

Abstract
We and others have recently demonstrated that cognitive and physical stimulation in form of environmental enrichment reduces cerebral β‐amyloid (Aβ) deposition in transgenic mouse models of Alzheimer’s disease. This effect was independent from amyloid precursor protein (APP) expression or processing and rather a consequence of enhanced clearance of Aβ. However, the detailed mechanisms remain unclear. In the present study, we show that environmental enrichment in TgCRND8 mice (carrying human APPSwedish+Indiana) affect the neurovascular unit by increased angiogenesis and differential regulation of Aβ receptor/transporter molecules, namely up‐regulation of LRP1, ApoE and A2M as well as down‐regulation of RAGE so that brain to blood Aβ clearance is facilitated. These results suggest a hitherto unknown effect of environmental enrichment counteracting the vascular dysfunction in Alzheimer diseased brain.
INTRODUCTION
Epidemiological evidence suggests that education, occupational attainment and participation in leisure activities are protective against the development of Alzheimer’s disease (AD) (17). Until recently, the most common explanation for this effect was that such lifestyle enhances the cognitive reserve and enables the patients to compensate the clinical expression of AD without affecting AD‐related neuropathology (27). We and others have demonstrated that cognitive and physical stimulation in form of environmental enrichment in transgenic mice does interfere with AD neuropathology and results in significant reduction of cerebral β‐amyloid (Aβ) plaques (1, 2, 26) and in a lower extent of amyloid angiopathy (2), without influencing human amyloid precursor protein (APP) transcription/translation or amounts of carboxy‐terminal fragments of APP, making alterations in APP expression or processing unlikely as a cause of reduced Aβ deposition (2, 26). More probably, reduced Aβ burden following environmental enrichment results from altered clearance of Aβ, as enhanced enzymatic activity of Aβ degrading endopeptidase, neprilysin (26) or increased phagocytic activity of microglia in “enriched” brains (2) suggests. However, the detailed mechanism remains obscure.
It is well known that physical stimulation increases cerebral angiogenesis in healthy adult rodents (6, 23) and antagonizes neurovascular dysfunction in diseases such as stroke (15, 19). On the other hand, a growing body of evidence suggests that neurovascular dysfunction in terms of aberrant angiogenesis and faulty clearance of Aβ peptide across the blood–brain barrier (BBB) contributes to neurodegeneration in AD (37). We therefore questioned whether environmental enrichment counteracts the cerebrovascular insufficiency also in AD.
After enriched housing (EH) of TgCRND8 mice (10) from day 30 until the age of 5 months we examined the cerebral expression of angiogenesis‐associated genes and proteins, determined cerebral vessel proliferation and density, and studied alterations of Aβ exchange processes across the BBB by determination of Aβ content in brain and blood plasma and quantification of Aβ‐transporters. We here demonstrate that environmental stimulation leads to increased cerebral angiogenesis in TgCRND8 mice, indicated by up‐regulation of pro‐angiogenic genes and proteins, increased proliferation of vessel endothelial cells, and a tighter vessel network. Up‐regulation of transporters promoting Aβ efflux across the BBB and down‐regulation of molecules reversing this process further support the model that environmental enrichment decreases AD‐related pathology by enhanced Aβ clearance.
MATERIALS AND METHODS
Animals and housing conditions.
We examined 18 female TgCRND8 mice carrying a double‐mutated form of the human APP 695 (APP695), the “Swedish” and “Indiana” mutations, controlled by the Syrian hamster prion promoter, on a hybrid C57BL/6‐C3H/HeJ background. At 30 days of age, animals were transferred to the experimental housing conditions. Nine mice were kept in groups of three or four in standard housing (SH) conditions, while nine were housed in equally composed groups in EH. SH consisted of transparent polycarbonate cages (38 × 22 × 15 cm) including sawdust as bedding material. A wooden scaffolding, a plastic inset and further nesting material supplemented the so‐called enriched “home cages” (Figure 1, EH left side). During the dark phase, EH animals additionally gained access to a second “stimulus cage” (Figure 1, EH right side) that was connected to the home cage by a plexiglass tunnel. The “stimulus cage” contained different objects divided in five categories. (i) Permanently, a gnawing wood and sisal rope were available. Accessorily, objects of the categories, (ii) tunnels, (iii) balls, (iv) soft materials and (v) varied locomotive substrates including wooden ramps and ladders, plastic stairs as well as running‐wheels, were included. Every day, one stimulus object of a daily switching category was exchanged to expose EH mice to novel environmental stimulation. A photoperiod of 12 h light/dark cycle was sustained. All experimental procedures were accepted by the district government of Münster/Germany.
Figure 1.

Animal Housing. Female TgCRND8 mice were transferred at 30 days of age for 120 days into an enriched housing (EH, n = 9), equipped with diverse physically and cognitively stimulating objects. Control mice were kept in standard housing (SH, n = 9).
