

Abstract
Autosomal dominant Emery–Dreifuss muscular dystrophy is caused by mutations in the LMNA gene that code for the nuclear membrane protein lamin A/C. We investigated skeletal muscle fibers from several muscles for cytoplasmic degenerative changes in three patients with genetically confirmed Emery–Dreifuss muscular dystrophy. Methods included quantitative light and electron microscopy and PCR‐based mutational analysis. Results: The degenerative pathway was characterized by the gradual replacement of individual myofibers by connective tissue. Early stages of degeneration typically involved only a segment of the cross‐sectional area of a myofiber. Intermediate stages consisted of myofiber shrinkage due to “shedding” of peripheral cytoplasmic portions into the endomysial space, and fragmentation of the myofibers by interposed collagen fibrils. Empty basement membrane sheaths surrounded by abundant deposits of extracellular matrix marked the end stage of the degenerative process. The nuclear number‐to‐cytoplasmic area in myofibers of one patient increased with increasing cross‐sectional area, suggesting that satellite cell fusion with myofibers may have compensated for myofiber shrinkage. The pattern of degeneration described herein differs from muscular dystrophies with plasma membrane defects (dystrophinopathy, dysferlinopathy) and explains the frequently found absence of highly elevated serum creatine kinase levels in autosomal dominant Emery–Dreifuss muscular dystrophy.
INTRODUCTION
The LMNA gene encodes lamins A and C (2), which arise from alternative splicing introducing alternative 3′ ends in the LMNA transcript. Lamins A and C belong to the intermediate filament family and form the nuclear lamina, a mesh‐like structure associated with the nucleoplasmic side of the inner nuclear membrane (25, 28). “Laminopathies” caused by mutations of the LMNA gene encompass nine diseases that specifically affect skeletal and/or cardiac muscle, adipose tissue, and peripheral nervous tissue. In addition, LMNA has been implicated in premature aging syndromes (1, 25, 28). Three laminopathies affect skeletal muscle: autosomal dominant Emery–Dreifuss muscular dystrophy (AD‐EDMD), a rare autosomal recessive form (AR‐EDMD) and autosomal dominant limb girdle muscular dystrophy associated with cardiac conduction defects (LGMD1B). Mutations result in misfolding of the protein or failure to assemble correctly, leading to partial or complete loss of protein function (1, 25, 28).
The mechanism underlying these diseases remains to be established. The proposed pathophysiologic mechanisms of laminopathies include nuclear organization, nuclear fragility due to mechanical stress, combination of structural and signaling mechanisms, regulation of transcription, functions of the endoplasmic reticulum, altering cell cycle and replication, and effects on differentiation of stem cell lineages (13, 25, 28).
The pathomechanism leading to myofiber degeneration has not been previously elucidated. In the present study, we investigated skeletal muscle fibers from mildly to severely affected muscles for degenerative changes in three patients with genetically confirmed AD‐EDMD.
METHODS
Patient histories
Case 1. The patient’s history was in accordance with the consensus diagnostic criteria established by the European Neuromuscular Center (ENMC)‐supported International Consortium on EDMD (4, 16). His family history was negative. When first examined at 4 years of age, generalized weakness was noted. No contractures were noted at that time. An electrocardiogram (ECG) was normal, and the electromyogram (EMG) showed pathological changes which were interpreted to be neurogenic. Laboratory studies showed a mild creatine kinase (CK) increase (91 U/L; normal ≤70 U/L).
Later in the course of his disease, contractures of the Achilles tendons and elbow joints developed along with rigidity of the spine. Right septal atrial fibrillation and a third degree AV block with bradycardia were detected, which required implantation of a ventricular demand, nonphysiologic pacemaker at age 23. CK and CK‐MB levels were slightly elevated (85 U/L and 15 U/L, respectively). Death occurred at the age of 26 following cardiac arrest.
