

Abstract
The first tau transgenic mouse model was established more than a decade ago. Since then, much has been learned about the role of tau in Alzheimer’s disease and related disorders. Animal models, both in vertebrates and invertebrates, were significantly improved and refined as a result of the identification of pathogenic mutations in Tau in human cases of frontotemporal dementia. They have been instrumental for dissecting the cross‐talk between tau and the second hallmark lesion of Alzheimer’s disease, the Aβ peptide‐containing amyloid plaque. We discuss how the tau models have been used to unravel the pathophysiology of Alzheimer’s disease, to search for disease modifiers and to develop novel treatment strategies. While tau has received less attention than Aβ, it is rapidly acquiring a more prominent position and the emerging view is one of a synergistic action of Aβ and tau in Alzheimer’s disease. Moreover, the existence of a number of neurodegenerative diseases with tau pathology in the absence of extracellular deposits underscores the relevance of research on tau.
INTRODUCTION
Histopathologically, the Alzheimer’s disease (AD) brain is characterized by abundant amyloid plaques, neurofibrillary lesions and the loss of nerve cells and synapses. This review focuses on tau, a microtubule‐associated protein (MAP) and the principal component of the neurofibrillary lesions (41). They are found in nerve cell bodies and apical dendrites as neurofibrillary tangles (NFTs), in distal dendrites as neuropil threads and in the abnormal neurites that are associated with some amyloid plaques (neuritic plaques). In the absence of plaques, tau inclusions are abundant in a range of neurodegenerative diseases, which include Pick’s disease (PiD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease, sporadic frontotemporal dementia (FTD) and the inherited FTD and Parkinsonism linked to chromosome 17 (FTDP‐17) (48, 86).
Tau is expressed predominantly in neurons and at lower levels in astrocytes and oligodendrocytes (143). Moreover, in some diseases, tau also forms aggregates in glial cells and these can outnumber neurons with aggregates (48). Tau contains a particularly high content of serines and threonines, many of which are phosphorylated under physiological conditions (42). Under pathological conditions, tau becomes hyperphosphorylated, which means a higher degree of phosphorylation at physiological sites, as well as de novo phosphorylation at additional sites (15, 43). Phosphorylation decreases the binding of tau to microtubules. This increases the pool of soluble tau and is thought to trigger the disintegration of microtubules (43). In addition to phosphorylation, tau is subject to ubiquitination, nitration, truncation, prolyl isomerization, association with heparan sulphate proteoglycans, glycosylation, glycation and modification by advanced glycation end‐products (AGEs) (20).
Mutations in Tau have not been found in AD. Instead, mutations have been identified in the amyloid precursor protein (APP) gene, from which Aβ is derived by proteolytic cleavage, and in the presenilin‐1 (PS1) and presenilin‐2 (PS2) genes, both of which encode proteins involved in Aβ formation. These pathogenic mutations account for less than 1% of AD cases (27). In 1998, research into tau was spurred on by the identification of exonic and intronic mutations in the Tau gene in FTDP‐17, a familial dementia related to AD (69, 119, 137). These findings established that dysfunction of tau can cause neurodegeneration and lead to dementia. In addition to FTD, Tau mutations can give rise to clinical syndromes that resemble PSP, CBD, PiD and progressive subcortical gliosis (16, 44, 86, 153). So far, 39 different Tau mutations have been identified in over 100 families.
ANIMAL MODELS
Over the past decade, many tau transgenic animal models have been developed that reproduce aspects of AD and FTD (Figure 1). As outlined below, they have been used to identify pathomechanisms, disease modifiers and differentially expressed genes and proteins. This has been followed by validation in tissues from patients with AD and FTD. In addition, these models have also been used to develop novel therapeutic strategies (55).
Figure 1.
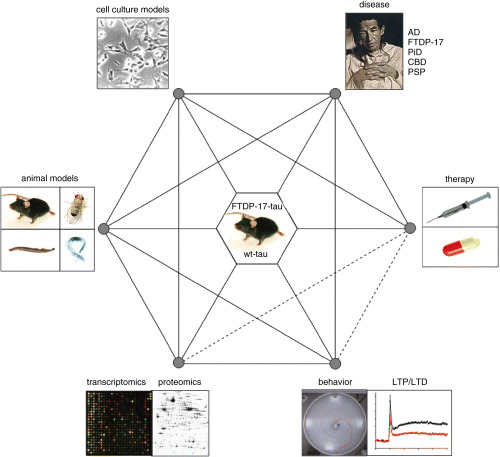
Interaction model illustrating the potential of human wild‐type and mutant tau transgenic animal models. AD = Alzheimer’s disease; CBD = corticobasal degeneration; FTDP‐17 = frontotemporal dementia and Parkinsonism linked to chromosome 17; PiD = Pick’s disease; PSP = progressive supranuclear palsy.
Wild‐type tau transgenic mice. The first transgenic models used expression of wild‐type four‐repeat (49) and three‐repeat (13) human tau. These mice reproduced aspects of human pathology, such as the somatodendritic localization and hyperphosphorylation of tau. Subsequently, stronger promoters were used to drive expression and this resulted in more pronounced phenotypes (71, 120, 138). Non‐filamentous tau aggregates formed, which could be stained with phosphorylation‐ and conformation‐dependent anti‐tau antibodies [reviewed in (48, 53)]. However, despite an age‐dependent decrease in tau solubility, NFTs did not form, unless the mice reached a very old age (72).
Signs of Wallerian degeneration, including axonal breakdown and segmentation of myelin into ellipsoids (so‐called “digestion chambers”), were observed in these mice. Furthermore, neurogenic muscle atrophy, with groups of small angular muscle fibres, was present in the hindleg musculature of transgenic, but not control, mice (120). A prominent characteristic of the transgenic mice was the presence of large numbers of axonal spheroids in brainstem and spinal cord, displaying accumulations of neurofilaments, mitochondria and vesicles (71, 120, 138). Axonal spheroids are a characteristic of most cases of amyotrophic lateral sclerosis (64, 103, 123). They have also been reported in cases of FTD of unknown etiology (155). In one transgenic line, microglial cells with phagocytosed myelin debris and myelin ovoids were present (138). These findings demonstrated that overexpression of human tau can lead to a central and peripheral axonopathy that results in nerve cell dysfunction and amyotrophy. Interestingly, similar axonal swellings have been described in Aβ plaque‐forming transgenic mice (140).
Axonopathy is also found in AD brain, as evidenced by choline acetyl transferase (ChAT)‐immunoreactive swellings (140). Interestingly, this axonopathy is dependent on the Braak stage (used to stage the neurofibrillary pathology of AD), as stages I–III revealed many ChAT‐reactive swellings, whereas these were only half as frequent at stages IV–VI. In contrast, phosphotau immunoreactivity, although present at stages I–III, became much more significant at stages IV–VI. It was concluded that axonal swellings occur before detectable Aβ deposits and pathological tau phosphorylation, and may lead to the formation of dystrophic neurites (140). How spheroids form and further implications of the axonal transport failure hypothesis of neurodegeneration are discussed elsewhere (54).
Mutant tau transgenic mice. Following the identification of pathogenic mutations in Tau in FTDP‐17, several groups reported on the formation of NFTs in neurons (1, 50, 88, 91, 141, 144, 145) and glial cells (36, 52, 62, 63, 109, 125, 129) in mice transgenic for mutant human tau, with a tetracycline‐regulated transactivator system being used in two models (52, 125). The first published NFT‐forming model expressed human P301L tau under the control of the murine PrP promoter (88). These mice developed motor disturbances by 10 months of age, which were more pronounced than those observed in wild‐type tau transgenic lines (71, 120, 138). NFTs were present in brain and spinal cord and the number of motor neurons in spinal cord was reduced by 50% (88). The P301L mice also showed abnormal tau filaments in astrocytes and oligodendrocytes, reflecting regulation of transgene expression by the PrP promoter (93). Using a different human tau isoform, mutation P301L was expressed under the control of the mThy1.2 promoter, which also led to the formation of NFTs, as identified by Gallyas silver staining and thioflavin S fluorescence (50) (Figure 2). In humans, the P301S mutation causes an earlier onset of clinical signs of FTD than mutation P301L (16). The shortest human four‐repeat tau isoform with the P301S mutation was expressed in mice using the mThy1.2 promoter (1). In brain and spinal cord, the levels of human tau were twofold higher than those of mouse tau. Human tau was expressed in neocortex and hippocampus, but levels were highest in brainstem and spinal cord. A twofold reduction in the number of spinal cord motor neurons was reported, indicating that motor neurons were either especially vulnerable to tau aggregation, or that expression of tau was particularly high in these cells. When antibodies specific for activated MAP kinase, JNK and p38 were used, tau‐positive neurons in P301S tau transgenic mice, but not in mice expressing wild‐type tau, were labeled (1). Interestingly, mice with a tau phenotype due to reduced protein phosphatase 2A (PP2A) activity also showed activation of these signaling pathways, suggesting that tau phosphorylation may have been brought about, at least in part, by activation of MAP kinase and JNK (78, 79).
Figure 2.
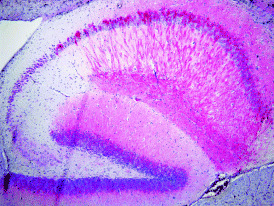
Phosphotau staining of the hippocampus of a P301L mutant human tau transgenic mouse.
