

Abstract
Most adult glioblastoma multiformes (GBMs) present in patients 45–70 years old; tumors occurring at the extremes of the adult age spectrum are uncommon, and seldom studied. We hypothesized that young‐adult GBMs would differ from elderly‐adult and from pediatric GBMs. Cases were identified from years 1997 to 2005. Demographic and histological features, MIB‐1 and TP53 immunohistochemical findings and epidermal growth factor receptor (EGFR) amplification status by fluorescence in situ hybridization were compiled and correlated with survival. Twenty‐eight (74%) of our 38 young‐adult GBM patients had primary de novo tumors, two of which occurred in patients with cancer syndromes. Two additional GBMs were radiation‐induced and eight (21%) were secondary GBMs. Seven patients were identified as long‐term (>3 years) survivors. Six of 38 cases manifested unusual morphological features, including three epithelioid GBMs, one rhabdoid GBM, one gliosarcoma and one small cell GBM containing abundant, refractile, eosinophilic inclusions. MIB‐1 index emerged as the most important prognosticator of survival (P < 0.005). Although there was a trend between extent of necrosis, TP53 immunohistochemical expression, and EGFR amplification status and survival, none reached statistical significance. GBMs in young adults are a more inhomogeneous tumor group than GBMs occurring in older adult patients and show features that overlap with both pediatric and adult GBMs.
INTRODUCTION
Age represents the most important prognostic determinant in patients with glioblastoma multiforme (GBM), the most frequent and lethal brain tumor type in adults (15, 35). Overall median survival time remains dismal at one year, but subsets of GBM patients with better survival times exist and may represent distinctive molecular subtypes (40). In addition, in most studies of long‐term survivors (defined variably, but most commonly, as those who live 3 years or more) (4, 5, 34, 35, 38, 39), young‐aged adult GBM patients are overrepresented, suggesting that fundamental biological differences may exist within GBMs occurring in this age range (7, 22, 26, 27, 34, 36, 38, 39, 40, 47).
While GBM patients less than age 40 years generally enjoy longer survival times as a whole (34), few studies have addressed what features might be further predictive of survival for individual patients within this already favorably aged cohort. Nor has the question generally been asked, “If you are a young adult with a GBM, what exactly is your chance of being a long‐term survivor?”
In part, this paucity of reports may be due to the fact that bona fide young age, non‐pediatric GBM patients represent a distinct minority of overall GBM cases, constituting less than 10% of the total GBMs in a large European hospital cohort (16) and only 2.6% of all GBMs in the latest Central Brain Tumor Registry of the United States report covering the years 1998–2002 (http://www.cbtus.org), although the latter may be a slight underestimate based on the fact that 6% of all gliomas in the database are listed as “not otherwise specified”. Whatever the exact percentage of GBMs that younger‐adult GBMs constitute, fully two‐thirds of all adult GBM patients present between the ages of 45 and 70 years, at a median age of 53 years (16). Clearly, only small percentages of GBM cases occur at either extreme end of the adult age spectrum (19, 34).
Conversely, even though brain tumors are relatively uncommon in younger adults, GBMs are still a very uncommon tumor type within this age range. GBMs constituted only 5.9% of all primary brain and central nervous system (CNS) tumors among young adults, ages 20–34 years, in the most recent CBTRUS statistical report (http://www.cbtus.org). Surveillance, Epidemiology and End Results (SEER) databases for the years 1973–2003 list 15% of all CNS neoplasms in young adults ages 20–35 years as being “astrocytoma, grade IV” (http://www.seer.cancer.gov/SEER*Stat Database: Incidence).
An added known confounding feature to studying GBMs occurring in young‐aged adults is that histological review is mandatory to exclude other similar tumor types that frequently occur within these same age ranges and are known to “contaminate” databases of GBMs, such as anaplastic oligodendroglioma, anaplastic mixed oligoastrocytoma and pleomorphic xanthoastrocytoma (PXA) (24, 26, 27). Inadvertent inclusion of cases with these other diagnoses favorably skews prognosis of GBMs in young adults and may incorrectly overestimate the numbers of long‐term GBM survivors (26, 27).
Despite these limitations, we posited that GBMs in young‐adult, non‐pediatric patients (defined as > age 16 years and < or = 35 years) might represent a particularly interesting subset to study, and especially to contrast with our previous studies on GBM patients at the other extreme of the age spectrum, that is, patients 75 years and older (18, 19). We also were interested to see if features within this group of GBMs overlapped with those seen in pediatric GBMs.
Given the paucity of information on GBMs in young adults, we chose to study several different aspects in the cohort: demographic features, including ethnicity and gender (41); secondary vs. primary vs. radiation‐induced origin (9, 20, 37); and histological features, including presence of necrosis and predominant cell type of the tumors (37). These features have previously been among the most frequently studied factors in relationship to prognosis in GBM patients across all age ranges.
We also investigated several parameters that have been reported to have a variable prognostic effect, specifically in conjunction with patient age. These include epidermal growth factor receptor (EGFR) amplification status (19, 42, 43, 44), TP53 protein expression (27, 30) and cell cycle labeling index (6, 13, 25). Although neither p53 mutational status nor TP53 protein expression has been found to be of prognostic significance in adult patients with GBMs (23), high TP53 protein expression, but not p53 gene mutational status, has been shown to impart a particularly adverse prognosis in malignant pediatric gliomas (30). Hence, to investigate any possible overlap in this area between young‐adult GBMs and pediatric GBMs, we assessed the effects of TP53 protein expression on prognosis.
METHODS
Case accrual and characterization of the cohort. The study was conducted with the approval of the University of Colorado Institutional Review Board. A computer‐based search was conducted from the Department of Pathology database using birth date to identify all patients in our system that were age 17–35 years, inclusive. All patients within this age range who had undergone brain or spinal cord neurosurgical procedures between 1/1/1997 and 12/31/2005 and were diagnosed with GBM (astrocytoma Grade IV) were eligible for this study, including referral (consultation) cases from outside hospitals, denoted by C in Table 2. Pediatric patients (defined as age 16 years or less) are treated at our allied, free‐standing pediatric hospital and were not included in this study.
Table 2.
Copy. Abbreviations: GBM = glioblastoma multiforme; EGFR = epidermal growth factor receptor; FISH = fluorescence in situ hybridization; M = male; N/A = not applicable; Ch. = chromosome; BCNU = carmustine; 7cen = 7 centromere; LOH = loss of heterozygosity; PCV = procarbazine, lomustine, and vincristine; F = female; LTF = lost to follow‐up; PNET = primitive neuroectodermal tumor; F = female; C = consultation case.
