

Abstract
Detailed analysis of the structural changes that follow human clinical spinal cord injury is limited by difficulties in achieving adequate tissue fixation. This study bypasses this obstacle by examining the spinal cord from paraplegic domestic animals, enabling us to document the ultrastructural changes at different times following injury. In all but one case, injury resulted from a combination of contusion and compression. There was infarction and hemorrhage, followed by gray matter destruction and the rapid development of a variety of white matter changes including axon swelling and myelin degeneration. Axons greater than 5 µm in diameter were more susceptible to degenerative changes, whereas smaller axons, particularly those in the subpial region, were relatively well preserved. Demyelinated axons were seen within 2 weeks after injury and, at later time points, both Schwann cell and oligodendrocyte remyelination was common. More subtle white matter abnormalities were identified by examining sagittal sections, including focal accumulation of organelles in the axoplasm and partial and paranodal myelin abnormalities. These observations serve to validate observations from experimental models of spinal contusion but also highlight the complexity of naturally occurring (ie, clinical) spinal injury. They also raise the possibility that focal abnormalities such as paranodal demyelination may contribute to early axonal dysfunction and possibly to progressive tissue damage.
INTRODUCTION
Survival and recovery following spinal cord injury (SCI) have improved over recent years, yet many victims suffer persistent somatic and autonomic disabilities. Experimental models have permitted great strides to be made in elucidating the pathophysiology of SCI, leading to a sophisticated understanding of the nature and progression of the secondary autodestructive events that exacerbate the initial injury. As a result, new treatments have emerged that encourage tissue preservation following experimental SCI, promote axonal regeneration or stimulate plastic responses (35). The question now arises as to whether the interventions proven to work in experimental models can be translated into effective clinical therapies.
As a first step in this translation to the clinic it is imperative to understand the nature of the naturally occurring lesion and how well it is reflected in the available SCI models. SCI is generally modeled by acute contusion of the exposed dorsal aspect of the spinal cord 1, 10, 13, 31, 45). This differs from the most frequent causes of SCI—fracture/luxations—in which there is a mixture of contusion and persistent compression, often caused by structures lying ventral to the cord. Studies carried out by The Miami Project to Cure Paralysis over the last decade have revealed features of human SCI that are well recognized in experimental animals, including apoptosis soon after injury, a variety of myelin abnormalities and cavitation of the injured cord 14, 17, 22). However, histological analysis of the injured human spinal cord is limited because it is often only available many years after the injury and is rarely optimally preserved (by perfusion‐fixation) as is routine in experimental animals. Detailed ultrastructural studies have therefore not been possible, making it difficult to be certain of the precise pathological outcome of real‐life human SCI and how closely it is modeled by experimental injuries.
Here we introduce an analysis of perfusion‐fixed naturally occurring SCI that is available from veterinary patients. Domestic dogs and cats frequently suffer SCI as a result of road traffic accidents or through explosive intervertebral disc extrusion—a common problem in specific susceptible breeds 24, 32). The causes of SCI in this patient group are therefore similar to those encountered in the human population and comprise a mixture of contusion and compression. As animals that suffer severe SCI are frequently euthanized soon after the incident and can be perfusion‐fixed they provide a bridge between the pure experimental lesion in animals and naturally occurring SCI in humans. Our analysis provides a comparison between experimental and naturally occurring SCI and our focus on ultrastructural features of the injury to white matter highlights some additional important features that may aid in interpretation of the clinical signs after human SCI. Such information is invaluable in the future planning of novel therapeutic interventions, as it is essential to understand the lesion environment that any putative treatment is attempting to modify.
MATERIALS AND METHODS
Tissue preparation.
This study was performed under the jurisdiction and ethical guidance of the Royal College of Veterinary Surgeons (UK) and with the consent of the owners of the animals. Seven paraplegic animals (see Table 1) were euthanized at their owner’s request and immediately perfused with 4% glutaraldehyde (Cases 2–7) or 4% paraformaldehyde (Case 1) to ensure optimal tissue fixation. The section of spinal cord containing the injury site was then removed and immersed in fixative for a minimum of 24 h before being cut into both transverse and sagittal blocks. These were immersed in 2% osmium tetroxide overnight at 4°C, then dehydrated using sequential ethanol washes (70% ethanol for 15 minutes; 95% for 15 minutes; 100% for 15 minutes, repeated twice) and propylene oxide (twice for 15 minutes). Tissue was impregnated with TAAB resin (Taab Laboratories Equipment Ltd, Aldermaston, UK) by immersing first in propylene oxide/resin (equal measures, for 3 h) and then 100% resin (twice, for six hours each); the tissue blocks were then embedded in fresh resin and hardened at 60°C over 24 h. Semithin sections (1 µm) were cut and stained using toluidine blue (5% in a 1% Borax solution). Areas of the lesion that warranted more detailed examination were identified using light microscopy; 90 nm sections were then cut, collected onto a 400 mesh copper grid (3 mm diameter) and stained with uranyl acetate (14% in 50% ethanol) and lead citrate (Merck—1 g in 100 mL 0.1 M NaOH). Sections were viewed under a Hitachi Model H600 transmission electron microscope and photographs taken using Kodak electron microscope film 4489 (Kodak, Rochester, USA).
Table 1.
Clinical details of paraplegic animals. Abbreviations: M− = motor function absent; S− = sensory function absent; S+ = sensory function intact (deep pain only); T = thoracic vertebra; L = lumbar vertebra; y = years; m = months.
| Animal | Species | Age | Duration of paraplegia | Cause of cord injury | Site of cord injury | Clinical state |
|---|---|---|---|---|---|---|
| Case 1 | Dog | 5 years | 18 h | Disc extrusion | T13‐L1 | M−; S− |
| Case 2 | Cat | 8 months | 36 h | Fracture | T12/T13 | M−; S− |
| Case 3 | Dog | 8.2 years | 48 h | Fracture with subluxation | L1/L2 | M−; S− |
| Case 4 | Cat | 7 months | 11 days | Fracture | T12/T13 | M−; S− |
| Case 5 | Dog | 4.5 years | 17 days | Subluxation | T13/L1 | M−; S− |
| Case 6 | Cat | 3 years | 9 weeks | Fracture | L3/L4 | M−; S− |
| Case 7 | Dog | 9 years | 12 weeks | Chronic disc compression | Multi‐site. Centered L1/L2 | M−; S+ |
Measurement of cord area.
Transverse sections of the spinal cord were examined using a very low power objective lens and the images were digitized. The outline of the cord was traced on a computer screen and the area measured, after calibration, using an MCID analysis system (MCID, Model M4, Toronto, Canada). The region of the cord containing the lesion and an adjacent region of structurally normal tissue cranial to the lesion was measured.