Case 2. This patient’s family history was negative for neuromuscular diseases. Contractures were observed in both elbows starting age 12. At 16, the neurological examination showed incipient scapular winging and an incipient pelvic contracture. ECG showed frequent polymorphic extrasystoles (Lown IIIb) and frequent supraventricular extrasystoles. Transthoracic echocardiography at age 16 gave normal results. The CK was normal or slightly elevated (2.5 µmol/L × s and 6.5 µmol/L × s (normal: ≤2.6)) (12).
Case 3. This patient is the son of healthy unrelated parents with two healthy siblings. Neurological examination at age 8 revealed proximal weakness of the legs, lumbar lordosis, and marked scapular winging. Slight ankle and elbow contractures were noted, tendon reflexes were reduced. There was a slight increase of CK activity (100 U/L, normal: ≤70). EMG of the right anterior tibial muscle and the left deltoid muscle revealed a neurogenic pattern. ECG and echocardiography were normal. A muscle biopsy specimen was taken from the right quadriceps muscle when the boy was 8 years old. In the following 3 years, muscle weakness was slowly progressive. At age 11, contractures of elbows were more marked, while the Achilles tendons were only mildly shortened. There was slight hyperlordosis. The patient was slim; however, his muscle bulk appeared to be preserved. The muscle strength (Medical Research Council) level of shoulder and pelvic girdle muscles was reduced to 4–5. This was the first time that ECG showed a I° AV block.
Mutational analysis. The PCR‐based mutational analysis was essentially performed as previously described (3).
Light and electron microscopy
Case 1. A biopsy specimen from the right vastus medialis muscle taken at the age of 4 was held at rest length and fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.2) and was prepared for electron microscopy. Post‐mortem samples from the midbelly regions of the right deltoid, left biceps and right vastus medialis muscles were processed in the same way.
The nuclei of 200 myofibers from each of the three muscles sampled post‐mortem were counted at the light microscopical level using a 60× objective. We measured the smallest diameter of each myofiber in order to calculate the myofiber’s cross‐sectional area using the KS 400 Imaging System (Release 3.0, Carl Zeiss Vision GmbH, Oberkochen, Germany).
Satellite cells were identified electron microscopically based on their location between the basement membrane and the plasma membrane of the adjacent myofiber. The numbers of satellite cells and myonuclei were counted at 10 000× magnification. The ratio of satellite cells was assessed according to this formula: number of satellite cells/total number of nuclei (nuclei of satellite cells plus myonuclei).
Case 2. A biopsy specimen was taken from the right biceps brachii muscle and processed for enzyme histochemistry and electron microscopy (12).
Case 3. A biopsy specimen was taken from the right quadriceps muscle and processed for enzyme histochemistry and electron microscopy.
RESULTS
Mutational analysis
Case 1. A LMNA base substitution c.746G>A in codon 249 was found leading to an amino acid exchange p.R249Q in the second rod domain of both lamins A and C. This mutation was not detected in the patient’s parents. The disease‐related significance of this mutation was validated in other sporadic cases with the same base substitution (3, 5, 6, 7, 9, 14, 15).
Case 2. This patient showed a paternal nonsense mutation in addition to a genetic variant in the maternal allele of the LMNA gene. The pathogenic mutation is an insertion, c.447insC which generates a deduced premature stop c.459X resulting in a putatively truncated lamin A/C peptide (p.Ser149_Thr150delinsThrfsX4). The patient’s father and brother were heterozygotic for the c.447insC mutation.
The patient’s father suffered a sudden cardiac death without skeletal muscle involvement, while the brother was still unaffected when last contacted at 19 years.
The maternal base substitution c.1201C>T in codon 401 led to an amino acid exchange p.R401C in the tail domain of lamins A and C and was classified as a genetic variant of unknown significance, because the patient’s clinically unaffected mother and sister also harbored the p.R401C mutation (12, 27).
Case 3. The patient was a heterozygote for a hitherto undescribed missense mutation of the LMNA gene. A base substitution c.254T>C in codon 85 of exon 1 was found leading to a deduced amino acid exchange L85P in the α‐helical rod domain of both lamins A and C. This mutation was not detected in the patient’s parents, underlining the pathogenic nature of the de novo mutation. A similar missense mutation (c.254T>G) of the same codon 85 was previously reported in a family with cardiac conduction disturbances and dilated cardiomyopathy but without muscle involvement (10).
