

Abstract
Chemokines regulate lymphocyte trafficking under physiologic and pathologic conditions. In this study, we have investigated the role of CXCR3 and CXCR4 in the activation of T lymphocytes and their migration to the central nervous system (CNS) using novel mutant chemokines to antagonize CXCR3 and CXCR4 specifically. A series of truncation mutants of CXCL11, which has the highest affinity for CXCR3, were synthesized, and an antagonist, CXCL11(4–79), was obtained. CXCL11(4–79) strongly inhibited the migration of activated mouse T cells in response to all three high‐affinity CXCR3 ligands, CXCL9, 10 and 11. CXCL12(P2G2), while exhibiting minimal agonistic activity, potently inhibited the migration of activated mouse T cells in response to CXCL12. Interfering with the action of CXCR3 and CXCR4 with these synthetic receptor antagonists inhibited experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis and reduced the accumulation of CD4+ T cells in the CNS. Further investigation demonstrated that CXCL12(P2G2) inhibited the sensitization phase, whereas CXCL11(4–79) inhibited the effector phase of the immune response. Our data suggest that simultaneous targeting of CXCR4 and CXCR3 may be of benefit in the treatment of the CNS autoimmune disease.
Keywords: autoimmune diseases, CD4+ T cells, chemokines, chemotaxis, multiple sclerosis
INTRODUCTION
Experimental autoimmune encephalomyelitis (EAE) is an inflammatory demyelinating disease of the central nervous system (CNS), which serves as an experimental model of multiple sclerosis (MS) 2, 34. The pathophysiology and pathogenesis of EAE are still not clearly understood. However, following the active or passive induction of the disease, there is a substantial cellular infiltrate into the CNS that comprises both CD4+and CD8+ T lymphocytes 46, 47, as well as B cells (20) and macrophages (21) that can be found in the perivascular space, meninges and parenchyma of the CNS. Polymorphonuclear cells (neutrophils) may also be present (29). There is an absolute requirement for CD4+T cells to initiate disease, but how the cascade of other cells is orchestrated, the factors regulating the recruitment of activated T cells to the CNS and which cells or cell products are responsible for CNS damage and clinical signs have not been clearly defined. Accumulating evidence implicates chemokines and their receptors in this process (9).
Within T‐dependent areas of the peripheral lymphoid tissue, naive T cells respond to antigenic peptides bound to MHC molecules on the surface of dendritic cells (DC) 16, 19. Following activation, expansion and differentiation, effector T cells migrate to the periphery to carry out their effector function. The processes driving both the coordinated colocalization of naive T cells and DC, and the peripheral homing of effector cells during inflammation involve different adhesion molecules as well as the large family of chemokines and their receptors.
The chemokine receptor CCR7 and its ligands CCL19/ELC and CCL21/SLC play a central role in maintaining the homeostatic trafficking and positioning of both DC and T cells within the peripheral lymphoid tissue 8, 30, 36. In contrast, receptors including CCR1‐6, CXCR3 and CXCR6 are up‐regulated on effector cells during an immune response and respond to chemokines produced during inflammation 10, 11, 24, 44. CXCR3, a prototypic inflammatory receptor, is the receptor for the chemokines CXCL9/mig, CXCL10/IP‐10 and CXCL11/I‐TAC (5). Its expression has been documented on short‐lived effector T cells 3, 44 as well as long‐lived memory T cells (45), in keeping with in vitro observations that T cells acquire CXCR3 upon antigen‐specific activation. Numerous studies have also demonstrated the preferential expression of CXCR3 on Th1 cells 3, 41, 43. In inflammatory diseases such as MS, myelin‐reactive T cells in the CSF and brain lesions express CXCR3 1, 48 and CXCR3 ligands are expressed in the CNS and CSF 48, 49.
Certain receptors and their ligands cannot be unambiguously affiliated with homeostatic or inflammatory trafficking (35). Included in this group is CXCR4, the most ubiquitously expressed chemokine receptor. Together with its ligand CXCL12/SDF‐1, CXCR4 is constitutively expressed in a wide variety of tissues and is the only chemokine receptor that has been shown to be essential for life 50, 52. CXCR4 is expressed on naive T cells and is widely acknowledged to play an important role in the homeostasis of the immune system (11). More recent studies indicate that it is also expressed on effector T cells and has been implicated in the accumulation of these cells in the rheumatoid synovium 4, 38.
Understanding the precise roles of CXCR3 and CXCR4 in regulating T cell migration under homeostatic vs. inflammatory conditions is an important objective as such knowledge is likely to enable us to more specifically control the immune response. Generally, the roles of chemokine receptors in diseases have been elucidated using transgenic and knockout mice, but specifically for CXCR4, this approach presents complications as deletion of CXCR4 or CXCL12 is perinatally lethal 50, 52. Additionally, the absence of a receptor from conception may induce compensation by one or more nontargeted receptors, further confounding data interpretation (51). For this reason, chemokine receptor antagonism, in which the receptor is blocked for only a defined time in a normally developed immune system, is an alternative strategy for studying various aspects of chemokine receptor biology.
The development of synthetic chemokine antagonists is built around the observation that chemokines have two main sites of interaction with their receptors—one in the N‐terminal region, which is responsible for the activating signaling event, and another within an exposed loop of the backbone, which is responsible for the initial binding event (7). Thus, N‐terminal truncations may still bind to the receptor efficiently, without activating signal transduction. This principle has been used to successfully develop several different chemokine receptor antagonists 7, 14, 40, some of which have proven useful in elucidating the role of specific chemokines and their receptors in various aspects of the immune response 15, 33, 39. In line with this methodology, we showed here that N‐terminal modifications of the chemokines CXCL12 and CXCL11, targeting CXCR4 and CXCR3 respectively, are capable of inhibiting the migration of mouse lymphocytes toward cognate ligands of the receptors both in vitro and in vivo. Furthermore, using EAE as a model of T‐dependent inflammation of the CNS, we have identified different roles for CXCR4 and CXCR3 in the priming and effector phases of the immune response respectively, and have demonstrated that interfering with either receptor inhibits CNS pathology in EAE.
MATERIALS AND METHODS
Reagents
Proteolipid protein (PLP) peptide139–151 (HSLGKWLGHPDKF) was produced by solid phase synthesis and HPLC purified. Pertussigen, a crude extract of Bordetella pertussis cells, was a gift from Associate Professor David Willenborg (Neurosciences Research Unit, Canberra Hospital, Australia). The chemokine antagonists were chemically synthesized as described previously 7, 14. The antagonists were synthetic peptides based on the sequence of the wild‐type (WT) chemokine, with a modified NH2terminal, and the CCL2(4Ala) control peptide was an analog of CCL2 that had all four cysteines (residue numbers 11, 12, 36 and 52) replaced with alanine.
Mice
Female SJL/J mice (H‐2s) aged 6–9 weeks were obtained from the Central Animal House at the University of Adelaide, South Australia. The mice were kept under standard temperature and light conditions, and afforded food and water ad libitum. Severely paralyzed mice were hand fed and watered.
Induction of active EAE
Female SJL/J mice were immunized s.c. in the hind flanks with 50 µg of PLP(139–151) in complete Freund's adjuvant (CFA) containing 0.5 mg/mL Mycobacterium butyricum (Difco Laboratories, Detroit, MI, USA) and 8.33 mg/mL Mycobacterium tuberculosis H37Ra (Difco Laboratories). On days 0 and 2, the mice received pertussigen i.v. Clinical EAE was evaluated on a scale of 0–5 as previously described (23).