Axon counting. To compare the effect of spinal injury on axons of different sizes, the diameter of intact axons was measured in four different regions of the spinal cord: the ventral, dorsal, lateral and dorsolateral funiculi. Axons were selected for measurement by drawing a straight line from the edge of the gray matter to the pial surface (see Figure 1) and the diameter of any morphologically normal axon touched by the line was recorded. Axons whose myelin sheath only was touched by the line were not counted and axons which appeared abnormal or whose myelin sheaths were abnormal were also excluded. Measurement was performed using a Nikon Eclipse E600 Microscope (Nikon, Kingston Upon Thames, UK) linked to an MCID analysis system: tissue sections were examined at an optical magnification of ×400 and the field of view was projected onto a 17″ computer screen for further magnification. Morphologically normal axons were defined as those that had an apparently normal distribution of organelles (axons with a glassy appearance or with unusually dense staining were excluded) and which had a myelin sheath of appropriate thickness for the size of axon, with no splitting of the myelin membrane and without a large increase in the space between the axon and myelin sheath.
Figure 1.
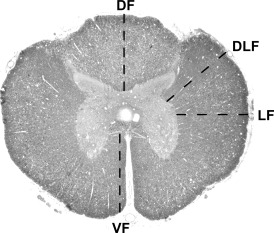
Axon counting. Transverse section of the spinal cord showing the four regions in which axon counting was carried out. A line was drawn as indicated and the diameter of each morphologically normal axon in contact with the line was measured. VF = ventral funiculus; DF = dorsal funiculus; LF = lateral funiculus; DLF = dorsolateral funiculus.
Measurements were performed at the lesion epicenter in all cases. In addition, to provide an approximate comparison of the extent to which measurements differed from normal tissue, control axon measurements were taken from the same regions in undamaged tissue segments remote from the lesion. Distortion of the cord at the lesion site made consistent placement of the line difficult and a detailed comparison of measurements from lesion and control tissue was therefore impossible. For the same reason, figures obtained from different animals could not reliably be compared. (It is recognized that a line method inherently favors selection of larger axons, as it is more likely to transect a large object than a small object. In this study, this error was minimized by excluding axons if only the myelin sheath lay on the line: as larger axons have thicker myelin, they also have an increased chance of being rejected).
RESULTS
Spinal injury occurred through either intervertebral disc extrusion or following spinal trauma subsequent to a road traffic accident (Table 1) and therefore resulted from a combination of contusion and a variable degree of compression (Figure 2). Only in Case 4 was there no apparent residual compression.
Figure 2.
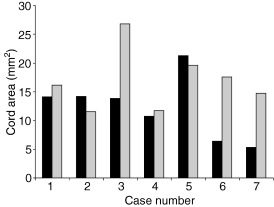
Cross‐sectional area of the spinal cord. Graph shows the cross‐sectional area of the spinal cord at the lesion epicenter and in a section of structurally normal cord cranial to the lesion. There was marked compression of the cord in Cases 3, 6 and 7; although the cord area was not reduced in Cases 1, 2 and 5, there was clear distortion of the cord outline, indicative of persistent compression. There was no compression in Case 4. (▪) Lesion area, () control area.
Histology of the lesion epicenter. In Case 1 (18 h post‐injury; mild compression), small areas of edema and hemorrhage were scattered throughout the cord parenchyma and there were lucent areas in the white matter, particularly in the ventral part of the cord (Figure 3A,B). In the gray matter, many of the neurons showed condensation of chromatin and had irregular nuclear membranes; astrocytes in the gray matter were commonly swollen and had pale staining nuclei with peripheral clumping of chromatin (Figure 3C). Many of the myelin sheaths in the ventral and lateral funiculi were split or vacuolated and there was frequently an increase in the peri‐axonal space. The axons were commonly darker staining than normal and appeared to have lost intra‐axoplasmic organelles; some axons were swollen and a proportion had completely lost their myelin sheath (Figure 3D).
Figure 3.
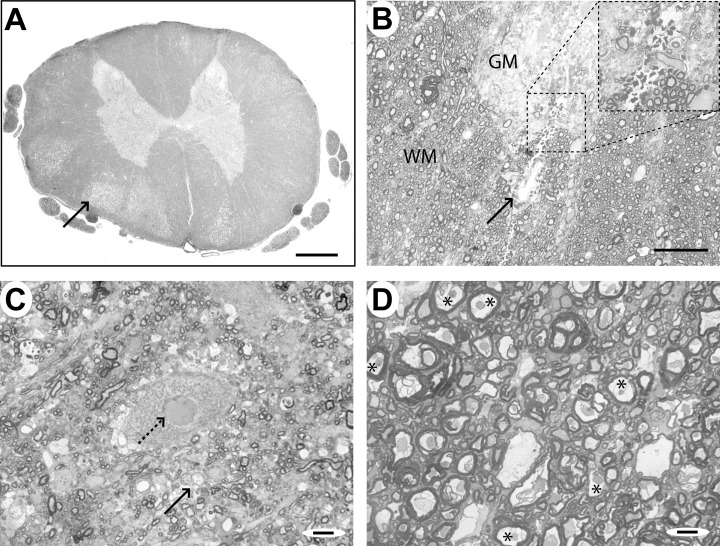
Case 1 (18 h), lesion epicenter. A. Transverse section of the spinal cord, showing a clear distinction between gray and white matter and areas of lucency in the white matter (arrow), particularly in the ventral part of the cord. There is little gross distortion of the cord. B. Transverse section of the spinal cord showing a region of perivascular hemorrhage (arrow) at the border between the gray matter (GM) of the ventral horn and the white matter (WM). C. A motor neuron in the ventral horn of the gray matter showing an irregular nuclear membrane (dotted arrow). Adjacent glial cells (arrow) appear swollen and pale staining. D. White matter from a lucent area in the ventral spinal cord, showing dark staining axons with condensed axoplasm, expanded peri‐axonal space and splitting of myelin sheaths (*). Some myelin sheaths contain no discernible axon. Scale bars: A: 1 mm; B: 100 µm; C&D: 10 µm.
At later time points (Case 2: 36 h post‐injury, mild compression; Case 3: 48 h post‐injury, marked compression), there was extensive hemorrhage and edema throughout the white matter and the gray matter was necrotic and partially cystic (Figure 4A). There was also perivascular cuffing of blood vessels and infiltration of the meninges with leucocytes (Figure 4B). In the white matter, there were few normal axons, although in the periphery of the cord and the dorsal funiculus a few well‐preserved axons persisted (Figure 4C–F). There were large numbers of swollen axonal spheroids, interspersed with loops of degenerating myelin membrane and occasional debris‐laden macrophages (Figure 4B,D). Ultrastructural examination revealed that these spheroids comprised accumulations of axonal organelles, especially mitochondria (Figure 5A,B). Some axons were a normal size and had intact myelin sheaths but contained mitochondria that were slightly swollen and had lost their cristae (Figure 5C,D).
