

Abstract
Deposition of conformationally altered proteins prominently characterizes pathogenesis and pathomorphology of a number of neurodegenerative disorders. 2‐(1‐{6‐[(2‐[F‐18]fluoroethyl) (methyl)amino]‐2‐naphthyl} ethylidene) malononitrile ([F‐18]FDDNP), a hydrophobic, viscosity‐sensitive, solvent‐sensitive, fluorescent imaging probe has been used with positron emission tomography to visualize brain pathology in the living brain of Alzheimer disease (AD) patients. Its non‐radiofluorinated analog FDDNP was shown to label senile plaques and neurofibrillary tangles (NFTs) in brain tissue sections. This work aimed at evaluating FDDNP labeling of various protein deposits in fixed, paraffin‐embedded brain tissue sections of selected neurodegenerative disorders: AD, cerebral amyloid angiopathy (CAA), transmissible spongiform encephalopathies, progressive supranuclear palsy (PSP), Pick disease (PiD), Parkinson disease, dementia with Lewy bodies, multiple system atrophy (MSA). Cerebral hypertensive vascular hyalinosis (HVH) was used as negative control. Significant agreement between amyloid histochemical properties and FDDNP labeling of the deposits was established. FDDNP labeling showed high positive predictive value for birefringence in senile plaques and NFTs in AD, prion plaques and amyloid deposits in CAA. No FDDNP labeled structures were observed in HVH, PSP, PiD or MSA tissue sections. Our findings may be of significant value for the detection of neuropathological aggregates with [F‐18]FDDNP in some of these disorders in the living brain of human subjects.
INTRODUCTION
Formation and accumulation of aggregated protein deposits is a common feature of a number of neurodegenerative diseases (20, 34). They are increasingly recognized as “conformational” disorders (8) because of the process of protein misfolding, which precedes self‐assembly of aberrant proteins into insoluble aggregates (9, 39). These can form characteristic microscopic structures in the brain tissue that aid the pathomorphologic diagnosis of neurodegenerative disorders.
A variety of different proteins are involved in formation of deposits in these disorders. Certain misfolded proteins [eg, amyloid‐β peptide (Aβ) and aberrant isoform of prion protein] deposit in the form of amyloid, extracellular aggregates formed by 7–13 nm‐wide fibrils of variable length (11). Fibrils are composed of several protofilaments, each of them consisting of continuous hydrogen bonded β‐structure formed by self‐assembly of these amyloidogenic proteins (15, 33). This characteristic ultrastructure appears responsible for the uniform tinctorial properties of amyloid, which is histochemically characterized by the apple‐green birefringence pattern, seen in cross‐polarized light microscopy of Congo red stained tissue sections (10, 31). Some other proteins (eg, α‐synuclein) can form intracellular deposits with ultrastructural and histochemical properties, similar to those of amyloid [eg, Lewy bodies (LBs) (27)], which are therefore called amyloid‐like aggregates. On the other hand, some tau protein deposits [neurofibrillary tangles (NFTs) with tau straight filaments, globose neurofibrillary tangles (GNFTs), Pick bodies (PiBs)] show few or none of the structural properties of amyloid (12). Deposition of protein aggregates is believed to prominently characterize disease pathogenesis (34). Starting well before the onset of clinical illness (26), it often seems to be in correlation with severity of neurological or psychiatric dysfunction (4, 14). The ability to detect such protein deposits in the brain in vivo would thus aid the clinical diagnosis, enable disease progression studies and facilitate search for effective treatment options.
2‐(1‐{6‐[(2‐[18‐F]Fluoroethyl)(methyl) amino]‐2‐naphthyl}ethylidene)malononitrile ([18‐F]FDDNP) has been established as the first molecular imaging probe for successful detection of amyloid deposits with positron emission tomography (PET) in the living brain of patients with Alzheimer disease (AD) (2, 3, 35). Unlike Congo red, FDDNP is not charged and therefore rapidly crosses the blood–brain barrier allowing in vivo assessment of these brain pathologies in human subjects. FDDNP also displays fluorescent properties suitable for fluorescence microscopy (3, 17) with excitation in the visible range (440–490 nm) thus minimizing tissue auto‐fluorescence (17, 30). Upon binding to protein aggregates, FDDNP fluorescence emission yield increases because of the hydrophobic microenvironment on the aggregate surface and peaks at about 500 nm when bound in vitro to Aβ (2). Because of its outstanding histochemical properties, confocal fluorescent microscopy has been used to observe FDDNP labeling of two most prominent histological structures in AD—amyloid senile plaques and NFTs (2). Recently, FDDNP has also been shown to successfully label prion plaques (PPs) in brain tissue sections from patients with transmissible spongiform encephalopathies (TSEs): variant Creutzfeldt‐Jakob disease (vCJD), Gerstmann‐Sträussler‐Scheinker disease (GSS) and sporadic CJD (sCJD) (5). Aiming at better defining the FDDNP diagnostic potential the present paper describes assessment of fluorescent FDDNP labeling of protein deposits in brain tissue sections of various neurodegenerative disorders with regard to the principal aggregated misfolded proteins and the related microscopic structures.
