

Abstract
Calcific aortic valve disease (CAVD) sits at the confluence of multiple world-wide epidemics of aging, obesity, diabetes, and renal dysfunction, and its prevalence is expected to nearly triple over the next three decades. This is of particularly dire clinical relevance, as CAVD can progress rapidly to aortic stenosis, heart failure, and eventually premature death. Unlike in atherosclerosis, and despite the heavy clinical toll, so far no pharmacotherapy has proven effective to halt CAVD progression, with invasive and costly aortic valve replacement representing the only treatment option currently available. This substantial gap in care is largely due to our still-limited understanding of both normal aortic valve biology and the key regulatory mechanisms that drive disease initiation and progression. Drug discovery is further hampered by the inherent intricacy of the valvular microenvironment: a unique anatomical structure, a complex mixture of dynamic biomechanical forces, and diverse and multipotent cell populations collectively contributing to this currently-intractable problem. One promising and rapidly-evolving tactic is the application of multi-omics approaches to fully define disease pathogenesis. Herein, we summarize the application of (epi)genomics, transcriptomics, proteomics, and metabolomics to the study of valvular heart disease. We also discuss recent forays towards the omics-based characterization of valvular (patho)biology at single-cell resolution; these efforts promise to shed new light on cellular heterogeneity in healthy and diseased valvular tissues and represent the potential to efficaciously target and treat key cell sub-populations. Lastly, we discuss systems biology- and network medicine-based strategies to extract meaning, mechanisms, and prioritized drug targets from multi-omics datasets.
Keywords: Calcific Aortic Valve Disease, Aortic Stenosis, Multi-Omics, Systems Biology, Network Medicine
Subject Codes: Valvular Heart Disease, Proteomics, Epigenetics, Gene Expression and Regulation
Aortic Stenosis – A Poorly Understood Disease without Therapeutics
Calcific aortic valve disease (CAVD) is the most common of all valvular heart diseases, with aortic sclerosis (ASc) affecting 25% of those over age 65 and aortic stenosis (AS) occurring in nearly 2%. Those who develop symptomatic AS have an average further lifespan of less than two years without valve replacement,1 and three-quarters of these patients will develop heart failure, undergo valve replacement, or die within five years.2 Presently, there are no approved pharmaceutical therapies for CAVD, as statins successful in atherosclerosis have failed to provide benefit in prospective clinical trials.3 Due to the complete lack of pharmaceutical therapy, over 370,000 aortic valve replacement surgeries are performed worldwide each year,4 with costs exceeding $10.2 billion/year in the USA alone.5 Worldwide CAVD rates will rise 240% by 2040, and will grow disproportionally in regions without access to cardiac surgery or interventional cardiology.6 Though revolutionary in the developed world, transcatheter aortic valve replacement (TAVR) will be unable to meet this growing critical need elsewhere, and as a result there is an urgent and growing need for therapeutics aimed at early, tailored, and targeted intervention against the initiation and progression of CAVD.
This lack of effective pharmacotherapy is mainly due to our poor understanding of the molecular mechanisms that direct valvular homeostasis and disease progression, which is exacerbated by the aortic valve’s complex structure, cellular composition, and (micro-)environment. Adult human valve leaflets are ~1 mm in thickness7 and composed of three stacked layers: facing into the left ventricle, the ventricularis is rich in radially-oriented elastin, the spongiosa contains a variety of glycosaminoglycans and proteoglycans, and the disease-prone fibrosa sits closest to the ascending aorta, and is composed of dense bundles of circumferentially-oriented collagen fibers.8 The normal aortic valve contains two main and rather distinct cell types: valvular interstitial cells (VICs) populate all three leaflet layers and are believed to consist primarily of quiescent fibroblast-like cells with multipotent progenitor capacity.9 Valvular endothelial cells (VECs) cover both the fibrosa and ventricularis sides of the valve leaflets, and are phenotypically distinct from endothelial cells found throughout the rest of the arterial circulation.10 VICs and VECs are also mechanosensitive, and are responsive to the cyclic stretch, compression, and blood shear forces that valve leaflets experience throughout the cardiac cycle (reviewed in11).
In this complex environment, disease initiation and progression thus appears to be multifactorial, as aspects of lipid accumulation, inflammation, myofibrogenesis, osteogenesis, extracellular vesicle biosynthesis and aggregation, platelet activation, osteochondrogenesis, extracellular matrix turnover, phosphate handling, cellular senescence, and biomechanical forces have all been implicated in disease development.12–18 Furthermore, for reasons that remain largely unclear, those born with bicuspid aortic valves (BAV) are ~25 times more prone to severe CAVD and suffer from accelerated progression of disease when compared to those with the standard configuration of a tricuspid aortic valve (TAV).19,20 After disease initiation, the early stages of CAVD are marked by sclerotic/fibrotic thickening of the leaflets,7 followed by dystrophic and ectopic leaflet calcification that predominantly affects the fibrosa layer.21 As valvular fibrosis and calcification worsens, the narrowed aortic orifice hampers cardiac output, leading to angina, chest pain and reduced exercise tolerance, ultimately resulting in left ventricular remodeling, which almost inexorably proceeds towards a need for replacement of the aortic valve or death (Figure 1). Recently, a new paradigm of investigation has led the field to begin to embrace an integrative physiological approach where omics strategies potentiate the holistic study of this multifaceted disease in order to define healthy baselines, characterize molecular signatures of disease, identify putative drug targets, and validate their therapeutic potential and efficacy. When paired with novel systems biology techniques, multi-omics studies of the (epi)genome, transcriptome, proteome, and metabolome have the potential to enable rapid headway towards revolutionizing the standard-of-care for CAVD. The high degree of sensitivity of this latest generation of omics instruments has placed even more emphasis on the importance of standardized and uniform sample collection, processing, loading, normalization, batch correction, and statistical analysis of differential enrichment. Multiple omics methods, or modalities, can be employed to study a host of diverse valve-related sample types from a variety of sources (Figure 2). These may include the isolation of DNA, RNA, non-coding RNA, protein, extracellular vesicles, or metabolites directly from valve tissue or whole blood, plasma, or serum. In a different approach, VICs and VECs are often isolated and cultured in a variety of formats, including standard 2D culture (plates/flasks) or in more complex 3D structures that mimic aspects of the native valve architecture or microenvironment (e.g. anatomical layers/stiffnesses, VIC/VEC co-culture). Lastly, valve-derived cells may be cultured in a variety of bioreactor designs that recapitulate aspects of valvular biomechanics (e.g. cyclic stretch/compression, oscillatory shear stresses). Other recent tactics have included the usage of induced pluripotent stem cell (iPSC)-derived cells as a standardizable alternative to primary VIC/VEC cultures,22,23 though relevant challenges of IPSC-based approaches include variable efficiency of differentiation, difficulty in modeling polygenic/sporadic/late-onset disease, variability based on cellular source/differentiation methodology, and impact of epigenetic state (reviewed in24,25). Again, these various cell culture models allow one to extract sample material from both the cells themselves or the cell culture supernatant, the latter of which contains the secretome. The choice of tissue/blood-derived vs. cell cultured-derived omics often reflects a trade-off between the direct study of native in vivo phenotypes vs. that of a highly-controlled experimental system, which enables fine manipulation of variables, molecules, or mechanisms of interest.
Figure 1: Pathogenesis of calcific aortic valve disease (CAVD).
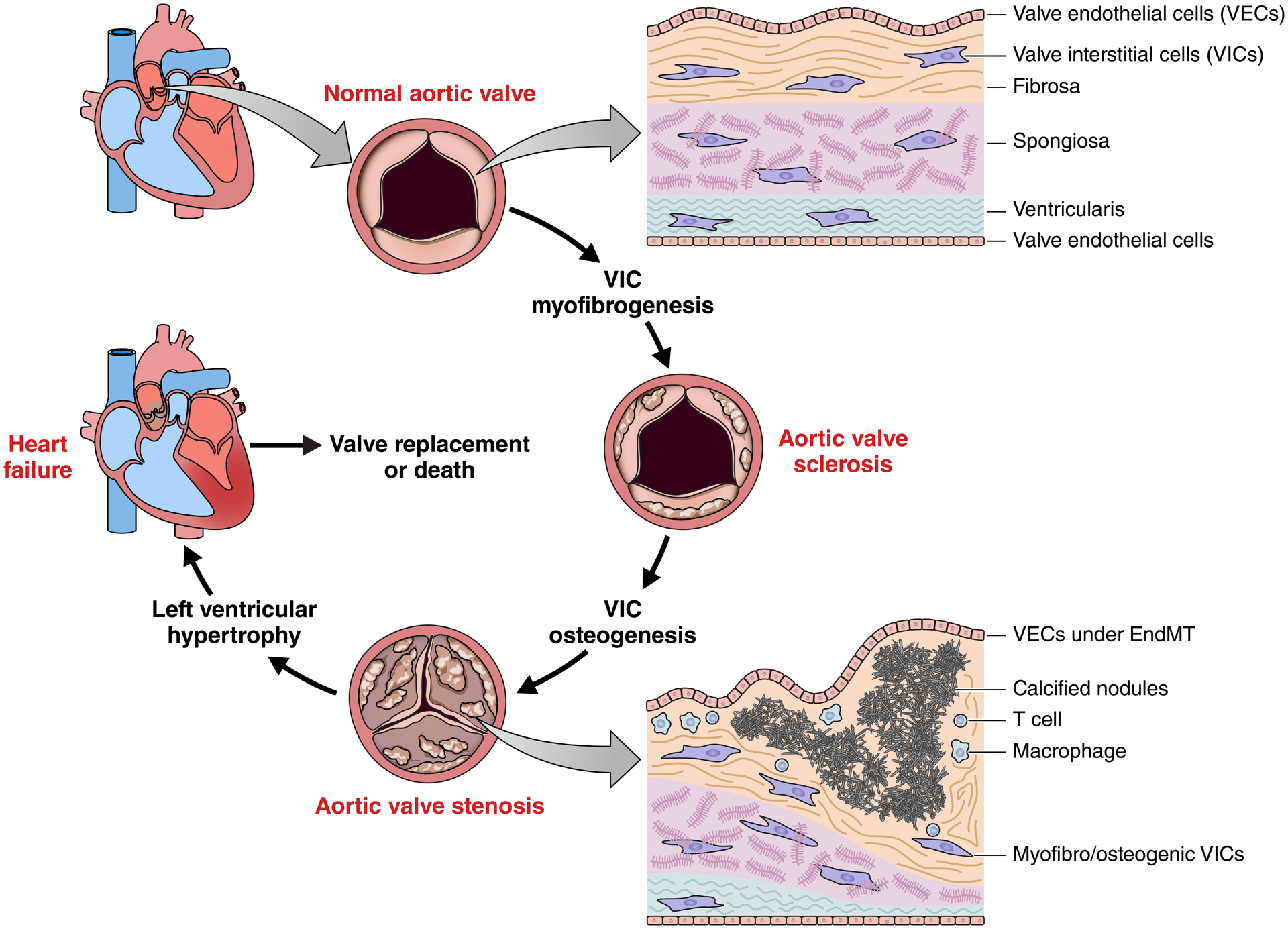
The aortic valve sits between the left ventricle and ascending aorta, and enables efficient pumping of blood in the systemic circulation. Valve leaflets are ~1mm thick, lined with valve endothelial cells (VECs), and composed of three layers with distinct extracellular matrix composition, structure, and biomechanics. During disease initiation and progression, resident valve interstitial cells (VICs) undergo myofibrogenesis, resulting in sclerotic thickening of the aortic valve. Osteogenic differentiation of VICs leads to calcification and mineralization of valvular tissues and eventual impairment of leaflet opening/closure (stenosis). The left ventricle hypertrophies as a result of this increased resistance, leading towards heart failure. Surgical/transcatheter replacement of the aortic valve is currently the only therapeutic strategy and untreated aortic stenosis suffers from very high mortality rates.
Figure 2: Sample sources for omics studies of CAVD.
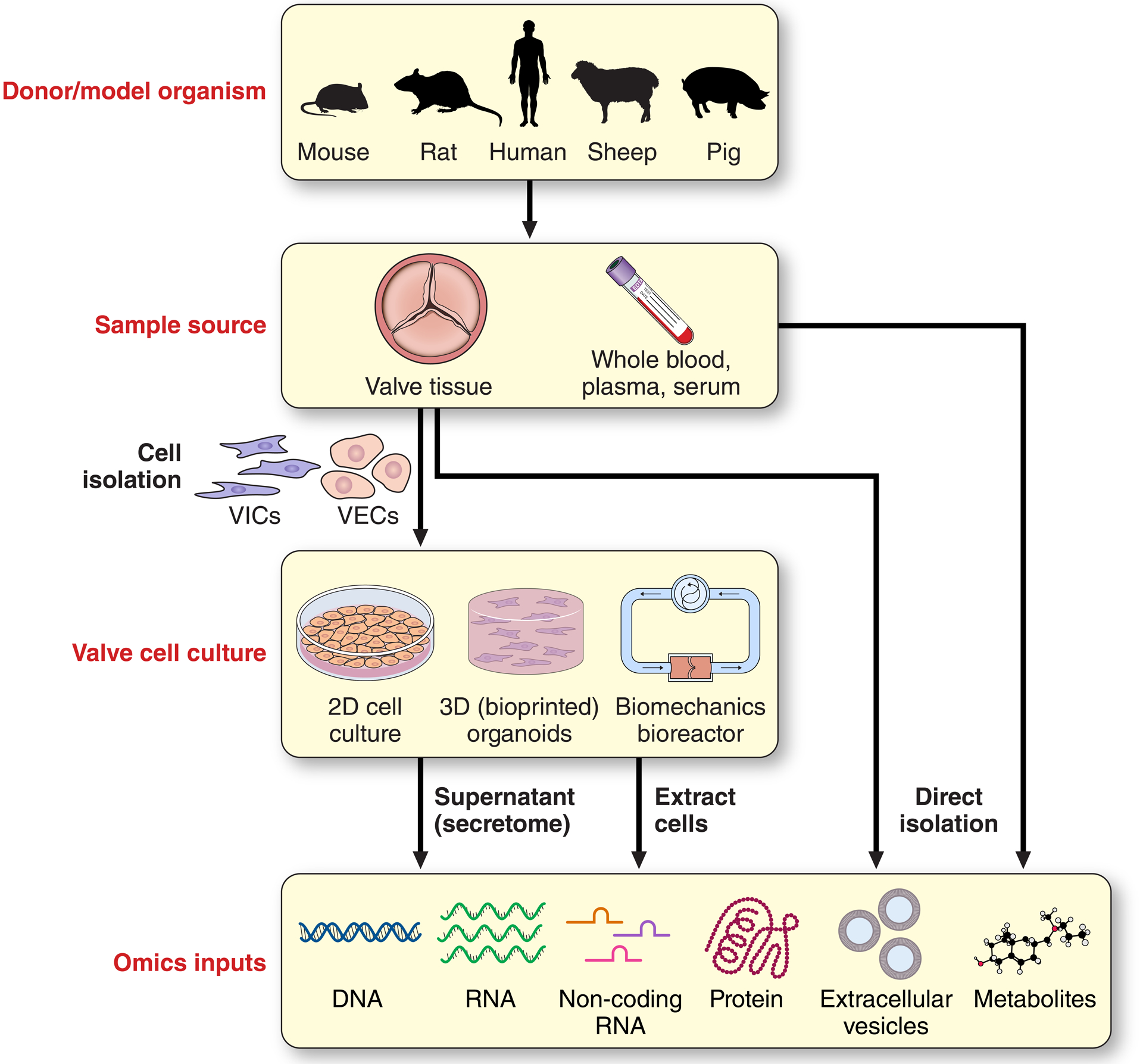
Along with human studies, a number of well-defined animal models of calcific aortic valve disease (CAVD) exist with differing (dis)advantages.217 Collection of valvular tissues or biofluids such as whole blood, plasma, or serum from human donors or model organisms can act as both direct inputs into multi-omics studies, or can be used for isolation and subsequent culture of valve-derived cells (e.g. valve interstitial cells [VICs] or valve endothelial cells [VECs]). Depending on experimental aims/context, these valve-derived cells can be maintained in standard 2D in vitro cultures, 3D organoid-style constructs, or in bioreactors that mimic aspects of native biomechanics (e.g. cyclic shear, stretch, or compression). Subsequently, DNA, RNA (mRNA or non-coding), protein, extracellular vesicles (EVs), or metabolites derived from cell culture supernatants or the cells themselves can be input into omics pipelines.
Here, we detail a comprehensive discussion of multi-omics approaches towards atlas development, the study of pathogenic mechanisms of CAVD, and discovery of novel drug targets and treatments, all grounded in a practical consideration of the utility, strengths, and applications of each omics modality (Table 1).