| Patient age, gender, race, year(s) of neurosurgical procedure | Relevant history; primary, secondary or radiation‐induced GBM | Tumor location/ Surgical procedure | Histological features/ Extent of necrosis | EGFR amplification status, as assessed by FISH | TP53; MIB‐1 immunostaining, % positive nuclei | Clinical outcome/ Survival |
|---|---|---|---|---|---|---|
| 1. 18 M Hispanic 1997 C | Primary GBM | Brainstem Biopsy | Fibrillary and pleomorphic 1+ | N/A | N/A | N/A LTF |
| 2. 24 M Caucasian 1997 C 1997 (2 surgical procedures same year) | Primary GBM Presented with cerebral hemorrhage, thought to have metastasis | Right temporal Biopsy Resection (well‐circumscribed) | Epithelioid 3+ | Negative for EGFR amplification Polysomy for Ch. 7 (3–6 copies in 62.8% of cells) | 2+ TP53 29.6% MIB‐1 | Developed cerebrospinal fluid dissemination (+CSF cytology) Died 25 weeks |
| 3. 26 M Caucasian 1997 1997 (two outside procedures; slides unavailable for review) 1998 (GBM) | Secondary GBM Grade II oligodendroglioma 1997; radiation therapy; recurrence 1997, resection with BCNU wafers; secondary GBM 1998 | Right parietal Resection Resection Resection | Fibrillary with focal small cell and pleomorphic giant cell component 2+ | Negative for EGFR amplification Polysomy for Ch. 7, 1, 19 (3–4 copies 7cen in 56.6% of cells) Negative for LOH 1p, 19q | 4+ TP53 21.8% MIB‐1 | Alive as of 2/4/1998 2 weeks LTF |
| 4. 27 M Caucasian 1988 (slides reviewed) 1996 1998 (GBM) | Secondary GBM Grade II astrocytoma 1987; radiation therapy; recurrence 1996, resection with BCNU wafers, PCV; secondary GBM 1998 | Left fronto‐parietal Stereotactic biopsy Resection Resection | Small cell 2+ | Negative for EGFR amplification Polysomy for Ch. 7, 1, 19 (3–4 copies 7cen in 40.8% of cells) Negative for LOH 1p, 19q | 4+ TP53 23.3% MIB‐1 | Developed left neck lymph node metastases Died 48 weeks |
| 5. 30 M Race not specified 1998 C | Primary GBM Presented with hematoma, thought to have vascular lesion | Occipital Biopsy | Fibrillary 3+ | N/A | N/A | Died 57 weeks |
| 6. 29 M Race not specified 1997 C 1998 | Primary GBM | Right frontal lobe | Fibrillary with gemistocytic and pleomorphic giant cells 1997 2+ 1998 0 | N/A | 1+ TP53 19.3% MIB‐1 | Died 85 weeks |
| 7. 18 M Hispanic 1999 | Primary GBM | Right frontal Resection | Rhabdoid 3+ | Negative for EGFR amplification Polysomy for Ch. 7 (3–4 copies in 19.2% of cells) | 1+ TP53 26.3% MIB‐1 | Died 22 weeks Autopsy showed residual tumor in right frontal lobe, adjacent leptomeninges Implantation metastasis in right fronto‐parietal scalp, microscopic metastases in both lungs |
| 8. 32 M Caucasian 1998 1999 (GBM) 2000 (GBM) | Secondary GBM Anaplastic mixed oligoastrocytoma, 1998 GBM, 1999 | Right temporal lobe Resection Resection | Fibrillary 2+ | Negative for EGFR amplification Polysomy for Ch. 7 (3–4 copies in 64.2% of cells) Negative for LOH 1p, Positive for LOH 19q | 1+ TP53 8.2% MIB‐1 | Died 90 weeks |
| 9. 21 M Race not specified 2000 C | Secondary GBM Myoclonic seizures for 2 years preceding tumor discovery; neuroimaging showed lesion of left motor strip but thought to be cortical dysplasia and not biopsied. Seizures resolved, medications discontinued, pt. OK until rapid development of right hemiparesis one month before biopsy | Left fronto‐parietal Stereotactic biopsy | Fibrillary with scattered multinucleated giant cells 1+ | Positive high level EGFR amplification (10 to >20 copies per cell in 19.5% of cells, Ratio = 2.7) Negative for polysomy of Ch. 7 | 1+ TP53 28.9% MIB‐1 | Died 42 weeks |
| 10. 31 F Korean 1999 (not reviewed) (GBM) 2000 (GBM + abscess) 2000 C (GBM) | Primary GBM | Right parieto‐occipital Resection | Fibrillary Necrotic abscess on first surgery of 2000, positive for actinomyces | N/A | 4+ TP53 2.2% MIB‐1 | Alive as of 8/18/2005 297 weeks |
| 11. 29 M Race not specified 2000 C | Primary GBM | Thalamus (side not specified) Biopsy | Fibrillary 0 | N/A | N/A | Died 25 weeks |
| 12. 35 M Irish National 1998 1999 2000 (all surgeries in Ireland, slides unavailable for review) 2000 (GBM) | Secondary GBM Oligoastrocytoma 1998; recurred in 1999; received radiation therapy and temozolomide, underwent reoperation in 2000 but tumor could not be resected. Came to USA for surgery the next month | Left frontal Resection | Small cell 3+ | Positive for high level EGFR amplification (Ratio = 6.6) Polysomy for Ch. 1, 7, 19 (3–4 copies 7cen in 47.7% of cells) Negative for LOH 1p, 19q | 1+ TP53 31.6% MIB‐1 | Alive as of 12/31/2005 295 weeks |
| 13. 27 F Caucasian 2001 | Primary GBM Sharply demarcated but deep in the white matter | Left occipital Resection | Epithelioid 2+ | Positive for cells with EGFR amplification (Ratio = 2.0, 6–20 copies in 44.1% of cells) Polysomy for Ch. 7 (3–10 copies in 39.8% of cells) | 4+ TP53 42.2% MIB‐1 | Alive as of 12/31/2005 234 weeks |
| 14. 24 F Caucasian 2001 C 2001 C | Primary GBM | Right frontal Stereotactic biopsy Resection | N/A | N/A | N/A | Died 122 weeks |
| 15. 32 F Hispanic 2001 | Probable secondary GBM History of seizures for 6 years; symptoms had worsened recently and had delivered baby 3 months prior to surgery | Right frontal lobe Biopsy | Gemistocytic 2+ | N/A | N/A | Alive as of 3/8/2002 but LTF 28 weeks |
| 16. 30 M Caucasian 2001 | Secondary GBM | Right occipital Resection | Fibrillary 1+ | Positive for EGFR amplification Polysomy for Ch. 7 (3 copies) | N/A | Alive as of 12/6/2004 but LTF 175 weeks |
| 17. 21 M Race not specified 2002 | Primary GBM Presented with thalamic hemorrhage | Right thalamus Stereotactic biopsy | Fibrillary 3+ | Negative for EGFR amplification Polysomy for Ch. 7 (3–4 copies in 55.7% of cells) | 3+ TP53 25.6% MIB‐1 | Died 5 weeks |
| 18. 24 M Race not specified 2002 C | Primary GBM | Right frontal | Fibrillary 3+ | N/A | N/A | LTF |
| 19. 22 M Caucasian 1984 (ependymoma) 2002 (GBM) 2002 (GBM) | Radiation‐induced GBM Original tumor posterior fossa ependymoma, WHO grade II 1984 (slides reviewed) | Left cerebellum Resection Resection | Fibrillary, with prominent mucinous degeneration, profuse microvascular proliferation 0 | Negative for EGFR amplification Polysomy for Ch. 7 (4–10 copies in 49.2% of cells) | 1+ TP53 10.6% MIB‐1 | Alive as of 12/31/2005 174 weeks |
| 20. 18 M 2002 C Race not specified | Primary GBM Germline short arm chromosome 22 abnormality, lower limb amputation for developmental anomaly, recurrent Burkitt lymphoma of gastrointestinal tract | Right fronto‐parietal Resection | Epithelioid admixed with pleomorphic cells 1+ | Negative for EGFR amplification Polysomy for Ch. 7 (3–4 copies in 30.3% of cells) | 4+ TP53 19.1% MIB‐1 | Alive as of 12/31/2005 173 weeks 2 years out from stem cell rescue has no residual brain tumor, receives IV/IgG for low immunoglobulin levels |
| 21. 27 M Hispanic 2002 | Radiation‐induced GBM Acute lymphoblastic leukemia 5 years previous | Cerebellum Biopsy | Fibrillary 3+ | Negative for EGFR amplification Negative for polysomy of Ch. 7 | 1+ TP53 46.8% MIB‐1 | Died 47 weeks |
| 22. 24 F Caucasian 2002 C | Primary GBM Turcot syndrome Poorly differentiated colon carcinoma 2002, metastatic to liver | Right temporal parietal | N/A | N/A | N/A | Died 165 weeks |
| 23. 31 M Hispanic 2002 | Primary GBM | Left frontal parietal | Fibrillary and small cell 3+ | N/A | N/A | Alive as of 6/7/2002 but LTF 3 weeks |
| 24. 34 M Hispanic 2001 C (grade II astrocytoma) 2002 (GBM) | Secondary GBM Astrocytoma grade II left frontal lobe 2001 (slides reviewed); recurred as bifrontal GBM 2002 | Bifrontal Resection | Small cell with scattered pleomorphic giant cells 2+ | N/A | 4+ TP53 46.3% MIB‐1 | Alive as of 3/4/2004 but LTF 31 weeks |
| 25. 22 F Race not specified 2003 C | Primary GBM (gliosarcoma) | Right temporal Resection | Gliosarcoma 3+ | Negative for EGFR amplification Polysomy for Ch. 7 (3 copies in 42.5% of cells) | 1+ TP53 25.9% MIB‐1 | Died 34 weeks |