MATERIAL AND METHODS
Tissue sections. We used representative, 10 µm thick, paraformaldehyde‐fixed, paraffin‐embedded brain tissue sections from ten cases of neuropathologically confirmed neurodegenerative diseases (Table 1). Brain tissue section from a patient with hypertensive vascular hyalinosis (HVH) has been included as a negative control. As a standard step in CJD brain tissue processing, the sCJD and vCJD (but not GSS) tissue blocks were immersed in 96% formic acid for 1 h after fixation in paraformaldehyde (6).
Table 1.
Studied neurodegenerative disorders, along with their corresponding pathohistological structures and major deposited proteins.
| Disease | Microscopic structure | Deposited protein | Ultrastructure |
|---|---|---|---|
| Alzheimer disease | Neuritic senile plaques: with amyloid core, without amyloid core* | Aβ | 7–10 nm wide amyloid fibrils (33) |
| Neurofibrillary tangle | Tau | Paired helical filaments, periodicity 80 nm, 8–20 nm wide, also straight filaments (12) | |
| Cerebral amyloid angiopathy | Vascular amyloid | Aβ | 7–10 nm wide amyloid fibrils (33) |
| Sporadic Creutzfeldt‐Jakob disease | Prion plaque Primitive prion plaque | PrPsc | 8–10 nm wide fibrils in amyloid cores (13) |
| Variant Creutzfeldt‐Jakob disease | |||
| Gerstmann‐Sträussler‐Scheinker disease | |||
| Progressive supranuclear palsy | Globose neurofibrillary tangle | Tau | 15–18 nm wide straight filaments (12) |
| Pick disease | Pick body | 9.91–14.19 nm wide straight filaments, some PHF‐like, 11.65–20.49 nm wide, with long (81.91–153.79 nm) periodicity (21) | |
| Parkinson disease | Lewy body | α‐synuclein | 5–10 nm filaments (36) |
| Dementia with Lewy bodies | Cortical Lewy body | ||
| Multiple system atrophy | Glial cytoplasmic inclusion | 30–50 nm filaments (1) | |
| Hypertensive vascular hyalinosis | Vascular hyaline | Plasma proteins |
Based on their haematoxylin and eosin and immunohistochemistry appearance, neuritic senile plaques were subdivided into two groups: (i) classic senile plaques with well‐formed amyloid core; and (ii) neuritic senile plaques without amyloid core (11). Only a few diffuse plaques were identified in our Alzheimer disease samples and were therefore excluded from the statistical analysis.
Histochemical procedures. Each tissue section was sequentially labeled using four histochemical procedures (Figure 1). The sequence of labeling steps used in this study had been designed and tested to eliminate interference between consecutive labeling steps. Photomicrographs of the selected loci (see the methods below) were taken after each labeling step with a Nikon DXM2000 camera attached to Olympus AX‐81 fluorescent microscope also equipped with light polarizing optics. FDDNP fluorescence was observed using a FITC filter. Glycerol was used for cover‐slipping.
Figure 1.
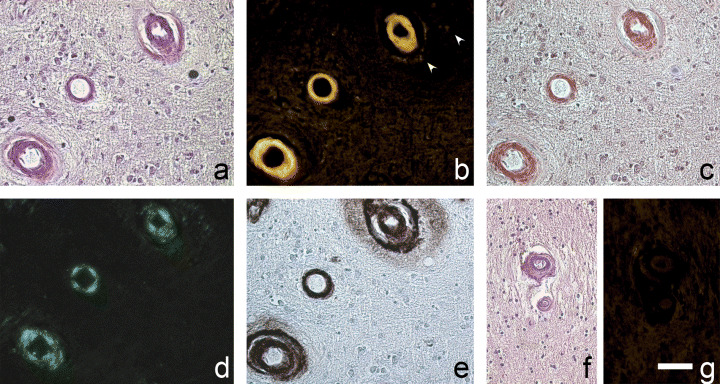
Cerebral amyloid angiopathy tissue section labeling sequence (A–E), and hypertensive vascular hyalinosis (HVH, F, G). Blood vessels with marked Aβ deposition, shown in haematoxylin and eosin (A), labeled with FDDNP (B, FDDNP is also labeling less dense Aβ deposit spreading from vascular wall, arrowheads), Congo red in light (C) and cross‐polarized light microscopy (D), and anti‐Aβ immunohistochemistry (E). Vascular hyaline in HVH (shown in haematoxylin and eosin, F) is not labeled with FDDNP (G). Green fluorescent signal in B was pseudocolored yellow. Scale bar: A–E, 50 µm; F, G, 60 µm.