Figure 4.
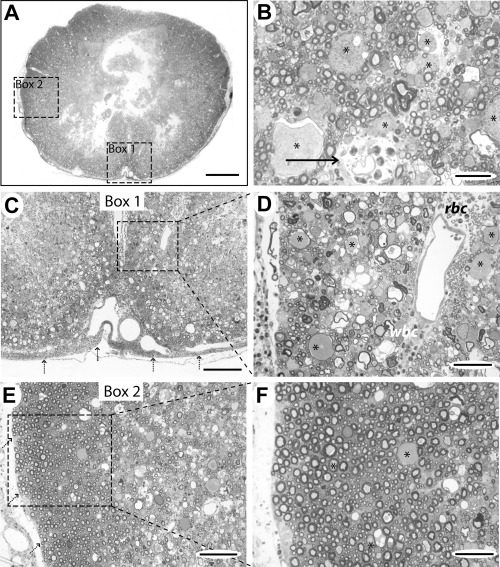
Case 2 (36 h), lesion epicenter. A. Transverse section showing necrosis of the central part of the cord and relative preservation of the subpial tissue. The boxed areas 1 and 2 are shown at higher magnification in (C) and (E). B. Higher power view of the white matter showing infiltration of the perivascular space with polymorphonuclear cells (arrow). Many axons are normal in this field but there are also several large swollen axons (*). C. Boxed area 1 from (A), showing extensive disruption of the normal cord anatomy and cellular infiltration of the meninges (dotted arrows). D. Higher magnification of the boxed area from (C), showing perivascular erythrocytes (rbc) and leucocytes and large numbers of swollen axons (*). A large number of the myelin sheaths are thin and some loops of myelin contain no axon but many smaller axons appear structurally normal. E. Boxed area 2 from (A), showing a rim of intact white matter around the periphery of the cord (pial margin indicated by dotted arrows). F. High magnification of the boxed area from (E), showing that most axons in this region are structurally normal, comprising a uniform‐staining axon surrounded by myelin with no splitting of lamellae. There are some abnormal axons (*) and occasional loops of degenerating myelin. Scale bars: A: 1 mm; B: 25 µm; C: 200 µm; D: 50 µm; E: 100 µm; F: 50 µm.
Figure 5.
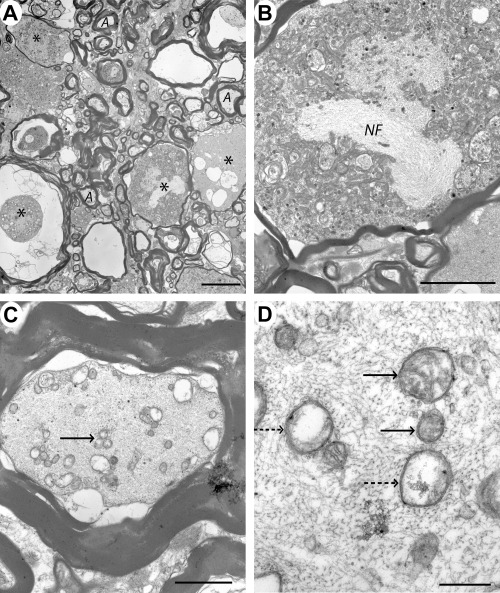
Case 3 (48 h), lesion epicenter. A. Electron micrograph of part of the ventral funiculus showing several large swollen axons (*) with abnormal myelin sheaths. Occasional smaller axons appear normal and have intact myelin sheaths (A). B. Higher magnification showing a swollen axon containing densely packed organelles, predominantly mitochondria and a bundle of neurofilament (NF), within the axoplasm. C. Transverse section through an axon which has a relatively normal myelin sheath but vacuolated axoplasm; in addition, some of the mitochondria appear swollen and have lost detail of their internal structure (arrow). D. Transverse section through a similar axon showing some mitochondria which appear normal (arrows) and others which have lost their cristae (dotted arrows). Scale bars: A: 10 µm; B: 5 µm; C: 2 µm; D: 0.5 µm.
In Case 4 (11 days following injury, no apparent compression) there was edema throughout the cord parenchyma and cystic dilatation of the ventral part of the spinal cord. Large swollen axons were interspersed among loops of degenerating myelin and there were large numbers of debris‐filled macrophages. Case 5 (17 days) was similar, though there was moderate compression of the ventral cord on one side and there was no cyst present. Axon and myelin degeneration were widespread and there were clusters of demyelinated axons in the ventral part of the cord adjacent to the site of compression (Figure 6A,B). Although no remyelination was visible using light microscopy, ultrastructural examination revealed occasional axons which had thin myelin sheaths typical of oligodendrocyte remyelination (Figure 6C,D).
Figure 6.
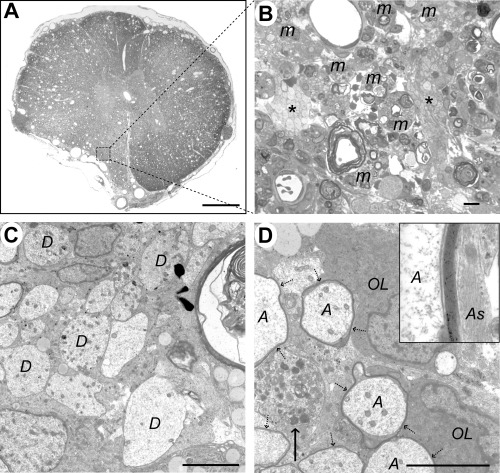
Case 5 (17 days), lesion epicenter. A. Transverse section showing distortion of the left ventrolateral funiculus, loss of the distinction between gray and white matter and widespread vacuolation. B. Boxed area from (A), showing debris‐laden macrophages (m) adjacent to two clusters of demyelinated axons (*). C. Electron micrograph showing a group of demyelinated axons (D). D. Two oligodendrocyte cell bodies (OL) are adjacent to several axons (A) surrounded by thin myelin sheaths (dotted arrows). A demyelinated axon containing an accumulation of organelles, predominantly mitochondria, is also present (arrow). The inset shows a high power view of the myelin sheath; note the adjacent astrocyte processes (As) containing intermediate filaments, with no intervening basement membrane. The calculated interperiod distance for this sheath was 10 nm, a figure consistent with oligodendrocyte myelin (34). Scale bars: A: 1 mm; B: 10 µm; C&D: 5 µm.