Table 1:
Examplar Studies Employing Omics to Define CAVD Pathogenesis
| Tissue/Model | Species/Sample size | Data | Methods/Analytical tools | Major findings | Ref. |
|---|---|---|---|---|---|
| Stenotic aortic valves/spatiotemporal segmentation | Human 9/3 |
Yesa | LC-MS/MS, RNA-seq Qlucore Omics Explore, DESeq2, Proteome Discoverer, ConsensusPathDB |
First spatially/temporally resolved multi-omics study of human CAVD tissue; identification of molecular regulatory networks in CAVD, demonstration of side-specific VIC calcification potential | 26 |
| Stenotic aortic valves/spatiotemporal segmentation | Human 9 |
Yesb | MS/MS (targeted PRM) Proteome Discoverer, Qlucore Omics Explorer, ConsensusPathDB |
ApoC-III is increased in fibrotic and calcific tissues; induces calcification via mitochondrial dysfunction/inflammation | 13 |
| Stenotic aortic valves/bulk tissue (RNA); buffy coat (DNA) | Human 233 (TWAS) 5,115/354,072 cases/controls (GWAS) |
Yesc | GWAS/TWAS (eQTL) PLINK, SNPTest |
Implicates IL6, ALP and NAV1 as new genetic loci in CAVD pathogenesis | 27 |
| Stenotic aortic valves/bulk tissue; EDTA-plasma | Human 96/106 |
Yesd | MS (non-targeted metabolomics and targeted lipidomics) MetaboAnalyst, Cytoscape |
Initial metabolomic map of human CAVD; LsyoPA is associated with CAVD severity and accelerated disease progression | 28 |
| Cultured VICs | Pig 7,037 cells |
Yese | Single-cell RNA-seq Cell Ranger, DESeq2, Seurat |
NO rescues aortic valve calcification by a S-nitrosylation of USP9X to activate NOTCH signaling | 29 |
| Calcific/non-diseased aortic valves | Human 6/6 |
Yesf | Bulk and single-cell RNA-seq CellRanger, STAR, Monocle |
Identification of 14 cell subtypes; endothelial-to-mesenchymal transition contributes to CAVD pathogenesis | 30 |
ProteomeXchange Consortium; dataset identifier PXD021858
Available from the corresponding authors on reasonable request
European Molecular Biology Laboratory–European Bioinformatic Institute MetaboLights database; identifier MTBLS1267
GEO; accession number GSE161123
BioProject database of NCBI; PRJNA562645 (single-cell sequencing raw data), PRJNA552159 (bulk sequencing raw data)
(Epi)Genomics and the Genetic Regulation of Valvulopathy
CAVD and AS demonstrate significant familial aggregation, clustering, and heritability – a study of 6,117,263 Swedish siblings found nearly 5% of those with AS had a sibling history of CAVD as well, and having one or more siblings with AS was associated with a hazard ratio of 3.41.31 It is thus well understood and widely accepted that genetics play a significant role in the onset and progression of CAVD. The advent of genome-wide association studies (GWAS) heralded a new era in our understanding of genetic contributions to CAVD. Simply put, GWAS aim to associate single-nucleotide polymorphism mutations (SNPs) in genomic DNA sequences with the presence of disease traits or phenotypes. Typically, such studies leverage DNA microarrays of known, high-frequency SNPs. In a groundbreaking study, Thanassoulis et. al. identified a SNP (rs10455872) in LPA, the lipoprotein(a) (Lp(a)) gene to be significantly associated with the presence of aortic valve calcification and AS in European cohorts, and was subsequently replicated in additional cohorts of white European, African-American, and Hispanic-American descent.32 Circulating Lp(a) levels were also correlated with valvular calcification, further supporting a causal role in CAVD pathogenesis. Two SNPs in the pro-inflammatory gene IL1F9 also reached genome-wide significance for mitral annular calcification in the discovery cohort, but could not be reliably replicated in other cohorts. More recent work identified genome-wide AS risk loci near PALMD (rs7543130) and TEX41 (rs1830321) in Icelandic cases and a European validation cohort, and also replicated the earlier association of LPA variant rs10455872. The mechanistic basis underlying the contribution of PALMD and TEX41 to disease remains a largely open question, though there is some evidence that PALMD modulates apoptotic responses in vitro, along with being implicated in myofibroblastic differentiation, perhaps through its co-localization to the actin cytoskeleton and resultant control of cell morphology.33–35 The latter two functions may explain, in part, its putative association with atrial fibrillation and stroke. Most recently, two additional novel loci for AS reached genome-wide significance in a GWAS meta-analysis of 5115 cases and 354 072 controls (along with replication of LPA and PALMD): IL6 and ALPL (encoding tissue non-specific alkaline phosphatase, or TNAP). Both IL-6 and TNAP are well-described modulators of VIC calcification in vitro: IL-6 expression is dramatically elevated in VICs cultured under phosphate-rich conditions via the P2Y2 receptor, and in turn drives expression of the pro-osteogenic genes RUNX2 and BMP.36 TNAP is a key and well-described protein that controls biomineralization in a host of tissue types by hydrolysis of organic phosphate to promote the formation of hydroxyapatite crystals, and is itself elevated in calcified vs. non-calcified portions of diseased valve leaflets.26 The genetic data derived from such studies may form the basis for the future development of polygenic risk scores (PRS) for CAVD – as is the case for coronary artery disease37 – which will undoubtedly improve the management of patients with high susceptibility for the development of CAVD. Such PRS may also benefit future interventional trials targeting druggable pathways to prevent late stage CAVD. Given that multiethnic PRS improve risk prediction in various populations,38 a high diversity in GWAS participants may help to tailor treatment options more accurately and, eventually, to mitigate existing healthcare disparities.
As noted earlier, the presence of a BAV is associated with dramatically increased risk of CAVD and accelerated deficits in valvular function. Importantly, BAVs are highly heritable, and determination is thought to be mainly genetic, although no single-gene model clearly describes its inheritance and congenital abnormalities (e.g. hypoplastic left heart syndrome, aortic coarctation) can also be associated with a bicuspid phenotype.39,40 Whole exome sequencing (WES) on 17 members of a family with a history of BAV failed to find high-effect variants, implying that common WES filtering approaches may be insufficient for a complex trait like BAV.41 GWAS of 466 BAV cases and 4,660 controls has recently identified a non-coding variant (rs6601627) near the cardiac-specific transcription factor GATA4 with genome-wide significance for incidence of BAV, along with a common missense variant in GATA4 itself (rs3729856) that was also associated with BAV (p = 8.8 × 10−8).42 In addition, 14 variants in closely-related GATA6 are nominally associated with BAV, though not to locus-wide significance.43 In the above AS GWAS, PALMD (rs7543130) was also associated with presence of BAV.44 GWAS studies against BAV have also implicated genetic variants in non-motile primary cilia, cell surface organelles with important roles in mechano/chemosensation and resultant signal transduction.45 Fulmer and colleagues discovered that ~8–10% of annotated cilia-associated genes sat in close proximity to nearly 1,900 of the most-significant BAV GWAS SNPs in a discovery cohort of 452 individuals with BAV and 1,834 controls. Though combined p-values in a validation cohort did not reach genome-wide significance for exocyst (cilial biogenesis) genes, further knockout studies in zebrafish and mice of exocyst genes yielded animals with BAVs, dysmorphic valves, and stenosis and calcification with age. The potential linkage between defects in cilial shear-sensation, BAV incidence, and accelerated onset of BAV-CAVD in humans is worthy of additional study. Interestingly, while the earliest evidence of a specific genetic cause of BAV was found in families with inherited NOTCH1 mutations,46 and mice with mutated Notch1 or Notch1 effectors develop BAV as well,47,48 GWAS studies have yet to identify NOTCH1 or associated molecules as a significant risk locus for BAV. A number of other genes associated with guanylate cyclase signaling have been demonstrated to induce BAV, CAVD, and ascending aortic aneurysms in mice, and while these putative drivers of congenital malformation have not been validated by GWAS hits, administration of the soluble guanylate cyclase agonist ataciguat slows the progression of valvular calcification in humans with mild-to-moderate fibrocalcific AS.49–51 As is the case with GWAS against incidence, there remains a disconnect between our understanding of causative gene mutations in animal models and discovery/replication/validation of those genes in humans. These efforts are hampered by the relatively low incidence of valvular diseases in the general population, the complex interaction of an array of CAVD contributors, and etiological (e.g., lipid vs. renal failure-driven CAVD) as well as spatiotemporal disease heterogeneity. In the valve, rare allele variants may have an outsized impact on pathogenesis, thus requiring the incorporation of whole genome/exome sequencing or copy number variant analyses in discovery pipelines focused on genomic causes of CAVD.
Importantly, changes to the underlying DNA sequence are not necessary for a heritable change in phenotype. Epigenetics encompasses the study of gene regulation via both inheritance and in response to environmental factors via a number of complementary means. Epigenetic regulation of gene expression typically involves alteration of chromatin accessibility (and thus transcription), either by methylation of CpG nucleotides or post-translational modifications of histone proteins that alter how tightly histones cling to DNA, and thus how available that DNA is for transcription and gene expression. Epigenetic modifications can be assayed in a high-throughput means using techniques such as bisulfite sequencing (methylation patterns), chromatin immunoprecipitation sequencing (ChIP-seq; binding sites of DNA-associated proteins, e.g., transcription factors), or ATAC-seq (assay for transposase-accessible chromatin using sequencing; chromatin accessibility). There has so far been relatively little study of epigenetic regulation in the context of CAVD, though interest in this area of the field has exploded of late. Early work demonstrated the promise of epigenetic regulation of valvular calcification, as differential expression of the leukotriene-synthesizing enzyme 5-lipoxygenase in human AS was correlated with the presence of altered methylation density in the gene’s promoter region. Hypomethylation of DNA in cultured VICs by treatment with 5-aza-2′-deoxycytidine recapitulated these alterations in 5-lipoxygenase promoter methylation and expression.52 Hypermethylation of the epidermal growth factor receptor (EGFR) appears to be involved in the induction of CAVD (thickened leaflets, formation of cartilage and calcification, inflammatory infiltration) and left ventricular hypertrophy in mice with the hypomorphic Egfrwa2 allele.53 In a seminal human study, Hadji et. al. linked epigenetic regulation of non-coding RNA expression with aortic valve calcification. Total RNA sequencing revealed significant increases of the long noncoding RNA H19 in stenotic human aortic valves, and modestly but statistically increased in earlier disease stages. Hypomethylation of the H19 promoter was present in the leaflets of humans with CAVD, and correlated to elevated expression of H19. Chemical manipulation of global methylation levels in VIC cultures confirmed these findings, and showed that H19 induced a pro-osteogenic transcriptional program (increases in osteocalcin, BMP2, RUNX2) and production of mineralization. Mechanistically, elevated H19 was tied to reduced NOTCH1 transcription via impaired recruitment of p53. Together, these data shed light on the potential of a novel therapeutic approach (pharmaceutical manipulation of the epigenome) and represented an elegant integration of epigenetic effects on valvular phenotypes. Other epigenetic studies have been motivated by GWAS hits at the genomic level: Lp(a) carries a number of oxidized phospholipids, from which lysophosphatidic acid (lysoPA) is produced by autotaxin and goes on to induce VIC osteogenesis.54 Phospholipid phosphatases degrade lysophosphatidic acid, and their expression is downregulated in human CAVD. This reduction is inversely correlated with cytosine methylation on an intronic enhancer of the phospholipid phosphatase PLPP3, as determined by whole-genome quantification of DNA methylation by Illumina arrays and knockdown of PLPP3 in cultured VICs drove osteogenesis and calcification.55 Intriguingly, methylation fingerprints appear to vary between normal human aortic and mitral valves: following bisulfite sequencing of over 1,600 promoters from across the genome, nearly 40% were significantly differentially methylated, including those of genes involved in endothelial-to-mesenchymal transition, TGF-beta signaling, fluid shear stress sensation, mechanical force generation, and ECM remodeling.56 Collectively, these findings imply epigenetic contributions to the regulation of valvular tissue homeostasis during both embryogenesis and disease development.
Gene Expression: Quantification of the Coding- and Non-Coding Transcriptome
Work to understand valvular global tissue states in health and disease in an unbiased manner began with and has until recently largely focused on transcriptomics, the study of the complete set of RNA transcripts under production. Early transcriptomics approaches to study CAVD relied on microarrays and relatively small sample sizes – the first large-scale measurements of gene expression in normal and stenotic valves utilized tissue-derived RNA microarrays of five non-diseased controls and five leaflets with AS, and identified 715 genes that were up- or down-regulated in the diseased state.57 Manual pathway analyses revealed functionality associated with lipid retention, inflammation, calcification, ECM remodeling, angiogenesis, and blood-derived cell adhesion/infiltration, with the top-most upregulated genes the macrophage-specific matrix metalloprotease MMP12, the ECM remodeler CHI3L1, which drives proteogycanogenesis, and the integrin-binding sialoprotein, which acts as a significant component of bone ECM. RNA microarrays have also been employed to study sex differences in porcine-derived VICs that could underlie the elevated occurrence of CAVD in men, and detected elevations in genes associated with proliferation, apoptosis, calcification, and inflammation.58 With advances in next-generation sequencing technology, bulk RNA-sequencing became widely available and more affordable, it revolutionized the study of the valvular transcriptome. Importantly, RNA-seq enables the identification of novel transcripts, has a substantially lower limit of detection and wider dynamic range, and demonstrates significantly higher specificity and sensitivity than do microarrays in the detection of differentially-expressed genes.59,60 By combining total RNA-sequencing of a pair of calcified human aortic valves with RNA-seq datasets available for 13 human tissues from the Human Body Map Project, Wang and colleagues constructed a quantified valvular transcriptome of 19,505 protein-coding genes and an additional 4,948 long noncoding RNAs (lncRNAs). They observed significant tissue specificity of the valvular transcriptome, and a substantial reduction of expression of numerous genes in the valve shown to be universally and constitutively expressed in other tissue types.61 Comprehensive transcriptomic characterizations have also been applied to define global trends in fetal development of the human aortic valve via RNA-seq, with tissue from between 14–22 weeks gestation demonstrating reductions in cell cycle and cellular metabolism, while pathways associated with cell-cell and cell-matrix adhesion, cell-cell communication, and adaptive immunity were upregulated.62 Bulk temporal expression analyses based on co-abundance63 revealed early fetal activation of resident VICs, while quiescence and apoptosis were elevated midway through the second trimester. Others have shown the presence of distinctive transcriptional profiles associated with proliferation and ECM biosynthesis in postnatal development via bulk valvular RNA-sequencing of newborn and 4 month old mice.64 Transcriptomics approaches have most recently been applied to identify causes of drug-induced valvular heart disease and disease-correcting drug targets in vitro. RNA-sequencing of valvular tissue from rabbits treated with pergolide, dexfenfluramine, or high-dose serotonin revealed a deficiency of cellular metabolism-associated processes and increased growth factor responses indicative of metabolic reprogramming,65 and microarrays have previously been used to dissect the impact of cyclic stretch and serotonin signaling on porcine valvulopathy.66 Using a groundbreaking combination of iPSC disease modeling, machine learning, and targeted RNA-sequencing, Theodoris and colleagues identified XCT790, a compound with predicted anti-osteogenic potential, as a putative corrector of gene network dysregulation of 119 signature genes induced by haploinsufficiency of NOTCH1 in iPSC-derived endothelial cells from a donor with CAVD.23 In effect, 1,595 small molecules were screened for their ability to revert the gene network produced by RNA-seq of these 119 signature genes back towards that of isogenic control cells. They went on to confirm by RNA-seq of cultured primary human VECs that XCT790 broadly corrected gene dysregulation in VECs from BAV donors back to that of VECs from TAV donors.
While coding RNA transcripts that eventually will be translated into protein represent a small fraction of the transcriptome, non-coding RNAs can regulate key steps of protein synthesis at different levels such as protein translation (rRNAs, tRNAs) or post-transcriptional gene expression (miRNAs, lncRNAs). To date, the valvular non-coding RNAome has largely been explored only by a miRNA microarray-based approach, including differentiation between human rheumatic and calcified aortic valve disease miRNAomes.67 Of note, a dual mRNA and miRNA microarray approach revealed chemokine-miRNA target pairs across highly-calcified and healthy human aortic valve leaflets, with disease initiating changes related to inflammatory processes and immune responses via miR-125b-mediated modulation of CCL3 and CCL4.68 This dual mRNA/miRNA array approach has also illuminated the impact of shear- and leaflet side-specific changes to the transcriptome of human VECs and intact porcine leaflets.69,70 Other work has pointed towards miR-26a and miR-195 downregulation in the aortic valves of those undergoing aortic valve replacement for AS or aortic insufficiency, and concomitant lifting of suppression of pro-calcifying genes, including BMP2, ALPL, and RUNX2 in human VIC cultures.71 Recent microarray-based work in human normal and CAVD leaflets expanded these sets of disease-dysregulated miRs to a total of 92, with 8,717 putative target genes involved in such disease-associated pathways as p53 signaling, ECM interactions, and Wnt and TGF-β signaling.72,73 miRNA microarrays of cultured human VICs have also been utilized to implicate miR-486 and miR-204 in regulation of valvular myofibrogenesis and osteogenesis.74 Given that these studies were largely based on microarray-based approaches, the application of lnc-RNA-seq or miRNA-seq to examine the valvular non-coding RNAome for novel or lowly-expressed transcripts remains an area of need. Importantly, unlike the lncRNA H19 (mentioned earlier), the majority of lncRNAs are not evolutionarily conserved between mice and humans, and so care must be taken when studying their effects in non-human models of disease.75
Several studies have also attempted to tease apart distinctions in the presumably differential pathogenesis of CAVD in aortic valves with tricuspid or bicuspid morphology. Using five diseased BAV and three diseased TAV donors, Padang et. al. identified 236 differentially-enriched genes by bulk total RNA-seq, and found heterogeneity within the diseased BAV transcriptome that correlated with mildly- or heavily-calcified BAVs. Transcriptomes of the latter BAV subset resembled those of calcified TAVs, and shared elevated inflammatory and reduced NOTCH1-related signaling.76 Guauque-Olarte and colleagues closely followed this report with RNA-seq data from 10 calcified human BAVs, nine calcified TAVs, and eight non-calcified TAVs from heart transplant recipients, all males.77 In contrast to the report by Padang, and despite doubling of sample sizes, this study identified only two genes (IGF1, RSPO2; both upregulated) with differential expression between calcified BAVs and TAVs, implying that, even if there were pre-existing differences in the tissue transcriptome, valvular morphotypes may converge to a common phenotype during the advanced stages of disease. Notably, inhibition of insulin-like growth factor 1 (IGF1) by dipeptidyl peptidase-4 drives human VIC osteogenesis and CAVD in rabbits,78 while RSPO2 encodes a regulator of pro-osteogenic Wnt/β-catenin signaling. This absence of substantial detected alterations in the BAV/TAV transcriptome was not due to any inherent power issues with the study itself, as 420 genes were differentially expressed between normal TAVs and diseased BAVs, along with 499 altered between normal and diseased TAVs – 84% of which were common to both comparisons, and were most-strongly associated with fibrosis-related functionality by pathway analysis, further strengthening the notion that ECM metabolism is tightly linked to CAVD. Importantly, these studies did not assess differences in early-stage disease, nor whether there were underlying alterations in the BAV/TAV transcriptome prior to disease initiation (it should be noted this is exceedingly difficult, as the low prevalence of BAV makes it extremely challenging to collect non diseased BAV tissues from humans). In the context of the non-coding RNA transcriptome, one early study employed a miRNA microarray approach on eight diseased TAV and eight BAV human donors to demonstrate differential expression of 35/1583 miRs. Among those 35 was miR-141, whose expression was reduced nearly 15-fold in BAVs, and which represses TGF-β-induced myofibrogenesis, BMP2/Runx2 signaling, and calcification in cultured porcine VICs.79 While this miR showed promise as a putative drug target, its specific gene target requires further study, as no high-confidence predicted targets could be identified. Microarrays have also been utilized to study circulating miRNA profiles in the plasma of patients with AS in an attempt to develop diagnostic and prognostic biomarkers for both BAV and its frequently co-existing aortic dilatation in 24 individuals with BAV or healthy TAV controls.80 This assessment of the plasma miRNAome identified 260 miRs, and found significant upregulation of miR-122 and miR-486 in those patients with BAVs, and a further two miRs (−130a and −718) were altered in those with dilated ascending aortas and BAVs. Functional analyses of these miRs and their predicted gene targets implicated altered control of TGF-β1 signaling via modulation of expression of the TGFβR1/2 receptor subunits and downstream SMAD2/4 effectors.