Priming of donor lymphocytes, cell culture and transfer of EAE
Donor SJL/J mice were primed by s.c. immunization with 25 µg PLP139–151emulsified in CFA. Ten days later, the draining lymph nodes (LNs) were collected, and single cell suspensions were prepared and restimulated in vitro in the presence of 50 µg/mL PLP139–151. After 96 h of culture, the cells were harvested and 5 × 107 cells were transferred i.v. into SJL/J recipients, as previously described (23). The recipient mice were scored as for active EAE.
Treatment of mice with antagonists
In the experiments involving CXCL12(P2G2), the mice were injected into the peritoneal cavity (i.p.) with 250 µL of endotoxin‐free PBS containing 100 µg of CXCL12(P2G2)or the control peptide CCL2(4Ala). In the experiments involving CXCL11(4–79), the mice were injected i.p. with 250 µL of endotoxin‐free PBS containing 250 µg of either CXCL11(4–79) or CCL2(4Ala). The antagonist was administered on days 1, 3, 5, 7, 9, 11, 13 and 15 post‐EAE induction (PI), or on days 0, 2, 4, 6 8 and 12 post‐transfer. In the experiments involving the combined treatment of CXCL12(P2G2) and CXCL11(4–79), the mice were injected i.p. with 250 µL of PBS containing 100 µg of CXCL12(P2G2) + 250 µg of CXCL11(4–79) or with 350 µg of CCL2(4Ala). In these experiments, the antagonists were administered on days 1, 3, 5, 7, 9, 11 and 13 PI.
Collection of lymphoid and CNS tissue
The mice were euthanased by CO2 asphyxiation, perfused through the left ventricle with PBS to remove the circulating leukocytes, then the draining LNs and the spinal cords were extracted. Tissue samples were either snap frozen in liquid nitrogen for RNA analysis or embedded in Tissue‐Tek® OCT embedding medium (Sakura Finetek, Torrence, CA, USA), frozen, sectioned and stained with hematoxylin/eosin or used for dual immunofluorescence (described below). In some instances, the spinal cord was passed through a cell strainer. The resultant cell suspension was collected in 30 mL of RPMI + 1% FCS and 20 mL of 90% Percoll (Amersham Pharmacia Biotech, Little Chalfont, UK) was added, before centrifuging at 1000 g for 25 minutes at 4°C, with no brake. Following centrifugation, the myelin cake was discarded, and the cells were washed twice, then resuspended in PBS + 1% BSA.
RT‐PCR
Following removal from the animals, the spinal cords were snap frozen and stored at −70°C until required. The samples were thawed and homogenized in TrizolTM (Invitrogen Corp, Carlsbad, CA, USA), and total RNA was extracted according to the manufacturer's instructions. The extracted RNA was then treated with DNAase I (Promega, Madison, WI, USA) to remove contaminating genomic DNA. cDNA was reverse transcribed from DNAase‐treated RNA using Superscript II reverse transcriptase (Invitrogen), and then used as template in PCR for CXCR3, CXCR4 and GAPDH using the following specific oligonucleotide primers (5′−3′): CXCR3 forward—GTGCTAGATGCCTCGGACTT; CXCR3 reverse—GAGGCGCTGATCGTAGTTGG; CXCR4 forward—ACCACGGCTGTAGAGCGAGT; CXCR4 reverse—GCTGATGAAGGCCAGGATGA; GAPDH forward—TCCTT GGAGGCCATGTAGGCCAT; GAPDH reverse—TGATGACAT CAAGAAGGTGGTGAAG. PCR reactions were amplified using Amplitaq Gold (Perkin Elmer Life Sciences, Waltham, MA, USA) in 1.5 mM MgCl2 and the following therocycling conditions: 95°C 1 minute, then cycles of 95°C 30 s 55°C 1 minute, 72°C 1 minute. A range of cycle numbers were tested for each PCR to ensure that the amplification of PCR products was in the linear phase when analyzed. After the completion of cycling, an extension step at 72°C for 5 minutes was performed. PCR products were separated by gel electrophoresis and visualized using a Molecular Imager FX and Quantity One software package following staining with SYBR‐gold (Invitrogen).
Dual immunofluorescence
The sections were rehydrated in PBS for 5 minutes, and nonspecific binding was blocked using Powerblock® (InnoGenex, San Ramon, CA, USA) at RT for 1 h then washed in PBS and the appropriate primary antibodies added and incubated overnight at 4°C. Monoclonal FITC‐labeled antibodies used were anti‐CD4 (GK1.5) and rat IgG2b both from BD Pharmingen (San Diego, CA, USA). Polyclonal antibodies used were goat anti‐mCXCR3, goat anti‐mCXCR4 and control goat IgG, all from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The sections were then washed twice in PBS before the addition of the anti‐goat secondary antibody conjugated to Alexa 594 preblocked with both rat and mouse gamma globulins. Following incubation for an hour at 4°C, the sections were washed thrice in PBS, and coverslips were mounted in Mowiol mounting medium + PPD (Calbiochem, La Jolla, CA, USA). All incubations were carried out in a humid chamber. The slides were viewed and photographed using an Olympus Provis AX reflected light microscope equipped with a Photometrics CE 200A digital camera, using V for Windows software (Photometrics, Tuscon, AR, USA) for image capture.
Flow cytometric analysis
The cells (2 × 105) were resuspended in PBS + 1% BSA + 0.04% sodium azide (PBA). Fc receptors were blocked by incubating for 30 minutes at room temperature with 50 µg of mouse gamma globulin (Rockland Immunochemicals, Gilbertsville, PA, USA)/106 cells. The cells were mixed with saturating concentrations of primary antibodies (Ab) for 30 minutes at room temperature. R‐Phycoerythrin‐labeled monoclonal antibodies to CD4 (H129.19) and control rat IgG2a were from BD Pharmingen. Polyclonal goat Abs directed against CXCR4, CXCR3 and the control goat IgG were obtained from Santa Cruz Biotechnology. After washing in staining buffer, the cells were incubated with biotin‐conjugated anti‐goat Ab (Rockland) for 45 minutes at 4°C followed by a 30‐minute incubation with streptavidin conjugate. The lymphocytes were gated using forward and side scatter characteristics.
Transwell chemotaxis assays
For dose–response and inhibition studies, the cells were fluorescently labeled with Calcein‐AM (Molecular Probes, Eugene, OR, USA; 40 nM final concentration in RPMI 0.5% BSA) for 30 minutes at 37°C, followed by three washes in RPMI–BSA. The cells were resuspended to 107 viable cells/mL in RPMI–BSA. Chemotaxis assays were conducted using the Transwell cell culture chambers (Costar, Corning, NY, USA) as previously described (23) with the agonists and antagonists added to the bottom chamber.
Lymphocyte proliferation assay
The mice immunized with 25 µg PLP139–151 were euthanased 12 days PI and the Draining Lymph Nodes (DLNs) were taken for proliferation assays using a modification of a previously published protocol (28). Briefly, single‐cell suspensions (2 × 107viable cells/mL) were prepared, and the cells were fluorescently labeled by incubating with carboxyfluorescein diacetate succinimidyl ester [(CFSE) Molecular Probes] (2.5 µM final concentration in RPMI/0.1%BSA) for 10 minutes at 37°C. The staining reaction was quenched by the addition of a large volume of complete media for 5 minutes at room temperature. The cells were washed twice and resuspended at a concentration of 2.5 × 106 viable cells/mL. In a final volume of 200 µL, 2.5 × 105 cells were cultured in 96‐well round‐bottom plates with added peptide at a concentration of 25 or 50 µg/mL, or with 1.5 µg/mL concanavalin A (Con A). After 4 days of culture, the cells were harvested, labeled for CD4 (Pharmingen) and analyzed by flow cytometry. Cell division (proliferation) was determined as progressive halving of CFSE fluorescence intensity.