| 26. 30 M Hispanic 2003 | Primary GBM | Left frontal Resection | Small cell and focal epithelioid 3+ | High level of EGFR amplification (Ratio = 7.1) Polysomy for Ch. 7 (3–4 copies in 47.6% of cells) | 4+ TP53 53.8% MIB‐1 | Died 3 weeks |
| 27. 29 M American Indian 2003 | Primary GBM | Left temporal Stereotactic biopsy | Small cell 0 | Negative for EGFR amplification Polysomy for Ch. 1, 7, 19 (3–4 copies 7cen in 50.8% of cells) No LOH 1p, 19q | 1+ TP53 42.0% MIB‐1 | Died 67 weeks |
| 28. 31 M Caucasian 2003 2003 | Primary GBM | Right frontal Stereotactic biopsy Resection | Small cell with scattered pleomorphic giant cells 2+ | EGFR not performed Negative for deletion of 1p, Positive for deletion of 19q | 4+ TP53 24.5% MIB‐1 | Alive as of 12/31/2005 117 weeks |
| 29. 25 F Race not specified 2003 C | Primary GBM Thought to be a brain abscess | Left parietal Biopsy | Fibrillary 3+ | Positive for EGFR amplification (20 copies in 85% of cells) | 1+ TP53 54.3% MIB‐1 | Died 61 weeks |
| 30. 28 F Caucasian 2004 2004 | Primary GBM | Right temporal Resection Resection | Small cell 2+ | Negative for EGFR amplification Polysomy for Ch. 7 (3–4 copies in 37.6% of cells) | 1+ TP53 37.6% MIB‐1 | Died 61 weeks |
| 31. 31 M Caucasian 2004 | Primary GBM | Left medulla Biopsy | Fibrillary and small cell 3+ | Negative for EGFR amplification Polysomy for Ch. 7 (3–4 copies in 26.8% of cells) | 2+ TP53 28.9% MIB‐1 | Died 48 weeks |
| 32. 26 M Race not specified 2004 C | Primary GBM Presented with cerebral hemorrhage | Right frontal Resection | Fibrillary and small cell 2+ | Negative for EGFR amplification Polysomy for Ch. 7 (3–4 copies in 54.2% of cells) Negative for LOH 1p, 19q | 1+ TP53 21.4% MIB‐1 | Died 53 weeks |
| 33. 29 M Caucasian 2004 | Primary GBM | Thoracic cord, T7‐T11 Resection | Fibrillary and small cell 0 | Negative for EGFR amplification Negative for polysomy of Ch. 7 | 1+ TP53 29.8% MIB‐1 | Alive as of 12/31/2005 63 weeks |
| 34. 34 F Caucasian 2004 C | Primary GBM | Left temporal parietal | Fibrillary and gemistocytic 1+ | N/A | N/A | Died 91 weeks |
| 35. 29 M Race not specified 2005 C | Primary GBM | Right fronto‐parietal Biopsy | Small cell with unusual eosinophilic filamentous inclusions 0 | Negative for EGFR amplification Polysomy for Ch. 7 (3–4 copies in 42.5% of cells) | 4+ TP53 31.7% MIB‐1 | Died 49 weeks |
| 36. 17 M Caucasian 2005 | Primary GBM Presented with cerebral hemorrhage | Left temporal Resection | Small cell 3+ | Positive for cells with EGFR amplification (8.4% of cells, Ratio = 1.4) Polysomy for Ch. 1, 7, 19 (3–6 copies of 7cen in 63.4% of cells) Negative for LOH 1p, 19q | 4+ TP53 69.3% MIB‐1 | Alive as of 12/31/2005 39 weeks (patient treated on PNET chemotherapy regimen) |
| 37. 29 F Caucasian 2005 | Primary GBM | Left frontal Resection | Fibrillary 1+ | Negative for EGFR amplification Polysomy for Ch. 1, 7, 19 (3–4 copies 7cen in 44.2% of cells) Negative for LOH 1p, 19q | 3+ TP53 28.8% MIB‐1 | Alive as of 12/31/2005 23 weeks |
| 38. 34 M Caucasian 2005 | Primary GBM Cystic mass thought to be abscess, or less likely tumor | Left frontal | Small cell 2+ | Negative for EGFR amplification Polysomy for Ch. 1, 7, 19 (3–6 copies 7cen in 76.8% of cells) Negative for LOH 1p, 19q | 1+ TP53 52.8% MIB‐1 | Alive as of 12/31/2005 3 weeks |
File review generated the 38 young‐aged GBM patients for this study, 10 of whom (26.3%) were neuropathology consultation cases, compared with the 38% of our overall neuropathology practice that derives from consultation cases. With the exception of five consultation cases where all slides and blocks had been returned to the originating hospital, all slides were able to be re‐reviewed at the time of this study. The few consultation cases where slides had been returned were included because a detailed microscopic description was available and/or had been seen by the lead author originally. Re‐review ensured accurate classification, with particular attention paid to eliminating any examples of anaplastic oligodendroglioma, anaplastic mixed glioma or PXA with anaplastic features from incorrect inclusion as GBM.
The 38 young adults, aged 17–35 years inclusive, included 10 females and 28 males. These 38 GBMs represented 10.9% of all GBMs in our files for the years 1997–2005, paralleling the incidence in the World Health Organization (WHO) book (16). Median age was 28.5 years (mean = 27 years), with 13 patients being ages 17–25 years and 25 being in the upper age ranges, ages 26–35 years. The majority of patients were Caucasian; seven patients were Hispanic. The latter is reflective of the population treated in general at hospitals within our University system, given the demographics of Denver County and the state, where 31.7% and 17.1%, respectively, of the population is of Hispanic origin (http://quickfacts.census.gov/qfd/states/08/08031.html). One patient each had furnished his or her ethnicity as American Indian, Korean or a foreign national. No African‐American individuals were known to be included in the cohort. In comparison, census data for Denver County and the state, respectively, for individuals of American Indian, Asian and African‐American ethnicity is 1.3%/1.0%; 2.8%/2.2% and 11.1%/3.8% (http://quickfacts.census.gov/qfd/states/08/08031.html).
Tumors were classified as primary or secondary GBMs and graded according to WHO criteria (16, 17). Twenty‐eight patients (74%) had primary GBMs, eight patients (21%) had secondary GBMs, and two patients (5%) developed radiation‐induced GBMs. Both radiation‐induced GBMs developed within the posterior fossa, at intervals of 18 and 5 years following radiation therapy given for an ependymoma and acute lymphocytic leukemia, respectively. These cases have been previously reported in abbreviated format (20).
Medical records, the patient’s neurosurgeon and the referring pathologist were consulted to obtain relevant clinical history. Five patients presented with cerebral hemorrhages within their tumors; in these patients, ruptured vascular malformations were often higher in the original differential diagnostic list than GBM. In the youngest patient in the series (Case 36), age 17 years, the sharp demarcation, cystic change and intratumoral hemorrhage prompted neuroimaging consideration of primitive neuroectodermal tumor, not GBM (Figure 1). The operative report was utilized to identify site of neoplasms. Almost all tumors were supratentorial, with only two brainstem, two thalamic and one spinal cord tumor. Survival data were gathered with the assistance of Ms. Amy Kendall at the Colorado Tumor Registry.
Figure 1.
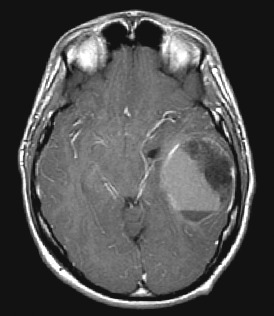
Magnetic resonance imaging scan (MRI) of Case 36, the youngest patient in the series, demonstrating a relatively well‐demarcated mass with enhancement and a fluid–fluid level because of intratumoral hemorrhage.
Methods for histological classification and staining. Sections were fixed in 10% formalin, cut at 4–6 microns, and stained with Harris hematoxylin and eosin (H&E). Immunostaining was conducted as specified in Table 1. Immunostaining employed for the rhabdoid GBM has been previously published (49).
Table 1.
Antibodies used in study. Abbreviations: EMA = epithelial membrane antigen; GFAP = glial fibrillary acidic protein; M = monoclonal; P = polyclonal.
| Antibody | Source* | Clonality | Dilution | Retrieval |
|---|---|---|---|---|
| Vimentin | Ventana | M | Prediluted | Yes |
| EMA | Dako | M | 1:00 | No |
| Pancytokeratin | Ventana | M | Prediluted | Yes |
| GFAP | Dako | M | 1:100 | Yes |
| S100 protein | Dako | P | 1:500 | No |
| Desmin | Dako | M | 1:50 | Yes |
| Muscle‐specific actin | ENZO Diagnostics | M | 1:40 | No |
| Synaptophysin | Cell Marque | M | 1:400 | No |
| INI‐1 protein (BAF47) | BD Transduction | M | 1:250 | Yes |
| P53 | Dako | M | 1:200 | Yes |
| MIB‐1 | Dako | M | 1:50 | Yes |
Suppliers = Biogenex, San Ramon, CA, USA; Dako Corporation, Carpinteria, CA, USA; ENZO Diagnostics, Farmingdale, NY, USA; Signet, Dedham, MA, USA; Immunotech/Coulter, Westbrook, ME, USA; BD Transduction Laboratories, San Jose, CA, USA.
