

Abstract
The 4th edition of the WHO Classification of Tumours of the Nervous System (WHO 2007) introduces changes that reflect both the recognition of new brain tumour types and a better understanding of neoplastic behavior. Three new tumours, angiocentric glioma (AG), pilomyxoid astrocytoma (PMA), and pituicytoma are added to the section on gliomas. AG is a slowly growing cerebral tumour that typically presents with seizures in children and young adults. It is characterized by monomorphous, bipolar tumour cells with a striking perivascular growth pattern. Although the ‘cell of origin’ of AG is not clear, ultrastructural evidence points to an ependymal derivation. Typically, AG can be cured by total resection, and is designated WHO grade I. PMA is a solid, circumscribed tumour occurring mainly in the hypothalamic region of young children. It is composed of a monomorphous population of bipolar tumour cells within a rich myxoid background, with a conspicuous anglocentric arrangement. While PMA is considered a more aggressive variant of pilocytic astrocytoma, this relationship awaits further clarification. The PMA has been designated WHO grade II. The pituicytoma, involves the posterior pituitary and/or its stalk and affects adults. It is solid in architecture, composed of spindle cells and presumably derived from pituicytes. Pituicytomas are indolent tumours, and are designated WHO grade I.
INTRODUCTION
The newly revised WHO Classification of Tumours of the Nervous System (WHO 2007) has introduced a number of substantial changes compared with the previous edition (WHO 2000) (25). These reflect both the recognition of new brain tumor types and a better understanding of neoplastic behavior. These changes include additions and shifts of entities as well as modest alterations in diagnostic criteria. For example, gliomatosis cerebri (GC), previously considered to be a glial tumor of uncertain origin and an entity sui generis, has been redefined as a diffuse glioma growth pattern, most commonly a presentation form of diffuse astrocytoma, but which occasionally can be a growth pattern exhibited by oligodendroglioma or mixed glioma (15).
Three new tumor types, which constitute the focus of this review, have been added to the glioma section of the 2007 Classification. Two arise predominantly in children, adding to the diversity of an already complex set of pediatric brain tumors. The first of these, angiocentric glioma (AG), has been codified as a distinct entity and is now included under the category of “Other Neuroepithelial Tumors” (formerly “Glial Tumors of Uncertain Origin”) (4). The second, pilomyxoid astrocytoma (PMA), has been added as a formally recognized variant of pilocytic astrocytoma (30). The third, pituicytoma, affects the neurohypophysis and stalk in adults, and is thought to derive from the specialized glia indigenous to this site (39).
ANGIOCENTRIC GLIOMA (AG)
AG was introduced into the literature in 2005 by two independent and almost simultaneous reports 23, 38). These two papers, together with an additional report in abstract form (28), describe a total of 26 patients that represent the extent of experience with this neoplasm. The unique clinical, radiologic and pathologic features of AG as described in these reports have led to acceptance and codification as a new diagnostic entity in the 2007 WHO Classification. The neoplasm has been placed in the same category as chordoid glioma and astroblastoma (25). All three tumors in this category are currently regarded as having uncertain histogenesis; however, it is of note that some degree of ependymal differentiation characterizes all three entities.
Definition. AGs are slowly growing, cerebral hemispheric tumors occurring in children and young adults that typically present with seizures. They are characterized histologically by monomorphous, bipolar glial tumor cells oriented along vascular structures. Their precise derivation has not been firmly established, but ependymal features are conspicuous, particularly at the immunohistochemical and ultrastructural levels. AG is placed in the category of “Other Neuroepithelial Tumors” and has been designated WHO grade I.
Clinical features. AG occurs within a wide age range (2.3–70 years) but is most commonly encountered in childhood and adolescence (mean age, 17 years). Nearly all patients have a long‐standing (1–17 years) history of epilepsy, with intractable partial seizures.
Neuroimaging. AGs occur in the cerebral hemispheres. The most common location is the frontal lobe, followed by temporal and parietal lobes. Although typically centered in cortex, they may extend into the underlying white matter. Cortical involvement results in cortical gyral expansion and effacement of sulci. It has been suggested that a “stalk‐like” extension of the lesion to the ventricular surface might be pathognomonic (23). On MR imaging, AGs are T2‐ and FLAIR‐hyperintense lesions that lack enhancement following contrast administration (Figure 1). Radiologically well defined, AG expands affected parenchyma rather than forming a discrete mass. Calcifications are rare.
