

Abstract
Lamins A/C, the major constituent of the nuclear lamina, confer mechanical stability to nuclei. We examined the myonuclei of LMNA null mice at the myotendinous junctions (MTJ), the site of longitudinal force transmission from contractile proteins to extracellular proteins. The right soleus and rectus femoris muscles of five null mutants aged 5–7 weeks and two wild‐type animals aged 5 weeks and 6 months were examined by electron microscopy. The myofibers merging into the tendons were assessed for nuclear disintegration and cytoplasmic degeneration. The myofibers of the wild‐type rectus femoris and soleus muscles revealed 19–27 nuclei/50 myofibers and 5–8/20, respectively, with no signs of degeneration. The rectus femoris muscle fibers of the null mice contained 75–117 myonuclei/50 myofibers, the soleus muscle, 13–36 nuclei/20 myofibers. Eleven to twenty‐one per 50 myonuclei of the rectus femoris myonuclei showed chromatin clumping, nuclear fragmentation, nuclear inclusions and invaginations, and intranuclear filaments. The values were 12–19/50 for the soleus myonuclei. Moreover, 5–12/50 rectus femoris myofibers and 5–14/20, of the soleus myofibers showed cytoplasmic degeneration. None of these changes was found distal to the MTJ. These results favor the notion that myonuclei lacking a functional lamina are susceptible to mechanical stress in vivo. These alterations may contribute to the development of early joint contractures, a feature of ADEDMD.
Keywords: Electron microscopy, myotendinous junction, nucleo‐cytoskeletal integrity
INTRODUCTION
The autosomal dominant form of Emery‐Dreifuss muscular dystrophy (ADEDMD) is caused by mutations in the LMNA gene. The clinical phenotype is characterized by contractures at the elbows, Achilles' tendons, and spine and progressive muscle wasting in a humero‐peroneal distribution (5). In contrast with muscular dystrophies such as dystrophinopathies and dysferlinopathies, the contractures appear early in the disease. The pathogenetic mechanism(s) underlying this process is not clear.
The LMNA gene that is mutated in ADEDMD encodes for lamins A and C, the A‐type lamins which are generated from the same gene by alternate splicing, and which are major constituents of the nuclear lamina 3, 4. The proteins contain chromatin binding sites and also bind to transcription factors and to RNA processing enzymes. This has led to the hypothesis that lamins A/C have a role in organizing chromatin, regulating transcription, and controlling differentiation 3, 4.
Lamins A/C are also attached to the inner nuclear membrane through interactions with integral membrane proteins. This has led to the additional hypothesis that LMNA mutations result in fragile and mechanically unstable nuclei 3, 4. In effect, lamins A/C are essential for the maintenance of normal nuclear architecture: More than 80% of the embryonic fibroblast nuclei from LMNA null mice revealed herniation of the nuclear membranes (12). When mechanical strain was placed on nuclei of embryonic fibroblasts in vitro, LMNA−/− nuclei showed significantly larger deformations than wild‐type cells (7). In a different approach, in which single mouse embryonic fibroblast nuclei were compressed in vitro, most −/− nuclei became damaged, resulting in strands of DNA protruding into the cytoplasm, while the majority of +/+ nuclei remained largely intact (1). Taken together, there is compelling in vitro evidence for the A‐type lamins having a role in the mechanical stabilization of nuclei.
Data from in vivo studies are not as straightforward as in vitro. Muscle biopsies from patients with genetically confirmed ADEDMD showed ultrastructural myonuclear alterations. Two out of three muscle biopsies showed clumping of heterochromatin and detachment of chromatin from the nuclear membrane (11). Reichart et al found nuclear membrane invagination in 10% and peripheral chromatin condensation in 20% of the myonuclei studied (9). Myonuclei of three biopsies from genetically confirmed ADEDMD cases revealed changes in chromatin distribution as well as pseudoinclusions. The percentage of affected myonuclei was not specified (6). Sabatelli et al studied the biopsies from three patients with genetically confirmed ADEDMD and found reduced peripheral heterochromatin, condensed clumps detached from the inner nuclear membrane, and sharp borders between residual heterochromatic areas and interchromatin in 10% of the myofibers (10).
