

Abstract
The neuronal ceroid lipofuscinoses (NCLs, Batten disease) are fatal inherited lysosomal storage diseases of children characterized by increasing blindness, seizures and profound neurodegeneration but the mechanisms leading to these pathological changes remain unclear. Sheep with a CLN6 form that have a human‐like brain and disease progression are invaluable for studying pathogenesis. A study of preclinical pathology in these sheep revealed localized glial activation at only 12 days of age, particularly in cortical regions that subsequently degenerate. This has been extended by examining fetal tissue from 60 days of gestation onwards. A striking feature was the presence of reactive astrocytes and the hypertrophy and proliferation of perivascular cells noted within the developing white matter of the cerebral cortex 40 days before birth. Astrocytic activation was evident within the cortical gray matter 20 days before birth, and was confined to the superficial laminae 12 days after birth. Clusters of activated microglia were detected in upper neocortical gray matter laminae shortly after birth. Neuronal development in affected sheep was undisturbed at these early ages. This prenatal activation of non‐neuronal cells within the affected brain indicates the onset of pathogenesis during brain development and that an ordered sequence of glial activation precedes neurodegeneration.
INTRODUCTION
The neuronal ceroid lipofuscinoses (NCLs, Batten disease) are a group of fatal inherited lysosomal storage diseases 5, 9, 10, 32). Most forms are progressive neurodegenerative diseases of children, characterized by blindness, seizures and a relentless psychomotor and cognitive decline, ending in a premature death. A number of disease causing mutations have been found in a separate gene for each form, resulting in a range of ages of onset and rates of disease progression 30, 32; http://www.ucl.ac.uk/ncl). Despite considerable efforts to determine mechanistic links to pathological changes, it is still unclear how the mutations in these genes lead to the devastating effects of these diseases.
Naturally occurring forms of NCL have been found in a number of animals, including sheep and dogs which have been bred and studied as models of the human disease 16, 17, 18, 19, 20, 24, 26). Genetically accurate mouse models of a number of forms are now available (29) and a number of invertebrate models are being studied. The naturally occurring sheep models are especially valuable for neuropathology studies, because their larger and more complex brain more closely resembles the human brain, and the disease progression more accurately reflects the human pathology, particularly the severe cortical atrophy, profound neuronal loss and retinal degeneration. Most extensively characterized is the form in New Zealand South Hampshire sheep, which present with clinical symptoms that resemble human CLN6 16, 19, 26). Linkage analysis localized the disease‐associated gene to a region on sheep chromosome 7 orthologous with the 15q21–23 human region, containing the human CLN6 gene, FLJ 20561 4, 8, 50).
A common feature of all but one human form of NCL is the specific lysosomal storage of subunit c of mitochondrial adenosine triphosphate synthase, first described in South Hampshire sheep 7, 38), and subsequently in human CLN2, CLN3, CLN5, CLN8 and a range of animal forms 18, 24, 25, 37, 48). Storage body accumulation is scattered throughout the affected South Hampshire sheep brain from early ages (36). In contrast early neurodegeneration displays a remarkable regional selectivity 35, 36). These neuronal changes are preceded by marked astrocytosis and microglial activation in discrete focal clusters in superficial laminae of occipital and somatosensory cortical regions already evident in presymptomatic sheep at only 12 days of age. Perivascular cells (perivascular macrophages) are also activated (36). These findings were surprising, particularly as myelination is still in progress during this early postnatal period, and prompted us to investigate affected fetal sheep brains to determine the onset and progression of reactive changes. A succession of prenatal reactive changes was observed.
METHODS
Animals. Animal procedures accorded to National Institutes of Health guidelines and the New Zealand Animal Welfare Act, 1999. Affected embryos were generated by super‐ovulation and artificial insemination of affected South Hampshire sheep maintained under New Zealand pastoral conditions, and surgically transferred to normal surrogate Coopworth ewes 6 days later (21). Pregnancies were confirmed by ultrasound scanning after 50 days (51). Control Coopworth breed fetuses were generated by artificial insemination of synchronized ewes. Fetal ages were calculated from the day of insemination.
Tissue processing and sampling of sections. Fetuses were obtained after 60, 80, 100, 110, 130, and 150 days of gestation (within 24 h of birth) and 12 days after birth, exsanguinated, the brains removed, weighed, divided down the midline and immersion fixed in 10% buffered formalin (1 : 10 dilution of 37% formaldehyde), pH 7.0. After at least 2 weeks’ fixation, the right cerebral hemispheres were cut sagittally into 10 mm thick slices, equilibrated at 4°C in cryoprotectant [15% sucrose, 30% ethylene glycol, in 0.05 M phosphate buffered saline (PBS), pH 6.8] and stored at −130°C. Subsequently, 50 µm serial sagittal sections were cut on a freezing microtome and collected sequentially in 96‐well plates in cryoprotectant and stored at −20°C, as previously described (36). Based on the relative mediolateral adult positions of structures (http://www.msu.edu/user/brains/sheepatlas), sections from each age were selected that included the occipital and frontal lobes, corpus callosum and cerebellum.