PSP, CBD and some cases of FTDP‐17 are characterized by a substantial glial tau pathology, which has been reproduced in animal models. The first model expressed G272V mutant tau by combining a PrP‐driven expression system with an autoregulatory transactivator loop that resulted in high tau expression in a subset of neurons and oligodendrocytes. Electron microscopy established that tau filament formation was associated with hyperphosphorylation of tau. Thioflavin S‐positive fibrillary inclusions were identified in oligodendrocytes and motor neurons of the spinal cord (52), but the clinical phenotype was only subtle. In contrast, when human wild‐type tau was overexpressed in neurons and glial cells under the control of the mouse Tα1 α‐tubulin promoter, a glial pathology was found that resembled the astrocytic plaques of CBD and the coiled bodies of PSP and CBD (62). In contrast, no neuronal inclusions were found. The solubility of tau decreased with age, while its phosphorylation increased in parallel. A significant age‐related loss of neurons was only found in 18‐month‐old mice, whereas oligodendrocytes were already lost at 6 months of age. As Gallyas‐positive tau filaments in oligodendrocytes were only observed after 24 months, this does suggest that the accumulation of tau led to cell death well before the formation of abnormal filamentous aggregates. Subsequently, using the 2′,3′‐cyclic nucleotide 3′‐phosphodiesterase promoter, transgenic mice were generated that expressed human P301L tau exclusively in oligodendrocytes (63). Interestingly, structural disruption of myelin and axons preceded the emergence of thioflavin‐S reactive tau inclusions in oligodendrocytes, as did impaired axonal transport, which occurred even earlier, preceding motor deficits (63). The question of whether and to what extent glial pathology contributes to the clinical features of PSP and CBD may best be addressed by behavioral studies using mice with a pronounced glial tau pathology.
To reproduce the Aβ plaque and NFT pathologies in a single model without the need for crossings, triple transgenic mice carrying the PS1 M146V, APP K670N/M671L (Swedish) and P301L tau transgenes were developed. In these mice, synaptic dysfunction, including deficits in long‐term potentiation, were reported to precede Aβ plaque and NFT formation (109). In order to allow a better side‐by‐side comparison of the effects of wild‐type and P301L mutant human tau expression, an approach was chosen whereby two strains were generated, both expressing the longest human tau isoform at similar levels, with one bearing the P301L mutation and the other being wild‐type (145). The two strains developed very different phenotypes. Mice expressing wild‐type human tau developed early motor impairement at around 6–8 weeks of age and an axonopathy in the absence of tau aggregates. However, their survival rates were normal. In contrast, the P301L mutant mice started to develop NFTs at 6 months of age, in the absence of an axonal phenotype, but despite having only minor motor problems, all the mice died before 13 months of age. It was concluded that excessive binding of wild‐type human tau to microtubules, as opposed to reduced binding of P301L tau, may have been responsible for the development of axonopathy and tauopathy, respectively. In addition, a conformational change in P301L mutant tau may have been a determinant in triggering tauopathy. A third line was a knock‐in replacing the mouse gene with a single copy of the wild‐type human tau expression construct used above. These mice had normal survival rates without any obvious pathology and with only minor motor problems later in life (145). When compared with results obtained in the other tau models, it can be concluded that the range of phenotypes encountered was likely due to the use of different promoters for transgene expression, the integration sites of the transgenes, the expression levels of human tau and/or the mouse strains used, without necessarily shedding any light on the underlying mechanisms (53). At the molecular level, much remains to be done to understand how tau impairs neuronal and glial function and causes nerve cell death.
To determine whether NFTs are central to the neurotoxic cascade in AD or represent a protective response, P301L mice were generated in which transgene overexpression could be reduced (125). Mice expressing doxycycline‐repressible human P301L mutant tau developed progressive age‐related NFTs, remarkable neuronal cell loss (70% in the CA1 region) and behavioral impairment. Following a reduction of transgenic tau from 13‐ to 2.5‐fold overexpression, memory function recovered and the number of neurons stabilized, but NFTs continued to accumulate. These findings have shown that dysfunction of tau impairs memory, when mutant tau is massively overexpressed. They further imply that NFTs per se (as entities of fibrillar accumulation that are visible at the light microscopic level) are not sufficient to cause cognitive decline or neuronal death in this model (125). In agreement with this, cognitive impairment in our P301L tau transgenic mice occurred in the absence of NFT formation (112, 114). NFTs are found in only a small percentage of neurons in the animal models published so far and are outnumbered by dysfunctional neurons containing tau aggregates, but lacking NFTs. Therefore, it is not surprising that functional impairment in these mice did not correlate with NFTs, but rather with the larger number of cells containing pre‐NFT aggregates.
Behavioral studies have also been performed with the V337M tau mutant mice, which expressed transgenic tau only in the hippocampus. These mice showed increased locomotor activity and memory deficits in the elevated plus maze, increased spontaneous locomotion in the open‐field, but no significant impairment in the Morris water maze (142). R406W tau mutant mice expressed highest tau levels in the hippocampus and, to a lesser extent, other cortical and subcortical brain areas (144). A slight decrease in locomotor activity was shown in these mice during the first 5 minutes of the open‐field test and, in addition, a significant impairment in the contextual and cued fear conditioning test was present.
Another approach is to take advantage of virus‐based expression to address the role of tau in disease. In one study Sindbis virus was used to express human tau in hippocampal slices. Tau acquired a pathological conformation, as evidenced by Alz50/MC1‐reactivity. It became hyperphosphorylated and insoluble, resulting in region‐specific neurodegeneration. Live imaging demonstrated that tau‐dependent degeneration was associated with the development of a “ballooned cell” phenotype. Surprisingly, spine densities and morphologies were not altered, suggesting that synaptic integrity was remarkably stable in the presence of tau‐dependent degeneration (131).
Crosses onto a tau null background. The first tau knock‐out model was established in 1994 by insertion of a neomycin resistance cassette into exon 1 of Tau (60). In large‐calibre axons, an increase in MAP1A appeared to compensate for the loss of tau. In some small‐calibre axons, however, microtubule stability was reduced and microtubule organization was significantly changed (60). Mice lacking tau developed muscle weakness, as determined by the wire‐hanging test, hyperactivity in a novel environment and impairment of contextual fear conditioning. They also had a tendency to fall off the rod more easily in the narrow beam test (70). In cases of sporadic and familial forms of FTD without tau pathology, reduced levels of tau protein, but not mRNA, have been reported (6, 156). It suggests that the loss of tau protein itself may lead to some of the neurological characteristics observed in FTD and that a loss of physiological function of tau in the presence of FTDP‐17 mutations may contribute to disease in mutation carriers. A reduction in tau levels may be a cause of mental retardation in humans, as suggested by the recent reports of heterozygous microdeletions on chromosome 17q21.31 (80, 95, 132, 133, 149).
A second tau knock‐out strain was established by inserting a neomycin resistance cassette into exon 1 of Tau (26). In contrast to the above strain, primary hippocampal cultures from these mice showed a significant delay in maturation, as judged by axonal extension. The technique of selectively enhancing axonal growth on laminin substrates failed to restore normal neuronal maturation of the tau knock‐out neurons. By crossing tau transgenic with knock‐out mice, tau‐deficient neurons were reconstituted with human tau, thus restoring a normal pattern of axonal growth and neuronal maturation. The ability of human tau protein to rescue tau‐deficient mouse neurons confirms that tau expression can affect the rate of neurite extension (26). The third tau model is a knock‐in strain generated by targeting a cDNA encoding enhanced green fluorescent protein (EGFP) to exon 1 of Tau (147). These mice did not show any abnormalities in either central or peripheral nervous system.
In separate work, the entire wild‐type human tau gene was expressed at levels that were twofold to threefold higher than those observed in mice transgenic for a single isoform of wild‐type human tau (32). Surprisingly, these mice did not develop any obvious histopathological or neurological phenotype. However, when they were crossed with the EGFP knock‐in mice (147), they developed tau filaments and pathological tau accumulated in cell bodies and dendrites of neurons in a spatiotemporally relevant fashion (2). This may suggest that endogenous mouse tau can inhibit filament formation of human tau. In P301L and P301S tau transgenic mice, murine tau has been shown to be excluded from the filaments (1, 88). Similarly, in transgenic mouse models of Parkinson’s disease, insoluble inclusions were reported which were immunoreactive for human, but not mouse, α‐synuclein (84, 96).
Invertebrate models. Although the mouse has been the major species used for modeling AD and FTD, other organisms, such as the fruit fly Drosophila melanogaster, the nematode Caenorhabditis elegans and the sea lamprey Petromyzon marinus have also been used [reviewed in (53, 87)] (Figure 1).
Drosophila offers the advantage of low cost, small size and short lifespan, which makes it an attractive organism for drug and modifier screens. When wild‐type and FTDP‐17 mutant forms of human tau were expressed in Drosophila, flies exhibited key features of the human disorder, including adult‐onset, progressive neurodegeneration, early death, enhanced toxicity of mutant tau, accumulation of abnormal tau and anatomical selectivity. However, neurodegeneration occurred without NFT formation (152), which is not entirely contradictary to findings in human AD brain, where around 15% of the neuronal loss cannot be explained by NFTs (39, 45). Interestingly, when expression of wild‐type human tau was combined with that of the Drosophila GSK‐3 homolog “Shaggy”, neurofibrillary pathology with tau filaments was observed (73). In a related study, expression of wild‐type human tau caused impaired axonal transport with vesicle aggregation and associated loss of locomotor function, in the absence of neuronal loss (102). Interestingly, co‐expression of a constitutively active form of GSK‐3β enhanced both axonal transport and locomotor phenotype and two GSK‐3 inhibitors, lithium and AR‐A014418, reversed these changes. Both studies have suggested that GSK‐3 may be an attractive drug target.
Directed expression of wild‐type human and Drosophila tau in adult mushroom body neurons, which are centres of olfactory learning and memory, strongly impaired associative olfactory learning and memory, but olfactory conditioning‐relevant osmotactic and mechanosensory responses remained intact (99). For comparison, studies in P301L tau transgenic mice revealed an impairment of conditioned taste aversion memory, a specific type of olfactory learning in mice (14, 112, 128). In another Drosophila model (102), wild‐type human tau was expressed in larval motor neurons. This caused significant morphological and functional disruption of the neuromuscular junction. Mitochondrial numbers were reduced in presynaptic nerve terminals. Collapse of the mitochondrial membrane potential in wild‐type larvae phenocopied the effects of tau overexpression on synaptic transmission. The authors suggested that overexpression of tau caused synaptic dysfunction, which may have resulted from a reduction in the number of functional mitochondria (19). Mitochondrial dysfunction has also been reported in human P301L tau transgenic mice, based on a proteomics screen (24).