Tissue preparation.
At 150 days of age mice were decapitated. One brain hemisphere was fixed in 4% buffered formaldehyde for 24 h followed by dehydration and paraffin embedding; the other hemisphere was immediately snap‐frozen in liquid nitrogen. Total RNA and subsequently protein was extracted from the same homogenized tissue of the whole cerebral hemisphere (without cerebellum and brain stem) using TRIzol Reagent (Invitrogen, Karlsruhe, Germany) following manufacturer’s instructions. RNA was DNase treated, cleaned and its quality assessed by Agilent Bioanalyzer 2100 (Agilent Technologies, Inc., Palo Alto, CA, USA). Proteins were dissolved in 1% sodium dodecyl sulfate (SDS). Nine SH vs. nine EH mice were used for morphometrical analysis. For RNA and protein analyses the number of EH mice was nine and the number of SH mice (caused by loss of one pellet) eight. Trunk blood was collected in heparinized capillaries directly after decapitation. Blood samples were centrifuged and blood plasma separated. Because of material limitation blood plasma analyses were performed in five SH and four EH mice.
Angiogenesis profiler PCR arrays.
Expression profiling of 84 genes modulating biological processes of angiogenesis was performed utilizing mouse angiogenesis RT2 profiler polymerase chain reaction (PCR) arrays APM‐024A (SuperArray, Frederick, MD, USA) based on SYBR‐Green real‐time PCR. Therefore, isolated RNA from animals of the same housing condition was pooled, receiving four pools consisting of two biological duplicates termed standard 1 (four animals), standard 2 (four animals), enriched 1 (four animals) and enriched 2 (five animals). cDNA was synthesized from 2 µg RNA per pool using Omniscript RT kit (Qiagen, Hilden, Germany) following manufacturer’s instructions. cDNA quality was determined by conventional Gapdh PCR. SYBR‐Green real‐time PCR and fold‐changes calculation was accomplished according to manufacturer’s instructions: calculated Ct values were averaged for biological duplicates. ΔCt for each gene was calculated by subtraction of averaged Ct of five housekeeping genes from Ct values of angiogenic genes. ΔΔCt for each gene was calculated by subtraction of ΔCt value of the control group from ΔCt value of the experimental group (enriched). Fold‐changes for each gene were determined by exponentiation of 2 with −ΔΔCt. Assays were run in duplicate.
TaqMan assay.
Transcriptional regulation of the genes receptor‐associated protein (Rap), apolipoprotein J [Apoj, also known as clusterin (Clu)], apolipoprotein E (Apoe) and alpha‐2‐macroglobulin (A2m) was determined by TaqMan assay. Therefore, cDNA samples of each mouse were synthesized separately (Omniscript RT kit, Qiagen). Primer Express software (version 2.0, Applied Biosystems, Foster City, CA, USA) was applied for designing PCR primers and TaqMan probes. A BLAST search was conducted to guarantee amplicon specifity. Gapdh was availed for normalization. Assays were run in triplicate. Primers and cycling conditions are available upon request.
Immunohistochemistry.
For the detection of cerebral blood vessels two pairs of 2 µm sagittal brain sections were preprocessed with a pepsin solution (Sigma‐Aldrich, Munich, Germany, 0.4% dissolved in 10 mM HCl) for 30 minutes and automatically tagged with anti‐laminin polyclonal antibody (Sigma‐Aldrich, 1:3000) in a TechMate instrument (DAKO, Hamburg, Germany) followed by the DAKO StreptABC complex‐horseradish peroxidase conjugated duet anti‐rabbit antibody kit and development with 3,3′‐diaminobenzidine. For labeling of proliferating cells sections adjacent to laminin stained slices were pretreated with an antigen retrieval solution (0.1 M citric acid solution, pH 6.0) and boiled for 35 minutes in a humid chamber followed by automated tagging with anti‐Ki‐67 monoclonal antibody (Epitomics, Burlingame, CA, USA, 1:300). Secondary antibody and chromogen were the same as for laminin staining. Counterstaining was performed with hematoxylin. The pairs of sections (10 µm distance) were located between 100 and 300 µm lateral from the mid‐sagittal fissure. Each staining was performed in two successive procedures making sure that brains of both groups were equally distributed in all procedures.
Quantitative evaluation of laminin and Ki‐67 immunoreactivity.
Blood vessel density quantification was achieved by digitization of the neocortices and hippocampi of all stained sections (Olympus BX50, ColorView II, charge‐coupled device camera, Olympus, Hamburg, Germany) under constant light and filter settings. A constant color threshold was chosen for all images to detect immunoreactive staining (analySIS 5, Soft Imaging System, Münster, Germany). The total blood vessel area was determined in the neocortex and hippocampus. Absolute values were related to the investigated area (vessel area proportion). Furthermore, the number of blood vessel bifurcations was determined by counting vessel ends and branch points and dividing the sum by two as previously described (8). The absolute number of vessel branches was set in relation to the tested area. The observer was blind to experimental conditions. For the quantification of Ki‐67 positive endothelial cell nuclei, stained sections were digitized and images were merged with laminin images (Photoshop CS, Adobe, San Jose, CA, USA). Ki‐67 positive nuclei co‐localizing with laminin positive vessels were counted and set in relation to the examined area. The observer was blind to experimental conditions.
