


Abstract
RNase Y and RNase E are disparate endoribonucleases that govern global mRNA turnover/processing in the two evolutionary distant bacteria Bacillus subtilis and Escherichia coli, respectively. The two enzymes share a similar in vitro cleavage specificity and subcellular localization. To evaluate the potential equivalence in biological function between the two enzymes in vivo we analyzed whether and to what extent RNase E is able to replace RNase Y in B. subtilis. Full-length RNase E almost completely restores wild type growth of the rny mutant. This is matched by a surprising reversal of transcript profiles both of individual genes and on a genome-wide scale. The single most important parameter to efficient complementation is the requirement for RNase E to localize to the inner membrane while truncation of the C-terminal sequences corresponding to the degradosome scaffold has only a minor effect. We also compared the in vitro cleavage activity for the major decay initiating ribonucleases Y, E and J and show that no conclusions can be drawn with respect to their activity in vivo. Our data confirm the notion that RNase Y and RNase E have evolved through convergent evolution towards a low specificity endonuclease activity universally important in bacteria.
INTRODUCTION
The adaptation of gene expression to a changing environment requires the instability of messenger RNA (mRNA). In bacteria, mRNA decay generally follows first-order kinetics. Control of mRNA degradation, if it is to be efficient, must thus occur at the generally rate-limiting initiating step of the process. Most fundamental knowledge in this respect stems from studies in Escherichia coli and Bacillus subtilis, two organisms separated by an evolutionary stretch of several billion years. For quite some time, mRNA decay in these two model organisms was thought to be radically different, but now can probably best be summarized by ‘different enzymes – similar strategies’. Indeed, the major ribonucleases RNase E (E. coli) and RNase Y (B. subtilis) involved in the initiation of mRNA decay are completely different proteins but functionally similar endoribonucleases with relaxed sequence specificity. Their crucial role in producing short-lived decay intermediates is now clearly recognized (1). First described >40 years ago in E. coli (2,3) RNase E is a large 1061 residue protein. It is composed of a catalytic N-terminal domain and a natively unstructured C-terminal region containing small microdomains that can associate with the 3′ exoribonuclease PNPase, the DEAD box RNA helicase RhlB and the glycolytic enzyme enolase to form a multienzyme complex known as the RNA degradosome (4–8). A 15 residue membrane targeting sequence (MTS) that can fold an amphipathic α-helix is located at the beginning of the non-catalytic region and is necessary for the attachment of RNase E to the inner cytoplasmic membrane (9,10). This subcellular localization is important for the stability of the enzyme, optimal rates of global mRNA degradation and protection of ribosome-free transcripts from premature interactions with RNase E in the nucleoid (11). In live cells RNase E rapidly diffuses over the inner membrane forming short-lived foci which have been proposed to arise from the transient clustering of RNase E into cooperative degradation bodies (10).
In B. subtilis, RNase Y affects global mRNA stability; the protein has endonucleolytic activity with an RNase E-like single strand specific cleavage specificity on preferably 5′ monophosphorylated substrates in vitro (12). Similar to RNase E, RNase Y is tethered to the inner cell membrane via a single-pass N-terminal helix (13), RNase Y is called YmdA in this reference) which fits well with the observation that translating ribosomes are distributed predominantly around the cell periphery (14,15). The intracellular levels of a majority of transcripts is affected in RNase Y depleted strains (16–18). A degradosome-like complex centered on RNase Y has been proposed (19,20) but cannot be isolated in the absence of crosslinking agents, unlike the RNase E-based degradosome in E. coli (4,21). The formation of meaningful interactions between RNase Y and other ribonucleases in vivo remains an open question (22–24). However, three small proteins, YlbF, YmcA and YaaT forming the so-called Y-complex (25,26) were shown to alter RNase Y activity in vivo (27), being notably required for the efficient maturation of operon mRNAs and affecting the abundance of certain riboswitches (28).
Similar to RNase E, RNase Y was found to diffuse rapidly across the membrane in the form of dynamic short-lived foci but unlike RNase E, formation of RNase Y foci is not dependent on the presence of RNA substrates; they become more abundant and increase in size following transcription arrest (29). Y-complex mutations have a similar but much stronger effect suggesting that the Y-complex can shift the assembly status of RNase Y towards fewer and smaller complexes leading to increased cleavage of complex substrates like polycistronic mRNAs (29).
Considering the evolutionary distance between E. coli and B. subtilis it is not surprising that, despite a similar enzymatic activity in vitro, RNase E and RNase Y are susceptible to interact with a variety of completely different co-factors in their respective hosts to tune their activity in vivo. This might explain why complementation of RNase E by RNase Y was not very successful; survival of E. coli depleted of RNase E and expressing RNase Y could only be observed in special media and growing the cells for up to 2 weeks (30).
We have previously tried to replace RNase Y in B. subtilis by E. coli RNase E and these earlier experiments encouraged us to carry out a comprehensive study on how and to what extent RNase E might be able to make up for RNase Y in B. subtilis. We observed a surprisingly efficient complementation of an RNase Y null mutant by RNase E, with respect to recovering growth as well as restoring transcription profiles of individual genes and on a global scale. Full-length RNase E is clearly the most successful in replacing RNase Y over shorter forms of the enzyme. However, it is the membrane localization of RNase E that turned out to be the single most relevant parameter. Our data provide clear in vivo evidence for the convergent evolution of RNases E and Y and support the notion ‘different enzymes - similar strategies’ characterizing bacterial mRNA degradation and processing. It will also now be possible to compare major critical parameters that determine substrate specificity for both enzymes in vivo.
MATERIALS AND METHODS
Bacterial strains and growth conditions
The B. subtilis strains used in this work are derivatives of strain SSB1002, a wild-type laboratory stock strain derived from strain 168. E. coli strain JM109 was used for plasmid constructions, and E. coli XL1-Blue for site-directed mutagenesis experiments. B. subtilis and E. coli strains were grown at 37°C in LB medium with aeration. When required, the following antibiotics were added to the medium: chloramphenicol (5 μg/ml), spectinomycin (100 μg/ml) for B. subtilis and ampicillin (200 μg/ml) for E. coli. The B. subtilis rny deletion strain SSB503 was constructed by replacing the rny ORF in phase with the ORF encoding the chloramphenicol acetyltransferase from the S. aureus plasmid pC194. B. subtilis strains SSB500, SSB498, SSB502 and SSB508 contained xylose-inducible copies of wild type (pHMK7), aa 1-688 (pHMK5) and aa 1–529 (pHMK8) versions of E. coli RNase E or the empty pDR160T plasmid, respectively, integrated in single copy at the amyE locus and were transformed in a second step with chromosomal DNA of strain SSB503 to delete the rny gene. SSB507 is wild-type for rny and contains the empty pDR160T vector integrated at amyE. Expression of E. coli RNase E was induced by the addition of 50 mM xylose to the medium.
Growth of strains was monitored on solid media (LB or SMS, containing 50 mM xylose). Fresh overnight colonies were resuspended in PBS buffer (137 mM NaCl, 10 mM phosphate, 2.7 mM KCl, pH 7.4) to an OD600 = 0.5 and spotted on the plates in serial 10-fold dilutions. LB plates were incubated O/N and SMS plates for at least 48h at 37°C.