The in vivo mechanism by which LMNA mutations contribute to these myonuclear alterations is at present unknown. It is in particular open to discussion if and in which way the observed ultrastructural nuclear changes are related to mechanical stress. In the present study, we examined the myonuclear and cytoplasmic ultrastructure at the MTJs of LMNA−/− null mice as the MTJs are the site of longitudinal force transmission from contractile proteins to extracellular proteins. The MTJ is particularly vulnerable to damage (13). Susceptibility of myonuclei to mechanical stress in vivo is expected to be revealed at this site.
MATERIAL AND METHODS
Animals
The derivation of LMNA − / − mice has been previously described (12). Five knockout animals ranging in age from 5–7 weeks were used as well as two wild‐type animals aged 5 weeks and 6 months (Table 1). The animals were killed by CO2 asphyxiation and skinned. They were then immersion‐fixed in 2% glutaraldehyde and 4% freshly prepared paraformaldehdyde (PFA) (the 5‐week‐old null mutant and its wild‐type litter mate) or 0.5% glutaraldehyde and 3% PFA (the 7‐week‐old null mice and the 6‐month‐old wild‐type animal) in 0.1M cacodylate buffered at pH 7.2.
Table 1.
Numbers of altered myonuclei and myofibers.
| Age | Mutation | Muscle | Myonuclei/fibers merging into the tendon | Myonuclei disintegrating* | Myofibers degenerating† |
|---|---|---|---|---|---|
| 5 weeks | LMNA−/− | Rectus femoris‡ | 75/50 | 17/50 | 12/50 |
| Soleus§ | 28/20 | 18/50 | 10/20 | ||
| 7 weeks | LMNA−/− | Rectus femoris | 91/50 | 15/50 | 10/50 |
| Soleus | 13/20 | 13/50 | 7/20 | ||
| 7 weeks | LMNA−/− | Rectus femoris | 117/50 | 21/50 | 11/50 |
| Soleus | 36/20 | 12/50 | 14/20 | ||
| 7 weeks | LMNA−/− | Rectus femoris | 82/50 | 21/50 | 13/50 |
| Soleus | 31/20 | 19/50 | 5/20 | ||
| 7 weeks | LMNA−/− | Rectus femoris | 79/50 | 12/50 | 5/50 |
| Soleus | 22/20 | 13/50 | 10/20 | ||
| 5 weeks | LMNA+/+ | Rectus femoris | 27/50 | 0/50 | 0¶ |
| Soleus | 8/20 | 0/50 | 0¶ | ||
| 6 months | LMNA+/+ | Rectus femoris | 19/50 | 0/50 | 0¶ |
| Soleus | 5/20 | 0/50 | 0¶ |
Number of disintegrating myonuclei per 50 nuclei counted.
†Number of degenerating myofibers per 50 (rectus femoris muscle) and 20 (soleus muscle) fibers counted, respectively.
‡Of a total of 50 fibers per muscle.
§Of a total of 20 fibers per muscle.
¶Number of degenerating myofibers across the whole section.
Electron microscopy
The right soleus and rectus femoris muscles were excised from the fixed animals and prepared for electron microscopy. The myonuclei of 50 myofibers merging into the tendon of the rectus femoris and 20 myofibers of the soleus muscle merging into the Achilles tendon were counted at the ultrastructural level in order to assess the ratio of myonuclei per myofiber at these sites. Furthermore, myonuclei undergoing disintegration were assessed in a total of 50 myonuclei of every muscle studied. Finally, the number of myofibers showing signs of degeneration was assessed from 50 (rectus femoris muscle) or 20 fibers (soleus muscle) merging into the respective tendons.