Histology and immunohistochemistry. Nissl staining, immunohistochemical and lectin histochemical procedures were carried out as previously described (36). Sections mounted on gelatine chrome alum coated glass slides were stained with the Nissl dye cresyl fast violet, dehydrated and mounted in DePeX (DPX, NWR, Poole, UK). Free‐floating sections were washed and stained with rabbit anti‐cow antibodies to glial fibrillary acidic protein (GFAP) (1 : 25 000, Dako, Carpinteria, CA, USA), the class II major histocompatibility complex (MHC‐II) antigen (monoclonal 1 : 200, Veterinary Preclinical Centre, Parkville, Victoria, Australia) or with biotinylated α‐D‐galactose specific isolectin I‐B4 from Griffonia simplicifolia (GSB4) (1 : 500, Vector Laboratories, Peterborough, UK). Immunoreactivity was detected using appropriate biotinylated secondary antibodies and Extravidin‐horse radish peroxidase (HRP) (1 : 1000, Sigma, St Louis, MO, USA) and diaminobenzidine tetrahydrochloride (DAB), in 0.1 M acetate buffer at pH 6.0. Washing and incubation of sections were carried out at 4°C in PBS containing 1% normal goat serum and 0.2% Triton X100. Bound lectin was detected using Extravidin‐HRP and DAB (36). Sections were then mounted onto slides, air‐dried, dehydrated and coverslips attached with DPX. Appropriate negative controls were included in each staining run, ie, omission of primary antisera (immunohistochemistry) or GSB4 lectin (histochemistry).
RESULTS
General organization and development of the CLN6 sheep brain. The marked differences in the activation of a variety of cell types between control and affected brains in the prenatal period studied (60–150 days gestation, E60–E150) noted below were not accompanied by any gross differences in the rapid growth, increasing structural organization or apparent maturation of the developing brain. At E60 both affected and normal brains weighed only a little over 2 g (Figure 1), approximately 2% of the average adult brain weight, and the brain surface was smooth. The brains grew rapidly from then on, to 60% of the adult mass 12 days after birth (Figure 1), and the adult brain structure emerged over this period. Gyral patterning of the cerebrum noted around E100 became more complex and the hippocampus was first evident from E80. The laminar and spatial organization of the cerebral cortex seen in Nissl stained sections of affected brains appeared to be similar to that of control brains. Neuronal migration was well underway in both control and affected brains by E60, neocortical layers were clearly defined by E100 and large pyramidal neurons could be distinguished from this age onwards. The gross and cellular development of the affected cerebellum was also normal.
Figure 1.
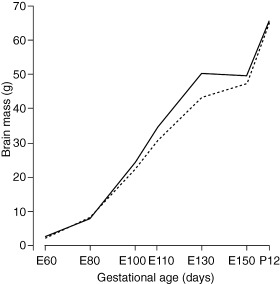
Progressive increase in brain weight during fetal development, and 12 days after birth (P12). The weights of CLN6 affected brains taken for this set of experiments (———), and of control brains (‐‐‐‐‐‐‐‐‐‐).
Cellular reactivity within the cerebral cortex. In contrast to apparently normal neuronal development, significant changes were apparent in non‐neuronal cells within the developing affected sheep brain. Activation of GFAP‐positive astrocytes (intensely stained GFAP‐positive cell bodies and thickened processes) was evident within the developing white matter of affected brains from E110–E130 days (Figure 2E–H,M–P) compared with control brains (Figure 2A–D,I–L). A proportion of astrocytes surrounding blood vessels were also intensely labeled within the white matter of affected fetuses (arrowheads, Figure 2E–H). In contrast GFAP staining of cerebellar white matter astrocytes and Bergmann glia was similar in affected and control animals at all ages.
Figure 2.