The lesions in the two remaining cases were different from one another, with the cord from Case 6 (9 weeks following injury) both cystic and compressed whereas that from Case 7 (12 weeks following injury) was markedly compressed without a cyst. The cystic center of the lesion in Case 6 was filled with a network of interconnecting cell processes and large numbers of macrophages. Axons in the cyst were frequently remyelinated by Schwann cells but many had thin myelin sheaths typical of oligodendrocyte remyelination (Figure 7A,B). A small proportion appeared to have oligodendrocyte myelin of normal thickness and there were a few demyelinated axons. Surrounding this cystic region, more densely packed axons were present, interspersed among macrophages and groups of fibroblast/meningeal cells. A high proportion of these axons were remyelinated by Schwann cells (Figure 7C,D), though occasional demyelinated axons were also seen. In Case 7, there was dramatic compression of the spinal cord and the cross‐sectional area at the lesion epicenter was markedly reduced compared with adjacent sections (Figure 2). Nonetheless, there were numerous surviving axons, a large number of which had thin myelin sheaths typical of oligodendrocyte remyelination (Figure 8A,B). There was also marked gliosis, particularly towards the periphery of the cord (Figure 8C,D).
Figure 7.
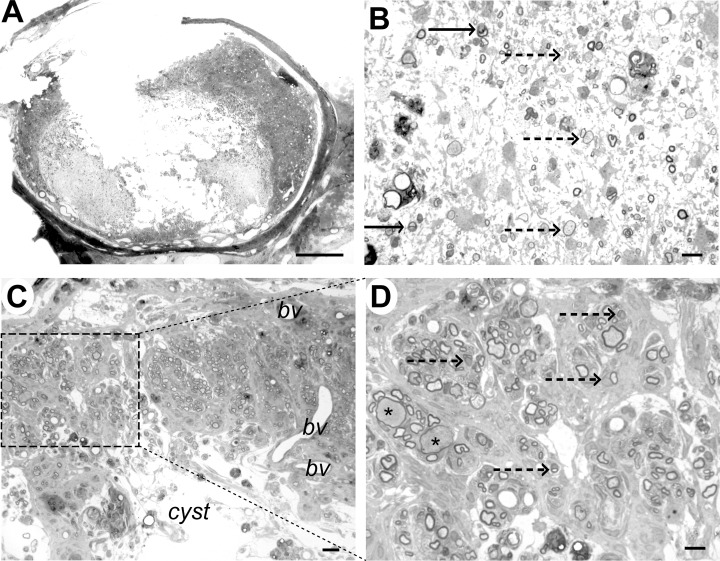
Case 6 (9 weeks), immediately adjacent to lesion epicenter. A. Transverse section showing marked disruption of the cord structure with cystic cavitation; some tissue structure remains in the dorsal funiculus. B. Within the cyst cavity, cells and myelinated axons can be identified. Some axons are myelinated by Schwann cells (arrow) and some have thin myelin sheaths that could represent oligodendrocyte remyelination (dotted arrow). C. Dorsal part of the cord showing tissue adjacent to the cyst cavity. Whorls of myelinated fibres are present, surrounded by densely packed cells, most likely fibroblasts or meningeal cells. Blood vessels are also evident (bv). D. Higher magnification of the boxed area from (C) showing that the myelin sheaths are relatively thick, densely stained and frequently intimately associated with a cell nucleus (dotted arrows), features that are typical of Schwann cell myelin. Myelin sheaths on two axons appear thin for their diameter (*), suggesting pathological axonal swelling. Scale bar: A: 1 mm; B: 10 µm; C: 20 µm; D: 10 µm.
Figure 8.
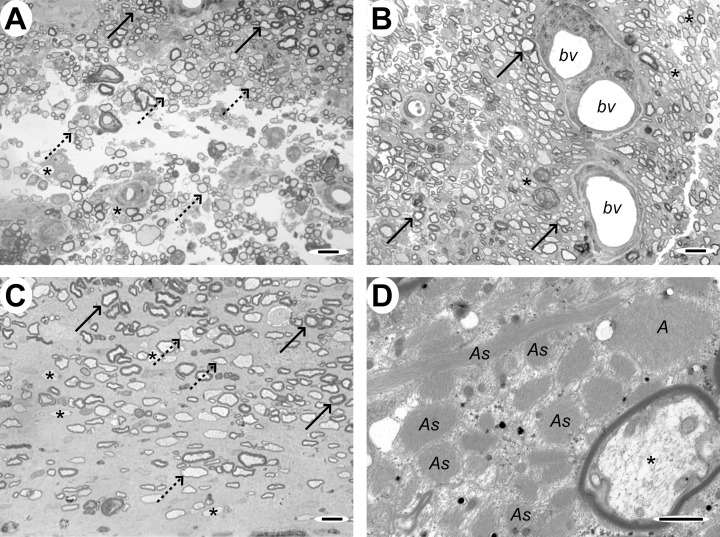
Case 7 (12 weeks), lesion epicenter. A&B. Transverse section of the cord showing large numbers of axons with thin myelin sheaths typical of oligodendrocyte remyelination. In (A) there are some normally myelinated axons at the top of the micrograph (solid arrows) which serve as a reference for distinguishing the remyelinated axons that comprise the majority of those in these sections (dotted arrows). Occasional demyelinated axons are also present (*). In (B), the remyelinated axons surround three small blood vessels (bv). C. Transverse section adjacent to the edge of the cord, showing both normally myelinated (solid arrows), oligodendrocyte remyelinated (dotted arrows) and demyelinated axons (*) in a markedly gliotic background. D. Electron micrograph showing many densely packed cell processes containing bundles of intermediate filaments and glycogen granules (As), features typical of astrocytes, and an adjacent oligodendrocyte‐remyelinated axon (*). Scale bars: A&B: 10 µm; C: 5 µm; D: 1 µm.
Variation in axon damage. Axon damage was most severe in the ventral funiculus of all cases, immediately overlying the site of compression, whereas axons in the dorsal funiculus were relatively well preserved. Axons were also relatively well preserved in the periphery of the cord, immediately beneath the pia (Figure 4). There were few morphologically normal large diameter axons in any region; random counts revealed that intact axons greater than 5 µm in diameter were very rare, whereas those less than 5 µm in diameter were numerous, particularly in the dorsal funiculus (Figure 9). In the ventral funiculus, there were few normal axons of any size in any of the animals (Figure 9).
Figure 9.
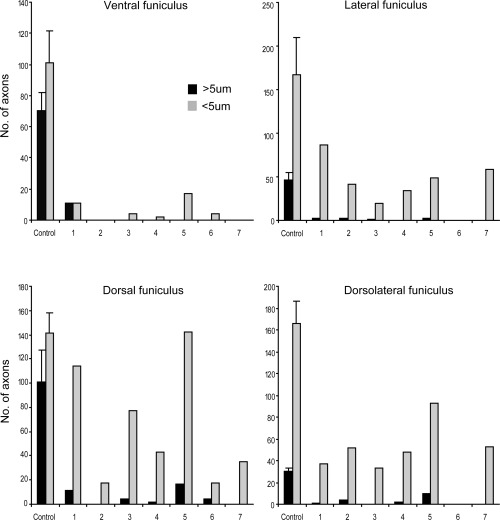
Variation in the severity of white matter damage in different regions. Counts of morphologically normal axons remaining in each animal in different regions of the cord. The diameter of axons in the ventral (A), lateral (B), dorsal (C) and dorsolateral funiculi (D) was measured and the number of axons greater and less than 5 µm in diameter was calculated. In the ventral funiculus, where the most severe injury occurred, very few normal axons of any size remained in any of the animals (A). However, in other regions, there was a relative preservation of axons less than 5 µm diameter (B–D).