Figure 1.
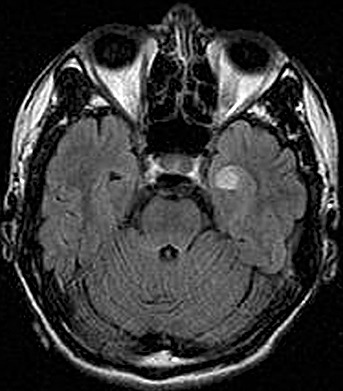
Angiocentric glioma (AG). Axial fluid attenuation inversion recovery image of AG in a young adult male. The lesion partially expands the cortex, and is hyperintense on T2‐weighted and hypointense on T1‐weighted images. This lesion did not exhibit contrast enhancement.
Microscopic features. The defining histologic feature of AG is the presence of monomorphous, bipolar tumor cells intimately associated with vessels of the involved cortex and white matter (Figure 2). The elongate, slender cells are often oriented parallel to vessels, sometimes expanding perivascular spaces with streaming arrays of either single or multilayered cells. In some examples, tumor cells are radically oriented about to vessels in a pattern reminiscent of ependymal or astroblastomatous rosettes. All sizes of vessel are involved, including large‐caliber arteries near the cortical surface as well as more deeply situated capillaries. A similar tendency to accumulate beneath the pia mater with a perpendicular orientation of the elongated cells, thus imparting a palisading appearance, is seen in a small subset of tumors. In most examples, however, the principal pattern is angiocentric. Less frequently, small numbers of cells or clusters are arranged in solid nests or nodules within brain parenchyma between affected blood vessels.
Figure 2.
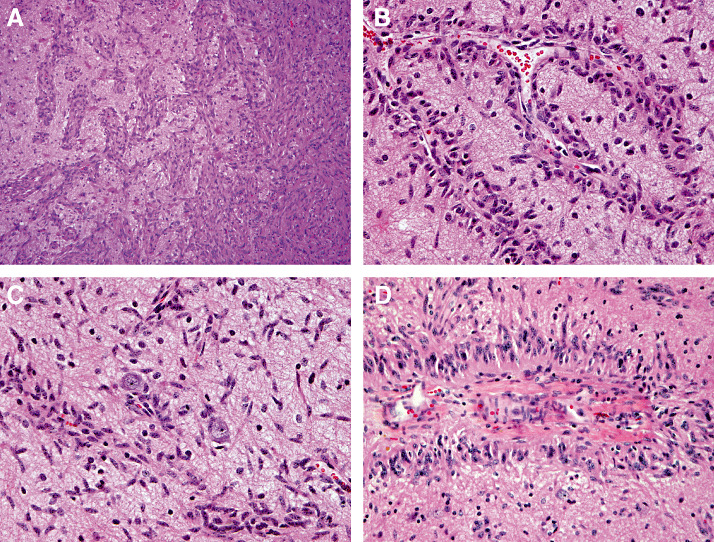
Angiocentric glioma (AG). AG exhibits solid and perivascular growth patterns (A). The latter is seen at higher power in (B). In addition to solid and perivascular growth, a highly characteristic feature is diffuse infiltration (C). In some areas, the perivascular growth takes the form of perivascular pseudorosettes, similar to those seen in ependymoma (D).
Cytologically, the tumor cells orienting along vessels are uniform and spindle‐shaped, with conspicuous oval or elongated nuclei, speckled chromatin and pink, tapering cytoplasm. Anaplasia is lacking. Mitoses are usually only rarely encountered, and MIB‐1 proliferation indices range from 1% to 5%, with the majority at the lower end of this spectrum. The most consistently reported immunohistochemical findings support inclusion of AG in the glial category. Immunoreactivity is consistently strong for glial fibrillary acidic protein (GFAP), S‐100 and vimentin. Tumor cells have also been found to show surface and paranuclear/intercellular “dot‐like” staining for epithelial membrane antigen (EMA)—a pattern typical of ependymoma. Ependymal differentiation has also been identified at the ultrastructural level, which demonstrates microlumen formation, microvilli, cilia and complex, zipper‐like intermediate junctions.
