and late stages of Alzheimer’s disease. Exp Neurol 156:100–110. [DOI] [PubMed] [Google Scholar]
- 13. DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel J‐P, Aronin N (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277:1990–1993. [DOI] [PubMed] [Google Scholar]
- 14. Everett CM, Wood NW (2004) Trinucleotide repeats and neurodegenerative disease. Brain 127:2385–2405. [DOI] [PubMed] [Google Scholar]
- 15. Fujigasaki H, Martin J‐J, De Deyn PP, Camuzat A, Deffond D, Stevanin G, Dermaut B, Van Broeckhoven C, Dürr A, Brice A (2001) CAG repeat expansion in the TATA box‐binding protein gene causes autosomal dominant cerebellar ataxia. Brain 124:1939–1947. [DOI] [PubMed] [Google Scholar]
- 16. Gómez‐Tortosa E, MacDonald ME, Friend JC, Taylor SAM, Weiler LJ, Cupples LA, Srinidhi J, Gusella JF, Bird ED, Vonsattel J‐P, Myers RH (2001) Quantitative neuropathological changes in presymptomatic Huntington’s disease. Ann Neurol 49:29–34. [PubMed] [Google Scholar]
- 17. Gourfinkel‐An I, Cancel G, Duyckaerts C, Faucheux B, Hauw J‐J, Trottier Y, Brice A, Agid Y, Hirsch EC (1998) Neuronal distribution of intranuclear inclusions in Huntington’s disease with adult onset. NeuroReport 9:1823–1826. [DOI] [PubMed] [Google Scholar]
- 18. Greco CM, Berman RF, Martin RM, Tassone F, Schwartz PH, Chang A, Trapp BD, Iwahashi C, Brunberg J, Grigsby J, Hessl D, Becker EJ, Papazian J, Leehey MA, Hagerman RJ, Hagerman PJ (2006) Neuropathology of fragile X‐associated tremor/ataxia syndrome (FXTAS). Brain 129:243–255. [DOI] [PubMed] [Google Scholar]
- 19. Gutekunst C‐A, Li S‐H, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li X‐J (1999) Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J Neurosci 19:2522–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hayashi Y, Kakita A, Yamada M, Koide R, Igarashi S, Takano H, Ikeuchi T, Wakabayashi K, Egawa S, Tsuji S, Takahashi H (1998) Hereditary dentatorubral‐pallidoluysian atrophy: detection of widespread ubiquitinated neuronal and glial intranuclear inclusions in the brain. Acta Neuropathol 96:547–552. [DOI] [PubMed] [Google Scholar]
- 21. Jackson M, Gentleman S, Lennox G, Ward L, Gray T, Randall K, Morrel K, Lowe J (1995) The cortical neuritic pathology of Huntington’s disease. Neuropathol Appl Neurobiol 21:18–26. [DOI] [PubMed] [Google Scholar]
- 22. Kakita A, Oyanagi K, Nagai H, Takahashi H (1997) Eosinophilic intranuclear inclusions in the hippocampal pyramidal neurons of a patient with amyotrophic lateral sclerosis. Acta Neuropathol 93:532–536. [DOI] [PubMed] [Google Scholar]
- 23. Kambouris M, Bohlega S, Al‐Tahan A, Meyer BF (2000) Localization of the gene for a novel autosomal recessive neurodegenerative huntington‐like disorder to 4p15.3. Am J Hum Genet 66: 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Katsuse O, Dickson DW (2005) Ubiquitin immunohistochemistry of frontotemporal lobar degeneration differentiates cases with and without motor neuron disease. Alzheimer Dis Assoc Disord 19:S37–S43. [DOI] [PubMed] [Google Scholar]
- 25. Litvan I, Hauw JJ, Bartko JJ, Lantos PL, Daniel SE, Horoupian DS, McKee A, Dickson D, Bancher C, Tabaton M, Jellinger K, Anderson DW (1996) Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol 55:97–105. [DOI] [PubMed] [Google Scholar]
- 26. Maat‐Schieman MLC, Dorsman JC, Smoor MA, Siesling S, Van Duinen SG, Verschuuren JJGM, Den Dunnen JT, Van Ommen G‐J, Roos RAC (1999) Distribution of inclusions in neuronal nuclei and dystrophic neurites in Huntington disease brain. J Neuropathol Exp Neurol 58:129–137. [DOI] [PubMed] [Google Scholar]
- 27. Mackenzie IR, Baker M, West G, Woulfe J, Qadi N, Gass J, Cannon A, Adamson J, Feldman H, Lindholm C, Melquist S, Pettman R, Dessa Sadovnick A, Dwosh E, Whiteheart SW, Hutton M, Pickering‐Brown SM (2006) A family with tau‐negative frontotemporal dementia and neuronal intranuclear inclusions linked to chromosome 17. Brain 129:853–867. [DOI] [PubMed] [Google Scholar]
- 28. Margolis RL, Rudnicki DD, Holmes SE (2005) Huntington’s disease like‐2: review and update. Acta Neurol Taiwan 14:1–8. [PubMed] [Google Scholar]
- 29. McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez‐Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM‐Y, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova‐Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M (2005) Diagnosis and management of dementia with Lewy bodies. Third report of the DLB consortium. Neurology 65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 30. Meenakshi‐Sundaram S, Arun Kumar MJ, Sridhar R, Rani U, Sundar B (2004) Neuroacanthocytosis misdiagnosed as Huntington’s disease: a case report. J Neurol Sci 219:163–166. [DOI] [PubMed] [Google Scholar]