Drosophila models have been used to address the mitosis failure hypothesis of AD. It is based on the concept that the mechanisms underlying synaptic plasticity are largely based on external morphoregulatory cues and internal signaling pathways. Non‐neuronal cells use these mechanisms to sense their relationship to the local neighborhood and to control proliferation and differentiation. This puts nerve cells at risk of erroneously converting signals derived from plastic synaptic changes into positional cues that will activate the cell cycle. Cell cycle activation potentially links synaptic plasticity to cell death, as shown in mice expressing human tau on a mouse tau null background (4). Both apoptotic and non‐apoptotic modes of neurodegeneration have been described in these mice. Data from wild‐type and mutant tau‐expressing Drosophila support a role for cell cycle activation downstream of tau phosphorylation and upstream of apoptosis. It was shown that ectopic cell‐cycle activation leads to apoptosis of postmitotic neurons in vivo. TOR (target of rapamycin) kinase activity was increased and required for neurodegeneration (77). Apoptosis was confirmed by TUNEL staining, visualization of fragmented nuclei and membrane blebbing, but non‐apoptotic forms of cell death may also have been present. Furthermore, evidence for a role of cell cycle‐regulated genes in neurodegeneration has been obtained in mice transgenic for wild‐type human tau (3) and in tau‐expressing human neuroblastoma cells (Hoerndli et al, unpublished). However, in human P301S tau transgenic mice, none of the tested activators and co‐activators of the cell cycle was overexpressed and levels of two inhibitors of cell cycle progression, p21Cip1 and p27Kip1, were increased in brain and spinal cord (28). As these inhibitors accumulated in the cytoplasm of nerve cells, the majority of which contained tau aggregates, this implies that reactivation of the cell cycle was not involved in tau hyperphosphorylation or filament formation. Taking all experimental models into consideration, the role played by cell cycle re‐entry and apoptosis in tauopathies remains controversial.
In contrast to Drosophila, the sea lamprey Petromyzon marinus is not routinely used as an experimental organism. It is a parasitic, eel‐like fish, with a set of six giant neurons in the hindbrain; these so‐called anterior bulbar cells, or ABCs, are readily accessible for manipulation (56). They resemble most large vertebrate neurons, in that they have extensively branched, tapered dendrites, which receive a large number of synaptic inputs (57). Self‐replicating mRNAs derived from Semliki Forest Virus were microinjected into ABCs to overexpress tau. It was possible to reproduce the type of degeneration observed in AD, with the earliest and most severe changes occurring in distal dendrites. Degeneration correlated with the appearance of AD‐related phosphoepitopes of tau (58). A lipid‐soluble, low‐molecular weight proprietary compound, was shown to retard the progressive degeneration of human tau‐expressing ABCs (59).
The nematode C. elegans has also been used as a model organism in tau research. It can be grown in microtiter dishes, screens to identify gene knock‐outs can be automated and large‐scale set‐ups have been devised that allow inactivation of thousands of genes in parallel through RNA interference (10). Moreover, small single‐stranded regulatory RNAs, called microRNAs, capable of interfering with intracellular mRNAs that contain either complete or partial complementarity, were first identified in C. elegans (92) and have potential for AD research. C. elegans and Drosophila are particularly well suited for non‐biased screens for suppressors and enhancers of neurodegeneration (37). Expression of wild‐type and mutant human tau in C. elegans led to similar behavioral, synaptic and pathological abnormalities, although expression of mutant tau caused an earlier and more severe phenotype. Moreover, an uncoordinated movement (Unc) phenotype was noted prior to the accumulation of insoluble tau, suggesting that, while aggregation may contribute to neuronal loss, it is not required for neuronal dysfunction (81). Expression in mechanosensory neurons revealed differences for wild‐type and mutant forms of tau, in that worms expressing wild‐type tau showed a smaller reduction in response to touch than those expressing mutant tau. As neurons started to degenerate, they accumulated tau in nerve cell bodies and processes, in the absence of apoptosis (100).
POTENTIAL OF ANIMAL MODELS
Dissection of pathophysiology. Transgenic mice have been used to address the question of cross‐talk between tau inclusions and amyloid plaques. The major plaque component is the 39–43 amino acid long Aβ (mainly Aβ40 and Aβ42), which is derived from APP through proteolytic cleavage (40, 97). In cerebral cortex of aged rhesus monkeys, injection of plaque‐equivalent concentrations of fibrillar, but not soluble, Aβ resulted in neuronal loss, tau phosphorylation and microglial proliferation. As the same preparations were not toxic to younger monkeys, this suggested that Aβ toxicity may be age‐dependent. Similar effects were not observed in rodents following the injection of fibrillar Aβ (38). These results in primates have suggested that Aβ neurotoxicity may be more pronounced in the aging brain. Therefore, longevity could contribute to the susceptibility to AD by rendering the brain more vulnerable to Aβ neurotoxicity.
To determine whether Aβ can affect tau pathology, preparations of synthetic fibrillar Aβ42 were injected into the somatosensory cortex and the hippocampal CA1 region of human wild‐type and P301L tau transgenic mice and non‐transgenic controls. This caused a fivefold increase in the number of NFTs in the amygdala of P301L tau mice, but not in wild‐type tau transgenic mice or control mice (51). In contrast, when the non‐fibrillogenic reverse peptide Aβ42–1 was injected, the number of NFTs was not altered. A different approach was adopted by Lewis et al, who crossed their P301L tau transgenic line with Aβ plaque‐forming APP mutant mice (89). They found that double transgenic mice had a more than sevenfold increase in NFTs in selected brain areas, as compared with P301L tau transgenic mice. When triple transgenic mice were injected intracerebrally with anti‐Aβ antibodies or a γ‐secretase inhibitor to prevent Aβ formation, this resulted in the disappearance of somatodendritic tau staining in young, but not old, mice, again demonstrating an interaction between Aβ and tau (110).
An interaction between Aβ and tau has also been demonstrated in cell lines (35, 113, 115, 151) and through the functional validation of proteomics findings (24). Transcriptomics and proteomics, collectively known as “Functional Genomics”, are being used increasingly to dissect the pathogenesis of neurodegenerative diseases (23, 68) (Figure 1). Using proteomics, mitochondrial proteins, antioxidant enzymes and synaptic proteins have been found to be differentially expressed in the brain of human P301L tau transgenic mice. Interestingly, the reduction in the levels of mitochondrial complex V was confirmed in human carriers with the P301L mutation. Functional analysis demonstrated mitochondrial dysfunction in P301L tau mice, together with reduced NADH‐ubiquinone oxidoreductase activity and, with age, impaired mitochondrial respiration and ATP synthesis. Mitochondrial dysfunction was associated with higher levels of reactive oxygen species in aged transgenic mice. The increased tau pathology in aged homozygous P301L tau mice revealed modified lipid peroxidation and an up‐regulation of antioxidant enzymes in response to oxidative stress. Therefore, it may be that changes in the respiratory chain linked to the appearance of oxidative damage lead to neuronal degeneration in AD brain.
To investigate whether brain cells from P301L tau mice are more susceptible to Aβ than controls, the mitochondrial membrane potential of isolated cortical brain cells was measured in the presence and absence of Aβ (24). Previous experiments using PC12 cells had shown that addition of Aβ leads to a significant decrease in mitochondrial membrane potential (76). Interestingly, it was found that in nerve cells from P301L tau mice the basal mitochondrial membrane potential was normal. A secondary insult with Aβ42 resulted in a greater reduction in the membrane potential of mitochondria from P301L tau mice than from controls. Importantly, this effect was region‐specific and therefore probably dependent on the presence of P301L mutant tau, as cells from the cerebrum (high P301L tau) were more susceptible than cells from the cerebellum (low P301L tau). These data have suggested a synergistic action of Aβ and tau on mitochondrial function. Moreover, it can be concluded that the tau pathology involves a mitochondrial and oxidative stress disorder distinct from that caused by Aβ (24).
In a subsequent study, both pathologies were combined by injecting Aβ42 into the brain of P301L tau mutant mice or by incubating neuroblastoma cells expressing human P301L tau with Aβ42 (25). By using proteomics, a significant fraction of the proteins altered in each system was found to be involved in responses to stress or to play a role in metabolic control. Model‐specific effects were also identified, such as differences in cell signaling proteins in the cellular model and cytoskeletal and synaptic proteins in the case of the animal model. By western blotting and immunohistochemistry, 72% of tested candidates were also altered in AD brain, with a major emphasis on proteins involved in the stress‐related unfolded protein response. These processes may therefore be important initiators of the Aβ42‐mediated pathogenic cascade in AD (25). In a separate study, proteomics was used in a line of mice transgenic for wild‐type human tau (146).
In addition to proteomics, transcriptomics has been applied to tau transgenic animal models. Using UniGem mouse microarray chips, inflammatory mediators and inhibitors of apoptosis were down‐regulated in human P301L tau transgenic brains (65). A second P301L tau transgenic line was analyzed using Affymetrix microarrays (21). A single up‐regulated gene, glyoxalase I, was identified. This enzyme plays a crucial role in the detoxification of dicarbonyl compounds by reducing the formation of AGEs. Levels of glyoxalase I mRNA and protein were significantly elevated in the brains of human P301L tau mice. Moreover, a glyoxalase I‐specific antiserum revealed many intensely stained, flame‐shaped neurons in AD brain, compared with control brains, suggesting a previously unidentified role for glyoxalase I (21). The finding that anti‐AGE antibodies reacted predominantly with intracellular NFTs, as opposed to extracellular NFTs, has suggested that AGE modifications may be involved in the early stages of disease and may not just be secondary consequences of tau deposition (15, 126).