Light and electron microscopy
Case 1. The vastus medialis muscle biopsy showed a chronic myopathic pattern with abnormal variation of fiber size and mildly increased endomysial and perimysial connective tissue. Necrosis, myophagocytosis and basophilic fibers were absent (not shown).
Less than 1% of the myofibers showed a loss of peripheral cytoplasmic material when viewed at the cross‐sectional level (Figure 1A,F). Peripheral cytoplasmic fragments were separated from their parent myofiber by extracellular matrix. Hence, the myofiber appeared to “shed” cytoplasmic portions into the endomysial space (Figure 1A,F).
Figure 1.
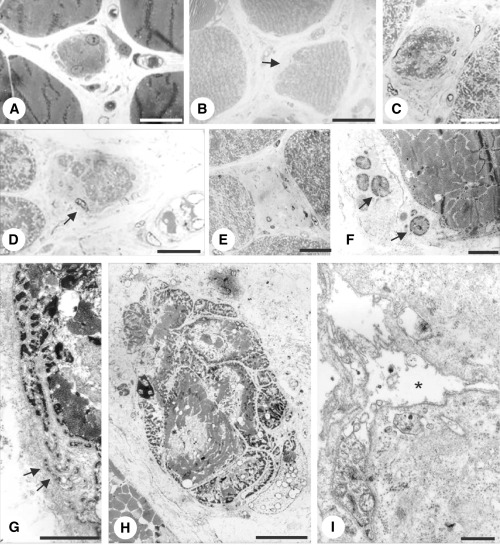
Case 1. Myofibers in early (A,B,F,G), intermediate (C,D,H) and late (E,I) stages of degeneration. A,F. Biopsy at 4 years. Right vastus medialis muscle. B–E,G–I. Autopsy specimens. B,G–I. Left biceps muscle. C–E. Right deltoid muscle. A. The cytoplasm of the myofiber is fragmented at the periphery (arrow). B. The myofiber’s surface is indented on the left side and replaced by extracellular matrix. C,D. Intermediate stages. The cross‐sectional area is reduced. The myofibers are surrounded by deposits of extracellular matrix. Note small cytoplasmic fragments separated from the parent myofiber by extracellular matrix in (D) (arrow). E. End stage. Deposits of collagen and fibroblasts occupy endomysial space among myofibers. F,G.“Shedding” of cytoplasmic fragments (arrowheads). H. Myofiber fragmented by streaks of extracellular matrix. I. End stage. Basement membrane sheath containing few cytoplasmic fragments is surrounded by deposits of extracellular matrix.
Muscles sampled at autopsy also showed variable pathological features consisting of a chronic myopathic pattern. The deltoid muscle was more severely affected than the biceps or the vastus medialis muscle (not shown). Loss of peripheral cytoplasmic portions and deposits of extracellular matrix characterized the early degenerative stages (Figure 1B). Intermediate stages of degeneration consisted of a significant reduction of the myofiber’s cross‐sectional area (Figure 1C,D). The residual portions of the fiber were normal. The myofiber’s cytoplasm eventually disappeared (Figure 1E). “Shedding” of peripheral cytoplasmic portions was observed in early stages of degeneration (Figure 1G) similar to the biopsied specimen. Occasionally, the cross‐sectional area of a myofiber was intersected by streaks of extracellular matrix (Figure 1H). Deposits of extracellular matrix surrounded the atrophic myofibers (Figure 1C,D). In the end stage of degeneration the myofiber disappeared and was replaced by deposits of extracellular matrix (Figure 1E,I). The end stage of myofiber degeneration was marked by basement membrane sheaths surrounded by deposits of extracellular matrix (Figure 1I). The percentage of degenerating myofibers (as in Figure 1B–D) or degenerated ones (as in Figure 1E) was 7.5%, 1% and 0.5% in the right deltoid, left biceps and right vastus medialis muscles, respectively.
