

Abstract
In Alzheimer's disease (AD), there is abnormal accumulation of Aβ and tau proteins in the brain. There is an associated immunological response, but it is still unclear whether this is beneficial or harmful. Inflammation in AD, specifically in the form of microglial activation, has, for many years, been considered to contribute to disease progression. However, two types of evidence suggest that it may be appropriate to revise this view: first, the disappointing results of prospective clinical trials of anti‐inflammatory agents and, second, the observation that microglia can clear plaques in AD following Aβ immunization. Although Aβ immunization alters AD pathology, there is limited evidence so far of benefit to cognitive function. Immunization against microorganisms is almost always used as a method of disease prevention rather than to treat a disease process that has already started. In animal models, immunotherapy at an early age can protect against Aβ accumulation and it will be interesting to see if this can usefully be applied to humans to prevent AD.
Keywords: Alzheimer's disease, immunization, inflammation, immune system, therapy
INTRODUCTION
Alzheimer's disease (AD) is an age‐related chronic neurodegenerative disease characterized by memory loss and severe cognitive decline. The key pathological features of AD relate to accumulation of Aβ protein and tau protein (72). Aβ accumulates in the form of plaques in the cortical grey matter and in the walls of cortical and leptomeningeal small arteries and arterioles in the form of cerebral amyloid angiopathy (CAA). Tau protein accumulates within neurons in cell bodies (tangles), fine calibre neuronal processes in the grey matter neuropil (neuropil threads) and in distorted and swollen neuronal processes in the region of Aβ plaques (dystrophic neurites). Detailed studies have also documented neuronal and synaptic loss (64, 114).
THE AMYLOID HYPOTHESIS
Evidence of a key role for Aβ peptide in AD pathogenesis gives rise to the “amyloid hypothesis” of AD (46). Aβ is a peptide predominantly 40 or 42 amino acids in length derived from the transmembrane protein amyloid precursor protein (APP) after cleavage by β and γ secretases (44, 67). Several lines of evidence from the human disease support a pivotal role for abnormalities of Aβ in the pathogenesis of AD, leading to neuronal dysfunction and cell death (99): (i) APP point mutations can cause familial AD; (ii) familial AD, whether because of point mutations of PSEN1, PSEN2 or APP, have in common increased production of Aβ42 which is particularly prone to aggregation; and (iii) Down's syndrome, which is usually due to trisomy 21 on which the APP gene is located, is associated with AD at an early age. It is worth noting that this evidence comes from the very rare genetically determined forms of AD, and whether the role of Aβ applies equally to the common sporadic form of AD is not yet known with certainty. A crucial test of the amyloid hypothesis would be whether prevention of Aβ aggregation prevents the neurodegenerative decline.
There is now good evidence that AD that has a genetic cause is due to an increase in the production of Aβ, resulting in Aβ accumulation. In the much more common sporadic AD, there is not good evidence for this and, instead, evidence has been emerging for an age‐related impairment of elimination of Aβ as the cause of Aβ accumulation. It seems likely that there is a dynamic equilibrium between the production and elimination of Aβ protein in the human brain. Evidence for several potential mechanisms by which Aβ is normally eliminated from the brain has emerged, including (i) by cellular mechanisms involving microglia (85, 90); (ii) by enzymatic degradation (eg, neprilysin and insulin degrading enzyme) (54, 91); (iii) by transport across the blood–brain barrier mediated by the low density lipoprotein receptor‐related protein‐1 (LRP) receptor (101); and/or (iv) by bulk flow with interstitial fluid along the perivascular drainage pathway (97, 119).
Transgenic mice which over‐express familial AD APP point mutations develop Aβ plaques as they age providing a valuable animal model (25, 36), although lacking tau pathology. Recent evidence in a triple transgenic animal model of AD, with point mutations in APP, PSEN1 and tau genes, indicates that Aβ accumulation precedes the tau pathology in a cascade of events that ultimately leads to the cognitive alterations (81).
Apolipoprotein E
Genetic studies in the early 1990s clearly identified polymorphism of the apolipoprotein E gene (APOE) as the major genetic risk factor for sporadic AD (110). APOE molecules are lipid carriers (15) involved in the redistribution of cholesterol within the brain to maintain the structural and functional integrity of membranes and synapses (63). APOE also acts as an Aβ‐scavenging molecule that regulates Aβ concentration through internalization of APOE receptors by the endosomal/lysosomal pathway (89). In addition, recently, it has been revealed that Aβ has an essential physiological role in lipid homeostasis (43). This evidence suggests that clearance of Aβ is likely to be regulated by the APOE–Aβ interactions. Three different isoforms of APOE occur within the brain (APOE E2, E3 and E4), which differ in amino acids at position 112 and 158, according to the genotype of the individual (63). The increased risk of developing AD associated with APOE E4 may be due to its inability to internalize, and therefore clear, extracellular Aβ to endosomes/lysosomes. The evidence for this comes from a failure to develop plaques in transgenic mice that both over‐express human APP and are APOE deficient (48). In addition, the biochemical difference of APOE E4 may induce the promotion of lipid rafts that have a suitable environment for the amyloidogenic processes (19). APOE genotype has also been associated with differences in microglia; both in the degree of microglial activation in AD brains (27) and in the microglial expression of inflammatory molecules (59, 62).
EVIDENCE FOR INVOLVEMENT OF THE IMMUNE SYSTEM IN AD
The immune system has evolved to protect the body against invasion by foreign microorganisms. The efficiency of the immune system is the result of two different but complementary forms of activation—innate and adaptive. Innate immunity is the first response to infection and plays a major role in controlling the infection during the gestation of adaptive immunity. The macrophage is a central component of innate immunity. If innate immunity is overcome by pathogens, adaptive immunity operating via dendritic cells, lymphocytes and antibodies, will build a specific response to the infection. The key property of adaptive immunity is to recognize pathogens specifically and to provide enhanced protection against re‐infection.
A growing number of studies in AD have reported alterations in the immune system, including: the presence of circulating auto‐antibodies; the presence in the brain of proteins from the complement system; abnormal production of cytokines; and changes in the distribution and activation of microglia. This implies that, in a sense, the immune system is capable of recognizing the proteins that aggregate in the brain in AD as abnormal or “foreign” proteins that should be disposed of. This raises the question as to whether this involvement of the immune system can help to ameliorate the progress of AD or simply adds to the damage.
Auto‐antibodies
An increase in auto‐antibodies, defined as antibodies to “self‐tolerant” proteins, in the blood of healthy elderly humans has been observed (35, 45, 95). This led to two different hypotheses: (i) auto‐antibodies contribute to the diseases associated with ageing, as occurs in autoimmune disease (116); or (ii) auto‐antibodies play a role in eliminating senescent cells to maintain the integrity of the host (37). More recently, a number of studies have reported the presence of specific anti‐Aβ antibodies in the blood and cerebrospinal fluid (CSF) of healthy humans and AD patients (26, 50, 73, 77, 118). More than 20 years ago, immunoglobulin (Ig)‐G was seen by light and electron microscopy in the AD brain to be co‐localized with neuritic plaques (28, 52, 53); however, the role of IgG in the AD process, including in plaque formation, remains unclear. Indeed, the findings in relation to antibodies in AD are somewhat inconsistent, with the level of anti‐Aβ antibodies being either increased or decreased, possibly because of differences in the methodology employed. However, most of the studies have identified a decrease of anti‐Aβ antibodies in AD patients compared with age‐matched healthy controls (26, 73, 118), raising the possibility that some people are, in a sense, able to immunize themselves against Aβ and therefore protect themselves against AD.