Histological tumor type was classified as small cell, fibrillary, gemistocytic, giant cell, or epithelioid based on the predominant (greater than 50%) cell type. Assessment of the type was made using both H&E and glial fibrillary acidic protein immunohistochemical staining. Epithelioid GBM was defined as a tumor composed of cells with rounded cytoplasmic contours, large eccentric nuclei and prominent nucleoli, but no cytoplasmic ball‐like filaments and no polyphenotypic immunohistochemical expression. The cells often displayed discohesion. Epithelioid GBMs were immunostained for the INI‐1 protein (BAF47) and, by definition, were negative. The definition used for “small cell” histology was that of Burger et al (4). Rhabdoid GBM was defined by a combination of morphological, immunohistochemical, electron microscopic and genetic features. Rhabdoid GBM manifested cells with not only rounded cytoplasmic contours, but also prominent cytoplasmic ball‐like filaments. Nuclei were large, eccentric, and contained prominent nucleoli. There was polyphenotypic expression of neuronal markers, vimentin, smooth muscle actin and epithelial membrane antigen. The single case with this rhabdoid phenotype has been previously published in detail (49). Immunohistochemical staining for the INI‐1 protein (BAF47) was conducted for the first time on this case for the purposes of the current study and the previously unpublished results are reported for the first time in this paper.
Necrosis was semiquantitatively assessed on a 0–3+ scale, with 0 indicating no necrosis present on the biopsy/resection specimen, 1+ indicating focal small areas of necrosis identified, and 2+ indicating multifocal broad zones of necrosis or pseudopalisading necrosis present. A score of 3+ was reserved for cases with extensive necrosis, estimated to occupy 10% or more of the sampled specimen.
Cell cycle labeling indices were assessed by immunostaining for MIB‐1 (Dako Corp., Carpinteria, CA, USA), with 1000 cell counts performed in highest labeled areas of tumor. Counts in all cases were done by a single author (BKK‐D). Extent of nuclear TP53 immunostaining was estimated semiquantitatively, with a score of 1+ given for 0–5%, 2+ for 6–25%, 3+ for 26–50% and 4+ for >50% of tumor nuclei showing immunoreactivity.
Whenever paraffin tissue blocks or appropriate blank slides were still available in our files, cases in this study could be further studied for the above‐mentioned immunohistochemical markers, and if sufficient tissue existed, by fluorescence in situ hybridization (FISH) analyses for EGFR status. Gains of chromosomes 1, 7, 19 were also noted. In many cases with an original diagnosis of oligodendroglioma or mixed oligoastrocytoma, or small cell histology, assessment by FISH for possible loss of heterozygosity (LOH) of chromosomes 1p, 19q had been conducted at the time of the patient’s initial routine diagnostic workup. Data from this testing were recorded whenever available, but study of LOH 1p, 19q was not conducted on each case retrospectively specifically for the purposes of this study.
Statistical analyses were performed, using the SPSS 14 program software for Windows. A Student’s t test was used for statistical analyses. Correlation coefficients were also determined to approximate whether parameters were informative for individual patients. P < 0.05 was considered the statistical level of significance.
Methods for FISH. FISH analysis was performed on formalin‐fixed, paraffin‐embedded tissue sections. Three dual color FISH probe sets, manufactured by Vysis (Abbott Laboratories Inc., Des Plaines, IL, USA), were used for LOH studies of chromosome 1p36 and 19q13, and amplification status of EGFR. The probe sets were: chromosome 1p36 (Spectrum Orange) and 1q25 (Spectrum Green); chromosome 19p13 (Spectrum Green) and 19q13 (Spectrum Orange); EGFR (7p12—Spectrum Orange) and CEP7 (D7Z1—Spectrum Green).
H&E‐stained sections were used to select tumor areas, and a sequential blank section of each specimen was used for FISH. The slides were incubated for 4 h at 60°C, deparaffinized in Xylene and rinsed in 100% ethanol for 5 minutes. Pretreatment included incubation in 2× SSC at 75°C for 20 minutes and digestion with 0.25 mg/mL Proteinase K/2× SSC at 45°C for 20 minutes. Slides were then washed in 2× SSC for 5 minutes and dehydrated in an ethanol series. Probe and chromosomal DNAs were co‐denatured for 10 minutes at 80°C, and incubated at 37°C overnight. A post‐hybridization wash was performed with 0.4× SSC at 75°C for 1–2 minutes. After air drying, chromatin was counterstained with DAPI [4′,6‐diamidino‐2‐phenylindole‐dihydrochloride (Boehringer Mannheim, Indianapolis, IN, USA) in a p‐phenylenediamine dihydrochloride (Sigma, St. Louis, MO)/glycerol antifade solution].
Analysis was performed on an epifluorescence microscope using single interference filter sets for green (Spectrum Green, Vysis), red (Spectrum Orange, Vysis) and blue (DAPI), as well as dual (red, green) and triple (blue, red, green) band pass filters. Image capture was performed with the Applied Imaging Cytovision System (San Jose, CA, USA). The quality of the FISH signals was excellent in all specimens and at least 100 nuclei were scored in each tumor area selected.
In the LOH studies for 1p36 and 19q13, a ratio of relative loss of the probe of interest (1p36 and 19q13) compared with the control probe (1q25 and 19p13, respectively) was calculated for each experiment. A normal ratio is approximately 1.0. Any ratio <0.80 is consistent with deletion of the region of interest.
The copies of EGFR gene were enumerated relative to the copies of chromosome 7 centromere per cell. Polysomy for chromosome 7 was reported if equal numbers of 7 centromere and EGFR signals were observed in greater than a diploid copy number. Amplification of EGFR was reported if the overall ratio of EGFR to 7 centromere signals was greater than 2.0, or if the section contained a major cell population with >10 copies of EGFR in a signal pattern consistent with the presence of double minutes (29).
RESULTS
Characterization of histological features. Histological features in young‐adult GBMs were typical of GBMs in general. Given several reports suggesting more favorable prognosis in patients with giant cell GBMs (33, 41), it is worth noting that no giant cell GBMs were identified in the group. GBMs with predominantly small cell histological features constituted about 1/4 of cases (n = 10). Two of these small cell GBMs (Cases 4 and 12) were secondary GBMs arising from previous oligodendrogliomas; the alternate diagnoses of anaplastic oligoastrocytoma or GBM with oligodendroglial component were carefully excluded by us (BKK‐D). The histological features of the secondary small cell GBM from Case 12 are shown to illustrate the point that oligodendroglial features were not present in the GBM (Figure 2A). One of the GBMs with small cells admixed with pleomorphic giant cells (Case 9, Figure 2B) had prompted consideration of a possible PXA with anaplastic features by the original pathologist. The youngest patient in the group had a well‐demarcated mass showing enhancement on neuroimaging studies and a fluid–fluid level because of intratumoral hemorrhage (Figure 1); this was also a small cell GBM with extensive necrosis (Figure 2C).
Figure 2.