Almost all myofibers of the gastrocnemius muscle were replaced by connective tissue and fat (not shown).
Case 2. The biopsy revealed a mild myopathic pattern with the main findings consisting of fiber diameters ranging from 10 to 80 µm and a slight increase of endomysial connective tissue (12). Examination of the resin‐embedded material demonstrated degeneration of approximately 1% of the myofibers. As noted in case 1, early degeneration was characterized by indentations over a small segment of the fibers’ circumference (Figure 2A,B). Partial cytoplasmic loss resulted in empty basement membrane sheaths early in the course of degeneration (Figure 2C). Intermediate stages showed a fragmentation of myofibers (Figure 2D,E). The deposits of extracellular matrix predominated over myofiber remnants in late stages of degeneration (Figure 2F–H).
Figure 2.
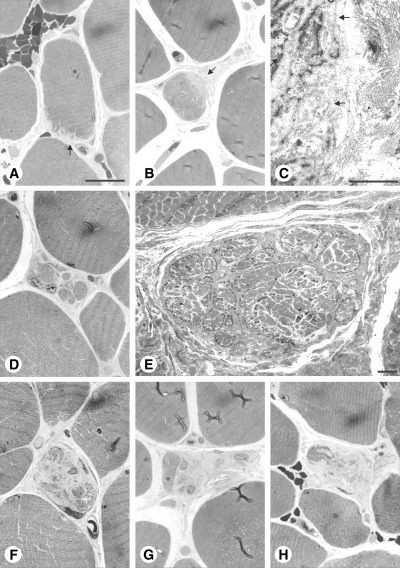
Case 2. Right biceps brachii muscle. Early (A–C), intermediate (D–E) and late stages (F–H) of myofiber degeneration. A,B. The cytoplasm of the myofibers is fragmented at the periphery over a small segment of the fibers’ circumference (arrows). C. Higher magnification of (B). Note empty basement membrane sheaths in (C) (arrows). D,E. The cross‐sectional area of these myofibers is fragmented by interposed collagen fibrils. F–H. Collagen fibrils have replaced most of the myofibers’ cross‐sectional area. Scale bars 30 µm (A) and 3 µm (C,E).
Case 3. The biopsy showed a mild mixed myopathic and neurogenic pattern with few angular atrophic and round hypotrophic fibers. Examination of the resin‐embedded material revealed degeneration of two myofibers. The ultrastructural findings of the two fibers represented an intermediate type of degeneration with “shedding” of a substantial number of fragments (Figure 3A) and fragmentation (Figure 3B). Spaced serial sections of the myofiber depicted in Figure 3A demonstrates the gradual replacement of the myofiber by connective tissue (Figure 4).
Figure 3.
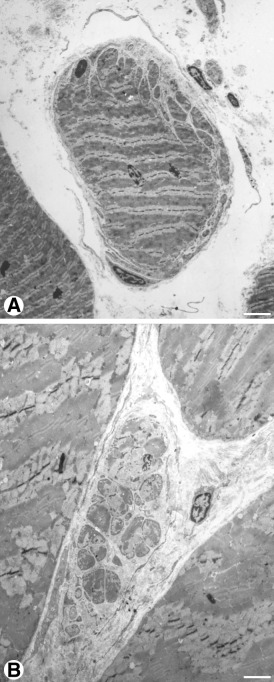
Case 3. Right quadriceps muscle. Intermediate stage of myofiber degeneration. A. There is abundant “shedding” of cytoplasmic fragments in this myofiber. B. This myofiber is fragmented. Scale bars 5 µm (A) and 3 µm (B).
Figure 4.