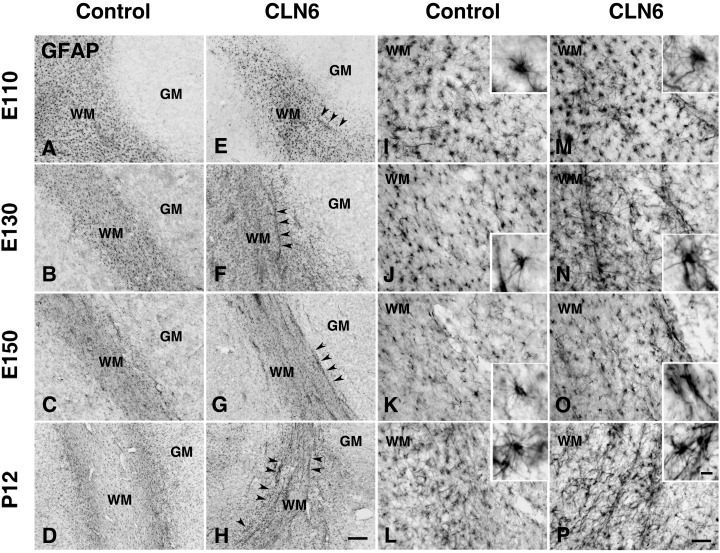
Astrocytic activation in the cortical white matter of CLN6 sheep. Immunohistochemical staining for GFAP reveals progressive astrocytosis in the cortical white matter (WM) of CLN6 sheep (arrowheads E–H arrowheads, M–P) compared with age matched controls (A–D,I–L) and more detailed morphology shown in higher magnification insets. At gestational day 110 (E110), GFAP immunoreactivity was already more prominent in affected sheep (E,M) than controls (A,I) with astrocytes exhibiting numerous thickened GFAP positive processes (M). This morphology was maintained in the white matter of affected sheep during subsequent development with intense GFAP immunoreactivity evident in hypertrophied astrocytes at 130 days (E130, F,N), at birth (E150, G,O) and postnatal day 12 (P12, H,P), compared with age matched controls (E130, B,J; E150, C,K; P12, D,L). At these ages prominent GFAP‐positive astrocytes were frequent around blood vessels within the cerebral white matter of affected sheep (F–H,N–P), but not in age matched controls (B–D,J–L). Scale bars: 200 µm (A–H), 50 µm (I–P), 10 µm (insets in I–P).
Astrocytic activation was detected within the cortical gray matter of affected sheep 20 days before birth (E130) and was well established at birth (E150) (Figure 3A,B). This astrocytosis in affected gray matter rapidly intensified and became most prominent in the upper cortical layers by 12 days after birth (Figure 3C vs. D) and was more pronounced in occipital and somatosensory cortices than other regions. GFAP immunoreactivity was detected at the pial surface in both control and affected animals over the range of ages (Figure 3A–D), whereas radial glial processes were weakly labeled.
Figure 3.
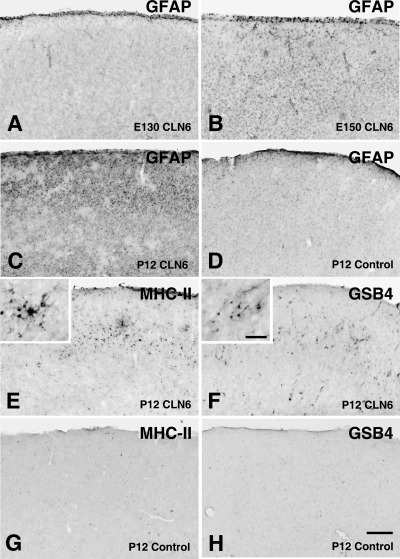
Prenatal reactive changes in the cortical gray matter of CLN6 sheep. Immunohistochemical staining for the astrocyte marker glial fibrillary acidic protein (GFAP) (A–D), the microglial marker the class II major histocompatibility complex (MHC‐II) (E,G) or α‐D‐galactose specific isolectin I‐B4 (GSB4) lectin immunohistochemistry (F,H) in the occipital cortex of control (D,G–H) and affected (A–C,E–F) sheep brains. GFAP immunoreactivity was initially confined to the pial surface of affected CLN6 sheep brains at 130 days gestation (E130, A). Subsequently GFAP immunoreactivity was detected particularly within the occipital cortex of affected sheep at birth (E150, B) and was prominent within superficial laminae of affected brain by postnatal day 12 (P12, C), but not in the age matched control (D). Microglial activation within the cortical gray matter (MHC II and GSB4 lectin reactivity) was relatively delayed. Clusters of MHC‐II immunoreactive (E) and GSB4 positive (F) microglia were first detected in the occipital cortex of affected sheep at 12 days of age, but not were present in age matched controls (G,H). Scale bar: 250 µm (A–H), 50 µm (higher magnification insets in E,F).
MHC class II immunoreactivity was largely absent within both affected and control brains up to birth (E150), but was prominent in microglia in affected brains 12 days later (Figure 3E vs. G), particularly within the upper neocortical layers II and III in the occipital and somatosensory cortex. A proportion of these cells were ramified, whereas others lacked processes, and clusters of MHC class II reactive cells were scattered throughout these upper layers of the cerebral cortex (inset of Figure 3E). MHC class II staining of microglia was not observed in the control sheep (Figure 3G). The pattern of GSB4 lectin staining was similar. Foci of activated microglia clearly evident in the upper layers of the affected neocortex 12 days after birth (Figure 3F) were not found in control cortical gray matter (Figure 3H).
