

Abstract
In focal brain lesions, alterations in blood flow and cerebral metabolism can be detected in brain areas remote from the primary injury. The cellular consequences of this phenomenon, originally termed diaschisis, are not fully understood. Here, we report that in two distinct models of forebrain injury, neuronal death in the cerebellum, a site distant to the primary injury, results as consequence of neuronal loss in the forebrain. Fourteen‐day‐old rats were subjected to unilateral forebrain injury, achieved by either hypoxia‐ischemia (right carotid artery ligation and hypoxia) or direct needle injury to brain tissue. At defined times after injury, the presence of apoptosis was investigated by cell morphology, in situ end labeling, electron microscopy and poly‐ADP‐ribose polymerase (PARP) cleavage. Injury to the rat forebrain following hypoxia‐ischemia increased apoptosis in the internal granular and Purkinje cell layers of the cerebellum, a site distant to that of the primary injury. The number of apoptotic cells in the cerebellum was significantly related to cell death in the hippocampus. Similarly, direct needle injury to the forebrain resulted in extensive apoptotic cell death in the cerebellum. These results emphasize the intimate relationship between defined neuronal populations in relatively distant brain areas and suggest a cellular basis for diaschisis.
In models of perinatal hypoxic‐ischemic injury (HII; 18), acute neuronal loss in the infarct region is followed by diffuse cell death in the ischemic penumbra and throughout much of the affected hemisphere and can be observed several days after the initial insult (12). The mechanisms of delayed cerebral injury are poorly understood, but cells with the morphological features of apoptosis are widely distributed throughout the brain after HII (2, 12, 17, 18, 21, 33). These observations point to an intimate cellular relationship between defined neuronal populations relatively distant from each other.
Focal cerebral ischemia can also result in alterations in blood flow and cerebral metabolism in brain areas remote from the primary injury (19). This phenomenon, originally termed diaschisis (29), has been described in several human central nervous system pathologies (9, 11, 19). Such changes are not only widespread but can even occur in the hemisphere contralateral to injury, excluding the possibility that they arise either from partial ischemia, as in the penumbra (10), or from spreading depressions (23). Several days after cerebral infarction in humans it has been reported that blood flow decreases in the hemisphere contralateral to the primary lesion (14) and even several weeks following injury, cerebral metabolism is still impaired in the contralateral hemisphere (30). Neuronal hyperexcitability has also been reported in structurally intact brain areas remote from the lesion, including in the contralateral hemisphere (5). As with changes in blood flow, neuronal hyperexcitability can also develop several hours after injury and may persist for several months (31).
Early studies showed that cerebral ischemia can induce cell damage in projection areas unaffected by the insult and suggested retrograde degeneration of neurons distant from the site of injury (13). The survival of neurons may depend on specific neurotrophic factors secreted by cells that synapse or innervate them; this dependence on target derived survival factors is displayed by many neurons with apoptosis as the default fate of deprived neurons (22). It follows that the elimination of such target cells may also result in the apoptotic death of dependent neurons (21). These observations suggest an intimate relationship exists between neurons within different regions of the brain and led us to hypothesize that neuronal death in the forebrain as a result of HII would lead to neuronal death in brain regions distant from the primary injury. In the present study we investigated the nature of cell death in the cerebellum following injury to the developing rat forebrain. We demonstrate that unilateral forebrain injury, produced by HII or direct needle injury to forebrain tissue, induces and correlates with cell death in the cerebellum. This suggests an intimate cellular relationship between neuronal populations relatively distant from each other.
MATERIALS AND METHODS
Materials. All chemicals used were obtained from Sigma (Poole, UK) unless stated otherwise.
In vivo models of forebrain injury. Wistar dams and their litters were maintained on a 12 h cycle of light and dark with food and water freely available. Fourteen‐day‐old pups of either sex and weighing 25–34 g were removed from the litters for preparation and study, and returned to be suckled by their dams in the intervals between. All animal procedures used were in accordance with the Home Office guidelines and specifically licensed under the Animals (Scientific Procedures) Act, 1986 (UK).