- 31. Moore RC, Xiang F, Monaghan J, Han D, Zhang Z, Edström L, Anvret M, Prusiner SB (2001) Huntington disease phenocopy is a familial prion disease. Am J Hum Genet 69:1385–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Paviour DC, Lees AJ, Josephs KA, Ozawa T, Ganguly M, Strand C, Godbolt A, Howard RS, Revesz T, Holton JL (2004) Frontotemporal lobar degeneration with ubiquitin‐only‐immunoreactive changes: broadening the clinical picture to include progressive supranuclear palsy. Brain 127:2441–2451. [DOI] [PubMed] [Google Scholar]
- 33. Persichetti F, Srinidhi J, Kanaley L, Ge P, Myers RH, D’ Arrigo K, Barnes GT, MacDonald ME, Vonsattel J‐P, Gusella JF, Bird ED (1994) Huntington’s disease CAG trinucleotide repeats in pathologically confirmed post‐mortem brains. Neurobiol Dis 1:159–166. [DOI] [PubMed] [Google Scholar]
- 34. Piao Y‐S, Wakabayashi K, Kakita A, Yamada M, Hayashi S, Morita T, Ikuta F, Oyanagi K, Takahashi H (2003) Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis: 102 autopsy cases examined between 1962 and 2000. Brain Pathol 12:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Richfield EK, Vonsattel J‐P, MacDonald ME, Sun Z, Deng Y‐PP, Reiner A (2002) Selective loss of striatal preprotachykinin neurons in a phenocopy of Huntington’s disease. Mov Disord 17:327–332. [DOI] [PubMed] [Google Scholar]
- 36. Rinne JO, Daniel SE, Scaravilli F, Pires M, Harding AE, Marsden CD (1994) The neuropathological features of neuroacanthocytosis. Mov Disord 9:297–304. [DOI] [PubMed] [Google Scholar]
- 37. Sapp E, Penney J, Young A, Aronin N, Vonsattel J‐P, DiFiglia M (1999) Axonal transport of N‐terminal huntingtin suggests early pathology of corticostriatal projections in Huntington disease. J Neuropathol Exp Neurol 58:165–173. [DOI] [PubMed] [Google Scholar]
- 38. Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR (1999) Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N‐terminal fragment of huntingtin. Hum Mol Gen 8:397–407. [DOI] [PubMed] [Google Scholar]
- 39. Seilhean D, Takahashi J, El Hachimi KH, Fujigasaki H, Lebre A‐S, Biancalana V, Dür A, Salachas F, Hogenhuis J, De Thé H, Hauw J‐J, Meininger V, Brice A, Duyckaerts C (2004) Amyotrophic lateral sclerosis with neuronal intranuclear protein inclusions. Acta Neuropathol 108:81–87. [DOI] [PubMed] [Google Scholar]
- 40. Sieradzan KA, Mechan AO, Jones L, Wanker EE, Nukina N, Mann DMA (1999) Huntington’s disease intranuclear inclusions contain truncated ubiquitinated huntingtin protein. Exp Neurol 156:92–99. [DOI] [PubMed] [Google Scholar]
- 41. Siesling S, Vegter‐van de Vlis M, Losekoot M, Belfroid RD, Maat‐Kievit JA, Kremer HP, Roos RA (2000) Family history and DNA analysis in patients with suspected Huntington’s disease. J Neurol Neurosurg Psychiatry 69:54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stevanin G, Fujigasaki H, Lebre A‐S, Camuzat A, Jeannequin C, Dodé C, Takahshi J, Sân C, Bellance R, Brice A, Durr A (2003) Huntington’s disease‐like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain 126:1599–1603. [DOI] [PubMed] [Google Scholar]
- 43. Stevenson VL, Hardie RJ (2001) Acanthocytosis and neurological disorders. J Neurol 248:87–94. [DOI] [PubMed] [Google Scholar]
- 44. Takahashi‐Fujigasaki J (2003) Neuronal intranuclear hyaline inclusion disease. Neuropathology 23:351–359. [DOI] [PubMed] [Google Scholar]
- 45. The American College of Medical Genetics/American Society of Human Genetics Huntington Disease Genetic Testing Working Group (1998) Laboratory guidelines for Huntington disease genetic testing. Am J Hum Genet 62:1243–1247. [PMC free article] [PubMed] [Google Scholar]
- 46. The Huntington’s Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72:971–983. [DOI] [PubMed] [Google Scholar]
- 47. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease (1997) Consensus recommedations for the postmortem diagnosis of Alzheimer’s disease. Neurobiol Aging 18:S1–S2. [PubMed] [Google Scholar]
- 48. Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW (2000) Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington’s disease. Proc Natl Acad Sci USA 97:8093–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vonsattel J‐P, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr (1985) Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol 44:559–577. [DOI] [PubMed] [Google Scholar]
- 50. Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE (2001) Accumulation of mutant huntingtin fragments in aggresome‐like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell 12:1393–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xuereb JH, MacMillan JC, Davies P, Harper PS (1996) Neuropathological diagnosis and CAG repeat expansion in Huntington’s disease. J Neurol Neurosurg Psychiatry 60:78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]