Clear differences in morphology and labeling intensity of GSB4 labeled perivascular cells in control and affected brains became apparent from an early age. These cells were first detected in cortical white matter around E100–E110 days in both affected and control animals (Figure 4A,E,I). GSB4 positive perivascular cells continued to increase in size and labeling intensity in the affected brains as they developed, and also appeared to proliferate (Figure 4F–H,J–L), whereas they became less conspicuous in developing control brains (Figure 4B–D).
Figure 4.
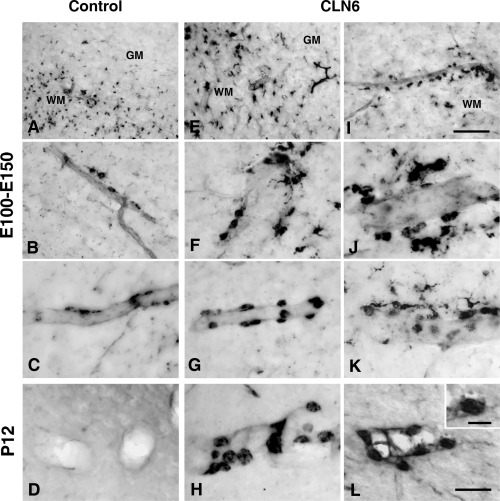
Perivascular cell activation in the cortical white matter of affected sheep.α‐D‐galactose specific isolectin I‐B4 lectin histochemistry revealed numerous perivascular macrophages (perivascular cells) in the cortical white matter of both affected (E–L) and control (A–D) sheep from gestational day 100 (E100) until postnatal day 12 (P12). These cells maintained a flattened morphology in control animals between E100 and E130 (A, E130, B, E100 and C, E110). In affected brains they had the morphology of hypertrophied perivascular macrophages and were more numerous at these ages (E–G, E110, I, E100 and J–K, E110) and continued to increase in number, size and labeling intensity (H–L, P12) whereas they were no longer present in age matched control brains (D, P12). Scale bar: 100 µm (A, E, I), 50 µm (B–D,F–H,J–L), 20 µm (inset in L).
GSB4 lectin also labeled vesicle‐laden macrophages within the leptomeninges covering the cerebral hemispheres, vascular endothelium, amoeboid microglia and perivascular cell populations in control and affected brains. The numbers of GSB4 positive leptomeningeal macrophages declined between E60 and E150, but showed a marked increase 12 days after birth (P12) in animals of both genotypes. Vascular endothelia in both control and affected animals were labeled with GSB4 to varying degrees from around E60. Capillaries and newly formed vessels were more intensely labeled. This staining of endothelial cells also decreased with development and was virtually absent by postnatal day 12.
Amoeboid microglia were present within sub‐ependymal regions of the lateral ventricles, corpus callosum (Figure 4A,E), internal and external capsules between E60–E110 in both affected and control animals. The number of amoeboid microglia subsequently declined and they disappeared by the time of birth. Ramifying microglia could be detected particularly towards the boundaries between the white and gray matter in animals of both genotypes.
DISCUSSION
The prenatal activation of non‐neuronal cells within the ovine CLN6 brain indicates that the onset of pathogenesis occurs earlier than previously considered. Reactive changes in non‐neuronal cells are usually interpreted as a response to neurodegeneration so the precocity of the cascade of reactive changes observed was unexpected. Changes were discrete and regionally defined, markers of astrocytosis, perivascular cell and microglial activation becoming apparent at different times. Activated astrocytes appeared in developing white matter 40–20 days before birth and astrocytic activation within the gray matter 20 days before birth. Hypertrophy and proliferation of perivascular cells in white matter was noticeable 40 days before birth. Clusters of activated microglia were detected in upper cortical gray matter layers shortly after birth.
These reactive changes began during the period of most rapid brain growth and development. Consistent with previous studies (27), the brain of the sheep showed a 25x increase in mass from E60 to birth (Figure 1), at which time the brain is close to 50% of adult brain mass. The adult brain architecture and lamellar structure emerges during this period and the vascular network is formed.
There is growing evidence for early neuro‐inflammatory responses in a variety of lysosomal storage disorders 15, 34, 49). As we recently reported, presymptomatic CLN6 affected South Hampshire sheep display reactive changes within the brain during the early postnatal period (36), spreading from discrete foci during disease progression. The distribution of microglial clusters at 12 days of age accurately predicts the cortical regions subsequently most vulnerable to neurodegeneration. A low level activation of astrocytes and microglia preceding widespread loss of neurons by many months has also been observed in a murine juvenile NCL (JNCL) model (40), suggesting that early glial activation may be a common feature of the NCLs. A re‐examination of these and other models is warranted, to determine if activation during brain development is a common feature.