Hypoxic‐ischemic injury. Anaesthesia was induced and maintained with halothane (5% and 1%–2%, respectively) in oxygen : air (1 : 1). Animals underwent ventral midline cervical incision, and permanent right carotid artery ligation, the procedure lasting 5–6 minutes. Animals were allowed to regain consciousness and were then placed in a perspex box within a standard neonatal incubator (Vickers Medical Ltd, Sidcup, UK) at 34 ± 0.5°C with a relative humidity of >80% in air. After 60 minutes, hypoxia was induced by rapidly changing the air in the incubator to 8% oxygen/92% nitrogen for 90 minutes at the same temperature and humidity, after which animals were removed and returned to their dam. At 6, 12, 24 h and 5 days following completion of hypoxia animals were sacrificed. Control animals were sham‐operated (i.e. halothane anesthesia and isolation but not ligation of the carotid artery). Further control experiments were performed in which animals were subjected to either ischemia (carotid artery ligation) or hypoxia alone (with sham ligation). These controls confirmed the lack of contribution of either ischemia alone or hypoxia alone to remote injury in the cerebellum and the validity of this model.
Needle injury to the forebrain and cerebellum. Under halothane anesthesia the skull was exposed and the interaural line visualized. A 23‐g needle was inserted through a small burr hole either into the right forebrain (3.0 mm anterior to the interaural line, 4.0 mm laterally, 3.0 mm deep) or into the right cerebellum (5.0 mm posterior to the interaural line, 2.5 mm laterally, 2.8 mm deep), and the tissue was gently aspirated. After wound closure, animals were returned to their dam. Animals were sacrificed at 24 h and 5 days after the injury. Six animals were used at each time point, with six animals as untreated controls. Brains were subsequently examined by light microscopy following in situ end labeling (ISEL) of apoptotic cells, prepared for electron microscopy, or analyzed for poly‐ADP‐ribose polymerase (PARP) cleavage by Western blotting.
Histology and in situ end labeling. Following hypoxia‐ischemia (HI) or needle injury, animals were euthanized with pentabarbitone (intraperitoneal 30 mg/kg) and transcardially perfusion‐fixed with 25–30 mL 0.9% NaCl followed by 25–30 mL paraformaldehyde (PFA, 4% w/v in 0.9% NaCl). Brains were then removed and fixed overnight in 4% PFA at 4°C and then transferred to 15% (w/v) sucrose in phosphate‐buffered saline (PBS) (4°C). Specimens were routinely dehydrated in serial alcohols and paraffin embedded before sectioning. Sections were cut at a point corresponding to between 2.6 and 3.2 mm anterior to the interaural line to show the dorsal hippocampus and neocortex, and between 2.0 and 6.0 mm posterior to interaural line to show the cerebellum (25).
ISEL was performed as described by Ansari et al (1) with minor modifications. Briefly, 5 µm coronal sections were dewaxed, followed by digestion with proteinase K (20 µg/mL) for 15 minutes at room temperature. After washing, slides were incubated in 0.3% hydrogen peroxide for 15 minutes at room temperature and then washed in PBS. Labeling mix was added (containing deoxynucleotides, biotinylated dATP and the Klenow fragment of DNA polymerase) for 60 minutes at 37°C. After further washing, slides were treated with avidin‐biotin complex (Vector Laboratories, Peterborough, UK) for 1 h at room temperature then exposed to Diaminobenzidine (DAB) and counterstained with Cole’s hematoxylin. For quantitative analysis, ISEL‐positive nuclei were counted in the right and left hippocampus (dentate gyrus) and in each lateral hemisphere of the cerebellum. Only ISEL‐positive cells with clearly labeled pyknotic or karyorrhectic nuclei and reduced cytoplasmic volume were counted as apoptotic. Cells without features of apoptosis but with cytoplasmic staining by ISEL were excluded from the quantitative analysis.
Electron microscopy. Animals were sacrificed and transcardially perfused with 0.9% NaCl. The brains were removed, the cerebellum micro‐dissected, and 1 mm3 samples were fixed in glutaraldehyde (2% in PBS) for 2 h. After washing in phosphate buffer, tissues were osmicated and dehydrated in acidified 2,2 dimethoxypropane before routine embedding in Taab® resin (TAAB Laboratories, Aldermaston, UK). Thin sections (1 µm) were cut and stained toluidine blue for block selection at the light microscope level. Sections of 100 nm thickness were then cut and collected on nickel grids, stained with uranyl acetate and lead citrate and examined by electron microscopy (CM‐10, Phillips, Netherlands).
Western blot analysis of PARP cleavage. For Western blotting analysis of brain tissue, animals were sacrificed at defined time points after the end of HI or after needle injury. The brains were removed and the hippocampus and cerebellum micro‐dissected, divided into left and right sides, and then immediately snap frozen in liquid nitrogen. Tissue was homogenized in 1% sodium dodecyl sulphate by trituration through a 21 g needle and heated to 90°C for 5 minutes. Lysates were clarified by centrifugation (15 000 g for 5 minutes) and the supernatants stored at −80°C until analysis.