One of the original reports described a well‐differentiated neuronal component in AG (23). These authors indicated that the large neurons were nearly always cytologically unremarkable or only slightly dysplastic, and could represent normal neurons. Not surprisingly, the neurons show immunoreactivity for neurofilament protein, synaptophysin, Neu‐N and chromogranin. Wang et al considered this element to be non‐neoplastic entrapped neurons (38). The current WHO working group consensus is that the neoplastic cells of AG do not stain with neuronal markers.
Cytogenetics and molecular genetics. The molecular genetic properties of AG have yet to be fully studied. One tumor showed loss at 6q24‐q25 by classic CGH, while another showed a gain at 11p11.2 by high resolution array CGH (28). These findings await corroboration.
Clinical outcome. The original descriptions of AG suggest indolent behavior, a stable clinical course and probable cure by surgical resection alone. The majority of patients undergoing subtotal resections have shown stable, residual tumor on MRI at mean follow‐up periods of 2.5 and 4 years in the two published series 23, 38). One tumor reportedly recurred as a more aggressive lesion, and proved ultimately fatal 5 years after clinical presentation. Longer follow‐up of additional cases will be required for a better understanding of the clinical course of AG.
PILOMYXOID ASTROCYTOMA
PMA was introduced as a distinctive entity less than 10 years ago (35). Subsequent reports have rounded out the clinicopathologic definition and underscored their relatively aggressive behavior as compared with the closely related pilocytic astrocytoma 8, 12, 21). Precise diagnostic criteria for PMA, as well as its nosologic relationship to pilocytic astrocytoma, ependymoma and the glioneuronal tumors, remain to be defined.
Definition. PMA is a monomorphous neoplasm composed of bipolar (piloid) tumor cells lying within a rich myxoid background. Tumor cells often display a striking angiocentric arrangement. Although largely solid in architecture, the tumor may, like pilocytic astrocytoma, show limited parenchymal infiltration at its periphery. Currently, PMA has been assigned WHO Grade II and is formally codified in the 2007 WHO Classification as a variant of pilocytic astrocytoma.
Clinical features. PMA is typically a tumor of early childhood, but has been reported in older children. The hypothalamic/chiasmal region is the most characteristic location. Signs and symptoms of PMA relate to mass effect. Clinical features in the young include failure to thrive, developmental delay, vomiting and feeding difficulties. Some patients manifest impaired head movement or weakness. In older children, headaches, nausea, disorientation and/or diplopia are more frequent symptoms (21).
Neuroimaging. On imaging, PMA often appears as a well‐circumscribed mass in the hypothalamic region (Figure 3). Its most distinctive imaging feature is a largely solid appearance with nearly homogenous contrast‐enhancement. Other MR features that may be seen include secondary hydrocephalus, extension of T2‐weighted signal hyperintensity into the deep gray matter, cystic change and occasionally CSF dissemination (1). A recent proton magnetic resonance spectroscopic study suggests differences in metabolite concentrations between PMA and pilocytic astrocytoma (9).
Figure 3.
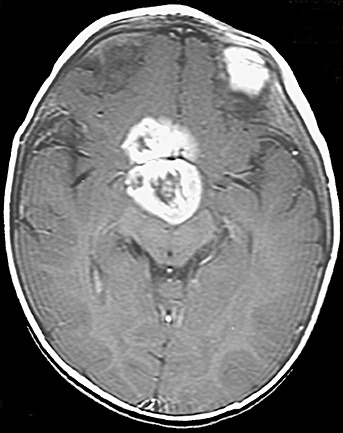
Pilomyxoid astrocytoma. Axial, contrast‐enhanced T1‐weighted image of pilomyxoid astrocytoma in a 2‐year‐old girl. The tumor demonstrates avid enhancement as well as central hypointense areas.
Microscopic features. The histologic appearance of PMA is dominated by monomorphous bipolar (piloid) cells, which lie within a rich myxoid matrix and often show an angiocentric arrangement (Figure 4). Because these features may be focally observed in typical pilocytic astrocytoma, infiltrating astrocytoma, or AG, caution is in order when making the diagnosis of PMA based on focal myxoid or angiocentric cell arrangement. PMA is a hypercellular and strikingly monomorphous neoplasm, in contrast to the often biphasic appearance that is characteristic of ordinary pilocytic astrocytoma. PMAs exhibit a compact architecture aside from the limited peripheral infiltration of brain parenchyma that may be seen. Tumor cells are characteristically bipolar, moderate in size, feature hyperchromatic nuclei and only rarely show nuclear pleomorphism. Unlike ordinary pilocytic astrocytoma, PMA typically lacks Rosenthal fibers and only exceptionally displays eosinophilic granular bodies. Mitotic figures may be seen but are not abundant.