Described in brief, the steps in the labeling sequence were as follows: (i) automated haematoxylin and eosin staining (Tissue Stainer TST 30, Medizintechnik, Germany). Sections were analyzed and photographed and then de‐stained by 4 h incubation in 2% HCl solution in ethanol. (ii) Tissue was carefully and thoroughly rinsed with water and incubated in the dark at room temperature for 20 minutes with freshly prepared 10 µM FDDNP in aqueous 1% ethanol (v/v) (2). (iii) Congo red labeling was subsequently performed by immersion of rinsed tissue into 1% aqueous Congo red solution for 1 h, followed by brief differentiation in 2% solution of KOH in 80% ethanol (v/v). After rinsing with water, haematoxylin (5 minutes) was used for nuclear counterstaining. (iv) Finally, immunohistochemical or immunofluorescent identification of deposited protein was performed. All immunohistochemical procedures were carried out on an automated machine (Ventana Medical Systems, Tucson, AZ, USA) using Ventana kits and manufacturer‐prescribed protocols at every step of the procedure. We used monoclonal mouse anti‐human (Aβ antibodies (Dako, Glostrup, Denmark; 200 mg/L), clone 6F/3D (in 1:10 dilution) after 3 minutes pre‐treatment of tissue with 95% formic acid at room temperature; mouse monoclonal anti‐PrP antibodies, clone 3F4 (Senetec, Maryland Heights, MO, USA; 1 mg/mL) diluted 1:50, after 30 minutes autoclaving in distilled water followed by 5 minutes immersion in 95% formic acid at room temperature; polyclonal rabbit anti‐human tau antibodies (Dako; 6.9 g/L) diluted 1:250 used without tissue pre‐treatment; mouse anti‐α‐synuclein antibodies, clone LB509 (Zymed Laboratories, San Francisco, CA, USA; 142 µg/L) diluted 1:50 used after epitope retrieval procedure in citrate buffer at 96C for 15 minutes followed by 1 minutes incubation in 96% formic acid. FITC‐labeled antisera to human IgG and IgM were used for direct IF labeling of vascular hyaline.
Data collection and analysis. Loci containing visible microscopic structures of interest (10–15 per microscopic slide) were selected after haematoxylin and eosin staining. They were photomicrographed and their coordinates were recorded. Each labeling step (as well as haematoxylin and eosin de‐stainig procedure) was followed by digital photography of these same sites using either light (Congo red, immunohistochemistry), fluorescence (prior to and after FDDNP, immunofluorescence) or cross‐polarized light (Congo red) microscopy. Microscopic structures formed by deposition of protein aggregates (identified in haematoxylin and eosin and immunohistochemistry or immunofluorescence photomicrographs) were assessed by three independent raters for their FDDNP labeling (rated 0, 1, 2 : 0—no detectable labeling, 1—moderate labeling, discernible from background, 2—intense labeling) and for the presence of amyloid histochemical property: apple‐green birefringence in cross‐polarized light on sections stained with Congo red (in short, birefringence; rated 0—absent or 1—present).
Statistical analysis. Because of subjective nature of FDDNP fluorescence rating, inter‐rater agreement was assessed using intraclass correlation coefficient (ICC). For detailed description of ICC, please refer to McGraw and Wong (24). As both raters and targets were random samples from their respective populations, the appropriate model for our case was the two‐way random model for absolute agreement which includes between‐raters variance in the ICC denominator. In McGraw and Wong (24), the exact model is designated Case 2 ICC (A, 3), while in SPSS it is referred to as Reliability procedure ICC (2, 3) for absolute agreement.
In order to compare FDDNP and birefringence ratings, the symmetric version of Somer’s D coefficient of ordinal correlation was calculated. In addition, the assessment of FDDNP labeling was dichotomized (0—no labeling, 1—labeling present) and agreement between FDDNP labeling and presence of birefringence (pairs of dichotomous variables) was assessed using Cohen’s Kappa coefficient. To compare the agreement across structures, we performed the Mantel‐Haenszel test for conditional independence of odds ratios.
Because of differences in number of microscopic structures in groups, the proportion of a given structure labeled by FDDNP was estimated along with exact binomial 90% confidence interval, which was calculated in order to estimate the range which holds the true value for the structure group with 90% probability.
The positive and negative predictive values of FDDNP labeling for birefringence were calculated for each microscopic structure group. The probability that FDDNP labeled structures would display birefringence after Congo red staining was expressed as positive predictive value. Negative predictive value for a structure group denotes the probability that structures not labeled with FDDNP would also not display birefringence.
Statistical analyses were carried out using the SPSS for Windows 11.0 (SPSS Inc., Chicago, IL, USA) and StatXact 4.01 (Cytel Software Corp., Cambridge, MA, USA) software.
RESULTS
Validity of the assessment procedure. As the procedure for the assessment of presence of FDDNP labeling and birefringence depended on subjective judgment, we tested the validity of the observations by assessing inter‐rater agreement. Inter‐rater agreement proved to be high and statistically significantly different from that expected by chance at P < 0.0001 for both staining methods and criteria used. Inter‐rater agreement regarding FDDNP labeling of observed microscopic structures (ICC 0.788) slightly exceeds that of birefringence after Congo red staining (ICC 0.758). Comparing the two dichotomous criteria for identifying histological structures, the inter‐rater agreement was practically identical.
FDDNP labeling of protein deposits. In AD, FDDNP labeled all observed classic senile plaques (cSPs, n = 12) and 33% of neuritic senile plaques (nSPs, n = 12, Figure 2). Central localization of FDDNP fluorescence in cSPs coincided with Aβ core. Tau protein deposits contributed little to the extent of plaque labeling (Figure 3). Sixty‐four percent of all NFTs (n = 45) were birefringent and 31% displayed FDDNP fluorescence (Figure 2). Seventy‐five percent of FDDNP labeled NFTs were also birefringent.
Figure 2.