Total cellular proteins (50 µg/lane) were separated on a 7.5% polyacrylamide gel and electro‐transferred onto nitrocellulose membrane (Hybond ECL, Amersham International, Bucks, UK). Membranes were blocked for 2 h at room temperature in blocking buffer (T‐TBS plus 5% non‐fat dried milk). Membranes were incubated with mouse monoclonal anti‐PARP antibody C‐2‐10 (Transduction Laboratories, Lexington, Kentucky, USA) diluted 1 : 3000 for 60 minutes at room temperature, then horseradish peroxidase‐conjugated anti‐mouse antibody (1 : 1000) under the same conditions. Blots were developed by enhanced chemiluminescence detection (Supersignal; Perbio Science, Northumberland, UK) and scanned for densitometrical analysis.
Statistical analysis. All results are expressed as mean ± SEM. Data were inspected and the appropriate transformation was applied to achieve normality (log in every case), and multiple comparisons were performed using ANOVA with Bonferroni test. Significance P‐values are <0.01, <0.001 and <0.0001.
RESULTS
We have previously shown that cerebral HII in the 14‐day‐old rat results in apoptosis in the hippocampus followed by infarction of forebrain tissue in the region of the middle cerebral artery on the side of carotid artery ligation (12, 27). In the present study, analysis of cell death in the ipsilateral dentate gyrus by ISEL confirmed that HII significantly increased the number of apoptotic cells in a time dependent manner (data not shown; 12). In the same tissues, we analyzed the effects of forebrain HII on cell death in the cerebellum, a site significantly distant from the primary insult. Forebrain HII increased the number of cells in the internal granular layer of the cerebellum displaying the morphological characteristics of apoptosis. Moreover, the majority of the nuclei in these dying cells were positively stained by ISEL. Apoptosis was confirmed at the electron microscopic level with cerebellar granule neurons (CGN) ipsilateral to forebrain HII greatly reduced in size compared to healthy CGN (Figure 1A). Their nuclei were typically small with condensed chromatin and the cytoplasmic volume of these cells was noticeably diminished with aggregations of ribosomes and evidence of vacuolation (Figure 1A, arrowhead).
Figure 1.
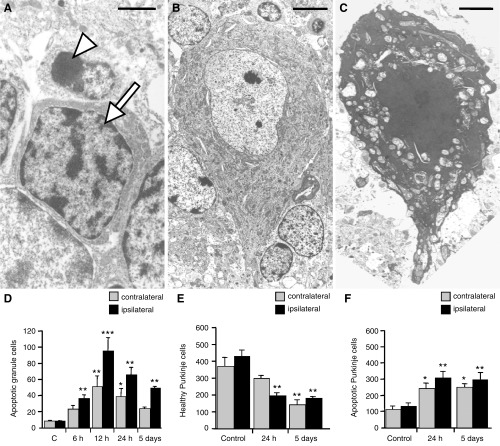
A–C. Electron micrographs of cells in the ipsilateral cerebellar hemisphere 24 h following forebrain hypoxic‐ischemic injury (HII). Healthy (arrow) cerebellar granule neurons (CGN) had large, heterochromatin‐rich nuclei, while apoptotic (arrowhead) CGN typically showed a reduced cell volume with highly condensed nuclear chromatin (A). Healthy Purkinje cells (PC) (B) contained opaque euchromatic nuclei in electron dense, organelle‐rich cytoplasm. In apoptotic PC (C) the plasma membrane and the mitochondria remained intact and nuclei were shrunken. Bars represent 0.1 µm (A), 0.5 µm (B) and 0.2 µm (C). Note that the apoptotic PC is significantly shrunken compared with its healthy counterpart. D–F. Bar graphs showing the number of apoptotic cells in the ipsilateral and contralateral cerebellum at defined times following forebrain HII. in situ end labeling staining allowed quantification of apoptotic cells in the cerebellar granule layer (D), and healthy (E) and apoptotic (F) cells in the PC layer. Results are expressed as mean ± SEM, n = 5 (controls) or n = 6 (24 h or 5 days post HII). Significance levels are shown compared with respective hemisphere of sham‐operated controls unless indicated otherwise: *P < 0.01; **P < 0.001; ***P < 0.0001.