A. Photomicrograph of one of the two small cell glioblastoma multiformes (GBMs) that were secondary GBMs. Note absence of recognizable oligodendroglioma component. Case 12 illustrated. Hematoxylin and eosin (H&E) 200×. B. Photomicrograph of one of the small cell GBMs containing a few interspersed pleomorphic multinucleated giant cells; these were too few to meet criteria for giant cell GBM. Case 9 illustrated. H&E, 200×. C. Photomicrograph of the small cell GBM from the 17‐year‐old male, whose magnetic resonance imaging (MRI) scan is illustrated in Figure 1C. This tumor was relatively homogeneous, although glial areas were also present. Case 36 illustrated. H&E, 200×. D. High power photomicrograph of one of the epithelioid GBMs illustrates the characteristic lack of cell‐to‐cell cohesion that may prompt consideration of metastatic malignancies and the epithelioid morphology, with rounded cytoplasmic contours and absence of fibrillary cell processes. Case 2 illustrated. H&E, 600×. E. High power photomicrograph of the epithelioid GBM illustrating focal vacuolization and probable lipidization of the tumor cells. H&E, 600×. This patient subsequently developed cerebrospinal fluid (CSF) dissemination and CSF cytology was positive for the same tumor cells. Case 2 illustrated. Inset, 600×. F. High power photomicrograph of the rhabdoid GBM demonstrating the prominent nucleoli and glassy, ball‐like cytoplasm. H&E, 600×. G. High power photomicrograph of the rhabdoid GBM, immunostained for BAF47 (INI1 protein), showed retention of nuclear staining in much of the tumor, but focally the most rhabdoid‐appearing cells with large eccentric nuclei, prominent nucleoli and ball‐like cytoplasm showed nuclear loss of immunostaining (arrows). Case 7 illustrated. Immunostaining for INI‐1 protein, with light hematoxylin counterstain, 600×. H. Another unusual, but single, example was a GBM containing abundant eosinophilic, refractile, cytoplasmic inclusions. Case 35 illustrated. H&E, 600×. I. These refractile inclusions were strongly positive for both periodic acid Schiff reaction (top panel) and trichrome stains (bottom panel). Case 35 illustrated. Both, 600×.
Three epithelioid GBMs were identified in the group (Figure 2D,E). In none of these three epithelioid GBM cases was there evidence of polyphenotypic immunohistochemical staining for synaptophysin or epithelial membrane antigen. All three epithelioid GBMs were immunostained for INI‐1 protein (BAF47), and all three showed retention of nuclear staining throughout all cells of the tumor.
This contrasted with the polyphenotypic immunohistochemical expression seen in the one rhabdoid GBM in this series (Figure 2F), which has been previously published, as noted above (49). Recent application of immunohistochemical staining for the INI‐1 protein (BAF47 antibody) to this case now has shown (on four different sections, immunostained at two different times) retention of immunostaining for INI‐1 protein in the nucleus of most of the tumor cells but focal loss in a subset of the cells with the most rhabdoid features (Figure 2G).
The other GBM with highly unusual histology seen in this cohort was Case 35, which possessed unusual filamentous‐like, refractile, cytoplasmic inclusions (Figure 2H) which were positive on both periodic acid Schiff (Figure 2I, top panel) and trichrome stains (Figure 2I, bottom panel). The morphology and staining pattern of these cytoplasmic inclusions did not match Rosenthal fibers or eosinophilic granular bodies.
Results of survival analyses. The median survival time of the group was 59 weeks (mean = 82 weeks) for those in whom the death date could be confirmed or for whom the follow‐up time was complete to the close of the study (12/31/2005). The median survival time was not significantly different at 61 weeks (mean = 90 weeks), when the three patients who were eventually lost to follow‐up, but had had at least a 30‐week follow‐up interval, were also included in the survival analyses (see Table 2, Cases 10, 16, 24) (n = 31 of 38 patients, 82% of entire cohort). The two most recent cases in our cohort, with follow‐up times of less than 30 weeks to the close of the study (Cases 37, 38) were excluded from all survival analyses, as were the five patients for whom follow‐up data had been lost (usually because of the patient having left the state or country) and follow‐up time was limited to 0–29 weeks after diagnosis (see Table 2, Cases 1, 3, 15, 18, 23).
All subsequent survival assessments and comparisons were conducted only on the 31 patients with a known death date or a minimal follow‐up time of 30 weeks. Using these criteria, the median survival time for assessable patients with primary GBMs was 61 weeks (mean = 84 weeks) and for assessable patients with secondary GBMs was 69 weeks (mean = 114 weeks). Seven patients (Cases 10, 12, 13, 16, 19, 20, 22) were known to be long‐term survivors, defined as survivors of 3 years (156 weeks), or more. Long‐term survivors composed 23% of the assessable cohort. One of these was a patient with Turcot syndrome (Case 22), a condition with previously reported longer survival for GBM patients (46). One long‐term survivor had an intratumoral abscess develop (Case 10), an occurrence of uncertain prognostic effect. One of the two radiation‐induced GBMs enjoys long‐term survival, while the other died at 47 weeks. Two of the epithelioid GBMs, including one with an undefined tumor syndrome associated with a chromosome 22 defect (Case 20) were also long‐term survivors. But the third epithelioid GBM (Case 2) succumbed quickly following cerebrospinal fluid (CSF) dissemination of his tumor (see Table 2 and Figure 2I). Hence, several of the long‐term survivors had interesting etiological causes or histological features, but long‐term survival was generally unpredictable.
Histological features, extent of necrosis and TP53 immunostaining were compared with survival data. Assessing patients with a minimal follow‐up time of 30 weeks, the median survival of the nine assessable small cell GBMs was 49 weeks (mean = 79 weeks). In contrast, GBMs with fibrillary, gemistocytic or mixed features (17 assessable cases) had a median survival of 57 weeks (mean = 79 weeks). Other histological types—epithelioid (three cases), rhabdoid (one case) and a gliosarcoma (one case)—were too infrequent to provide meaningful data.
The extent of necrosis in the cohort varied considerably, with 11 assessable cases showing very extensive (3+) necrosis. An example of an unusual tumor with small cell phenotype and extensive (3+) necrosis was the 17‐year‐old patient, also pictured in the magnetic resonance imaging (MRI) in Figure 1. The median survival of these patients with 3+ necrosis was 48 weeks (mean = 69 weeks), which can be contrasted with the median survival time for the whole cohort of 61 weeks (mean = 90 weeks).
Comparing the 20 assessable patients with 2+ or 3+ necrosis vs. the 10 assessable patients with 0 or 1+ necrosis, the median survivals were 48 weeks (mean = 69 weeks) for those with more necrosis vs. 79 weeks (mean = 116) for those with less necrosis. Although there was an obvious trend between the presence of more extensive necrosis and shorter patient survival, the correlation coefficient was –0.0256 (t = 1.35; P = 0.29). These numbers fail to reach statistical significance and illustrate that extent of necrosis could not predict outcome for either individual patients or across the entire cohort.
TP53 immunostaining was grouped into 1–2+ vs. 3–4+ categories; for these two categories the median survival times of the assessable patients were 61 weeks (mean = 78 weeks) vs. 49 weeks (mean = 99 weeks), respectively; correlation coefficient was 0.118 (t = 0.597; P = 0.556). These numbers fail to reach statistical significance and illustrate that extent of TP53 immunostaining could not predict outcome for either individual patients or across the entire cohort.
MIB‐1 cell cycle labeling indices were able to be performed on 28 cases and showed a median of 28.9% (mean = 31.5%). The highest labeling index was found in the 17‐year‐old patient (69.3%). Using the median (29.3%) as a dividing point, and utilizing only those cases with assessable survival data, as defined above, there were 27 assessable cases. The mean survival time for the group with low labeling indices below the median was 89 weeks. The mean survival time for the group with high labeling indices above the median was 75 weeks. The MIB‐1 indices correlation with survival times reached statistical significance (t = 3.05; P < 0.005).
By FISH studies, none of the patients was found to have LOH for chromosome 1p or combined 1p, 19q (see Table 2), although as noted above, not all patients had had this study done during their routine work‐up, and this feature was not specifically studied in the current investigation. Seven of the 26 assessable cases (36%) had EGFR amplification by FISH analyses. Polysomy for chromosomes 1, 7 and 19 were observed in the majority of patients for whom FISH was performed. The median survival time of the patients with EGFR amplification was 61 weeks (mean = 121 weeks, range 3–295 weeks) vs. those without EGFR amplification, in which the median survival was 49 weeks (mean = 65 weeks, range 5–174 weeks). The correlation coefficient for EGFR amplification status and its relationship to patient survival was 0.349 (t = 1.28; P = 0.242). Using several different methods of statistical assessment, these numbers fail to reach statistical significance and illustrate that the presence or absence of EGFR amplification could not predict outcome for either individual patients or across the entire cohort.
DISCUSSION
The strengths of this report are the relatively large number of young‐aged GBM patients, accrued from a single institution and the ability to directly compare our results in these young patients with those for GBMs in the elderly, which we have previously reported (19). The number of patients in this study (n = 38) is comparable to those of recent and/or oft‐cited studies on pediatric GBMs (1, 2, 21, 45). GBMs in young adults were found to comprise 10.9% of the GBM population at our institution, identical to the 10% cited in the WHO fascicle (16) and similar to the equally small percentage of GBMs that occur in children (8.8%) (8). Hence, the ability to conduct larger studies on young adults with GBMs is limited. A second strength of the report is that the GBMs in our young adults were characterized for the same histological, MIB‐1 cell cycling, and EGFR amplification status as was done for GBM patients over age 75 years, allowing direct comparisons (19). A third strength of this paper is the documentation of several unique histological variants of GBMs, reported here within the context of a study on young‐adult GBMs, but unlikely to be limited to this age cohort. A potential weakness of the study was that adequate follow‐up could be obtained on only 82% of the cohort and all cases could not be studied for all parameters.