Figure 4.
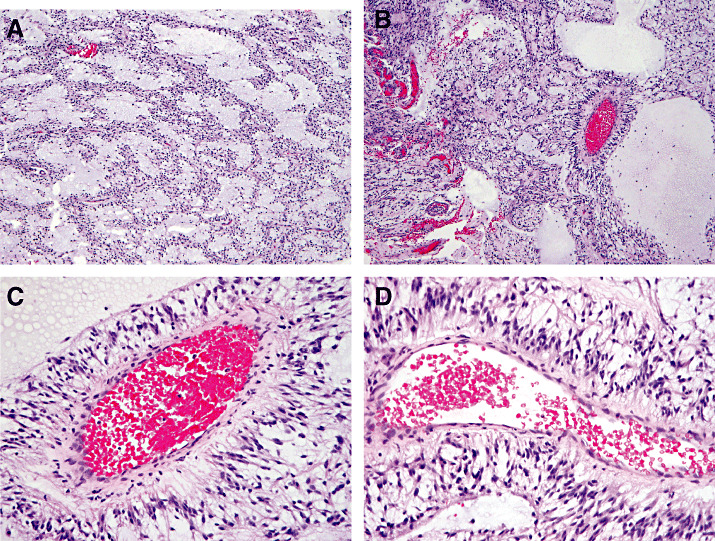
Pilomyxoid astrocytoma. Characteristic histologic features include an overall monomorphic appearance, prominent myxoid microcystic background (A), spindled astrocytes with delicate, elongated cytoplasmic processes (B), and perivascular pseudorosettes (C,D).
Immunohistochemically, PMA stains strongly and diffusely for GFAP and vimentin, but is negative for neuronal markers such as synaptophysin, neurofilament proteins and chromogranin. Significant staining for synaptophysin or other neuronal markers should prompt consideration of other diagnostic possibilities, especially some form of glioneuronal tumor. MIB‐1 labeling indices are often around 5%, but no detailed analysis of proliferation index relative to tumor behavior has been reported. Rare examples are immunoreactive for p53 protein.
Cytogenetics and molecular genetics. No large studies of the cytogenetic and molecular genetic characteristics of PMA have been published. One report found no abnormalities in a series of PMAs examined by comparative genomic hybridization (21). One recent case study reported a PMA in an NF1 patient, but the significance of this finding is unclear (20). Most reports have shown no evidence of TP53 gene alterations, but anecdotal observations have noted increased nuclear p53 immunostaining (35). A recent study of one case revealed an insertion on chromosome 17 that involved disruption of the BCR gene, a finding that requires further study (26).
Historical considerations. Early reports of PMA contain a number of relevant observations and deserve credit for the emergence of this entity. Jaenisch et al were first to draw attention to the relationship between young age, tumor location and clinical behavior, noting that a subset of tumors exhibited aggressive clinical behavior (17). The authors concluded that such tumors had a similar, if not identical, histologic appearance to that of pilocytic astrocytoma. Cottingham et al suggested that some histological features are more typical in “infantile” pilocytic astrocytomas, regardless of location (10). It is unclear whether the tumors examined in this study, reported in abstract form, were PMAs. Later studies supported the conclusion that the PMA histology was associated with a more aggressive clinical course than that of pilocytic astrocytoma 8, 12, 21).
The histogenesis of PMA and its relation to pilocytic astrocytoma remains controversial. Certainly, pilocytic astrocytoma is considered to be astrocytic in nature. Although most studies of PMA note the resemblance to pilocytic astrocytoma and suggest an astrocytic histogenesis, others have proposed an ependymal origin (14). A third hypothesis points to a possible origin from the radial glia (7), and a recent fourth study suggests a glioneuronal nature (11). There is no conclusive evidence that strongly supports any of these possibilities, yet most studies conclude that PMA has an astrocytic origin. Reports of “hybrid tumors” containing elements of both conventional pilocytic astrocytoma and PMA, together with anecdotal reports of a “maturation” of a PMA to typical pilocytic astrocytoma following treatment and on recurrence 6, 8, 12), suggest that the two tumors are related and belong in the same category. The latter concept is reflected in the 2007 WHO Classification.