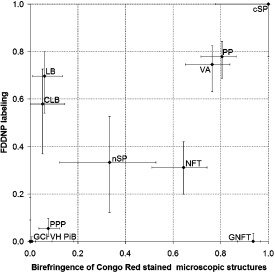
Scatter plot of Congo red birefringence vs. FDDNP labeling of studied types of microscopic structures. Horizontal axis shows the proportion of microscopic structures (identified in haematoxylin and eosin and immunohistochemistry photomicrographs), displaying Congo red birefringence and vertical axis shows the proportion of microscopic structures displaying FDDNP fluorescence. Good agreement between Congo red birefringence and FDDNP fluorescence is observed in all but three structure types analyzed: Lewy bodies (LBs) and cortical Lewy bodies (CLBs) displayed moderate FDDNP fluorescence but very little Congo red birefringence, whereas globose neurofibrillary tangles (GNFT) displayed Congo red birefringence to a high degree but showed no FDDNP fluorescence. Abbreviations: GCI = glial cytoplasmic inclusion; NFT = neurofibrillary tangle; nSP = neuritic senile plaques; PP = prion plaque; PPP = primitive prion plaque; PiB = Pick body; VA = vascular amyloid; VH = vascular hyaline.
Figure 3.
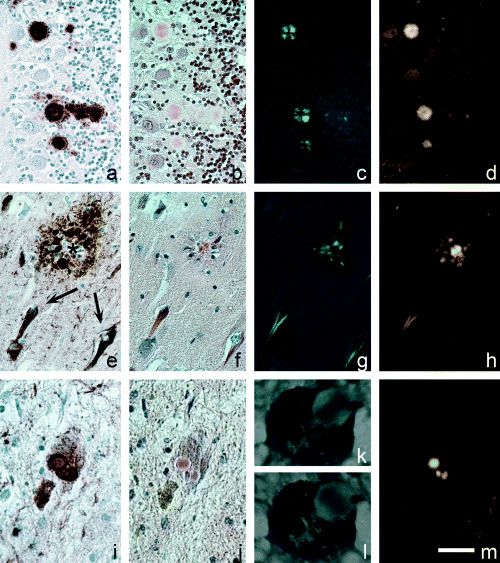
Prion plaques in variant Creutzfeldt‐Jakob disease (A–D), labeled with anti‐PrP immunohistochemistry (immunohistochemistry, A), stained with Congo red (B), display birefringence (C) and FDDNP labeling (D). Aβ core of a classic senile plaque in Alzheimer Disease (AD) (E–H) remains unlabeled in anti‐tau immunohistochemistry (E, asterisk), but displays marked congophilia (F), birefringence (G) and FDDNP fluorescence (H). Neurofibrillary tangles in AD are labeled with anti‐tau antibodies (E, arrows), are congophilic (F), display birefringence (G), and are labeled with FDDNP (H). Lewy bodies in Parkinson disease (I–M) are α‐synuclein immunopositive deposits (I), that display congophillia (J) and weak birefringence, which is best seen with 90 degree shift of plane of light polarization (K, L), and display FDDNP fluorescence (M). Green fluorescent signal in D, H, M was pseudocolored yellow. Scale bar: A–H, 50 µm; I, J, M, 30 µm and K, L, 10 µm.
Vascular amyloid (VA) in cerebral amyloid angiopathy (CAA) (n = 55) was labeled with FDDNP in 75% of cases (1, 2). Intensity of FDDNP fluorescence varied with vessel diameter and became undetectable in Aβ deposits of small vessels. Vascular hyaline (VH) in HVH (n = 12) showed neither apple green birefringence nor FDDNP labeling (1, 2).
Out of 77 amyloid PPs examined, 60 (78%) were FDDNP positive (2, 3). Of those, 88% displayed birefringence and 12% did not. Five out of 92 (5.4%) primitive prion plaques (PPPs) studied were FDDNP labeled (Figure 2). Those primitive plaques that were not labeled with FDDNP displayed birefringence in only 5.7%.
LBs in Parkinson disease (PD, n = 33) were labeled with FDDNP in 70%, and 58% of cortical LBs (CLBs) in dementia with Lewy bodies (DLB, n = 19) showed FDDNP fluorescence. Notably, all LBs and CLBs, displaying birefringence were also FDDNP labeled (3, 4).
Figure 4.
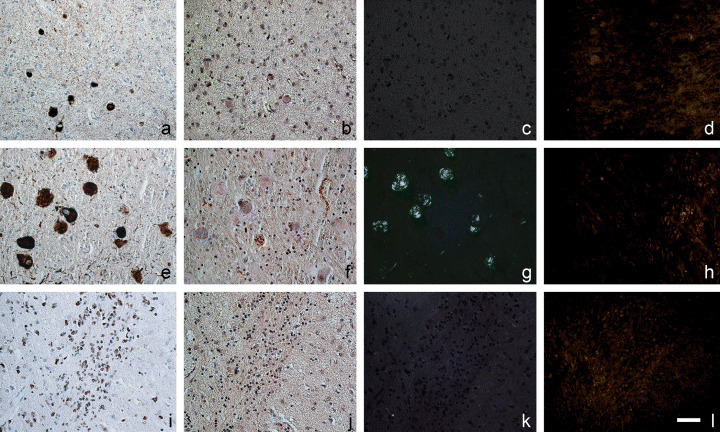
Pick bodies (A–D) and glial cytoplasmic inclusions (I–L), labeled with anti‐tau (A) or anti‐α‐synuclein (I) in immunohistochemistry, display no birefringence (C, K), when stained with Congo red (B, J). Only background fluorescence can be seen with FDDNP staining (D, L). Globose neurofibrillary tangles (E–H) are tau‐immunopositive (E), display congophillia (F) and birefringence (G), yet consistently show no detectable FDDNP fluorescence (L). Green fluorescent signal in D, H, L was pseudocolored yellow. Scale bar: 50 µm.