Chaperones have been proposed to play a role in tau pathology (31, 134, 154), as supported by proteomics findings (25). The E3 ligase CHIP interacts with both Hsp90 and Hsc70 and targets the associated unfolded proteins to the proteasome (104). Hsc70 and CHIP have both been demonstrated to mediate tau degradation by the proteasome and Hsp90 has been found to inhibit tau aggregation (31, 134). To determine a role for CHIP in suppressing NFT formation in vivo, a quantitative analysis of CHIP levels was performed in human and mouse brains (124). Increased levels of CHIP and Hsp70 were found in AD compared with controls. In AD brain, the amount of CHIP was inversely proportional to that of sarkosyl‐insoluble tau. In human P301L tau transgenic mice, CHIP was widely distributed, but it was only weakly expressed in spinal cord, the region with the most tau inclusions and neuronal loss. In cerebellum from transgenic mice, CHIP levels were significantly higher than in controls. Human tau was more highly expressed in this region than in spinal cord, but only moderate levels of sarkosyl‐insoluble tau were detected. To further determine its role in tau aggregation, CHIP was deleted in mice (29). This led to the accumulation of non‐aggregated, ubiquitin‐negative and hyperphosphorylated tau. When P301L mutant tau was expressed on the CHIP null background, neither pre‐NFTs nor NFTs developed, despite an increased accumulation of phosphorylated tau. These findings, in conjunction with RNAi studies in C. elegans, have led to the suggestion that polyubiquitination of tau by CHIP may facilitate the formation of NFTs (29). In line with this, recent mass spectrometry studies of paired helical filament (PHF) tau (22) have suggested that ubiquitination of PHF‐tau may be an earlier pathological event than previously thought (8).
In search of modifiers. Tau protein is hyperphosphorylated in human neurodegenerative diseases. Consequently, much work has gone into the mapping of phosphorylation sites and the identification of candidate protein kinases and phosphatases. PP2A is the major tau phosphatase in brain (94). Its activity has been reported to be reduced in AD brain and dephosphorylation of tau can be blocked in cells by okadaic acid, an inhibitor of PP1 and PP2A (5, 46, 117). When rat brain slices were incubated with okadaic acid, tau became phosphorylated at several sites (111). Moreover, transgenic mice with neuronal expression of a dominant‐negative mutant form of the catalytic subunit of PP2A developed a pre‐NFT phenotype with hyperphosphorylation of murine tau (78). Subsequent analysis of these mice revealed that chronic inhibition of PP2A activity resulted in the activation of ERK and JNK, in the absence of apoptosis (79).
Tau phosphorylation has also been studied using transgenic expression of GSK‐3β and cyclin‐dependent kinase 5 (cdk5)/p25. Phosphorylation of serine 9 inactivates GSK‐3β and mice expressing the constitutively active S9AGSK‐3β displayed increased kinase activity, in the absence of neurofibrillary pathology. When they were crossed with mice transgenic for wild‐type human tau, axonopathy and motor deficits markedly improved (139). These findings have suggested that GSK‐3β may be protective. However, other studies using different models have reached the opposite conclusion.
One approach has been to use a tetracycline/doxycycline‐driven transactivation system. Transgenic mice overexpressing doxycycline‐regulatable GSK‐3β (Tet/GSK‐3β mice) were crossed with mice expressing tau with three FTDP‐17 mutations (VLW mice), giving rise to prefibrillar tau aggregates. They developed thioflavin S‐positive tau aggregates and filaments. Moreover, the atrophy of the dentate gyrus of the hippocampus, which was present in Tet/GSK‐3β mice, developed much faster in the Tet/GSK‐3β/VLW mice (33). To explore whether the phenotype resulting from increased GSK‐3 activity could revert following restoration of normal GSK‐3 levels, transgene expression was shut down in symptomatic mice. This led to normal phosphotau levels, diminished neuronal cell death and suppression of the cognitive deficits (34).
In another study, P301L tau transgenic mice were given lithium chloride, a reasonably specific GSK‐3 inhibitor. This resulted in significant inhibition of GSK‐3 activity, accompanied by a reduction in tau phosphorylation and a decrease in the levels of aggregated, sarkosyl‐insoluble tau. Administration of a separate GSK‐3 inhibitor also resulted in a reduction in sarkosyl‐insoluble tau, supporting the notion that lithium chloride exerted its effects through the inhibition of GSK‐3. Levels of aggregated tau correlated with the degree of axonal degeneration, which was ameliorated temporarily when lithium chloride was administered during early stages of NFT formation (107). In C. elegans, co‐expression of GSK‐3 and wild‐type tau was reported to bring about a deterioration in the touch response which was greater than that present in worms transgenic for tau alone. Co‐expression of the chaperone Hsp70 led to some improvement in the touch response (100).
A second protein kinase investigated through transgenic expression in mice is cdk5 and its activator p25. When human P301L tau transgenic mice were crossed with p25‐overexpressing mice, a fivefold increase in the number of NFTs was observed, alongside increased phosphorylation of tau at several sites (106). A similar phenotype has also been obtained when P301L tau transgenic mice were crossed with mice expressing a dominant‐negative mutant form of PP2A (Deters et al, unpublished). In a separate study, however, transgenic mouse lines that co‐expressed cdk5 and its activator p35, as well as wild‐type human tau protein, failed to exhibit increased phosphorylation of tau in nerve cells (148).
Cdk5, GSK‐3, and PP2A have been reported to co‐immunoprecipitate, suggesting a functional association (118). Overactivation of cdk5 has been shown to inhibit GSK‐3 activity in young mice. This effect was lost in older mice, resulting in increased GSK‐3 activity and phosphorylation of tau. The authors concluded that GSK‐3 is a key mediator of tau hyperphosphorylation, with cdk5 acting as modulator (118). More recently, a potential role for tyrosine phosphorylation of tau has emerged and a correlation between tyrosine phosphorylation and tau aggregation has been demonstrated (150). Disease‐related modifications of tau, that is, isoforms and FTD mutations, affected the interaction between tau and fyn, a non‐receptor tyrosine kinase of the Src family (12).
Parkin is a ubiquitin ligase, loss‐of‐function mutations of which cause familial forms of Parkinson’s disease (84). Some of these patients exhibit significant tau pathology (47). When parkin null mice were crossed with the VLW human tau transgenic mice, a number of abnormalities were observed that were not present in the single transgenic lines (98). They included progressive gait abnormalities, loss of dopamine and dopamine markers in striatum, nuclear tau‐immunoreactive deposits in motor neurons, abnormal expression of glial markers and enhanced levels of pro‐apoptotic proteins. In addition, the mice had reduced levels of CHIP‐Hsc70 and pSer9GSK‐3β. These findings suggest that interactions between tau and parkin could be relevant for the pathogenesis of at least some tauopathies.
Drosophila has been used in modifier screens (Figure 1). In a model tauopathy, protein kinases and phosphatases comprised the major classes of modifiers and several candidate kinases (GSK‐3 and cdk5 among them) enhanced tau toxicity in vivo (18, 135). Overall, increased tau phosphorylation correlated with an increase in toxicity. One exception was MARK/PAR‐1, which behaved as a genetic suppressor (135). MARK is a kinase that phosphorylates the KXGS motifs in the microtubule‐binding region of tau (7). In contrast to these findings, in a separate study, MARK/PAR‐1 enhanced tau toxicity by triggering an ordered phosphorylation process. Through loss‐of‐function and overexpression studies, it was shown that PAR‐1 plays a central role in regulating tau phosphorylation and toxicity in Drosophila, but without promoting NFT formation (105). In a third modifier screen, puromycin‐sensitive aminopeptidase (PSA) was identified and shown to protect against tau‐induced neurodegeneration in vivo, with loss‐of‐function of PSA exacerbating neurodegeneration (75).
In C. elegans, neuronal expression of human tau resulted in the accumulation of insoluble phosphorylated tau, neurodegeneration and uncoordinated movements. In a genome‐wide RNA interference screen, 16 757 genes were tested and 75 were found to enhance the transgene‐induced behavioral phenotype (82). Forty‐six of these genes had sequence similarities to known human genes and fell into a number of broad classes, including kinases, phosphatases, chaperones and proteases. They comprised some of the proteins discussed above, such as MARK, GSK‐3β, PP2A and CHIP. The other 29 modifiers only had sequence similarities with other nematode genes.
In conclusion, the tau transgenic animal studies carried out to date support a role for serine/threonine kinases (in particular ERK, JNK, p38, GSK3‐β, cdk5/p25 and MARK) and phosphatases (PP2A), as well as the chaperone system in diseases with filamentous tau deposits.
Treatment strategies. The search for novel therapeutic approaches to AD (Figure 1) has been stimulated by studies in Aβ plaque‐forming transgenic mice, in which active and passive immunization strategies have led to a reduction or the prevention of Aβ deposition in brain (9, 127, 136). Further work revealed that immunization can also reduce cognitive deficits in mouse models with Aβ accumulation (30, 74, 101) and, likewise, in AD patients (66, 67). Oddo et al demonstrated that a single intracerebral injection of anti‐Aβ antibodies into the hippocampus of triple transgenic mice significantly reduced extracellular and intracellular accumulation of Aβ and resulted in the clearance of early tau pathology (110). When the selective M1 muscarinic receptor agonist AF267B was assessed in this model, it was found to rescue the cognitive deficits in a spatial task, but not the deficits in contextual fear conditioning (17). These effects predicted the neuropathological findings, as Aβ and tau pathologies were reduced in cerebral cortex and hippocampus, but not in the amygdala. The reduction in tau pathology may have been mediated through a decrease in GSK‐3β activity. This study has suggested that selective M1 agonists may be efficacious for the treatment of AD.
To determine if the formation of NFTs could be reduced or even prevented using Aβ‐based vaccination, P301L tau transgenic mice were immunized with pre‐aggregated Aβ42, until high serum anti‐Aβ42 titers were reached (83). NFT formation was then induced through bilateral intrahippocampal injection of Aβ42 aggregates (51). To determine whether the previously reported Aβ42‐mediated increase in NFTs could be blocked by active immunization, the animals were analyzed 28 days after the injection. The intracerebral injection of Aβ42 caused a significant increase in NFTs, but this was not affected by immunization. It was concluded that active immunization was not sufficient to prevent the effects of Aβ42 on tau aggregation in this model system. Further studies are needed to determine whether modifications of the protocol used could affect the Aβ42‐mediated induction of NFT formation (83).