Plasmid constructs
pDR160T
Plasmid pDR160T is based on pDR160 [amyE::Psweet-xylR (spec)], an ectopic integration vector containing the xylose-inducible Psweet promoter (31), D. Rudner, unpublished). The transcription terminator of the B. subtilis rnjA gene was inserted at the BamHI site of pDR160 in order to terminate the transcripts transcribed from the xylose inducible promoter. The BamHI site upstream of the terminator remains functional for cloning. The inserted sequence at the BamHI site (underlined) is GGATCCACAGAAAAAGAGGCACTCCCTAAGGGAGTGCCTCTTTTTATTTGATCC.
pHMK5
Plasmid pHMK5 contains a shortened version of the E. coli rne gene, encoding a truncated RNase E (aa 1–688) under the control of the xylose-inducible promoter and the Shine-Dalgarno sequence of the B. subtilis thrZ gene. A 2 kb PCR fragment (oligonucleotides HP1568–HP1569) was cleaved with PacI and BamHI and inserted into the vector pDR160T cleaved with PacI–BamHI.
pHMK7
Plasmid pHMK7 contains the full-length E. coli rne gene, encoding wild type RNase E (aa 1–1061) under the control of the xylose-inducible promoter and the Shine-Dalgarno sequence of the B. subtilis thrZ gene. A 3.2 kb PCR fragment (oligonucleotides HP1568-HP1590) was cleaved with PacI and NheI and inserted into the ectopic integration vector pDR160T cleaved with PacI-NheI.
pHMK7-ΔMTS
This plasmid corresponds to pHMK7 but the E. coli rne gene lacks the MTS sequence. Using inverse PCR on pHMK7, 16 amino acids (residus 567–582) were deleted from the rne ORF.
pHMK8
Plasmid pHMK8 contains a shortened version of the E. coli rne gene, encoding a truncated RNase E (aa 1–529) under the control of the xylose-inducible promoter and the Shine-Dalgarno sequence of the B. subtilis thrZ gene. A 1.6 kb PCR fragment (oligonucleotides HP1568-HP1673) was cleaved with PacI and BamHI and inserted into the ectopic integration vector pDR160T cleaved with PacI–BamHI.
Oligonucleotides
| HP857 | AGAATTCTAATACGACTCACTATAGGAGATTAAGAAAGACACACG |
| HP1165 | AAAACCCCGCCCCCTATGAAAG |
| HP1568 | AATGATTAATTAAACAACAATGAATAAGGAGTGTTCTAAAATGAAAAGAAT |
| GTTAATCAACGCAACT | |
| HP1569 | GATAGGATCCAATAAAAAGAGGCACTCCCTTAGGGAGTGCCTCTTTTTCT |
| GTTTAACGTTTATCATCATTACGGCGGCGG | |
| HP1590 | GATAGCTAGCAATAAAAAGAGGCACTCCCTTAGGGAGTGCCTCTTTTTCT |
| GTTTACTCAACAGGTTGCGGACGCG | |
| HP1673 | GATAGGATCCTTTACAGCGCAGGTTGTTCCGGACGCTTACG |
Epi-fluorescence microscopy
GFP fluorescent images were taken with the Zeiss Axio Imager M1 microscope equipped with an AxioCam MRm camera (Zeiss) using filter set 10 (Zeiss). For the visualization of cells from exponentially growing cultures, overnight cultures in LB medium were diluted to OD600 ∼0.1 and grown at 37°C in fresh LB medium for at least three generations. Cells were mounted on 1% (w/v) agarose pads and images acquired by an AxioCam camera MRm (Zeiss) using a 1.3 NA ×100 oil objective on a phase-contrast/fluorescence microscope (Zeiss Axio Imager M1).
RNase cleavage assays
As substrate we used a 279 nt T7 RNA polymerase in vitro transcript of the thrS leader mRNA generated from a PCR template obtained with oligonucleotides HP857 and HP1165. The 3′ end of the transcript corresponds to the fourth U residue at the end of the leader terminator structure. Cleavage reactions were carried out in 10 μl reactions as described previously (32) using approximately 0.2 pmoles 5′ [32P]-end-labeled monophosphorylated thrS leader mRNA and 10 pmol of the respective enzymes. RNase Y and RNase J1 were purified as 6xHis-tagged proteins as described previously (32) and unmodified RNase E (aa 1–528) was purified from a C-terminal intein fusion construct according to the manufacturer (Biolabs, plasmid pTYB2). Control reactions were performed by incubating the substrate with the reaction buffer alone. Samples were extracted with phenol/chloroform and reprecipitated prior to gel analysis to avoid retention of RNA by the ribonuclease. The samples were analyzed on 5% denaturing polyacrylamide urea gels. Radioactive signals were visualized by a Typhoon FLA 9500 biomolecular imager (GE Healthcare). Images were treated with Image J software.
Northern blot
RNA blot analysis was carried out using 5 μg of total RNA separated either on a 1% formaldehyde-agarose or 10% acrylamide-urea gels (10% PAA, 0.5 g/ml urea, 1x Tris– borate) and electroblotted onto Hybond N+ membranes (GE Healthcare). RNA was cross-linked to the membrane at 120 mJ cm–2 for 1 min. RNA probes were denatured at 80°C for 3 min, kept on ice for 2 min and then added to the prehybridized membranes (Amersham Rapid-hyb buffer) for hybridization at 70°C for 2 hours. Membranes were washed at 68°C with washing solutions I (2 × SSC and 0.1% SDS), II (1 × SSC and 0.1% SDS) and III (0.5 × SSC and 0.1% SDS) for 15 min each. The signals were detected with a GE Healthcare Typhoon FLA 9500 imaging system. 5S rRNA (5S) was used as loading control.
Western blot
For Western blot analysis, 20 μg of protein extract prepared as described previously (33) was separated by SDS-PAGE (10%). After electrophoretic transfer of the proteins, the nitrocellulose membrane (GE Healthcare) was stained with amido black to check for equal transfer across all lanes. The membrane was blocked for 1 h with 5% BSA in phosphate-buffered saline (PBS)–Tween buffer (100 mM NaH2PO4–Na2HPO4, pH 7.4, 100 mM NaCl, 0.1% Tween) and incubated with polyclonal RNase E antiserum or a monoclonal RNase Y antibody diluted in PBS-Tween for at least 4 h. Signals were detected by ECL chemiluminescence (BIO-RAD, ClarityTM Western ECL) associated with a CCD camera (BIO-RAD ChemiDoc XR System+). When necessary, ECL detected proteins were quantified by ImageLab software (Bio-Rad).
Transcriptome analysis
RNA samples were treated with the Ribo-Zero Kit for Bacteria to remove ribosomal RNA (rRNAs). Multiplex RNA-Seq libraries were prepared with the Illumina TruSeq Stranded Total RNA Sample Preparation and sequenced (HiSeq2000). The complete genome sequence of the Bacillus subtilis subsp. subtilis str. 168 (NCBI accession number for chromosome: NC_000964.3) and its annotation were retrieved from the NCBI: (https://www.ncbi.nlm.nih.gov/genome/?term=Bacillus±subtilis) and from Subtiwiki (34). Reads processing, mapping and differential gene expression analysis were performed according to (35). Briefly, single-end 100-nt reads were mapped to the reference genome using bwa and allowing soft-clipping in the alignment parameters (bwa mem) (36). Analysis of the mappings used samtools for processing, sorting and indexing of the alignment files; BEDtools for computing coverage; the IGV browser for displaying the alignment files (36–38). Reads counts were normalized either as reads per million (RPM) or as transcripts for million (TPM). Differential gene expression analysis between strains was performed with the EdgeR package (39) using the Trimmed Mean of M-values (TMM) scaling factor as normalization method. Gene features were considered as significantly up- or down-regulated in the mutants versus WT samples if the log2 fold-change (FC) ratio was ≥1 or ≤–1 and the p-value adjusted for multiple testing False Discovery Rate (FDR) calculated using the Benjamini-Hochberg (BH) method in edgeR was equal or lower than 5% (FDR ≤ 0.05). Multi-dimensional scaling (MDS) and MA plots (visualizing measurement differences between two samples by transforming the data onto M (log ratio) and A (mean average) scales) were generated, respectively, through the ‘plotMDS.dge’ and the plotSmear functions of the edgeR package. Raw sequencing data are deposited at the NCBI Sequence Read Archive accession number (SRA) SUB8491357.