RESULTS
Myonuclear degeneration
The number of myonuclei in myofibers merging into the tendon was elevated in knockout mice when compared with those in the wild‐type animals ( Figure 1; Table 1). Many of the nuclei were internalized. The numbers of myonuclei of the rectus femoris muscles ranged from 75/50 to 117/50, whereas they ranged from 13/20 to 36/20 in soleus muscles. The wild‐type mice showed 27/50 and 19/50 myonuclei in the rectus femoris muscle and 8/20 and 5/20 myonuclei in the soleus muscle.
Figure 1.
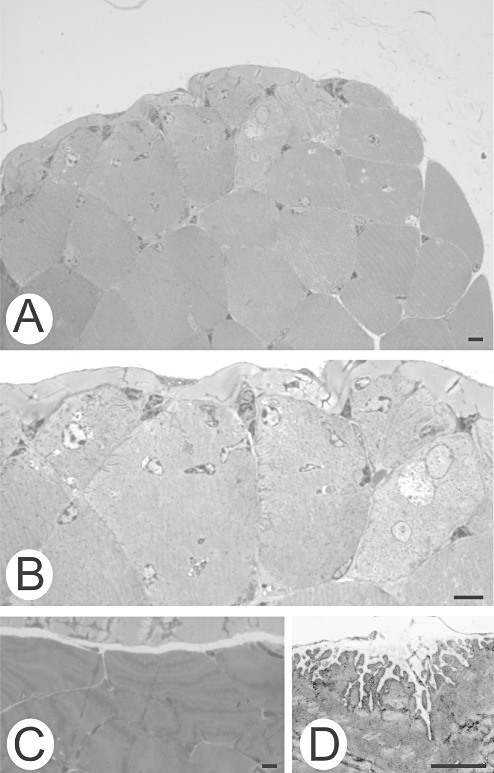
Myofibers of the rectus femoris muscle facing the tendon. A, B. Null mouse. A. The number of myonuclei per myofiber is increased in muscle adjacent to tendon. B. Higher magnification. C, D. Wild‐type mouse. C. This was not seen in the wild‐type mouse. D. The electron micrograph shows the junctional folds of a myofiber merging with the tendon. Bars, 10 µm.
Many myonuclei of the LMNA null rectus femoris and soleus muscles showed signs of degeneration ( Figure 2; Table 1). These consisted of chromatin clumping (Figure 2B–F), nuclear disintegration (Figure 2D–F), intranuclear inclusions or invaginations (Figure 2D,G–I), and/or intranuclear filaments measuring 10–12 nm in diameter (Figure 2J). Chromatin clumping occasionally occurred in conjunction with the perinuclear arrangement of felt‐like filaments (Figure 2B) or perinuclear concentric tubules (Figure 2C).
Figure 2.
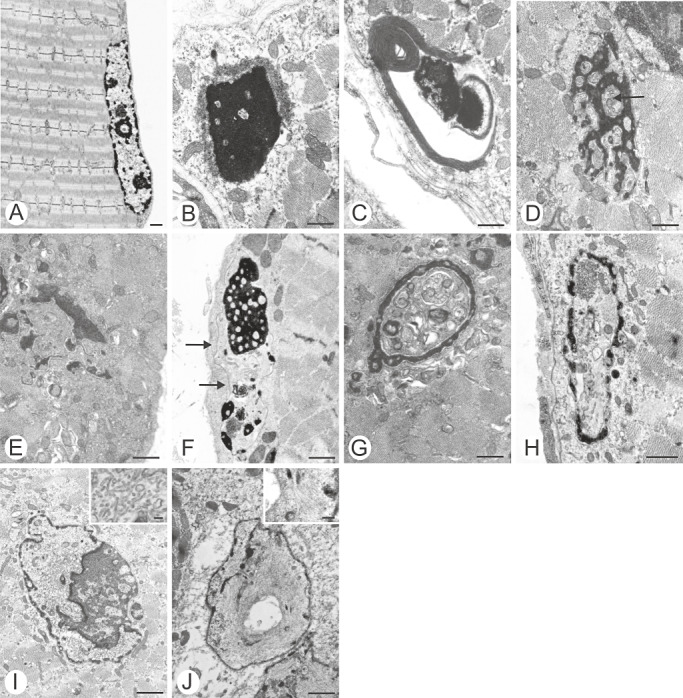
A. Normal myonucleus of the wild‐type mouse. B–J. The spectrum of myonuclear changes in null mutants. B. Condensed chromatin, surrounded by a felt‐like rim. C. Condensed chromatin, surrounded by concentric lamellae. D. Fragmented nucleus containing tubular profiles (arrow). E. Condensed fragmented chromatin. F. Condensed fragmented chromatin with inclusions. Note nuclear membrane (arrows). G, H. Pseudoinclusions. I. Intranuclear tubular profiles. J. Intranuclear filaments. Bars, 1 µm.