Reactive changes in cortical gray matter. The prenatal activation of astrocytes and microglia in the gray matter occurs predominantly within superficial laminae (Figure 3), MHC class II and GSB4 positive microglia becoming evident after the appearance of GFAP positive activated astrocytes. These changes lead on to postnatal reactive changes (36), first evident within superficial laminae of occipital and somatosensory cortex, and spread to successively deeper laminae. It is not clear why these events should originate within the more superficial laminae, but localized glial responses are typically considered a sensitive indicator of neuronal damage or dysfunction 23, 41, 45). An investigation of other pathological changes within these superficial laminae may reveal the triggering events.
Reactive changes in developing white matter. In addition to labeling microglia in their various morphological states 44, 46), GSB4 lectin histochemistry also labels perivascular cells and other mononuclear phagocytes associated with the central nervous system (39).
Perivascular macrophages are strategically placed, being intimately close to both the systemic circulation and the brain parenchyma (41). The increased numbers of hypertrophied GSB4 positive perivascular cells (Figure 4), and perivascular macrophage and astrocytic activation within the white matter, may occur in response to systemic inflammation or as part of an attempted neuro‐immune response (22). Nevertheless, we found no evidence for microglial activation within developing white matter tracts and the perivascular cells, although hypertrophied and more numerous, did not express MHC class II antigens more typically associated with an “immune”—activated phenotype.
An alternative hypothesis is that the activation of perivascular cells reflects some other process that occurs early in CLN6 pathogenesis. These cells appear to invest blood vessels around the time that a blood‐brain barrier is established (42) but little is known about the specific roles they play during brain development.
Development of the ovine CLN6 brain. It has been assumed that development of the NCL brain is relatively normal and that the characteristic disease phenotypes become evident later. The brain weight increase, (Figure 1), gross morphological structure and anatomy of the developing ovine CLN6 brain support this notion. The histological assessment of cortical development revealed no overt differences between affected and control animals, and the formation of the laminae followed descriptions in the literature 1, 2, 13). Normal migration of cortical neurons from the germinal layer on radial glia (12) was seen in both genotypes in the early stages of development, the development of laminae in the cerebellum was the same and the distribution of amoeboid microglia within the developing white matter was in keeping with previous descriptions (12). These cells are considered to participate in axonal guidance (3) and represent a developmental rather than a pathologically activated state of microglia. None of these data suggest that neuronal development is significantly disturbed at early ages nevertheless more subtle pathological changes may be underway.
Progressive reactive events early in CLN6 pathogenesis. Collectively our data provide novel evidence for successive and localized reactive events during the prenatal development of ovine CLN6. These events are not directly associated with storage. Storage of subunit c of mitochondrial ATPase has been detected shortly before birth in murine JNCL(6), and prenatal lysosomal storage in South Hampshire sheep reported (19), but these storage bodies are sparse, even at birth. Unlike the prenatal activation of glial cell populations (2, 3, 4), or activation in the immediate postnatal period (36), the distribution of this storage material is not focal. This lack of a temporal or regional correlation between storage body accumulation and glial activation argues against the idea that activation occurs in a direct response to the presence of storage bodies or to neuronal damage caused by excessive amounts of subunit c.
The identity and cellular origin of the underlying molecular cues responsible for these events remain unknown, and it is unclear how they may be linked. Perivascular cell activation in developing white matter could be a separate event to glial activation within the gray matter and may be initiated by blood borne cues. Nor do these studies indicate any link to the biochemical lesion, absence of an endoplasmic reticulum resident protein 11, 31), which may affect pre‐lysosome vesicular transport, as indicted by its affect on arylsulfatase A endocytosis via the plasma membrane 300 kDa mannose‐6‐phosphate receptor (11). It is also possible that the apparent initial activation is not signaled by any external event but arises intracellularly as a direct consequence of the genetic defect.
The spread of glial activation through the affected brain (36) may signify an ongoing inflammatory response initiated early during pathogenesis. A chronic inflammatory response is damaging to neurons 41, 43, 47), and given that glial responses are evident as early as 40 days before birth (2, 4), it is surprising how long the development of the affected sheep brains follows a normal path. Glial activation has been reported in various other neurodegenerative conditions including multiple sclerosis, Alzheimer disease, Parkinson’s disease, HIV‐associated dementia, scrapie, trauma and ischemia 14, 33). These are mainly acute disease studies and it has been assumed that the responses are initiated by dying neurons or some ligand such as abnormally deposited β‐amyloid protein 28, 43), but there is mounting evidence that inflammation plays a pathogenic role in other storage diseases. For instance, suppression of microglial activation, as a side effect of bone marrow transplantation, suppressed neurodegeneration in a mouse model of Sandhoff disease (49). Deletion of a macrophage inflammatory protein, MIP‐1α, had the same effect, directly implicating inflammation in pathogenesis (52). These studies suggest that suppression of inflammation may be a valuable adjunct to other therapies based on replacing the mutated gene product with a functional protein. Our data argue that inflammatory events are initiated before birth, but follow an orderly cascade. Determining when this cascade becomes fatally damaging to neurons will have important implications for the timing and delivery of therapeutic interventions in this profoundly disabling disorder.