RESULTS
E. coli RNase E can restore growth of a B. subtilis rny mutant
We used a B. subtilis RNase Y knockout mutant to analyze whether E. coli RNase E can productively replace the Bacillus enzyme. Since a Δrny strain could not be transformed we first inserted genes encoding different versions of E. coli RNase E under the control of a xylose-inducible promoter in single copy at the amyE locus. This includes four gene constructs that express wild type RNase E (1061 amino acids (aa)), a full-length enzyme lacking the membrane tethering sequence (MTS, Δ567–582), a 688 aa truncated version that retains the MTS sequence but lacks the C-terminal scaffold required for degradosome formation and a 529 aa construct that corresponds to the catalytic domain. A schematic of the RNase E and RNase Y domain structures is shown in Figure 1. A complete deletion of the rny open reading frame between the start and stop codons was introduced in a second step as described in Materials and Methods.
Figure 1.
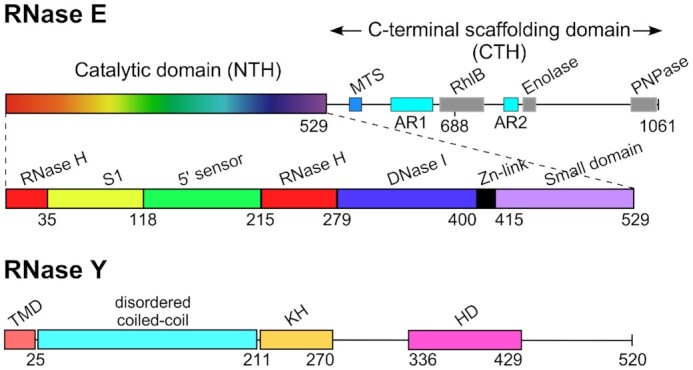
Domain composition of E. coli RNase E (1061 aa) and B. subtilis RNase Y (520 aa) monomers. RNase E is composed of the N-terminal catalytic region (NTH, aa 1–529) and the C-terminal noncatalytic region (aa 530–1061). The catalytic region contains a large globular domain which is a composite of recurrent structural subdomains (50) and a small folded domain (aa 415–529). The C-terminal half (CTH) of the protein is predicted to be unfolded but contains microdomains that mediate interactions with the inner plasma membrane (MTS) and other components of the degradosome (the helicase RhlB, enolase, and PNPase). AR1 and AR2 are arginine-rich segments probably involved in RNA binding (51). B. subtilis RNase Y (520 aa) has a completely different domain structure including an N-terminal transmembrane domain (aa 1–25) anchoring the protein at the inner membrane, followed by a large region predicted to be disordered (aa ∼30–210), an RNA binding KH domain (aa 211–270) and a metal-chelating HD domain (aa 336–429) containing the conserved His/Asp motif required for RNase activity (1).
The B. subtilis Δrny strain (SSB503) is viable but has a severe growth defect. Even in rich medium (LB) at 37°C the cells reached a maximal cell density of 0.4–0.5 that slowly decreases upon further incubation and growth in SMS minimal medium was difficult to monitor (data not shown). We therefore compared growth of the various strains on solid media. Expression of E. coli RNase E from the xylose-inducible promoter can almost completely alleviate the growth defect caused by the absence of RNase Y. Indeed, both wild type RNase E (1061 aa) and the 688 aa truncated version lacking the degradosome scaffold can restore growth to almost wild type levels (Figure 2A). Even the shortest tested version of RNase E (529 aa) corresponding to the catalytic N-terminal half of the enzyme can significantly restore growth of the Δrny mutant. Interestingly, full-length RNase E lacking the MTS sequence and thus located in the cytoplasm (see below) was the least efficient in compensating for the loss of RNase Y (Figure 2A). Compensation of the rny mutant growth defect depended on the induced expression of the plasmid encoded RNase E proteins (Figure 2A, compare left and right panels).
Figure 2.
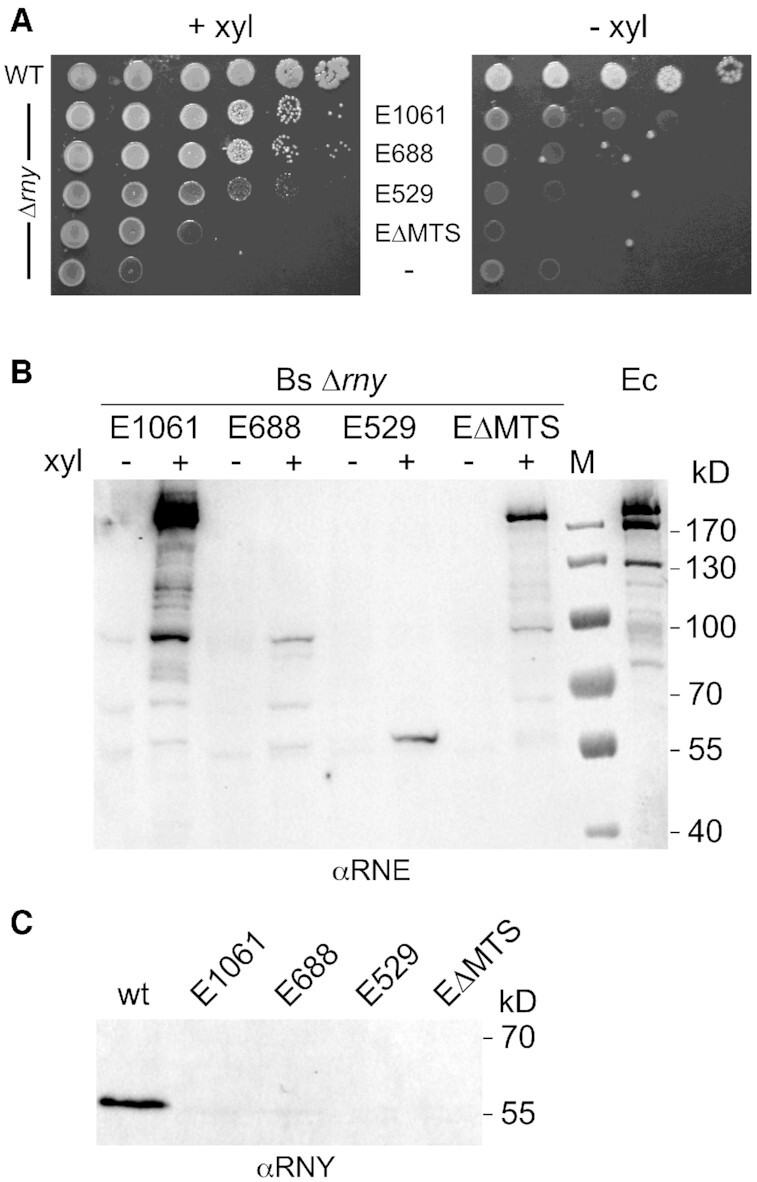
Functional complementation of B. subtilis Δrny by E. coli RNase E. (A) Growth of a B. subtilis Δrny mutant strain (SSB508) expressing different versions of E. coli RNase E. Cells from fresh colonies were resuspended and spotted in serial 10-fold dilutions on SMS medium agar plates containing 50 mM xylose to induce RNase E expression. Plates were incubated at 37°C for 48 h. (B) In vivo expression levels of RNase E variants in B. subtilis Δrny. A polyclonal antibody directed against E. coli RNase E was used to estimate expression of the various RNase E proteins from a xylose inducible promoter in a B. subtilis rny null mutant strain grown in LB medium with (+) or without xylose (−). E1061 : wild type full-length RNase E ; E688 : RNase E truncated after aa 688 ; E529 : RNase E truncated after aa 529; EΔMTS: full-length RNase E lacking the membrane tethering sequence (Δ567–582). The last lane (Ec) is a positive control that contains a total extract of E. coli strain JM109 grown in LB medium. (C) Western analysis of RNase Y expression in the B. subtilis wild type and the Δrny strain expressing the various forms of RNase E using an RNase Y specific antibody. The extracts analyzed were the same as those probed with anti-RNase E antibodies (panel B). M: marker proteins.