Statistical tests
Statistical tests were performed using GraphPad InStat software (GraphPad Software Inc., San Diego, CA, USA). Unless otherwise stated, the two‐tailed, unpaired Student's t‐test was used for statistical analysis. In the analyses of the EAE experiments, the analyses of variance were performed in order to allow for variability between individual experiments. For all analyses, P‐values of <0.05 were considered significant.
RESULTS
CXCR4 and CXCR3 are expressed by CNS infiltrating CD4+ cells during EAE
The initial experiments centered on the evaluation of the expression and function of CXCR3 and CXCR4 in the DLNs and the CNS during EAE. Single‐cell suspensions were prepared from the LNs, draining the site of immunization with PLP in CFA as previously shown (23). The cells were restimulated with antigen in vitro, and flow cytometry was conducted to evaluate the level of expression of CXCR4 or CXCR3 on the CD4+T cell population (Figure 1A). Both activated (divided) and unactivated (undivided) CD4+ T cells expressed the receptors, although a larger proportion of activated than unactivated CD4+ T cells expressed CXCR3 (Figure 1B), and the cells responded chemotactically to CXCL12 and the CXCR3 ligands CXCL11 and CXCL10 (Figure 1C), although the relative level of expression of the two receptors did not correlate directly with the level of response. In order to examine the expression of CXCR3 and CXCR4 in the CNS during EAE, we analyzed the levels of mRNA for these receptors in the spinal cord both before and after immunization of the SJL/J mice with either PLP/CFA or PBS/CFA. Relative expression of CXCR4 in the spinal cord was significantly increased in PLP/CFA‐immunized mice on day 12 and on day 21 compared with unimmunized mice or mice immunized with PBS/CFA (Figure 2A). Similarly, the expression of CXCR3 was also elevated in the spinal cord at the same stages of the disease. To follow up these observations, and because CD4+ T cells have been implicated as the primary effector cells causing pathology in EAE, we examined the expression of CXCR4 and CXCR3 protein by CD4+ T cells in the spinal cord using immunofluorescent staining. EAE was induced as described, and the sections of the spinal cord taken during the peak disease (day 12) were analyzed using antibodies specific for CD4, CXCR3 and CXCR4. CD4+ cells could be clearly visualized around the lesions within the spinal cords of the diseased mice (Figure 2B) but not in the spinal cords of unimmunized control mice (data not shown). Both CXCR3+ and CXCR4+ cells could also be visualized within the spinal cords of the mice with EAE, and there was a significant colocalization between CD4 and both CXCR3 and CXCR4 (Figure 2B), indicating that these two chemokine receptors are expressed by CD4+ cells, which infiltrate the CNS during EAE. To further characterize the T cell infiltrate, the proportion of CD4+ T cells accumulating in the spinal cord that were CXCR3+ or CXCR4+ was determined. The results of these experiments showed that maximal accumulation of CD4+ T cells expressing CXCR3 or CXCR4 at day 12 PI, corresponding with peak disease (Figure 3A). Finally, the activation status of the CD4+ T cells accumulating in the spinal cord was evaluated by determining the proportion of CXCR3+ and CXCR4+ cells that had undergone division. To achieve this, the mice were fed the DNA precursor BrdU by gavage during the induction of EAE, and the proportion of T cells in the spinal cord on day 12 postinduction that had incorporated BrdU was determined by flow cytometry. The results of these experiments indicate that the majority of CD4+ T cells accumulating in the spinal cord had divided (Figure 3B), and the majority of CXCR3+ or CXCR4+ CD4+ T cells had divided (Figure 3C).
Figure 1.

Characterization of chemokine receptor expression and function in vitro. The cells from the lymph nodes isolated from the proteolipid protein (PLP)/complete Freund's adjuvant (CFA)‐immunized mice on day 9 postimmunization were loaded with carboxyfluorescein diacetate succinimidyl ester (CFSE) and cultured for 4 days in the presence of PLP139–151. The cells were then harvested and analyzed by flow cytometry. A. Representative density plots of divided and undivided CD4+ T cell populations expressing CXCR4 or CXCR3. B. Within the CD4+ T cell population, the percentage of cells expressing CXCR4 or CXCR3 within the divided (low CFSE fluorescence intensity) and undivided (high CFSE fluorescence intensity) cell populations was determined. *Significantly different from undivided cells (P < 0.001), n = 3. C. Dose–response of chemotaxis of activated lymphocytes in response to wild‐type (WT) CXCL12, WT CXCL11 or WT CXCL10. Data represent mean ± standard error of the mean (n = 4, from four independent experiments).
Figure 2.

Characterization of chemokine receptor expression in the central nervous system (CNS) during experimental autoimmune encephalomyelitis (EAE). EAE was induced as described previously (23). The control mice were subjected to complete Freund's adjuvant (CFA) injection and pertussigen treatment, without proteolipid protein (PLP) in the CFA emulsion (PBS/CFA). A.Comparative PCR analysis of the chemokine receptor mRNA levels in the CNS. On days 0, 9, 12 and 21 postimmunization, the spinal cords were removed and total RNA was extracted, and PCR was performed to produce CXCR4, CXCR3 and GAPDH amplicons. The volume of receptor bands was normalized according to the GAPDH band volume for the same sample template and cycle number. Statistically significant different from day 0 or the PBS/CFA value at the same time point: *P < 0.05; **P < 0.01; ***P < 0.001. Data are presented as mean ± standard error of the mean (n = 3–6, from two independent experiments). B. Expression of CXCR4 and CXCR3 by CD4+ T cells within EAE spinal cords. At peak disease (day 12), the mice were sacrificed and perfused to remove the circulating leukocytes; the spinal cord was removed and was frozen in OCT, and 6 µM cryostat sections were prepared. The sections were fixed and stained with appropriate antibodies and analyzed by fluorescent microscopy. Two images were collected per sample using different filters (left and center panels), and the images overlayed (right panels). The cells stained with anti‐CD4 only appear green, and the cells stained with anti‐chemokine receptor antibody appear red. Colocalization in overlayed images appears yellow. A representative image of each is shown, captured using the ×20 objective (×10 for isotype control images).
Figure 3.
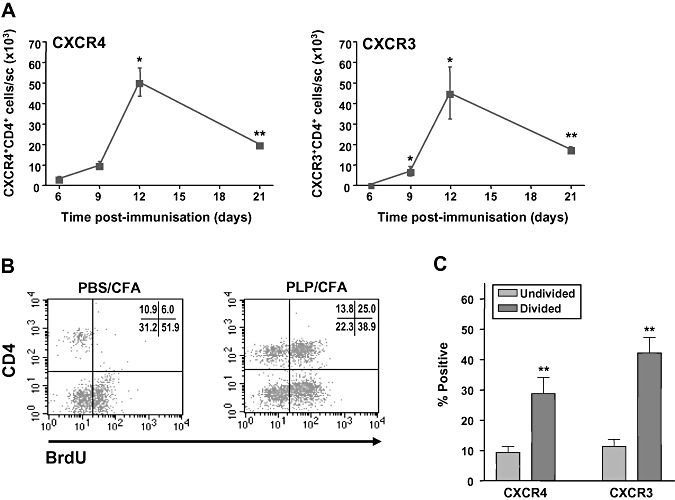
Accumulation of chemokine receptor‐positive CD4+ T cells in the spinal cord during experimental autoimmune encephalomyelitis (EAE). A. EAE was induced in SJL/J mice; the spinal cords were removed after the indicated times, and the single‐cell suspensions were prepared for flow cytometric analysis. *Statistically significant from day 6 values (P < 0.05). **Statistically significant from day 6 values (P < 0.01). Data are presented as mean ± standard error of the mean (SEM) (n = 4). B,C. BrdU was administered by gavage and 12 days later (peak disease), the spinal cords were removed, the single‐cell suspensions were prepared and the divided cells were evaluated by flow cytometry. **Statistically significant from undivided cell values (P < 0.01). Data are presented as mean ± SEM (n = 8). Abbreviations: PLP = proteolipid protein; CFA = complete Freund's adjuvant.