Questions remain regarding the “sufficient” criteria for PMA diagnosis. The vast majority of PMAs occur in the hypothalamic /chiasmatic region of children under the age of 4 years. The most relevant diagnostic features of PMA include a monomorphous cytologic appearance, abundant myxoid matrix and angiocentric cell arrangement. Because most of these features can be observed, at least focally, in typical pilocytic astrocytoma, current practice restricts the diagnosis of PMA to those tumors that exhibit the characteristics in a uniform fashion.
Summary. The status of PMA as a separate tumor entity remains under debate. Reports to date appear to describe a distinctive constellation of histopathologic and clinical features. The current categorization of PMA as a variant of pilocytic astrocytoma in the 2007 WHO Classification emphasizes the features shared with the larger tumor class, but also acknowledges a possible differing biology that warrants further investigation and clarification.
PITUICYTOMA
Historical consideration. The term “pituicytoma” has been variously used to refer to granular cell tumor as well as pilocytic astrocytoma affecting the suprasellar region, infundibulum, or posterior pituitary. At present, the designation is restricted to an astrocytic tumor that is distinct from both of these entities. The synonym “infundibuloma” has not found acceptance.
Definition. Pituicytoma is defined as a solid, circumscribed, low‐grade spindle cell astrocytic tumor of adults having an origin in the posterior pituitary or its stalk. The tumor is considered WHO grade I.
Clinical features. Pituicytomas are rare. The largest single series included nine cases (3). To date, 26 examples have been described 2, 3, 5, 7, 13, 16, 19, 22, 27, 31, 32, 36, 37). All tumors have occurred in adults, with a male predilection of 1.6:1. As pituicytomas originate in the posterior pituitary or its stalk, they may be intrasellar, suprasellar, or both.
Clinical features. The signs and symptoms of pituicytoma are secondary to mass effect and are thus similar to those of other non‐endocrine tumors of the sellar region. These include visual disturbances because of compression of the optic chiasm, hypopituitarism because of compression of the pituitary gland and headache. Infundibular compression results in interference of hypothalamic dopamine delivery, with subsequent hyperprolactinemia and the accompanying amenorrhea and decreased libido 3, 5). Rarely, pituicytoma has been reported as an incidental autopsy finding (33).
Imaging. On CT and MRI scan, pituicytomas are architecturally solid and are demarcated as homogenous in appearance. Uniform contrast enhancement is the rule. Heterogeneous enhancement and cystic change are rarely seen (3).
Macroscopic findings. At surgery, pituicytomas are circumscribed and architecturally solid. Their texture is variably firm. Cystic change is infrequent 3, 37). Although grossly discrete, pituicytomas may adhere to surrounding structures in the suprasellar space.
Histopathology. Being solid in nature, pituicytomas consist almost exclusively of bipolar spindle cells arranged in interlacing fascicles or in a somewhat storiform pattern (Figure 5A). Their fusiform to plump cells possess moderate to abundant eosinophilic cytoplasm with discrete borders. Nuclei are moderate in size, often oval or elongate, and generally show little atypia. Mitoses are rare to absent. No appreciable intercellular reticulin is seen, staining being limited to vessels. In contrast to pilocytic astrocytomas, pituicytomas lack Rosenthal fibers and eosinophilic granular bodies. Lack of cytoplasmic granularity attributable to lysosomes or mitochondria distinguishes pituicytoma from granular cell tumor and spindle cell oncocytoma, respectively. Unlike normal posterior pituitary tissue, pituicytomas are more cellular and lack both axons and axonal swellings (Herring bodies).
Figure 5.
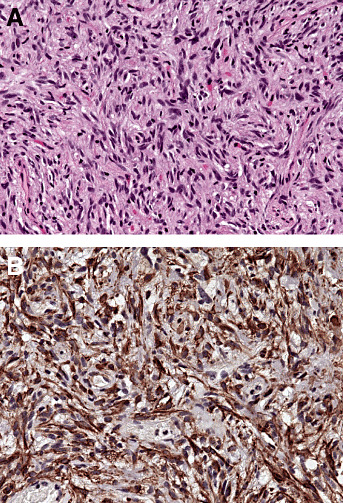
Pituicytoma. The histology varies from streaming bundles of cells to a somewhat storiform pattern (A). Strong staining for glial fibrillary acidic protein is seen (B).