Despite these limitations, several interesting epidemiological features emerge. First, while the majority of patients (74%) were found to have sporadic, primary de novo GBMs unassociated with any known underlying tumor‐predisposing condition, there was some overlap between young‐adult and pediatric GBM patients in the fact that at least four patients had predisposing factors. Two young‐adult GBMs were radiation‐induced; in comparison one radiation‐induced tumor was identified among our elderly GBM cohort (19). An additional two cases were associated with either a known tumor syndrome (Turcot) or a germline chromosomal mutation. Secondary GBMs were relatively frequent in the young‐aged cohort (21%), in contrast to their incidence of 5% within all age groups in a recent large population‐based study (28) and their absence among elderly patients with GBMs (19). Secondary GBMs typically occur in patients around the age of 40 years (9) so the high proportion found within patients in these younger age ranges was an expected finding. The male‐to‐female ratio for GBMs across all ranges is 1.5:1 (16). In our cohort, the male‐to‐female ratio overall was even higher at 2.8:1, and all six of our secondary GBMs were male (see Table 2). Male‐to‐female ratios for pediatric GBM patients in recent studies have ranged from 1.2:1 to 2.2:1 (1, 2, 11, 45). The male predominance we identified in our young‐aged adult GBM patients appears to be more similar to that seen in pediatric GBM patients than to that found in older adults. Interestingly, in our elderly GBM cohort the male‐to‐female ratio had nearly equalized (19).
Second, median survival overall for young‐adult GBM patients was 61 weeks. The median survival time for assessable patients with primary GBMs was 61 weeks and for secondary GBMs was 69 weeks. Hence, patients with secondary GBMs did not show longer median survival times once their tumor had upgraded to a GBM. This is a similar observation to that of Dropcho and Soong (9), who found no difference in median survival time for secondary vs. primary GBM patients, but contrasts with that of Winger et al (48).
While other investigators have suggested that young‐aged adults with GBMs in general enjoy a longer median survival time (34, 37, 38), in our study the median survival was just over 1 year, nearly identical to that in GBMs across all adult age ranges. Median and mean survival times in our previously published elderly GBM cohort (20 patients, aged 75–87 years) were strikingly shorter at 2.5 months and 4.6 months, respectively (19). No long‐term survivors were identified in the elderly adult GBM cohort, and the longest survival time in a patient over age 75 years was only 13 months. In the current study of young‐adult GBMs, in contrast, 23% were identified as long‐term (>3 years) survivors. However, it should be emphasized that several of the remaining 77% of the young‐aged adult GBM patients manifested a prognosis just as dismal as that found in our elderly adults (see Table 2) (19). Thus, there appear to be at least two distinct and dichotomous subsets of young‐adult GBM patients in terms of survival: those with prognosis that is equally poor as for GBMs in older adults and those at the other extreme that survive 3 years or longer. Few of our patients demonstrated intermediate length of survival; with the exception of the patient with Turcot syndrome, none died beyond the 2‐year time span from diagnosis.
Third, histological subtypes seen in the young aged adults were fairly typical and in most cases paralleled those seen in elderly GBM patients (19). Tumors with a predominantly small cell phenotype were not rare among young‐aged adults (n = 10; 26% of the current cohort). As in our previous study (19), we did not find a one‐to‐one correspondence between small cell histology and the presence of EGFR amplification (see Table 2). The median survival time for patients with small cell GBMs was considerably shorter, at 49 weeks, than the median for the cohort as a whole, at 61 weeks. The small numbers of cases in this study preclude firm conclusions about this adverse effect of small cell histology on survival, however, and mandate further validation by others of this finding specifically in young‐adult GBM patients.
The youngest patient in this series (age 17 years) had a tumor with predominantly small cell histology, and a consideration of a supratentorial primitive neuroectodermal tumor (sPNET) was raised by the patient’s clinicians. The diagnosis of GBM was confirmed by an outside consultant and further verified by the positive EGFR amplification testing by FISH. Cases such as this illustrate the inherent problems in categorizing and distinguishing one poorly differentiated tumor type from another, especially in pediatric patients (see editorial by Peter Burger, 3).
GBMs in young adults are sufficiently uncommon in daily neurosurgical and pathology practice that, not surprisingly, several of these had prompted pre‐biopsy consideration of other conditions that are more frequently seen in younger‐aged patients, such as vascular malformations, melanoma metastases, or abscesses. And post biopsy, diagnoses of other glioma tumor types (anaplastic oligodendroglioma, PXA) were often higher on the differential diagnostic list than was GBM for the original pathologist who saw the case.
Most GBMs in young adults manifested relatively unremarkable histological features; unusual histological types of tumors, including rhabdoid, epithelioid, gliosarcoma, and filament‐bearing examples (n = 6) were exceptions. The epithelioid GBMs appeared similar to six cases described in abstract format by Dr. Greg Fuller and colleagues at the 1998 meeting of the American Association of Neuropathologists (10). Although our three cases are described herein in the context of a study of young‐aged GBM patients, we have personally seen three other examples in older patients (data not shown). Hence, we concur with Dr. Fuller and colleagues that this epithelioid GBM phenotype is not restricted to young‐aged patients; his six patients were ages 11–71 years. Unlike the series by Dr. Fuller and colleagues, only one of our three epithelioid GBMs was superficially located and mimicked a meningioma. This patient died after he developed CSF tumor dissemination. The other two epithelioid GBMs were, however, circumscribed according to the clinicians who reviewed their neuroimaging studies, albeit more deep‐seated in white matter. Our three epithelioid GBMs overlap to a lesser degree with the lipid‐rich epithelioid GBMs described by Dr. M. Rosenblum and colleagues (32). Although two of our epithelioid GBMs are long‐term survivors, and although there have been reports of long‐term survival in patients with giant cell GBMs (33, 41), none of our epithelioid GBMs meet criteria for giant cell GBM (16) or PXA with anaplastic features (12, 31).
Fourth, we assessed a number of standard features for possible correlation with survival in this group, including MIB‐1 cell cycle labeling indices, TP53 immunohistochemical staining, and extent of necrosis. MIB‐1 labeling index showed a significant correlation with prognosis within this young‐aged cohort (P < 0.005). McKeever et al have previously shown that proliferation index, as assessed by MIB‐1 immunostaining, is related to patient age and prognosis in adult GBMs (25). They concluded that younger patient age was a significant predictor of a low MIB‐1 proliferation index in GBMs (25). Within our young‐adult GBM cohort, MIB‐1 labeling index was strongly predictive of prognosis (P < 0.005), concurring with other previous studies that MIB‐1 is one of the best prognostic indicators of survival time (13). Ho et al have also shown the prognostic utility of MIB‐1 labeling index in predicting survival in pediatric patients, with labeling indices being a better stratifying parameter for prognosis in pediatric patients with anaplastic astrocytoma than grading criteria (14). Hence, MIB‐1 labeling indices prove to be a good predictor of survival for young‐aged adults with GBMs, as well as for pediatric patients and older adults with GBMs.
Both extent of necrosis and TP53 immunohistochemical staining showed a trend toward correlation with median survival time, with extensively necrotic tumors showing worse prognosis and tumors with strong TP53 immunostaining showing better prognosis than the mean. However, the numerical correlation coefficient was not high in either case, due to the fact that there were too many cases that were individual exceptions within these categories and P‐values all failed to reach statistical significance. Hence, these features cannot reliably predict prognosis for a given patient within this young‐aged cohort or across the entire cohort. Some studies of long‐term adult survivors with GBMs have shown a higher prevalence of high TP53 immunostaining, but not TP53 mutations, in this group (27). Our findings in young adults contrast with those studies in pediatric gliomas which show an adverse prognosis associated with high TP53 immunostaining (11, 30).