None of the 30 glial cytoplasmic inclusions (GCIs) studied showed either birefringence or FDDNP labeling. Similarly, none of the 138 PiBs displayed birefringence or FDDNP labeling. GNFTs (n = 30) exhibited consistent congophilia and birefringence (93%), but no detectable FDDNP fluorescence (2, 4).
FDDNP—birefringence matching. Agreement between methods is reported in Table 2. All Kappa and Sommer’s D values are statistically significantly different from zero at P < 0.0001, thus indicating preferential FDDNP labeling of birefringent structures.
Table 2.
Agreement between methods overall and within each rater. See Results section for details.
| Overall | Rater 1 | Rater 2 | Rater 3 | |
|---|---|---|---|---|
| Kappa (FDDNP (0/1) and birefringence) | 0.385 | 0.396 | 0.416 | 0.399 |
| Sommer’s D (FDDNP (0–2) and birefringence) | 0.404 | 0.403 | 0.427 | 0.412 |
Types of microscopic structures differed regarding agreement between FDDNP labeling and birefringence, which was confirmed by the Mantel‐Haenszel test. It showed that the odds ratio for the FDDNP vs. birefringence cross‐classification was different across structures (P < 0.001).
FDDNP labeling showed high positive predictive value for birefringence when used in labeling of cSPs, PPs, VA and NFTs. The probability that FDDNP‐unlabeled structures were also non‐birefringent was high in LBs, nSPs, PPs, VH, GCIs and PiBs (Table 3).
Table 3.
Positive and negative predictive values of FDDNP labeling for birefringence. Abbreviations: GNFT = globose neurofibrillary tangle; PiB = Pick body; GCI = glial cytoplasmic inclusion; VH = vascular hyaline; cSP = classic senile plaque; LB = Lewy body; CLB = cortical Lewy body; NFT = neurofibrillary tangle; nSP = neuritic senile plaques; PP = prion plaque; PPP = primitive prion plaque; VA = vascular amyloid; NA, not applicable.
| Histological structure | cSP | nSP | NFT | VA | PP | PPP | GNFT | PiB | LB | CLB | GCI | VH |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Positive predictive value | 1.0 | 0.50 | 0.71 | 0.86 | 0.88 | 0.40 | NA† | NA† | 0.87 | 0.09 | NA† | NA† |
| Negative predictive value | NA* | 0.75 | 0.39 | 0.57 | 0.47 | 0.94 | 0.06 | 0.99 | 1.0 | 1.0 | 1.0 | 1.0 |
FDDNP labeled all cSPs, therefore negative prediction value of FDDNP labeling can not be calculated.
FDDNP did not detectably label any of GNFTs, PiBs, GCIs or VH, therefore positive prediction value of FDDNP labeling can not be calculated.
DISCUSSION
The aim of this study was to assess fluorescent histochemical dye FDDNP labeling of protein aggregates in brain tissue sections of a range of neurodegenerative disorders. Our results show that FDDNP preferentially labeled structures displaying amyloid histochemical properties. The proportion of FDDNP labeled microscopic structures was highest in AD (senile plaques, and NFTs), TSEs (PPs) and CAA (VA). LBs were labeled in a somewhat smaller percentage. Apart from NFTs in AD, tau protein deposits in other taupathies studied showed no FDDNP fluorescence and neither did GCI in multiple system atrophy (MSA), nor VH in malignant hypertension.
A number of probes intended for PET imaging of amyloid in AD have been developed in recent years (28). In vitro, the thioflavin T analog 2‐(4′‐methylaminophenyl) benzothiazole (BTA‐1) was reported to predominantly label senile plaques, but also NFTs (23). Predominant senile plaque labeling was reported for BF‐145, a styryl‐fluorobenzoxazole derivative (29). (Trans,trans),‐1‐bromo‐2,5‐bis‐(3‐hydroxycarbonyl‐4‐hydroxy) styrylbenzene (BSB) is a Congo red derived fluorescent probe that has not yet been used in human neuroimaging; it was, however, systematically studied on human postmortem neurodegenerative disease brain sections (32). Both probes, FDDNP and BSB, label Aβ and NFTs in AD tissue, but they differ substantially in labeling of other protein deposits. FDDNP labeled high proportion of LBs in PD, while “only occasional LBs [were] weakly stained with BSB” (32). On the other hand, unlike FDDNP, BSB is reported to extensively label GNFTs and GCIs and to also weakly stain PiBs (32). BSB has also recently been shown to label prion protein aggregates in brain tissue sections of scrapie infected hamsters (16). However, for adequate comparison of protein aggregates labeling, both probes, BSB and FDDNP, should be used under identical conditions.