ACKNOWLEDGMENTS
We would like to thank Nigel Jay, Gretchen McGuire, Nadia Mitchell and John Wynyard for expert technical assistance, and Dr Ming Lim and Dr Alison Barnwell for constructive comments on the manuscript. These studies were supported by the New Zealand Neurological Foundation (GWK, DNP), the US National Institutes of Health, NINDS grants NS40297 (GWK, DNP) Lysosomal Diseases New Zealand (DNP), and NS41930 (JDC); The Wellcome Trust, UK, Biomedical Research Collaboration Grant 023360 (JDC, DNP); and grants to JDC from the Natalie Fund, Batten Disease Support and Research Association, Batten Disease Family Association, Batten Disease Support and Research Trust.
REFERENCES
- 1. Åström K‐E (1967) On the early development of the isocortex in fetal sheep. Prog Brain Res 26:1–59. [PubMed] [Google Scholar]
- 2. Barlow RM (1969) The foetal sheep: morphogenesis of the nervous system and histochemical aspects of myelination. J Comp Neurol 135:249–262. [DOI] [PubMed] [Google Scholar]
- 3. Bass WT, Singer GA, Liuzzi FJ (1998) Transient lectin binding by white matter tract border zone microglia in the foetal rabbit brain. Histochem J 30:657–666. [DOI] [PubMed] [Google Scholar]
- 4. Broom MF, Zhou C, Broom JE, Barwell KJ, Jolly RD, Hill DF (1998) Ovine neuronal ceroid lipofuscinosis: a large animal model syntenic with the human neuronal ceroid lipofuscinosis variant CLN6. J Med Genet 35:717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cooper JD (2003) Progress towards understanding the neurobiology of Batten disease or neuronal ceroid lipofuscinosis. Curr Opin Neurol 16:121–128. [DOI] [PubMed] [Google Scholar]
- 6. Cotman SL, Vrbanac V, Lebel LA, Lee RL, Johnson KA, Donahue LR, Teed AM, Antonellis K, Bronson RT, Lerner TJ, MacDonald ME (2002) Cln3Δex7/8 knock‐in mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth. Hum Mol Genet 11:2709–2721. [DOI] [PubMed] [Google Scholar]
- 7. Fearnley IM, Walker JE, Martinus RD, Jolly RD, Kirkland KB, Shaw GJ, Palmer DN (1990) The sequence of the major protein stored in ovine ceroid lipofuscinosis is identical with that of the dicyclohexylcarbodi‐imide‐reactive proteolipid of mitochondrial ATP synthase. Biochem J 268:751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gao H, Boustany RM, Espinola JA, Cotman SL, Srinidhi L, Antonellis KA, Gillis T, Qin X, Liu S, Donahue LR, Bronson RT, Faust JR, Stout D, Haines JL, Lerner TJ, MacDonald ME (2002) Mutations in a novel CLN6‐encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in man and mouse. Am J Hum Genet 70:324–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goebel HH, Mole SE, Lake BD (eds) (1999) Neuronal Ceroid Lipofuscinoses (Batten Disease), Vol. 33. IOS Press: Amsterdam. [Google Scholar]
- 10. Haltia M (2003) The neuronal ceroid‐lipofuscinoses. J Neuropathol Exp Neurol 62:1–13. [DOI] [PubMed] [Google Scholar]
- 11. Heine C, Koch B, Storch S, Kohlschütter A, Palmer DN, Braulke T (2004) Defective endoplasmic reticulum‐resident membrane protein CLN6 affects lysosomal degradation of endocytosed arylsulfatase A. J Biol Chem 279:22347–22352. [DOI] [PubMed] [Google Scholar]
- 12. Hewicker‐Trautwein M, Schultheis G (1994) Lectin labelling of amoeboid and ramified microglial cells in the telencephalon of ovine fetuses with the B4 isolectin from Griffonia simplicifolia . J Comp Pathol 111:21–31. [DOI] [PubMed] [Google Scholar]
- 13. Hewicker‐Trautwein M, Trautwein G (1993) An immunohistochemical study of the fetal sheep neocortex and cerebellum with antibodies against nervous system‐specific proteins. J Comp Pathol 109:409–421. [DOI] [PubMed] [Google Scholar]
- 14. Hunot S, Hirsch EC (2003) Neuroinflammatory processes in Parkinson’s disease. Ann Neurol 53:s49–58. [DOI] [PubMed] [Google Scholar]