The inducible expression of RNase E and the absence of RNase Y in the B. subtilis strains was verified by Western blot (Figure 2B/C). Despite expression under identical conditions from the same transcriptional and translational signals the absolute intracellular levels of the different RNase E versions varied considerably. The full-length wild type protein migrating with an apparent MW of 180 kDa was expressed most strongly including some degradation intermediates similar to what we observed in E. coli (Figure 2B). The lowest levels of expression were observed for the truncated 688 aa and 529 aa RNase E proteins (Figure 2B) even if their absolute levels are underestimated by a factor of ∼2-fold (see Discussion and Supplementary Figure S1). We also measured the transcript levels of the RNase E versions from RNA-Seq data generated from the mutant strains. Since differences in gene length generate unequal read counts for genes expressed at the same level (e.g. longer gene have more reads), we normalized read counts for gene length and sequencing depth, generating TPM (transcript per million). The rne (ΔMTS), rne (688) and rne (529) transcript levels were about 20, 29 and 30% of that measured for wild type rne (1061), respectively.
The effective deletion of the rny gene in the complemented strains was confirmed by PCR (data not shown), absence of rny transcripts as revealed by lack of RNA-Seq coverage (Supplementary Figure S2) and the absence of RNase Y in total cell extracts as judged by Western blot (Figure 2C).
Functional complementation of RNase Y by E. coli RNase E was not dependent on the medium used to cultivate the bacteria. In defined minimal medium (SMS-fructose) we observed a very similar pattern of growth recovery (data not shown).
The N-terminal transmembrane domain (aa 1–20) anchors RNase Y to the inner membrane (Figure 3A). In E. coli, RNase E tethers to the inner membrane via a 15 amino acid sequence (MTS) that forms an amphipathic helix. We wanted to know whether RNase E behaves in a similar way when expressed in B. subtilis. In order to visualize the protein in the cell we fused a green fluorescent protein (GFP) to the C-terminal end of the four RNase E constructs used for complementation. As expected, both wild type RNase E and the 688 aa version located primarily to the cell periphery (Figure 3B and D). In contrast, the catalytic N-terminal half of RNase E (529 aa) and the full-length enzyme lacking the MTS are dispersed uniformly in the cytoplasm (Figure 3C and E). Wild type B. subtilis cells grow as single or dividing cells (Figure 3A) while the Δrny mutant formed spirals interspersed with long chains (Figure 3F). The mutant complemented by RNase E grows as chains whether RNase E is expressed as a GFP fusion protein or as a wild type protein (compare Figure 3B and G).
Figure 3.
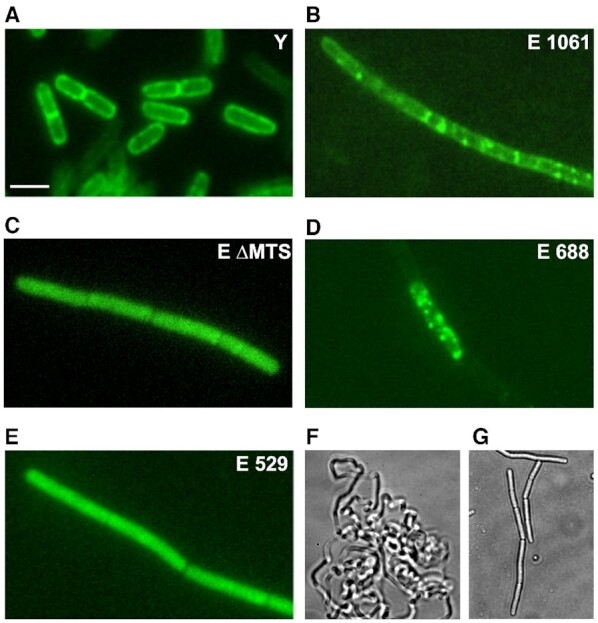
Localization of E. coli RNase E variants in B. subtilis by fluorescence microscopy. B. subtilis strains expressing GFP fusion proteins of RNase Y or various RNase E versions expressed from a xylose inducible promoter (panels A–E) were grown in LB medium to mid-log phase and imaged. Panels F and G show phase contrast images of the Δrny mutant (F) and the same mutant expressing wild-type RNase E without the GFP moiety (strain SSB500).
RNase Y and RNase E can cleave at the same site in vitro
Depletion of RNase Y leads to an increase of many 5′ UTR encoded riboswitches including the thrS leader (17) which contains the elements required for tRNA mediated antitermination (40). We have previously shown that an E. coli RNase E degradosome preparation as well as B. subtilis RNases J1 and J2 can cleave in vitro upstream of the thrS leader terminator at the same site observed in vivo (41–43).
This prompted us to use the thrS 5′ leader sequence (279 nt) to compare the in vitro cleavage specificity of all three enzymes, RNase Y, the catalytic N-terminal half of RNase E (aa 1–529) and RNase J1. As shown in Figure 4, all three enzymes can cleave at the same site, in a single stranded sequence 6–9 nucleotides upstream of the leader terminator structure (Figure 4B). This corroborates previous observations that suggested a similar cleavage specificity for RNase Y and RNase E on the one hand (12) and RNase E and RNase J on the other hand (35,43).
Figure 4.
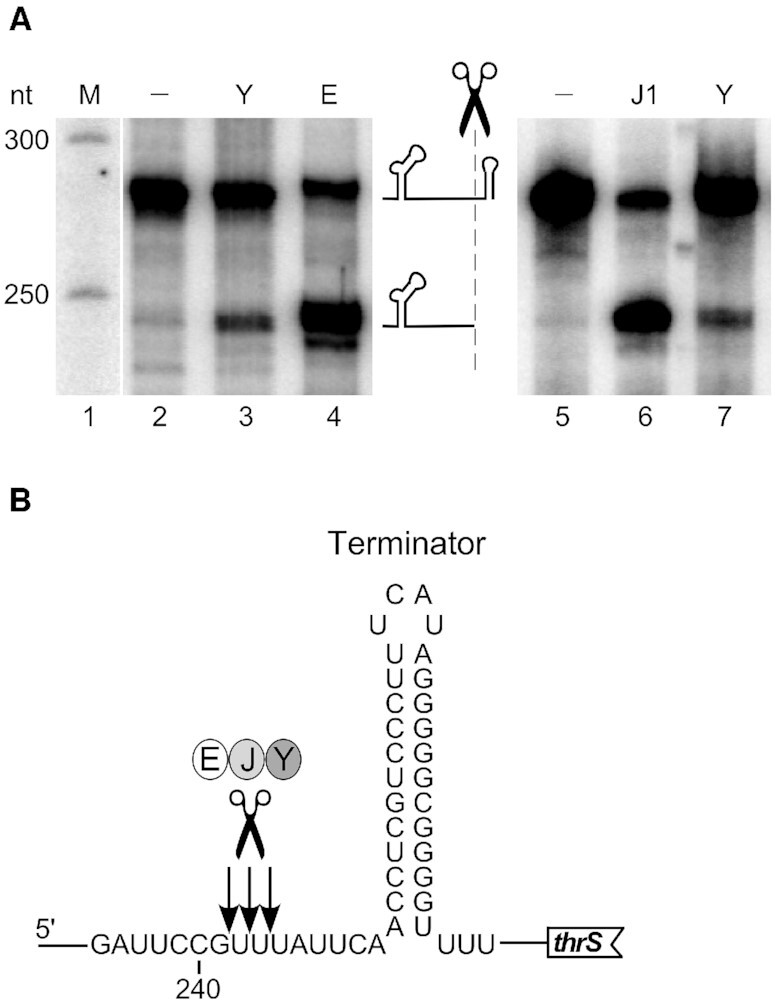
In vitro cleavage specificity of RNases Y, E and J. (A) Cleavage of 5′ end-labeled monophosphorylated thrS leader mRNA by purified B. subtilis RNase Y (Y), E. coli RNase E 529 (E) and B. subtilis RNase J1 (J1). The major cleavage site upstream of the leader terminator is indicated by a scissors symbol. Products were resolved on a 5% PAGE. M = marker. (B) Sequence context around the major cleavage site. Nucleotide numbering is with respect to the transcription start of the thrS gene. Arrows indicate the precise processing sites previously determined for RNase J1 (43).