FDDNP has been shown to bind to Aβ fibrils in both radioactive and fluorescence titration assays with high affinity. Furthermore, FDDNP labeling of SPs in AD brain sections has been observed using autoradiography and fluorescence microscopy (2, 3). All Aβ‐positive cores of cSPs were labeled with FDDNP in our study, thus not only confirming high sensitivity of this fluorescent probe for Aβ, but also adding quantitative information about FDDNP cSPs labeling in AD. FDDNP, like Congo red, labeled “disordered centers” of cSPs amyloid cores, as described by Jin et al (18) in the structural study of Aβ and prion amyloid plaques using linear birefringence and dichroism imaging. Vascular Aβ labeling was qualitatively similar to that of cSPs. nSPs with less formed amyloid probably labeled in proportion to their amyloid content.
Extensive prion amyloid FDDNP labeling in this study confirmed our previous findings (5). All types of PPs visible in haematoxylin and eosin stained sections were consistently labeled, whereas labeling of PPPs apparently depended upon the presence of amyloid histochemical characteristic (apple green birefringence of Congo red stained material) in the deposit. It has been shown that incubation of tissue sections with formic acid reduces congophilia and birefringence of prion plaques in CJD and GSS (38) and eliminates thioflavin S and X‐34 (1,4‐bis(3‐carboxy‐4‐hydroxyphenylethenyl)‐benzene) staining of Aβ deposits in AD (37). Prion plaque birefringence was preserved in our CJD tissue sections. Thus, the effect of formic acid was not extensive in our study, probably because of the fact that tissue blocks (as opposed to individual sections) of vCJD and sCJD were immersed in formic acid. However, if attenuation of birefringence did occur in our studied samples because of formic acid, our calculated predictive values actually underestimated the matching of FDDNP labeling and birefringence in TSEs.
The three synucleinopathies studied differed in the ultrastructural organization of their α‐synuclein deposits. LBs have amyloid ultrastructure (27, 36), but based on current criteria their intracellular localization prevents them from being classified as amyloid (22). Small size of LBs (Figure 3I–M) in comparison to classical extracellular amyloid deposits could be the reason for weak (Figure 3K,L) or absent birefringence in cross‐polarized light microscopy after Congo red staining, for it has been established that the birefringence phenomenon of Congo red stained tissue sections depends on section thickness, thus implying a requirement for “critical” amyloid quantity in the observed sample (10). FDDNP labeled a majority of both types of LBs: CLBs in DLB and well formed LBs in PD. In contrast to the labeling of these amyloid‐like structures, no labeling of GCIs [which consist of 30–50 nm filaments (1)] could be observed in MSA. It should be noted, that AD‐associated pathology, namely SPs and NFTs, were also present in the DLB tissue included in the study. The tissue was classified as DLB based on current Consortium on DLB recommendations (25).
FDDNP labeled numerous NFTs in AD, as previously reported (2). Interestingly, FDDNP labeling also correlated with observation of birefringence in a large majority of NFTs after Congo red staining, as can be seen from the high positive predictive value of FDDNP labeling for birefringence (Table 3). This might imply the existence of related structural conditions for both, FDDNP binding fluorescence and birefringence in cross‐polarized light after Congo red staining. The existence of two distinct ultrastructural patterns (paired helical filaments and straight filaments) of hyperphosphorilated tau aggregation in the process of NFT formation (12) makes such implication even more attractive. However, this structural consideration for the target protein aggregate does not imply a common binding site for both FDDNP and Congo red for NFT, as was clearly shown earlier for Aβ (3).
Apart from NFTs in AD, no other tau deposits displayed detectable FDDNP fluorescence in our study. GNFTs in progressive supranuclear palsy, that consist of 15–18 nm straight filaments (7, 12), were the only histological structures with frequent display of birefringence after Congo red staining but consistent non‐detectable FDDNP labeling, which could indicate that straight filaments in tissue sections lack the “motif” for FDDNP binding.
Random distribution of straight filaments and paired helical filaments—like filaments with long periodicity (19, 21) could explain the absence of birefringence of PiBs. In our study, performed on samples from a single case of Pick disease (PiD), none of the 138 studied PiBs displayed any of the histochemical amyloid characteristics and we also did not detect FDDNP fluorescent labeling of these intracellular inclusions. Earlier, the radioactive FDDNP analog, [F‐18]FDDNP was shown to label PiBs using microautoradiography (3). In addition to possible absence of structural conditions for FDDNP labeling in PiBs of our single case of PiD, the different sensitivity of histofluorescence compared with microautoradiography cannot be discounted.
Control tissues used in our study were brain samples of HVH with plasma protein imbibition into the vessel walls. The proteins comprising vascular wall hyaline are not misfolded, do not stack up into fibrils and do not form amyloid, thus being unlikely to provide appropriate microenvironment for FDDNP binding. The results of our study confirmed this prediction.
In conclusion, we have demonstrated that FDDNP labeling of protein deposits is closely related to the histochemical amyloid properties of these aggregates. FDDNP labeling predicts birefringence in Congo red stained histological sections with high reliability. Microscopic structures in classical amyloid forming disorders, namely AD, CAA and TSEs are most reliably labeled. Our findings may be of significant value for the detection of neuropathological aggregates with [F‐18]FDDNP in some of these disorders in the living brain of human subjects.