How can transcriptomics data be leveraged to identify causative drivers or risk/susceptibility alleles? In a broadly similar manner to GWAS, TWAS (transcriptome-wide association studies) search for correlations between gene expression and traits (reviewed in detail in81). Whereas GWAS examine correlations between SNPs and a particular phenotype, expression quantitative trait loci (eQTL) mapping tests whether a particular locus explains a fraction of the genetically-determined variance of a gene expression phenotype. TWAS leverage eQTL cohorts that contain gene expression and genotypes to identify associations between gene expression and traits. For example, SNPs in RUNX2 and the voltage-dependent calcium channel CACNA1C were found to be significantly correlated with elevated expression and valvular calcification using an eQTL of 22 human aortic valves.82 By leveraging an eQTL dataset comprised of 233 human aortic valves, TWAS have reinforced the significant association of PALMD with AS, and confirmed that risk alleles in PALMD result in reduced expression in the aortic valve, but not 44 other tissue types.34,35 The same eQTL, in concert with the GWAS that identified IL6 and ALPL, revealed NAV1 (neuron navigator 1) as a new candidate gene for CAVD, though the presumed mechanism of action is unknown.27 The NAV1 risk allele was associated with higher expression, but only in those with TAVs. While NAV1 is predominantly found in the nervous system, it is also expressed in the aorta, likely has a strong actin-binding potential, is also involved in blood pressure regulation, and associated with carotid artery stenosis. Besides eQTL mapping, other molecular quantitative trait loci such as methylation, chromatin accessibility, or protein QTL and their integration with GWAS data have gained increasing attention recently to dissect their association with cardiovascular diseases,83–85 opening an exciting avenue for future research to refine candidate drug target identification in CAVD. Finally, the availability of different layers of omics information (eg, TWAS and GWAS) allows for the application of sophisticated strategies to infer causality, including summary-data-based Mendelian randomization, a method increasingly used to define causative disease drivers and whose application is not limited to eQTLs.86
Rapidly-changing factors, including the affordability of next-generation sequencing, technical advances in sample collection and reproducible sequencing results despite low input quality, increases in computational power, and substantial improvements in computational analyses to reliably detect differential expression, have dramatically changed standard practices in the field of transcriptomics in recent years. Moving forward, the importance of expanding sample sizes and the careful, appropriate, standardized, and fully-disclosed selection of bioinformatics pipelines for calculation of differential expression is undeniably essential. State-of-the-art approaches to transcript normalization, modeling of variance, and correction for multiple comparisons have never been more crucial, and elegant open-source solutions are currently readily available (reviewed in87).
Evaluating Functional Biomolecules by Mass Spectrometric Proteomics
Though genetics and gene expression are clearly fundamental contributors to disease phenotypes, comparisons of transcriptomics and proteomics data have recently established the presence of a common and widespread discordance in transcription and translation in at least 29 human tissues (e.g. brain, colon, heart, kidney, liver, lung, spleen, smooth muscle) that may be caused by numerous regulatory mechanisms or differential degradation/turnover kinetics.88 This is particularly the case in the aortic valve, where, in comparison to other organs and tissues, relatively low cell densities and high ECM content creates the potential for a further disconnect between transcript and protein levels, and thus a clear motivation to study large-scale protein abundances.26 Standard measurements of protein quantification by Western blotting or ELISAs suffer from i) requirements for relatively high protein input, ii) limited throughput, and iii) low dynamic range. Proteomics has been performed using immunoassay arrays, Edman degradation, or protein microarrays, though current high-throughput studies rely heavily on mass spectrometry. Here, protein is extracted from cultured cells, cell culture supernatant, biofluids, or tissues, and then digested into short peptides using proteolytic enzymes. These heterogeneous mixtures are then separated by differential gel electrophoresis (DIGE), or liquid or gas chromatography, then ionized (typically by electrospray ionization) and injected into a mass spectrometer where they or their fragments are selected and analyzed based on their mass-to-charge ratio (m/z) or mass alone (in time-of-flight instruments). These resultant mass spectra are matched against databases of known proteins or protein sequences. After identification, quantification is performed either through stable isotope labeling, or label-free workflows. When performing unlabeled experiments, peptides are injected without further pre-processing, and relative differences in abundance of peptides (and the protein from which they originate) are determined directly from the intensity of their mass spectra. Label-based techniques involve the incubation of peptides with isotope-labeled tags (e.g., tandem mass tags [TMT], isobaric tags [iTRAQ]) that are sample-specific and allow pooling/multiplexing of 2–16 samples into a single injection. When these labeled peptides are fragmented in the mass spectrometer, their tags encode detectable information on the sample of origin, and tag intensities are proportional to the abundances of that peptide in the multiplexed samples. The label-free approach offers straightforward sample preparation, easy comparison of all samples against all others, and highly-flexible study design where new samples can be added at any time. Label-based techniques offer two- to 16-fold reductions in instrument time and reduced sample-to-sample variation, though they suffer from much more complex sample preparation, difficulty in comparing samples that were not multiplexed together, and a rather non-adaptable study design. The techniques mentioned so far aim to identify and quantify all proteins in a sample in an unbiased manner. In contrast, targeted proteomics can be leveraged to analyze a pre-specified subset of proteins of interest – consider novel proteins without commercially-available antibodies, or clinical settings where high sensitivity and reproducibility are necessary. Common workflows for targeted proteomics include selected or parallel reaction monitoring (SRM/PRM), where the initial intact peptide ion (precursor ion) is filtered based on a targeted m/z range, then fragmented (to yield product ions), which are carefully analyzed for sequence specificity to the protein of interest.
Early work to apply these mass spectrometric approaches to the study of CAVD began a decade ago, when a 2D-DIGE-based method was devised to minimize the interference of abundant calcium on protein extraction and mass spectrometry.89 This group went on to apply this approach to surgically-derived CAVD tissue from 20 human donors and 20 necropsy-sourced controls, and identified alterations in 43 proteins associated with fibrosis, coagulation, cholesterol transport, inflammation, and ECM composition.90 Subsequent refinements to this approach moved away from differential gel electrophoresis and towards liquid chromatography mass spectrometry (LC-MS), and utilized iTRAQ-based labeling for sample multiplexing and quantification of 712 proteins, followed by validation by SRM.91 Of those, 56 were significantly altered between surgically removed CAVD and necropsy controls, a considerable number of which were associated with proteoglycan-associated ECM (e.g., biglycan, periostin, prolargin). A pioneering study assessed levels of over 1,800 proteins from 9 human donors with CAVD via single-shot, global unlabeled and label-based tandem-mass-tagged proteomics, along with paired transcriptomics by RNA-seq (Figure 3).26 Stenotic aortic valves that had undergone SAVR were separated into spatial regions (fibrosa, spongiosa, ventricularis layers) as well as temporal stages of disease (non-diseased, fibrotic, calcified). This early protein atlas also made a number of key contributions to our understanding of CAVD pathobiology. Myofibrogenic processes and oxidative stress-related pathways marked the fibrotic stage, while significant enrichment of proteins involved in regulation of mineralization (e.g., fetuin A, TNAP) characterized regions of advanced calcification. Layer-specific TMT proteomics identified glial fibrillary acidic protein (GFAP) as a putative spongiosa-specific VIC marker almost exclusively expressed in the spongiosa, and revealed that contrary to the popularly-held notion that fibrosis precedes and drives calcification, the ventricularis was most-enriched in markers of myofibrogenesis, while the disease-prone fibrosa held abundant levels of proteoglycans and apolipoproteins, implicating osteochondrogenic processes and lipid-mediated mechanisms in the initiation of CAVD. Recent follow-on work employed targeted proteomics using PRM on those same 9 donors to monitor 12 apolipoproteins across stages of disease progression aimed at identifying the lipid promoters of CAVD.13 Eight apolipoproteins were enriched in calcified valvular tissue, while apo(a), apoB, apoC-III, apoE and apoJ were co-localized with calcific regions and the disease-prone fibrosa layer. In vitro validation studies in cultured VICs confirmed that apoC-III promotes calcification via induction of mitochondrial dysfunction. While circulating apoC-III was known to bind Lp(a), and its serum levels are elevated in patients with ASc,92 this work led to the discovery of a novel mechanistic link between apoC-III and CAVD. Although bulk tissue-based mass spectrometry has enabled substantial insights into mechanisms of valvular homeostasis and disease, we have noted earlier that the normal structure of the valve leaflet is complex, anisotropic, and heterogeneous, and CAVD susceptibility varies widely between specific regions (fibrosa layer, leaflet base). Bulk proteomics cannot capture this heterogeneity without difficult, expensive, and time-consuming microdissection or laser-capture isolation techniques.26 One solution to this is matrix-assisted laser desorption ionization imaging mass spectrometry, or MALDI-IMS. In MALDI, tissue sections are placed on an arrayed crystalline matrix and laser-irradiated in a spatially-controlled, step-wise manner. This vaporization of tissue and matrix results in ionization of the sample, which is then assessed by mass spectrometry. A spatial proteome is deconvolved and reconstructed in silico for every ablation point to a resolution of ~50 μm. When applied to stenotic aortic valve sections, MALDI has revealed strong enrichment and segregation of collagen VI and NDRG-2 to areas around calcific nodules in three human donors.93 These proteins are known to be responsible for accumulation of calcium and hydroxyproline on human osteoblasts and in the progression of p53-mediated apoptosis, thereby implicating these processes in the advanced stages of CAVD.
Figure 3: Case study: a spatiotemporal multi-omics atlas of the aortic valve.
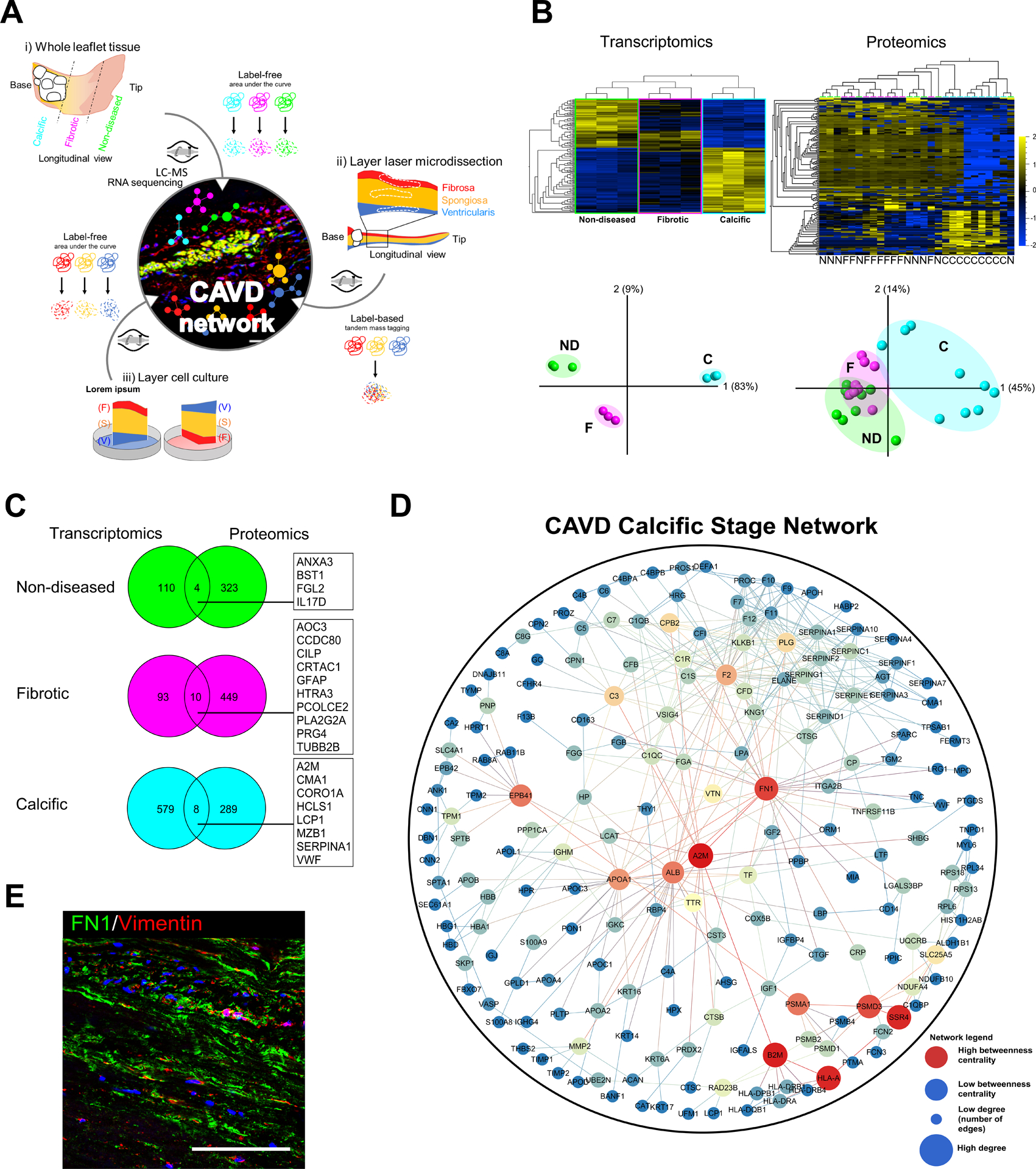
(A) Schlotter et al. employed proteomics and transcriptomics studies to investigate layer-specific pathobiology and temporal pathogenesis of human calcific aortic valve disease (CAVD). (B) Transcriptomics and proteomics leveraged to assess differences in molecular phenotypes across stages of disease (ND: non-diseased, F: fibrotic, C: calcified) demonstrate pathobiological differential enrichment and stage-specific clustering by principle component analyses (PCA). (C) Multi-omics integration in the aortic valve must be approached with care and sophistication, as the low cell density (and resultant abundance of proteinaceous extracellular matrix) and other regulatory or pathological mechanisms can lead to low correlation between tissue-based transcript and protein levels. (D) Network medicine approaches can be subsequently employed for target prioritization: protein-protein interaction (PPI) networks build from stage-specific proteomics of human aortic valves revealed key “hub node” proteins (red) implicated in valvular calcification. (E) Immunofluorescence staining in calcified regions of human aortic valves to validate the network-derived target FN1 (in green, nuclei in blue, VIC marker vimentin in red); scale bar = 100 μm. Adapted from26.
Proteomics have also been applied with some regularity to the study of cultured valve-derived primary cells. Early work on bovine-derived VICs detected 34 cytosolic and 10 membrane-associated proteins that were altered by pro-inflammatory culture conditions (lipopolysaccharide (LPS) stimulation), and which are associated with the notion that induction of oxidative stress underlies inflammation-driven VIC calcification.94 Side-specific proteomics of primary human VIC cultures from the fibrosa and ventricularis were also included in the above CAVD atlas by Schlotter et. al.,26 and revealed an absence of known plasma-derived drivers of calcification (e.g., Lp(a), apoC-III, etc.) in response to osteogenic media-induced calcification – implying that simple VIC culture models do not mimic the entirety of pro-calcifying mechanisms at play in vivo. Through global unlabeled proteomics on whole-cell, nuclear, and cytoplasmic fractions, the innate passage-dependency of cultured VIC calcification potential was revealed to be associated with passage-dependent reductions of TNAP abundance and activity under culture conditions containing organic phosphate sources.95 Importantly, the provision of inorganic phosphate in culture media abrogated passage-dependent calcification potential, along with alterations in TNAP abundance/activity, highlighting the importance of VIC culture condition standardization and careful selection of the relevant culture model (e.g., altered phosphate metabolism present in patients with chronic kidney disease).