Complement system
The complement system is a sophisticated system evolved to destroy pathogens and to assist in the phagocytosis of waste materials. Four main functions are carried out by complement: recognition, opsonization, activation of inflammation and killing of the pathogen. Fibrillar Aβ is a strong stimulator of the complement system (94) and can activate the classical (antibody‐dependent) (2, 20, 57, 69, 117) and alternative (antibody‐independent) (13, 111) pathways. Activation of complement by Aβ appears to be highly specific to fibrillar Aβ, as other peptides of similar size are unable to activate the complement system (13). Hyperphosphorylated tau contained in neurons can also activate the classical complement cascade (68, 100) as demonstrated by the staining of tau‐positive neurons by anti‐complement antibodies. Therefore, in AD, there is evidence that the complement system is strongly activated and could participate either in the exacerbation or amelioration of the pathology. In APP transgenic mice which have inhibition of the complement system at the level of C3 (the central component of complement), neurodegeneration is increased compared with non complement‐inhibited controls (123). This finding suggests that the complement system may have a protective role in AD.
Cytokines
Four main cytokines have been investigated extensively in AD: interleukin 1 (IL‐1), IL‐6, tumor necrosis factor α (TNF‐α) and transforming growth factor β1 (TGF‐β1). Immunoreactivity for IL‐1, IL‐6 and TGF‐β1 has been reported in association with Aβ plaques (41, 49, 115). Elevated TNF‐α and TGF‐β1 levels have been detected in the serum and CSF of AD patients (17, 18, 32, 113). It has been proposed that cytokines secreted by glia interact with neurons to form a positive feedback loop or “cytokine cycle” which, once initiated, leads to progressive neurodegeneration (42, 76). Initial studies found some evidence for associations between specific cytokine gene polymorphisms and AD, for example, in the IL‐1 gene (78), but subsequent studies suggest these are not of major importance (92) with the possible exception of TNF gene polymorphism (9). Overall, the role of the cytokines in AD, and particularly whether on balance they are beneficial or harmful, remains uncertain.
Microglia
A key component of the innate immune system in the brain is the microglial cell which is the representative of the monocyte/macrophage lineage. Microglia are activated in AD but, despite much interest in the subject, the role that microglia play and whether they are harmful or helpful, remains unclear. In non‐neural tissues, the state of cell activation and the nature of the activating stimulus play a significant role in determining the spectrum of molecules that are secreted by a macrophage (1, 40). Furthermore, the initial state of activation of the macrophage is an important determinant of the magnitude of a particular response (40). Microglia are highly sensitive to disturbance of CNS homeostasis (58). They respond to signals which arise from injured neurons, and the presence of activated microglia seems to be limited spatially to those regions occupied by injured neurons and neuronal processes. However, the damage does not have to be lethal to the neurons in order to elicit a reactive microgliosis. This is illustrated by traumatic and ischemic lesions where peri‐necrotic areas contain surviving neurons and reactive microglia (75, 107) or as observed following intra‐hippocampal injection of bacterial component in a mouse brain (10). According to the type of injury or insult, activation of microglia can be either tolerated or, alternatively, harmful leading to a destructive profile of microglia associated with neuronal loss (88). A third phenotype of microglia has also been suggested in which microglia become dysfunctional with ageing, characterized by structural deterioration and increased apoptosis (108). As a result, microglia may lose their neuroprotective properties with advancing age (109) leading to chronic neurodegeneration such as AD.
It is important to consider the origin of the microglia that responds to brain pathology. There is now good evidence that two types of microglia coexist within the brain: first, resident microglia which are derived from the mesoderm and migrate to the brain during the embryogenesis (87) and, second, bone marrow‐derived microglia which are characterized by recruitment from the blood during life in response to an appropriate stimulus from the brain (87, 103). Recent evidence from animal models suggests that the activated microglia that surround plaques have been recruited from the blood and are specifically attracted to the accumulated Aβ peptide (104). The authors postulated that blood‐derived microglia are more efficient at presenting antigen and may be more efficient than resident microglia in phagocytosing Aβ (103). Study of mice lacking the chemokine receptor 2, a microglial cell‐surface receptor that mediates recruitment of blood‐derived microglia, supports the idea that bone marrow‐derived macrophages infiltrate the brain and can clear Aβ from the brain (29). In human AD, despite the presence of abundant activated microglia, Aβ is rarely observed within microglia. When it is, the microglia seem unable to degrade Aβ as observed by electron microscopy (34, 85) unless, as demonstrated in an AD model, they express Toll‐like receptors, receptors usually observed during a bacterial attack (39, 112). Nonetheless, an exception is observed when an infarct occurs in AD, the microglia are then able to phagocytose all of the tissue components, including Aβ plaques in the vicinity of the infarct (3).
Study of a prion mouse model, an analogous chronic neurodegenerative disease with accumulation of an amyloid protein, shows that (i) microglia may be activated by neurodegeneration rather than protein deposition (21, 122); and (ii) conversely, the microglia express TGF‐β1 which down‐regulates scavenger receptors hampering phagocytosis (11, 21). These events may also occur during the neurodegenerative process of AD, in which TGF‐β1 is also expressed (65).
These observations highlight the point that, rather than simply demonstrating that microglia have been activated, it is the specific way in which microglia are activated that is important in determining the influence of microglia on neurodegeneration (88).
MANIPULATION OF THE IMMUNE SYSTEM AS A THERAPEUTIC TOOL
Anti‐inflammatory therapy in Alzheimer's disease
Modulation of the immune system as a therapy for AD became of great interest in the 1990s. Early observations indicated that patients with rheumatoid disease who were on long‐term anti‐inflammatory therapy had a lower prevalence of AD than controls, raising the possibility that their medication may have been protective (71). The main mechanism postulated was that down‐regulation of microglia would have a beneficial effect in preventing or slowing neurodegeneration in AD, with supporting evidence from animal models (60). However, subsequent prospective trials of anti‐inflammatory agents have, to date, proved disappointing (51, 71). It has been suggested that key questions relating to this approach remain unanswered, for example (i) the type of anti‐inflammatory agent to use; (ii) the dose at which use them; and (iii) the duration of the treatment (4).
Aβ immunization in animal models
Different approaches to immunization
Approaches have been developed using anti‐Aβ antibodies, either injected directly (ie, passive immunization) or induced by active immunization with the Aβ peptide, in order to reduce Aβ formation or to facilitate clearance of Aβ from the brain. Initially, it was observed that aggregated Aβin vitro was dissolved and its formation prevented in the presence of anti‐Aβ antibodies (105, 106). Subsequently, in an APP transgenic mouse model of AD, active immunization with Aβ42 peptide demonstrated in vivo that it was possible to prevent or reverse Aβ accumulation in the brain (96). Studies of peripheral immunization with Aβ antibodies showed the presence of Aβ antibodies within the brain (6, 56). That antibodies could cross the blood–brain barrier was already known (93); however, the passive Aβ immunization experiments showed that the IgG antibodies to Aβ are directly involved in amyloid plaque removal (6). The specificity of the Aβ antibodies has been also investigated and it has been shown that only Aβ antibodies directed against the N‐terminal part of the peptide were efficient to remove amyloid (7) and that the Fc part of the antibody was fundamental for the clearance of amyloid (7). IgM antibodies, produced by active vaccination before IgG antibodies, might also act in the clearance of Aβ despite their inability to cross the blood–brain barrier because of their large size. Immunization of mice with a prototype vaccine containing a modified Aβ peptide that induced only IgM antibodies has shown a decrease of Aβ load in the brain (102). The IgM effect is known as the “sink hypothesis”, defined by immunization modifying the balance between Aβ peptide and Aβ antibodies in the periphery and consequently attracting Aβ from the brain to the periphery (23, 24) (Table 1).