Protein analysis.
Protein concentration was determined by the DC Protein Assay (Bio‐Rad, Munich, Germany). Cerebral Aβ40 and Aβ42 peptide levels of each animal were determined using enzyme‐linked immunosorbent assay (ELISA) (Biosource, Solingen, Germany). Aβ40 peptide levels in blood plasma of animals were assigned individually using ELISA (Biosource). All assays were run in duplicate. Receptor for advanced glycation end products (RAGE), low‐density lipoprotein receptor‐related protein 1 (LRP1), Ephrin B2 (EFNB2) and FMS‐like tyrosine kinase 1 (FLT1) protein levels were defined in each SH vs. EH mice by quantitative Western Blot analysis. A total of 20 µg protein of each animal was loaded on a 12.5%, 10% or 7.5% SDS‐polyacrylamide gel for RAGE, EFNB2 and LRP1/FLT1, respectively. Electrophoresis and wet‐blotting preceded membrane blocking with 5% non‐fat milk in TST buffer (10 mM Tris‐HCl pH 7.6, 150 mM NaCl, 0.05% Tween 20) for 1 h at room temperature and subsequent incubation with RAGE antibody (SP5151P, 1:500, Acris Antibodies, Hiddenhausen, Germany), LRP1 antibody (5A6, 1:1000, Merck KGaA, Darmstadt, Germany), EFNB2 antibody (AF496, 1:500, R&D Systems, Minneapolis, MN, USA) or anti‐FLT1 (1303‐1, 1:5000, Epitomics) overnight at 4°C followed by secondary antibody (A2074, 1:2000, Sigma‐Aldrich, for RAGE; A0168, 1:10 000, Sigma‐Aldrich, for LRP1; A9452, 1:5000, Sigma‐Aldrich, for EFNB2; 111‐035‐003, 1:5000, Jackson ImmunoResearch, West Grove, PA, USA for FLT1) and peroxidase‐catalyzed enhanced chemiluminescence (ECL‐Plus, Amersham Biosciences, Freiburg, Germany). Normalization was conducted by β‐actin control immunoblotting (primary and secondary antibody 1:10 000 each, Sigma‐Aldrich). Evaluation of protein expression levels was achieved by densitometry software Gel‐Pro Analyzer (Media Cybernetics, Silver Spring, MD, USA). All samples were analyzed individually and in duplicate.
Statistical analysis.
One‐sample Kolmogorov–Smirnov test validated normal distribution of all data sets. As all data were normally distributed, SH and EH groups were compared using an unpaired Student’s t‐test. All tests were applied two‐tailed except for the morphometrical analysis of vessel density and for EFNB2 and FLT1 Western blot analyses. All tests were performed using the software package SPSS (version 12.0.1). Differences were considered significant at P < 0.05.
RESULTS
Induction of angiogenesis.
To assess angiogenic activity, expression profiling of genes modulating biological processes of angiogenesis was performed on whole cerebral tissue utilizing Mouse Angiogenesis RT2 Profiler PCR arrays. These arrays contain a mixture of pro‐/anti‐angiogenic genes. In our experiments seven genes of 84 were up‐regulated (and none down‐regulated) following EH (P < 0.05, Table 1), six of them being unambiguously pro‐angiogenic molecules, one with a putative angiogenesis‐modulating function, and none with an anti‐angiogenic function (Table 1). To validate these results, we conducted quantitative Western blotting against the pro‐angiogenic proteins EFNB2 (Figure 2A) and FLT1 (alias: VEGFR‐1, Figure 2B). Both molecules were up‐regulated following EH (Figure 2C,D) (EFNB2: +64%, SH: 1 ± 0.34, mean ± SD, EH: 1.64 ± 0.78, P = 0.02; FLT1: +618%, SH: 1 ± 1.87, EH: 7.18 ± 25.34, P = 0.049). Furthermore, the number of proliferating endothelial cells per mm2 neocortical and hippocampal area detected by Ki‐67 immunohistochemistry increased significantly by 153% (Figure 2E–H) (SH: 0.6 ± 0.06, mean ± SD, EH: 1.51 ± 0.55, P = 0.04). Morphometry analysis using immunohistochemical staining for laminin (Figure 2I,J) revealed higher vessel density in EH mice as compared with SH mice. The number of cerebral vessel branches per mm2 tested brain area increased by 29% (Figure 2K) (SH: 48.64 ± 15.83, mean ± SD, EH: 62.77 ± 7.39, P = 0.04) and total area covered by vessels by 23% (Figure 2L) (SH: 2.5% ± 0.39%, mean ± SD, EH: 3.08% ± 0.11%, P = 0.005).