As we note in Figure 2, samples of interest to the study of CAVD are not limited to only bulk tissue, plasma, or cultured cells: the secretome (comprised of biomolecules expressed into the extracellular space) is a particularly inviting source of biomarkers and potential drug targets. Early attempts to assay the valvular secretome involved the five-day culture of intact stenotic aortic valve leaflets, followed by collection of cell culture supernatant and mass spectrometry.96 Newly-secreted proteins were quantified by the metabolic incorporation of labeled amino acids found in the culture media in order to rule out those that originated from the blood. Significant enrichment of ECM-associated functionality was found in the 61 labeled secreted proteins that were detected, including several glycoproteins (e.g., PEDF and clusterin) implicated in regulation of oxidative stress and regulation of inflammation. In an opposite manner to its known reduction in atherosclerosis, the actin-binding protein Gelsolin was elevated in both the secretome of stenotic leaflets and plasma samples of an independent validation cohort and holds promise as a differentiating biomarker between vascular and valvular calcification. Certain subsets of the secretome appear to be particularly relevant to the initiation and progression of cardiovascular calcification: extracellular vesicles (EVs) are membrane-bound particles (~50–500 nm in diameter) that are actively released by the body’s cells, and which are loaded with bioactive non-coding RNA, protein, lipid, and metabolite cargoes.97,98 These EVs form the physical building blocks of cardiovascular calcification99, and proteomics offers a unique means to identify and quantify protein cargoes from these tiny and low-abundance structures. Label-free quantitative proteomics of EVs derived from cultured VICs and vascular SMCs identified the shared presence of the calcium binding protein annexin A1 (ANXA1), which was then determined to modulate EV-EV tethering, aggregation, and the formation of microcalcifications in atherosclerotic plaques and stenotic aortic valves. What role do EV cargoes play in driving disease progression? Ongoing studies have leveraged bacterial-derived collagenase to digest fibrocalcific atherosclerotic plaques and aortic valves, then applied mass spectrometry to develop a method for extracting tissue-entrapped EVs directly from these human tissue samples.100 Integrated proteomics and miRNA-sequencing of the resultant EV vesiculome revealed unique roles for valve-derived EVs in modulating p53-mediated transcriptional regulation, a known driver of valvular calcification.101,102 Attractively, the approaches developed therein to remove bacterial collagenase components after mass spectrometry of mixed samples also represented the first application of metaproteomics (the study of microbial proteins by mass spectrometry) to cardiovascular disease, and acts as proof-of-principle for novel methods to study potential infections contributors to CAVD (e.g., endocarditis103).
One rapidly growing area of focus in proteomics is the study of protein post-translational modifications (PTMs), where protein structures are covalently (and typically enzymatically) modified after ribosomal synthesis in a manner that can alter protein function, localization, or degradation. Examples of common PTMs include phosphorylation, ubiquitination, SUMOylation, palmitoylation, and ribosylation; proteomics has become a technique of choice for the detection and quantification of global or targeted PTM levels (reviewed in104,105). However, these proteomics techniques remain largely unexplored in the context of CAVD and represent an area that is ripe for further investigation. In one example, Angel and colleagues applied MALDI to survey valvular ECM for N-glycosylation PTMs.106 They found that in normal pediatric human valves, the N-glycome was spatially organized by anatomical valve layer, while N-glycanated proteins were localized to hypertrophic commissures and sialylated N-glycans were elevated in pediatric congenital AS. Others have recently utilized LC-MS to identify nitric oxide-mediated S-nitrosylation of 217 proteins in cultured VICs, and subsequently demonstrated that S-nitrosylation of the deubiquitinase USP9X modulates NOTCH signaling in VICs, prevents murine valvular calcification, and is reduced in human valves with CAVD.29 The continued expansion of CAVD proteomics studies to include PTMs promises to reveal additional novel pathobiology, regulatory cascades, and putative drug targets.
Metabolomics: Closing in on Measurement of Molecular Phenotype
The penultimate link in the genome-to-phenome chain is the metabolome, which consists of small molecules (including specific lipid species, as in lipidomics) that originate from the body’s metabolism. These molecules can have endogenous (e.g. sugars, fatty acids, organic acids, amino acids, nucleic acids, vitamins, pigments) or exogenous (e.g. pharmaceuticals, environmental contaminants, food additives, toxins, xenobiotics) origins, and, as afore-mentioned, represent the molecular species that are most closely tied to the biochemistry and phenotype of an organism.107 Typically, metabolomics studies leverage one of two technological platforms: 1) nuclear magnetic resonance (NMR), or 2) mass spectrometry (MS). In NMR, samples are exposed to a magnetic field that, simplistically, induces resonance of isotopes (e.g., 1H, 13C, 14N) at frequencies which depend on the location of those isotopes within the molecular structure. Peaks in the ensuing frequency spectrum produce an identifiable and unique profile for every compound. NMR offers the advantage of being directly quantifiable, non-destructive, and highly reproducible (reviewed in108), and has therefore often been used for large-scale clinical studies, such as the UK Biobank and Million Veteran Program. In contrast, mass spectrometry offers a limit-of-detection that is typically 10–100x lower than NMR, and is applied to metabolomics in largely the same way as it is for proteomics: metabolites are extracted from tissues, biofluids, cells, or cell culture supernatants, then purified and injected by gas or liquid-based chromatography. Their molecular mass-to-charge ratios and fragmentation patterns are then collected and compared against databases of known and predicted molecules.109 Despite its widespread use in the field of cardiovascular biomarker discovery and a number of investigations into the plasma metabolome associated with coronary artery disease110 or carotid artery calcification111 or the tissue metabolome of varied arterial disease,112 there has been relatively limited work at the nexus of CAVD and metabolomics. In fact, many efforts to date have focused on identifying diagnostic or prognostic biomarkers to assess efficacy and outcomes of aortic valve replacement.
Over the last five years, several studies have probed the question of diagnosing the presence of a BAV or of CAVD through plasma- or urine-based biomarkers. At the moment, such diagnoses are completed almost uniformly via ultrasound imaging, which is relatively inexpensive and widely available in the developed world, but sorely lacking everywhere else. Simple blood tests for these conditions could revolutionize screening initiatives and the provision of care for huge swaths of the developing world. Early work employing LC-MS and serum samples from 50 donors identified 41 metabolites that were significantly differentially enriched between those with normal TAVs and diseased BAVs.113 Metabolite pathway analyses were utilized to annotate this altered metabolome with functionality related to purine metabolism, fatty acid biosynthesis, redox imbalance, and deficient energy production. Using levels of monoglyceride (18:2) and glycerophospho-n-oleoyl ethanolamine, a dual-metabolite model predicted diseased BAVs with a sensitivity of 76.7%, specificity of 90%, and AUC of 0.9. One important caveat to this approach is the notion that it differentiated between normal aortic valves with tricuspid morphology, and diseased aortic valves with bicuspid morphology – this model did not predict bicuspid morphology in isolation. Later, Martinez-Micaelo and colleagues assessed the plasma metabolome in 212 participants with aortic dilatation or stenosis, and determined that those with BAVs displayed reduced levels of α-tocopherol, paraoxonase 1, and high-density lipoprotein, and elevated c-reactive protein.114 Together, these molecules implicated increased levels of inflammation and endothelial dysfunction along with reduced antioxidant capacity, and the authors developed multivariable biomarker models with sensitivities of 0.91–0.93, specificities of 0.86–1.00, and AUCs of 0.92–0.99 for the prediction of BAV. Pre- vs. post-TAVR metabolomics in 57 TAV and BAV recipients has also shed light on both native differences in TAV- vs. BAV-CAVD, but also on the differential responses of these valvular morphotypes to valve replacement.115 Both before and after surgical valve replacement, BAV patients showed reductions in arginine and proline metabolism (particularly l-glutamine, l-proline, hydroxyproline, pyrrole-2-carboxylic acid, n2-succinyl-l-ornithine and spermine) vs. those with TAVs, thereby implicating impaired nitric oxide generation (via endothelial nitric oxide synthase) and elevated oxidative stress and inflammation. In addition, subgroup enrichment analysis of those patients with BAVs whose left ventricular mass and transvalvular velocity recovered well or poorly after TAVR found that elevated arachidonic acid metabolism (5-KETE, 15(S)-HETE, arachidonic acid, prostaglandin G2, thromboxane B2, leukotriene A4 and leukotriene B4) was present in patients with poor recovery responses. Interestingly, arachidonic acid metabolism is centered around the COX-2 enzyme, whose role in valvular calcification remains uncertain: COX-2 expression is elevated in calcified human aortic valves and its inhibition prevents valvular calcification in some mouse models,116 though the nonsteroidal anti-inflammatory drug celecoxib is a COX-2 inhibitor, and was recently shown to be associated with dystrophic VIC calcification and AS in humans.117
Plasma-based LC- and GC-mass spectrometry have also been employed to assess short-term changes in the circulating metabolome pre-to-post-TAVR, with the long-term goal of identifying prognostic biomarkers of surgical outcomes.118 Rapid changes in the metabolism of purines, bile acid biosynthesis, and glycerolipid metabolism were detected, while sphingolipid metabolism, characteristic of heart failure and ventricular remodeling, was dominantly enriched post-TAVR. Several hubs associated with arginine and proline metabolism were identified that correlated with left ventricular regression (resolution of cardiac hypertrophy) after one-year follow-up that may hold promise as biomarkers for prediction of surgical valve replacement efficacy. Again, such arginine and proline metabolites may be functionally associated with nitric oxide synthesis, reduced vascular resistance, and improved endothelium-dependent vasorelaxation. Other work in a total of 50 donors with either severe AS (pre-/post-TAVR) or healthy controls detected 50 plasma metabolites that were significantly altered between those with AS and controls, while 19 were altered by the TAVR procedure. Reductions in long-chain phosphatidylcholines/sphingomyelins (membrane components involved in cell signaling), and elevations of acylcarnitines (inflammation, impaired fatty acid oxidation, and deficient mitochondrial uptake), biogenic amines, and amino acids (associated with a hypercatabolic state and heart failure) were present in those with AS. In contrast, valve replacement was associated with reversal and normalization of lysophosphatidylcholines, which drive pro-ostegenic gene expression in human VICs and are generated by phospholipase A2 – an enzyme known to be elevated in patients with AS.119 Notably, autotaxin, an enzyme that is enriched in Lp(a) and has been shown to promote VIC-mediated CAVD, produces pro-osteogenic lysoPA from lysophosphatidylcholines.15,54
Most recently, metabolomics has begun to be leveraged for the study of disease pathogenesis and discovery of drug targets. In an attempt to characterize CAVD pathobiology and risk of progression, concurrent urine and plasma metabolomics in 92 human donors found 14 urine metabolites and 15 plasma metabolites that were significantly different between those with AS and healthy controls.120 Of these, only trans-aconitic acid and 2,4-di-tert-butylphenol were significantly altered in the same direction in both biofluids, the remainder of significant metabolites were regulated in contrasting directions in urine and plasma (with most elevated in urine and downregulated in plasma, indicative of excretion).121,122,123,124,103,125In a related finding, targeted mass spectrometric quantification of dietary or gut microbiota-derived metabolite levels in the plasma of patients with moderate-severe AS, ASc, or normal controls revealed elevations of plasma choline, but not trimethylamine-n-oxide or betaine that correlated with both AS, valvular calcification, transvalvular velocity, and a number of histological measures of CAVD pathogenesis.126 Surendran et al.28 employed untargeted global metabolomics of valve tissue-derived samples to identify a total of 72 metabolites and lipids that were differentially enriched along the progression from mild to moderate to severe CAVD in 96 human donors, and describe associations with glycerophospholipid metabolism, linoleic acid metabolism, and bile acid biosynthesis therein. Notably, despite the earlier studies described above that identified blood-based metabolites associated with presence of BAV, they reported that the tissue metabolome did not differ between stenotic TAV vs. BAV. However, lysoPA was highly correlated to measurements of impaired valve function regardless of valve morphology. Subsequent targeted lipidomics (a subset of metabolomics; the large-scale study of lipids) against seven lysoPA species on valve leaflets from that cohort confirmed these findings, and revealed that lysoPA levels correlated strongly with rapid worsening of aortic jet velocity, but were independent of other traditional risk factors (e.g., age, sex, lipid profile, hypertension, BAV). Plasma proteomics in a small subset of donors revealed significant correlations between tissue and plasma levels of two lysoPA species, and offers an interesting opportunity for novel biomarker or drug development.15,54 MALDI-based mass spectrometry has also been leveraged to perform untargeted valvular lipidomics in healthy pediatric and adult ovine aortic valve leaflets, as well as human aortic valve tissue from pediatric patients with congenital AS and healthy controls.127 Importantly, the spatial aspect of MALDI enabled the authors to distinguish layer-specific patterns in the valvular lipidome. This work also found a distinct association between ECM components, ECM organization, and lipid profiles, along with altered fatty acid content in human pediatric AS and acted as an important precursor to future proteomics-based work by others that identified a driving role for ECM-bound ApoC-III in layer-specific VIC calcification.13
Moving forward, a further focus on tissue-based metabolomics is primed to identify those biomolecules that most closely follow or drive valvular phenotypes, and which may therefore be among the most-promising targets for therapeutic intervention. The expansion of this technique into mouse studies, in vitro cell culture, and perhaps even down to the level of extracellular vesicle cargoes (reviewed in128) will likely be potentiated as technical advances in instrumentation and sample processing require lower amounts of input. This field is rapidly improving the accuracy of mass spectrometric annotation and feature filtration129, and establishing guidelines that prevent, for example, otherwise-easy and problematic contamination by exogenous metabolites, rapid degradation of endogenous metabolites, or the temperature-induced activation of platelets and resultant contamination of the serum/plasma metabolome.130,131 This is of particular relevance to plasma- or serum-based metabolomic studies of CAVD, as activated platelets have recently been demonstrated to play key roles in the progression of this disease via generation of pro-osteogenic lysoPA.15 As is evident above, studies of the CAVD metabolome have to date been applied to only relatively small cohorts of patients and only in isolation – the inclusion of metabolomics in large, multi-omics studies will be key to definitively link phenotype with causative molecular drivers of disease. Care must also be taken when performing metabolomics in preclinical animal models of disease, as similar to the case of lncRNAs, a number of key species-specific differences exist that can alter metabolic drivers of CAVD. Such differences include phosphate-mediated mineralization, where CD73-deficient mice do not mimic the phenotype of cardiovascular calcification found in humans with CD73 deficiency as a result of alterations to murine adenosine catabolism (reviewed in132). In the context of lipid metabolism, Lp(a) is a major carrier of oxidized phospholipids, but apo(a) is notably absent in mice or rabbits and so these animals do not naturally form Lp(a). Humanized animals (engrafted with human cells, tissues, organs, or transgenic material) are therefore essential tools to decipher this important aspect of lipid-driven CAVD pathogenesis and other human-specific modes of pathogenesis (reviewed in133). In short, caution should be exercised to account for species-specificity in all stages of multi-omics studies, including during screening and validation. Computational methods to increase translatability of omics datasets between preclinical models and humans are under active development that leverage orthology, machine learning, and signaling networks (reviewed in134) to develop tools for cross-species omics translation.135–137 Fundamental biological understanding must be paired with technical omics expertise in order to make translatable discoveries.
Handling Heterogeneity: Omics Approaches at Single-Cell Resolution
The complex nature of the aortic valve and its surrounding microenvironment have long complicated the field’s search for pathobiological understanding. Outstanding questions around whether certain cellular subsets play active roles in disease prevention/progression or are simple onlookers, the tissue-resident or circulating origins of disease-driving cell populations, the putative differentiation of resident VECs or multipotent VICs, the degree and type of infiltration of inflammatory cells, and their relative contributions and mechanism of action to the onset and progression of CAVD are poorly understood and remain difficult to study quantitatively. Single-cell RNA-sequencing (scRNA-seq) represents a transformative capability in this area (reviewed in138). Here, single-cell droplet suspensions, typically through microfluidic processes, are created and individually barcoded. Subsequently, library production and sequencing are performed much in the way as bulk RNA-sequencing, with deconvolution of the resultant transcriptome down to that of individual cells by leveraging the cell-specific barcoding. The ability to work at single-cell resolution offers a means to identify small (low-incidence) but highly relevant cell populations that may be masked by more abundant cells or ECM in bulk omics, enables the identification of novel differentiating markers that could assist targeted drug delivery, and provides the opportunity to discover novel cell types or cellular transitions during disease initiation and progression.139 Through the usage of inferred pseudotime analyses,140 cells can be computationally ordered along a spectrum of transcriptome similarity – this structure could serve as a suitable proxy for cellular lineage tracing. How do we visualize, analyze, and interpret the measurement of thousands of genes/transcripts/metabolites in hundreds or thousands of donors (in bulk multi-omics) or cells (in single-cell approaches)? Dimensionality reduction is a key aspect of cluster identification, whereby high-dimensional data is transformed into lower-dimensional data that retains important aspects of the original dataset. Principle component analysis (PCA) has become a popular means of dimensionality reduction, and relies on identification of those linear combinations of original variables that best preserve variance in the data when moving from high- to low-dimensional space. This approach is rapid, but assumes linearity and orthogonality, and can be biased towards high-variance variables. Recently, non-linear means of dimensionality reduction have been developed (t-distributed stochastic neighborhood embedding, t-SNE; uniform manifold approximation and projection, UMAP) which instead attempt to identify a lower-dimensional projection that optimizes for similarity with the original dataset (reviewed in141). Both t-SNE and UMAP dramatically outperform PCA, with UMAP demonstrating lower computational burden than t-SNE, as well as the highest reproducibility and most subtle/biologically-relevant clustering of single-cell data vs. both PCA and multiple forms of t-SNE.142 Overall, single-cell omics represent a powerful, new, and rapidly-progressing tool whose promise is only beginning to be attained.