- 15. Jeyakumar M, Thomas R, Elliot‐Smith E, Smith DA, Van Der Spoel AC, D’Azzo A, Perry VH, Butters TD, Dwek RA, Platt FM (2003) Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain 126:974–987. [DOI] [PubMed] [Google Scholar]
- 16. Jolly RD, Janmaat A, West DM, Morrison I (1980) Ovine ceroid‐lipofuscinosis: a model of Batten’s disease. Neuropathol Appl Neurobiol 6:195–209. [DOI] [PubMed] [Google Scholar]
- 17. Jolly RD, Martinus RD, Palmer DN (1992) Sheep and other animals with ceroid‐lipofuscinoses: Their relevance to Batten disease. Am J Med Genet 42:609–614. [DOI] [PubMed] [Google Scholar]
- 18. Jolly RD, Palmer DN, Studdert VP, Sutton RH, Kelly WR, Koppang N, Dahme G, Hartley WJ, Patterson JS, Riis RC (1994) Canine ceroid‐lipofuscinoses: A review and classification. J Small Anim Pract 35:299–306. [Google Scholar]
- 19. Jolly RD, Shimada A, Dopfmer I, Slack PM, Birtles MJ, Palmer DN (1989) Ceroid‐lipofuscinosis (Batten’s disease): pathogenesis and sequential neuropathological changes in the ovine model. Neuropathol Appl Neurobiol 15:371–383. [DOI] [PubMed] [Google Scholar]
- 20. Katz ML, Khan S, Awano T, Shahid SA, Siakotos AN, Johnson GS (2005) A mutation in the CLN8 gene in English Setter dogs with neuronal ceroid‐lipofuscinosis. Biochem Biophys Res Commun 327:541–547. [DOI] [PubMed] [Google Scholar]
- 21. Kay GW, Hughes SM, Palmer DN (1999) In vitro culture of neurons from sheep with Batten disease. Mol Genet Metab 67:83–88. [DOI] [PubMed] [Google Scholar]
- 22. Kida S, Steart PV, Zhang ET, Weller RO (1993) Perivascular cells act as scavengers in the cerebral perivascular spaces and remain distinct from pericytes, microglia and macrophages. Acta Neuropathol 85:646–652. [DOI] [PubMed] [Google Scholar]
- 23. Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. TINS 19:312–318. [DOI] [PubMed] [Google Scholar]
- 24. Lingaas F, Mitchison HM, Mole SE, Koppang N, Goebel HH, Lake BD (1999) Animal models of NCL. In: Neuronal Ceroid Lipofuscinoses (Batten Disease). Goebel HH, Mole SE, Lake BD (eds), pp. 152–167. IOS Press: Amsterdam. [Google Scholar]
- 25. Martinus RD, Harper PAW, Jolly RD, Bayliss SL, Midwinter GG, Shaw GJ, Palmer DN (1991) Bovine ceroid‐lipofuscinosis (Batten’s disease): the major component stored is the DCCD‐reactive proteolipid, subunit c, of mitochondrial ATP synthase. Vet Res Commun 15:85–94. [DOI] [PubMed] [Google Scholar]
- 26. Mayhew IG, Jolly RD, Pickett BT, Slack PM (1985) Ceroid‐lipofuscinosis (Batten’s disease): pathogenesis of blindness in the ovine model. Neuropathol Appl Neurobiol 11:273–290. [DOI] [PubMed] [Google Scholar]
- 27. McIntosh GH, Baghurst KI, Potter BJ, Hetzel BS (1979) Foetal brain development in the sheep. Neuropathol Appl Neurobiol 5:103–114. [DOI] [PubMed] [Google Scholar]
- 28. Minagar A, Shapshak P, Fujimura R, Ownby R, Heyes M, Eisdorfer C (2002) The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV‐associated dementia, Alzheimer disease, and multiple sclerosis. J Neurol Sci 202:13–23. [DOI] [PubMed] [Google Scholar]
- 29. Mitchison HM, Lim MJ, Cooper JD (2004) Selectivity and types of cell death in the neuronal ceroid lipofuscinoses. Brain Pathol 14:86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mole SE (2004) The genetic spectrum of human neuronal ceroid‐lipofuscinoses. Brain Pathol 14:70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mole SE, Michaux G, Codlin S, Wheeler RB, Sharp JD, Cutler DF (2004) CLN6, which is associated with a lysosomal storage disease, is an endoplasmic reticulum protein. Exp Cell Res 298:399–406. [DOI] [PubMed] [Google Scholar]
- 32. Mole SE, Williams RE, Goebel HH (2005) Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics 6:107–126. [DOI] [PubMed] [Google Scholar]
- 33. Neumann H (2001) Control of glial immune function by neurons. Glia 36:191–199. [DOI] [PubMed] [Google Scholar]