Table 1.
Mechanisms of Aβ removal following Aβ immunization.
Intracranial injection of anti‐Aβ antibodies induces two phases of Aβ removal: first, a rapid decrease starting 24 h after the administration and before cellular activation, and then after 3 days, a further decrease in Aβ associated with activation of microglia. This observation suggests that there are two different mechanisms involved in removal of Aβ, respectively, independent and dependent of microglial activation (120) (Table 1). Immunization appears to alter the activation state of microglia so that they are able to efficiently phagocytose the aggregated amyloid, although the precise mechanisms remain unclear. The Fc part of the antibody has been suggested to be essential to mediate the phagocytosis of amyloid through binding to the microglial Fc receptor (6, 7). On the other hand, Aβ42 immunization of APP transgenic mice crossed with FcRγ−/− mice and therefore lacking the possibility of activation of microglia by immune complexes also showed a decrease of amyloid plaques (22). Furthermore, by using in vivo multiphoton microscopy, intracranial application of F(ab')2 fragments of anti‐Aβ antibodies have been found sufficient to decrease amyloid load (5). These two experiments suggest the existence of an Fc‐independent mechanism which results in phagocytosis of amyloid by microglia. It seems clear that the mechanisms by which the activated microglia are prompted to phagocytose amyloid plaques are not exclusive. The role of complement in the removal of Aβ has been explored relatively little, but it seems not to be essential for plaque clearance to occur (7). However, it has been suggested that these findings may reflect differences in complement activation, being relatively weak in the transgenic mouse models compared with AD patients (70).
In summary, the key pathological changes following Aβ immunization in animal models include: (i) prevention of Aβ deposits if the immunotherapy is administered before the onset of Aβ accumulation (96); (ii) clearance of Aβ plaques if the immunotherapy is started at an age at which plaques are already present in the brain (6, 96, 102); (iii) presence of Aβ in microglia (5); and (iv) removal of dystrophic neurites, defined in APP‐transgenic mice as swollen axons and dendrites surrounded amyloid plaques which contain APP but not tau (14, 61, 96).
Immunization prevents neurodegeneration and functional decline
Removal of Aβ plaques in APP transgenic mice has been shown associated with an improvement of cognitive function in the treated animals by comparison with the untreated transgenic mice after active immunization (56, 74), passive immunization (121) or by using the “sink” mechanism (102). Interestingly, improvement of cognitive function in immunized APP‐transgenic mice is observed with only an attenuated immune response (102) and does not need a complete clearance of Aβ plaques from the brain (56). Quantification of the synaptic density in immunized transgenic mice showed a prevention of the synaptic degeneration following active or passive procedures in association with the Aβ clearance (16).
Limitations of the animal models
Some caution is necessary in translating these impressive findings in animal models into expectations for the effects of immunotherapy in human AD. Since the first APP transgenic mouse model (36), numerous animal models have been designed based on the different APP point mutations that can cause familial AD. It is important to recognize that the APP transgenic mice are models of Aβ pathophysiology which lack some of the key features of AD, particularly tau pathology. Therefore, the state of chronic microglial activation in these animals may not simulate the inflammation in AD.
Immunization of the triple transgenic mice, characterized by the Aβ and tau accumulation, have shown: Aβ removal, clearance of the early but not late‐hypersphosphorylated tau aggregates and improvement of cognitive function (82, 83). Investigation of inflammation in the triple transgenic mice has detected early inflammatory processes in relation with the Aβ and tau deposits (55); however, the consequences of immunization on the inflammatory compounds have still not been studied.
Aβ immunization in human AD
The first clinical trials of Aβ immunization
As a consequence of the finding that active Aβ42 immunization of APP transgenic mice results in plaque removal (96), a human clinical trial was initiated in 2000. This first trial was conducted in the UK involving 80 patients with mild to moderate AD (8) and was designed to assess the antigenicity and tolerability of multiple dose immunization with full length Aβ42 peptide with adjuvant (AN1792 + QS‐21). No adverse events were identified and 53% of the immunized patients developed anti‐Aβ antibodies at varying titers. A subsequent larger clinical trial (n = 372) designed to assess efficacy was halted when 18/298 (6%) of the patients developed a subacute neurological deterioration accompanied by cerebral white matter abnormalities on imaging and lymphocytes in the CSF (84).
Effects of Aβ immunization on the neuropathology of AD
Post‐mortem neuropathology of patients who were immunized in these studies, and subsequently died for incidental reasons, has shown remarkable evidence of modification of the AD pathology (12, 31, 66, 79, 80) (Figure 1). Most importantly, there was evidence that the Aβ immunization had resulted in removal of Aβ plaques in a manner similar to that seen in the animal models, providing “proof of principle” for the rationale of the studies (Table 1). Comparison of the histological patterns of Aβ shows remarkably close modeling of the immunized AD patients by the APP transgenic mice (Table 2). The characteristic set of features shared by the immunized AD patients and the APP transgenic mice include: extensive areas of cerebral cortex cleared of plaques; a “moth‐eaten” appearance of some of the residual plaques; the presence of “naked” dense plaque cores in otherwise plaque‐free areas; persistence of CAA and association of Aβ with capillaries in plaque‐free areas; and localization of Aβ in microglia (Figure 2), confirmed by confocal microscopy as representing Aβ phagocytosed by microglia (80). It is important to note that not all of these features were present in each of the cases examined, but together they can be considered as the defining alterations in the histological pattern of Aβ in the brain following active immunization with Aβ42 peptide.
Figure 1.
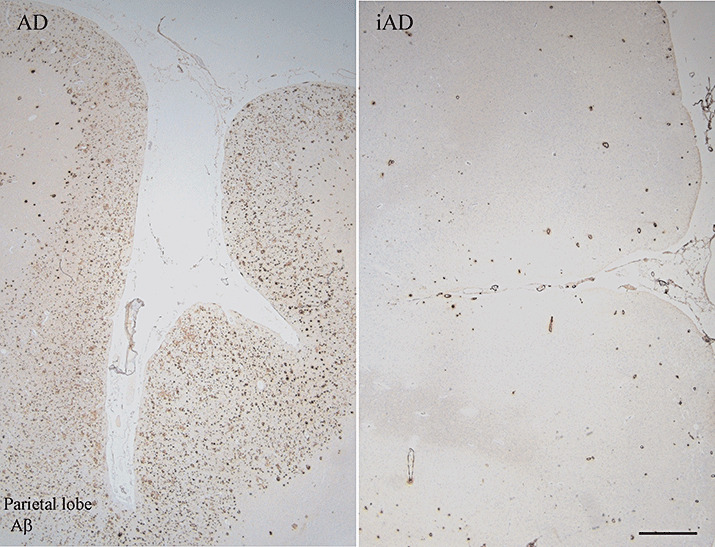
Aβ immunohistochemistry in unimmunized Alzheimer's disease (AD) and after Aβ42 immunization. Pattern of Aβ immunoreactivity (pan‐Aβ antibody, residues 8–17) in the parietal lobe of an unimmunized AD case (AD) and an immunized AD case (iAD). The AD case reveals the presence of numerous Aβ plaques in the cerebral cortex, but whereas in the iAD case, the area is virtually devoid of plaques with a persistence of Aβ in the cerebral vasculature. Scale bar = 1 mm.
Table 2.