Transcriptome analysis of Δrny mutants expressing different RNase E versions
In order to obtain a global view of how RNase E expression might compensate for the altered transcription patterns of an RNase Y null mutant we sequenced the transcriptome of the Δrny, WT, Δrny + Pxyl-rne(1061), Δrny + Pxyl-rne(ΔMTS), Δrny + Pxyl-rne(688) and Δrny + Pxyl-rne(529) strains (three replicates for each strain), using stranded RNA-Seq. A total of ∼386 million raw sequencing reads was obtained from the transcriptomic analysis of 18 samples, with an average per sample of ∼20-million reads (Supplementary Table S1) mapped to the B. subtilis genome.
The presence of reads corresponding to the various E. coli rne gene constructs integrated at the amyE locus confirmed the successful expression of the full-length and shortened versions of RNase E (Figure 5A).
Figure 5.

Effect of RNase E expression on the transcriptome of the Δrny strain. (A) Browser view of RNA-Seq reads showing the integration and expression of the E. coli rne gene versions inserted at the amyE locus on the B. subtilis genome. Reads from a single replicate of each strain are shown as representative examples. The arrow represents the E. coli rne coding sequence and its orientation on the genome, the red rectangle indicates the position of the MTS domain. (B) Multidimensional scaling (MDS) plot of Δrny, Δrny + Pxyl-rne(1061), Δrny + Pxyl-rne(ΔMTS), Δrny + Pxyl-rne(688), Δrny + Pxyl-rne(529) and WT triplicate libraries. Distance between samples is based on the leading log2 fold-change (FC) of the top 500 most differentially regulated genes. (C) MA plots of differentially expressed genes (red dots) at FDR ≤ 0.05 identified in the mutant and complemented strains compared to WT. The ordinate represents log2-fold change.
To explore differential gene expression between all strains, we measured the distance of gene expression values between any pair of samples. Our datasets included the analysis of 3932 RNA features including open reading frames (ORFs) and untranslated regions (UTRs, riboswitches, small RNAs) derived from the chromosome and retrieved from Subtiwiki (34). On a multidimensional scaling (MDS) plot (Figure 5B), the triplicates within each group (strain) clustered closely, indicating similar gene expression patterns and consistency between biological replicates. A certain degree of dissimilarity was instead observed among the different groups. The Δrny and Δrny + Pxyl-rne(529) strains were very close on the MDS plot indicating similar expression levels (Figure 5B). Together with the Δrny + Pxyl-rne(ΔMTS) strain they were clearly separated from the WT samples. By contrast, the Δrny mutants expressing Pxyl-rne(1061) and Pxyl-rne(688) clustered separately but close to the WT (Figure 5B). This suggested that localization to the membrane mediated by the MTS sequence is important for RNase E when substituting for RNase Y in vivo. These observations are corroborated by the MA plots of the expression data (Figure 5B) which relate the ratio of level counts for each RNA feature between WT and mutant strains against the average level counts for each feature from all libraries. We applied a cutoff of log2-FC ≥1 and ≤−1 at a FDR ≤0.05 to call for differentially expressed gene (DEGs) features (red dots above and below the blue line in Figure 5C) and we categorized them into functional groups according to Gene Ontology (GO) annotation (Supplementary Tables S2 and S3). Compared to WT, the Δrny and the Δrny strains expressing RNase E ΔMTS or RNase E 529 had very similar numbers of DEGs (Table I): 1089, 1130 and 1149 upregulated features and 575, 500 and 789 downregulated features, respectively. Among the upregulated transcripts identified the majority were in common with the Δrny mutant, 73% for RNase E ΔMTS and 63% for RNase 529.
By contrast, expression of RNase E 1061 and RNase E 688 strongly reduced the number of upregulated features caused by the Δrny phenotype to 368 and 537, of which 27% and 54%, respectively, were common to the Δrny mutant (Table 1).
Table 1.
Number of up- and downregulated transcripts with a log2 fold-change (FC) ≥ 1 or ≤–1 (FDR ≤ 0.05) in mutants strains compared to WT
| Strain, relevant genotype | No. upregulated transcripts | No. downregulated transcripts |
|---|---|---|
| SSB508 Δrny | 1089 | 575 |
| SSB500 Δrny + rne(1061) | 368 | 662 |
| SSB498 Δrny + rne(688) | 536 | 537 |
| SSB502 Δrny + rne(529) | 1130 | 500 |
| SSB572 Δrny + rne(1061-ΔMTS) | 1149 | 789 |
To take subsets of the most relevant genes within this initial set of DEGs, we focused on the up-regulated genes of the Δrny mutant. An increased mRNA level in the mutant is often an indication of the direct action of the RNase on the substrate RNA while down-regulated mRNAs are in large part caused by indirect effects as observed in most RNase related transcriptome studies. Of the 1089 RNA features up-regulated in the Δrny mutant, 759 transcripts were restored to WT levels in the strain expressing E. coli wild type RNase E (Supplementary Table S1). 43% of them (326 in total) belonged to the GO categories ‘undefined’, ‘unknown,’ and ‘general function prediction’. Other well represented functional categories concern ‘amino acids transport and metabolism’ (∼6%), ‘transcription’ (∼6%) and ‘cell wall/membrane’ (∼5%). Genes involved in the initiation of DNA replication (dnaA, dnaB, dnaE,dnaX), in biosynthesis of teichoic acid (tagG, tagB, tagH, tagO) and in translation (rpsO, rpsB) were among 52 essential genes that were upregulated in the Δrny mutant. Of these 32, 30, 15 and 13 transcripts were restored to WT levels upon expression of wild-type RNase E, RNase 688, RNase E ΔMTS and RNase E 529, respectively. Even though there is no direct link, these results are consistent with the capacity of the individual proteins to restore growth (Figure 2A).
Interestingly, among the ‘complemented’ 759 mRNAs, the levels of 342 transcripts depended on the presence of the MTS domain only, since they were up-regulated in the ΔMTS strain and the E529 strain. RNase E (688) which lacks the degradosome scaffold was also quite efficient in replacing RNase Y by restoring more than half (553) of the upregulated RNAs in the Δrny mutant. This contrasts with the two cytoplasmically located RNase E variants (ΔMTS and 529) whose global transcript profiles resemble that of the Δrny mutant (Figure 5C and Table 1).