To date, we are aware of only five published studies that have applied single-cell techniques to the study of heart valves. Initial work in normal C57BL/6J mice lead the charge to down to single-cell resolution, where pooled cells from collagen-dissociated mitral and aortic valves at both 7 days (n = 13 mice) and 30 days (n = 10 mice) post-natal.143 A total of 2,840 cells were sequenced by Drop-seq, a droplet and bead-based barcoding method. t-SNE dimensional reduction identified the presence of four primary cell types at P7 and P30: immune, melanocyte, endothelial, and interstitial. Of note, the interstitial cell type contained substantially-increased heterogeneity: six distinct sub-clusters of VICs were identified at a clustering resolution that detected only one type each of immune, melanocyte, and endothelial cells. While the transcriptomes of non-VIC cell types were largely static between P7 and P30, VICs matured from a pair of collagen- and proteoglycan-producing populations into four further-differentiated cell types with gene signatures reminiscent of matrifibrocytes (involved in responses to cardiac injury), fibroblastic VICs (collagen and ECM organization), complement-associated VICs (expression of genes associated with the defense response), and antigen-presenting VICs (associated with non-professional adaptive immune responses). Higher-resolution clustering then revealed three spatially-distinct endothelial sub-populations that were located in proximity to i) lymph-like VECs on the fibrosa surface, ii) VECs associated with expression of growth factors and ECM genes, and iii) migratory-like VECs marked by Wnt-β catenin signaling and located in areas where leaflet coaptation occurs. In a similar manner, five sub-populations of immune cells were discovered, including mast cells, dendritic cells, T cells, and two macrophage sub-sets. During post-natal maturation, mast and T-cell populations were reduced, dendritic cell numbers remained relatively unchanged, and macrophage populations shifted dramatically towards a chemokine-expressing subset. While these studies did not directly address the pathogenesis of CAVD, the notion that developmental processes are aberrantly re-activated during disease pathogenesis (reviewed in144) lends added disease relevance to this study, as cellular sub-types involved in early leaflet maturation may also play key roles in protecting against or initiating valvular inflammation, fibrosis, and calcification. Pre-processing to perform cell-type-specific enrichment or depletion is also commonly used. In a broader study of fibroblast heterogeneity in four organs,145 enzymatic digestion of 24 C57Bl6 murine hearts from 10–20-week-old mice, enrichment of fibroblasts via sorting for PDGFRα+ or PDGFRβ+ and CD31− cells followed by plate-/Smart Seq2-based single-cell sequencing of a total of 1,279 heart-derived cells. UMAP dimensional reduction identified eight clusters, of which the smallest was tentatively identified as skeletal muscle perimysial cell-like VICs positive for WIF1, COMP, and CYP2E1 and which expressed a number of ECM-modulating genes.
In humans, the first characterization of valvular single-cell transcriptomes occurred after enzymatic digestion and Drop-seq on the hearts of 18 human embryos between 5–25 week gestation. Of the 4,948 cells captured, 427 were identified as valvular-derived cells with elevated SOX9 expression, and a cluster-specific transcriptome rich in gene ontology terms associated with the regulation of cell death and apoptosis, two key aspects of leaflet maturation.146 Sub-clustering revealed distinct early VIC- and VEC-like populations, as well as separation between cells of the atrioventricular (mitral and tricuspid) and semilunar (aortic and pulmonary) valves. In the first study of postnatal tissue-derived human aortic valves, Xu et. al. used six human donors (two non-diseased TAVs, four diseased TAVs) and the 10X Chromium v2 chemistry, they sequenced 34,632 cells after mechanical and enzymatic tissue dissociation.30 They describe three low-incidence VIC sub-types, two VEC populations, and small populations of monocytes, macrophages, and lymphocytes (one cluster each). The vast majority of sequenced cells were ascribed to six “valve-derive stromal cell” types. Non-diseased specimens were composed primarily of the 3 VIC subsets, which appeared to each be present in every layer of the non-diseased human valve. Incidence of valve-derived stromal cells was noticeably elevated in specimens with CAVD, while the VIC populations present in non-diseased were reduced. In addition, the VEC population present primarily in healthy controls shrank dramatically, and a second VEC sub-type appeared in diseased samples. This is presumably a sign of VIC and VEC differentiation during disease progression, as pseudotime analysis in Monocle suggested that the three VIC subsets differentiated into stromal cell subtypes. In concert, these pseudotime analyses revealed that the CAVD-associated VEC sub-type appeared to act as a transition state between normal VECs and VICs, indicated the potential onset of pathogenic EMT.147 Roles and functions of the non-VIC/VEC/stromal cell populations remain to be determined. Of note, this study reported substantial difficulty during sample processing, as 16 specimens were prepared with only 6/16 producing usable datasets (37.5%). Failures were ascribed to the presence of substantial mineralized debris along with digested ECM and other contaminating material that interacted poorly with the 10X microfluidic-based system. It should be remembered that the application of single-cell transcriptomics to solid tissues requires the release of individual cells, which was accomplished in the above studies by enzymatic dissociation – typically at 37°C for up to several hours after tissue is removed from the human or animal. Recently, comprehensive assessments of systematic bias in solid-tissue scRNA-seq have determined that warm enzymatic dissociation leads to the presence of heterogenous artifactual changes to the transcriptome in response to the dissociation itself and other environment stresses, including cell death processes (as transcriptional machinery is active during enzymatic incubation), with a particular impact on immune and endothelial cell types.148 These techniques can also be prone to loss of large cells (e.g. macrophage sub-types) due to the physical dimensions of the 10X chips, and inflation of immune cell fractions. Other concerns revolve around incomplete or biased extraction of cells from heterogeneous tissues (e.g., samples with a mixture of non-diseased, fibrotic, or calcified tissues).
Most recently, scRNA-seq techniques have been expanded to cultured VICs and other species besides mice and humans: single-cell RNA-seq (10X Chromium) of 7,037 cultured porcine VICs29 showed distinct clustering with/without treatment with a nitric oxide donor, revealing a uniform yet transient de-activation of VICs and suppression of the myofibroblast markers ACTA2, TAGLN, VIM, and CNN1 – all indicative of impaired VIC myofibrogenesis in response to nitric oxide. This de-activation was mirrored by treatment with a compound that induced the formation of s-nitrosylation post-translational modifications. In an elegant application of value-added multi-omics, this group went on to demonstrate the dramatic enrichment of proteins carrying s-nitrosylation post-translational modifications after treatment of rat VICs with nitric oxide donor, and identified the deubiquitinase USP9X as a s-nitrosylated activator of NOTCH1 signaling whose inhibition promoted calcification of cultured VICs and whose endothelial-specific deletion induced AS in mice.
As this rapidly-changing field matures, a number of new technologies are permitting an improved assessment of cellular phenotypes.138 Single-nuclei RNA-sequencing (sNuc-seq) may avoid a number of the artifactual pitfalls present with tissue-based scRNA-seq studies, and has been successfully utilized to map cellular and transcriptional diversity in the human heart.149 While powerful, single-cell sequencing data is often of much lower quality than that of bulk RNA-seq. scRNA-seq, sNuc-seq. and other sequencing-based barcoding approaches are affected by the “dropout” phenomenon, where genes may be detected in one cell but not another of the same type – this is typically due to technical limitations associated with inefficient mRNA capture or reverse transcription and stochastic mRNA expression. Existing single-cell computational methods, including those highlighted in Table 2, attempt to address this issue through a pre-processing combination of normalization, imputation, and dimensionality reduction to account for the high sparsity and zero-inflated nature of single-cell sequencing data. Recent bioinformatic efforts have attempted to leverage dropouts as a useful complementary tool for cell clustering based on the co-occurrence of gene dropout within cell types,150 and may re-purpose this otherwise-problematic event as a means to refine cellular clustering algorithms. Spatial transcriptomics employs spatial rather than cellular barcoding in order to perform array-based transcriptomics on tissue sections with encoded coordinate data for each transcript,151 and its potential use in concert with MALDI offers a tantalizing opportunity for spatial multi-omics of regions around calcific nodules or between valvular layers or disease stages. Mass-spectrometric single-cell proteomics has been developed thus far via mass cytometry (CyTOF). CyTOF is essentially mass spectrometric flow cytometry, where cells incubated with heavy-metal-conjugated antibodies are nebulized and antibodies are detected by a time-of-flight mass spectrometer instead of by fluorescence.152 Significant improvements in spectral overlap allow quantification of protein abundance across up to 135 channels on current instruments, vs. the 20 or so color channels that the most advanced fluorescence-based flow cytometry can achieve. Other avenues towards single-cell proteomics leverage isobaric labeling and a spiked-in carrier proteome to enable peptide identifications from low-input samples.153 Novel chemistries and informatics tools have enabled the dual assessment of gene expression and chromatin accessibility via combined scRNA-seq and scATAC-seq,154 while the advent of CITE-seq allowed concurrent measurement of protein (via barcoded antibody-oligonucleotide conjugates) and transcripts (via prototypical scRNA-seq workflows). CITE-seq has been successfully combined with CyTOF and scRNA-seq to identify distinct T cell and macrophage subsets that contribute to symptomatic disease in human atherosclerotic plaques.155 On the other hand, single-cell metabolomics remains a major technical challenge (reviewed in156), though advances in instrument sensitivity and throughput show promise. In the future, the changing economics of these techniques may enable the routine and widespread application of these novel single-cell approaches to a host of ex vivo and in vitro samples.
Table 2:
Selected Bioinformatics/Systems Biology Resources for Multi-Omics Studies.
| Tool | Description | Ref. |
|---|---|---|
| Search | ||
| Eagle2 | Open-source, imputation/haplotype phasing | 178 |
| Bowtie2 | Open-source; sequence alignment to reference genome | 179 |
| SAMtools | Open-source; SNP/variant calling | 180 |
| STAR/HTSeq | Open-source; RNA-seq genome annotation/alignment, read mapping, counting | 181,182 |
| Kallisto/Sailfish/Salmon | Open-source; transcriptome pseudocounts; sample-specific bias models | 183–185 |
| seqcluster | Open-source; miRNA alignment and annotation | 186 |
| Proteome Discoverer | Commercial; protein/peptide sequence ID; label-free, MS1-/isobaric labeling, PTMs | 187 |
| MaxQuant | Free; quantitative shotgun proteomics; label-free, MS1-/isobaric labeling, PTMs | 188 |
| Skyline | Open source; targeted proteomics/metabolomics; selected/multiple/parallel reaction monitoring | 189 |
| Compound Discoverer | Commercial; small-molecule structural identification; visualization; stable isotope labeling (untargeted); metabolite pathway analysis | 190 |
| XCMS Online | Free; untargeted metabolomics; retention time matching; compound ID; multiple reaction monitoring; differential statistics; visualization; integration; metabolite pathway analysis | 191 |
| Cell Ranger | Free; single-cell RNA-seq output processing; read alignment, generation of feature-barcode matrices, clustering, gene expression analysis | 192 |
| Statistical Analysis/Differential Expression | ||
| PLINK | Open source; whole-genome association analyses; linkage disequilibrium; stratification | 193 |
| SNPTest | Free; single SNP association in genome-wide studies; binary and multiple phenotypes; Bayesian and Frequentist tests | 194 |
| FastQTL | Open-source; cis-QTL mapping; beta approximation-based permutation scheme; internal quantile normalization of phenotypes | 195 |
| QTLtools | Open-source; molecular QTL discovery/analysis; sequencing data QC; cis/trans QTL mapping; functional annotation density; QTL enrichment; covariate correction | 196 |
| DESeq2 | Open-source; transcript quantification/differential expression; normalization (median of ratios) | 197 |
| EdgeR | Open-source; transcript quantification/differential expression; normalization (mean of Ms) | 198 |
| Perseus | Free; protein quantification, interaction, PTMs; high-dimensional omics; normalization, time-series, multiple-hypothesis tests; patient classification | 199 |
| MSstats | Open-source; label-free and label-based proteomics; statistical relative quantification; visualization; differential protein abundance | 200 |
| MetaboAnalyst | Free; high-throughput metabolomics; differential metabolite abundance; classification; visualization, compound ID; ontology | 201 |
| Qlucore Omics Explorer | Commercial, multi-omics (RNA-seq, DNA-seq, DNA methylation, single-cell, proteomics, metabolomics, flow); visualization; differential enrichment; clustering | 202 |
| Seurat | Open-source; single-cell; QC/normalization; clustering; dimensionality reduction; differential expression, highly-variable genes; integration; visualization | 203 |
| Monocle | Open-source; single-cell; clustering; dimensionality reduction; differential expression, visualization; time-series/pseudo-time | 140 |
| Loupe Browser | Free; single-cell; filtering, clustering; differential expression; integrated V(D)J and gene expression analysis; visualization | 204 |
| Multi-Omics Integration | ||
| MOFA | Open-source; heterogenous/sparse datasets; bulk/single-cell; latent factors of variation | 174 |
| PaintOmics | Free; transcriptomics/proteomics/metabolomics/ATAC-seq/ChIP-seq; KEGG pathway-based integration; visualization; pathway analysis | 205 |
| Escher | Open-source; genomics/transcriptomics/proteomics/metabolomics; gene/reaction/metabolite pathway visualization |
206 |
| Cross-Species Translation | ||
| sbv IMPROVER | Open-source; crowdsourcing; biological networks; automated literature curation | 136 |
| diseaseQUEST | Open-source; integration of human GWAS and model organism network models of tissue/cell-type-specific function, machine learning, candidate gene prioritization | 137 |
| FIT | Free; cross-species extrapolation of gene expression between mice and humans | 135 |
| Interactions, Ontologies, Networks, and Pathways | ||
| ConsensusPathDB | Molecular function interaction database; gene/metabolite enrichment; multi-level gene ontology and multi-database pathway enrichment; protein interactions; visualization | 207 |
| Enrichr | Gene set enrichment analyses; gene ontology; multi-database pathways; protein complexes; small molecule perturbagens; human phenotypes; tissue/cell type expression; visualization | 208 |
| STRING | Protein-protein interaction networks; functional enrichment analyses; visualization | 209 |
| MetaCore | Commercial; curated; protein-protein/DNA-protein/protein-compound interactions; transcription factors; metabolic/signaling pathway analyses; ontologies; visualization | 210 |
| DAVID | Free; ontology/pathway enrichment; BioCarta & KEGG pathway visualization; protein interaction; gene-disease associations | 211 |
| Cytoscape | Open-source; integration and visualization of multi-omics molecular interaction networks; ontology/pathway enrichment; network analyses; clustering | 212 |
| Ingenuity | Commercial; multi-omics integration, pathway analyses; network visualization/causal analyses | 213 |
| WGCNA | Open-source; network construction, module detection, gene selection, calculation of topological properties, data simulation, visualization | 214 |
Extraction of Insight: Bioinformatics, Systems Biology, and Multi-Omics Integration
Though omics approaches hold immense promise in furthering our understanding of complex tissues, microenvironments, and disease states, the extraction of molecular insights from large-scale omics data is a critical step. In effect, careful and thoughtful application of bioinformatics and systems biology are necessary to effectively pair holistic data-driven studies with classical hypothesis-driven science. In Figure 4, we outline a prototypical multi-omics pipeline for study of pathobiology and discovery of drug targets. After sample collection, processing, and the actual performance of sequencing or mass spectrometry, there are a multitude of crucial stages of analysis: the first, and arguably most important, is the accurate identification of detected molecules, including alignment and annotation of raw sequencing reads to the correct region of the genome or transcriptome, and peptide or compound identification from raw mass spectra. These steps are inherently specific to each omics modality and the software package(s) being employed, and are beyond the scope of this current article. So too are the critical normalization schema that must be employed against raw quantified datasets in order to account for variability in sample preparation, instrument calibration, sequencing depth, gene length, RNA composition, signal intensity, batch-to-batch variation, or sources of physiological or clinical co-variation (e.g., age, sex, co-morbidities, etc.); these aspects are reviewed elsewhere.157,158 After these steps, statistical treatments to rigorously identify molecules that are differentially-enriched between sample groups are performed. Importantly, it should be noted that utilizing and/or reporting per-molecule un-adjusted p-values, perhaps with the addition of fold-change cutoffs, is no longer recognized as broadly sufficient when dealing with large datasets containing thousands of compounds. This is due to the multiple testing problem – for example, a microarray of 10,000 spots leads to 10,000 separate hypothesis tests. At a typical p-value cutoff of 0.05, this leads to 500 genes to be considered as statistically significantly differentially-expressed, purely by chance. This inflated false-positive rate is exacerbated with datasets that are continuously growing in size. State-of-the-art differential statistical analysis utilizes various forms of p-value adjustment (typically Benjamini-Hochberg159, also reported as the q-value or false discovery rate (FDR)) to account for these hundreds or thousands of multiple comparisons; this functionality is built into a number of popular software packages (Table 2), many of which are user-friendly and free or open-sourced.
Figure 4: Multi-omics pipelines for discovery of pathobiology and drug targets.

DNA, RNA (mRNA or non-coding), protein, extracellular vesicles (EVs), or metabolites derived from tissues, blood, or cell culture experiments undergo a variety of high-throughput next-generation approaches to obtain “-omes” of interest. Bioinformatics, systems biology, and network approaches are then employed to quantify differential expression/enrichment of molecules between populations or experimental conditions, to integrate information from multiple “-omes” together, and to probe them for insights into initiation and progression of pathobiology. In silico methods of target/biomarker prioritization provide molecules for validation in 2D/3D culture or in/ex vivo disease models.