Pathological features following Aβ42 immunization. Comparison between observations in the animal models and the human immunized AD cases. Abbreviations: AD = Alzheimer's disease; APP = amyloid precursor protein; CAA = cerebral amyloid angiopathy. N/A = not applicable; ✗ = observed.
| Animal models | Immunized AD cases | |
|---|---|---|
| Aβ pathology | ||
| Removal of Aβ plaques | ✓ | ✓ |
| Aβ in microglia | ✓ | ✓ |
| Increased CAA | ✓ | ✓ |
| Tau pathology | ||
| Removal of dystrophic neurites | ✓ (APP‐containing only) | ✓ |
| Persistence of tangles | N/A | ✓ |
| Persistence of neuropil threads | N/A | ✓ |
| Unexpected events | ||
| T lymphocytes | ✗ | ✓ (some cases only) |
| Leucoencephalopathy | ✗ | ✓ (some cases only) |
Figure 2.
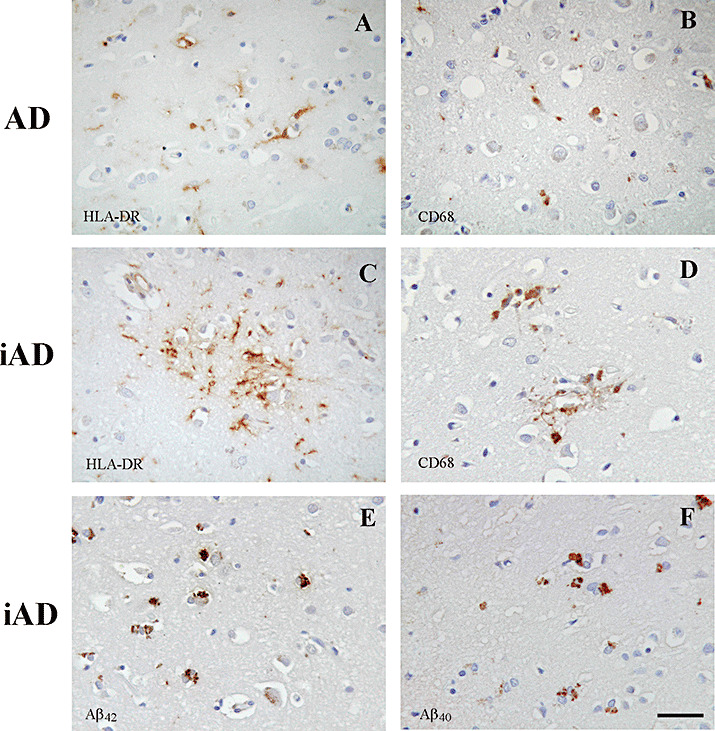
Activated microglia in Alzheimer's disease (AD) vs. immunized AD (iAD). Detection of HLA‐DR (A) and CD68 (B) positive microglia in an unimmunized AD case. After Aβ42 immunization, HLA‐DR (C) and CD68 (D) positive microglia are observed particularly clustered around residual plaques. Using isoform specific antibodies, Aβ42 (E) and Aβ40 (F) peptides are detected within microglia after Aβ42 immunization, rarely seen in unimmunized AD cases. Scale bar = 40 µm.
Of particular interest, in view of the longstanding uncertainty of the causal inter‐relationship between extracellular accumulation of Aβ and intracellular accumulation of tau protein, is the observation that in regions where plaques have been removed, tau‐containing plaque‐associated dystrophic neurites are absent. This implies that when plaques are removed, the dystrophic neurites are also removed, presumably either by resolution or phagocytosis. The APP transgenic mice do not have tau pathology but they do have APP‐immunoreactive plaque‐associated dystrophic neurites and these resolve after plaque removal, again closely modeling the situation in human AD (14, 80). Studies in one case also provided evidence of local inhibition of the stress‐activated protein kinase/ c‐Jun N‐terminal kinase and p38 kinase, enzymes involved in tau phosphorylation, where Aβ had been removed (31). Nevertheless, quantification of tau immunoreactive tangles and neuropil threads does not show any clear evidence that they are reduced in regions of cortex where plaques have been removed (79, 80) (Table 2). The pathological changes observed in immunized AD are summarized in the Figure 3.
Figure 3.
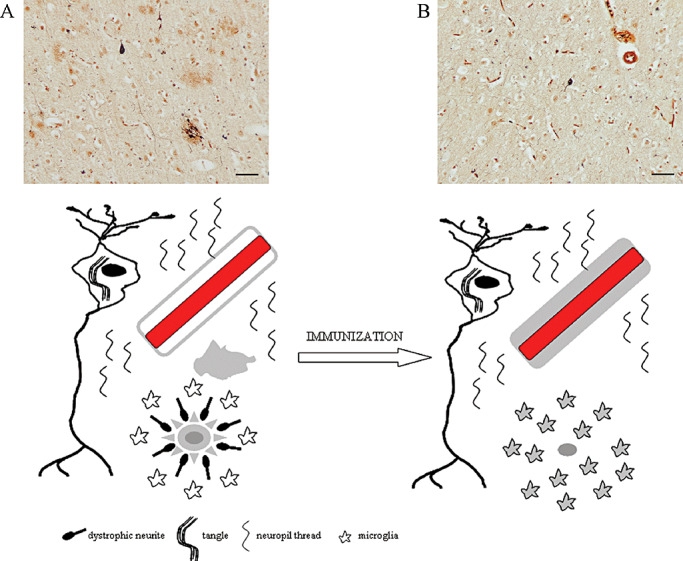
Summary of the pathological changes observed in Alzheimer's disease (AD) after Aβ42 immunization. AD pathology is characterized by the presence of Aβ plaques, dystrophic neurites, intraneuronal tangles and neuropil threads with activated microglia and cerebral amyloid angiopathy, as illustrated with modified Bielschowsky staining and in diagrammatic form (A). After Aβ42 immunization, the Aβ plaques and dystrophic neurites are removed, microglia have phagocytosed Aβ, Aβ is increased in the vasculature and the neuropil threads and tangles are still observed (B). Scale bar = 50 µm.
What pathophysiological events underlay the meningoencephalitis?
A striking difference between the animal models and the human studies was the development of “meningoencephalitis” in a small proportion of the patients which was not predicted in the preclinical studies (84). Two of the humans studied neuropathologically had this clinically defined side effect (31, 79); however, they both survived many months after this event, so study of the neuropathology can only provide limited information about the nature of the responsible pathological process. Nevertheless, important pathological correlates of this side effect are: (i) the presence of T lymphocytes in the leptomeninges, particularly near to blood vessels severely affect by CAA, but almost entirely absent from the cerebral cortex; and (ii) widespread changes in the deep cerebral white matter including rarefaction of myelinated fibers and abundant macrophages (Figure 4). To link these two features must be a matter of speculation but previous studies of severe CAA affecting cortical and leptomeningeal blood vessels have identified degenerative changes in the deep cerebral white matter as a common feature (98). Presumably, this unwanted side effect has potentially identifiable risk factors and AD patients with these factors could be deemed unsuitable for this form of therapy. Such risk factors might include, for example, a previously primed immune system, severe CAA and genetic variation, including possibly APOE genotype. A further risk factor for meningoencephalitis is likely to be the presence of Aβ in the brain, strengthening the argument for use of Aβ immunization as a preventative therapy. However, it is important to note that the meningoencephalitis is not required for the clearance of Aβ (66). The potentially beneficial and/or harmful effects of Aβ42 immunization are summarized in Table 3. Possible causes of the unwanted side effect in addition to the T lymphocyte infiltrate include an over‐exuberant activation of microglia prompted by opsonization of Aβ and alterations in fluid balance in the brain triggered by interaction of antibody‐Aβ immune complexes and the cerebral vasculature. Clear understanding of the pathophysiology of this phenomenon has important implications for future immunization trials as many of the current protocols have been specifically designed to avoid T lymphocyte activation. If T lymphocytes are responsible then it should not raise its head again as a problem, but if not then it may cloud the results of future immunization trials.