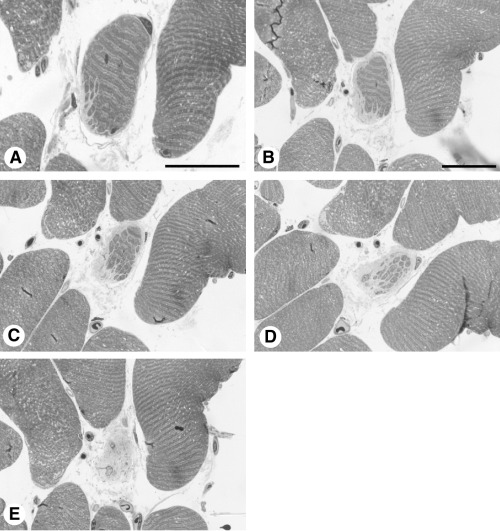
Case 3. Spaced serial sections of the myofiber depicted in Figure 3A to demonstrate gradual replacement of the myofiber by connective tissue. Scale bars 30 µm.
Myonuclei
Case 1. The number of myonuclei was assessed in the autopsied specimens of this patient because thick sections showed an excess of nuclei within many myofibers. Most nuclei were in a subsarcolemmal position; only a small number was centrally located (not shown). Myonuclear counts of the post‐mortem muscle samples revealed an increase of the nuclear number‐to‐cytoplasmic area with increasing cross‐sectional area of a myofiber (Figure 5).
Figure 5.
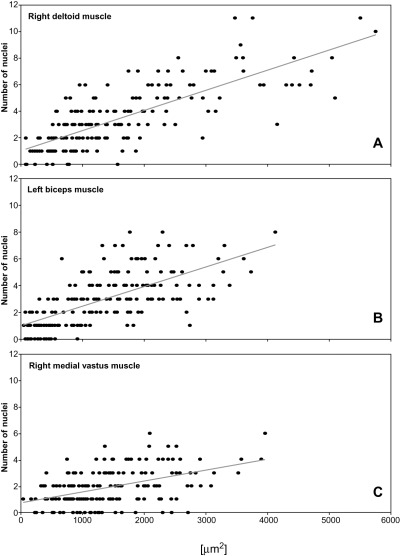
Case 1. The cross‐sectional area of an individual myofiber is plotted against the total number of its nuclei to compare the nuclear number‐to‐cytoplasmic area between myofibers of different sizes. Each dot represents a muscle fiber (n = 200). Regression lines indicate the slope trend in each muscle. The nuclear number‐to‐cytoplasmic area increases with increasing myofiber cross‐sectional area.
Satellite cells. The ratio of satellite cells was assessed to determine whether myofiber nuclei or satellite cells accounted for the increase in nuclei. Three percent of the total nuclei (ie, satellite cells plus myonuclei) belonged to satellite cells in the biceps muscle. Accordingly, the ratio of satellite cells was in the normal range (22). The same was true for the vastus medialis muscle (2%). This indicates that an increased number of myonuclei rather than satellite cells accounted for the increased nuclear number‐to‐cytoplasmic area. The ratio of satellite cells was elevated in the deltoid muscle (16%). Hence, in this muscle both satellite cells and myonuclei contributed to the increased nuclear number‐to‐cytoplasmic area.
DISCUSSION
Cytoplasmic degeneration. The degenerative pathway resulted in excess deposits of extracellular matrix at the expense of the myofibers’ cytoplasmic portions (1, 2, 3, 4). The basement membrane was left intact (1, 2). This process started at the myofiber’s surface (1, 2, 4). The atrophic process was marked by “shedding” of fragments (1, 3) and fragmentation (1, 2, 3, 4). We used the term “shedding” to describe cytoplasmic fragments separated from the parent myofiber by interposed collagen fibrils at the level studied (1, 3). This pattern does not rule out the presence of cytoplasmic bridges at other levels of the specimen. As there were no plasma membrane defects and no exocytosis to account for the loss of cytoplasmic material, we suggest that the myofiber in AD‐EDMD is not able to generate sufficient protein turnover necessary to maintain its structural integrity.