Immunohistochemistry. As expected, pituicytomas are immunoreactive for vimentin, S‐100 protein and GFAP (Figure 5B) 3, 13). Staining varies, particularly with respect to GFAP. No neuronal or neuroendocrine marker reactivity, such as neurofilament protein, synaptophysin or chromogranin, is evident, nor are pituitary hormones demonstrable. With respect to delimitation from surrounding tissue, neurofilament protein immunoreactivity is limited to peritumoral axons and, unlike pilocytic astrocytoma, is not found deep within the tumor. Cytoplasmic EMA positivity is occasionally seen but, unlike in ependymoma, neither membrane nor dot‐like patterns are encountered.
Proliferation. As a rule, MIB‐1 labeling indices are low, ranging from 0.5% to 2%3, 22). No studies have shown a correlation of proliferation indices and tumor behavior.
Histogenesis. Pituicytomas are thought to originate from pituicytes, specialized glial cells of the posterior lobe 3, 18, 24). This explanation is in keeping with the localization of pituicytomas in the stalk and posterior lobe. Pituicytomas are linked to granular cell tumors by their common cytogenesis—the pituicyte. Interestingly, heterogeneity of pituicyte subtypes has been reported (34). The nature of pituicytes varies among different species, as does their morphology in different physiologic states (29).
Prognosis. Pituicytomas are indolent neoplasms, enlarging only slowly. Their demarcated nature permits effective surgical resection. Even subtotal resection is accompanied by only slow regrowth over years. Pituicytomas have not been shown to undergo malignant transformation or craniospinal spread.
SUMMARY
These three additions to the astrocytoma category expand its spectrum in a meaningful fashion. None are common, but each has a fascinating biology of its own; AG in the way in which a patently infiltrative tumor exhibits unexpected ependymal features and behaves in a favorable manner, PMA in providing a diagnostic niche for disseminating tumors mimicking pilocytic astrocytoma, and pituicytoma in being a tumor derived from functionally specialized glial cells that in various species are linked to either ependymal or astrocytic differentiation.
REFERENCES
- 1. Arslanoglu A, Cirak B, Horska A, Okoh J, Tihan T, Aronson L, Avellino AM, Burger PC, Yousem DM (2003) MR imaging characteristics of pilomyxoid astrocytomas. AJNR Am J Neuroradiol 24:1906–1908. [PMC free article] [PubMed] [Google Scholar]
- 2. Benveniste RJ, Purohit D, Byun H (2006) Pituicytoma presenting with spontaneous hemorrhage. Pituitary 9:53–58. [DOI] [PubMed] [Google Scholar]
- 3. Brat DJ, Scheithauer BW, Staugaitis SM, Holtzman RN, Morgello S, Burger PC (2000) Pituicytoma: a distinctive low‐grade glioma of the neurohypophysis. Am J Surg Pathol 24:362–368. [DOI] [PubMed] [Google Scholar]
- 4. Burger PC, Jouvet A, Preusser M, Hans VH, Rosenblum MK, Lellouch‐Tubiana A (2007) Angiocentric glioma. In: World Health Organization Classification of Tumors of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), IARC: Lyon. In press. [Google Scholar]
- 5. Cenacchi G, Giovenali P, Castrioto C, Giangaspero F (2001) Pituicytoma: ultrastructural evidence of a possible origin from folliculo‐stellate cells of the adenohypophysis. Ultrastruct Pathol 25:309–312. [DOI] [PubMed] [Google Scholar]
- 6. Ceppa EP, Bouffet E, Griebel R, Robinson C, Tihan T (2007) The pilomyxoid astrocytoma and its relationship to pilocytic astrocytoma: report of a case and a critical review of the entity. J Neurooncol 81:191–196. [DOI] [PubMed] [Google Scholar]