Figure 4.
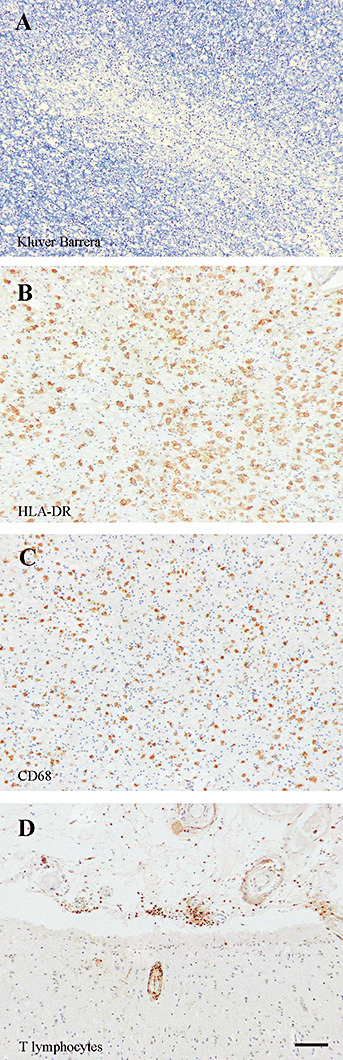
Meningoencephalitis. Features observed in one immunized patient with meningoencephalitis who came to autopsy more than 1 year later: rarefaction of white matter as detected by Kluver–Barrera staining (A), marked increase of HLA‐DR (B) and CD68 (C) positive microglia/macrophages and the presence of T lymphocytes in the leptomeninges and in relation to a cerebral blood vessel (D, anti‐CD45RO antibody). Scale bar = 50 µm.
Table 3.
Effects of Aβ immunization on the neuropathology of AD. Abbreviations: AD = Alzheimer's disease; CAA = cerebral amyloid angiopathy.
| Potentially beneficial effects | Potentially harmful effects |
|---|---|
| Removal of plaques | Increased soluble/oligomeric Aβ |
| Microglial activation | Microglial activation |
| Decreased CAA | Increased CAA |
| Resolution of dystrophic neurites | Presence of T lymphocytes |
| Leucoencephalopathy |
Intriguingly, the constellation of features identified in AD patients who have been immunized with Aβ as described above, and including an inflammatory infiltrate, has been identified in specific subgroups of unimmunized patients (30, 98). This raises the question as to whether Aβ pathology in natural disease may be in a dynamic state of flux, with episodic or progressive deposition and removal, possibly mediated at least in part by the immune system.
Effects on cognitive function
Overall, the data from the AD immunization trials so far do not seem to show evidence of a substantial effect on cognitive function, either in improving function or in preventing progressive decline (8, 38) (Holmes, pers. comm.). However, it is important to bear in mind that the active immunization resulted in antibody production in only about half of the immunized patients and those patients produced antibodies at varying titers (8). Studies performed so far in subgroups of the initial trials, dividing patients into responders and non‐responders, have suggested there may be some beneficial effect (38, 47). The results of passive immunization, in which the antibody levels available to the brain are controlled, are awaited with interest.
Intriguingly, and perhaps counter‐intuitively, sequential in vivo imaging studies have shown evidence of accelerated cerebral atrophy in antibody responders compared with non‐responders (33). The reasons for this reduction in brain volume are unclear, but might include a removal of plaque‐associated proteins, a reduction in the glial cell reaction and fluid redistribution.
CONCLUSIONS
The amyloid hypothesis predicts that removal of Aβ from the brain, or prevention of its accumulation, will ameliorate the Alzheimer process. In transgenic animal models of Aβ accumulation, both passive and active Aβ immunization can result in removal of Aβ or prevent its accumulation, resulting in functional benefits. Studies of Aβ immunization in humans with AD have provided “proof of principle” that Aβ accumulation can be reversed. However, there is limited effect on some aspects of AD pathology, particularly tau accumulation, and limited evidence of functional benefit. Current concern with the Aβ immunization approach is centered on an unpredicted inflammatory complication that occurred in a minority of AD patients, and therefore, immunization protocols aimed to circumvent this problem are currently in clinical trials. The long‐term consequences of activation of the immune system on cognitive function in AD may depend on the net balance of potentially beneficial and harmful effects on the complex array of pathological changes that are induced. Consequently, if it can be done safely, Aβ immunization at an earlier stage an preferably before irreversible AD‐related brain damage has occurred will be of interest.
ACKNOWLEDGMENTS
The authors are funded by the Alzheimer Research Trust (JARN and DB: ART/PG2006/4) and the Medical Research Council (DB: G0501033). We thank Dr Peter Seubert from Elan Pharmaceuticals for the Aβ42 and Aβ40 antibodies.
REFERENCES
- 1. Adams DO, Hamilton TA (1992) Molecular basis of macrophage activation: diversity and origins. In: The Macrophage, Lewis CE, McGee JOD (eds), pp. 73–102. JRL Press Oxford: Oxford. [Google Scholar]
- 2. Afagh A, Cummings BJ, Cribbs DH, Cotman CW, Tenner AJ (1996) Localization and cell association of c1q in Alzheimer's disease brain. Exp Neurol 138:22–32. [DOI] [PubMed] [Google Scholar]
- 3. Akiyama H, McGeer PL (2004) Specificity of mechanisms for plaque removal after a beta immunotherapy for Alzheimer disease. Nat Med 10:117–118; author reply 118–119. [DOI] [PubMed] [Google Scholar]
- 4. Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM et al (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21:383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT (2002) Non‐Fc‐mediated mechanisms are involved in clearance of amyloid‐beta in vivo by immunotherapy. J Neurosci 22:7873–7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H et al (2000) Peripherally administered antibodies against amyloid beta‐peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6:916–919. [DOI] [PubMed] [Google Scholar]
- 7. Bard F, Barbour R, Cannon C, Carretto R, Fox M, Games D et al (2003) Epitope and isotype specificities of antibodies to beta – amyloid peptide for protection against Alzheimer's disease‐like neuropathology. Proc Natl Acad Sci USA 100:2023–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L et al (2005) Evaluation of the safety and immunogenicity of synthetic Abeta42 (AN1792) in patients with AD. Neurology 64:94–101. [DOI] [PubMed] [Google Scholar]
- 9. Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE (2007) Systematic meta‐analyses of Alzheimer's disease genetic association studies: the AlzGene database. Nat Genet 39:17–23. [DOI] [PubMed] [Google Scholar]
- 10. Boche D, Cunningham C, Gauldie J, Perry VH (2003) Transforming growth factor‐beta 1‐mediated neuroprotection against excitotoxic injury in vivo. J Cereb Blood Flow Metab 23:1174–1182. [DOI] [PubMed] [Google Scholar]
- 11. Boche D, Cunningham C, Docagne F, Scott H, Perry VH (2006) TGFbeta1 regulates the inflammatory response during chronic neurodegeneration. Neurobiol Dis 22:638–650. [DOI] [PubMed] [Google Scholar]
- 12. Bombois S, Maurage CA, Gompel M, Deramecourt V, Mackowiak‐Cordoliani MA, Black RS et al (2007) Absence of beta‐amyloid deposits after immunization in Alzheimer disease with Lewy body dementia. Arch Neurol 64:583–587. [DOI] [PubMed] [Google Scholar]
- 13. Bradt BM, Kolb WP, Cooper NR (1998) Complement‐dependent proinflammatory properties of the Alzheimer's disease beta‐peptide. J Exp Med 188:431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brendza RP, Bacskai BJ, Cirrito JR, Simmons KA, Skoch JM, Klunk WE et al (2005) Anti‐Abeta antibody treatment promotes the rapid recovery of amyloid‐associated neuritic dystrophy in PDAPP transgenic mice. J Clin Invest 115:428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brown MS, Goldstein JL (1986) A receptor‐mediated pathway for cholesterol homeostasis. Science 232:34–47. [DOI] [PubMed] [Google Scholar]