Development and characterization of CXCR4 and CXCR3 receptor antagonists
The role of CXCR3 and CXCR4 in EAE was investigated using the antagonists of CXCL12 and mCXCL11. The amino acid sequences of the chemokines and chemokine antagonists used in this study are shown in Figure 4. The CXCL12 mutant [CXCL12(P2G2)] was identical to functional human CXCL12(1–67), except that the proline in position two was substituted with glycine (7). The mCXCL11 mutants were developed based on previously characterized hCXCL11 truncation series (5). CCL2(4Ala), a truncated nonfunctional peptide of CCL2 with cysteine to alanine substitutions disrupting the structure (15) was used as a negative control.
Figure 4.

Sequences of wild‐type chemokines and mutated analogs used in this study. All chemokines were chemically synthesized, folded in vitro and HPLC purified as previously described 7, 14.
The initial tests determined the effects of these mutants on activated T cells prepared as described in the section above. CXCL12(P2G2) was negligibly chemotactic for activated cells compared with WT CXCL12 (Figure 5A). CXCL11(3–79) elicited a chemotactic response at 1 µg/mL, while CXCL11(4–79) and CXCL11(5–79) were poorly chemotactic compared with WT CXCL11 (Figure 5A). CCL2(4Ala) was completely inactive over the same dose range (data not shown).
Figure 5.
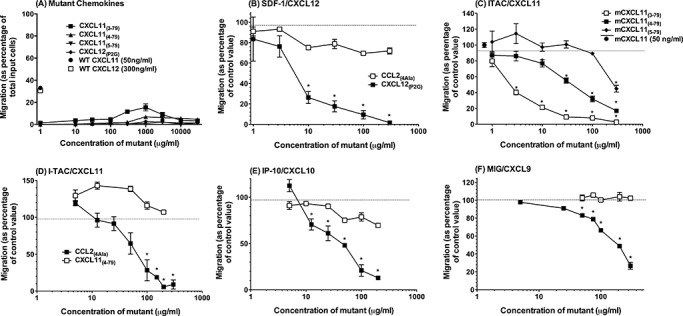
Characterization of chemokine receptor antagonists in vitro. Lymph nodes were removed from the proteolipid protein (PLP)/complete Freund's adjuvant (CFA)‐immunized mice, and single‐cell suspensions were prepared. The cells were cultured for 4 days in the presence of PLP139–151, loaded with calcein‐AM, and chemotaxis was assessed in response to either (A) the chemokine mutants used in this study, (B) 100 ng/mL CXCL12 and the indicated concentrations of either CXCL12(P2G2)or CCL2(4Ala), (C) 50 ng/mL CXCL11 and the indicated concentrations of or CXCL11(3–79), CXCL11(4–79) or CXCL11(5–79), (D–F) 50 ng/mL CXCL11 (D), 50 ng/mL CXCL10 (E) or 100 ng/mL CXCL9 (F) and the indicated concentrations of either CXCL11(4–79) or CCL2(4Ala). Migration was converted to a percentage of migration in response to wild‐type (WT) chemokine alone. *Significantly different from WT chemokine alone and corresponding concentration of CCL2(4Ala), P < 0.05. All data represent mean ± standard error of the mean (n = 4, from four independent experiments).
The potential of mutants to antagonize WT chemokine‐mediated chemotaxis of activated T cells was then examined. These experiments verified the antagonist activity of CXCL12(P2G2) (Figure 5B) and identified both CXCL11(3–79) and CXCL11(4–79) as the antagonists of WT CXCL11 (Figure 5C). However, CXCL11(4–79) was chosen for use in subsequent experiments as it demonstrated limited agonistic activity compared with CXCL11(3–79). In these experiments, CXCL11(4–79) dose dependently and specifically prevented the migration of activated T cells in response to all three known CXCR3 ligands, respectively (Figure 5D–F).
CXCR4 and CXCR3 antagonists inhibit EAE
To examine the antagonist function in vivo, we examined the effect of the chemokine antagonists on the development of EAE. Following the induction of EAE, the antagonists were administered every second day by intraperitoneal injection using a maximal dose of 100–250 µg/mouse (∼2–5 mg/kg/2 days), a dose shown to produce maximal effects in vivo for other modified chemokine peptide antagonists 15, 31, 33. This prophylactic treatment of mice with either CXCL12(P2G2)or CXCL11(4–79) significantly inhibited the development of EAE in three independent experiments, the results of which have been combined (Figure 6A,B). Over the course of EAE, the mice in the antagonist‐treated groups exhibited less severe neurologic symptoms of EAE compared with those treated with the control peptide, resulting in reduced mean cumulative disease scores (Table 1). It should also be noted that antagonist treatment afforded some mice a complete protection from the disease, in contrast to CCL2(4Ala) treatment (Table 1).
Figure 6.
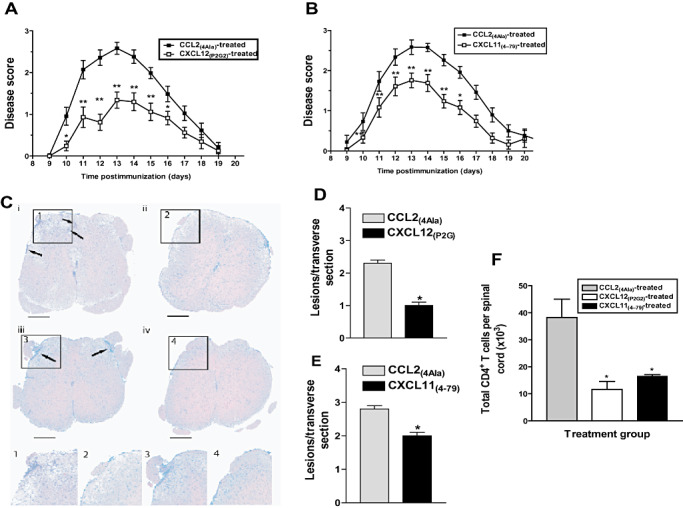
CXCL12(P2G2) and CXCL11(4–79) block development of experimental autoimmune encephalomyelitis (EAE). EAE was induced in SJL/J mice, as described. On days 1, 3, 5, 7, 9, 11, 13 and 15 postimmunization, the mice received an i.p. injection of (A) 100 µg of CXCL12(P2G2) or 100 µg of CCL2(4Ala), or (B) 250 µg of CXCL11(4–79) or 250 µg of CCL2(4Ala). The mice were scored for clinical signs of the disease over a period of 20 days postimmunization. Data are presented as mean clinical disease score ± standard error of the mean (SEM) as a function of days postimmunization (n = 22–26 from three independent experiments, n ≥ 8 per group). Significantly different from CCL2(4Ala) treatment (analysis of variance): *P < 0.05; **P < 0.005. Twelve days postimmunization, the mice were sacrificed and perfused to remove circulating leukocytes. C. Hematoxylin & Eosin (H&E)‐stained lumbar spinal cord sections (×4 objective) from CCL2(4Ala)‐treated mice (panels i and iii), CXCL12(P2G2)‐treated mice (panel ii) or CXCL11(4–79)‐treated mice (panel iv) were assessed for mononuclear cell infiltration (arrows). Panels 1–4 show higher power images of the areas marked in insets. Photomicrographs are representative of the 36 sections from three mice per treatment group. At the time of sacrifice, the mean disease scores ± SEM from the CCL2(4Ala)‐, CXCL12(P2G2)‐ and CXCL11(4–79)‐treated mice were 2.75 ± 0.32, 1.66 ± 0.37 and 1.93 ± 0.11, respectively. D,E. Histopathology in the central nervous system of CCL2(4Ala)‐, CXCL12(P2G2)‐ or CXCL11(4–79)‐treated mice. The total number of lesions were enumerated from the thoracic lumbar region of the spinal cords from three representative animals from each treatment group. Cell infiltration was assayed in 36 sections from each mouse by H&E staining at day 12 postimmunization. *Significantly different from CCL2(4Ala)‐treated mice (P < 0.001). F. Single‐cell suspensions were prepared from the spinal cords and the number of CD4+ T cells present was determined using flow cytometry. *Significantly different from CCL2(4Ala)‐treated mice (P < 0.05). Data represent mean ± SEM (n = 6).