Table 1.
Up‐regulation of pro‐angiogenic genes in EH mice measured by angiogenesis RT2 profiler PCR arrays (P < 0.05). Abbreviation: PCR = polymerase chain reaction.
| Gene symbol | Gene name | Accession unigene | Accession gene bank | P‐value | n‐fold up‐regulation | Reference |
|---|---|---|---|---|---|---|
| Cxcl1 | Chemokine (C‐X‐C motif) ligand 1 | Mm.21013 | NM_008176 | 0.026 | 2.7 | 28 |
| Efnb2 | Ephrin B2 | Mm.209813 | NM_010111 | 0.023 | 23 | 21 |
| Il6 | Interleukin 6 | Mm.1019 | NM_031168 | 0.014 | 3.4 | 36 |
| Tbx4 | T‐box 4 | Mm.275336 | NM_011536 | 0.004 | 2 | 29 |
| Flt1 * | FMS‐like tyrosine kinase 1 | Mm.3464 | NM_010228 | 0.008 | 11.7 | 25 |
| Itgav | Integrin alpha V | Mm.227 | NM_008402 | <0.001 | 4.5 | 18 |
| Thbs2 † | Thrombospondin 2 | Mm.26688 | NM_011581 | 0.04 | 10.3 | 31 |
Alias: VEGFR‐1 (vascular endothelial growth factor receptor).
Thbs2 is discussed to show angiogenesis‐modulating and anti‐inflammatory function.
Figure 2.
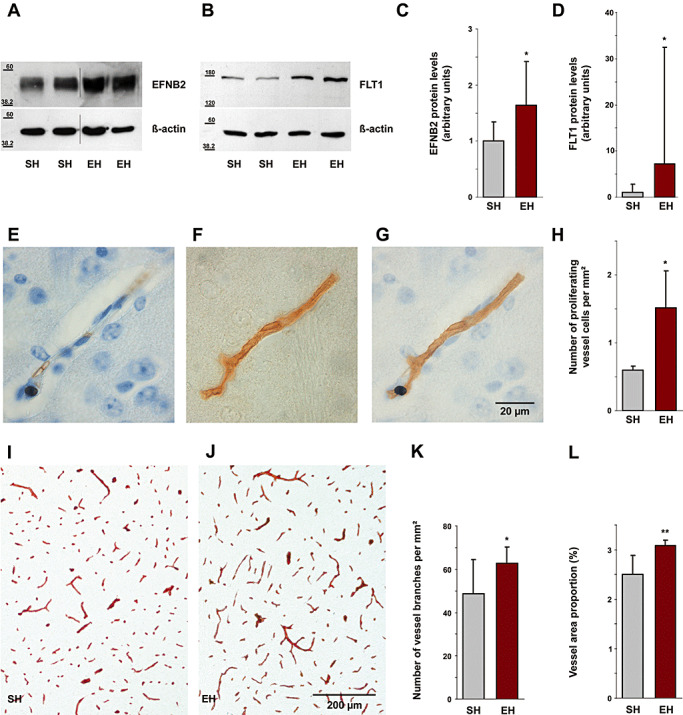
Angiogenesis induced by environmental enrichment. Immunoblot analysis showed an increase in cerebral EFNB2 (A) and FLT1 (B) levels following enrichment (cropped images). C,D. Densitometry analysis of immunoblots identified a significant increase of cerebral EFNB2 by 64% (P = 0.02) and of FLT1 by 618% (P = 0.049). Immunohistochemically stained Ki‐67 positive cell nucleus (E) and laminin labeled blood vessel (F) are co‐localized (G), indicating proliferation of a blood vessel. Scale bar, 20 µm. H. The number of proliferating vessel cells per mm2 in the neocortex and hippocampus increased by 153% (P = 0.04). I,J. Labeling of cerebral blood microvessels by immunohistochemical staining for laminin shows increased vessel density in EH mice as compared with SH mice. Scale bar, 200 µm. K. The number of cerebral vessel branches per mm2 in the neocortex and hippocampus increased by 29% (P = 0.04). L. Vessel area proportion (total area covered by vessels related to brain area) was also increased in same brain regions by 23% (P = 0.005). EFNB2 = Ephrin B2; FLT1 = FMS‐like tyrosine kinase 1; EH = enriched housing; SH = standard housing.
Differential regulation of Aβ carriers.