- 16. Buttini M, Masliah E, Barbour R, Grajeda H, Motter R, Johnson‐Wood K et al (2005) Beta‐amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer's disease. J Neurosci 25:9096–9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chao CC, Ala TA, Hu S, Crossley KB, Sherman RE, Peterson PK, Frey WH 2nd (1994) Serum cytokine levels in patients with Alzheimer's disease. Clin Diagn Lab Immunol 1:433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chao CC, Hu S, Frey WH 2nd, Ala TA, Tourtellotte WW, Peterson PK (1994) Transforming growth factor beta in Alzheimer's disease. Clin Diagn Lab Immunol 1:109–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chauhan NB (2003) Membrane dynamics, cholesterol homeostasis, and Alzheimer's disease. J Lipid Res 44:2019–2029. [DOI] [PubMed] [Google Scholar]
- 20. Chen S, Frederickson RC, Brunden KR (1996) Neuroglial‐mediated immunoinflammatory responses in Alzheimer's disease: complement activation and therapeutic approaches. Neurobiol Aging 17:781–787. [DOI] [PubMed] [Google Scholar]
- 21. Cunningham C, Wilcockson DC, Boche D, Perry VH (2005) Comparison of inflammatory and acute‐phase responses in the brain and peripheral organs of the ME7 model of prion disease. J Virol 79:5174–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Das P, Howard V, Loosbrock N, Dickson D, Murphy MP, Golde TE (2003) Amyloid‐beta immunization effectively reduces amyloid deposition in FcRgamma‐/‐ knock‐out mice. J Neurosci 23:8532–8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM (2001) Peripheral anti‐A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 98:8850–8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM (2002) Brain to plasma amyloid‐beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science 295:2264–2267. [DOI] [PubMed] [Google Scholar]
- 25. Dodart JC, Mathis C, Bales KR, Paul SM (2002b) Does my mouse have Alzheimer's disease? Genes Brain Behav 1:142–155. [DOI] [PubMed] [Google Scholar]
- 26. Du Y, Dodel R, Hampel H, Buerger K, Lin S, Eastwood B et al (2001) Reduced levels of amyloid beta‐peptide antibody in Alzheimer disease. Neurology 57:801–805. [DOI] [PubMed] [Google Scholar]
- 27. Egensperger R, Kosel S, Von Eitzen U, Graeber MB (1998) Microglial activation in Alzheimer disease: Association with APOE genotype. Brain Pathol 8:439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eikelenboom P, Stam FC (1982) Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study. Acta Neuropathol 57:239–242. [DOI] [PubMed] [Google Scholar]
- 29. El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD (2007) Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer‐like disease. Nat Med 13:432–438. [DOI] [PubMed] [Google Scholar]
- 30. Eng JA, Frosch MP, Choi K, Rebeck GW, Greenberg SM (2004) Clinical manifestations of cerebral amyloid angiopathy‐related inflammation. Ann Neurol 55:250–256. [DOI] [PubMed] [Google Scholar]
- 31. Ferrer I, Boada Rovira M, Sanchez Guerra ML, Rey MJ, Costa‐Jussa F (2004) Neuropathology and pathogenesis of encephalitis following amyloid‐beta immunization in Alzheimer's disease. Brain Pathol 14:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fillit H, Ding WH, Buee L, Kalman J, Altstiel L, Lawlor B, Wolf‐Klein G (1991) Elevated circulating tumor necrosis factor levels in Alzheimer's disease. Neurosci Lett 129:318–320. [DOI] [PubMed] [Google Scholar]
- 33. Fox NC, Black RS, Gilman S, Rossor MN, Griffith SG, Jenkins L, Koller M (2005) Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 64:1563–1572. [DOI] [PubMed] [Google Scholar]
- 34. Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC (1992) Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta‐amyloid fibrils. Acta Neuropathol 84:225–233. [DOI] [PubMed] [Google Scholar]
- 35. Franceschi C, Monti D, Sansoni P, Cossarizza A (1995) The immunology of exceptional individuals: the lesson of centenarians. Immunol Today 16:12–16. [DOI] [PubMed] [Google Scholar]
- 36. Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C et al (1995) Alzheimer‐type neuropathology in transgenic mice overexpressing V717F beta‐amyloid precursor protein. Nature 373:523–527. [DOI] [PubMed] [Google Scholar]
- 37. Garbar P (1983) Autoantibodies and the physiological role of immunoglobulins. Immunol Today 4:337–340. [DOI] [PubMed] [Google Scholar]
- 38. Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC et al (2005) Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64:1553–1562. [DOI] [PubMed] [Google Scholar]
- 39. Gordon S (2002) Pattern recognition receptors: doubling up for the innate immune response. Cell 111:927–930. [DOI] [PubMed] [Google Scholar]
- 40. Gordon S (2003) Alternative activation of macrophages. Nat Rev Immunol 3:23–35. [DOI] [PubMed] [Google Scholar]
- 41. Griffin WS, Sheng JG, Roberts GW, Mrak RE (1995) Interleukin‐1 expression in different plaque types in Alzheimer's disease: significance in plaque evolution. J Neuropathol Exp Neurol 54:276–281. [DOI] [PubMed] [Google Scholar]
- 42. Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI et al (1998) Glial‐neuronal interactions in Alzheimer's disease: the potential role of a “cytokine cycle” in disease progression. Brain Pathol 8:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grimm MO, Grimm HS, Patzold AJ, Zinser EG, Halonen R, Duering M et al (2005) Regulation of cholesterol and sphingomyelin metabolism by amyloid‐beta and presenilin. Nat Cell Biol 7:1118–1123. [DOI] [PubMed] [Google Scholar]
- 44. Haass C, Selkoe DJ (1993) Cellular processing of beta‐amyloid precursor protein and the genesis of amyloid beta‐peptide. Cell 75:1039–1042. [DOI] [PubMed] [Google Scholar]
- 45. Hallgren HM, Buckley CE 3rd, Gilbertsen VA, Yunis EJ (1973) Lymphocyte phytohemagglutinin responsiveness, immunoglobulins and autoantibodies in aging humans. J Immunol 111:1101–1107. [PubMed] [Google Scholar]
- 46. Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356. [DOI] [PubMed] [Google Scholar]
- 47. Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller‐Tillmanns B et al (2003) Antibodies against beta‐amyloid slow cognitive decline in Alzheimer's disease. Neuron 38:547–554. [DOI] [PubMed] [Google Scholar]
- 48. Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM et al (1999) Expression of human apolipoprotein E reduces amyloid‐beta deposition in a mouse model of Alzheimer's disease. J Clin Invest 103:R15–R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hull M, Berger M, Volk B, Bauer J (1996) Occurrence of interleukin‐6 in cortical plaques of Alzheimer's disease patients may precede transformation of diffuse into neuritic plaques. Ann N Y Acad Sci 777:205–212. [DOI] [PubMed] [Google Scholar]
- 50. Hyman BT, Smith C, Buldyrev I, Whelan C, Brown H, Tang MX, Mayeux R (2001) Autoantibodies to amyloid‐beta and Alzheimer's disease. Ann Neurol 49:808–810. [DOI] [PubMed] [Google Scholar]
- 51. In 'T Veld BA, Ruitenberg A, Hofman A, Launer LJ, Van Duijn CM, Stijnen T et al (2001) Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Eng J Med 345:1515–1521. [DOI] [PubMed] [Google Scholar]