Table 1.
The effect of antagonist treatment on the development of clinical EAE. SJL/J mice were induced with EAE and then treated with either CXCL12(P2G2), CXCL11(4‐79), combined CXCL12(P2G2) & CXCL11(4‐79) or CCL2(4Ala). Upon disease completion, various parameters of disease were analysed, and are presented below.
| Parameter | CCL2(4Ala) treatment | CXCL12(P2G2) treatment | CCL2(4Ala) treatment | CXCL11(4‐79) treatment | CCL2(4Ala) treatment | CXCL12(P2G2) & CXCL11(4‐79) treatment |
|---|---|---|---|---|---|---|
| Disease incidence (No. Sick/Total) | 22/22 | 22/25 | 26/26 | 25/26 | 16/16 | 12/16 |
| Mean score per mouse a , c | 1.4 ± 0.2 | 0.7 ± 0.1* | 1.6 ± 0.1 | 0.9 ± 0.1* | 1.3 ± 0.1 | 0.5 ± 0.1* |
| Mean maximum clinical score a , b | 2.8 ± 0.2 | 1.9 ± 0.2* | 3.1 ± 0.1 | 2.4 ± 0.2* | 2.7 ± 0.2 | 1.4 ± 0.2* |
| Mean cumulative score a , c | 13.8 ± 1.1 | 7.2 ± 0.9* | 16.6 ± 0.9 | 9.8 ± 0.9* | 14.6 ± 1.3 | 5.1 ± 1.1* |
| Mean day of onset a , b | 10.4 ± 0.1 | 11.9 ± 0.4* | 10.7 ± 0.2 | 11.7 ± 0.3* | 10.8 ± 0.2 | 12.5 ± 0.4* |
| Mean length of disease (days) a , c | 8.1 ± 0.3 | 5.4 ± 0.3* | 8.7 ± 0.3 | 5.9 ± 0.4* | 7.9 ± 0.4 | 4.8 ± 0.5* |
Mean ± SEM.
Excluding asymptomatic mice.
Including asymptomatic mice (mice that never showed clinical manifestation were classified as showing a maximum clinical score of 0).
Significantly different from the CCL2(4Ala) treatment group at p < 0.01 (students t test).
Histologic analyses showed that reduced disease severity correlated with a reduced number of inflammatory infiltrates in the spinal cords of the antagonist‐treated mice compared with the control peptide‐treated mice (Figure 6C–E). The spinal cord sections from CCL2(4Ala)‐treated mice showed extensive meningeal and perivascular, and some parenchymal mononuclear cell infiltration (Figure 6C, arrows). In contrast, the spinal cord sections from either CXCL12(P2G2)‐ or CXCL11(4–79)‐treated mice showed little or no mononuclear cell infiltration (Figure 6C) and significantly fewer lesions (Figure 6D,E). The observed reduction in disease severity and lesion burden may be attributed to decreased numbers of CD4+ T cells in the spinal cord of the antagonist‐treated mice (Figure 6F), indicating the importance of CXCR3 and CXCR4 in T cell accumulation in the CNS during EAE.
Involvement of CXCR4 and CXCR3 in the priming and effector phases of the immune response, respectively
The ability to separate the EAE disease process into sensitization and effector phases provides a means to investigate points in the immune response at which antagonism of chemokine receptors may regulate EAE pathology. CD4+ T cells from the mice treated with CXCL12(P2G2) proliferated less robustly ex vivo than those from the control‐treated mice in response to varying doses of the neuroantigen PLP139–151 (Figure 7A), indicating a role for CXCL12/CXCR4 interactions in the generation of encephalitogenic T cells. This proliferation was antigen specific as CD4+ T cells from the mice immunized with PBS/CFA did not proliferate in response to any tested dose of PLP139–151, and the cells from the PLP/CFA‐immunized mice did not proliferate in response to an irrelevant peptide (data not shown). The observed reduction in the proliferation of cells from CXCL12(P2G2)‐treated mice was also antigen specific, as the proliferation of CD4+ lymphocytes from CCL2(4Ala)‐ and CXCL12(P2G2)‐treated mice in response to the mitogen Con A was not significantly different. In contrast, the treatment of mice with CXCL11(4–79) during the sensitization phase of EAE had no effect on the capacity of CD4+ T cells to proliferate in response to PLP139–151 or Con A (Figure 7B). Instead, adoptive transfer experiments revealed a role for CXCR3, but not CXCR4 in the effector phase of EAE. The treatment of the recipient mice with CXCL12(P2G2) failed to prevent the passive transfer of EAE in two separate experiments; the results of which have been combined (Figure 7C). In contrast, prior treatment of the recipients with CXCL11(4–79) significantly reduced disease transfer in two separate experiments (Figure 7D).
Figure 7.

Involvement of CXCR4 and CXCR3 in the sensitization and effector phases of experimental autoimmune encephalomyelitis (EAE), respectively. A,B. EAE was induced in SJL/J mice. On days 1, 3, 5 and 7 postimmunization, the mice received an i.p. injection of 100 µg of CXCL12(P2G2) or CCL2(4Ala), or 250 µg of CXCL11(4–79) or CCL2(4Ala). On day 9 postimmunization, the cells from the Draining Lymph Nodes (DLNs) were harvested and analyzed by flow cytometry to determine the effect of antagonist treatment on lymphocyte division upon restimulation in vitro. *Significantly different from CCL2(4Ala)‐treated group at P < 0.05; n = 4–6, from two independent experiments. C,D.EAE was adoptively transferred to recipient SJL/J mice that were treated on the day of transfer and days 2, 4, 6, 8, 10, 12 and 14 post‐transfer with 100 µg of CXCL12(P2G2) or CCL2(4Ala), or 250 µg of CXCL11(4–79) or CCL2(4Ala). The mice were scored for clinical signs of the disease over a period of 20 days post‐transfer. Data are presented as mean clinical disease score ± SEM, as a function of days after transfer (n = 14). Significantly different from CCL2(4Ala) treatment (analysis of variance): *P < 0.05; **P < 0.005. Abbreviations: Con A = concanavalin A; PLP = proteolipid protein; NS = not significant.
In light of the above observations showing that CXCR4 plays an important role in the generation of encephalitogenic CD4+ T cells, whereas CXCR3 is involved in the localization of the encaphalitogenic T cells in the CNS, it was of interest to examine the effect of combined antagonist treatment on the development of EAE. Two separate experiments showed that the combination of CXCL12(P2G2) and CXCL11(4–79) significantly inhibited clinical EAE and the accumulation of leukocytes, including CD4+ lymphocytes in the spinal cord (Figure 8A,B) and that disease incidence (Table 1) was markedly lower than that seen when mice were treated with single antagonists. The combined antagonist treatment resulted in a significant reduction in disease severity beginning at day 10 post immunization that lasted until day 16 PI. During this time, the mice treated with control peptide exhibited complete tail paralysis and limited mobility, whereas the antagonist‐treated mice had near perfect motor skills and no signs of paralysis. These observations correlated with a significant decrease in the mean cumulative score of the CXCL12(P2G2)‐ and CXCL11(4–79)‐treated mice compared with the control mice.