A similar pattern emerged when we analyzed 40 non coding RNAs, mostly riboswitches and small RNAs, that were upregulated in the Δrny mutant (Supplementary Table S4). A majority of them (22) were reduced to wild type levels in the presence of full-length RNase E, among which the levels of 12 transcripts depended on the presence of the MTS domain only and 9 on both the degradosome scaffold and the MTS. RNase E 688 and cytoplasmic RNase E ΔMTS and 529 restored normal levels only for 14, 9 and 7, respectively, of the upregulated RNAs (Supplementary Table S4).
The impact of RNase Y depletion on antisense RNAs was generally less important compared to translated RNAs, in agreement with previous transcriptomic work (17). When we examined the RNA-seq coverage of antisense RNAs (Supplementary Table S5), we found that 211, 217 and 224 transcripts were increased in the Δrny, the RNase E ΔMTS and RNase E 529 strains, respectively, but only 89 and 151 antisense RNAs were upregulated in the mutant expressing membrane bound wild-type RNase E or RNase E 688.
Compensation of RNase Y by RNase E in modulating specific transcripts levels in vivo
We carried out Northern blots of specific transcripts to confirm the RNA-Seq data and to obtain a more precise picture to what extent E. coli RNase E can restore the patterns and levels of specific transcripts altered by the absence of RNase Y (Figure 6). In the first example, we looked at the thrS leader also used for the in vitro activity assays. As expected, the prematurely terminated leader transcript is almost undetectable in the wild type strain but accumulated in the Δrny strain. In good agreement with the RNA-Seq profile (Figure 6, right panel) full-length RNase E was very efficient in compensating RNase Y and reduce the 279 nt leader transcript to a very low level. The other three RNase E versions tested were also able to reduce the high thrS leader transcript level caused by the absence of RNase Y, but less efficiently. Cleavage of the thrS leader mRNA has previously been used as an assay to identify RNases J1/J2 (43) and generally RNase J is more efficient in cleaving this substrate in vitro than RNase Y (Figure 4A). We therefore analyzed which of the two enzymes contributes most to the cleavage of the prematurely terminated 5′ UTR in vivo. Quantification of the thrS leader levels showed a 29-fold increase in the Δrny mutant compared to a wild type strain (Figure 6, thrS, lower panel). By contrast, in a rnjA/rnjB double mutant and a rnjB mutant strain expressing no RNase J or only RNase J1 the thrS leader is only induced 4- and 3-fold, respectively (Figure 6, thrS). This indicates that in vivo RNase Y is the major enzyme involved in the decay initiating cleavage of the terminated thrS 5′ UTR (> 85%).
Figure 6.

Effect of E. coli RNase E expression on transcription profiles of coding and non-coding RNAs in a Δrny mutant. The left panels show a Northern analysis of total RNA isolated from WT, Δrny and Δrny strains expressing full-length E. coli RNase E (E FL), a cytoplasmic version of the full-length enzyme (E ΔMTS) and shorter RNase E proteins (E 688 and E 529). The blots were hybridized to specific riboprobes complementary to the RNA regions indicated by a green arrow below the corresponding RNAseq profiles (right panels). The curves representing the RNA-Seq coverage expressed as RPM of the various strains follow the color code presented on top of the first graph (thrS).
The znuA (adcA) gene encodes a zinc-binding lipoprotein which together with the products of the two downstream genes znuC and znuB constitutes an ABC transporter responsible for zinc uptake (44). The 400 3′ proximal nucleotides of the znuA mRNA are transcribed as a non coding RNA from an internal sigma A promoter which, as a direct substrate of RNase Y, is only detected in the absence of RNase Y (17). Similarly, to what we observed for the thrS leader, full-length RNase E can completely replace RNase Y and restore the transcription pattern of the wild type strain (Figure 6, znuA). RNase E 688 lacking the degradosome scaffold is also very efficient in cleaving this RNA, significantly better than the two cytoplasmic RNase E proteins (ΔMTS and 529).
A similar pattern was also observed for the gltA-gltB bi-cistronic mRNA encoding the two subunits of glutamate synthase (Figure 6, gltA), the tagD mRNA (Figure 6, tagD) and the SAM riboswitch leader transcript of the yitJ gene (Figure 6, yitJ). However, the accumulation of the 2.2 kb cggR-gapA transcript caused by the absence of RNase Y processing could only be reversed by the full-length cytoplasmic version of RNase E and to some extent by the RNase E 688 construct (Figure 6, gapA).
DISCUSSION
A principal contribution of this study is the observation that E. coli RNase E can effectively replace RNase Y in B. subtilis. The extent of this compensation is quite surprising. We have shown that wild type RNase E can rescue the poor growth phenotype of a mutant lacking RNase Y. The presence of the RNase E degradosome scaffold is not crucially important for the capacity of RNase E to complement for RNase Y. RNase E 688 restored growth almost as efficiently as full-length RNase E and while the latter reduced 70% of the upregulated transcripts in the rny mutant to wild type levels, RNase E 688 still compensated for RNase Y in over 50% of these transcripts. We do not know whether an E. coli like degradosome can form in B. subtilis. However, the high variability and apparently independent acquisition of microdomains serving as binding sites for e.g. PNPase in different bacteria (45,46) are not in favor of the formation of such a complex in B. subtilis.
On the other hand, membrane localization of RNase E is likely the single most important parameter that determines the overall efficiency with which the E. coli enzyme compensates for RNase Y in B. subtilis. RNase E ΔMTS and RNase E 529 clearly localize in the cytoplasm indicating that the amphipathic helix of the MTS is required and sufficient to tether E. coli RNase E to the inner membrane in B. subtilis as in E. coli. Full-length RNase E ΔMTS is very inefficient in restoring growth of the rny mutant, even more than RNase E 529 which contains only the catalytic N-terminal half of RNase E. Corroborating this observation, almost half of the upregulated transcripts restored to wild type levels in the presence of RNase E are not responsive to RNase E ΔMTS. The presence or absence of the MTS sequence does not significantly impact the enzymatic parameters of E. coli RNase E in vivo and in vitro (11). The strong decrease in biological activity of cytoplasmic RNase E in vivo is somewhat surprising but reflects a recent similar observation made in the natural host E. coli (11). We also found no correlation between increased mRNA levels and the function or cellular location of encoded proteins. In general, upregulation of translated mRNA, non-coding RNAs and antisense transcripts alike caused by the absence of RNase Y was less efficiently corrected by the cytoplasmic versions of RNase E.
In order to compare the functional activity of the different RNase E versions in vivo we took care to express the genes from identical transcription and translation signals. Nevertheless, the four RNase E proteins were present at very different levels in B. subtilis. From the RNA-Seq data it appears that the mRNA levels of the ΔMTS and the two shorter RNase E forms are significantly lower than for the full-length enzymes. However, we cannot exclude differential protein stabilities in the heterologous host, even in E. coli cytoplasmic RNase E is unstable compared to the wild type membrane tethered protein (11). The high levels of wild-type RNase E that accumulate in B. subtilis could be related to the clearly visible formation of higher order structures (foci) that might confer greater stability to the protein. This accumulation critically depends on the presence of the C-terminal degradosome scaffold as the membrane tethered RNase E688 protein appears highly unstable. We also carried out Western blots comparing anti-RNase E and anti-GFP antibodies on protein extracts from strains expressing the various GFP fusion proteins (Supplementary Figure S1). By comparing the expression levels measured by probing with RNase E and GFP antibodies, respectively, we could determine that the RNase E antibodies detected the truncated RNase E versions with reduced efficacy, but less than ∼2-fold. The presence of the GFP moiety greatly affected protein abundance when compared to the RNase E constructs without GFP. The levels of the full-length GFP constructs were reduced while those of the truncated versions were strongly increased (Supplementary Figure S1). This is not faithfully reflected in the fluorescence microscopy images which were variably exposed to obtain images of comparable intensity to better reveal their cellular localization. Growth of strains containing the GFP constructs grew similarly to strains containing constructs without GFP (data not shown). The fluorescence microscopy images also show that the filamentous phenotype caused by the rny mutation cannot be reversed by the expression of wild type RNase E or any of the truncated versions. This phenotype is not due to the presence of the GFP moiety as cells expressing wild-type RNase E without GFP also grow in chains (Figure 3G). Similarly, natural competence lost in the Δrny mutant is not recovered by expressing RNase E (data not shown).