- 52. Ishii T, Haga S (1975) Identification of components of immunoglobulins in senile plaques by means of fluorescent antibody technique. Acta Neuropathol 32:157–162. [DOI] [PubMed] [Google Scholar]
- 53. Ishii T, Haga S (1976) Immuno‐electron microscopic localization of immunoglobulins in amyloid fibrils of senile plaques. Acta Neuropathol 36:243–249. [DOI] [PubMed] [Google Scholar]
- 54. Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E et al (2000) Identification of the major Abeta1‐42‐degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med 6:143–150. [DOI] [PubMed] [Google Scholar]
- 55. Janelsins MC, Mastrangelo MA, Oddo S, LaFerla FM, Federoff HJ, Bowers WJ (2005) Early correlation of microglial activation with enhanced tumor necrosis factor‐alpha and monocyte chemoattractant protein‐1 expression specifically within the entorhinal cortex of triple transgenic Alzheimer's disease mice. J Neuroinflammation 2:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD et al (2000) Abeta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature 408:979–982. [DOI] [PubMed] [Google Scholar]
- 57. Jiang H, Burdick D, Glabe CG, Cotman CW, Tenner AJ (1994) beta‐Amyloid activates complement by binding to a specific region of the collagen‐like domain of the C1q A chain. J Immunol 152:5050–5059. [PubMed] [Google Scholar]
- 58. Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19:312–318. [DOI] [PubMed] [Google Scholar]
- 59. Laskowitz DT, Matthew WD, Bennett ER, Schmechel D, Herbstreith MH, Goel S, McMillian MK (1998) Endogenous apolipoprotein E suppresses LPS‐stimulated microglial nitric oxide production. Neuroreport 9:615–618. [DOI] [PubMed] [Google Scholar]
- 60. Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B et al (2000) Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci 20:5709–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lombardo JA, Stern EA, McLellan ME, Kajdasz ST, Hickey GA, Bacskai BJ, Hyman BT (2003) Amyloid‐beta antibody treatment leads to rapid normalization of plaque‐induced neuritic alterations. J Neurosci 23:10879–10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lynch JR, Morgan D, Mance J, Matthew WD, Laskowitz DT (2001) Apolipoprotein E modulates glial activation and the endogenous central nervous system inflammatory response. J Neuroimmunol 114:107–113. [DOI] [PubMed] [Google Scholar]
- 63. Mahley RW (1988) Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240:622–630. [DOI] [PubMed] [Google Scholar]
- 64. Masliah E, Terry RD, Alford M, DeTeresa R, Hansen LA (1991) Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer's disease. Am J Pathol 138:235–246. [PMC free article] [PubMed] [Google Scholar]
- 65. Masliah E, Ho G, Wyss‐Coray T (2001) Functional role of TGFbeta in Alzheimer's disease microvascular injury: lessons from transgenic mice. Neurochem Int 39:393–400. [DOI] [PubMed] [Google Scholar]
- 66. Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C et al (2005) Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology 64:129–131. [DOI] [PubMed] [Google Scholar]
- 67. Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA 82:4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. McGeer PL, Akiyama H, Itagaki S, McGeer EG (1989) Immune system response in Alzheimer's disease. Can J Neurol Sci 16:516–527. [DOI] [PubMed] [Google Scholar]
- 69. McGeer PL, Akiyama H, Itagaki S, McGeer EG (1989) Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett 107:341–346. [DOI] [PubMed] [Google Scholar]
- 70. McGeer PL, McGeer E (2003) Is there a future for vaccination as a treatment for Alzheimer's disease? Neurobiol Aging 24:391–395. [DOI] [PubMed] [Google Scholar]
- 71. McGeer PL, McGeer EG (2007) NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol Aging 28:639–647. [DOI] [PubMed] [Google Scholar]
- 72. Mirra SS, Hyman BT (2002) Ageing and dementia. In: Greenfield's Neuropathology. Graham DI, Lantos PL (eds), pp. 195–272. Arnold: London. [Google Scholar]
- 73. Moir RD, Tseitlin KA, Soscia S, Hyman BT, Irizarry MC, Tanzi RE (2005) Autoantibodies to redox‐modified oligomeric Abeta are attenuated in the plasma of Alzheimer's disease patients. J Biol Chem 280:17458–17463. [DOI] [PubMed] [Google Scholar]
- 74. Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J et al (2000) A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 408:982–985. [DOI] [PubMed] [Google Scholar]
- 75. Morioka T, Kalehua AN, Streit WJ (1993) Characterization of microglial reaction after middle cerebral artery occlusion in rat brain. J Comp Neurol 327:123–132. [DOI] [PubMed] [Google Scholar]
- 76. Mrak RE, Griffin WS (2005) Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging 26:349–354. [DOI] [PubMed] [Google Scholar]
- 77. Mruthinti S, Buccafusco JJ, Hill WD, Waller JL, Jackson TW, Zamrini EY, Schade RF (2004) Autoimmunity in Alzheimer's disease: increased levels of circulating IgGs binding Abeta and RAGE peptides. Neurobiol Aging 25:1023–1032. [DOI] [PubMed] [Google Scholar]
- 78. Nicoll JA, Mrak RE, Graham DI, Stewart J, Wilcock G, MacGowan S et al (2000) Association of interleukin‐1 gene polymorphisms with Alzheimer's disease. Ann Neurol 47:365–368. [PMC free article] [PubMed] [Google Scholar]
- 79. Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO (2003) Neuropathology of human Alzheimer's disease after immunization with amyloid‐beta peptide: a case report. Nat Med 9:448–452. [DOI] [PubMed] [Google Scholar]
- 80. Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P et al (2006) Abeta species removal after Abeta42 immunization. J Neuropathol Exp Neurol 65:1040–1048. [DOI] [PubMed] [Google Scholar]
- 81. Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM (2003) Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging 24:1063–1070. [DOI] [PubMed] [Google Scholar]
- 82. Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM (2004) Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43:321–332. [DOI] [PubMed] [Google Scholar]
- 83. Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM (2006) Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem 281:39413–39423. [DOI] [PubMed] [Google Scholar]
- 84. Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC et al (2003) Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 61:46–54. [DOI] [PubMed] [Google Scholar]
- 85. Paresce DM, Chung H, Maxfield FR (1997) Slow degradation of aggregates of the Alzheimer's disease amyloid beta‐protein by microglial cells. J Biol Chem 272:29390–29397. [DOI] [PubMed] [Google Scholar]
- 86. Patton RL, Kalback WM, Esh CL, Kokjohn TA, Van Vickle GD, Luehrs DC et al (2006) Amyloid‐beta peptide remnants in AN‐1792‐immunized Alzheimer's disease patients: a biochemical analysis. Am J Pathol 169:1048–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Perry VH, Hume DA, Gordon S (1985) Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience 15:313–326. [DOI] [PubMed] [Google Scholar]
- 88. Perry VH, Cunningham C, Holmes C (2007) Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol 7:161–167. [DOI] [PubMed] [Google Scholar]
- 89. Poirier J (2000) Apolipoprotein E and Alzheimer's disease. A role in amyloid catabolism. Ann N Y Acad Sci 924:81–90. [DOI] [PubMed] [Google Scholar]