Figure 8.
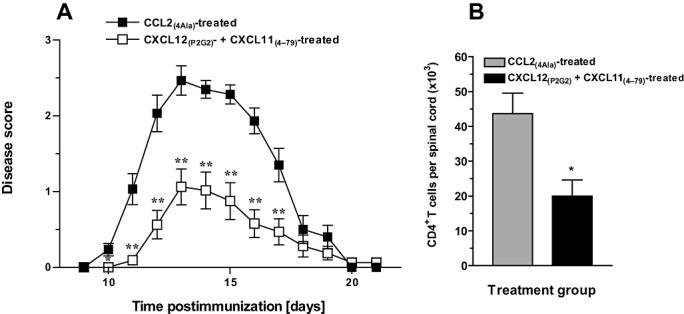
Effect of combined antagonism of CXCR4 and CXCR3 on experimental autoimmune encephalomyelitis (EAE). EAE was induced in SJL/J mice. On days 1, 3, 5, 7, 9 and 11 postimmunization, the mice received an i.p. injection of 100 µg of CXCL12(P2G2) and 250 mg of CXCL11(4–79) or 350 µg of CCL2(4Ala). A. The mice were scored for the clinical signs of disease over a period of 21 days postimmunization. The data presented were obtained from two independent experiments, [mean clinical disease score ± standard error of the mean (SEM); n = 16] Significantly different from CCL2(4Ala) treatment (analysis of variance): *P < 0.05; **P < 0.005. B. Single cell suspensions were prepared from spinal cords and the number of leukocytes and CD4+ T cells present was determined using flow cytometry. *Significantly different from CCL2(4Ala)‐treated mice (P < 0.05). Data represent mean ± SEM (n = 6).
DISCUSSION
The results of this study provided several important novel pieces of information. First, they demonstrated that CXCR4 and CXCR3 play an important role in the accumulation of activated T lymphocytes in EAE, an animal model of MS, via effects on the priming and effector phases of the adaptive immune response, respectively. Second, our data provided proof of concept that small molecule CXCR antagonists may be useful in controlling T cell‐dependent pathologies such as MS, particularly if treatment can be tailored to inhibit both the priming and effector phases of the immune response.
An antagonistic role for CXCL12(P2G2) has previously been described in vitro in terms of binding capacity and chemotactic inhibition of cultured human cells (7). In the present study, in vitro tests of CXCL12(P2G2) mutant clearly demonstrated that it dose dependently inhibited CXCL12‐mediated chemotaxis of PLP139–151‐stimulated murine lymphocytes.
In light of the findings that CXCL11 is the dominant ligand for CXCR3, (6) and that N‐terminal truncations of CXCL10 (27) and CXCL9 (unpublished data) lose their capacity to bind CXCR3, three N‐terminal truncation mutants of mCXCL11 based on a previous study of human CXCL11 (5) were synthesized and tested for their ability to antagonize CXCR3. The agonistic properties of the truncation mutants correlated inversely with an increase in the number of amino acids removed. The opposite was evident for antagonism. However, while mCXCL11(4–79) dose dependently inhibited CXCL11‐, CXCL10‐ and CXCL9‐mediated chemotaxis of activated T cells, the analyses revealed that it was unable to completely inhibit CXCL10‐ and CXCL9‐mediated migration. It is possible that the first three amino acids are involved in the high‐affinity binding of CXCL11 to CXCR3, and that loss of these results in a decreased ability to fully compete with CXCL10 and CXCL9 binding. In any case, by all our previous criteria 5, 7, 14, 15, 33, CXCL11(4–79)appears to be a good CXCR3 antagonist.
Our initial experiments clearly demonstrate and increase in level of expression of mRNA for both CXCR3 and CXCR4 and the accumulation of activated (divided) CXCR3+ and CXCR4+ CD4+ T cells in the spinal cord during the development of active EAE. The treatment of mice with antagonists against either receptor inhibited the pathology of active EAE, in terms of both clinical disease score and accumulation of leukocytes including CD4+ T cells. These data unequivocally prove that both CXCR3 and CXCR4 play an important role in the pathogenesis of EAE.
The treatment of recipient mice with CXCL12(P2G2) prior to transfer of encephalitogenic T cells had no effect on the development of EAE, suggesting against a critical role for CXCR4 in T cell accumulation in the CNS, despite the responsiveness of these cells to CXCL12 in vitro. In contrast, restimulation assays demonstrated that CD4+ lymphocytes from CXCL12(P2G2)‐treated mice proliferated less robustly in response to the immunizing antigen ex vivo compared with lymphocytes from control‐treated mice. Together, these observations suggest that the inhibition of active EAE in CXCL12(P2G2)‐treated mice is because, at least in part, of a reduced antigen‐specific CD4+ T cell response, consistent with the described costimulatory role for CXCL12 during human T cell activation in vitro (38).
However, these data contrast with a report, which emerged during the course of this study, in which the administration of a specific small molecule antagonist of CXCR4, AMD3100, exacerbated EAE (32). The authors speculated that this was because of the inhibition of CXCR4‐mediated retention of mononuclear cells within the perivascular cuffs of the CNS, an event that normally prevents the widespread accumulation of leukocytes in the parenchyma. The contrast between our results and those of McCandless et al, are probably because of the differences in the experimental systems utilized in which the genetic background of the mice studied (SJL/J vs. C57Bl/6), the immunizing peptide (PLP139–151vs. MOG35–55), the type of disease induced (relapsing remitting vs. chronic EAE) and potentially the nature and dosage of antagonist used are all different. Further studies will be required to clarify these issues.
In contrast to our results with CXCL12(P2G2), T cells isolated from the draining LN of CXCL11(4–79)‐treated mice proliferated to the same extent as those from CCL2(4Ala)‐treated counterparts in response to PLP139–151, while treatment of the recipient mice with CXCL11(4–79) prior to the transfer of encephalitogenic cells significantly reduced the clinical symptoms of EAE. These data are consistent with the results of previous in vitro studies suggesting a role for CXCR3 and its ligands in T cell entry into the peripheral tissues (17) and the CNS (12). However, the situation with CXCR3 and its ligands is clearly complex. Recent observations in CXCR3−/−mice show that CXCR3‐deficient C57Bl/6 mice immunized with MOG peptide develop more severe EAE compared with WT mice 26, 37 and, in contrast to the findings of the present study, point to CXCR3 essentially suppressing rather than promoting EAE. The mechanisms underlying this have been attributed to alterations in the localization of effector T cells, macrophages and regulatory T cells within the CNS parenchyma (37) and to a defect in IFNγ production (26), although it is not yet clear whether these mechanisms are causal. As is the case with CXCR4, it is important to consider the differences in the models used and the approaches taken when interpreting these data together. The analysis of CXCR3−/−mice demonstrates the effect of chronic deficiency in responses mediated by this receptor, and does not indicate the effects of blocking CXCR3 during active immune responses in a normally developed immune system. Therefore, the apparently conflicting data may reflect the difference in approach—genetic deletion with possible compensatory or nonspecific effects vs. acute/transient pharmacologic antagonism. Of relevance, both CXCR3−/− and CXCL10−/− mice display a defective Mixed Lymphocyte Reaction (MLR) 17, 18, indicating an inability to mount a normal immune response. Furthermore, the model of EAE used by Liu et al (26) and Muller et al (37) was chronic EAE induced in C57Bl/6 mice by immunization with MOG(33–55). In contrast, the present study analyzed the relapsing–remitting EAE in SJL/J mice. There are several pieces of previously published evidence which may point to the disparate roles for CXCR3 and its ligands between these two models of EAE. Notably, and similarly to that observed in CXCR3−/− mice, C57Bl/6 mice deficient in the CXCR3 ligand CXCL10 also develop more severe MOG‐induced EAE (22). However, several studies using SJL/J mice have implicated CXCL10 in the pathogenesis of EAE. For instance, the attenuation of EAE using altered peptide ligands was associated with reduced CXCL10 and CXCR3 expression in SJL/J mice (13). Furthermore, the treatment with a neutralizing CXCL10 antibody inhibited the disease following the adoptive transfer of encephalitogenic cells, demonstrating that CXCL10 plays an important role in the recruitment and accumulation of T cells, which cause CNS pathology (12). Our data (also in the SJL/J mouse strain) would fit well with the notion that interactions between CXCR3 and its ligands are important for driving the recruitment of pathogenic mononuclear cells into the CNS parenchyma. However, taken as a whole, these studies clearly demonstrate inherent complexity in this system whereby signaling through CXCR3 may drive the recruitment of both pathogenic and regulatory cells of the immune response into the CNS. This must be considered carefully if CXCR3 is to be targeted therapeutically in human MS, as has been suggested in a recent review of this area (25).