Similarly, electron microscopy revealed significant differences in the appearance of Purkinje cells (PC) following forebrain HII. Healthy PC had large, euchromatic nuclei in a cytoplasm that was densely packed with organelles, particularly mitochondria (Figure 1B). Following cerebral HII, the nuclei of apoptotic PC appeared condensed and intensely stained, while the chromatin was highly compacted (Figure 1C). The cytoplasm was shrunken and highly electron dense, and organelles were packed more closely together because of a reduced cell volume. Significantly, while the mitochondria were dilated, most remained relatively intact (Figure 1C), a characteristic feature of cells actively undergoing apoptosis. As with the apoptotic CGN, PC cells exhibiting characteristics of apoptosis were also ISEL‐positive.
At the molecular level, apoptosis in the cerebellum after forebrain HII was investigated by measuring PARP cleavage to an 85 kDa fragment, indicative of caspase activation (12, 15, 24). In untreated animals no PARP cleavage was evident, but 12 h post HII, PARP cleavage increased in the ipsilateral hemisphere to 74.4 ± 8.2% of total PARP protein while in the contralateral hemisphere it was 36.8 ± 9.4% (12). In parallel with this observation, quantitation of the effect of forebrain injury on CGN in the ipsilateral cerebellar hemisphere indicated a tenfold increase in apoptosis 12 h post HII (Figure 1D). The number of apoptotic CGN in the contralateral hemisphere increased with time, but was lower than on the side of injury at every time point tested (Figure 1D). Healthy PC accounted for around 80% of the total population and decreased at each time after forebrain HII (Figure 1E). In addition, PC apoptosis increased on both the ipsilateral and contralateral sides at 24 h following HII, with no further increase at 5 days (Figure 1F).
Regression analysis (n = 23) revealed that at all time points following HII the number of apoptotic CGN in the ipsilateral cerebellar hemisphere was linearly related to apoptosis in the ipsilateral dentate gyrus, the contralateral dentate gyrus and to CGN apoptosis in the contralateral cerebellar hemisphere (Table 1). These results indicate that forebrain HII significantly increased apoptosis in the cerebellum, which was greater on the ipsilateral side. It should be stressed that neither ischemia alone (carotid artery ligation) nor hypoxia alone (with sham ligation) caused a significant increase in cerebellar apoptosis (data not shown).
Table 1.
Regression analysis of cell death in the cerebellum and forebrain following hypoxic‐ischemic injury (HII). Cerebellar granule neuron (CGN) apoptosis in the cerebellum was linearly related to apoptosis in the dentate gyrus (DG) at all times following HII to the forebrain (n = 23).
| CGN contralateral | DG ipsilateral | DG contralateral | |
|---|---|---|---|
| CGN ipsilateral | 0.927 (P < 0.001) | 0.768 (P < 0.001) | 0.859 (P < 0.001) |
| CGN contralateral | — | 0.068 (P < 0.01) | 0.728 (P < 0.001) |
To further investigate the possibility that cell death in the cerebellum was a consequence of neuronal death in the forebrain, we used a different model of forebrain injury. Traumatic needle injury to the right forebrain in the 14‐day‐old rat produced tissue disruption in the ipsilateral hemisphere and was associated with a significant increase in the number of apoptotic CGN in the ipsilateral cerebellar hemisphere only (Figure 2A). Quantitative analysis of the PC layer revealed a fivefold increase in the number of apoptotic PC in the ipsilateral cerebellum (523 ± 22 cells/field; P < 0.001 compared with control) (Figure 2B) which was accompanied by a corresponding decrease in the number of healthy PC. After 5 days the number of apoptotic PC in the ipsilateral side had decreased (256 ± 44 cells/field; P < 0.01 compared with 24 h), consistent with the clearance of apoptotic cells. At this time holes were apparent in the parenchyma of the ipsilateral cerebellum at 5 days post injury where PC were previously detected (data not shown).
Figure 2.
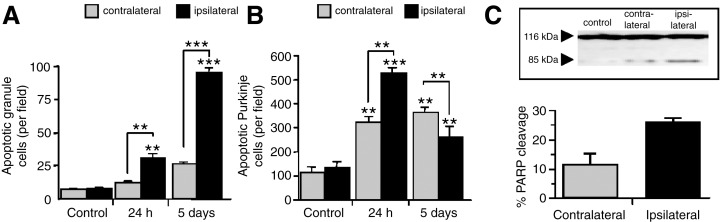
Apoptosis in rat cerebellum after traumatic needle injury to the right forebrain. Apoptotic cerebellar granule neurons (A) and Purkinje cells (B) were quantitated in the contralateral and ipsilateral cerebellar hemispheres. Results are presented as mean ± SEM (n = 6 for each group); control animals were uninjured. Representative immunoblot of poly‐ADP‐ribose polymerase (PARP) (116 kDa band) and its cleaved fragment (85 kDa) in contralateral and ipsilateral cerebellar hemispheres 24 h after right forebrain needle injury (C inset). Quantitative densitometric analysis of the 85 kDa cleaved PARP fragment PARP cleavage (C). Significantly higher PARP cleavage was observed in the cerebellar hemisphere ipsilateral to forebrain injury. Results are mean ± SD (n = 3) at each data point. Significance levels are shown compared with respective hemisphere of untreated controls unless indicated otherwise (A and B) and contralateral hemisphere (C), **P < 0.001; ***P < 0.0001.