- 34. Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF (2003) Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc Natl Acad Sci USA 100:1902–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oswald MJ, Kay GW, Palmer DN (2001) Changes in GABAergic neuron distribution in situ and in neuron cultures in ovine (OCL6) Batten disease. Eur J Paediatr Neurol 5:S135–S142. [DOI] [PubMed] [Google Scholar]
- 36. Oswald MJ, Palmer DN, Kay GW, Shemilt SJA, Rezaie P, Cooper JD (2005) Glial activation spreads from specific cerebral foci and precedes neurodegeneration in presymptomatic ovine neuronal ceroid lipofuscinosis (CLN6). Neurobiol Dis 20:49–63. [DOI] [PubMed] [Google Scholar]
- 37. Palmer DN, Fearnley IM, Walker JE, Hall NA, Lake BD, Wolfe LS, Haltia M, Martinus RD, Jolly RD (1992) Mitochondrial ATP synthase subunit c storage in the ceroid‐lipofuscinoses (Batten disease). Am J Med Genet 42:561–567. [DOI] [PubMed] [Google Scholar]
- 38. Palmer DN, Martinus RD, Cooper SM, Midwinter GG, Reid JC, Jolly RD (1989) Ovine ceroid lipofuscinosis. The major lipopigment protein and the lipid‐binding subunit of mitochondrial ATP synthase have the same NH2‐terminal sequence. J Biol Chem 264:5736–5740. [PubMed] [Google Scholar]
- 39. Pennell N, Hurley S, Streit W (1994) Lectin staining of sheep microglia. Histochemistry 102: 483–486. [DOI] [PubMed] [Google Scholar]
- 40. Pontikis CC, Cella CV, Parihar N, Lim MJ, Chakrabarti S, Mitchison HM, Mobley WC, Rezaie P, Pearce DA, Cooper JD (2004) Late onset neurodegeneration in the Cln3 −/− mouse model of juvenile neuronal ceroid lipofuscinosis is preceded by low level glial activation. Brain Res 1023:231–242. [DOI] [PubMed] [Google Scholar]
- 41. Raivich G, Bohatschek M, Kloss CUA, Werner A, Jones LL, Kreutzberg GW (1999) Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Brain Res Rev 30:77–105. [DOI] [PubMed] [Google Scholar]
- 42. Rezaie P, Dean A, Male D, Ulfig N (2005) Microglia in the cerebral wall of the human telencephalon at second trimester. Cereb Cortex 15:938–949. [DOI] [PubMed] [Google Scholar]
- 43. Stoll G, Jander S (1999) The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol 58:233–247. [DOI] [PubMed] [Google Scholar]
- 44. Streit WJ (2001) Microglia and macrophages in the developing CNS. Neurotoxicology 22:619–624. [DOI] [PubMed] [Google Scholar]
- 45. Streit WJ (2002) Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 40:133–139. [DOI] [PubMed] [Google Scholar]
- 46. Streit WJ, Kreutzberg GW (1987) Lectin binding by resting and reactive microglia. J Neurocytol 16:249–260. [DOI] [PubMed] [Google Scholar]
- 47. Streit WJ, Mrak RE, Griffin WST (2004) Microglia and neuroinflammation: a pathological perspective. J Neuroinflam 1:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tyynelä J, Suopanki J, Santavuori P, Baumann M, Haltia M (1997) Variant late infantile neuronal ceroid‐lipofuscinosis: pathology and biochemistry. J Neuropathol Exp Neurol 56:369–375. [DOI] [PubMed] [Google Scholar]
- 49. Wada R, Tifft CJ, Proia RL (2000) Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc Natl Acad Sci USA 97:10954–10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wheeler RB, Sharp JD, Schultz RA, Joslin JM, Williams RE, Mole SE (2002) The gene mutated in variant late‐infantile neuronal ceroid lipofuscinosis (CLN6) and in nclf mutant mice encodes a novel predicted transmembrane protein. Am J Hum Genet 70:537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. White IR, Russel AJ, Fowler DG (1984) Real‐time ultrasonic scanning in the diagnosis of pregnancy and the determination of fetal numbers in sheep. Vet Rec 115:140–143. [DOI] [PubMed] [Google Scholar]
- 52. Wu YP, Proia RL (2004) Deletion of macrophage‐inflammatory protein 1 retards neurodegeneration in Sandhoff disease mice. Proc Natl Acad Sci USA 101:8425–8430. [DOI] [PMC free article] [PubMed] [Google Scholar]