It is noteworthy that neither antagonist completely inhibited EAE. There are several possible explanations for this. First, it is likely that receptor antagonism does not completely inhibit receptor activity. Second, it is likely that other chemokine receptors are involved in the response and are still active during the antagonism of CXCR3 or CXCR4. Indeed, other studies indicate that CCR6 plays an important role in the priming phase of EAE (Liston et al, in preparation), and that receptors for CCL5 (CCR1 and CCR5) also play a role in the effector phase [(42) and McColl, unpublished observations].
The knowledge that CXCR3 and CXCR4 were operating in the priming and effector phases of EAE respectively, provided a conceptual framework for testing the hypothesis that simultaneous treatment with CXCL12(P2G2)and CXCL11(4–79) would be more beneficial than targeting either receptor alone. Indeed, we observed that both maximal disease score and disease incidence were lower in the dual treatment cohort. The results of prior studies indicate that targeting a chemokine receptor in conjunction with an immunosuppressive drug results in enhanced protection from allograft rejection (17), but our study is the first to show that targeting more than one chemokine receptor leads to a better outcome than targeting receptors individually.
Collectively, our data provided important novel insights into the spatial and temporal aspects of CXCR4 and CXCR3 interactions with their ligands during the generation of T cell‐dependent pathology in the CNS. Both receptors appear to play an important role in the accumulation of activated CD4+T cells in the spinal cord during EAE by two distinct mechanisms. The novel reagents developed for this study should also prove useful in defining the role of these two important receptors in numerous other pathologies.
This work was supported by the National Health and Medical Research Council of Australia and the Multiple Sclerosis Society. R.E.K. is the recipient of an Australian Postgraduate Award.
REFERENCES
- 1. Balashov KE, Rottman JB, Weiner HL, Hancock WW (1999) CCR5(+) and CXCR3(+) T cells are increased in multiple sclerosis and their ligands MIP‐1alpha and IP‐10 are expressed in demyelinating brain lesions. Proc Natl Acad Sci USA 96:6873–6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baxter AG (2007) The origin and application of experimental autoimmune encephalomyelitis. Nat Rev Immunol 7(11):904–912. [DOI] [PubMed] [Google Scholar]
- 3. Bonecchi R, Bianchi G, Bordignon PP, D'Ambrosio D, Lang R, Borsatti A et al (1998) Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med 187:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buckley CD, Amft N, Bradfield PF, Pilling D, Ross E, Arenzana‐Seisdedos F et al (2000) Persistent induction of the chemokine receptor CXCR4 by TGF‐beta 1 on synovial T cells contributes to their accumulation within the rheumatoid synovium. J Immunol 165:3423–3429. [DOI] [PubMed] [Google Scholar]
- 5. Clark‐Lewis I, Mattioli I, Gong JH, Loetscher P (2003) Structure‐function relationship between the human chemokine receptor CXCR3 and its ligands. J Biol Chem 278:289–295. [DOI] [PubMed] [Google Scholar]
- 6. Cole KE, Strick CA, Paradis TJ, Ogborne KT, Loetscher M, Gladue RP et al (1998) Interferon‐inducible T cell alpha chemoattractant (I‐TAC): a novel non‐ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med 187:2009–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana‐Seisdedos F et al (1997) Solution structure and basis for functional activity of stromal cell‐derived factor‐1; dissociation of CXCR4 activation from binding and inhibition of HIV‐1. EMBO J 16:6996–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cyster JG (2000) Leukocyte migration: scent of the T zone. Curr Biol 10:R30–R33. [DOI] [PubMed] [Google Scholar]
- 9. Dogan RN, Karpus WJ (2004) Chemokines and chemokine receptors in autoimmune encephalomyelitis as a model for central nervous system inflammatory disease regulation. Front Biosci 9:1500–1505. [DOI] [PubMed] [Google Scholar]
- 10. Ebert LM, McColl SR (2001) Coregulation of CXC chemokine receptor and CD4 expression on T lymphocytes during allogeneic activation. J Immunol 166:4870–4878. [DOI] [PubMed] [Google Scholar]
- 11. Ebert LM, Schaerli P, Moser B (2005) Chemokine‐mediated control of T cell traffic in lymphoid and peripheral tissues. Mol Immunol 42:799–809. [DOI] [PubMed] [Google Scholar]
- 12. Fife BT, Kennedy KJ, Paniagua MC, Lukacs NW, Kunkel SL, Luster AD et al (2001) CXCL10 (IFN‐gamma‐inducible protein‐10) control of encephalitogenic CD4+ T cell accumulation in the central nervous system during experimental autoimmune encephalomyelitis. J Immunol 166:7617–7624. [DOI] [PubMed] [Google Scholar]
- 13. Fischer FR, Santambrogio L, Luo Y, Berman MA, Hancock WW, Dorf ME (2000) Modulation of experimental autoimmune encephalomyelitis: effect of altered peptide ligand on chemokine and chemokine receptor expression. J Neuroimmunol 110:195–208. [DOI] [PubMed] [Google Scholar]
- 14. Gong JH, Clark‐Lewis I (1995) Antagonists of monocyte chemoattractant protein 1 identified by modification of functionally critical NH2‐terminal residues. J Exp Med 181:631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gong JH, Ratkay LG, Waterfield JD, Clark‐Lewis I (1997) An antagonist of monocyte chemoattractant protein 1 (MCP‐1) inhibits arthritis in the MRL‐lpr mouse model. J Exp Med 186:131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S (2002) Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol 20:621–667. [DOI] [PubMed] [Google Scholar]
- 17. Hancock WW, Lu B, Gao W, Csizmadia V, Faia K, King JA et al (2000) Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med 192:1515–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hancock WW, Gao W, Csizmadia V, Faia KL, Shemmeri N, Luster AD (2001) Donor‐derived IP‐10 initiates development of acute allograft rejection. J Exp Med 193:975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heath WR, Carbone FR (2001) Cross‐presentation, dendritic cells tolerance and immunity. Annu Rev Immunol 19:47–64. [DOI] [PubMed] [Google Scholar]
- 20. Hjelmstrom P, Juedes AE, Fjell J, Ruddle NH (1998) B‐cell‐deficient mice develop experimental allergic encephalomyelitis with demyelination after myelin oligodendrocyte glycoprotein sensitization. J Immunol 161:4480–4483. [PubMed] [Google Scholar]
- 21. Huitinga I, Van Rooijen N, De Groot CJ, Uitdehaag BM, Dijkstra CD (1990) Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J Exp Med 172:1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klein RS, Izikson L, Means T, Gibson HD, Lin E, Sobel RA et al (2004) IFN‐inducible protein 10/CXC chemokine ligand 10‐independent induction of experimental autoimmune encephalomyelitis. J Immunol 172:550–559. [DOI] [PubMed] [Google Scholar]