Modulation of tau phosphorylation and cleavage is another therapeutic strategy. It could benefit patients with AD and those with diseases characterized only by tau deposits. Several candidate tau kinases have been targeted in vivo. A major problem is that these kinases have many other functions besides phosphorylating tau (61, 90, 116). A recent study has evaluated this concept further by using an orally bioavailable and blood‐brain barrier‐penetrating analog of the relatively non‐specific protein kinase inhibitor K252a (85). This compound prevented motor deficits in the P301L tau transgenic mouse line JNPL3 (88) and reduced the levels of soluble aggregated, hyperphosphorylated tau. However, NFT numbers were not reduced, suggesting that the main cytotoxic effects of tau were not exerted by NFTs, but by as yet ill‐defined aggregates, named tau*, in analogy with Aβ*56 (85). This is in line with studies where a reduction in the expression of transgenic human P301L tau led to a recovery of memory and the stabilization of neuronal numbers, despite continued accumulation of NFTs (125). The protein kinase inhibitor rapamycin has been reported to reduce toxicity in Drosophila expressing wild‐type or mutant forms of tau, probably by reducing the amount of insoluble tau (11). Rapamycin induces autophagy through inhibition of TOR (108). It had a similar effect in a Drosophila model of Huntington’s disease (121, 122). It remains to be seen whether the beneficial effects of rapamycin may have had something to do with the TOR‐dependent cell cycle activation that has been described in Drosophila tauopathy models.
OUTLOOK
The identification of pathogenic mutations in AD and FTD has helped to understand disease pathogenesis and has paved the way for the development of animal models. These have been invaluable for dissecting pathogenic mechanisms, for identifying disease modifiers and for developing novel therapeutic strategies (Figure 1). Although AD is the most common dementia (48, 53, 86), tau‐containing lesions are also present in several other neurodegenerative diseases, where they form in the absence of extracellular plaques (130). It follows that Aβ‐directed therapies are likely to be ineffective in these diseases, unless they target common pathogenic mechanisms or structural characteristics shared between tau and Aβ (55).
ACKNOWLEDGMENTS
JG is a Medical Foundation Fellow and has been supported by the University of Sydney, the National Health and Medical Research Council (NHMRC), the Australian Research Council (ARC), the New South Wales Government through the Ministry for Science and Medical Research (BioFirst Program), the Nerve Research Foundation, the Medical Foundation (University of Sydney) and the Mason Foundation.
REFERENCES
- 1. Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M (2002) Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci 22:9340–9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andorfer C, Kress Y, Espinoza M, De Silva R, Tucker KL, Barde YA, Duff K, Davies P (2003) Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem 86:582–590. [DOI] [PubMed] [Google Scholar]
- 3. Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P (2005) Cell‐cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci 25:5446–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arendt T (2003) Synaptic plasticity and cell cycle activation in neurons are alternative effector pathways: the “Dr. Jekyll and Mr. Hyde concept” of Alzheimer’s disease or the yin and yang of neuroplasticity. Prog Neurobiol 71:83–248. [DOI] [PubMed] [Google Scholar]
- 5. Arendt T, Holzer M, Fruth R, Brückner MK, Gärtner U (1995) Paired helical filament‐like phosphorylation of tau, deposition of beta/A4‐amyloid and memory impairment in rat induced by chronic inhibition of phosphatase 1 and 2A. Neuroscience 69:691–698. [DOI] [PubMed] [Google Scholar]
- 6. Arnold SE, Han LY, Clark CM, Grossman M, Trojanowski JQ (2000) Quantitative neurohistological features of frontotemporal degeneration. Neurobiol Aging 21:913–919. [DOI] [PubMed] [Google Scholar]
- 7. Augustinack JC, Schneider A, Mandelkow EM, Hyman BT (2002) Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol (Berl) 103:26–35. [DOI] [PubMed] [Google Scholar]
- 8. Bancher C, Grundke‐Iqbal I, Iqbal K, Fried VA, Smith HT, Wisniewski HM (1991) Abnormal phosphorylation of tau precedes ubiquitination in neurofibrillary pathology of Alzheimer disease. Brain Res 539:11–18. [DOI] [PubMed] [Google Scholar]
- 9. Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson‐Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T (2000) Peripherally administered antibodies against amyloid beta‐peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6:916–919. [DOI] [PubMed] [Google Scholar]
- 10. Baumeister R, Ge L (2002) The worm in us—caenorhabditis elegans as a model of human disease. Trends Biotechnol 20:147–148. [DOI] [PubMed] [Google Scholar]
- 11. Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, Schmitt I, Wullner U, Evert BO, O’Kane CJ, Rubinsztein DC (2006) Rapamycin alleviates toxicity of different aggregate‐prone proteins. Hum Mol Genet 15:433–442. [DOI] [PubMed] [Google Scholar]
- 12. Bhaskar K, Yen SH, Lee G (2005) Disease‐related modifications in tau affect the interaction between Fyn and Tau. J Biol Chem 280:35119–35125. [DOI] [PubMed] [Google Scholar]
- 13. Brion JP, Tremp G, Octave JN (1999) Transgenic expression of the shortest human tau affects its compartmentalization and its phosphorylation as in the pretangle stage of Alzheimer’s disease. Am J Pathol 154:255–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Broggio E, Pluchon C, Ingrand P, Gil R (2001) Taste impairment in Alzheimer’s disease. Rev Neurol (Paris) 157:409–413. [PubMed] [Google Scholar]
- 15. Buée L, Bussière T, Buée‐Scherrer V, Delacourte A, Hof PR (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 33:95–130. [DOI] [PubMed] [Google Scholar]
- 16. Bugiani O, Murrell JR, Giaccone G, Hasegawa M, Ghigo G, Tabaton M, Morbin M, Primavera A, Carella F, Solaro C, Grisoli M, Savoiardo M, Spillantini MG, Tagliavini F, Goedert M, Ghetti B (1999) Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J Neuropathol Exp Neurol 58:667–677. [DOI] [PubMed] [Google Scholar]
- 17. Caccamo A, Oddo S, Billings LM, Green KN, Martinez‐Coria H, Fisher A, LaFerla FM (2006) M1 receptors play a central role in modulating AD‐like pathology in transgenic mice. Neuron 49:671–682. [DOI] [PubMed] [Google Scholar]
- 18. Chau KW, Chan WY, Shaw PC, Chan HY (2006) Biochemical investigation of Tau protein phosphorylation status and its solubility properties in Drosophila. Biochem Biophys Res Commun 346:150–159. [DOI] [PubMed] [Google Scholar]
- 19. Chee FC, Mudher A, Cuttle MF, Newman TA, MacKay D, Lovestone S, Shepherd D (2005) Over‐expression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Neurobiol Dis 20:918–928. [DOI] [PubMed] [Google Scholar]
- 20. Chen F, David D, Ferrari A, Götz J (2004) Posttranslational modifications of tau—role in human tauopathies and modeling in transgenic animals. Curr Drug Targets 5:503–515. [DOI] [PubMed] [Google Scholar]
- 21. Chen F, Wollmer MA, Hoerndli F, Münch G, Kuhla B, Rogaev EI, Tsolaki M, Papassotiropoulos A, Götz J (2004) Role for glyoxalase I in Alzheimer’s disease. Proc Natl Acad Sci USA 101:7687–7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cripps D, Thomas SN, Jeng Y, Yang F, Davies P, Yang AJ (2006) Alzheimer disease‐specific conformation of hyperphosphorylated paired helical filament‐Tau is polyubiquitinated through Lys‐48, Lys‐11, and Lys‐6 ubiquitin conjugation. J Biol Chem 281:10825–10838. [DOI] [PubMed] [Google Scholar]
- 23. David D, Hoerndli F, Götz J (2005) Functional Genomics meets neurodegenerative disorders Part I: transcriptomic and proteomic technology. Prog Neurobiol 76:153–168. [DOI] [PubMed] [Google Scholar]
- 24. David DC, Hauptmann S, Scherping I, Schuessel K, Keil U, Rizzu P, Ravid R, Dröse S, Brandt U, Müller WE, Eckert E, Götz J (2005) Proteomic and functional analysis reveal a mitochondrial dysfunction in P301L tau transgenic mice. J Biol Chem 280:23802–23814. [DOI] [PubMed] [Google Scholar]
- 25. David DC, Ittner LM, Gehrig P, Nergenau D, Shepherd C, Halliday G, Götz J (2006) β‐Amyloid treatment of two complementary P301L tau‐expressing Alzheimer’s disease models reveals similar deregulated cellular processes. Proteomics 6:6566–6577. [DOI] [PubMed] [Google Scholar]
- 26. Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP (2001) Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci 114:1179–1187. [DOI] [PubMed] [Google Scholar]
- 27. Delacourte A, Sergeant N, Champain D, Wattez A, Maurage CA, Lebert F, Pasquier F, David JP (2002) Nonoverlapping but synergetic tau and APP pathologies in sporadic Alzheimer’s disease. Neurology 59:398–407. [DOI] [PubMed] [Google Scholar]
- 28. Delobel P, Lavenir I, Ghetti B, Holzer M, Goedert M (2006) Cell‐cycle markers in a transgenic mouse model of human tauopathy: increased levels of cyclin‐dependent kinase inhibitors p21Cip1 and p27Kip1. Am J Pathol 168:878–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dickey CA, Yue M, Lin WL, Dickson DW, Dunmore JH, Lee WC, Zehr C, West G, Cao S, Clark AM, Caldwell GA, Caldwell KA, Eckman C, Patterson C, Hutton M, Petrucelli L (2006) Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho‐ and caspase‐3‐cleaved tau species. J Neurosci 26:6985–6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM (2002) Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci 5:452–457. [DOI] [PubMed] [Google Scholar]