Apoptosis was again confirmed as the mode of cell death by electron microscopy and biochemical evidence of apoptosis was obtained by analyzing PARP cleavage. Following needle injury, there was an increase in the 85 kDa PARP cleavage product in the ipsilateral cerebellum consistent with caspase activation (Figure 2C). Regression analysis of apoptosis in the cerebellum following forebrain needle injury revealed that CGN apoptosis in the ipsilateral hemisphere was significantly correlated with that in the contralateral side at all time points (r = 0.950, P < 0.001, n = 6). Similarly, the amount of PC death in the ipsilateral and contralateral cerebellar hemispheres was also linearly related (r = 0.750, P < 0.02, n = 6).
These results all point to the fact that injury to the forebrain, irrespective of the mechanism, induces a corresponding cell death in the cerebellum, emphasizing an intimate intercellular relationship between these distinct regions in the developing brain. To delineate this relationship further, a subset of animals were subjected to direct needle injury to the right cerebellar hemisphere which resulted in severe tissue disruption in this hemisphere only. Cells surrounding the site of injury were positively stained by ISEL and, significantly, unilateral cerebellar needle injury induced both CGN and PC apoptosis in the contralateral cerebellar hemisphere. The number of apoptotic CGN in this region increased to 19.8 ± 1.4 cells/field after 24 h and to 45.2 ± 3.7 cells/field after 5 days (Figure 3A). Similarly, PC apoptosis in the contralateral hemisphere increased 24 h after cerebellar injury with a corresponding reduction in the number of healthy cells (Figure 3B). Interestingly, histological examination of the forebrain of these animals revealed a small but significant increase in neuronal apoptosis in the contralateral dentate gyrus after 5 days (5 ± 0.8 cells/field; n = 6). Most significantly, CGN apoptosis in the contralateral cerebellum was significantly correlated with apoptosis in the contralateral dentate gyrus (r = 0.876, P < 0.05, n = 6).
Figure 3.
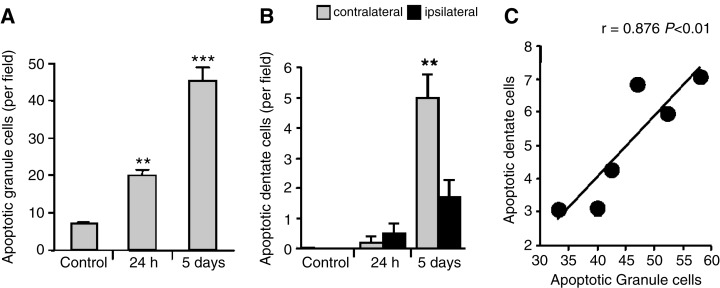
Bar graphs showing the number of apoptotic cells in the rat brain at defined times following unilateral cerebellar needle injury. Apoptotic cells were quantitated in the granular cell layer of the left cerebellar hemisphere (A), and in the contralateral and ipsilateral dentate gyrus (B) after needle injury to the right cerebellar hemisphere. Results are expressed as mean ± SEM (n = 6) in each group and significant differences were obtained between treated and control groups for each data set (P < 0.01) except for the 24 h time point. Regression analysis of the relationship between cerebellar granule neuron apoptosis in the contralateral cerebellum and the contralateral dentate gyrus following cerebellar needle injury (C); for the data presented, r = 0.876, P < 0.05 (n = 6). Significance levels are shown compared with untreated controls: **P < 0.001; ***P < 0.0001.
DISCUSSION
The present study shows that unilateral forebrain injury in the 14‐day‐old rat produces cell death with the biochemical and morphological characteristics of apoptosis in the cerebellum, a site distant from the primary injury. Apoptosis occurred in both the cerebellar granular and Purkinje cell layers, increased with time after forebrain injury, and was greater in the injured hemisphere than on the contralateral side. Moreover, direct injury to one cerebellar hemisphere induced apoptosis in the opposite hemisphere and in the forebrain. Taken together, our findings suggest that in the developing rat forebrain neurons communicate (either directly or indirectly) with both cerebellar hemispheres and that the survival of cell populations in the cerebellum can be specifically affected by the death of neurons in the hippocampus.