However, expression levels of RNase E are likely no crucial parameter as the RNase E 688 lacking the degradosome scaffold is expressed much weaker than the wild type and also the RNase E 529 protein (see Figure 2B), yet it supports growth of the Δrny mutant almost as efficiently as wild type RNase E.
Our in vitro data confirm that all three enzymes B. subtilis RNases J1/J2 and Y as well as E. coli RNase E can cleave the thrS 5′ UTR substrate at the same site (Figure 4B). This indicates that common cleavage parameters including a low sequence specificity and secondary structure context have evolved by convergent evolution and are a hallmark of the major RNA decay-initiating enzymes. However, in vivo the situation is likely not so clear-cut. Other factors, as intracellular localization, interaction with other proteins or RNAs and the translation process itself provide ample room to modulate their action in enzyme and/or species-specific ways.
Interestingly, the RNase E proteins containing the MTS sequence tether to the B. subtilis membrane as they do in E. coli and they form foci which appear to be much more prominent than those observed in E. coli (Khemici et al., 2008). It remains to be seen whether these foci also result from transient clustering of RNase E into cooperative degradation bodies as has been proposed in E. coli (10). The efficient replacement of RNase Y by RNase E in B. subtilis was first of all perceptible by recovering wild type growth of a Δrny mutant. This phenotype is accompanied by a quite impressive recovery of wild type levels for hundreds of transcripts. We have analyzed more than ten different mRNAs that are known substrates of RNase Y by Northern analysis. We found that, in almost all cases, induced expression of wild type RNase E was sufficient to restore the altered transcript profile observed in the Δrny strain to the wild type pattern. However, specific constraints exist that differentiate the accessibility of individual cleavage sites, e.g. the cleavage in the cggR mRNA (gapA operon) was only recovered by expressing full-length cytoplasmic RNase E (ΔMTS) but not the membrane tethered wild type enzyme nor the short cytoplasmic form (see Figure 6).
Small non-coding RNAs and many 5′ UTRs containing riboswitches require cleavage by RNase Y to induce their turn-over. Wild-type RNase E was clearly the most efficient in replacing RNase Y (22 out of 40) and about 60% of the restored cleavages required the MTS sequence and thus membrane localization. It is noteworthy that in general all the substrates cleaved by the less efficient RNase E versions ΔMTS, 688 and 529 are a subgroup of the substrates recognized by WT RNase E. This suggests that membrane tethering of RNase E is important for the cleavage of most untranslated RNAs and that cytoplasmic versions of the enzyme cannot recruit additional substrates. A future analysis of a cytoplasmic version of RNase Y will shed more light on how far the analogy with RNase E goes.
We have initially identified RNase Y as well as RNase J1/J2 as endoribonucleases with a cleavage specificity similar to E. coli RNase E (12,43). At present, we know that in B. subtilis RNase Y is undoubtedly the major enzyme initiating global mRNA decay and bulk mRNA is stabilized in its absence (12). However, this does not exclude a role for the endonucleolytic activity of RNase J. In order to directly compare the predicted RNase E-like activity of RNases Y, E and J we chose the thrS leader mRNA as a substrate. This allowed to show that all three ribonucleases can indeed cleave at the same site in a single substrate (Figure 4). We previously found that cyanobacterial orthologs of RNase E and RNase J have an endonucleolytic cleavage specificity that is very similar between them and also compared to the orthologous enzymes in E. coli and B. subtilis (35). Together, our in vitro data support the view that the three totally unrelated ribonucleases Y, E and J have evolved independently towards a common activity and been maintained in bacteria throughout evolution. The presence of more than one of these nucleases in a bacterium obviously complicates the analysis of the individual contribution of each enzyme to RNA metabolism. In the case of the prematurely terminated thrS leader transcript we could show that in vivo RNase Y is the principal enzyme initiating the degradation of this short RNA, whereas RNase J1/J2 accounts for less than 20% of this activity (Figure 6). We cannot yet conclude on which of the two enzymes is primarily involved in cleaving the same RNA in the context of the longer thrS read-through transcript which might occur in a different structural context, i.e. the antiterminator conformation.
The simple presence of two or more of the three major decay initiating enzymes RNase Y, E and J therefore does not allow to draw any conclusions on their respective importance in mRNA metabolism. For example, in S. aureus the global contribution of RNase Y in mRNA processing/degradation is much more limited than in B. subtilis (17,47,48). RNase J has a much greater effect than RNase Y on mRNA steady state levels in this organism (49).
There exists a variety of parameters that obviously play a role in coordinating and regulating initiation of mRNA decay, including RNA secondary structure, formation of degradosome-like complexes, intracellular localization of substrates and enzymes. The observation that RNase E can substitute quite efficiently for RNase Y in B. subtilis paves the way for further experiments that will allow to study and compare two completely unrelated key ribonucleases in vivo in the same cellular context. This should shed light on evolutionarily conserved parameters that govern initiation of bacterial mRNA decay.
DATA AVAILABILITY
The NCBI Sequence Read Archive (SRA) submission number is SUB8491357
Supplementary Material
ACKNOWLEDGEMENTS
We thank Lena Zig for technical assistance at the onset of this study.
Notes
Present address: Sylvain Roque, CNRS, UMR5237, Centre de Recherche en Biologie Cellulaire de Montpellier, 34293 Montpellier, France.
Contributor Information
Soumaya Laalami, CNRS, UMR8261, Institut de Biologie Physico-Chimique, Université de Paris, 75005 Paris, France.
Marina Cavaiuolo, CNRS, UMR8261, Institut de Biologie Physico-Chimique, Université de Paris, 75005 Paris, France.
Sylvain Roque, CNRS, UMR8261, Institut de Biologie Physico-Chimique, Université de Paris, 75005 Paris, France.
Carine Chagneau, CNRS, UMR8261, Institut de Biologie Physico-Chimique, Université de Paris, 75005 Paris, France.
Harald Putzer, CNRS, UMR8261, Institut de Biologie Physico-Chimique, Université de Paris, 75005 Paris, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Centre National de la Recherche Scientifique [UMR 8261]; Université de Paris; Agence Nationale de la Recherche [RNAJAY and IB-mRND]; ‘Initiative d’Excellence’ program from the French State [‘DYNAMO,’ ANR-11-LABX-0011-01]. Funding for open access charge: CNRS [UMR8261].
Conflict of interest statement. None declared.