To clarify whether Aβ receptor/transporter molecules are differentially regulated we performed Western blot analysis against LRP1, facilitating Aβ brain to blood efflux (33, 35), and against RAGE which promotes Aβ influx into the brain (13, 35) (Figure 3A,B). Following EH, RAGE was suppressed by 60% (Figure 3C) (SH: 2.48 ± 1.16, mean ± SD, EH: 1 ± 0.52, P = 0.004), whereas LRP1 was up‐regulated by 384% (Figure 3D, SH: 1 ± 1.29, EH: 4.84 ± 4.58, P = 0.04). Moreover, in TaqMan assays the mRNA expression levels of Apoe (+42%, SH: 1 ± 0.12, mean ± SD, EH: 1.42 ± 0.20, P = 0.006) as well as A2m (+50%, SH: 1 ± 0.14, EH: 1.5 ± 0.34, P = 0.02) were increased (Figure 3E). Both of these molecules are LRP1 ligands mediating Aβ transport via LRP1 across the BBB (5, 35). In contrast, no changes were seen in the steady state mRNA levels of Apoj, the major carrier molecule of Aβ in blood plasma (38) and Rap, a chaperone for LRP1 which binds to this receptor and antagonizes binding of other ligands (30) (Figure 3E, Apoj: SH: 1 ± 0.06, mean ± SD, EH: 1.05 ± 0.06, P = 0.25; Rap: SH: 1 ± 0.04, EH: 1.03 ± 0.04, P = 0.31).
Figure 3.
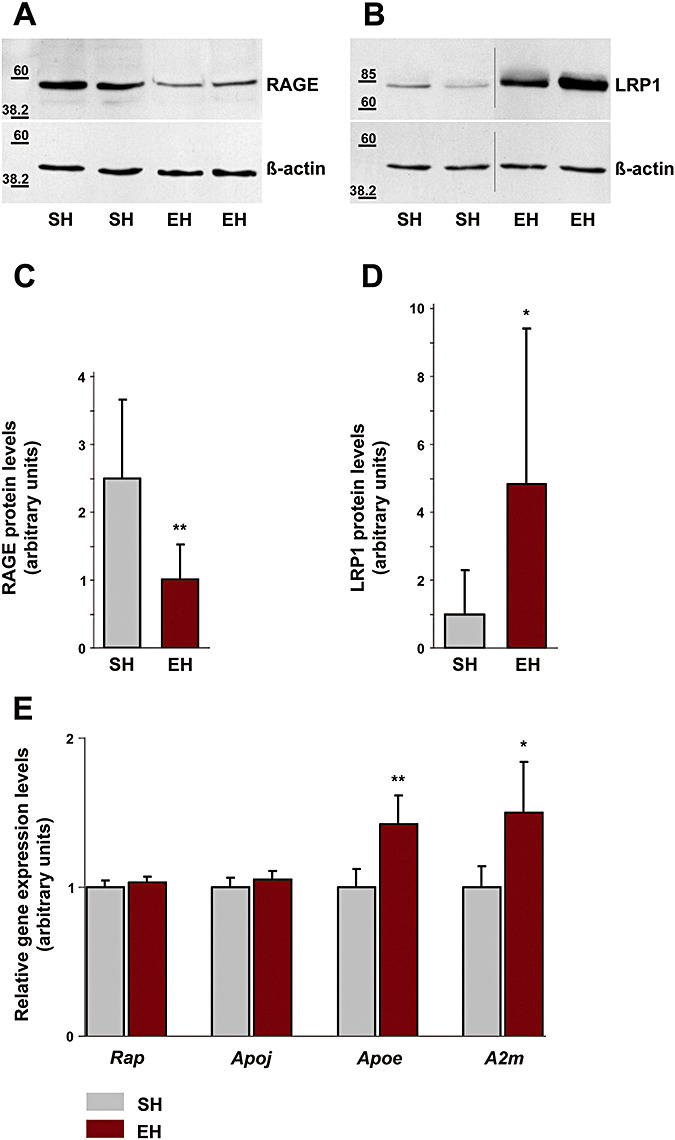
Differential regulation of Aβ receptor/transporter molecules following environmental enrichment. Western blot analysis revealed a decrease in cerebral RAGE levels (A) and an increase in LRP1 protein (B) following enrichment (cropped images). C. Densitometry analysis of immunoblots identified a significant decrease of cerebral RAGE by 60% (P = 0.004). D. Densitometry analysis of immunoblots identified a significant increase of LRP1 protein by 384% (P = 0.04). E. TaqMan quantitative real‐time PCR revealed enhanced gene expression of the Aβ binding proteins Apoe by 42% (P = 0.006) and A2m by 50% (P = 0.02) in enriched housed mice, whereas gene expression levels of Rap (P = 0.31) and Apoj (P = 0.25) remained unaltered. Data are given as mean ± SD, statistics unpaired Student’s t‐test. RAGE = receptor for advanced glycation end products; LRP1 = low‐density lipoprotein receptor‐related protein 1; PCR = polymerase chain reaction; EH = enriched housing; SH = standard housing.
Aβ levels in brain and blood.