Once a set of statistically differentially-enriched compounds has been rigorously selected, it can then i) undergo further statistical treatments to collect samples into coregulated groups via, typically, unsupervised dimensional reduction (e.g., principal component axis, hierarchical clustering), and ii) be probed for interpretation of biological understanding, mechanistic insight, and drug target prioritization. This latter stage aims to extract biological information, and primarily leverages a variety of databases where genes are annotated either by their cellular locations, molecular functions, or associated biological processes (gene ontology; GO160), or by their membership in pre-defined gene sets that are associated with specific metabolic, genetic, or signal transduction pathways (e.g., KEGG, Reactome, BioCarta161–163). GO or pathway enrichment analyses determine in which ontologies or pathways the omics-derived differential-enriched molecules are statistically over-represented vs. random chance and serve to append localization and functionality. From there, network medicine approaches can be utilized to identify dominant biological functions or mechanisms, and to assign topological order to these molecular data in an unbiased and quantifiable manner. Three primary network types have been employed in the study of CAVD thus far: i) pathway networks, where nodes are individual pathways, and edges (links) are formed by the molecules shared by any two pathways and which were detected in the omics layer(s) under analysis,26,100 ii) miRNA-target networks, where detected miRs or other regulatory non-coding RNAs are linked to their high-confidence gene targets,100 and iii) protein-protein interaction networks, where nodes are proteins and edges represent known physical interactions between proteins – typically with experimental support (e.g., protein complexes, enzyme-substrate reactions, signaling interactions, regulatory interactions).26,100,164 Regardless of the network type, quantitative aspects of network topology (order/arrangement) can be leveraged to prioritize molecules with important biological impact and or promising drug targets. Highly connected network hub nodes (those with the most edges) tend to hold special functional significance within a network, and have been shown to play essential and central roles – for this reason, they have been prioritized by traditional pharmacology, though there is growing concern that targeting hubs themselves may cause substantial side effects due to their high connectivity to the rest of the network.165 One popular alternative has been the selection of nodes with high betweenness-centrality – the number of shortest paths between network constituents that pass through a node of interest. Such nodes appear as bottlenecks and are associated with prediction of better drug candidates.166 We employed these approaches to identify fibronectin-1 and alpha-2-macroglobulin as central drivers of the advanced calcific stage of human CAVD.26 Other work has leveraged networks as a means to screen potential drug targets in CAVD by assessing the ability of various small molecules to restore a diseased network topology back to one that most closely resembles the appearance of the baseline, healthy network.23 Following the prioritization of network nodes as putative drug targets, validation studies on the efficacy of these molecules are performed in vitro or in vivo, often via targeted genetic knockdown/overexpression or small molecule-based activation/repression in already-established models of CAVD (e.g., hypercholesterolemic/diabetic,15 bicuspid,167 and wire-injury mouse models,168 and VIC/VEC culture under osteogenic conditions95,169,15,167,168).
As noted earlier, the large-scale and unbiased study of CAVD by omics approaches enables a holistic understanding of disease. Integrative omics/multi-omics/trans-omics approaches layer multiple omics modalities together (e.g., genomics and transcriptomics, proteomics and metabolomics) and analyze them simultaneously in order to decipher higher-order pathways of disease development, including complex molecular regulators of phenotype or cross-layer interactions. Multi-omics integration has already been applied to the study of AS and CAVD, though in a largely limited capacity. Indeed, rigorous integration of multiple omics modalities remains in its infancy throughout biomedical science. To assess differential drivers of AS and aortic valve regurgitation (aortic insufficiency, AI), Mourino-Alvarez and colleagues integrated plasma proteomics and metabolomics in 6 subjects (6 with AS, 6 with AI) in order to separate molecular alterations due to aortic valve calcification from those due to the cardiocirculatory or hemodynamic alterations associated with valvular dysfunction present in both AS and AI.170 Statistical analysis for differential enrichment was performed separately within each omics layer, and differentially-enriched molecules from both modalities were then classified according to their primary biological functions into four groups related to coagulation, inflammation and immune response, response to ischemia and lipid metabolism. The predictive power of these combined molecular panels was then assessed in a validation cohort and demonstrated potential diagnostic tools. Via bioinformatic integration of pre-existing publicly-available datasets, others have incorporated transcriptomics, proteomics and metabolomics data from several of the studies described earlier in this review. To account for sample, approach, study, donor, technical, and statistical heterogeneity, this meta-type analysis selected differentially-regulated molecules that were shared between studies, and performed direct pathway and network-based analyses for pathway enrichment and multi-omics layer integration. In doing so, a novel linkage between CAVD and amyloid-like deposits was identified and validated via immunohistochemistry of human aortic valve tissue.171 Other bioinformatic meta-analyses have integrated publicly-available transcriptomics datasets from multiple groups to increase sample size/statistical power and reveal additional differentially-expressed genes between normal and CAVD human tissues, marking functionality associated with transendothelial leukocyte migration, cell adhesion, and ALDH2-mediated detoxification.172,173 The application of network-based integration methods is growing rapidly in popularity, as these approaches offer a more elegant, sophisticated approach that goes well beyond the straightforward union of multiple unfiltered omics datasets or differentially-enriched molecules from multiple layers. Schlotter et. al. utilized networks of pathways enriched in valvular tissue transcriptomics and proteomics to combine mutually-exclusive genes and proteins that were uniquely elevated in specific stages of disease and certain anatomical layers. In a multi-omics study of valve-derived tissue-entrapped extracellular vesicles, Blaser and colleagues exploited a similar pathways-network approach to integrate miRNA-seq and proteomics performed on vesicle cargoes to describe the overall functional impact of these EVs on recipient cells.100 Other novel approaches to perform integrative multi-omics are under active development, and include a latent factors-based methodology that separates axes of heterogeneity which are shared across multiple omics modalities.174 This method infers hidden factors in a manner analogous to principal component axis analyses, and is well-suited to identify drivers of variation in clinically and biologically heterogeneous diseases such as CAVD. To date, many bioinformatics tools are available to identify co-expression networks, key driver genes or differential networks, such as WGCNA (weighted correlation network analysis), Mergeomics, ARACNe (algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context) or PANDA (passing attributes between networks for data assimilation), but only the former has been applied previously to define CAVD pathogenesis.118,175–177 Indeed, by employing WGCNA on three microarray-based gene expression datasets of normal versus calcific aortic valves, Liu and colleagues identified four CAVD-related gene modules with similar expression patterns, including genes involved in extracellular matrix turnover and angiogenesis, such as CTHRC1 and SCG2.177
In Table 2, we curate a number of well-documented bioinformatics, systems biology, and multi-omics toolsets that are particularly useful for the types of studies we have described herein. Some of these resources are open-source, many others are freely-available, and some are commercial products. The systems biology community is a rapidly-growing body that has embraced scientific openness, data-sharing, rapid iteration, and sound methodology. Moving forward, the practical success of omics-based studies of valvular biology and drug discovery will necessitate the formation of close relationships between diverse teams of researchers, including molecular biologists, clinicians, physicists, bioinformaticians, and network scientists.
Future Pitfalls and Promise
Paradoxically, as transcatheter valve replacement rapidly becomes a necessity for large groups of moderate- and low-risk patients in the developed world, these changes to the standard of care impact research that would otherwise positively impact those without access to cardiology or cardiac surgery. Research into valve pathobiology will suffer from the impending loss of surgically-derived diseased human tissue samples. Such studies are, in effect, currently in a race against time to obtain as much information from this limited tissue source in order to enable discovery of therapeutics that could be employed not only in regions with high access to interventional cardiology, but also world-wide.215 Development of novel multi-cellular organoids has gained acceptance as an alternative means to study other tissues by multi-omics approaches,216 and bioprinted valvular organoids with containing VICs could be integrated with existing bioreactor technologies to act as suitable models of CAVD, though modeling circulating or inflammatory sources of disease will remain challenging.18,169 The importance of benchmarking molecular signatures of human disease while this resource remains widely available cannot be overstated, and so we as researchers must learn to do more with less and better leverage existing tissue banks. When paired with traditional histopathology studies and primary cell culture, comprehensive multi-omics approaches arguably offer the best means to identify novel insights into valvular health and disease. Luckily, rapid developments in next-generation sequencing throughput, advanced mass spectrometry instrumentation, computational power, and systems biology analysis techniques are dramatically lowering the economic bar to entry for multi-omics studies, which are already becoming routinely applied to not just the most precious of clinical samples, but also to controlled experimentation in animal and cell culture models. As these techniques become democratized, the importance of standardized guidelines for the open-sourcing/sharing of sample quality control data, omics methods, raw data, analytical pipelines, and a priori target prioritization schema is essential to ensure reproducible and high-quality findings. Equally important will be a push to move omics approaches beyond characterization and atlas development, and towards definitive identification of key regulatory mechanisms, biomarkers, and druggable targets. CAVD appears to be a highly multifactorial disease, and the future integration of phenomics via electronic health records with large-N multi-omics datasets to leverage patient-specific metadata (demographics, clinical factors, valvular/cardiac functional/morphological measurements) may allow for fine sample population matching, deep covariate adjustment, clustering, phenotyping, and identification of novel disease sub-types or disease-contributing factors.
We envision a number of research areas in which dramatic progress could be made by leveraging a standardized, large-N, multi-center, comprehensive multi-omics approach. First, these studies may elucidate the molecular mechanisms that underpin key unanswered aspects of CAVD pathogenesis, such as why only a fraction of patients with ASc progress to stenosis, or why stenosis is much more rapid and prevalent in those with BAVs. Second, careful analyses of altered molecules, signaling pathways, and phenotypes through the use of multiple omics modalities and in combination with temporal studies may enable the currently-challenging differentiation of causal and correlative changes in omics datasets – in the complex valvular microenvironment, what variations cause, or are caused by, altered phenotypes (e.g., leaflet morphology, EMT, fibrotic ECM accumulation, pathological VIC differentiation). Third, rapid technological advancements in omics at single-cell resolution coupled with pseudotime and trajectory-based analysis have the potential to shed light on fundamental questions surrounding resident and infiltrating cellular origins, heterogeneity, differentiation, and contribution to disease initiation and progression. Knowledge gleaned from these studies could allow targeted delivery to key cellular subpopulations responsible for homeostasis or pathogenesis. Fourth, biomarker panels that predict the onset, severity, rate of progression, or transition from valvular sclerosis to stenosis may be identified by plasma-based multi-omics (e.g., circulating RNA, protein, metabolites, EVs) – such advances would revolutionize the prototypical watchful waiting approach to clinical care. Other biomarkers may be predictive of successful responses post-therapy or post-surgery or TAVR. Finally, integration of multi-species (e.g., human, mouse, pig) and in vitro model (2D/3D, static/sheared) multi-omics datasets would allow scientists to more accurately select relevant animal or in vitro models that best recapitulate global phenotypes or specific pathogenic mechanisms of interest in human disease. As this new paradigm of multi-omics and personalized medicine is brought to bear on CAVD, an over-reliance on these techniques must of course also be avoided – careful validation of omics-derived hits and drug targets remains utterly essential to the end goal of, finally, developing pharmacotherapies that can offer a reprieve from this insidious and potentially deadly illness.
Sources of Funding
This study was supported by NIH grants R01 HL114805, R01 HL119798, R01 HL136431, R01 HL141917, and R01 HL147095. SK and TFL are supported by funding from the Swiss Heart Foundation (FF19056 and FF20094).
Abbreviations
- AS
aortic stenosis
- BAV
bicuspid aortic valve
- CAVD
calcific aortic valve disease
- CyTOF
cytometry by time-of-flight; mass cytometry
- GWAS
genome-wide association study
- Lp(a)
lipoprotein(a)
- LC
liquid chromatography
- MS
mass spectrometry
- NMR
nuclear magnetic resonance
- PRS
polygenic risk score
- PTM
post-translational modification
- SMC
smooth muscle cell
- SAVR
surgical aortic valve replacement
- TAV
tricuspid aortic valve
- TAVR
transcatheter aortic valve replacement
- VIC
valvular interstitial cell
- VEC
valvular endothelial cell
Footnotes
Disclosures
None.
References
- 1.Clark MA, Arnold SV, Duhay FG, Thompson AK, Keyes MJ, Svensson LG, Bonow RO, Stockwell BT, Cohen DJ. Five-year clinical and economic outcomes among patients with medically managed severe aortic stenosis: Results from a medicare claims analysis. Circ Cardiovasc Qual Outcomes. 2012;5:697–704 [DOI] [PubMed] [Google Scholar]
- 2.Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, Kitzman DW, Otto CM. Clinical factors associated with calcific aortic valve disease. Cardiovascular health study. Journal of the American College of Cardiology. 1997;29:630–634 [DOI] [PubMed] [Google Scholar]
- 3.Chan KL, Teo K, Dumesnil JG, Ni A, Tam J, Investigators A. Effect of lipid lowering with rosuvastatin on progression of aortic stenosis: Results of the aortic stenosis progression observation: Measuring effects of rosuvastatin (astronomer) trial. Circulation. 2010;121:306–314 [DOI] [PubMed] [Google Scholar]
- 4.Zilla P, Brink J, Human P, Bezuidenhout D. Prosthetic heart valves: Catering for the few. Biomaterials. 2008;29:385–406 [DOI] [PubMed] [Google Scholar]
- 5.Moore M, Chen J, Mallow PJ, Rizzo JA. The direct health-care burden of valvular heart disease: Evidence from us national survey data. Clinicoecon Outcomes Res. 2016;8:613–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danielsen R, Aspelund T, Harris TB, Gudnason V. The prevalence of aortic stenosis in the elderly in iceland and predictions for the coming decades: The ages-reykjavik study. International journal of cardiology. 2014;176:916–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O’Brien KD. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 1994;90:844–853 [DOI] [PubMed] [Google Scholar]
- 8.Thubrikar M The aortic valve. Boca Raton: CRC Press, Inc; 1990. [Google Scholar]
- 9.Nomura A, Seya K, Yu Z, Daitoku K, Motomura S, Murakami M, Fukuda I, Furukawa K. Cd34-negative mesenchymal stem-like cells may act as the cellular origin of human aortic valve calcification. Biochemical and biophysical research communications. 2013;440:780–785 [DOI] [PubMed] [Google Scholar]
- 10.Young EW, Wheeler AR, Simmons CA. Matrix-dependent adhesion of vascular and valvular endothelial cells in microfluidic channels. Lab on a chip. 2007;7:1759–1766 [DOI] [PubMed] [Google Scholar]
- 11.Ayoub S, Ferrari G, Gorman RC, Gorman III JH, Schoen FJ, Sacks MS. Heart valve biomechanics and underlying mechanobiology. Comprehensive physiology.1743–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, Weissleder R. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation. 2007;115:377–386 [DOI] [PubMed] [Google Scholar]
- 13.Schlotter F, de Freitas RCC, Rogers MA, et al. Apoc-iii is a novel inducer of calcification in human aortic valves. The Journal of biological chemistry. 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogers MA, Buffolo F, Schlotter F, et al. Annexin a1-dependent tethering promotes extracellular vesicle aggregation revealed with single-extracellular vesicle analysis. Sci Adv. 2020;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bouchareb R, Boulanger MC, Tastet L, et al. Activated platelets promote an osteogenic programme and the progression of calcific aortic valve stenosis. Eur Heart J. 2019;40:1362–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wirrig EE, Hinton RB, Yutzey KE. Differential expression of cartilage and bone-related proteins in pediatric and adult diseased aortic valves. J Mol Cell Cardiol. 2011;50:561–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuevas RA, Hortells L, Boufford C, et al. Abstract 15843: A novel role for telomerase in calcific aortic valve disease. Circulation. 2020;142:A15843–A15843 [Google Scholar]
- 18.Heath JM, Fernandez Esmerats J, Khambouneheuang L, Kumar S, Simmons R, Jo H. Mechanosensitive microrna-181b regulates aortic valve endothelial matrix degradation by targeting timp3. Cardiovascular engineering and technology. 2018;9:141–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation. 2005;111:920–925 [DOI] [PubMed] [Google Scholar]