- 31. Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H (2003) Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci USA 100:721–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Duff K, Knight H, Refolo LM, Sanders S, Yu X, Picciano M, Malester B, Hutton M, Adamson J, Goedert M, Bürki K, Davies P (2000) Characterization of pathology in transgenic mice over‐expressing human genomic and cDNA tau transgenes. Neurobiol Dis 7:87–98. [DOI] [PubMed] [Google Scholar]
- 33. Engel T, Lucas JJ, Gomez‐Ramos P, Moran MA, Avila J, Hernandez F (2006) Co‐expression of FTDP‐17 tau and GSK‐3β in transgenic mice induce tau polymerization and neurodegeneration. Neurobiol Aging 27:1258–1268. [DOI] [PubMed] [Google Scholar]
- 34. Engel T, Hernandez F, Avila J, Lucas JJ (2006) Full reversal of Alzheimer’s disease‐like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase‐3. J Neurosci 26:5083–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ferrari A, Hoerndli F, Baechi T, Nitsch RM, Götz J (2003) Beta‐amyloid induces PHF‐like tau filaments in tissue culture. J Biol Chem 278:40162–40168. [DOI] [PubMed] [Google Scholar]
- 36. Forman MS, Lal D, Zhang B, Dabir DV, Swanson E, Lee VM, Trojanowski JQ (2005) Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J Neurosci 25:3539–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, Parks AL, Xu W, Li J, Gurney M, Myers RL, Himes CS, Hiebsch R, Ruble C, Nye JS, Curtis D (2002) aph‐1 and pen‐2 are required for Notch pathway signaling, gamma‐secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell 3:85–97. [DOI] [PubMed] [Google Scholar]
- 38. Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA (1998) Aging renders the brain vulnerable to amyloid beta‐protein neurotoxicity. Nat Med 4:827–831. [DOI] [PubMed] [Google Scholar]
- 39. Giannakopoulos P, Herrmann FR, Bussière T, Bouras C, Kovari E, Perl DP, Morrison JH, Gold G, Hof PR (2003) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60:1495–1500. [DOI] [PubMed] [Google Scholar]
- 40. Glenner GG, Wong CW (1984) Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120:885–890. [DOI] [PubMed] [Google Scholar]
- 41. Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A (1988) Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule‐associated protein tau. Proc Natl Acad Sci USA 85:4051–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goedert M, Jakes R, Crowther RA, Six J, Lübke U, Vandermeeren M, Cras P, Trojanowski JQ, Lee VMY (1993) The abnormal phosphorylation of tau protein at Ser‐202 in Alzheimer disease recapitulates phosphorylation during development. Proc Natl Acad Sci USA 90:5066–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goedert M, Spillantini MG, Jakes R, Crowther RA, Vanmechelen E, Probst A, Götz J, Bürki K, Cohen P (1995) Molecular dissection of the paired helical filament. Neurobiol Aging 16:325–334. [DOI] [PubMed] [Google Scholar]
- 44. Goedert M, Spillantini MG, Crowther RA, Chen SG, Parchi P, Tabaton M, Lanska DJ, Markesbery WR, Wilhelmsen KC, Dickson DW, Petersen RB, Gambetti P (1999) Tau gene mutation in familial progressive subcortical gliosis. Nat Med 5:454–457. [DOI] [PubMed] [Google Scholar]
- 45. Gomez‐Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT (1997) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol 41:17–24. [DOI] [PubMed] [Google Scholar]
- 46. Gong CX, Singh TJ, Grundke‐Iqbal I, Iqbal K (1993) Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem 61:921–927. [DOI] [PubMed] [Google Scholar]
- 47. Gosal D, Ross OA, Toft M (2006) Parkinson’s disease: the genetics of a heterogeneous disorder. Eur J Neurol 13:616–627. [DOI] [PubMed] [Google Scholar]
- 48. Götz J (2001) Tau and transgenic animal models. Brain Res Brain Res Rev 35:266–286. [DOI] [PubMed] [Google Scholar]
- 49. Götz J, Probst A, Spillantini MG, Schäfer T, Jakes R, Bürki K, Goedert M (1995) Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J 14:1304–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Götz J, Chen F, Barmettler R, Nitsch RM (2001) Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem 276:529–534. [DOI] [PubMed] [Google Scholar]
- 51. Götz J, Chen F, Van Dorpe J, Nitsch RM (2001) Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Abeta 42 fibrils. Science 293:1491–1495. [DOI] [PubMed] [Google Scholar]
- 52. Götz J, Tolnay M, Barmettler R, Chen F, Probst A, Nitsch RM (2001) Oligodendroglial tau filament formation in transgenic mice expressing G272V tau. Eur J Neurosci 13:2131–2140. [DOI] [PubMed] [Google Scholar]
- 53. Götz J, Streffer JR, David D, Schild A, Hoerndli F, Pennanen L, Kurosinski P, Chen F (2004) Transgenic animal models of Alzheimer’s disease and related disorders: histopathology, behavior and therapy. Mol Psychiatry 9:664–683. [DOI] [PubMed] [Google Scholar]
- 54. Götz J, Ittner LM, Kins S (2006) Do axonal defects in tau and amyloid precursor protein transgenic animals model axonopathy in Alzheimer’s disease? J Neurochem 98:993–1006. [DOI] [PubMed] [Google Scholar]
- 55. Götz J, Ittner LM, Schonrock N (2006) Alzheimer’s disease and frontotemporal dementia: prospects of a tailored therapy? Med J Aust 185:381–384. [DOI] [PubMed] [Google Scholar]
- 56. Hall GF, Cohen MJ (1983) Extensive dendritic sprouting induced by close axotomy of central neurons in the lamprey. Science 222:518–521. [DOI] [PubMed] [Google Scholar]
- 57. Hall GF, Chu B, Lee G, Yao J (2000) Human tau filaments induce microtubule and synapse loss in an in vivo model of neurofibrillary degenerative disease. J Cell Sci 113:1373–1387. [DOI] [PubMed] [Google Scholar]
- 58. Hall GF, Lee VM, Lee G, Yao J (2001) Staging of neurofibrillary degeneration caused by human tau overexpression in a unique cellular model of human tauopathy. Am J Pathol 158:235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hall GF, Lee S, Yao J (2002) Neurofibrillary degeneration can be arrested in an in vivo cellular model of human tauopathy by application of a compound which inhibits tau filament formation in vitro. J Mol Neurosci 19:253–260. [DOI] [PubMed] [Google Scholar]
- 60. Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato‐Yoshitake R, Takei Y, Noda T, Hirokawa N (1994) Altered microtubule organization in small‐calibre axons of mice lacking tau protein. Nature 369:488–491. [DOI] [PubMed] [Google Scholar]
- 61. Hernandez F, Lim F, Lucas JJ, Perez‐Martin C, Moreno F, Avila J (2002) Transgenic mouse models with tau pathology to test therapeutic agents for Alzheimer’s disease. Mini Rev Med Chem 2:51–58. [DOI] [PubMed] [Google Scholar]
- 62. Higuchi M, Ishihara T, Zhang B, Hong M, Andreadis A, Trojanowski J, Lee VMY (2002) Transgenic mouse model of tauopathies with glial pathology and nervous system degeneration. Neuron 35:433–446. [DOI] [PubMed] [Google Scholar]
- 63. Higuchi M, Zhang B, Forman MS, Yoshiyama Y, Trojanowski JQ, Lee VMY (2005) Axonal degeneration induced by targeted expression of mutant human tau in oligodendrocytes of transgenic mice that model glial tauopathies. J Neurosci 25:9434–9443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hirano A, Nakano I, Kurland LT, Mulder DW, Holley PW, Saccomanno G (1984) Fine structural study of neurofibrillary changes in a family with amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 43:471–480. [DOI] [PubMed] [Google Scholar]
- 65. Ho L, Xiang Z, Mukherjee P, Zhang W, De Jesus N, Mirjany M, Yemul S, Pasinetti GM (2001) Gene expression profiling of the tau mutant (P301L) transgenic mouse brain. Neurosci Lett 310:1–4. [DOI] [PubMed] [Google Scholar]
- 66. Hock C, Konietzko U, Papassotiropoulos A, Wollmer A, Streffer J, Von Rotz RC, Davey G, Moritz E, Nitsch RM (2002) Generation of antibodies specific for beta‐amyloid by vaccination of patients with Alzheimer disease. Nat Med 8:1270–1275. [DOI] [PubMed] [Google Scholar]
- 67. Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller‐Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, De Quervain DJ, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM (2003) Antibodies against beta‐amyloid slow cognitive decline in Alzheimer’s disease. Neuron 38:547–554. [DOI] [PubMed] [Google Scholar]
- 68. Hoerndli F, David D, Götz J (2005) Functional genomics meets neurodegenerative disorders. Part II: transcriptomic and proteomic technology. Prog Neurobiol 76:169–188. [DOI] [PubMed] [Google Scholar]
- 69. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering‐Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, De Graaff E, Wauters E, Van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, Van Swieten J, Mann D, Lynch T, Heutink P (1998) Association of missense and 5′‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature 393:702–705. [DOI] [PubMed] [Google Scholar]
- 70. Ikegami S, Harada A, Hirokawa N (2000) Muscle weakness, hyperactivity, and impairment in fear conditioning in tau‐deficient mice. Neurosci Lett 279:129–132. [DOI] [PubMed] [Google Scholar]
- 71. Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VMY (1999) Age‐dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron 24:751–762. [DOI] [PubMed] [Google Scholar]