ACKNOWLEDGMENTS
This work was supported by the Republic of Slovenia Ministry of Health and Ministry of Education, Science and Sport Grants 0381‐518, L3‐6006 and Slo‐US BI‐US/04‐05/4. We acknowledge the assistance of Dr. Herbert Budka (Institute of Neurology, University of Vienna, and Austrian Reference Centre for Human Prion Diseases, Vienna, Austria) and Dr. James Ironside (Neuropathology Laboratory, CJD Surveillance Unit, University of Edinburgh, Edinburgh, UK), who kindly provided the samples of GSS and vCJD cerebellar tissue. We are grateful to Gregor Repovs for valuable suggestions regarding the manuscript, as well as Marija Zupancic and Albina Stancic for help with tissue processing.
REFERENCES
- 1. Abe H, Yagishita S, Amano N, Iwabuchi K, Hasegawa K, Kowa K (1992) Argyrophilic glial intracytoplasmic inclusions in multiple system atrophy: immunocytochemical and ultrastructural study. Acta Neuropathol (Berl) 84:273–277. [DOI] [PubMed] [Google Scholar]
- 2. Agdeppa ED, Kepe V, Liu J, Flores‐Torres S, Satyamurthy N, Petric A, Cole GM, Small GW, Huang SC, Barrio JR (2001) Binding characteristics of radiofluorinated 6‐dialkylamino‐2‐naphthylethylidene derivatives as positron emission tomography imaging probes for beta‐amyloid plaques in Alzheimer’s disease. J Neurosci 21:RC189(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Agdeppa ED, Kepe V, Liu J, Small GW, Huang SC, Petric A, Satyamurthy N, Barrio JR (2003) 2‐Dialkylamino‐6‐acylmalononitrile substituted naphthalenes (DDNP analogs): novel diagnostic and therapeutic tools in Alzheimer’s disease. Mol Imaging Biol 5:404–417. [DOI] [PubMed] [Google Scholar]
- 4. Berg L, McKeel DW Jr, Miller JP, Storandt M, Rubin EH, Morris JC, Baty J, Coats M, Norton J, Goate AM, Price JL, Gearing M, Mirra SS, Saunders AM (1998) Clinicopathologic studies in cognitively healthy aging and Alzheimer’s disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol 55:326–335. [DOI] [PubMed] [Google Scholar]
- 5. Bresjanac M, Smid LM, Vovko TD, Petric A, Barrio JR, Popovic M (2003) Molecular‐imaging probe 2‐(1‐[6‐[(2‐fluoroethyl)(methyl) amino]‐2‐naphthyl]ethylidene) malononitrile labels prion plaques in vitro . J Neurosci 23:8029–8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Budka H, Aguzzi A, Brown P, Brucher JM, Bugiani O, Collinge J, Diringer H, Gullotta F, Haltia M, Hauw JJ (1995) Tissue handling in suspected Creutzfeldt‐Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathol 5:319–322. [DOI] [PubMed] [Google Scholar]
- 7. Buee L, Delacourte A (1999) Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP‐17 and Pick’s disease. Brain Pathol 9:681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carrell RW, Lomas DA (1997) Conformational disease. Lancet 350:134–138. [DOI] [PubMed] [Google Scholar]
- 9. Dobson CM (2004) Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol 15:3–16. [DOI] [PubMed] [Google Scholar]
- 10. Elghetany MT, Saleem A, Barr K (1989) The congo red stain revisited. Ann Clin Lab Sci 19:190–195. [PubMed] [Google Scholar]
- 11. Esiri M, Hyman B, Beyreuther K, Masters C (1997) Ageing and dementia. In: Greenfield’s Neuropathology, chapter 4. Graham D, Lantos P (eds), pp. 153–233. Arnold: London. [Google Scholar]
- 12. Forman MS, Lee VM, Trojanowski JQ (2000) New insights into genetic and molecular mechanisms of brain degeneration in tauopathies. J Chem Neuroanat 20:225–244. [DOI] [PubMed] [Google Scholar]
- 13. Giaccone G, Verga L, Bugiani O, Frangione B, Serban D, Prusiner SB, Farlow MR, Ghetti B, Tagliavini F (1992) Prion protein preamyloid and amyloid deposits in Gerstmann‐Straussler‐Scheinker disease, Indiana kindred. Proc Natl Acad Sci USA 89:9349–9353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gold G, Kovari E, Corte G, Herrmann FR, Canuto A, Bussiere T, Hof PR, Bouras C, Giannakopoulos P (2001) Clinical validity of a beta‐protein deposition staging in brain aging and Alzheimer disease. J Neuropathol Exp Neurol 60:946–952. [DOI] [PubMed] [Google Scholar]
- 15. Govaerts C, Wille H, Prusiner SB, Cohen FE (2004) Evidence for assembly of prions with left‐handed beta‐helices into trimers. Proc Natl Acad Sci USA 101:8342–8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoefert VB, Aiken JM, McKenzie D, Johnson CJ (2004) Labeling of the scrapie‐associated prion protein in vitro and in vivo . Neurosci Lett 371:176–180. [DOI] [PubMed] [Google Scholar]
- 17. Jacobson A, Petric A, Hogenkamp D, Sinur A, Barrio JR (1996) 1,1‐dicyano‐2‐[6‐(dimethylamino) naphthalen‐2‐yl]propene (DDNP): a solvent polarity and viscosity sensitive fluorophore for fluorescence microscopy. J Am Chem Soc 118: 5572–5579. [Google Scholar]