Ultrastructural studies
The cyst cavity in Case 6. Within the cyst described in Case 6 (9 weeks) there was a fine interlacing network of cell processes (Figure 10A). In some areas, moderate numbers of cells were interspersed among the cell processes and there were occasional demyelinated and myelinated axons (Figure 7B). Some of the myelinated axons were closely associated with a cell nucleus and had a relatively thick myelin sheath typical of Schwann cell myelin; others had thinner myelin sheaths than might be expected for their diameter, a finding suggestive of either partial demyelination or of oligodendrocyte‐mediated remyelination (Figure 7B). Ultrastructural examination confirmed the presence of Schwann cell and oligodendrocyte myelin and revealed that the interconnecting cell processes within the cyst contained intermediate filaments, indicating that they were astrocytes (Figure 10B,C). In addition, there were a large number of structures that comprised closely packed clusters of cell processes. These had appeared as ill‐defined cellular material using light microscopy but electron microscopic examination revealed two types of process in cross section, dark‐stained astrocyte processes containing densely packed intermediate filaments and small diameter processes which contained microtubules (Figure 10D).
Figure 10.
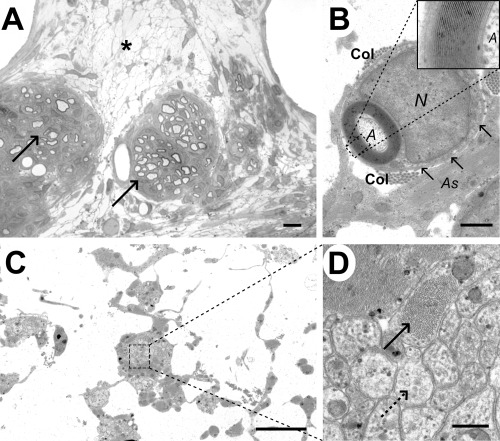
Analysis of the cyst cavity in case 6 (9 weeks). A. Light micrograph showing a diffuse network of cell processes within the cyst (*), with two adjacent clusters of Schwann cell remyelinated axons (arrows). B. Electron micrograph showing the typical appearance of a Schwann cell myelinated axon (A) within the cyst cavity, with a relatively thick myelin sheath surrounded in turn by a rim of cytoplasm. The Schwann cell nucleus (N) is adjacent to the myelin sheath and there is a basal lamina (arrows) and some collagen fibrils (Col) surrounding the Schwann cell; an adjacent astrocyte process (As) contains bundles of intermediate filaments. The inset shows a high power view of the myelin sheath, allowing the inter‐period distance to be calculated as 13 nm, a figure typical for peripheral type myelin (34). C. Electron micrograph of the cyst cavity, showing thin cell processes interlinking clusters of more densely packed cellular material. D. Higher magnification of the boxed area from (C), revealing that these clusters containing two types of cell process: one dark staining and packed with bundles of intermediate filament (solid arrow) and the other more light staining and containing microtubules (dotted arrow). Scale bars: A: 5 µm; B: 1 µm; C: 4 µm; D: 0.4 µm.
Focal axonal and myelin abnormalities. In sagittal tissue sections, some axons were identified having partial loss of their myelin sheath (Figure 11A). Nodes of Ranvier were occasionally seen in larger axons as a region of slightly reduced axon diameter adjacent to terminal loops of internodal myelin; some of these nodes appeared structurally abnormal, with apparent breaks in the continuity of the axon, while others appeared to have paranodal myelin disruption (Figure 11B,C). Under electron microscopy, long portions of individual axons were visible in sagittal sections and nodes and paranodes could be examined more closely. Although many axons were normal we also identified axons with focal accumulations of organelles within their axoplasm (Figure 11D,E) and a variety of paranodal myelin abnormalities (Figure 11D–I), often in axons that appeared morphologically normal over the remainder of their length. Paranodal abnormalities included increased nodal length, loss of the terminal loops of myelin on one or both sides of the node and splitting or detachment of the terminal loops from outer lamellae of the sheath. Although lesion variability prevented a systematic comparison of paranodal abnormalities in different cases, we counted the number of normal and abnormal paranodes in Cases 3–7 to examine whether there was variation in the frequency of nodal and paranodal abnormalities with time. In Case 3, only 3/12 nodes were considered normal, with the remainder showing some of the features illustrated in Figure 11. Cases 5 and 7 contained respectively 9/11 and 6/8 nodes that appeared normal and in Cases 4 and 6, there were relatively few intact axons and no complete paranodes were identified. Hemi‐nodes, in which only one side of the node of Ranvier was evident as the axon dipped out of the plane of the section, were seen almost as commonly as complete nodes in most sections and were not counted in this analysis.
Figure 11.
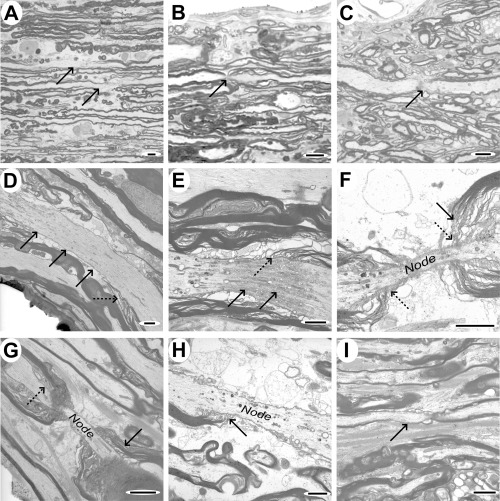
Sagittal sections showing paranodal and focal myelin and axonal abnormalities. A–C. Light micrographs of tissue from case 3, showing focal loss of myelin in two axons (indicated by arrows, A), asymmetric splitting of paranodal myelin (B; arrow) and an apparent loss of axon continuity at the node (C; arrow), though this last feature could arise because of the axon dipping out of the plane of the section. D–I. Electron micrographs illustrating a variety of features that were identified in sagittal sections. D. Axon demonstrating focal vacuolation at one point and an apparently elongated nodal region (solid arrows), with poorly defined terminal loops of myelin (dotted arrow). E. Focal accumulation of axoplasmic organelles and vacuoles at an elongated node (solid arrows), with abnormal terminal loops of myelin (dotted arrow). F&G. Elongated nodes of Ranvier, showing splitting of paranodal myelin (solid arrows) but relatively normal terminal loops of oligodendrocyte cytoplasm (dotted arrows). H. Node of Ranvier showing terminal loops of myelin (arrow) attaching to the axon on one side only. I. Small diameter axon showing an extended paranodal region (arrow) on one side of the node only. Scale bars: A–C: 5 µm; D–I: 2 µm.