ELISA analyses did not detect any significant differences in soluble Aβ40 or Aβ42 levels in the brain (Aβ40: SH: 21.2 ± 7.61 ng/mg protein, mean ± SD, EH: 20.37 ± 9.25 ng/mg, P = 0.85; Aβ42: SH: 84.06 ± 13.62 ng/mg, EH: 76.24 ± 17.83 ng/mg, P = 0.36) or blood (collected after decapitation) (Aβ40: SH: 8.03 ± 6.59 pg/mg, EH: 4.54 ± 3.08 pg/mg, P = 0.42) following EH.
DISCUSSION
Exposure of transgenic animals to EH results in a better cognitive performance [(1, 24), our own unpublished data] and a significant reduction of Aβ plaques (1, 2, 26) and amyloid angiopathy (2). Our data suggest that these effects are partly a consequence of changes in the neurovascular unit. Recent findings indicate that neurovascular dysfunction plays a pivotal role in cognitive decline and neurodegeneration in AD. Faulty clearance of Aβ across the BBB because of aberrant and insufficient angiogenesis (4) associated with low levels of Aβ clearance receptor LRP1 (16, 33) or increased levels of its influx receptor RAGE (13, 16) could lead to formation of vascular amyloid lesions and elevated fibrillar Aβ levels (12, 14). Four months of continuous and diversified environmental stimulation leads to increased cerebral angiogenesis indicated by up‐regulation of pro‐angiogenic genes and proteins (Table 1, Figure 2A–D), increased proliferation of endothelial vessel cells (Figure 2E–H) and a tighter vessel network (Figure 2I–L). Increased vessel density provides the brain with a larger surface area for molecular transport exchange with the periphery which is not only a supporting factor for a better supply of the brain with oxygen and nutrients but also for better Aβ clearance across the BBB. This effect is obviously underpinned by specific regulation of different Aβ carriers, namely up‐regulation of LRP1, (Figure 3B,D), down‐regulation of RAGE (Figure 3A,C) as well as induction of ApoE and A2M expression (Figure 3E), both of which serve as LRP1 ligands mediating Aβ transport across the BBB (5, 35). Furthermore, increase of LRP1 and decrease of RAGE seem to bear an anti‐atherosclerotic effect and indicate blockade of inflammatory processes and reduced oxidative stress (7, 20). Moreover, increased ApoE and A2M expression inhibits migration and proliferation of smooth muscle cells in the vasculature through interaction with LRP1, thereby antagonizing atherosclerotic lesion progression (7, 22, 34). In consideration of the high association between atherosclerosis and AD (9, 32) our results also suggest a possible anti‐atherosclerotic effect of EH on vasculature in Alzheimer diseased brain.
In spite of significant reduction of Aβ plaques and amyloid angiopathy following EH (2) the brain and blood soluble Aβ levels remained unchanged as measured by ELISA after decapitation. This finding may be related to a “sink” and degradation of Aβ at peripheral sites and an already established balance between soluble Aβ levels in the brain and plasma. Concededly, the low number of blood samples analyzed (because of material limitation five in SH and four in EH mice) limits the interpretation of this result.
It has been well documented that environmental stimulation in the form of physical activity increases angiogenesis in the normal adult brain (6, 23) and counteracts the vascular dysfunction in diseases such as stroke (15, 19). Our results—as far as results obtained in animal models are transferable to humans—extend this finding to brains with AD pathology.
Meanwhile there are other publications on the effects of environmental stimulation in transgenic AD models reporting partly contradictory results. Besides Lazarov et al (26) and Adlard et al (1) reporting also on reduction of cerebral Aβ deposition following EH, works made by Arendash et al failed to show any effects of EH on Aβ deposition in aged mice (3). Interestingly, later works by the same group revealed also a reduction of brain amyloid plaque content when they started with enrichment at weaning (11). An enhancement of amyloid deposition after EH has been reported by Jankowsky et al (24). However, their enrichment setting by keeping mice in large groups (with or without altering population) was likely to generate sustained social stress possibly blunting the beneficial effects of enrichment (26).
Previous work in our own lab has provided strong evidence that the effect of EH on reduction of cerebral Aβ deposition is independent from APP expression or processing and rather associated with reduced aggregation and enhanced clearance of Aβ (2). Moreover, works by Lazarov et al have shown the involvement of neprilysin, an Aβ‐degrading endopeptidase, in Aβ elimination from the brain following EH (26). The present work adds another argument for facilitated clearance by showing a hitherto unknown interplay between the environment and cerebrovascular unit in AD.
ACKNOWLEDGMENTS
This research was supported by a grant from Innovative Medical Research (KE 120517). We thank B. Heuer, S. Peetz‐Dienhart, M. Leisse, A. Wagner and R. Mersmann for technical assistance.