The morphologic features observed in these three patients is distinct from muscular dystrophies characterized by cycles of necrosis and regeneration such as dysferlinopathy and dystrophinopathy (18, 21, 23). In contrast to the findings in our patient, the degenerative pathway observed in early stages of dysferlinopathy is characterized by plasma membrane defects and exocytosis (23). Duchenne type of muscular dystrophy, the severe form of dystrophinopathy, also shows plasma membrane defects (18, 21). On the other hand, “shedding” and deposits of residual basement membranes were not found in dysferlinopathy or dystrophinopathy (18, 21, 23).
The frequency of changes in patients with mutations of the LMNA gene remains to be established. These changes are probably not seen in all biopsied cases of AD‐EDMD. Previous electron microscopic studies focusing on the nuclear pathology did not mention similar cytoplasmic changes (20, 24), and a recent case report described features of myofibrillar myopathy in a child with a de novo mutation of the LMNA gene (8).
Nuclear number‐to‐cytoplasmic area. Based on our observations, we found an elevated number of myonuclei in the autopsied muscles of the first patient. Quantification revealed up to 12 nuclei in individual myofibers. The number of myonuclei rose with increasing cross‐sectional area (Figure 5). The findings in our patient are different from normal fiber growth during myogenesis. In normal myofiber growth the nuclear number‐to‐cytoplasmic area decreases with increasing fiber diameter (19). The findings also differ from myofiber hypertrophy in healthy individuals. Strength‐trained athletes retain a constant nuclear number‐to‐cytoplasmic area when their myofibers undergo hypertrophy (14). LMNA gene knock‐out mice also showed an increased number of myonuclei (26), suggesting that the finding is related to the LMNA gene mutation. The pathomechanism leading to an increased number of myonuclei following LMNA loss of function remains to be established. As myonuclei in mature muscle fibers are not able to divide, it is suggested that satellite cells are incorporated into muscle fibers in an attempt to compensate for volume loss. A recent study using permanent myoblast cell lines of LMNA–/– mouse muscle demonstrated a delay in myoblast differentiation, resulting in impaired myotube formation in vitro (11). However, myogenesis in regenerating muscle of LMNA null mice is efficient (17), suggesting that satellite cells function normally in vivo.
In conclusion, we propose a degenerative pathway of muscular dystrophies characterized by an absence of high serum CK levels. Whether this is a direct or an indirect sequel of the mutation of the LMNA gene remains to be clarified.
ACKNOWLEDGMENTS
We wish to thank Elizabeth Rushing, Washington, DC, for her help with the English, and Suse Renkhold, for technical assistance. Financial help by the Brain‐Net Germany is acknowledged. AB and MW were supported by grants of the Deutsche Forschungsgemeinschaft (Bo 992/5‐1, 992/5‐2; We 1470/12‐3). GB and MW were supported by an EU‐grant (Euro‐Laminopathies contract ♯018690). MW and RK are members of the German network on muscular dystrophies (MD‐NET, 01GM0601) funded by the German Ministry of Education and Research (BMBF, Bonn, Germany). We also pay respect to patient 1 who in his lifetime made a conscious decision to contribute to the advancement of knowledge by dedicating his body to postmortal examination.
Dedicated to Hans Hilmar Goebel on the occasion of his 70th birthday.