DISCUSSION
This study details the pathological changes that occur following severe naturally occurring spinal injury, allowing comparison with those that occur following injuries in experimental animals. An important difference from the majority of experimental lesions is that most of the lesions examined here resulted from mixed contusive/compressive injuries, and therefore can be regarded as facsimiles of the most prevalent form of human SCI. In this study myelin and axonal abnormalities were common and severe at early time points and included segmental and paranodal myelin disruption that is easily overlooked if only transverse tissue sections are examined. Although remyelination was apparent at later time points, and in one case was extensive, persistently demyelinated axons were also seen. Our observations broadly support findings from experimental animal lesions in the period shortly after SCI but show differences at later times, when different lesion patterns were observed in different animals. Such heterogeneity is inevitable in any clinical group and highlights the extra dimension in understanding of human SCI that can be gained by study of this group of naturally occurring SCI patients.
Axonal lesions. Our observations show that axonal damage is not uniform across transverse sections of the lesion: larger axons were preferentially damaged and axons around the periphery of the cord better preserved. Previous experimental studies have recorded similar observations: although all spinal axons are destroyed by severe injuries 4, 10), milder contusion leaves subpial axons intact, with a decrease in axon density of approximately tenfold over the 500 µm beneath the pia (10). A similar pattern has been described following human SCI (14). One explanation for this selective damage is that the peracute displacement of tissue during contusion leads to longitudinal displacement of the spinal cord—the cord behaves like a viscous liquid with the central region undergoing the most marked displacement caused by laminar flow of the compressed tissue, while subpial tissue is distorted relatively little (10). We also found that axons larger than 5 µm in diameter were more likely to be morphologically abnormal, a finding that agrees with previous observations in experimental models 10, 37, 38). The reason for these differences is not wholly clear but might relate in part to the interdependency of oligodendrocytes and axons. Oligodendrocytes that provide internodes on large axons are calculated to support more myelin than those supporting several smaller internodes and this extra metabolic demand may render them more susceptible to energy deprivation following SCI (9).
Subcellular axonal abnormalities. Morphologically abnormal axons and degenerating myelin sheaths were readily identifiable using light microscopy; ultrastructural analysis confirmed these findings but also revealed that many of the axons that might be considered morphologically normal using light microscopy contained abnormal organelles. In particular, some mitochondria had lost the cristae from their inner membrane. Although similar changes can arise through poor tissue fixation, the presence of adjacent structurally normal organelles makes this an unlikely explanation and suggests that the mitochondrial abnormalities are genuine. Similar changes in axonal mitochondria have also been reported following experimental spinal contusion 2, 37) and this might have important implications for ongoing cell death following SCI. Mitochondrial dysfunction, leading to failure of the sodium/potassium pump and loss of calcium buffering, is one mechanism by which axons can become overloaded with calcium and degenerate 27, 44).
Axon regeneration. We identified small diameter neural processes intermeshed with astrocyte processes in the cystic lesion center of Case 6 (9 weeks post‐injury). These could be axons that survived the injury—they were small diameter and therefore more likely to survive—or they could be new axon sprouts beginning to extend across the lesion. Spontaneous sprouting at the site of injury has been identified in experimental models—both in segmental dorsal root fibres (30) and in ascending and descending white matter tracts 23, 25, 33). Such short distance sprouting might have beneficial effects on functional outcome, for instance by recruiting surviving axon tracts to perform new functions (3). However, uncontrolled sprouting of afferent fibres can be associated with the development of aberrant pain states (26) and may be detrimental in some circumstances.
Myelin lesions. Myelin abnormalities were common and observed at early post‐injury time points, frequently in association with degenerating axons [similar to findings in experimental injury 2, 25, 36, 37)]. At later time points (from 17 days onwards—Case 5), after myelin debris has been cleared by invading macrophages, morphologically normal, but demyelinated, axons became apparent. A few remyelinated axons were identified ultrastructurally at this time and at later time points but remyelination was not found in all regions of all cases. An interesting finding in Case 7 was the very extensive oligodendrocyte‐mediated remyelination that occurred in the face of persistent and marked cord compression (Figure 7A,B). Oligodendrocyte remyelination was also identified in the Cases 5 and 6, though less extensively, and in Case 6 Schwann cell myelin was present, both within the cyst cavity and in the surrounding rim of sub‐pial axons. In contrast to Case 7, in which there was no identifiable Schwann cell remyelination, Case 6 had suffered a severe injury that had disrupted the meninges, potentially allowing meningeal Schwann cells greater access to spinal axons.
Oligodendrocyte remyelination occurs through the proliferation of oligodendrocyte progenitors, which then differentiate into mature myelinating cells (46). The extensive remyelination seen in Case 7 (12 weeks post‐injury) indicates that a significant number of progenitor cells survived and proliferated following injury and that the environment of the compressed spinal cord was permissive for progenitor differentiation. By the same token, the absence of such extensive oligodendrocyte remyelination in Case 6 suggests few oligodendrocyte progenitor cells survived—perhaps because the injury was more severe—or that the environment was unsuitable for oligodendrocyte myelination. However, an alternative explanation is that the oligodendrocyte response is simply less vigorous than that of the invading Schwann cells: in experimental models of demyelination in which astrocytes are also damaged, Schwann cell predominates over oligodendrocyte remyelination (8). In addition to damaging the meninges, the severe injury experienced by Case 6 may have destroyed a large number of astrocytes, removing an intrinsic constraint on the formation of Schwann cell myelin in the central nervous system (CNS).
The extent to which demyelination persists after SCI has long been a matter of controversy. Some authors report that axons become demyelinated at early stages after experimental injury but then undergo spontaneous remyelination 5, 11, 20, 38); long standing demyelination 4, 11) and even chronic progressive demyelination have also been reported (41). The role that persistent demyelination plays in the clinical signs observed after human clinical SCI is similarly controversial: although it is a frequently cited cause of post‐traumatic dysfunction (e.g. 43) it has been detected in human material only infrequently (14). Our findings provide some support for both points of view, demonstrating that remyelination occurs and can be widespread but that a proportion of axons may be persistently demyelinated. It is well established that demyelinated axons do not conduct normally (29) and that remyelination restores conduction and functional parameters to normal limits 11, 28, 39). However, recent advances in understanding multiple sclerosis have further highlighted the importance of myelination status: in multiple sclerosis, axonal loss is now recognized as a prominent feature of both acute and chronic lesions 18, 42) thought to result, at least in part, from the loss of myelin ensheathment 6, 19). Much current research is directed at understanding the interaction between an axon and its myelinating cell and at promoting axonal survival through preservation of oligodendrocytes. Such advances in understanding clearly also have important implications in promoting axonal survival after SCI.