Myofiber degeneration
A number of myofibers facing the tendon showed one of two types of pathological changes: “Splitting” ( Figure 3A,B) or subsarcolemmal absence of myofibrils (Figure 4). Both patterns were considered to be signs of degeneration. The number of myofibers showing degeneration is indicated in Table 1. “Splitting” was accompanied by delicate connective tissue strands extending between the myofiber fragments (Figure 3A,B). This created cytoplasmic surfaces that were smooth (Figure 3B). This pattern differs from myofibers of normal mice that form junctional foldings at the tendon interface (Figure 3C).
Figure 3.
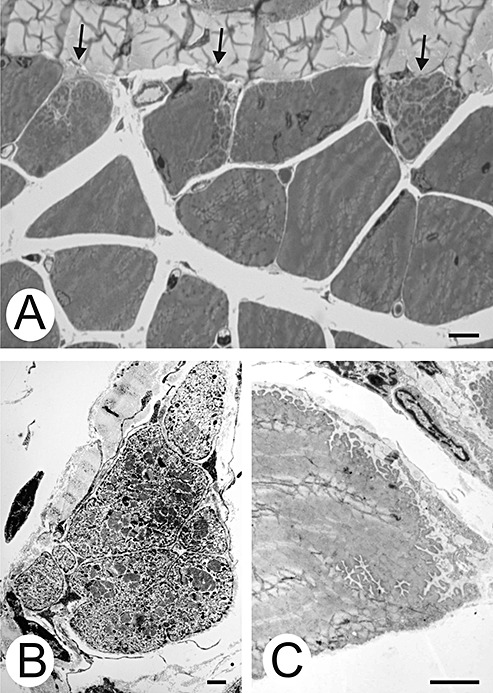
Cytoplasmic pathological changes. A. Three myofibers show “splitting” (arrows). Bar, 10 µm. B. The cytoplasmic surfaces abutting the extracellular matrix of a split myofiber are smooth. C. In contrast, this myofiber of the wild‐type mouse shows indentations wherever its surface contacts extracellular matrix. Bars, 5 µm.
Figure 4.
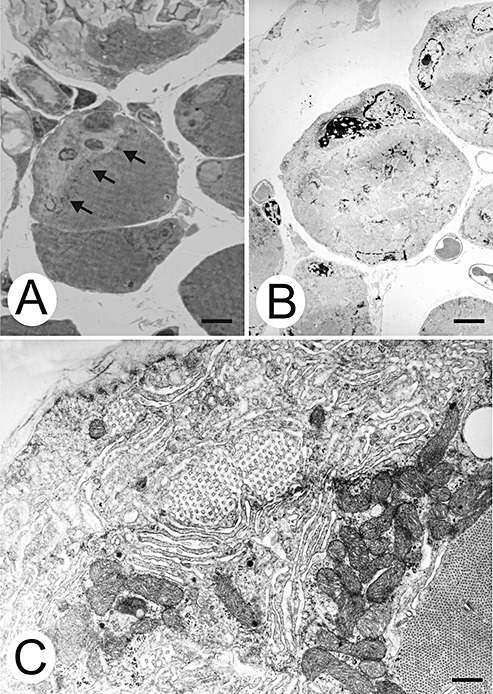
Subsarcolemmal myofibrillar loss. A. Myofibrils of the central myofiber do not reach the cell border. Arrows denote the border of the filamented cytoplasm. B. Electron micrograph of a parallel non‐serial section of the myofiber shown in A. One myonucleus in the myofibril‐free subsarcolemmal region shows clumped chromatin. Bars, 50 µm. C. Higher magnification of a subsarcolemmal area containing honeycomb structures, sarcotubular profiles, and polysomes. Bar, 5 µm.