Finally, we found an EGFR amplification frequency of 36% in this young‐aged adult GBM cohort, almost identical to the positive EGFR amplification seen in 1/3 or more of GBMs across all age ranges (16). This differs from the small minority (0–7%) of pediatric GBM patients in some studies with positive EGFR amplification (2, 45). Our previous study of elderly GBM patients demonstrated that 25% of elderly patients showed positive EGFR amplification (19). In contrast to the results of the current study on young adults, EGFR amplification status in the elderly cohort strongly correlated with survival (19). In that study we demonstrated a longer survival (mean = 8.3 months, median = 10.5 months) for elderly patients whose tumors were positive for EGFR amplification vs. a significantly shortened survival (mean = 3.2 months, median = 2.0 months) (P = 0.04) for those patients whose tumors were negative for EGFR amplification (19).
Previous studies in the literature have shown that the status of EGFR amplification has a variable impact on prognosis, depending on the age of the patient (19, 42, 43, 44). In our current study of young‐aged adult GBM patients, EGFR amplification status also impacted mean survival times. The median survival time of the patients with EGFR amplification was 61 weeks (mean = 121 weeks, range 3–295 weeks) vs. the median survival of 49 weeks (mean = 65 weeks, range 5–174 weeks) for those without EGFR amplification. The range was so broad, however, as to make EGFR amplification status of considerably lesser utility in predicting prognosis for individual young‐aged patients than for the elderly GBM patients. The polysomy of chromosomes 1, 7 and 19 that we found in most patients (see Table 2) likely relates to an overall polyploidy, a common finding in GBMs in general by standard cytogenetic studies.
In summary, this study represents an attempt to characterize a subset of GBMs for a variety of features not previously documented. Bona fide GBMs in young adults are uncommon but not rare. The original physicians involved with these young‐aged patients usually considered tumors or non‐neoplastic conditions other than GBM in their young patients. Males predominated, in a 2.8:1 M : F ratio. The majority of the tumors proved to be sporadic and primary and was unassociated with genetic syndromes or other identifiable risk factors. Patients with secondary GBMs did not enjoy longer survivals in our study. Most GBMs in young adults did not manifest unique histological features, and indeed small cell GBM constituted about 1/4 of tumors. Several unusual histological variants of GBMs were encountered but are unlikely to be confined to this aged cohort. MIB‐1 correlated best with prognosis. Long‐term (>3 years) survivors constituted about 1/4 of the group. The remaining 3/4 manifested a prognosis just as dismal as that found in older‐aged patients. There appear to be at least two dichotomous subsets of young‐adult GBM patients in terms of prognosis. GBMs in young adults are a more inhomogeneous group than GBMs in older, especially elderly, adults where outcome is more uniformly poor.
ACKNOWLEDGMENTS
The authors gratefully acknowledge Ms. Susan Peth for expert manuscript preparation, Ms. Amy Kendall in the University of Colorado Tumor Registry for obtaining survival data, Ms. Lisa Litzenberger for photographic assistance, Dr. Chun‐I Sze, Mr. Bob Silber and Jeanelle Sheeder, MSPH, for statistical analyses, and Mrs. Jeana Marks and Mr. David Davis for histotechnology support. This study was supported by funds from the Michele Plachy‐Rubin Foundation for Brain Tumor Research at UCHSC.
REFERENCES
- 1. Bhattacharjee MB, Bruner J (1997) p53 protein in pediatric malignant astrocytomas: a study of 21 patients. J Neuro-oncol 32:225–233. [DOI] [PubMed] [Google Scholar]
- 2. Bredel M, Pollack IF, Hamilton RL, James CD (1999) Epidermal growth factor receptor expression and gene amplification in high‐grade non‐brainstem gliomas of childhood. Clin Cancer Res 5:1786–1792. [PubMed] [Google Scholar]
- 3. Burger PC (2006) Supratentorial primitive neuroectodermal tumor (sPNET). Brain Pathol 16:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burger PC, Pearl DK, Aldape K, Yates AJ, Scheithauer BW, Passe SM, Jenkins RB, James CD (2001) Small cell architecture—a histological equivalent of EGFR amplification in glioblastoma multiforme? J Neuropathol Exp Neurol 60:1099–1104. [DOI] [PubMed] [Google Scholar]
- 5. Burton EC, Lamborn KR, Feuerstein BG, Prados M, Scott J, Forsyth P, Passe S, Jenkins RB, Aldape KD (2002) Genetic aberrations defined by comparative genomic hybridization distinguish long‐term from typical survivors of glioblastoma. Cancer Res 62:6205–6210. [PubMed] [Google Scholar]
- 6. Burton EC, Lamborn KR, Forsyth P, Scott J, O’Campo J, Uyehara‐Lock J, Prados M, Berger M, Passe S, Uhm J, O’Neill BP, Jenkins RB, Aldape KD (2002) Aberrant p53, mdm2, and proliferation differ in glioblastomas from long‐term compared with typical survivors. Clin Cancer Res 8:180–187. [PubMed] [Google Scholar]
- 7. Chandler KL, Prados MD, Malec M, Wilson CB (1993) Long‐term survival in patients with glioblastoma multiforme. Neurosurgery 32:716–720. [DOI] [PubMed] [Google Scholar]
- 8. Dohrmann GJ, Farwell JR, Flannery JT (1976) Glioblastoma multiforme in children. J Neurosurg 44:442–448. [DOI] [PubMed] [Google Scholar]
- 9. Dropcho EJ, Soong SJ (1996) The prognostic impact of prior low grade histology in patients with anaplastic gliomas: a case‐control study. Neurology 47:684–690. [DOI] [PubMed] [Google Scholar]
- 10. Fuller GN, Goodman JC, Vogel H, Ghorbani R (1998) Epithelioid glioblastoma: a distinct clinicopathologic entity. J Neuropathol Exp Neurol 57:501. [Google Scholar]
- 11. Ganigi PM, Santosh V, Anandh B, Chandramouli BA, Sastry Kolluri VR (2005) Expression of p53, EGFR, pRb and bcl‐2 proteins in pediatric glioblastoma multiforme: a study of 54 patients. Pediatr Neurosurg 41:292–299. [DOI] [PubMed] [Google Scholar]
- 12. Giannini C, Scheithauer BW, Burger PC, Brat DJ, Wollan PC, Lach B, O’Neill BP (1999) Pleomorphic xanthoastrocytoma: what do we really know about it? Cancer 85:2033–2045. [PubMed] [Google Scholar]
- 13. Ho DM, Hsu CY, Ting LT, Chiang H (2003) MIB‐1 and DNA topoisomerase II alpha could be helpful for predicting long‐term survival of patients with glioblastoma. Am J Clin Pathol 119:715–722. [DOI] [PubMed] [Google Scholar]
- 14. Ho DM, Wong TT, Hsu CY, Ting LT, Chiang H (1998) MIB‐1 labeling index in nonpilocytic astrocytoma of childhood: a study of 101 cases. Cancer 82:2459–2466. [DOI] [PubMed] [Google Scholar]
- 15. Hulshof MC, Koot RW, Schimmel EC, Dekker F, Bosch DA, Gonzalez Gonzalez D (2001) Prognostic factors in glioblastoma multiforme. 10 years experience of a single institution. Strahlenther Onkol 177:283–290. [DOI] [PubMed] [Google Scholar]
- 16. Kleihues P, Burger PC, Collins VP, Newcomb EW, Ohgaki H, Cavenee WK (2000) Glioblastoma. In: World Health Organization Classification of Tumours. Pathology & Genetics. Tumours of the Nervous System. Kleihues P, Cavenee WK (eds), pp. 29–39. IARC Press, Lyon. [Google Scholar]
- 17. Kleihues P, Ohgaki H (1999) Primary and secondary glioblastomas: from concept to clinical diagnosis. Neuro-oncol 1:44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kleinschmidt‐DeMasters BK, Lillehei KO, Breeze RE (2003) Neoplasms involving the central nervous system in the older old. Hum Pathol 34:1137–1147. [DOI] [PubMed] [Google Scholar]