REFERENCES
- 1. Ben Yaou BR, Muchir A, Arimura T, Massart C, Demay L, Richard P, Bonne G (2005) Genetics of laminopathies. Novartis Found Symp 264:81–97. [PubMed] [Google Scholar]
- 2. Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, Duboc D, Fardeau M, Toniolo D, Schwartz K (1999) Mutations in the gene encoding lamin A/C cause autosomal dominant Emery‐Dreifuss muscular dystrophy. Nat Genet 21:285–288. [DOI] [PubMed] [Google Scholar]
- 3. Bonne G, Mercuri E, Muchir A, Urtizberea A, Becane HM, Recan D, Merlini L, Wehnert M, Boor R, Reuner U, Vorgerd M, Wicklein EM, Eymard B, Duboc D, Penisson‐Besnier I, Cuisset JM, Ferrer X, Desguerre I, Lacombe D, Bushby K, Pollitt C, Toniolo D, Fardeau M, Schwartz K, Muntoni F (2000) Clinical and molecular genetic spectrum of autosomal dominant Emery‐Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol 48:170–180. [PubMed] [Google Scholar]
- 4. Bonne G, Ben Yaou RB, Beroud C, Boriani G, Brown S, De Visser M, Duboc D, Ellis J, Hausmanowa‐Petrusewicz I, Lattanzi G, Merlini L, Morris G, Muntoni F, Opolski G, Pinto YM, Sangiuolo F, Toniolo D, Trembath R, Van Berlo JH, Van der Kooi AJ, Wehnert M (2003) 108th ENMC International Workshop, 3rd Workshop of the MYO‐CLUSTER project EUROMEN, 7th International Emery‐Dreifuss Muscular Dystrophy (EDMD) Workshop, 13–15 September 2002, Naarden, The Netherlands. Neuromuscul Disord 13:508–515. [DOI] [PubMed] [Google Scholar]
- 5. Boriani G, Gallina M, Merlini L, Bonne G, Toniolo D, Amati S, Biffi M, Martignani C, Frabetti L, Bonvicini M, Rapezzi C, Branzi A (2003) Clinical relevance of atrial fibrillation/flutter, stroke, pacemaker implant, and heart failure in Emery‐Dreifuss muscular dystrophy: a long‐term longitudinal study. Stroke 2003:901–908. [DOI] [PubMed] [Google Scholar]
- 6. Brown CA, Lanning RW, McKinney KQ, Salvino AR, Cherniske E, Crowe CA, Darras BT, Gominak S, Greenberg CR, Grosmann C, Heydemann P, Mendell JR, Pober BR, Sasaki T, Shapiro F, Siimpson DA, Suchowersky O, Spence JE (2001) Novel and recurrent mutations in lamin A/C in patients with Emery‐Dreifuss muscular dystrophy. Am J Med Genet 102:359–367. [DOI] [PubMed] [Google Scholar]
- 7. Colomer J, Sabatelli P, Columbaro M, Nascimento A, Diaz A, Merlini L (2000) Clinical spectrum of Emery‐Dreifuss muscular dystrophy 2 (EDMD2): report of 6 patients. Neuromuscul Disord 14:591. Abstract. [Google Scholar]
- 8. D’Amico A, Benedetti S, Petrini S, Sambuughin N, Boldrini R, Menditto I, Ferrari M, Verardo M, Goldfarb L, Bertini E (2005) Major myofibrillar changes in early onset myopathy due to de novo heterozygous missense mutation in lamin A/C gene. Neuromuscul Disord 15:847–850. [DOI] [PubMed] [Google Scholar]
- 9. Di Barletta MR, Ricci E, Galluzzi G, Tonali P, Mora M, Morandi L, Romorini A, Voit T, Orstavik KH, Merlni L, Trevisan C, Biancalana V, Hausmanowa‐Petrusewicz I, Bione S, Ricotti R, Schwartz K, Bonne G, Toniolo D (2000) Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery‐Dreifuss muscular dystrophy. Am J Hum Genet 66:1407–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ Jr, Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Muehle G, Johnson W, McDonough B (1999) Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction system disease. N Engl J Med 341:1715–1724. [DOI] [PubMed] [Google Scholar]
- 11. Frock RL, Kudlow BA, Evans AM, Jameson SA, Hauschka SD, Kennedy BK (2006) Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev 20:486–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hanisch F, Neudecker S, Mehnert M, Zierz S (2002) Die Hauptmann‐Thannhauser‐Muskeldystrophie und Differenzialdiagnosen von Myopathien mit Kontrakturen. Nervenarzt 73:1004–1011. [DOI] [PubMed] [Google Scholar]