Paranodal abnormalities. We identified paranodal and segmental myelin abnormalities commonly in Case 2 (48 h post‐injury)—frequently in axons which had otherwise intact myelin sheaths—and less commonly in other cases. Paranodal abnormalities are potentially a highly important component of the injury, as such myelin disruption, and the associated aberrant exposure of paranodal and juxtaparanodal ion channels, is likely to have a major impact on impulse conduction. Indeed, experimental modeling of myelinated fibres suggests that paranodal demyelination in vivo will reduce excitability and conduction velocity (40). Furthermore, mice with null mutations in contactin, caspr or ceramide galactosyltransferase (cgt), all of which have structurally abnormal paranodes, have dramatically reduced nerve conduction velocity 7, 12, 15, 16).
Paranodal abnormalities were less commonly seen in animals at later time points following injury. Because of lesion variability, we were not able to examine analogous regions of the lesion from each case, so a direct comparison of different animals was not possible and interpretation of this observation must be cautious. Nonetheless, our observations suggest that paranodal myelin defects do not persist. In the peripheral nervous system, paranodal demyelination is followed by Schwann cell proliferation at the damaged paranode and by eventual re‐attachment of the outermost terminal myelin loops at their original site (21). This suggests that paranodal myelin injury can be repaired, though whether injury‐induced defects in paranodal CNS myelin are repaired so easily is not currently clear. Another possibility is that affected axons die, perhaps by the development of abnormal ion fluxes and the elevation of axoplasmic calcium as postulated in axons that completely lose their myelin sheath (6). Further investigations using experimental animals are necessary to identify the prevalence of paranodal demyelination under different circumstances and to establish to the fate of affected axons.
SUMMARY
Our observations suggest that the structural changes in the experimentally injured spinal cord are an accurate reflection of real‐life injury. However, the complexity of spontaneous injury means that the precise nature of the lesion varies from one case to another and suggests that any future treatment will need to be tailored to each individual. We have shown that paranodal and segmental abnormalities appear to be relatively commonplace in the aftermath of the injury, which is likely to contribute to functional impairment and may exacerbate progressive tissue damage. Because it forms a “translational model” between experimental rodent injuries and human clinical SCI, naturally occurring SCI in dogs has great value in assessing putative therapeutic interventions.
ACKNOWLEDGMENTS
The authors would like to thank Pet Plan Charitable Trust for funding much of the work carried out in this study and Mike Peacock for preparing tissue sections for electron microscopy. PS is part‐funded by Pfizer.
REFERENCES
- 1. Balentine JD (1978) Pathology of experimental spinal cord trauma. I. The necrotic lesion as a function of vascular injury. Lab Invest 39:236–253. [PubMed] [Google Scholar]
- 2. Balentine JD (1978) Pathology of experimental spinal cord trauma. II. Ultrastructure of axons and myelin. Lab Invest 39:254–266. [PubMed] [Google Scholar]
- 3. Bareyre FM, Kerschensteiner M, Raineteau O, Mettenleiter TC, Weinmann O, Schwab ME (2004) The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat Neurosci 7:269–277. [DOI] [PubMed] [Google Scholar]
- 4. Basso DM, Beattie MS, Bresnahan JC (1996) Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight‐drop device versus transection. Exp Neurol 139:244–256. [DOI] [PubMed] [Google Scholar]
- 5. Beattie MS, Bresnahan JC, Komon J, Tovar CA, Van Meter M, Anderson DK, Faden AI, Hsu CY, Noble LJ, Salzman S, Young W (1997) Endogenous repair after spinal cord contusion injuries in the rat. Exp Neurol 148:453–463. [DOI] [PubMed] [Google Scholar]
- 6. Bechtold DA, Smith KJ (2005) Sodium‐mediated axonal degeneration in inflammatory demyelinating disease. J Neurol Sci 233:27–35. [DOI] [PubMed] [Google Scholar]
- 7. Bhat MA, Rios JC, Lu Y, Garcia‐Fresco GP, Ching W, St Martin M, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JI, Bellen HJ (2001) Axon‐glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron 30:369–383. [DOI] [PubMed] [Google Scholar]
- 8. Blakemore WF (1982) Ethidium bromide induced demyelination in the spinal cord of the cat. Neuropathol Appl Neurobiol 8:365–375. [DOI] [PubMed] [Google Scholar]
- 9. Blakemore WF (1982) Myelination, demyelination and remyelination in the CNS. In: Rrecent Advances in Neuropathology, Chapter 3. Smith WT, Cavanagh JB (eds), pp. 53–82. Churchill Livingstone: New York. [Google Scholar]
- 10. Blight AR, DeCrescito V (1986) Morphometric analysis of experimental spinal cord injury in the cat: the relation of injury intensity to survival of myelinated axons. Neuroscience 19:321–341. [DOI] [PubMed] [Google Scholar]
- 11. Blight AR, Young W (1989) Central axons in injured cat spinal cord recover electrophysiological function following remyelination by Schwann cells. J Neurol Sci 91:15–34. [DOI] [PubMed] [Google Scholar]
- 12. Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B (2001) Contactin orchestrates assembly of the septate‐like junctions at the paranode in myelinated peripheral nerve. Neuron 30:385–397. [DOI] [PubMed] [Google Scholar]
- 13. Bresnahan JC, Beattie MS, Todd FD III, Noyes DH (1987) A behavioral and anatomical analysis of spinal cord injury produced by a feedback‐controlled impaction device. Exp Neurol 95:548–570. [DOI] [PubMed] [Google Scholar]
- 14. Bunge RP, Puckett WR, Becerra JL, Marcillo A, Quencer RM (1993) Observations on the pathology of human spinal cord injury. A review and classification of 22 new cases with details from a case of chronic cord compression with extensive focal demyelination. Adv Neurol 59:75–89. [PubMed] [Google Scholar]
- 15. Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, Suzuki K, Popko B (1996) Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell 86:209–219. [DOI] [PubMed] [Google Scholar]