- 19. Kleinschmidt‐DeMasters BK, Lillehei KO, Varella‐Garcia M (2005) Glioblastomas in the older old. Arch Pathol Lab Med 129:624–631. [DOI] [PubMed] [Google Scholar]
- 20. Kleinschmidt‐DeMasters BK, Kang JS, Lillehei KO (2006) The burden of radiation‐induced CNS tumors—a single institution’s experience. J Neuropathol Exp Neurol 65:204–216. [DOI] [PubMed] [Google Scholar]
- 21. Korshunov A, Sycheva R, Gorelyshev S, Golanov A (2005) Clinical utility of fluorescence in situ hybridization (FISH) in nonbrainstem glioblastomas of childhood. Mod Pathol 18:1258–1263. [DOI] [PubMed] [Google Scholar]
- 22. Kraus JA, Glesmann N, Beck M, Krex D, Klockgether T, Schackert G, Schlegel U (2000) Molecular analysis of the PTEN, TP53 and CDKN2A tumor suppressor genes in long‐term survivors of glioblastoma multiforme. J Neuro-oncol 48:89–94. [DOI] [PubMed] [Google Scholar]
- 23. Kraus JA, Wenghoefer M, Glesmann N, Mohr S, Beck M, Schmidt MC, Schroder R, Berweiler U, Roggendorf W, Diete S, Dietzmann K, Heuser K, Muller B, Fimmers R, Von Deimling A, Schlegel U (2001) TP53 gene mutations, nuclear p53 accumulation, expression of Waf/p21, Bcl‐2, and CD95 (APO‐1/Fas) proteins are not prognostic factors in de novo glioblastoma multiforme. J Neuro-oncol 52:263–272. [DOI] [PubMed] [Google Scholar]
- 24. Kraus JA, Wenghoefer M, Schmidt MC, Von Deimling A, Berweiler U, Roggendorf W, Diete S, Dietzmann K, Muller B, Heuser K, Reifenberger G, Schlegel U (2000) Long‐term survival of glioblastoma multiforme: importance of histopathological reevaluation. J Neurol 247:455–460. [DOI] [PubMed] [Google Scholar]
- 25. McKeever PE, Junck L, Strawderman MS, Blaivas M, Tkaczyk A, Cates MA, Yan M, Li L (2001) Proliferation index is related to patient age in glioblastoma. Neurology 56:1216–1218. [DOI] [PubMed] [Google Scholar]
- 26. McLendon RE, Halperin EC (2003) Is the long‐term survival of patients with intracranial glioblastoma multiforme overstated? Cancer 98:1745–1748. [DOI] [PubMed] [Google Scholar]
- 27. Morita M, Rosenblum MK, Bilsky MH, Fraser RA, Rosenfeld MR (1996) Long‐term survivors of glioblastoma multiforme: clinical and molecular characteristics. J Neuro-oncol 27:259–266. [DOI] [PubMed] [Google Scholar]
- 28. Ohgaki H, Kleihues P (2005) Population‐based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol 64:479–489. [DOI] [PubMed] [Google Scholar]
- 29. Okada Y, Hurwitz EE, Esposito JM, Brower MA, Nutt CL, Louis DN (2003) Selection pressures of TP53 mutation and microenvironmental location influence epidermal growth factor receptor gene amplification in human glioblastomas. Cancer Res 63:413–416. [PubMed] [Google Scholar]
- 30. Pollack IF, Finkelstein SD, Woods J, Burnham J, Holmes EJ, Hamilton RL, Yates AJ, Boyett JM, Finlay JL, Sposto R; Children’s Cancer Group (2002) Expression of p53 and prognosis in children with malignant gliomas. N Engl J Med 346:420–427. [DOI] [PubMed] [Google Scholar]
- 31. Prayson RA, Morris HH III (1998) Anaplastic pleomorphic xanthoastrocytoma. Arch Pathol Lab Med 122:1082–1086. [PubMed] [Google Scholar]
- 32. Rosenblum MK, Erlandson RA, Budzilovich GN (1991) The lipid‐rich epithelioid glioblastoma. Am J Surg Pathol 15:925–934. [DOI] [PubMed] [Google Scholar]
- 33. Sabel M, Reifenberger J, Weber RG, Reifenberger G, Schmitt HP (2001) Long‐term survival of a patient with giant cell glioblastoma. Case report. J Neurosurg 94:605–611. [DOI] [PubMed] [Google Scholar]
- 34. Salcman M, Scholtz H, Kaplan RS, Kulik S (1994) Long‐term survival in patients with malignant astrocytoma. Neurosurgery 34:213–219. [DOI] [PubMed] [Google Scholar]
- 35. Salmon I, Dewitte O, Pasteels JL, Flament‐Durand J, Brotchi J, Vereerstraeten P, Kiss R (1994) Prognostic scoring in adult astrocytic tumors using patient age, histopathological grade, and DNA histogram type. J Neurosurg 80:877–883. [DOI] [PubMed] [Google Scholar]
- 36. Salvati M, Cervoni L, Artico M, Caruso R, Gagliardi FM (1998) Long‐term survival in patients with supratentorial glioblastoma. J Neuro-oncol 36:61–64. [DOI] [PubMed] [Google Scholar]
- 37. Schmidt MC, Antweiler S, Urban N, Mueller W, Kuklik A, Meyer‐Puttlitz B, Wiestler OD, Louis DN, Fimmers R, Von Deimling A (2002) Impact of genotype and morphology on the prognosis of glioblastoma. J Neuropathol Exp Neurol 61:321–328. [DOI] [PubMed] [Google Scholar]
- 38. Scott JN, Rewcastle NB, Brasher PM, Fulton D, Hagen NA, MacKinnon JA, Sutherland G, Cairncross JG, Forsyth P (1998) Long‐term glioblastoma multiforme survivors: a population‐based study. Can J Neurol Sci 25:197–201. [DOI] [PubMed] [Google Scholar]
- 39. Scott JN, Rewcastle NB, Brasher PM, Fulton D, MacKinnon JA, Hamilton M, Cairncross JG, Forsyth P (1999) Which glioblastoma multiforme patient will become a long‐term survivor? A population‐based study. Ann Neurol 46:183–188. [PubMed] [Google Scholar]
- 40. Senger D, Cairncross JG, Forsyth PA (2003) Long‐term survivors of glioblastoma: statistical aberration or important unrecognized molecular subtype? Cancer J 9:214–221. [DOI] [PubMed] [Google Scholar]
- 41. Shinojima N, Kochi M, Hamada J, Nakamura H, Yano S, Makino K, Tsuiki H, Tada K, Kuratsu J, Ishimaru Y, Ushio Y (2004) The influence of sex and the presence of giant cells on postoperative long‐term survival in adult patients with supratentorial glioblastoma multiforme. J Neurosurg 101:219–226. [DOI] [PubMed] [Google Scholar]
- 42. Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, Makino K, Saya H, Hirano H, Kuratsu J, Oka K, Ishimaru Y, Ushio Y (2003) Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res 63:6962–6970. [PubMed] [Google Scholar]
- 43. Simmons ML, Lamborn KR, Takahashi M, Chen P, Israel MA, Berger MS, Godfrey T, Nigro J, Prados M, Chang S, Barker FG II, Aldape K (2001) Analysis of complex relationships between age, p53, epidermal growth factor receptor, and survival in glioblastoma patients. Cancer Res 61:1122–1128. [PubMed] [Google Scholar]
- 44. Smith JS, Tachibana I, Passe SM, Huntley BK, Borell TJ, Iturria N, O’Fallon JR, Schaefer PL, Scheithauer BW, James CD, Buckner JC, Jenkins RB (2001) PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst 93:1246–1256. [DOI] [PubMed] [Google Scholar]
- 45. Sung T, Miller DC, Hayes RL, Alonso M, Yee H, Newcomb EW (2000) Preferential inactivation of the p53 tumor suppressor pathway and lack of EGFR amplification distinguish de novo high grade pediatric astrocytomas from de novo adult astrocytomas. Brain Pathol 10:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Van Meir EG (1998) “Turcot’s syndrome”: phenotype of brain tumors, survival and mode of inheritance. Int J Cancer 75:162–164. [DOI] [PubMed] [Google Scholar]
- 47. Vertosick FT Jr, Selker RG (1992) Long‐term survival after the diagnosis of malignant gliomas: a series of 22 patients surviving more than 4 years after diagnosis. Surg Neurol 38:359–363. [DOI] [PubMed] [Google Scholar]
- 48. Winger MJ, Macdonald DR, Cairncross JG (1989) Supratentorial anaplastic gliomas in adults. The prognostic importance of extent of resection and prior low‐grade gliomas. J Neurosurg 71:487–493. [DOI] [PubMed] [Google Scholar]
- 49. Wyatt‐Ashmead J, Kleinschmidt‐DeMasters BK, Hill DA, Mierau GW, McGavran L, Thompson SJ, Foreman NK (2001) Rhabdoid glioblastoma. Clin Neuropath 20:248–255. [PubMed] [Google Scholar]