- 7. Chen KT (2005) Crush cytology of pituicytoma. Diagn Cytopathol 33:255–257. [DOI] [PubMed] [Google Scholar]
- 8. Chikai K, Ohnishi A, Kato T, Ikeda J, Sawamura Y, Iwasaki Y, Itoh T, Sawa H, Nagashima K (2004) Clinico‐pathological features of pilomyxoid astrocytoma of the optic pathway. Acta Neuropathol (Berl) 108:109–114. [DOI] [PubMed] [Google Scholar]
- 9. Cirak B, Horska A, Barker PB, Burger PC, Carson BS, Avellino AM (2005) Proton magnetic resonance spectroscopic imaging in pediatric pilomyxoid astrocytoma. Childs Nerv Syst 21:404–409. [DOI] [PubMed] [Google Scholar]
- 10. Cottingham SL, Boesel CP, Yates AJ (1996) Pilocytic astrocytoma in infants: a distinctive histological pattern. J Neuropathol Exp Neurol 55:654. [Google Scholar]
- 11. De Chadarevian JP, Halligan GE, Reddy G, Bertrand L, Pascasio JM, Faerber EN, Katsetos CD (2006) Glioneuronal phenotype in a diencephalic pilomyxoid astrocytoma. Pediatr Dev Pathol 9:480–487. [DOI] [PubMed] [Google Scholar]
- 12. Fernandez C, Figarella‐Branger D, Girard N, Bouvier‐Labit C, Gouvernet J, Paz Paredes A, Lena G (2003) Pilocytic astrocytomas in children: prognostic factors—a retrospective study of 80 cases. Neurosurgery 53:544–553; discussion 554–555. [DOI] [PubMed] [Google Scholar]
- 13. Figarella‐Branger D, Dufour H, Fernandez C, Bouvier‐Labit C, Grisoli F, Pellissier JF (2002) Pituicytomas, a mis‐diagnosed benign tumor of the neurohypophysis: report of three cases. Acta Neuropathol (Berl) 104:313–319. [DOI] [PubMed] [Google Scholar]
- 14. Fuller CE, Frankel B, Smith M, Rodziewitz G, Landas SK, Caruso R, Schelper R (2001) Suprasellar monomorphous pilomyxoid neoplasm: an ultastructural analysis. Clin Neuropathol 20:256–262. [PubMed] [Google Scholar]
- 15. Fuller GN, Kros JM (2007) Gliomatosis cerebri. In: World Health Organization Classification of Tumors of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), IARC: Lyon. In press. [Google Scholar]
- 16. Hurley TR, D’Angelo CM, Clasen RA, Wilkinson SB, Passavoy RD (1994) Magnetic resonance imaging and pathological analysis of a pituicytoma: case report. Neurosurgery 35:314–317; discussion 317. [DOI] [PubMed] [Google Scholar]
- 17. Janisch W, Schreiber D, Martin H, Gerlach H (1985) [Diencephalic pilocytic astrocytoma with clinical onset in infancy. Biological behavior and pathomorphological findings in 11 children. Zentralbl Allg Pathol 130:31–43. [PubMed] [Google Scholar]
- 18. Jenevein EP (1964) A Neurohypophyseal tumor originating from pituicytes. Am J Clin Pathol 41:522–526. [DOI] [PubMed] [Google Scholar]
- 19. Katsuta T, Inoue T, Nakagaki H, Takeshita M, Morimoto K, Iwaki T (2003) Distinctions between pituicytoma and ordinary pilocytic astrocytoma. Case report. J Neurosurg 98:404–406. [DOI] [PubMed] [Google Scholar]
- 20. Khanani MF, Hawkins C, Shroff M, Dirks P, Capra M, Burger PC, Bouffet E (2006) Pilomyxoid astrocytoma in a patient with neurofibromatosis. Pediatr Blood Cancer 46:377–380. [DOI] [PubMed] [Google Scholar]
- 21. Komotar RJ, Burger PC, Carson BS, Brem H, Olivi A, Goldthwaite PT, Tihan T (2004) Pilocytic and pilomyxoid hypothalamic/chiasmatic astrocytomas. Neurosurgery 54:72–79; discussion 79–80. [DOI] [PubMed] [Google Scholar]
- 22. Kowalski RJ, Prayson RA, Mayberg MR (2004) Pituicytoma. Ann Diagn Pathol 8:290–294. [DOI] [PubMed] [Google Scholar]