- 20.Michelena HI, Desjardins VA, Avierinos JF, Russo A, Nkomo VT, Sundt TM, Pellikka PA, Tajik AJ, Enriquez-Sarano M. Natural history of asymptomatic patients with normally functioning or minimally dysfunctional bicuspid aortic valve in the community. Circulation. 2008;117:2776–2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yabusaki K, Hutcheson JD, Vyas P, Bertazzo S, Body SC, Aikawa M, Aikawa E. Quantification of calcified particles in human valve tissue reveals asymmetry of calcific aortic valve disease development. Frontiers in cardiovascular medicine. 2016;3:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nachlas ALY, Li S, Jha R, Singh M, Xu C, Davis ME. Human ipsc-derived mesenchymal stem cells encapsulated in pegda hydrogels mature into valve interstitial-like cells. Acta Biomater. 2018;71:235–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Theodoris CV, Zhou P, Liu L, et al. Network-based screen in ipsc-derived cells reveals therapeutic candidate for heart valve disease. Science. 2021;371:eabd0724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doss MX, Sachinidis A. Current challenges of ipsc-based disease modeling and therapeutic implications. Cells. 2019;8:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Musunuru K, Sheikh F, Gupta RM, Houser SR, Maher KO, Milan DJ, Terzic A, Wu JC, American Heart Association Council on Functional G, Translational B, Council on Cardiovascular Disease in the Y, Council on C, Stroke N. Induced pluripotent stem cells for cardiovascular disease modeling and precision medicine: A scientific statement from the american heart association. Circ Genom Precis Med. 2018;11:e000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlotter F, Halu A, Goto S, et al. Spatiotemporal multi-omics mapping generates a molecular atlas of the aortic valve and reveals networks driving disease. Circulation. 2018;138:377–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Theriault S, Dina C, Messika-Zeitoun D, et al. Genetic association analyses highlight il6, alpl, and nav1 as 3 new susceptibility genes underlying calcific aortic valve stenosis. Circ Genom Precis Med. 2019;12:e002617. [DOI] [PubMed] [Google Scholar]
- 28.Surendran A, Edel A, Chandran M, et al. Metabolomic signature of human aortic valve stenosis. JACC. Basic to translational science. 2020;5:1163–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Majumdar U, Manivannan S, Basu M, et al. Nitric oxide prevents aortic valve calcification by s-nitrosylation of usp9x to activate notch signaling. Science Advances. 2020:2020.2008.2015.252171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu K, Xie S, Huang Y, et al. Cell-type transcriptome atlas of human aortic valves reveal cell heterogeneity and endothelial to mesenchymal transition involved in calcific aortic valve disease. Arterioscler Thromb Vasc Biol. 2020;40:2910–2921 [DOI] [PubMed] [Google Scholar]
- 31.Martinsson A, Li X, Zoller B, Andell P, Andersson C, Sundquist K, Smith JG. Familial aggregation of aortic valvular stenosis: A nationwide study of sibling risk. Circulation. Cardiovascular genetics. 2017;10:e001742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thanassoulis G, Campbell CY, Owens DS, et al. Genetic associations with valvular calcification and aortic stenosis. The New England journal of medicine. 2013;368:503–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dashzeveg N, Taira N, Lu ZG, Kimura J, Yoshida K. Palmdelphin, a novel target of p53 with ser46 phosphorylation, controls cell death in response to DNA damage. Cell Death Dis. 2014;5:e1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Theriault S, Gaudreault N, Lamontagne M, et al. A transcriptome-wide association study identifies palmd as a susceptibility gene for calcific aortic valve stenosis. Nature communications. 2018;9:988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Z, Gaudreault N, Arsenault BJ, Mathieu P, Bosse Y, Theriault S. Phenome-wide analyses establish a specific association between aortic valve palmd expression and calcific aortic valve stenosis. Commun Biol. 2020;3:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.El Husseini D, Boulanger MC, Mahmut A, Bouchareb R, Laflamme MH, Fournier D, Pibarot P, Bosse Y, Mathieu P. P2y2 receptor represses il-6 expression by valve interstitial cells through akt: Implication for calcific aortic valve disease. J Mol Cell Cardiol. 2014;72:146–156 [DOI] [PubMed] [Google Scholar]
- 37.Khera AV, Emdin CA, Drake I, et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. The New England journal of medicine. 2016;375:2349–2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marquez-Luna C, Loh PR, South Asian Type 2 Diabetes C, Consortium STD, Price AL. Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet Epidemiol. 2017;41:811–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. Journal of the American College of Cardiology. 2004;44:138–143 [DOI] [PubMed] [Google Scholar]
- 40.Prakash SK, Bosse Y, Muehlschlegel JD, et al. A roadmap to investigate the genetic basis of bicuspid aortic valve and its complications: Insights from the international bavcon (bicuspid aortic valve consortium). Journal of the American College of Cardiology. 2014;64:832–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin LJ, Pilipenko V, Kaufman KM, Cripe L, Kottyan LC, Keddache M, Dexheimer P, Weirauch MT, Benson DW. Whole exome sequencing for familial bicuspid aortic valve identifies putative variants. Circulation. Cardiovascular genetics. 2014;7:677–683 [DOI] [PubMed] [Google Scholar]
- 42.Yang B, Zhou W, Jiao J, et al. Protein-altering and regulatory genetic variants near gata4 implicated in bicuspid aortic valve. Nature communications. 2017;8:15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gharibeh L, Komati H, Bosse Y, et al. Gata6 regulates aortic valve remodeling, and its haploinsufficiency leads to right-left type bicuspid aortic valve. Circulation. 2018;138:1025–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Helgadottir A, Thorleifsson G, Gretarsdottir S, et al. Genome-wide analysis yields new loci associating with aortic valve stenosis. Nature communications. 2018;9:987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fulmer D, Toomer K, Guo L, et al. Defects in the exocyst-cilia machinery cause bicuspid aortic valve disease and aortic stenosis. Circulation. 2019;140:1331–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in notch1 cause aortic valve disease. Nature. 2005;437:270–274 [DOI] [PubMed] [Google Scholar]
- 47.LaHaye S, Majumdar U, Yasuhara J, Koenig SN, Matos-Nieves A, Kumar R, Garg V. Developmental origins for semilunar valve stenosis identified in mice harboring congenital heart disease-associated gata4 mutation. Disease models & mechanisms. 2019;12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koenig SN, Bosse K, Majumdar U, Bonachea EM, Radtke F, Garg V. Endothelial notch1 is required for proper development of the semilunar valves and cardiac outflow tract. Journal of the American Heart Association. 2016;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blaser MC, Wei K, Adams RLE, et al. Deficiency of natriuretic peptide receptor 2 promotes bicuspid aortic valves, aortic valve disease, left ventricular dysfunction, and ascending aortic dilatations in mice. Circ Res. 2018;122:405–416 [DOI] [PubMed] [Google Scholar]
- 50.Lee TC, Zhao YD, Courtman DW, Stewart DJ. Abnormal aortic valve development in mice lacking endothelial nitric oxide synthase. Circulation. 2000;101:2345–2348 [DOI] [PubMed] [Google Scholar]
- 51.Zhang B, Roos C, Hagler M, Verzosa G, Zhang H, Schaff H, Sarano M, Miller J. Abstract 123: Activation of oxidized soluble guanylate cyclase slows progression of aortic valve calcification. Arteriosclerosis, Thrombosis, and Vascular Biology. 2019;39:A123–A123 [Google Scholar]
- 52.Nagy E, Back M. Epigenetic regulation of 5-lipoxygenase in the phenotypic plasticity of valvular interstitial cells associated with aortic valve stenosis. FEBS Lett. 2012;586:1325–1329 [DOI] [PubMed] [Google Scholar]
- 53.Barrick CJ, Roberts RB, Rojas M, Rajamannan NM, Suitt CB, O’Brien KD, Smyth SS, Threadgill DW. Reduced egfr causes abnormal valvular differentiation leading to calcific aortic stenosis and left ventricular hypertrophy in c57bl/6j but not 129s1/svimj mice. Am J Physiol Heart Circ Physiol. 2009;297:H65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bouchareb R, Mahmut A, Nsaibia MJ, et al. Autotaxin derived from lipoprotein(a) and valve interstitial cells promotes inflammation and mineralization of the aortic valve. Circulation. 2015;132:677–690 [DOI] [PubMed] [Google Scholar]
- 55.Mkannez G, Gagne-Ouellet V, Jalloul Nsaibia M, et al. DNA methylation of a plpp3 mir transposon-based enhancer promotes an osteogenic programme in calcific aortic valve disease. Cardiovasc Res. 2018;114:1525–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Halawa S, Latif N, Tseng Y-T, Ibrahim AM, Chester AH, Moustafa A, Aguib Y, Yacoub M. Profiling genome-wide DNA methylation patterns in human aortic and mitral valves. medRxiv. 2020:2020.2009.2010.20190546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bosse Y, Miqdad A, Fournier D, Pepin A, Pibarot P, Mathieu P. Refining molecular pathways leading to calcific aortic valve stenosis by studying gene expression profile of normal and calcified stenotic human aortic valves. Circulation. Cardiovascular genetics. 2009;2:489–498 [DOI] [PubMed] [Google Scholar]
- 58.McCoy CM, Nicholas DQ, Masters KS. Sex-related differences in gene expression by porcine aortic valvular interstitial cells. PloS one. 2012;7:e39980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Z, Gerstein M, Snyder M. Rna-seq: A revolutionary tool for transcriptomics. Nature reviews. Genetics. 2009;10:57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang C, Gong B, Bushel PR, et al. The concordance between rna-seq and microarray data depends on chemical treatment and transcript abundance. Nature biotechnology. 2014;32:926–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang J, Wang Y, Gu W, Ni B, Sun H, Yu T, Gu W, Chen L, Shao Y. Comparative transcriptome analysis reveals substantial tissue specificity in human aortic valve. Evol Bioinform Online. 2016;12:175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gottlieb Sen D, Halu A, Razzaque A, Gorham JM, Hartnett J, Seidman JG, Aikawa E, Seidman CE. The transcriptional signature of growth in human fetal aortic valve development. Ann Thorac Surg. 2018;106:1834–1840 [DOI] [PubMed] [Google Scholar]
- 63.Lee LH, Halu A, Morgan S, Iwata H, Aikawa M, Singh SA. Xina: A workflow for the integration of multiplexed proteomics kinetics data with network analysis. J Proteome Res. 2019;18:775–781 [DOI] [PubMed] [Google Scholar]
- 64.Nordquist E, LaHaye S, Nagel C, Lincoln J. Postnatal and adult aortic heart valves have distinctive transcriptional profiles associated with valve tissue growth and maintenance respectively. Frontiers in cardiovascular medicine. 2018;5:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oury C, Marechal P, Donis N, et al. Dexfenfluramine and pergolide cause heart valve disease via valve metabolic reprogramming and ongoing matrix remodeling. Int J Mol Sci. 2020;21:4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Balachandran K, Bakay MA, Connolly JM, Zhang X, Yoganathan AP, Levy RJ. Aortic valve cyclic stretch causes increased remodeling activity and enhanced serotonin receptor responsiveness. Ann Thorac Surg. 2011;92:147–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Asulin N, Volinsky N, Grosman-Rimon L, et al. Differential micrornas expression in calcified versus rheumatic aortic valve disease. J Card Surg. 2020;35:1508–1513 [DOI] [PubMed] [Google Scholar]
- 68.Ohukainen P, Syvaranta S, Napankangas J, et al. Microrna-125b and chemokine ccl4 expression are associated with calcific aortic valve disease. Annals of medicine. 2015;47:423–429 [DOI] [PubMed] [Google Scholar]
- 69.Rathan S, Ankeny CJ, Arjunon S, et al. Identification of side- and shear-dependent micrornas regulating porcine aortic valve pathogenesis. Scientific reports. 2016;6:25397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holliday CJ, Ankeny RF, Jo H, Nerem RM. Discovery of shear- and side-specific mrnas and mirnas in human aortic valvular endothelial cells. Am J Physiol Heart Circ Physiol. 2011;301:H856–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nigam V, Sievers HH, Jensen BC, Sier HA, Simpson PC, Srivastava D, Mohamed SA. Altered micrornas in bicuspid aortic valve: A comparison between stenotic and insufficient valves. J Heart Valve Dis. 2010;19:459–465 [PMC free article] [PubMed] [Google Scholar]
- 72.Wang H, Shi J, Li B, Zhou Q, Kong X, Bei Y. Microrna expression signature in human calcific aortic valve disease. BioMed research international. 2017;2017:4820275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi J, Liu H, Wang H, Kong X. Microrna expression signature in degenerative aortic stenosis. BioMed research international. 2016;2016:4682172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Song R, Fullerton DA, Ao L, Zhao KS, Reece TB, Cleveland JC, Jr., Meng X. Altered microrna expression is responsible for the pro-osteogenic phenotype of interstitial cells in calcified human aortic valves. Journal of the American Heart Association. 2017;6:e005364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Washietl S, Kellis M, Garber M. Evolutionary dynamics and tissue specificity of human long noncoding rnas in six mammals. Genome Res. 2014;24:616–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Padang R, Bagnall RD, Tsoutsman T, Bannon PG, Semsarian C. Comparative transcriptome profiling in human bicuspid aortic valve disease using rna sequencing. Physiol Genomics. 2015;47:75–87 [DOI] [PubMed] [Google Scholar]
- 77.Guauque-Olarte S, Droit A, Tremblay-Marchand J, et al. Rna expression profile of calcified bicuspid, tricuspid, and normal human aortic valves by rna sequencing. Physiol Genomics. 2016;48:749–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Choi B, Lee S, Kim SM, et al. Dipeptidyl peptidase-4 induces aortic valve calcification by inhibiting insulin-like growth factor-1 signaling in valvular interstitial cells. Circulation. 2017;135:1935–1950 [DOI] [PubMed] [Google Scholar]
- 79.Yanagawa B, Lovren F, Pan Y, et al. Mirna-141 is a novel regulator of bmp-2-mediated calcification in aortic stenosis. The Journal of thoracic and cardiovascular surgery. 2012;144:256–262 [DOI] [PubMed] [Google Scholar]
- 80.Martinez-Micaelo N, Beltran-Debon R, Baiges I, Faiges M, Alegret JM. Specific circulating microrna signature of bicuspid aortic valve disease. J Transl Med. 2017;15:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wainberg M, Sinnott-Armstrong N, Mancuso N, et al. Opportunities and challenges for transcriptome-wide association studies. Nat Genet. 2019;51:592–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guauque-Olarte S, Messika-Zeitoun D, Droit A, et al. Calcium signaling pathway genes runx2 and cacna1c are associated with calcific aortic valve disease. Circulation. Cardiovascular genetics. 2015;8:812–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhao Q, Dacre M, Nguyen T, et al. Molecular mechanisms of coronary disease revealed using quantitative trait loci for tcf21 binding, chromatin accessibility, and chromosomal looping. Genome Biol. 2020;21:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huan T, Joehanes R, Song C, et al. Genome-wide identification of DNA methylation qtls in whole blood highlights pathways for cardiovascular disease. Nature communications. 2019;10:4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yao C, Chen G, Song C, et al. Genome-wide mapping of plasma protein qtls identifies putatively causal genes and pathways for cardiovascular disease. Nature communications. 2018;9:3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhu Z, Zhang F, Hu H,et al. Integration of summary data from gwas and eqtl studies predicts complex trait gene targets. Nat Genet. 2016;48:481–487 [DOI] [PubMed] [Google Scholar]
- 87.Corchete LA, Rojas EA, Alonso-Lopez D, De Las Rivas J, Gutierrez NC, Burguillo FJ. Systematic comparison and assessment of rna-seq procedures for gene expression quantitative analysis. Scientific reports. 2020;10:19737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Eraslan B, Wang D, Gusic M, et al. Quantification and discovery of sequence determinants of protein-per-mrna amount in 29 human tissues. Mol Syst Biol. 2019;15:e8513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gil-Dones F, Martin-Rojas T, Lopez-Almodovar LF, et al. Valvular aortic stenosis: A proteomic insight. Clin Med Insights Cardiol. 2010;4:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Martin-Rojas T, Gil-Dones F, Lopez-Almodovar LF, Padial LR, Vivanco F, Barderas MG. Proteomic profile of human aortic stenosis: Insights into the degenerative process. J Proteome Res. 2012;11:1537–1550 [DOI] [PubMed] [Google Scholar]
- 91.Martin-Rojas T, Mourino-Alvarez L, Alonso-Orgaz S, et al. Itraq proteomic analysis of extracellular matrix remodeling in aortic valve disease. Scientific reports. 2015;5:17290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gerber Y, Goldbourt U, Feinberg MS, Segev S, Harats D. Are triglyceride-rich lipoproteins associated with aortic valve sclerosis? A preliminary report. Atherosclerosis. 2003;170:301–305 [DOI] [PubMed] [Google Scholar]
- 93.Mourino-Alvarez L, Iloro I, de la Cuesta F,et al. Maldi-imaging mass spectrometry: A step forward in the anatomopathological characterization of stenotic aortic valve tissue. Scientific reports. 2016;6:27106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bertacco E, Millioni R, Arrigoni G, et al. Proteomic analysis of clonal interstitial aortic valve cells acquiring a pro-calcific profile. J Proteome Res. 2010;9:5913–5921 [DOI] [PubMed] [Google Scholar]
- 95.Goto S, Rogers MA, Blaser MC, et al. Standardization of human calcific aortic valve disease in vitro modeling reveals passage-dependent calcification. Frontiers in cardiovascular medicine. 2019;6:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Alvarez-Llamas G, Martin-Rojas T, de la Cuesta F, et al. Modification of the secretion pattern of proteases, inflammatory mediators, and extracellular matrix proteins by human aortic valve is key in severe aortic stenosis. Mol Cell Proteomics. 2013;12:2426–2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Blaser MC, Aikawa E. Differential mirna loading underpins dual harmful and protective roles for extracellular vesicles in atherogenesis. Circ Res. 2019;124:467–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Blaser MC, Aikawa E. Roles and regulation of extracellular vesicles in cardiovascular mineral metabolism. Frontiers in cardiovascular medicine. 2018;5:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hutcheson JD, Goettsch C, Bertazzo S, et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nature materials. 2016;15:335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Blaser MC, Buffolo F, Halu A, et al. Conserved and divergent modulation of calcification in atherosclerosis and aortic valve disease by tissue extracellular vesicles. bioRxiv. 2020 [Google Scholar]
- 101.Hadji F, Boulanger MC, Guay SP, et al. Altered DNA methylation of long noncoding rna h19 in calcific aortic valve disease promotes mineralization by silencing notch1. Circulation. 2016;134:1848–1862 [DOI] [PubMed] [Google Scholar]
- 102.Gao L, Ji Y, Lu Y, et al. Low-level overexpression of p53 promotes warfarin-induced calcification of porcine aortic valve interstitial cells by activating slug gene transcription. The Journal of biological chemistry. 2018;293:3780–3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kolltveit KM, Geiran O, Tronstad L, Olsen I. Multiple bacteria in calcific aortic valve stenosis. Microbial Ecology in Health and Disease. 2002;14:110–117 [Google Scholar]
- 104.Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nature biotechnology. 2003;21:255–261 [DOI] [PubMed] [Google Scholar]
- 105.Conibear AC. Deciphering protein post-translational modifications using chemical biology tools. Nature Reviews Chemistry. 2020;4:674–695 [DOI] [PubMed] [Google Scholar]