The second pattern of myofiber degeneration consisted of a loss of subsarcolemmal myofibrils from the surface directed toward the tendon (Figure 4). Myonuclei were frequently found in this myofibril‐free space (Figure 4A,B). These myonuclei often showed one of the degenerative features shown in Figure 2 (Figure 4B). The myofibril‐free space also contained sarcotubular profiles, honeycomb structures and polysomes (Figure 4C). None of these patterns was found distal to the MTJ.
DISCUSSION
We have demonstrated that LMNA null mice have both myonuclear disintegration and cytoplasmic degeneration at the MTJ. The MTJ is the site where force is transmitted from the myofibers to the tendon. Moreover, MTJs are affected in the Emery–Dreifuss phenotype of muscular dystrophy, resulting in joint contractures early in the course of the disease (5).
The null mice showed increased myonuclear numbers in the myofibers facing the tendons when compared with the wild‐type mice (Figure 1; Table 1). Elevated myonuclear numbers are not a constant finding at the MTJ. In a detailed in vivo study of mouse extensor digitorum longus muscle, Bruusgaard et al found that five out of 19 MTJ showed a nucleic density that was 80%–100% higher when compared with the length of the myofibers (2). The numbers of myonuclei found in myofibers facing the tendon ranged from 75 to 117 per 50 myofibers of the rectus femoris, and from 13 to 36 per 20 myofibers of the soleus muscle (Table 1). The values for the wild‐type mice were 27/50, 19/50, 8/20 and 5/20, respectively. This finding suggests that the number of myonuclei at the MTJ is increased in LMNA−/− mice when compared with normal animals. The reason for this is not clear. Myonuclei of healthy mice are immobile within the myofiber over periods of up to 4 days (2). It is suggested that myonuclei of LMNA−/− mice do in fact move, at least in the myotendinous region.
Many of the myonuclei of the MTJ showed signs of disintegration (Figure 2). Several of the features demonstrated here are similar to those shown in human biopsies from patients with genetically confirmed ADEDMD. Clumping of heterochromatin (Figure 2B–F) 9, 10, 11, detachment of chromatin from the nuclear membrane (Figure 2F) (11), pseudoinclusions (Figure 2D,G,H) (6) are features that are shared by human biopsies and LMNA−/− mice. Nuclear fragmentation, intranuclear filaments and intranuclear tubular structures illustrated in Figure 2I and J have so far not been reported in human biopsies of ADEDMD. Another feature hitherto unreported in human biopsies was perinuclear felt‐like filaments and concentric tubules as shown in Figure 2B and C. It is not clear whether these surrounding structures are cytoplasmic in origin, or whether they are derived from nuclear envelope structures.
We recently observed myofiber “splitting” as shown in Figure 3 in 0.5%–7% of myofibers in biopsy and autopsy tissue from three patients with ADEDMD (8). The percentages found in the human tissue were below most of the percentages detected in the LMNA−/− mice (Table 1). The paucity of affected fibers in human material might be explained by the distance of these sites from the MTJ of the respective muscles as diagnostic muscle biopsies are usually taken from the belly of the muscle rather than from the ends. The human condition of ADEDMD being heterozygous may also add to the milder phenotype.
The MTJ are specialized junctions for force transmission from contractile, cytoskeletal proteins to extracellular, structural proteins (13). Cytoplasmic “splitting” accompanies loss of the junctional folding (Figure 3B,C). Membrane folding has been identified as a mechanically important specialization of the MTJ because it reduces contractile stress generated by the myofiber by increasing its distribution over a larger junctional area. Loss of the junctional folding is associated with an increase in MTJ stress during loading (13). Hence, the accompanying cytoplasmic “splitting” and the blunting of the foldings may increase the mechanical stress at the MTJ, thereby aggravating the course of the disease.