Cell death in the forebrain after HII is progressive, developing over a period of several days. In contrast following forebrain needle injury cell death in the same region occurs immediately. Consequently, cell death in the cerebellum proceeds more rapidly after needle injury compared with HI. This may account for the eventual appearance of holes (indicative of cell loss) in the Purkinje cell layer 5 days after needle injury but not after HII.
Notably, both HII and forebrain needle injury increased apoptosis in the cerebellum, suggesting that the latter was in fact a consequence of neuronal death in the forebrain. This is further supported by the direct relationship between apoptosis in the forebrain and cerebellum after HII and needle injury, and the observation that direct injury to the right cerebellar hemisphere produced increased apoptosis in the left forebrain. Neuronal death in the forebrain following HII peaked at 12 h whereas cell death in the cerebellum increased up to 5 days post injury suggesting delayed neurodegeneration in this region. On‐going cell death is secondary to the loss of essential trophic support for regions with significant interconnections to the area of primary injury. Other studies have shown that cell death occurs in brain regions that are not directly affected by perinatal HII (19, 21, 32), suggesting that neuronal connectivity plays a role in neurodegeneration following HII to the immature brain.
Taken together, these findings suggest that there is an intimate relationship between the forebrain and cerebellum in the developing rat brain. In primates and humans, the cerebral cortex makes numerous neural connections with the cerebellum, via the pons (3, 4). It follows that mechanical or functional interruption of this corticopontocerebellar pathway will affect activity in the ipsilateral pons, and then the contralateral cerebellar cortex, a phenomenon known as crossed cerebellar diaschisis (11). In the rat brain, however, this pathway has not been demonstrated, and so the ipsilateral cortex may be linked to ipsilateral cerebellum rather than the contralateral (6). In this context, damage to the cerebral cortex would lead to damage in the ipsilateral cerebellum, and then in the contralateral hemisphere. Consistent with this possibility, we found that injury to the right forebrain induced apoptosis in both cerebellar hemispheres; however, apoptosis was greater (and occurred earlier) on the side of injury. Although each cerebral hemisphere communicates predominantly with the contralateral cerebellar hemisphere, many forebrain neurons connect via the ipsilateral cerebellum (28). One possible scenario is that damage to the forebrain leads to secondary damage in the ipsilateral cerebellum and then to tertiary damage in the contralateral hemisphere.
In addition to cell morphology, apoptosis in the cerebellum was demonstrated at a biochemical level by PARP cleavage to its signature 85 kDa fragment which is not a non‐specific feature of cell death, but a marker of caspase activation (12, 24). After forebrain injury, PARP cleavage was observed in both cerebellar hemispheres, but was greater on the side of injury, consistent with the increased apoptotic cell death in this region. Significantly, PARP cleavage in the hippocampus and cerebellum occurred immediately after HII, preceding the appearance of apoptotic cells in both regions (12).
The close relationship between apoptosis in the forebrain and the cerebellum confirms that these distinct neuronal populations may be connected and dependent on one another. Similarly, it has been demonstrated that unilateral intrahippocampal administration of the muscarinic receptor agonist pilocarpine also causes changes in the extracellular levels of excitatory amino acids in the cerebellum (26). In agreement with the results of the present study, kainate‐induced forebrain lesions in the rat lead to neuronal degeneration in the Purkinje cell layer of the ipsilateral cerebellar hemisphere (16).
Although the precise mechanisms involved in remote neuronal death in the developing brain following HII remain undefined, possible mechanisms could include (i) neuronal hyperexcitability, which has been observed in brain areas remote from the lesion, including in the contralateral hemisphere (5) and, like delayed cell death, can develop several hours after injury and persist for several months (31); (ii) decreased excitatory input to the cerebellar cortex which has been shown to decrease cerebellar blood flow and metabolism (8); or (iii) loss of survival signals secreted by cells that synapse or innervate them, as a dependence on target derived survival factors is displayed by many neurons with apoptosis as the default fate of deprived neurons (20, 22).
Our results also suggest that in the developing rat brain neural connections run in the reverse direction, from the cerebellum to the contralateral hippocampus. Similarly, it has been shown that stimulation of the fastigial nucleus in the cerebellum results in forebrain conditioning against injury, with protection of hippocampal pyramidal neurons from delayed neuronal death after global ischemia (7). This further highlights the complex cellular relationship between remote areas of the brain.