REFERENCES
- 1. Adlard PA, Perreau VM, Pop V, Cotman CW (2005) Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer’s disease. J Neurosci 25:4217–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ambree O, Leimer U, Herring A, Gortz N, Sachser N, Heneka MT, Paulus W, Keyvani K (2006) Reduction of amyloid angiopathy and Abeta plaque burden after enriched housing in TgCRND8 mice: involvement of multiple pathways. Am J Pathol 169:544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arendash GW, Garcia MF, Costa DA, Cracchiolo JR, Wefes IM, Potter H (2004) Environmental enrichment improves cognition in aged Alzheimer’s transgenic mice despite stable beta‐amyloid deposition. Neuroreport 15:1751–1754. [DOI] [PubMed] [Google Scholar]
- 4. Bailey TL, Rivara CB, Rocher AB, Hof PR (2004) The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol Res 26:573–578. [DOI] [PubMed] [Google Scholar]
- 5. Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV (2006) Transport pathways for clearance of human Alzheimer’s amyloid beta‐peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab 27:909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Black JE, Isaacs KR, Anderson BJ, Alcantara AA, Greenough WT (1990) Learning causes synaptogenesis, whereas motor activity causes angiogenesis, in cerebellar cortex of adult rats. Proc Natl Acad Sci USA 87:5568–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J (2003) LRP: role in vascular wall integrity and protection from atherosclerosis. Science 300:329–332. [DOI] [PubMed] [Google Scholar]
- 8. Casella GT, Marcillo A, Bunge MB, Wood PM (2002) New vascular tissue rapidly replaces neural parenchyma and vessels destroyed by a contusion injury to the rat spinal cord. Exp Neurol 173:63–76. [DOI] [PubMed] [Google Scholar]
- 9. Casserly I, Topol E (2004) Convergence of atherosclerosis and Alzheimer’s disease: inflammation, cholesterol, and misfolded proteins. Lancet 363:1139–1146. [DOI] [PubMed] [Google Scholar]
- 10. Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George‐Hyslop PS, Westaway D (2001) Early‐onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 276:21562–21570. [DOI] [PubMed] [Google Scholar]
- 11. Costa DA, Cracchiolo JR, Bachstetter AD, Hughes TF, Bales KR, Paul SM, Mervis RF, Arendash GW, Potter H (2007) Enrichment improves cognition in AD mice by amyloid‐related and unrelated mechanisms. Neurobiol Aging 28:831–844. [DOI] [PubMed] [Google Scholar]
- 12. Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, Zlokovic BV, Van Nostrand WE (2004) Early‐onset and robust cerebral microvascular accumulation of amyloid beta‐protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta‐protein precursor. J Biol Chem 279:20296–20306. [DOI] [PubMed] [Google Scholar]
- 13. Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B (2003) RAGE mediates amyloid‐beta peptide transport across the blood‐brain barrier and accumulation in brain. Nat Med 9:907–913. [DOI] [PubMed] [Google Scholar]
- 14. Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV (2004) LRP/amyloid beta‐peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43:333–344. [DOI] [PubMed] [Google Scholar]
- 15. Ding YH, Luan XD, Li J, Rafols JA, Guthinkonda M, Diaz FG, Ding Y (2004) Exercise‐induced overexpression of angiogenic factors and reduction of ischemia/reperfusion injury in stroke. Curr Neurovasc Res 1:411–420. [DOI] [PubMed] [Google Scholar]
- 16. Donahue JE, Flaherty SL, Johanson CE, Duncan JA 3rd, Silverberg GD, Miller MC, Tavares R, Yang W, Wu Q, Sabo E, Hovanesian V, Stopa EG (2006) RAGE, LRP‐1, and amyloid‐beta protein in Alzheimer’s disease. Acta Neuropathol 112:405–415. [DOI] [PubMed] [Google Scholar]
- 17. Friedland RP, Fritsch T, Smyth KA, Koss E, Lerner AJ, Chen CH, Petot GJ, Debanne SM (2001) Patients with Alzheimer’s disease have reduced activities in midlife compared with healthy control‐group members. Proc Natl Acad Sci USA 98:3440–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fukushima K, Miyamoto S, Tsukimori K, Kobayashi H, Seki H, Takeda S, Kensuke E, Ohtani K, Shibuya M, Nakano H (2005) Tumor necrosis factor and vascular endothelial growth factor induce endothelial integrin repertories, regulating endovascular differentiation and apoptosis in a human extravillous trophoblast cell line. Biol Reprod 73:172–179. [DOI] [PubMed] [Google Scholar]