In conclusion, we demonstrate myonuclear disintegration at the MTJ in LMNA−/− mice. This may be well be interpreted as an in vivo example of the increased nuclear vulnerability to mechanical stress in the absence of a functionally intact nuclear lamina. The accompanying cytoplasmic degeneration is most likely a result of the nuclear instability. The pathomechanism linking nuclear disintegration and cytoplasmic degeneration remains to be established. Loss of, or mutations within, the LMNA gene resulted in ADEDMD, which led to mechanical weakening of the nuclei; this pathology may be compounded by alterations to signaling pathways, together with effects on chromatin organization/transcription of specific genes.
ACKNOWLEDGMENTS
We thank Elisabeth Rushing for her help with the English, and Suse Renkhold for technical assistance. Financial help from the Deutsche Gesellschaft für Muskelkranke e. V. is gratefully acknowledged.
REFERENCES
- 1. Broers JLV, Peeters EAG, Kuijpers HJH, Endert J, Bouten CVC, Oomens CWJ et al (2004) Decreased mechanical stiffness in LMNA−/− cells is caused by defective nucleo‐cytoskeletal integrity: implications for the development of laminopathies. Hum mol Gen 13:2567–2580. [DOI] [PubMed] [Google Scholar]
- 2. Bruusgaard JC, Liestøl K, Edmark M, Kollstad K, Gundersen K (2003) Number and spatial distribution of nuclei in the muscle fibres of normal mice studied in vivo. J Physiol 551:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burke B, Stewart CL (2006) The laminopathies: the functional architecture of the nucleus and its contribution to disease. Annu Rev Genomics Hum Genet 7:369–405. [DOI] [PubMed] [Google Scholar]
- 4. Capell BC, Collins FS (2006) Human laminopathies: nuclei gone genetically awry. Nature Rev Gen 7:940–952. [DOI] [PubMed] [Google Scholar]
- 5. Emery AEH (2000) Emery‐Dreifuss muscular dystrophy—a 40‐year retrospective. Neuromuscul Disord 10:228–232. [DOI] [PubMed] [Google Scholar]
- 6. Fidzianska A, Hausmanowa‐Petrusewicz I (2003) Architectural abnormalities in muscle nuclei. Ultrastructural differences between X‐linked and autosomal dominant forms of EDMD. J Neurol Sci 210:47–51. [DOI] [PubMed] [Google Scholar]
- 7. Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD et al (2004) Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest 113:370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mittelbronn M, Hanisch F, Gleichmann M, Stötter M, Korinthenberg R, Wehnert M et al (2006) Myofiber degeneration in autosomal‐dominant Emery‐Dreifuss muscular dystrophy (AD‐EDMD) (LGMD1B). Brain Pathol 16:266–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reichart B, Klafke R, Dreger C, Krüger E, Motsch I, Ewald A et al (2004) Expression and localization of nuclear proteins in autosomal‐dominant Emery‐Dreifuss muscular dystrophy with LMNA R377H mutation. BMC Cell Biol 5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sabatelli P, Lattanzi G, Ognibene A, Columbaro M, Capanni C, Merlini L et al (2001) Nuclear alterations in autosomal dominant Emery‐Dreifuss muscular dystrophy. Muscle Nerve 24:826–829. [DOI] [PubMed] [Google Scholar]
- 11. Sewry CA, Brown SC, Mercuri E, Bonne G, Feng L, Camici G et al (2001) Skeletal muscle pathology in autosomal dominant Emery‐Dreifuss muscular dystrophy with lamin A/C mutations. Neuropathol Appl Neurobiol 27:281–290. [DOI] [PubMed] [Google Scholar]
- 12. Sullivan T, Escalante‐Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K et al (1999) Loss of A‐type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 147:913–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tidball JG (1991) Force transmission across muscle cell membranes. J Biomech 24(Suppl. 1):43–52. [DOI] [PubMed] [Google Scholar]