In summary, this study suggests that apoptosis in the cerebellum is a consequence of neuronal loss in the forebrain, and emphasize the fact that injury to one brain area can cause widespread neuronal loss in distant neuronal populations. The precise mechanism of this delayed cell death in brain areas remote to the site of injury deserves further investigation.
ACKNOWLEDGMENTS
We are grateful to Viv Eamons (Department of Histopathology, Imperial College London) for excellent technical assistance with electron microscopy. This work was funded by the Wellcome Trust UK (DLT, grant No. 046343/z/95), Action Research (Clinical Training Fellowship for UCJ) and the Garfield Weston Foundation (ADE and HM).
REFERENCES
- 1. Ansari B, Coates PJ, Greenstein BD, Hall PA (1993) In situ end‐labelling detects DNA strand breaks in apoptosis and other physiological and pathological states. J Pathol 179:1–8. [DOI] [PubMed] [Google Scholar]
- 2. Beilharz EJ, Williams CE, Dragunow M, Sirimanne ES, Gluckman PD (1995) Mechanisms of delayed cell death following hypoxic‐ischemic injury in the immature rat: evidence for apoptosis during selective neuronal loss. Brain Res Mol Brain Res 29:1–14. [DOI] [PubMed] [Google Scholar]
- 3. Brodal A (1981) Neurological Anatomy in Relation to Clinical Medicine. Oxford University Press: New York. [Google Scholar]
- 4. Brodal P (1978) The corticopontine projection in the monkey: origin and principles of organization. Brain 101:251–283. [DOI] [PubMed] [Google Scholar]
- 5. Buchkremer‐Ratzmann I, August M, Hagemann G, Witte OW (1996) Electrophysiological transcortical diaschisis after cortical photothrombosis in rat brain. Stroke 27:1105–1109. [DOI] [PubMed] [Google Scholar]
- 6. Feeney DM, Baron JC (1986) Diaschisis. Stroke 17:817–830. [DOI] [PubMed] [Google Scholar]
- 7. Golanov EV, Liu F, Reis DJ (1998) Stimulation of cerebellum protects hippocampal neurons from global ischemia. Neuroreport 9:819–824. [DOI] [PubMed] [Google Scholar]
- 8. Gold L, Lauritzen M (2002) Neuronal deactivation explains decreased cerebellar blood flow in response to focal ischemia or suppressed neocortical function. PNAS 99:7699–7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hamano S, Nara T, Nakanishi Y, Horita H, Kumagai K, Maekawa K (1993) Secondary changes in cerebellar perfusion (diaschisis) in hemiplegia during childhood: SPECT study of 55 children. Pediatr Neurol 9:435–443. [DOI] [PubMed] [Google Scholar]
- 10. Hossmann KA (1994) Viability thresholds and the penumbra of focal ischemia. Ann Neurol 36:557–565. [DOI] [PubMed] [Google Scholar]
- 11. Infeld B, Davis SM, Lichtenstein M, Mitchell PJ, Hopper JL (1995) Crossed cerebellar diaschisis and brain recovery after stroke. Stroke 26:90–95. [DOI] [PubMed] [Google Scholar]
- 12. Joashi UC, Greenwood K, Taylor DL, Kozma M, Mazarakis ND, Edwards AD, Mehmet H (1999) Poly(ADP ribose) polymerase cleavage precedes neuronal death in the hippocampus and cerebellum following injury to the developing rat brain. Eur J Neurosci 11:91–100. [DOI] [PubMed] [Google Scholar]
- 13. Kataoka K, Hayakawa T, Yamada K, Mushiroi T, Kuroda R, Mogami H (1989) Neuronal network disturbance after focal ischemia in rats. Stroke 20:1226–1235. [DOI] [PubMed] [Google Scholar]
- 14. Lagreze HL, Levine RL, Pedula KL, Nickles RJ, Sunderland JS, Rowe BR (1987) Contralateral flow reduction in unilateral stroke: evidence for transhemispheric diaschisis. Stroke 18:882–886. [DOI] [PubMed] [Google Scholar]
- 15. Lazebnik YA, Kaufmann SH, Sesnoyers S, Poirier GG, Earnshaw WC (1994) Cleavage of poly(ADP‐ribose) polymerase by a proteinase with properties like ICE. Nature 371:346–347. [DOI] [PubMed] [Google Scholar]
- 16. Liu HM, Lei DL, Yang DL (1996) Kainate‐induced brain lesion: similar local and remote histopathological and molecular changes as in ischemic brain infarct. J Neuropathol Exp Neurol 55:787–794. [PubMed] [Google Scholar]