- 13. Hutchison CJ, Worman HJ (2003) A‐type lamins: guardians of the soma? Nat Cell Biol 6:1062–1067. [DOI] [PubMed] [Google Scholar]
- 14. Kadi F, Eriksson A, Holmner S, Butler‐Browne GS, Thornell LE (1999) Cellular adaptation of the trapezius muscle in strength‐trained athletes. Histochem Cell Biol 111:189–195. [DOI] [PubMed] [Google Scholar]
- 15. Ki CS, Hong JS, Jeong GY, Ahn KJ, Choi KM, Kim DK, Kim JW (2002) Identification of lamin A/C (LMNA) gene mutations in Korean patients with autosomal dominant Emery‐Dreifuss muscular dystrophy and limb‐girdle muscular dystrophy 1B. J Hum Genet 47:225–228. [DOI] [PubMed] [Google Scholar]
- 16. Maraldi NM, Merlini L (2004) Emery‐Dreifuss muscular dystrophy. In: Myology, 3rd edn. Engel AG, Franzini‐Armstrong C (eds), chapter 35, pp. 1027–1037. McGraw‐Hill: New York. [Google Scholar]
- 17. Melcon G, Kozlov S, Cutler DA, Sullivan T, Hernandez L, Zhao P, Mitchell S, Nader G, Bakay M, Rottman JN, Hoffman EP, Stewart CL (2006) Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum Mol Genet 15:637–651. [DOI] [PubMed] [Google Scholar]
- 18. Mokri B, Engel AG (1975) Duchenne dystrophy: electron microscopic findings pointing to a basic or early abnormality in the plasma membrane of the muscle fiber. Neurology 25:1111–1120. [DOI] [PubMed] [Google Scholar]
- 19. Mozdziak PE, Schultz E, Cassens RG (1994) Satellite cell mitotic activity in posthatch turkey skeletal muscle growth. Poult Sci 73:547–555. [DOI] [PubMed] [Google Scholar]
- 20. Reichart B, Klafke R, Dreger C, Krüger E, Motsch I, Ewald A, Schäfer J, Reichmann H, Müller CR, Dabauvalle MC (2004) Expression and localization of nuclear proteins in autosomal‐dominant Emery‐Dreifuss muscular dystrophy with LMNA R377H mutation. BMC Cell Biol 5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmalbruch H (1975) Segmental fibre breakdown and defects of the plasmalemma in diseased human muscles. Acta Neuropathol 33:129–141. [DOI] [PubMed] [Google Scholar]
- 22. Schmalbruch H, Hellhammer U (1976) The number of satellite cells in normal muscle. Anat Rec 185:279–287. [DOI] [PubMed] [Google Scholar]
- 23. Selcen D, Stilling G, Engel AG (2001) The earliest pathologic alterations in dysferlinopathiy. Neurology 56:1472–1481. [DOI] [PubMed] [Google Scholar]
- 24. Sewry CA, Brown SC, Mercuri E, Bonne G, Feng L, Camici G, Morris GE, Muntoni F (2001) Skeletal muscle pathology in autosomal dominant Emery‐Dreifuss muscular dystrophy with lamin A/C mutations. Neuropathol Appl Neurobiol 27:281–290. [DOI] [PubMed] [Google Scholar]
- 25. Somech R, Shaklai S, Amariglio N, Rechavi G, Simon AJ (2005) Nuclear envelopathies—raising the nuclear veil. Pediatr Res 57:8R–15R. [DOI] [PubMed] [Google Scholar]
- 26. Sullivan T, Escalante‐Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B (1999) Loss of A‐type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 147:913–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vytopil M, Benedetti S, Ricci E, Galluzzi G, Dello Russo A, Merlini L, Boriani G, Gallina M, Morandi L, Politano L, Moggio M, Chiveri L, Hausmanova‐Petrusewicz I, Ricotti R, Vohanka S, Toman J, Toniolo D (2003) Mutation analysis of the lamin A/C gene (LMNA) among patients with different cardiomuscular phenotypes. J Med Genet 40:e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Worman HJ, Courvalin JC (2005) Nuclear envelope, nuclear lamina, and inherited disease. Int Rev Cyt 246:231–279. [DOI] [PubMed] [Google Scholar]