- 23. Kohler RE, Caon AC, Willenborg DO, Clark‐Lewis I, McColl SR (2003) A role for macrophage inflammatory protein‐3 alpha/CC chemokine ligand 20 in immune priming during T cell‐mediated inflammation of the central nervous system. J Immunol 170:6298–6306. [DOI] [PubMed] [Google Scholar]
- 24. Langenkamp A, Nagata K, Murphy K, Wu L, Lanzavecchia A, Sallusto F (2003) Kinetics and expression patterns of chemokine receptors in human CD4+ T lymphocytes primed by myeloid or plasmacytoid dendritic cells. Eur J Immunol 33:474–482. [DOI] [PubMed] [Google Scholar]
- 25. Liu L, Callahan MK, Huang D, Ransohoff RM (2005) Chemokine receptor CXCR3 an unexpected enigma. Curr Top Dev Biol 68:149–181. [DOI] [PubMed] [Google Scholar]
- 26. Liu L, Huang D, Matsui M, He TT, Hu T, Demartino J et al (2006) Severe disease, unaltered leukocyte migration, and reduced IFN‐gamma production in CXCR3−/− mice with experimental autoimmune encephalomyelitis. J Immunol 176:4399–4409. [DOI] [PubMed] [Google Scholar]
- 27. Loetscher P, Pellegrino A, Gong JH, Mattioli I, Loetscher M, Bardi G et al (2001) The ligands of CXC chemokine receptor 3, I‐TAC, Mig, and IP10, are natural antagonists for CCR3. J Biol Chem 276:2986–2991. [DOI] [PubMed] [Google Scholar]
- 28. Lyons AB, Parish CR (1994) Determination of lymphocyte division by flow cytometry. J Immunol Methods 171:131–137. [DOI] [PubMed] [Google Scholar]
- 29. Maatta JA, Sjoholm UR, Nygardas PT, Salmi AA, Hinkkanen AE (1998) Neutrophils secreting tumor necrosis factor alpha infiltrate the central nervous system of BALB/c mice with experimental autoimmune encephalomyelitis. J Neuroimmunol 90:162–175. [DOI] [PubMed] [Google Scholar]
- 30. Martin‐Fontecha A, Sebastiani S, Hopken UE, Uguccioni M, Lipp M, Lanzavecchia A, Salusto F (2003) Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med 198:615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matsui M, Weaver J, Proudfoot AE, Wujek JR, Wei T, Richer E et al (2002) Treatment of experimental autoimmune encephalomyelitis with the chemokine receptor antagonist Met‐RANTES. J Neuroimmunol 128:16–22. [DOI] [PubMed] [Google Scholar]
- 32. McCandless EE, Wang Q, Woerner BM, Harper JM, Klein RS (2006) CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J Immunol 177:8053–8064. [DOI] [PubMed] [Google Scholar]
- 33. McColl SR, Clark‐Lewis I (1999) Inhibition of murine neutrophil recruitment in vivo by CXC chemokine receptor antagonists. J Immunol 163:2829–2835. [PubMed] [Google Scholar]
- 34. McRae BL, Kennedy MK, Tan LJ, Dal Canto MC, Picha KS, Miller SD (1992) Induction of active and adoptive relapsing experimental autoimmune encephalomyelitis (EAE) using an encephalitogenic epitope of proteolipid protein. J Neuroimmunol 38:229–240. [DOI] [PubMed] [Google Scholar]
- 35. Moser B, Wolf M, Walz A, Loetscher P (2004) Chemokines: multiple levels of leukocyte migration control. Trends Immunol 25:75–84. [DOI] [PubMed] [Google Scholar]
- 36. Muller G, Lipp M (2003) Shaping up adaptive immunity: the impact of CCR7 and CXCR5 on lymphocyte trafficking. Microcirculation 10:325–334. [DOI] [PubMed] [Google Scholar]
- 37. Muller M, Carter SL, Hofer MJ, Manders P, Getts DR, Getts MT et al (2007) CXCR3 signaling reduces the severity of experimental autoimmune encephalomyelitis by controlling the parenchymal distribution of effector and regulatory T cells in the central nervous system. J Immunol 179:2774–2786. [DOI] [PubMed] [Google Scholar]
- 38. Nanki T, Hayashida K, El‐Gabalawy HS, Suson S, Shi K, Girschick HJ et al (2000) Stromal cell‐derived factor‐1‐CXC chemokine receptor 4 interactions play a central role in CD4+ T cell accumulation in rheumatoid arthritis synovium. J Immunol 165:6590–6598. [DOI] [PubMed] [Google Scholar]
- 39. Plater‐Zyberk C, Hoogewerf AJ, Proudfoot AE, Power CA, Wells TN (1997) Effect of a CC chemokine receptor antagonist on collagen induced arthritis in DBA/1 mice. Immunol Lett 57:117–120. [DOI] [PubMed] [Google Scholar]
- 40. Proudfoot AE, Power CA, Hoogewerf AJ, Montjovent MO, Borlat F, Offord RE et al (1996) Extension of recombinant human RANTES by the retention of the initiating methionine produces a potent antagonist. J Biol Chem 271:2599–2603. [DOI] [PubMed] [Google Scholar]
- 41. Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M et al (1998) The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest 101:746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rottman JB, Slavin AJ, Silva R, Weiner HL, Gerard CG, Hancock WW (2000) Leukocyte recruitment during onset of experimental allergic encephalomyelitis is CCR1 dependent. Eur J Immunol 30:2372–2377. [DOI] [PubMed] [Google Scholar]
- 43. Sallusto F, Lanzavecchia A, Mackay CR (1998) Chemokines and chemokine receptors in T‐cell priming and Th1/Th2‐mediated responses. Immunol Today 19:568–574. [DOI] [PubMed] [Google Scholar]
- 44. Sallusto F, Lenig D, Mackay CR, Lanzavecchia A (1998) Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med 187:875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sallusto F, Geginat J, Lanzavecchia A (2004) Central memory and effector memory T cell subsets: function, generation and maintenance. Annu Rev Immunol 22:745–763. [DOI] [PubMed] [Google Scholar]
- 46. Satoh J, Sakai K, Endoh M, Koike F, Kunishita T, Namikawa T et al (1987) Experimental allergic encephalomyelitis mediated by murine encephalitogenic T cell lines specific for myelin proteolipid apoprotein. J Immunol 138:179–184. [PubMed] [Google Scholar]
- 47. Sobel RA, Kuchroo VK (1992) The immunopathology of acute experimental allergic encephalomyelitis induced with myelin proteolipid protein. T cell receptors in inflammatory lesions. J Immunol 149:1444–1451. [PubMed] [Google Scholar]
- 48. Sorensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA et al (1999) Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest 103:807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sorensen TL, Trebst C, Kivisakk P, Klaege KL, Majmudar A, Ravid R et al (2002) Multiple sclerosis: a study of CXCL10 and CXCR3 co‐localization in the inflamed central nervous system. J Neuroimmunol 127:59–68. [DOI] [PubMed] [Google Scholar]
- 50. Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y et al (1998) The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 393:591–594. [DOI] [PubMed] [Google Scholar]
- 51. Wells TN, Power CA, Shaw JP, Proudfoot AE (2006) Chemokine blockers–herapeutics in the making? Trends Pharmacol Sci 27:41–47. [DOI] [PubMed] [Google Scholar]
- 52. Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR (1998) Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 393:595–599. [DOI] [PubMed] [Google Scholar]