- 16. Dupree JL, Coetzee T, Blight A, Suzuki K, Popko B (1998) Myelin galactolipids are essential for proper node of Ranvier formation in the CNS. J Neurosci 18:1642–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Emery E, Aldana P, Bunge MB, Puckett W, Srinivasan A, Keane RW, Bethea J, Levi AD (1998) Apoptosis after traumatic human spinal cord injury. J Neurosurg 89:911–920. [DOI] [PubMed] [Google Scholar]
- 18. Ferguson B, Matyszak MK, Esiri MM, Perry VH (1997) Axonal damage in acute multiple sclerosis lesions. Brain 120(Pt 3):393–399. [DOI] [PubMed] [Google Scholar]
- 19. Garbern JY, Yool DA, Moore GJ, Wilds IB, Faulk MW, Klugmann M, Nave KA, Sistermans EA, Van De Knaap MS, Bird TD, Shy ME, Kamholz JA, Griffths IR (2002) Patients lacking the major CNS myelin protein, proteolipid protein 1, develop length‐dependent axonal degeneration in the absence of demyelination and inflammation. Brain 125:551–561. [DOI] [PubMed] [Google Scholar]
- 20. Gledhill RF, Harrison BM, McDonald WI (1973) Demyelination and remyelination after acute spinal cord compression. Exp Neurol 38:472–487. [DOI] [PubMed] [Google Scholar]
- 21. Griffin JW, Drucker N, Gold BG, Rosenfeld J, Benzaquen M, Charnas LR, Fahnestock KF, Stocks EA (1987) Schwann cell proliferation and migration during paranodal demyelination. J Neurosci 7:682–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guest JD, Hiester ED, Bunge RP (2005) Demyelination and Schwann cell responses adjacent to injury epicenter cavities following chronic human spinal cord injury. Exp Neurol 192:384–393. [DOI] [PubMed] [Google Scholar]
- 23. Guizar‐Sahagun G, Grijalva I, Salgado‐Ceballos H, Espitia A, Orozco S, Ibarra A, Martinez A, Franco‐Bourland RF, Madrazo I (2004) Spontaneous and induced aberrant sprouting at the site of injury is irrelevant to motor function outcome in rats with spinal cord injury. Brain Res 1013:143–151. [DOI] [PubMed] [Google Scholar]
- 24. Hansen HJ (1952) A pathologic‐anatomical study on disc degeneration in dog, with special reference to the so‐called enchondrosis intervertebralis. Acta Orthop Scand Suppl 11:1–117. [DOI] [PubMed] [Google Scholar]
- 25. Hill CE, Beattie MS, Bresnahan JC (2001) Degeneration and sprouting of identified descending supraspinal axons after contusive spinal cord injury in the rat. Exp Neurol 171:153–169. [DOI] [PubMed] [Google Scholar]
- 26. Hofstetter CP, Holmstrom NA, Lilja JA, Schweinhardt P, Hao J, Spenger C, Wiesenfeld‐Hallin Z, Kurpad SN, Frisen J, Olson I (2005) Allodynia limits the usefulness of intraspinal neural stem cell grafts; directed differentiation improves outcome. Nat Neurosci 8:346–353. [DOI] [PubMed] [Google Scholar]
- 27. James AM, Murphy MP (2002) How mitochondrial damage affects cell function. J Biomed Sci 9:475–487. [DOI] [PubMed] [Google Scholar]
- 28. Jeffery ND, Blakemore WF (1997) Locomotor deficits induced by experimental spinal cord demyelination are abolished by spontaneous remyelination. Brain 120(Pt 1):27–37. [DOI] [PubMed] [Google Scholar]
- 29. McDonald WI, Sears TA (1969) Effect of demyelination on conduction in the central nervous system. Nature 221:182–183. [DOI] [PubMed] [Google Scholar]
- 30. McTigue DM, Horner PJ, Stokes BT, Gage FH (1998) Neurotrophin‐3 and brain‐derived neurotrophic factor induce oligodendrocyte proliferation and myelination of regenerating axons in the contused adult rat spinal cord. J Neurosci 18:5354–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Midha R, Fehlings MG, Tator CH, Saint‐Cyr JA, Guha A (1987) Assessment of spinal cord injury by counting corticospinal and rubrospinal neurons. Brain Res 410:299–308. [DOI] [PubMed] [Google Scholar]
- 32. Olby N, Harris T, Burr J, Munana K, Sharp N, Keene B (2004) Recovery of pelvic limb function in dogs following acute intervertebral disc herniations. J Neurotrauma 21:49–59. [DOI] [PubMed] [Google Scholar]
- 33. Olby NJ, Blakemore WF (1996) Primary demyelination and regeneration of ascending axons in the dorsal funiculus of the rat spinal cord following photochemically induced injury. J Neurocytol 25:465–480. [DOI] [PubMed] [Google Scholar]
- 34. Peters A, Palay SL, Webster HD (1991) The Fine Structure of the Nervous System, 3rd edn. Oxford University Press: New York. [Google Scholar]
- 35. Ramer LM, Ramer MS, Steeves JD (2005) Setting the stage for functional repair of spinal cord injuries: a cast of thousands. Spinal Cord 43:134–161. [DOI] [PubMed] [Google Scholar]
- 36. Reier PJ (2004) Cellular transplantation strategies for spinal cord injury and translational neurobiology. NeuroRx 1:424–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rosenberg LJ, Teng YD, Wrathall JR (1999) Effects of the sodium channel blocker tetrodotoxin on acute white matter pathology after experimental contusive spinal cord injury. J Neurosci 19:6122–6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Salgado‐Ceballos H, Guizar‐Sahagun G, Feria‐Velasco A, Grijalva I, Espitia L, Ibarra A, Madrazo I (1998) Spontaneous long‐term remyelination after traumatic spinal cord injury in rats. Brain Res 782:126–135. [DOI] [PubMed] [Google Scholar]
- 39. Smith KJ, Blakemore WF, McDonald WI (1979) Central remyelination restores secure conduction. Nature 280:395–396. [DOI] [PubMed] [Google Scholar]
- 40. Stephanova DI, Daskalova M (2005) Differences in potentials and excitability properties in simulated cases of demyelinating neuropathies. Part II. Paranodal demyelination. Clin Neurophysiol 116:1159–1166. [DOI] [PubMed] [Google Scholar]
- 41. Totoiu MO, Keirstead HS (2005) Spinal cord injury is accompanied by chronic progressive demyelination. J Comp Neurol 486:373–383. [DOI] [PubMed] [Google Scholar]
- 42. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L (1998) Axonal transection in the lesions of multiple sclerosis. N Engl J Med 338:278–285. [DOI] [PubMed] [Google Scholar]
- 43. Waxman SG (1992) Demyelination in spinal cord injury and multiple sclerosis: what can we do to enhance functional recovery? J Neurotrauma 9(Suppl. 1):S105–S117. [PubMed] [Google Scholar]
- 44. Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH (2001) Traumatic axonal injury induces calcium influx modulated by tetrodotoxin‐sensitive sodium channels. J Neurosci 21:1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wrathall JR, Pettegrew RK, Harvey F (1985) Spinal cord contusion in the rat: production of graded, reproducible, injury groups. Exp Neurol 88:108–122. [DOI] [PubMed] [Google Scholar]
- 46. Zhao C, Fancy SP, Kotter MR, Li WW, Franklin RJ (2005) Mechanisms of CNS remyelination—the key to therapeutic advances. J Neurol Sci 233:87–91. [DOI] [PubMed] [Google Scholar]