- 90. Qiu WQ, Ye Z, Kholodenko D, Seubert P, Selkoe DJ (1997) Degradation of amyloid beta‐protein by a metalloprotease secreted by microglia and other neural and non‐neural cells. J Biol Chem 272:6641–6646. [DOI] [PubMed] [Google Scholar]
- 91. Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB et al (1998) Insulin‐degrading enzyme regulates extracellular levels of amyloid beta‐protein by degradation. J Biol Chem 273:32730–32738. [DOI] [PubMed] [Google Scholar]
- 92. Rainero I, Bo M, Ferrero M, Valfre W, Vaula G, Pinessi L (2004) Association between the interleukin‐1alpha gene and Alzheimer's disease: a meta‐analysis. Neurobiol Aging 25:1293–1298. [DOI] [PubMed] [Google Scholar]
- 93. Reid DM, Perry VH, Andersson PB, Gordon S (1993) Mitosis and apoptosis of microglia in vivo induced by an anti‐CR3 antibody which crosses the blood‐brain barrier. Neuroscience 56:529–533. [DOI] [PubMed] [Google Scholar]
- 94. Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD et al (1992) Complement activation by beta‐amyloid in Alzheimer disease. Proc Natl Acad Sci USA 89:10016–10020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rowley MJ, Buchanan H, Mackay IR (1968) Reciprocal change with age in antibody to extrinsic and intrinsic antigens. Lancet 2:24–26. [DOI] [PubMed] [Google Scholar]
- 96. Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T et al (1999) Immunization with amyloid‐beta attenuates Alzheimer‐disease‐like pathology in the PDAPP mouse. Nature 400:173–177. [DOI] [PubMed] [Google Scholar]
- 97. Schley D, Carare‐Nnadi R, Please CP, Perry VH, Weller RO (2006) Mechanisms to explain the reverse perivascular transport of solutes out of the brain. J Theor Biol 238:962–974. [DOI] [PubMed] [Google Scholar]
- 98. Scolding NJ, Joseph F, Kirby PA, Mazanti I, Gray F, Mikol J et al (2005) Abeta‐related angiitis: primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain 128:500–515. [DOI] [PubMed] [Google Scholar]
- 99. Selkoe DJ (2002) Alzheimer's disease is a synaptic failure. Science 298:789–791. [DOI] [PubMed] [Google Scholar]
- 100. Shen Y, Lue L, Yang L, Roher A, Kuo Y, Strohmeyer R et al (2001) Complement activation by neurofibrillary tangles in Alzheimer's disease. Neurosci Lett 305:165–168. [DOI] [PubMed] [Google Scholar]
- 101. Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B et al (2000) Clearance of Alzheimer's amyloid‐ss(1‐40) peptide from brain by LDL receptor‐related protein‐1 at the blood‐brain barrier. J Clin Invest 106:1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sigurdsson EM, Knudsen E, Asuni A, Fitzer‐Attas C, Sage D, Quartermain D et al (2004) An attenuated immune response is sufficient to enhance cognition in an Alzheimer's disease mouse model immunized with amyloid‐beta derivatives. J Neurosci 24:6277–6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Simard AR, Rivest S (2004) Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. Faseb J 18:998–1000. [DOI] [PubMed] [Google Scholar]
- 104. Simard AR, Soulet D, Gowing G, Julien JP, Rivest S (2006) Bone marrow‐derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron 49:489–502. [DOI] [PubMed] [Google Scholar]
- 105. Solomon B, Koppel R, Hanan E, Katzav T (1996) Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer beta‐amyloid peptide. Proc Natl Acad Sci USA 93:452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Solomon B, Koppel R, Frankel D, Hanan‐Aharon E (1997) Disaggregation of Alzheimer beta‐amyloid by site‐directed mAb. Proc Natl Acad Sci USA 94:4109–4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Streit WJ (1998) Role of macrophages and microglia in the injured CNS. In: CNS Injuries: Cellular Responses and Pharmacological Strategies. Berry M, Logan A (eds), pp. 81–98. CRC Press: Boca Raton, FL, USA. [Google Scholar]
- 108. Streit WJ (2005) Microglia and neuroprotection: implications for Alzheimer's disease. Brain Res Brain Res Rev 48:234–239. [DOI] [PubMed] [Google Scholar]
- 109. Streit WJ (2006) Microglial senescence: does the brain's immune system have an expiration date? Trends Neurosci 29:506–510. [DOI] [PubMed] [Google Scholar]
- 110. Strittmatter WJ, Saunders AM, Schmechel D, Pericak‐Vance M, Enghild J, Salvesen GS, Roses AD (1993) Apolipoprotein E: high‐avidity binding to beta‐amyloid and increased frequency of type 4 allele in late‐onset familial Alzheimer disease. Proc Natl Acad Sci USA 90:1977–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Strohmeyer R, Shen Y, Rogers J (2000) Detection of complement alternative pathway mRNA and proteins in the Alzheimer's disease brain. Brain Res Mol Brain Res 81:7–18. [DOI] [PubMed] [Google Scholar]
- 112. Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K (2006) Role of toll‐like receptor signalling in Abeta uptake and clearance. Brain 129:3006–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Tarkowski E, Blennow K, Wallin A, Tarkowski A (1999) Intracerebral production of tumor necrosis factor‐alpha, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J Clin Immunol 19:223–230. [DOI] [PubMed] [Google Scholar]
- 114. Terry RD, Gonatas NK, Weiss M (1964) Ultrastructural studies in Alzheimer's presenile dementia. Am J Pathol 44:269–297. [PMC free article] [PubMed] [Google Scholar]
- 115. Van Der Wal EA, Gomez‐Pinilla F, Cotman CW (1993) Transforming growth factor‐beta 1 is in plaques in Alzheimer and Down pathologies. Neuroreport 4:69–72. [DOI] [PubMed] [Google Scholar]
- 116. Walford RL (1969) The Immunologic Theory of Aging. Williams & Wilkins: Philadelphia, PA. [Google Scholar]
- 117. Webster S, Bradt B, Rogers J, Cooper N (1997) Aggregation state‐dependent activation of the classical complement pathway by the amyloid beta peptide. J Neurochem 69:388–398. [DOI] [PubMed] [Google Scholar]
- 118. Weksler ME, Relkin N, Turkenich R, LaRusse S, Zhou L, Szabo P (2002) Patients with Alzheimer's disease have lower levels of serum anti‐amyloid peptide antibodies than healthy elderly individuals. Exp Gerontol 37:943–948. [DOI] [PubMed] [Google Scholar]
- 119. Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE (1998) Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer's disease. Am J Pathol 153:725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE et al (2003) Intracranially administered anti‐Abeta antibodies reduce beta‐amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci 23:3745–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D (2004) Passive immunotherapy against Abeta in aged APP‐transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation 1:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Williams AE, Lawson LJ, Perry VH, Fraser H (1994) Characterization of the microglial response in murine scrapie. Neuropathol Appl Neurobiol 20:47–55. [DOI] [PubMed] [Google Scholar]
- 123. Wyss‐Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E (2002) Prominent neurodegeneration and increased plaque formation in complement‐inhibited Alzheimer's mice. Proc Natl Acad Sci USA 99:10837–10842. [DOI] [PMC free article] [PubMed] [Google Scholar]