- 19. Gertz K, Priller J, Kronenberg G, Fink KB, Winter B, Schrock H, Ji S, Milosevic M, Harms C, Bohm M, Dirnagl U, Laufs U, Endres M (2006) Physical activity improves long‐term stroke outcome via endothelial nitric oxide synthase‐dependent augmentation of neovascularization and cerebral blood flow. Circ Res 99:1132–1140. [DOI] [PubMed] [Google Scholar]
- 20. Goldin A, Beckman JA, Schmidt AM, Creager MA (2006) Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 114:597–605. [DOI] [PubMed] [Google Scholar]
- 21. Hayashi S, Asahara T, Masuda H, Isner JM, Losordo DW (2005) Functional ephrin‐B2 expression for promotive interaction between arterial and venous vessels in postnatal neovascularization. Circulation 111:2210–2218. [DOI] [PubMed] [Google Scholar]
- 22. Herz J, Strickland DK (2001) LRP: a multifunctional scavenger and signaling receptor. J Clin Invest 108:779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Isaacs KR, Anderson BJ, Alcantara AA, Black JE, Greenough WT (1992) Exercise and the brain: angiogenesis in the adult rat cerebellum after vigorous Phys activity and motor skill learning. J Cereb Blood Flow Metab 12:110–119. [DOI] [PubMed] [Google Scholar]
- 24. Jankowsky JL, Melnikova T, Fadale DJ, Xu GM, Slunt HH, Gonzales V, Younkin LH, Younkin SG, Borchelt DR, Savonenko AV (2005) Environmental enrichment mitigates cognitive deficits in a mouse model of Alzheimer’s disease. J Neurosci 25:5217–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kearney JB, Kappas NC, Ellerstrom C, DiPaola FW, Bautch VL (2004) The VEGF receptor flt‐1 (VEGFR‐1) is a positive modulator of vascular sprout formation and branching morphogenesis. Blood 103:4527–4535. [DOI] [PubMed] [Google Scholar]
- 26. Lazarov O, Robinson J, Tang YP, Hairston IS, Korade‐Mirnics Z, Lee VM, Hersh LB, Sapolsky RM, Mirnics K, Sisodia SS (2005) Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell 120:701–713. [DOI] [PubMed] [Google Scholar]
- 27. Le Carret N, Auriacombe S, Letenneur L, Bergua V, Dartigues JF, Fabrigoule C (2005) Influence of education on the pattern of cognitive deterioration in AD patients: the cognitive reserve hypothesis. Brain Cogn 57:120–126. [DOI] [PubMed] [Google Scholar]
- 28. Mohsenin A, Burdick MD, Molina JG, Keane MP, Blackburn MR (2007) Enhanced CXCL1 production and angiogenesis in adenosine‐mediated lung disease. FASEB J 21:1026–1036. [DOI] [PubMed] [Google Scholar]
- 29. Naiche LA, Papaioannou VE (2003) Loss of Tbx4 blocks hindlimb development and affects vascularization and fusion of the allantois. Development 130:2681–2693. [DOI] [PubMed] [Google Scholar]
- 30. Pan W, Kastin AJ, Zankel TC, Van Kerkhof P, Terasaki T, Bu G (2004) Efficient transfer of receptor‐associated protein (RAP) across the blood‐brain barrier. J Cell Sci 117:5071–5078. [DOI] [PubMed] [Google Scholar]
- 31. Park YW, Kang YM, Butterfield J, Detmar M, Goronzy JJ, Weyand CM (2004) Thrombospondin 2 functions as an endogenous regulator of angiogenesis and inflammation in rheumatoid arthritis. Am J Pathol 165:2087–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Roher AE, Esh C, Rahman A, Kokjohn TA, Beach TG (2004) Atherosclerosis of cerebral arteries in Alzheimer disease. Stroke 35:2623–2627. [DOI] [PubMed] [Google Scholar]
- 33. Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV (2000) Clearance of Alzheimer’s amyloid‐ss(1‐40) peptide from brain by LDL receptor‐related protein‐1 at the blood‐brain barrier. J Clin Invest 106:1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Swertfeger DK, Bu G, Hui DY (2002) Low density lipoprotein receptor‐related protein mediates apolipoprotein E inhibition of smooth muscle cell migration. J Biol Chem 277:4141–4146. [DOI] [PubMed] [Google Scholar]
- 35. Tanzi RE, Moir RD, Wagner SL (2004) Clearance of Alzheimer’s Abeta peptide: the many roads to perdition. Neuron 43:605–608. [DOI] [PubMed] [Google Scholar]
- 36. Yao JS, Zhai W, Fan Y, Lawton MT, Barbaro NM, Young WL, Yang GY (2006) Interleukin‐6 upregulates expression of KDR and stimulates proliferation of human cerebrovascular smooth muscle cells. J Cereb Blood Flow Metab 27:510–520. [DOI] [PubMed] [Google Scholar]
- 37. Zlokovic BV (2005) Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci 28:202–208. [DOI] [PubMed] [Google Scholar]
- 38. Zlokovic BV, Martel CL, Matsubara E, McComb JG, Zheng G, McCluskey RT, Frangione B, Ghiso J (1996) Glycoprotein 330/megalin: probable role in receptor‐mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood‐brain and blood‐cerebrospinal fluid barriers. Proc Natl Acad Sci USA 93:4229–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]