- 17. MacManus JP, Buchan AM, Hill IE, Rasquinha I, Preston E (1993) Global ischaemia can cause DNA fragmentation indicative of apoptosis in rat brain. Neurosci Lett 164:89–92. [DOI] [PubMed] [Google Scholar]
- 18. Mehmet H, Yue X, Squier MV, Lorek A, Cady E, Penrice J, Sarraf C, Wylezinska M, Kirkbride V, Cooper C, Brown GC, Wyatt JS, Reynolds EOR, Edwards AD (1994) Increased apoptosis in the cingulate sulcus of newborn piglets following transient hypoxia‐ischaemia is related to the degree of high energy phosphate depletion during the insult. Neurosci Lett 181:121–125. [DOI] [PubMed] [Google Scholar]
- 19. Meyer JS, Obara K, Muramatsu K (1993) Diaschisis. Neurol Res 15:362–366. [DOI] [PubMed] [Google Scholar]
- 20. Northington FJ, Ferriero DM, Flock DL, Martin LJ (2001) Delayed neurodegeneration in neonatal rat thalamus after hypoxia‐ischemia is apoptosis. J Neurosci 21:1931–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Northington FJ, Ferriero DM, Graham EM, Traystman RJ, Martin LJ (2001) Early neurodegeneration after hypoxia‐ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol Dis 8:207–219. [DOI] [PubMed] [Google Scholar]
- 22. Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD (1993) Programmed cell death and the control of cell survival: lessons from the nervous system. Science 262:695–700. [DOI] [PubMed] [Google Scholar]
- 23. Schroeter M, Schiene D, Kraemer M, Hagemann G, Weigel H, Eysel UT, Witte OW, Stoll G (1995) Astroglial responses in photochemically induced focal ischemia of the rat cortex. Exp Brain Res 106:1–6. [DOI] [PubMed] [Google Scholar]
- 24. Shah GM, Shah RG, Poirier GG (1996) Different cleavage patttern for poly(ADP‐ribose) polymerase during necrosis and apoptosis in HL‐60 cells. Biochem Biophys Res Commun 229:838–844. [DOI] [PubMed] [Google Scholar]
- 25. Sherwood NM, Timiras PS (1970) A Stereotaxic Atlas of the Developing Rat Brain. University of California Press: Los Angeles. [Google Scholar]
- 26. Smolders I, Van Belle K, Ebinger G, Michotte Y (1997) Hippocampal and cerebellar extracellular amino acids during pilocarpine‐induced seizures in freely moving rats. Eur J Pharmacol 319:21–29. [DOI] [PubMed] [Google Scholar]
- 27. Taylor DL, Mehmet H, Cady EB, Edwards AD (2002) Improved neuroprotection with hypothermia delayed by 6 hours following cerebral hypoxia‐ischaemia in the 14‐day‐old rat. Pediatr Res 51:13–19. [DOI] [PubMed] [Google Scholar]
- 28. Thach WT, Goodkin HP, Keating JG (1992) The cerebellum and the adaptive coordination of movement. Annu Rev Neurosci 15:403–442. [DOI] [PubMed] [Google Scholar]
- 29. Von Monakow C (1895) Experimentelle und pathologisch‐anato‐mische Untersuchungen über die Haubenregion, Sehhügel und die Regio subthalamica, nebst Beiträgen zur Kenntnis früh erworbener Groß‐ und Kleinhirndefekten. Arch Psychtrie Nervenkrankheiten 27:1–128. [Google Scholar]
- 30. Wise R, Gibbs J, Frackowiak R, Marshall J, Jones T (1986) No evidence for transhemispheric diaschisis after human cerebral infarction. Stroke 17:853–861. [DOI] [PubMed] [Google Scholar]
- 31. Witte OW, Stoll G (1997) Delayed and remote effects of focal cortical infarctions, secondary damage and reactive plasticity. Adv Neurol 73:207–227. [PubMed] [Google Scholar]
- 32. Young C, Tenkova T, Dikranian K, Olney JW (2004) Excitotoxic versus apoptotic mechanisms of neuronal cell death in perinatal hypoxic/ischemia. Curr Mol Med 4:77–85. [DOI] [PubMed] [Google Scholar]
- 33. Yue X, Mehmet H, Penrice J, Cooper C, Cady E, Wyatt JS, Reynolds EOR, Edwards AD, Squier MV (1997) Apoptosis and necrosis in the newborn piglet brain following transient cerebral hypoxia‐ischaemia. Neuropathol Appl Neurobiol 23:16–25. [PubMed] [Google Scholar]