- 72. Ishihara T, Zhang B, Higuchi M, Yoshiyama Y, Trojanowski JQ, Lee VMY (2001) Age‐dependent induction of congophilic neurofibrillary tau inclusions in tau transgenic mice. Am J Pathol 158:555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jackson GR, Wiedau‐Pazos M, Sang T‐K, Wagle N, Brown CA, Massachi S, Geschwind DH (2002) Human wild‐type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 34:509–519. [DOI] [PubMed] [Google Scholar]
- 74. Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George‐Hyslop P, Westaway D (2000) A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 408:979–982. [DOI] [PubMed] [Google Scholar]
- 75. Karsten SL, Sang TK, Gehman LT, Chatterjee S, Liu J, Lawless GM, Sengupta S, Berry RW, Pomakian J, Oh HS, Schulz C, Hui KS, Wiedau‐Pazos M, Vinters HV, Binder LI, Geschwind DH, Jackson GR (2006) A genomic screen for modifiers of tauopathy identifies puromycin‐sensitive aminopeptidase as an inhibitor of tau‐induced neurodegeneration. Neuron 51:549–560. [DOI] [PubMed] [Google Scholar]
- 76. Keil U, Bonert A, Marques CA, Scherping I, Weyermann J, Strosznajder JB, Muller‐Spahn F, Haass C, Czech C, Pradier L, Muller WE, Eckert A (2004) Amyloid beta‐induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J Biol Chem 279:50310–50320. [DOI] [PubMed] [Google Scholar]
- 77. Khurana V, Lu Y, Steinhilb ML, Oldham S, Shulman JM, Feany MB (2006) TOR‐mediated cell‐cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr Biol 16:230–241. [DOI] [PubMed] [Google Scholar]
- 78. Kins S, Crameri A, Evans DR, Hemmings BA, Nitsch RM, Götz J (2001) Reduced PP2A activity induces hyperphosphorylation and altered compartmentalization of tau in transgenic mice. J Biol Chem 276:38193–38200. [DOI] [PubMed] [Google Scholar]
- 79. Kins S, Kurosinski P, Nitsch RM, Götz J (2003) Activation of the ERK and JNK signaling pathways caused by neuron specific inhibition of PP2A in transgenic mice. Am J Pathol 163:833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Koolen DA, Vissers LE, Pfundt R, De Leeuw N, Knight SJ, Regan R, Kooy RF, Reyniers E, Romano C, Fichera M, Schinzel A, Baumer A, Anderlid BM, Schoumans J, Knoers NV, Van Kessel AG, Sistermans EA, Veltman JA, Brunner HG, De Vries BB (2006) A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet 38:999–1001. [DOI] [PubMed] [Google Scholar]
- 81. Kraemer BC, Zhang B, Leverenz JB, Thomas JH, Trojanowski JQ, Schellenberg GD (2003) Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci USA 100:9980–9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kraemer BC, Burgess JK, Chen JH, Thomas JH, Schellenberg GD (2006) Molecular pathways that influence human tau‐induced pathology in Caenorhabditis elegans. Hum Mol Genet 15:1483–1496. [DOI] [PubMed] [Google Scholar]
- 83. Kulic L, Kurosinski P, Chen F, Tracy J, Mohajeri MH, Li H, Nitsch RM, Götz J (2005) Active immunization trial in Abeta(42)‐injected P301L tau transgenic mice. Neurobiol Dis 22:50–56. [DOI] [PubMed] [Google Scholar]
- 84. Kurosinski P, Guggisberg M, Götz J (2002) Alzheimer’s and Parkinson’s disease—overlapping or synergistic pathologies? Trends Mol Med 8:3–5. [DOI] [PubMed] [Google Scholar]
- 85. Le Corre S, Klafki HW, Plesnila N, Hubinger G, Obermeier A, Sahagun H, Monse B, Seneci P, Lewis J, Eriksen J, Zehr C, Yue M, McGowan E, Dickson DW, Hutton M, Roder HM (2006) An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc Natl Acad Sci USA 103:9673–9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lee VMY, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159. [DOI] [PubMed] [Google Scholar]
- 87. Lee VMY, Kenyon TK, Trojanowski JQ (2005) Transgenic animal models of tauopathies. Biochim Biophys Acta 1739:251–259. [DOI] [PubMed] [Google Scholar]
- 88. Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn‐Hardy K, Murphy PM, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M (2000) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25:402–405. [DOI] [PubMed] [Google Scholar]
- 89. Lewis J, Dickson DW, Lin W‐L, Chisholm L, Corral A, Jones G, Yen S‐H, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E (2001) Enhanced neurofibrillary degeneration in transgenic mice expressing mutant Tau and APP. Science 293:1487–1491. [DOI] [PubMed] [Google Scholar]
- 90. Li G, Faibushevich A, Turunen BJ, Yoon SO, Georg G, Michaelis ML, Dobrowsky RT (2003) Stabilization of the cyclin‐dependent kinase 5 activator, p35, by paclitaxel decreases beta‐amyloid toxicity in cortical neurons. J Neurochem 84:347–362. [DOI] [PubMed] [Google Scholar]
- 91. Lim F, Hernandez F, Lucas JJ, Gomez‐Ramos P, Moran MA, Avila J (2001) FTDP‐17 mutations in tau transgenic mice provoke lysosomal abnormalities and Tau filaments in forebrain. Mol Cell Neurosci 18:702–714. [DOI] [PubMed] [Google Scholar]
- 92. Lin SL, Ying SY (2006) Gene silencing in vitro and in vivo using intronic microRNAs. Methods Mol Biol 342:295–312. [DOI] [PubMed] [Google Scholar]
- 93. Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW (2003) Filamentous tau in oligodendrocytes and astrocytes of transgenic mice expressing the human tau isoform with the P301L mutation. Am J Pathol 162:213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Liu F, Grundke‐Iqbal I, Iqbal K, Gong CX (2005) Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci 22:1942–1950. [DOI] [PubMed] [Google Scholar]
- 95. Lupski JR (2006) Genome structural variation and sporadic disease traits. Nat Genet 38:974–976. [DOI] [PubMed] [Google Scholar]
- 96. Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L (2000) Dopaminergic loss and inclusion body formation in alpha‐synuclein mice: implications for neurodegenerative disorders. Science 287: 1265–1269. [DOI] [PubMed] [Google Scholar]
- 97. Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA 82:4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Menendez J, Rodriguez‐Navarro JA, Solano RM, Casarejos MJ, Rodal I, Guerrero R, Sanchez MP, Avila J, Mena MA, De Yebenes JG (2006) Suppression of Parkin enhances nigrostriatal and motor neuron lesion in mice over‐expressing human‐mutated tau protein. Hum Mol Genet 15:2045–2058. [DOI] [PubMed] [Google Scholar]
- 99. Mershin A, Pavlopoulos E, Fitch O, Braden BC, Nanopoulos DV, Skoulakis EM (2004) Learning and memory deficits upon TAU accumulation in Drosophila mushroom body neurons. Learn Mem 11:277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Miyasaka T, Ding Z, Gengyo‐ando K, Oue M, Yamaguchi H, Mitani S, Ihara Y (2005) Progressive neurodegeneration in C. elegans model of tauopathy. Neurobiol Dis 20:372–383. [DOI] [PubMed] [Google Scholar]
- 101. Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW (2000) A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature 408:982–985. [DOI] [PubMed] [Google Scholar]
- 102. Mudher A, Shepherd D, Newman TA, Mildren P, Jukes JP, Squire A, Mears A, Drummond JA, Berg S, MacKay D, Asuni AA, Bhat R, Lovestone S (2004) GSK‐3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol Psychiatry 9:522–530. [DOI] [PubMed] [Google Scholar]
- 103. Munoz DG, Greene C, Perl DP, Selkoe DJ (1988) Accumulation of phosphorylated neurofilaments in anterior horn motoneurons of amyotrophic lateral sclerosis patients. J Neuropathol Exp Neurol 47:9–18. [DOI] [PubMed] [Google Scholar]
- 104. Murata S, Minami Y, Minami M, Chiba T, Tanaka K (2001) CHIP is a chaperone‐dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep 2:1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Nishimura I, Yang Y, Lu B (2004) PAR‐1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell 116:671–682. [DOI] [PubMed] [Google Scholar]
- 106. Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, Gaynor K, LaFrancois J, Wang L, Kondo T, Davies P, Burns M, Veeranna, Nixon R, Dickson D, Matsuoka Y, Ahlijanian M, Lau LF, Duff K (2003) Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 38:555–565. [DOI] [PubMed] [Google Scholar]
- 107. Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K (2005) Inhibition of glycogen synthase kinase‐3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA 102:6990–6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Noda T, Ohsumi Y (1998) Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 273:3963–3966. [DOI] [PubMed] [Google Scholar]
- 109. Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple‐transgenic model of Alzheimer’s disease with plaques and tangles. Intracellular abeta and synaptic dysfunction. Neuron 39:409–421. [DOI] [PubMed] [Google Scholar]
- 110. Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM (2004) Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43:321–332. [DOI] [PubMed] [Google Scholar]
- 111. Pei JJ, Gong CX, An WL, Winblad B, Cowburn RF, Grundke‐Iqbal I, Iqbal K (2003) Okadaic‐acid‐induced inhibition of protein phosphatase 2A produces activation of mitogen‐activated protein kinases ERK1/2, MEK1/2, and p70 S6, similar to that in Alzheimer’s disease. Am J Pathol 163:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Pennanen L, Welzl H, D’Adamo P, Nitsch RM, Götz J (2004) Accelerated extinction of conditioned taste aversion in P301L tau transgenic mice. Neurobiol Dis 15:500–509. [DOI] [PubMed] [Google Scholar]
- 113. Pennanen L, Götz J (2005) Different tau epitopes define Abeta(42)‐mediated tau insolubility. Biochem Biophys Res Commun 337:1097–1101. [DOI] [PubMed] [Google Scholar]
- 114. Pennanen L, Wolfer DP, Nitsch RM, Götz J (2006) Impaired spatial reference memory and increased exploratory behavior in P301L tau transgenic mice. Genes Brain Behav 5:369–379. [DOI] [PubMed] [Google Scholar]