- 23. Lellouch‐Tubiana A, Boddaert N, Bourgeois M, Fohlen M, Jouvet A, Delalande O, Seidenwurm D, Brunelle F, Sainte‐Rose C (2005) Angiocentric neuroepithelial tumor (ANET): a new epilepsy‐related clinicopathological entity with distinctive MRI. Brain Pathol 15:281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liss L (1956) The cellular element of the human neurohypophysis; a study wtih silver carbonate. J Comp Neurol 106:507–525. [DOI] [PubMed] [Google Scholar]
- 25. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) (2007) World Health Organization Classification of Tumours of the Central Nervous System. IARC: Lyon. [Google Scholar]
- 26. Melendez B, Fiano C, Ruano Y, Hernandez‐Moneo JL, Mollejo M, Martinez P (2006) BCR gene disruption in a pilomyxoid astrocytoma. Neuropathology 26:442–446. [DOI] [PubMed] [Google Scholar]
- 27. Nakasu Y, Nakasu S, Saito A, Horiguchi S, Kameya T (2006) Pituicytoma. Two case reports. Neurol Med Chir (Tokyo) 46:152–156. [DOI] [PubMed] [Google Scholar]
- 28. Preusser M, Novak K, Czech T, Pannek HW, Tuxhorn IEB (2006) Angiocentric glioma: report of eight cases (Abstract P1064). Acta Neuropathologica 112:382–383. [Google Scholar]
- 29. Scheithauer BW, Horvath E, Kovacs K (1992) Ultrastructure of the neurohypophysis. Microsc Res Tech 20:177–186. [DOI] [PubMed] [Google Scholar]
- 30. Scheithauer BW, Hawkins C, Tihan T, VandenBerg SR, Burger PC (2007) Pilocytic astrocytoma. In: World Health Organization Classification of Tumors of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), IARC: Lyon. In press. [Google Scholar]
- 31. Schultz AB, Brat DJ, Oyesiku NM, Hunter SB (2001) Intrasellar pituicytoma in a patient with other endocrine neoplasms. Arch Pathol Lab Med 125:527–530. [DOI] [PubMed] [Google Scholar]
- 32. Shah B, Lipper MH, Laws ER, Lopes MB, Spellman MJ Jr (2005) Posterior pituitary astrocytoma: a rare tumor of the neurohypophysis: a case report. AJNR Am J Neuroradiol 26:1858–1861. [PMC free article] [PubMed] [Google Scholar]
- 33. Takei H, Goodman JC, Tanaka S, Bhattacharjee MB, Bahrami A, Powell SZ (2005) Pituicytoma incidentally found at autopsy. Pathol Int 55:745–749. [DOI] [PubMed] [Google Scholar]
- 34. Takei Y, Seyama S, Pearl GS, Tindall GT (1980) Ultrastructural study of the human neurohypophysis. II. Cellular elements of neural parenchyma, the pituicytes. Cell Tissue Res 205:273–287. [DOI] [PubMed] [Google Scholar]
- 35. Tihan T, Fisher PG, Kepner JL, Godfraind C, McComb RD, Goldthwaite PT, Burger PC (1999) Pediatric astrocytomas with monomorphous pilomyxoid features and a less favorable outcome. J Neuropathol Exp Neurol 58:1061–1068. [DOI] [PubMed] [Google Scholar]
- 36. Uesaka T, Miyazono M, Nishio S, Iwaki T (2002) Astrocytoma of the pituitary gland (pituicytoma): case report. Neuroradiology 44:123–125. [DOI] [PubMed] [Google Scholar]
- 37. Ulm AJ, Yachnis AT, Brat DJ, Rhoton AL Jr (2004) Pituicytoma: report of two cases and clues regarding histogenesis. Neurosurgery 54:753–757; discussion 757–758. [DOI] [PubMed] [Google Scholar]
- 38. Wang M, Tihan T, Rojiani AM, Bodhireddy SR, Prayson RA, Iacuone JJ, Alles AJ, Donahue DJ, Hessler RB, Kim JH, Haas M, Rosenblum MK, Burger PC (2005) Monomorphous angiocentric glioma: a distinctive epileptogenic neoplasm with features of infiltrating astrocytoma and ependymoma. J Neuropathol Exp Neurol 64:875–881. [DOI] [PubMed] [Google Scholar]
- 39. Wesseling P, Brat DJ, Fuller GN (2007) Pituicytoma. In: World Health Organization Classification of Tumors of the Central Nervous System. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), IARC: Lyon. In press. [Google Scholar]