- 18. Jin LW, Claborn KA, Kurimoto M, Geday MA, Maezawa I, Sohraby F, Estrada M, Kaminksy W, Kahr B (2003) Imaging linear birefringence and dichroism in cerebral amyloid pathologies. Proc Natl Acad Sci USA 100:15294–15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kato S, Nakamura H (1990) Presence of two different fibril subtypes in the Pick body: an immunoelectron microscopic study. Acta Neuropathol (Berl) 81:125–129. [DOI] [PubMed] [Google Scholar]
- 20. Kelly JW (1996) Alternative conformations of amyloidogenic proteins govern their behavior. Curr Opin Struct Biol 6:11–17. [DOI] [PubMed] [Google Scholar]
- 21. King ME, Ghoshal N, Wall JS, Binder LI, Ksiezak‐Reding H (2001) Structural analysis of Pick’s disease‐derived and in vitro‐assembled tau filaments. Am J Pathol 158:1481–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kisilevsky R (2004) The amyloidoses. In: Rubin’s Pathology: Clinicopathologic Foundations of Medicine, 4th edn, chapter 23. Rubin E, Gorstein F, Rubin R, Schwarting R, Strayer D (eds), pp. 1187–1200. Lippincott, Williams & Wilkins: Philadelphia. [Google Scholar]
- 23. Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Shao L, Hamilton RL, Ikonomovic MD, DeKosky ST, Mathis CA (2003) The binding of 2‐(4′‐methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci 23:2086–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McGraw K, Wong S (1996) Forming inferences about some intraclass correlation coefficients. Psychol Methods 1:30–46. [Google Scholar]
- 25. McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S (1996) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 47:1113–1124. [DOI] [PubMed] [Google Scholar]
- 26. Morris JC, Storandt M, McKeel DW Jr, Rubin EH, Price JL, Grant EA, Berg L (1996) Cerebral amyloid deposition and diffuse plaques in “normal” aging: evidence for presymptomatic and very mild Alzheimer’s disease. Neurology 46:707–719. [DOI] [PubMed] [Google Scholar]
- 27. Murray I, Lee V‐Y, Trojanowski J (2001) Synucleinopathies: a pathological and molecular review. Clin Neurosci Res 1:445–455. [Google Scholar]
- 28. Nordberg A (2004) PET imaging of amyloid in Alzheimer’s disease. Lancet Neurol 3:519–527. [DOI] [PubMed] [Google Scholar]
- 29. Okamura N, Suemoto T, Shiomitsu T, Suzuki M, Shimadzu H, Akatsu H, Yamamoto T, Arai H, Sasaki H, Yanai K, Staufenbiel M, Kudo Y, Sawada T (2004) A novel imaging probe for in vivo detection of neuritic and diffuse amyloid plaques in the brain. J Mol Neurosci 24:247–255. [DOI] [PubMed] [Google Scholar]
- 30. Petric A, Jacobson AF, Barrio JR (1998) Functionalization of a viscosity‐sensitive fluorophore for probing of biological systems. Bioorg Med Chem Lett 8:1455–1460. [DOI] [PubMed] [Google Scholar]
- 31. Puchtler H, Waldrop FS, Meloan SN (1985) A review of light, polarization and fluorescence microscopic methods for amyloid. Appl Pathol 3:5–17. [PubMed] [Google Scholar]
- 32. Schmidt ML, Schuck T, Sheridan S, Kung MP, Kung H, Zhuang ZP, Bergeron C, Lamarche JS, Skovronsky D, Giasson BI, Lee VM, Trojanowski JQ (2001) The fluorescent Congo red derivative, (trans, trans)‐1‐bromo‐2,5‐bis‐(3‐hydroxycarbonyl‐4‐hydroxy)styrylbenzene (BSB), labels diverse beta‐pleated sheet structures in postmortem human neurodegenerative disease brains. Am J Pathol 159:937–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Serpell LC, Blake CC, Fraser PE (2000) Molecular structure of a fibrillar Alzheimer’s a beta fragment. Biochemistry 39:13269–13275. [DOI] [PubMed] [Google Scholar]
- 34. Shastry BS (2003) Neurodegenerative disorders of protein aggregation. Neurochem Int 43:1–7. [DOI] [PubMed] [Google Scholar]
- 35. Shoghi‐Jadid K, Small GW, Agdeppa ED, Kepe V, Ercoli LM, Siddarth P, Read S, Satyamurthy N, Petric A, Huang SC, Barrio JR (2002) Localization of neurofibrillary tangles and beta‐amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry 10:24–35. [PubMed] [Google Scholar]
- 36. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) Alpha‐Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci USA 95:6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Styren SD, Hamilton RL, Styren GC, Klunk WE (2000) X‐34, a fluorescent derivative of Congo red: a novel histochemical stain for Alzheimer’s disease pathology. J Histochem Cytochem 48:1223–1232. [DOI] [PubMed] [Google Scholar]
- 38. Tashima T, Kitamoto T, Tateishi J, Sato Y (1986) Congophilia in cerebral amyloidosis is modified by inactivation procedures on slow transmissible pathogens. Brain Res 399:80–86. [DOI] [PubMed] [Google Scholar]
- 39. Zerovnik E (2002) Amyloid‐fibril formation. Proposed mechanisms and relevance to conformational disease. Eur J Biochem 269:3362–3371. [DOI] [PubMed] [Google Scholar]