- 115. Perez M, Hernandez F, Gomez‐Ramos A, Smith M, Perry G, Avila J (2002) Formation of aberrant phosphotau fibrillar polymers in neural cultured cells. Eur J Biochem 269:1484–1489. [DOI] [PubMed] [Google Scholar]
- 116. Phiel CJ, Wilson CA, Lee VM, Klein PS (2003) GSK‐3alpha regulates production of Alzheimer’s disease amyloid‐beta peptides. Nature 423:435–439. [DOI] [PubMed] [Google Scholar]
- 117. Planel E, Yasutake K, Fujita SC, Ishiguro K (2001) Inhibition of protein phosphatase 2A overrides tau protein kinase I/glycogen synthase kinase 3 beta and cyclin‐dependent kinase 5 inhibition and results in tau hyperphosphorylation in the hippocampus of starved mouse. J Biol Chem 276:34298–34306. [DOI] [PubMed] [Google Scholar]
- 118. Plattner F, Angelo M, Giese KP (2006) The roles of cyclin‐dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J Biol Chem 281:25457–25465. [DOI] [PubMed] [Google Scholar]
- 119. Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43:815–825. [DOI] [PubMed] [Google Scholar]
- 120. Probst A, Götz J, Wiederhold KH, Tolnay M, Mistl C, Jaton AL, Hong M, Ishihara T, Lee VMY, Trojanowski JQ, Jakes R, Crowther RA, Spillantini MG, Bürki K, Goedert M (2000) Axonopathy and amyotrophy in mice transgenic for human four‐repeat tau protein. Acta Neuropathol (Berl) 99: 469–481. [DOI] [PubMed] [Google Scholar]
- 121. Ravikumar B, Duden R, Rubinsztein DC (2002) Aggregate‐prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 11:1107–1117. [DOI] [PubMed] [Google Scholar]
- 122. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36:585–595. [DOI] [PubMed] [Google Scholar]
- 123. Rouleau GA, Clark AW, Rooke K, Pramatarova A, Krizus A, Suchowersky O, Julien JP, Figlewicz D (1996) SOD1 mutation is associated with accumulation of neurofilaments in amyotrophic lateral sclerosis. Ann Neurol 39:128–131. [DOI] [PubMed] [Google Scholar]
- 124. Sahara N, Murayama M, Mizoroki T, Urushitani M, Imai Y, Takahashi R, Murata S, Tanaka K, Takashima A (2005) In vivo evidence of CHIP up‐regulation attenuating tau aggregation. J Neurochem 94:1254–1263. [DOI] [PubMed] [Google Scholar]
- 125. Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309:476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Sasaki N, Fukatsu R, Tsuzuki K, Hayashi Y, Yoshida T, Fujii N, Koike T, Wakayama I, Yanagihara R, Garruto R, Amano N, Makita Z (1998) Advanced glycation end products in Alzheimer’s disease and other neurodegenerative diseases. Am J Pathol 153:1149–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson‐Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P (1999) Immunization with amyloid‐beta attenuates Alzheimer‐disease‐like pathology in the PDAPP mouse. Nature 400:173–177. [DOI] [PubMed] [Google Scholar]
- 128. Schiffman SS, Clark CM, Warwick ZS (1990) Gustatory and olfactory dysfunction in dementia: not specific to Alzheimer’s disease. Neurobiol Aging 11:597–600. [DOI] [PubMed] [Google Scholar]
- 129. Schindowski K, Bretteville A, Leroy K, Begard S, Brion JP, Hamdane M, Buée L (2006) Alzheimer’s disease‐like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol 169:599–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Shahani N, Brandt R (2002) Functions and malfunctions of the tau proteins. Cell Mol Life Sci 59:1668–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Shahani N, Subramaniam S, Wolf T, Tackenberg C, Brandt R (2006) Tau aggregation and progressive neuronal degeneration in the absence of changes in spine density and morphology after targeted expression of Alzheimer’s disease‐relevant tau constructs in organotypic hippocampal slices. J Neurosci 26:6103–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, Fitzpatrick CA, Segraves R, Richmond TA, Guiver C, Albertson DG, Pinkel D, Eis PS, Schwartz S, Knight SJ, Eichler EE (2006) Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet 38:1038–1042. [DOI] [PubMed] [Google Scholar]
- 133. Shaw‐Smith C, Pittman AM, Willatt L, Martin H, Rickman L, Gribble S, Curley R, Cumming S, Dunn C, Kalaitzopoulos D, Porter K, Prigmore E, Krepischi‐Santos AC, Varela MC, Koiffmann CP, Lees AJ, Rosenberg C, Firth HV, De Silva R, Carter NP (2006) Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat Genet 38:1032–1037. [DOI] [PubMed] [Google Scholar]
- 134. Shimura H, Schwartz D, Gygi SP, Kosik KS (2004) CHIP‐Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem 279:4869–4876. [DOI] [PubMed] [Google Scholar]
- 135. Shulman JM, Feany MB (2003) Genetic modifiers of tauopathy in Drosophila. Genetics 165:1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Sigurdsson EM, Scholtzova H, Mehta PD, Frangione B, Wisniewski T (2001) Immunization with a nontoxic/nonfibrillar amyloid‐beta homologous peptide reduces Alzheimer’s disease‐associated pathology in transgenic mice. Am J Pathol 159:439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 95:7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van Lommel A, Loos R, Van Leuven F (1999) Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four‐repeat human tau protein. Am J Pathol 155:2153–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Spittaels K, Van Den Haute C, Van Dorpe J, Geerts H, Mercken M, Bruynseels K, Lasrado R, Vandezande K, Laenen I, Boon T, Van Lint J, Vandenheede J, Moechars D, Loos R, Van Leuven F (2000) Glycogen synthase kinase‐3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four‐repeat tau transgenic mice. J Biol Chem 275:41340–41349. [DOI] [PubMed] [Google Scholar]
- 140. Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 307: 1282–1288. [DOI] [PubMed] [Google Scholar]
- 141. Tanemura K, Akagi T, Murayama M, Kikuchi N, Murayama O, Hashikawa T, Yoshiike Y, Park JM, Matsuda K, Nakao S, Sun X, Sato S, Yamaguchi H, Takashima A (2001) Formation of filamentous tau aggregations in transgenic mice expressing V337M human tau. Neurobiol Dis 8:1036–1045. [DOI] [PubMed] [Google Scholar]
- 142. Tanemura K, Murayama M, Akagi T, Hashikawa T, Tominaga T, Ichikawa M, Yamaguchi H, Takashima A (2002) Neurodegeneration with tau accumulation in a transgenic mouse expressing V337M human tau. J Neurosci 22:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Tashiro K, Hasegawa M, Ihara Y, Iwatsubo T (1997) Somatodendritic localization of phosphorylated tau in neonatal and adult rat cerebral cortex. Neuroreport 8:2797–2801. [DOI] [PubMed] [Google Scholar]
- 144. Tatebayashi Y, Miyasaka T, Chui DH, Akagi T, Mishima K, Iwasaki K, Fujiwara M, Tanemura K, Murayama M, Ishiguro K, Planel E, Sato S, Hashikawa T, Takashima A (2002) Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc Natl Acad Sci USA 99:13896–13901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van Haesendonck C, Borghgraef P, Van Leuven F (2005) Changed conformation of mutant Tau‐P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau‐4R/2N transgenic mice. J Biol Chem 280:3963–3973. [DOI] [PubMed] [Google Scholar]
- 146. Tilleman K, Van Den Haute C, Geerts H, Van Leuven F, Esmans EL, Moens L (2002) Proteomics analysis of the neurodegeneration in the brain of tau transgenic mice. Proteomics 2:656–665. [DOI] [PubMed] [Google Scholar]
- 147. Tucker KL, Meyer M, Barde YA (2001) Neurotrophins are required for nerve growth during development. Nat Neurosci 4:29–37. [DOI] [PubMed] [Google Scholar]
- 148. Van den Haute C, Spittaels K, Van Dorpe J, Lasrado R, Vandezande K, Laenen I, Geerts H, Van Leuven F (2001) Coexpression of human cdk5 and its activator p35 with human protein tau in neurons in brain of triple transgenic mice. Neurobiol Dis 8:32–44. [DOI] [PubMed] [Google Scholar]
- 149. Varela MC, Krepischi‐Santos AC, Paz JA, Knijnenburg J, Szuhai K, Rosenberg C, Koiffmann CP (2006) A 17q21.31 microdeletion encompassing the MAPT gene in a mentally impaired patient. Cytogenet Genome Res 114:89–92. [DOI] [PubMed] [Google Scholar]
- 150. Vega IE, Cui L, Propst JA, Hutton ML, Lee G, Yen SH (2005) Increase in tau tyrosine phosphorylation correlates with the formation of tau aggregates. Brain Res Mol Brain Res 138:135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Vogelsberg‐Ragaglia V, Bruce J, Richter‐Landsberg C, Zhang B, Hong M, Trojanowski JQ, Lee VM (2000) Distinct FTDP‐17 missense mutations in tau produce tau aggregates and other pathological phenotypes in transfected CHO cells. Mol Biol Cell 11:4093–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293:711–714. [DOI] [PubMed] [Google Scholar]
- 153. Yasuda M, Kawamata T, Komure O, Kuno S, D’Souza I, Poorkaj P, Kawai J, Tanimukai S, Yamamoto Y, Hasegawa H, Sasahara M, Hazama F, Schellenberg GD, Tanaka C (1999) A mutation in the microtubule‐associated protein tau in pallido‐nigro‐luysian degeneration. Neurology 53:864–868. [DOI] [PubMed] [Google Scholar]
- 154. Yoo BC, Kim SH, Cairns N, Fountoulakis M, Lubec G (2001) Deranged expression of molecular chaperones in brains of patients with Alzheimer’s disease. Biochem Biophys Res Commun 280:249–258. [DOI] [PubMed] [Google Scholar]
- 155. Zhou L, Miller BL, McDaniel CH, Kelly L, Kim OJ, Miller CA (1998) Frontotemporal dementia: neuropil spheroids and presynaptic terminal degeneration. Ann Neurol 44:99–109. [DOI] [PubMed] [Google Scholar]
- 156. Zhukareva V, Vogelsberg‐Ragaglia V, Van Deerlin VM, Bruce J, Shuck T, Grossman M, Clark CM, Arnold SE, Masliah E, Galasko D, Trojanowski JQ, Lee VM (2001) Loss of brain tau defines novel sporadic and familial tauopathies with frontotemporal dementia. Ann Neurol 49:165–175. [DOI] [PubMed] [Google Scholar]