- 106.Angel PM, Drake RR, Park Y, et al. Spatial n-glycomics of the human aortic valve in development and pediatric endstage congenital aortic valve stenosis. J Mol Cell Cardiol. 2021;154:6–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guijas C, Montenegro-Burke JR, Warth B, Spilker ME, Siuzdak G. Metabolomics activity screening for identifying metabolites that modulate phenotype. Nature biotechnology. 2018;36:316–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Emwas AH, Roy R, McKay RT, et al. Nmr spectroscopy for metabolomics research. Metabolites. 2019;9:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang JH, Byun J, Pennathur S. Analytical approaches to metabolomics and applications to systems biology. Seminars in nephrology. 2010;30:500–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fan Y, Li Y, Chen Y, et al. Comprehensive metabolomic characterization of coronary artery diseases. Journal of the American College of Cardiology. 2016;68:1281–1293 [DOI] [PubMed] [Google Scholar]
- 111.Vojinovic D, van der Lee SJ, van Duijn CM, et al. Metabolic profiling of intra- and extracranial carotid artery atherosclerosis. Atherosclerosis. 2018;272:60–65 [DOI] [PubMed] [Google Scholar]
- 112.Vorkas PA, Isaac G, Anwar MA, Davies AH, Want EJ, Nicholson JK, Holmes E. Untargeted uplc-ms profiling pipeline to expand tissue metabolome coverage: Application to cardiovascular disease. Anal Chem. 2015;87:4184–4193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang W, Maimaiti A, Zhao Y, et al. Analysis of serum metabolites to diagnose bicuspid aortic valve. Scientific reports. 2016;6:37023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Martinez-Micaelo N, Ligero C, Antequera-Gonzalez B, Junza A, Yanes O, Alegret JM. Plasma metabolomic profiling associates bicuspid aortic valve disease and ascending aortic dilation with a decrease in antioxidant capacity. J Clin Med. 2020;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xiong TY, Liu C, Liao YB, et al. Differences in metabolic profiles between bicuspid and tricuspid aortic stenosis in the setting of transcatheter aortic valve replacement. BMC Cardiovasc Disord. 2020;20:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wirrig EE, Gomez MV, Hinton RB, Yutzey KE. Cox2 inhibition reduces aortic valve calcification in vivo. Arterioscler Thromb Vasc Biol. 2015;35:938–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bowler MA, Raddatz MA, Johnson CL, Lindman BR, Merryman WD. Celecoxib is associated with dystrophic calcification and aortic valve stenosis. JACC. Basic to translational science. 2019;4:135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liao Y, Liu C, Xiong T, et al. Metabolic modulation and potential biomarkers of the prognosis identification for severe aortic stenosis after tavr by a metabolomics study. Cardiol Res Pract. 2020;2020:3946913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mahmut A, Boulanger MC, El Husseini D, Fournier D, Bouchareb R, Despres JP, Pibarot P, Bosse Y, Mathieu P. Elevated expression of lipoprotein-associated phospholipase a2 in calcific aortic valve disease: Implications for valve mineralization. Journal of the American College of Cardiology. 2014;63:460–469 [DOI] [PubMed] [Google Scholar]
- 120.Al Hageh C, Rahy R, Khazen G, Brial F, Khnayzer RS, Gauguier D, Zalloua PA. Plasma and urine metabolomic analyses in aortic valve stenosis reveal shared and biofluid-specific changes in metabolite levels. PloS one. 2020;15:e0242019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhao F, Wang P, Lucardi RD, Su Z, Li S. Natural sources and bioactivities of 2,4-di-tert-butylphenol and its analogs. Toxins (Basel). 2020;12:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nair RVR, Jayasree DV, Biju PG, Baby S. Anti-inflammatory and anticancer activities of erythrodiol-3-acetate and 2,4-di-tert-butylphenol isolated from humboldtia unijuga. Nat Prod Res. 2020;34:2319–2322 [DOI] [PubMed] [Google Scholar]
- 123.Altekar WW, Rao MR. Microbiological dissimilation of tricarballylate and trans-aconitate. J Bacteriol. 1963;85:604–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lewis GD, Wei R, Liu E, et al. Metabolite profiling of blood from individuals undergoing planned myocardial infarction reveals early markers of myocardial injury. The Journal of clinical investigation. 2008;118:3503–3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cohen DJ, Malave D, Ghidoni JJ, Iakovidis P, Everett MM, You S, Liu Y, Boyan BD. Role of oral bacterial flora in calcific aortic stenosis: An animal model. Ann Thorac Surg. 2004;77:537–543 [DOI] [PubMed] [Google Scholar]
- 126.Duygu K, Tokgozoglu L, Gurses KM, et al. Association of dietary and gut microbiota-related metabolites with calcific aortic stenosis. Acta Cardiol. 2020:1–9 [DOI] [PubMed] [Google Scholar]
- 127.Angel PM, Bayoumi AS, Hinton RB, Ru Su Y, Bichell D, Mayer JE, Scott Baldwin H, Caprioli RM. Maldi imaging mass spectrometry as a lipidomic approach to heart valve research. J Heart Valve Dis. 2016;25:240–252 [PubMed] [Google Scholar]
- 128.Williams C, Palviainen M, Reichardt NC, Siljander PR, Falcon-Perez JM. Metabolomics applied to the study of extracellular vesicles. Metabolites. 2019;9:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lu W, Xing X, Wang L, Chen L, Zhang S, McReynolds MR, Rabinowitz JD. Improved annotation of untargeted metabolomics data through buffer modifications that shift adduct mass and intensity. Anal Chem. 2020;92:11573–11581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Egidi MG, D’Alessandro A, Mandarello G, Zolla L. Troubleshooting in platelet storage temperature and new perspectives through proteomics. Blood Transfus. 2010;8 Suppl 3:s73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jóhannsson F, Yurkovich JT, Guðmundsson S, Sigurjónsson ÓE, Rolfsson Ó. Temperature dependence of platelet metabolism. bioRxiv. 2019:802660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Joolharzadeh P, St Hilaire C. Cd73 (cluster of differentiation 73) and the differences between mice and humans. Arterioscler Thromb Vasc Biol. 2019;39:339–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yeang C, Cotter B, Tsimikas S. Experimental animal models evaluating the causal role of lipoprotein(a) in atherosclerosis and aortic stenosis. Cardiovasc Drugs Ther. 2016;30:75–85 [DOI] [PubMed] [Google Scholar]
- 134.Brubaker DK, Lauffenburger DA. Translating preclinical models to humans. Science. 2020;367:742–743 [DOI] [PubMed] [Google Scholar]
- 135.Normand R, Du W, Briller M, Gaujoux R, Starosvetsky E, Ziv-Kenet A, Shalev-Malul G, Tibshirani RJ, Shen-Orr SS. Found in translation: A machine learning model for mouse-to-human inference. Nat Methods. 2018;15:1067–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Guryanova S, Guryanova A. Sbv improver: Modern approach to systems biology. In: Tatarinova TV, Nikolsky Y, eds. Biological networks and pathway analysis. New York, NY: Springer New York; 2017:21–29. [DOI] [PubMed] [Google Scholar]
- 137.Yao V, Kaletsky R, Keyes W, Mor DE, Wong AK, Sohrabi S, Murphy CT, Troyanskaya OG. An integrative tissue-network approach to identify and test human disease genes. Nature biotechnology. 2018;36:1091–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Iqbal F, Lupieri A, Aikawa M, Aikawa E. Harnessing single-cell rna sequencing to better understand how diseased cells behave the way they do in cardiovascular disease. Arterioscler Thromb Vasc Biol. 2020:ATVBAHA120314776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Blaser MC, Halu A, Saddic LA, Aikawa M, Aikawa E. Target discovery in calcification through omics and systems approaches. In: Aikawa E, Hutcheson JD, eds. Cardiovascular calcification and bone mineralization. Cham: Springer International Publishing; 2020:525–551. [Google Scholar]
- 140.Trapnell C, Cacchiarelli D, Grimsby J, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature biotechnology. 2014;32:381–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Saeys Y, Van Gassen S, Lambrecht BN. Computational flow cytometry: Helping to make sense of high-dimensional immunology data. Nat Rev Immunol. 2016;16:449–462 [DOI] [PubMed] [Google Scholar]
- 142.Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, Newell EW. Dimensionality reduction for visualizing single-cell data using umap. Nature biotechnology. 2018;37:38–44 [DOI] [PubMed] [Google Scholar]
- 143.Hulin A, Hortells L, Gomez-Stallons MV, et al. Maturation of heart valve cell populations during postnatal remodeling. Development (Cambridge, England). 2019;146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wirrig EE, Yutzey KE. Transcriptional regulation of heart valve development and disease. Cardiovascular pathology : the official journal of the Society for Cardiovascular Pathology. 2011;20:162–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Muhl L, Genove G, Leptidis S, et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nature communications. 2020;11:3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cui Y, Zheng Y, Liu X, et al. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell reports. 2019;26:1934–1950 e1935 [DOI] [PubMed] [Google Scholar]
- 147.Hjortnaes J, Shapero K, Goettsch C, Hutcheson JD, Keegan J, Kluin J, Mayer JE, Bischoff J, Aikawa E. Valvular interstitial cells suppress calcification of valvular endothelial cells. Atherosclerosis. 2015;242:251–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Denisenko E, Guo BB, Jones M, et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus rna-seq workflows. Genome Biol. 2020;21:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Litvinukova M, Talavera-Lopez C, Maatz H, et al. Cells of the adult human heart. Nature. 2020;588:466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Qiu P Embracing the dropouts in single-cell rna-seq analysis. Nature communications. 2020;11:1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Maniatis S, Aijo T, Vickovic S, et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science. 2019;364:89–93 [DOI] [PubMed] [Google Scholar]
- 152.Matos TR, Liu H, Ritz J. Research techniques made simple: Experimental methodology for single-cell mass cytometry. J Invest Dermatol. 2017;137:e31–e38 [DOI] [PubMed] [Google Scholar]
- 153.Cheung TK, Lee CY, Bayer FP, McCoy A, Kuster B, Rose CM. Defining the carrier proteome limit for single-cell proteomics. Nat Methods. 2021;18:76–83 [DOI] [PubMed] [Google Scholar]
- 154.Wang C, Sun D, Huang X, et al. Integrative analyses of single-cell transcriptome and regulome using maestro. Genome Biol. 2020;21:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fernandez DM, Rahman AH, Fernandez NF, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. 2019;25:1576–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Shrestha B Single-cell metabolomics by mass spectrometry. Methods Mol Biol. 2020;2064:1–8 [DOI] [PubMed] [Google Scholar]
- 157.Mertens BJA. Transformation, normalization, and batch effect in the analysis of mass spectrometry data for omics studies. In: Datta S, Mertens BJA, eds. Statistical analysis of proteomics, metabolomics, and lipidomics data using mass spectrometry. Cham: Springer International Publishing; 2017:1–21. [Google Scholar]
- 158.Liu X, Li N, Liu S, Wang J, Zhang N, Zheng X, Leung KS, Cheng L. Normalization methods for the analysis of unbalanced transcriptome data: A review. Front Bioeng Biotechnol. 2019;7:358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological). 1995;57:289–300 [Google Scholar]
- 160.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25:25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chowdhury S, Sarkar RR. Comparison of human cell signaling pathway databases--evolution, drawbacks and challenges. Database (Oxford). 2015;2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kanehisa M, Goto S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fabregat A, Jupe S, Matthews L, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2018;46:D649–D655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Menche J, Sharma A, Kitsak M, Ghiassian SD, Vidal M, Loscalzo J, Barabasi AL. Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science. 2015;347:1257601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Vitali F, Mulas F, Marini P, Bellazzi R. Network-based target ranking for polypharmacological therapies. J Biomed Inform. 2013;46:876–881 [DOI] [PubMed] [Google Scholar]
- 166.Yu H, Kim PM, Sprecher E, Trifonov V, Gerstein M. The importance of bottlenecks in protein networks: Correlation with gene essentiality and expression dynamics. PLoS Comput Biol. 2007;3:e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bosse K, Hans CP, Zhao N, et al. Endothelial nitric oxide signaling regulates notch1 in aortic valve disease. J Mol Cell Cardiol. 2013;60:27–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Honda S, Miyamoto T, Watanabe T, et al. A novel mouse model of aortic valve stenosis induced by direct wire injury. Arterioscler Thromb Vasc Biol. 2014;34:270–278 [DOI] [PubMed] [Google Scholar]
- 169.van der Valk D, van der Ven C, Blaser M, et al. Engineering a 3d-bioprinted model of human heart valve disease using nanoindentation-based biomechanics. Nanomaterials. 2018;8:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mourino-Alvarez L, Baldan-Martin M, Gonzalez-Calero L, et al. Patients with calcific aortic stenosis exhibit systemic molecular evidence of ischemia, enhanced coagulation, oxidative stress and impaired cholesterol transport. International journal of cardiology. 2016;225:99–106 [DOI] [PubMed] [Google Scholar]
- 171.Heuschkel MA, Skenteris NT, Hutcheson JD, et al. Integrative multi-omics analysis in calcific aortic valve disease reveals a link to the formation of amyloid-like deposits. Cells. 2020;9:2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhang Y, Ma L. Identification of key genes and pathways in calcific aortic valve disease by bioinformatics analysis. J Thorac Dis. 2019;11:5417–5426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Teng P, Xu X, Ni C, Yan H, Sun Q, Zhang E, Ni Y. Identification of key genes in calcific aortic valve disease by integrated bioinformatics analysis. Medicine (Baltimore). 2020;99:e21286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Argelaguet R, Velten B, Arnol D, Dietrich S, Zenz T, Marioni JC, Buettner F, Huber W, Stegle O. Multi-omics factor analysis-a framework for unsupervised integration of multi-omics data sets. Mol Syst Biol. 2018;14:e8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Duan C, Cao Z, Tang F, Jian Z, Liang C, Liu H, Xiao Y, Liu L, Ma R. Mirna-mrna crosstalk in myocardial ischemia induced by calcified aortic valve stenosis. Aging (Albany NY). 2019;11:448–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yang Y, Wang LQ, Yao BC, Guo ZG. Ubiquitin-specific protease as the underlying gene biomarker for aortic stenosis. Lipids Health Dis. 2020;19:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Liu M, Luo M, Sun H, Ni B, Shao Y. Integrated bioinformatics analysis predicts the key genes involved in aortic valve calcification: From hemodynamic changes to extracellular remodeling. Tohoku J Exp Med. 2017;243:263–273 [DOI] [PubMed] [Google Scholar]
- 178.Loh PR, Danecek P, Palamara PF, et al. Reference-based phasing using the haplotype reference consortium panel. Nat Genet. 2016;48:1443–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and samtools. Bioinformatics. 2009;25:2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. Star: Ultrafast universal rna-seq aligner. Bioinformatics. 2013;29:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Anders S, Pyl PT, Huber W. Htseq--a python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Patro R, Mount SM, Kingsford C. Sailfish enables alignment-free isoform quantification from rna-seq reads using lightweight algorithms. Nature biotechnology. 2014;32:462–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic rna-seq quantification. Nature biotechnology. 2016;34:525–527 [DOI] [PubMed] [Google Scholar]
- 186.Pantano L, Estivill X, Marti E. A non-biased framework for the annotation and classification of the non-mirna small rna transcriptome. Bioinformatics. 2011;27:3202–3203 [DOI] [PubMed] [Google Scholar]
- 187.Proteome discoverer. 2020;2020
- 188.Cox J, Mann M. Maxquant enables high peptide identification rates, individualized p.P.B.-range mass accuracies and proteome-wide protein quantification. Nature biotechnology. 2008;26:1367–1372 [DOI] [PubMed] [Google Scholar]
- 189.Pino LK, Searle BC, Bollinger JG, Nunn B, MacLean B, MacCoss MJ. The skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom Rev. 2020;39:229–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Compound discoverer. 2020;2020
- 191.Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G. Xcms online: A web-based platform to process untargeted metabolomic data. Anal Chem. 2012;84:5035–5039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cell ranger. 2021
- 193.Purcell S, Neale B, Todd-Brown K, et al. Plink: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–913 [DOI] [PubMed] [Google Scholar]
- 195.Ongen H, Buil A, Brown AA, Dermitzakis ET, Delaneau O. Fast and efficient qtl mapper for thousands of molecular phenotypes. Bioinformatics. 2016;32:1479–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Delaneau O, Ongen H, Brown AA, Fort A, Panousis NI, Dermitzakis ET. A complete tool set for molecular qtl discovery and analysis. Nature communications. 2017;8:15452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Robinson MD, McCarthy DJ, Smyth GK. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. The perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13:731–740 [DOI] [PubMed] [Google Scholar]
- 200.Choi M, Chang CY, Clough T, Broudy D, Killeen T, MacLean B, Vitek O. Msstats: An r package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics. 2014;30:2524–2526 [DOI] [PubMed] [Google Scholar]
- 201.Xia J, Psychogios N, Young N, Wishart DS. Metaboanalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009;37:W652–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Qlucore omics explorer. 2020;2020 [Google Scholar]
- 203.Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902 e1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Loupe browser. 2021
- 205.Hernandez-de-Diego R, Tarazona S, Martinez-Mira C, Balzano-Nogueira L, Furio-Tari P, Pappas GJ Jr., Conesa A. Paintomics 3: A web resource for the pathway analysis and visualization of multi-omics data. Nucleic Acids Res. 2018;46:W503–W509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.King ZA, Drager A, Ebrahim A, Sonnenschein N, Lewis NE, Palsson BO. Escher: A web application for building, sharing, and embedding data-rich visualizations of biological pathways. PLoS Comput Biol. 2015;11:e1004321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Kamburov A, Wierling C, Lehrach H, Herwig R. Consensuspathdb--a database for integrating human functional interaction networks. Nucleic Acids Res. 2009;37:D623–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: Interactive and collaborative html5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Szklarczyk D, Gable AL, Lyon D, et al. String v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Metacore. 2020;2020 [Google Scholar]
- 211.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat Protoc. 2009;4:44–57 [DOI] [PubMed] [Google Scholar]
- 212.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ingenuity. 2020;2020
- 214.Langfelder P, Horvath S. Wgcna: An r package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Atkins SK, Singh SA, Aikawa E. Calcific aortic valve disease “omics” is timely, but are we looking too late? JACC. Basic to translational science. 2020;5:1178–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Lindeboom RG, van Voorthuijsen L, Oost KC, et al. Integrative multi-omics analysis of intestinal organoid differentiation. Mol Syst Biol. 2018;14:e8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Sider KL, Blaser MC, Simmons CA. Animal models of calcific aortic valve disease. International journal of inflammation. 2011;2011:364310. [DOI] [PMC free article] [PubMed] [Google Scholar]