REFERENCES
- 1. Laalami S., Zig L., Putzer H.. Initiation of mRNA decay in bacteria. Cell. Mol. Life Sci.: CMLS. 2014; 71:1799–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Apirion D., Lassar A.B.. A conditional lethal mutant of Escherichia coli which affects processing of ribosomal RNA. J. Biol. Chem. 1978; 253:1738–1742. [PubMed] [Google Scholar]
- 3. Ono M., Kuwano M.. A conditional lethal mutation in an E. coli strain with a longer chemical lifetime of messenger RNA. J. Mol. Biol. 1979; 129:343–357. [DOI] [PubMed] [Google Scholar]
- 4. Carpousis A.J., Van Houwe G., Ehretsmann C., Krisch H.M.. Copurification of E. coli RNAse E and PNPase: evidence for a specific association between two enzymes important in mRNA processing and degradaion. Cell. 1994; 76:889–900. [DOI] [PubMed] [Google Scholar]
- 5. Py B., Higgins C.F., Krisch H.M., Carpousis A.J.. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature. 1996; 381:169–172. [DOI] [PubMed] [Google Scholar]
- 6. Hui M.P., Foley P.L., Belasco J.G.. Messenger RNA degradation in bacterial cells. Annu. Rev. Genet. 2014; 48:537–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mackie G.A. RNase E: at the interface of bacterial RNA processing and decay. Nature reviews. Microbiology. 2013; 11:45–57. [DOI] [PubMed] [Google Scholar]
- 8. Vanzo N.F., Li Y.S., Py B., Blum E., Higgins C.F., Raynal L.C., Krisch H.M., Carpousis A.J.. Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome. Genes Dev. 1998; 12:2770–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khemici V., Poljak L., Luisi B.F., Carpousis A.J.. The RNase E of Escherichia coli is a membrane-binding protein. Mol. Microbiol. 2008; 70:799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Strahl H., Turlan C., Khalid S., Bond P.J., Kebalo J.M., Peyron P., Poljak L., Bouvier M., Hamoen L., Luisi B.F.et al.. Membrane recognition and dynamics of the RNA degradosome. PLos Genet. 2015; 11:e1004961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hadjeras L., Poljak L., Bouvier M., Morin-Ogier Q., Canal I., Cocaign-Bousquet M., Girbal L., Carpousis A.J.. Detachment of the RNA degradosome from the inner membrane of Escherichia coli results in a global slowdown of mRNA degradation, proteolysis of RNase E and increased turnover of ribosome-free transcripts. Mol. Microbiol. 2019; 111:1715–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shahbabian K., Jamalli A., Zig L., Putzer H.. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J. 2009; 28:3523–3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hunt A., Rawlins J.P., Thomaides H.B., Errington J.. Functional analysis of 11 putative essential genes in Bacillus subtilis. Microbiology. 2006; 152:2895–2907. [DOI] [PubMed] [Google Scholar]
- 14. Lewis P.J., Thaker S.D., Errington J.. Compartmentalization of transcription and translation in Bacillus subtilis. EMBO J. 2000; 19:710–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mascarenhas J., Weber M.H., Graumann P.L.. Specific polar localization of ribosomes in Bacillus subtilis depends on active transcription. EMBO Rep. 2001; 2:685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Durand S., Gilet L., Bessieres P., Nicolas P., Condon C.. Three essential ribonucleases-RNase Y, J1, and III-control the abundance of a majority of Bacillus subtilis mRNAs. PLos Genet. 2012; 8:e1002520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Laalami S., Bessieres P., Rocca A., Zig L., Nicolas P., Putzer H.. Bacillus subtilis RNase Y activity in vivo analysed by tiling microarrays. PLoS One. 2013; 8:e54062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lehnik-Habrink M., Schaffer M., Mader U., Diethmaier C., Herzberg C., Stulke J.. RNA processing in Bacillus subtilis: identification of targets of the essential RNase Y. Mol. Microbiol. 2011; 81:1459–1473. [DOI] [PubMed] [Google Scholar]
- 19. Commichau F.M., Rothe F.M., Herzberg C., Wagner E., Hellwig D., Lehnik-Habrink M., Hammer E., Völker U., Stülke J.. Novel activities of glycolytic enzymes in Bacillus subtilis: interactions with essential proteins involved in mRNA processing. Mol. Cell. Proteomics. 2009; 8:1350–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lehnik-Habrink M., Pfortner H., Rempeters L., Pietack N., Herzberg C., Stülke J.. The RNA degradosome in Bacillus subtilis: identification of CshA as the major RNA helicase in the multiprotein complex. Mol. Microbiol. 2010; 77:958–971. [DOI] [PubMed] [Google Scholar]
- 21. Gao J., Lee K., Zhao M., Qiu J., Zhan X., Saxena A., Moore C.J., Cohen S.N., Georgiou G.. Differential modulation of E. coli mRNA abundance by inhibitory proteins that alter the composition of the degradosome. Mol. Microbiol. 2006; 61:394–406. [DOI] [PubMed] [Google Scholar]
- 22. Cascante-Estepa N., Gunka K., Stulke J.. Localization of components of the RNA-degrading machine in Bacillus subtilis. Front. Microbiol. 2016; 7:1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Newman J.A., Hewitt L., Rodrigues C., Solovyova A.S., Harwood C.R., Lewis R.J.. Dissection of the network of interactions that links RNA processing with glycolysis in the Bacillus subtilis degradosome. J. Mol. Biol. 2012; 416:121–136. [DOI] [PubMed] [Google Scholar]
- 24. Redder P. Molecular and genetic interactions of the RNA degradation machineries in Firmicute bacteria. Wiley Interdiscipl. Rev. RNA. 2018; 9:e1460-11. [DOI] [PubMed] [Google Scholar]
- 25. Adusei-Danso F., Khaja F.T., DeSantis M., Jeffrey P.D., Dubnau E., Demeler B., Neiditch M.B., Dubnau D.. Structure-function studies of the Bacillus subtilis Ric proteins identify the Fe-S cluster-ligating residues and their roles in development and RNA processing. mBio. 2019; 10:e01841-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carabetta V.J., Tanner A.W., Greco T.M., Defrancesco M., Cristea I.M., Dubnau D.. A complex of YlbF, YmcA and YaaT regulates sporulation, competence and biofilm formation by accelerating the phosphorylation of Spo0A. Mol. Microbiol. 2013; 88:283–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. DeLoughery A., Dengler V., Chai Y., Losick R.. Biofilm formation by Bacillus subtilis requires an endoribonuclease-containing multisubunit complex that controls mRNA levels for the matrix gene repressor SinR. Mol. Microbiol. 2016; 99:425–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DeLoughery A., Lalanne J.B., Losick R., Li G.W.. Maturation of polycistronic mRNAs by the endoribonuclease RNase Y and its associated Y-complex in Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E5585–E5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hamouche L., Billaudeau C., Rocca A., Chastanet A., Ngo S., Laalami S., Putzer H.. Dynamic membrane localization of RNase Y in Bacillus subtilis. mBio. 2020; 11:e03337-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tamura M., Kageyama D., Honda N., Fujimoto H., Kato A.. Enzymatic activity necessary to restore the lethality due to Escherichia coli RNase E deficiency is distributed among bacteria lacking RNase E homologues. PLoS One. 2017; 12:e0177915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bhavsar A.P., Zhao X., Brown E.D.. Development and characterization of a xylose-dependent system for expression of cloned genes in Bacillus subtilis: conditional complementation of a teichoic acid mutant. Appl. Environ. Microbiol. 2001; 67:403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mora L., Ngo S., Laalami S., Putzer H.. In vitro study of the major Bacillus subtilis ribonucleases Y and J. Methods Enzymol. 2018; 612:343–359. [DOI] [PubMed] [Google Scholar]
- 33. Putzer H., Brackhage A.A., Grunberg-Manago M.. Independent genes for two threonyl-tRNA synthetases in Bacillus subtilis. J. Bacteriol. 1990; 172:4593–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhu B., Stulke J.. SubtiWiki in 2018: from genes and proteins to functional network annotation of the model organism Bacillus subtilis. Nucleic Acids Res. 2018; 46:D743–D748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cavaiuolo M., Chagneau C., Laalami S., Putzer H.. Impact of RNase E and RNase J on global mRNA metabolism in the Cyanobacterium synechocystis PCC6803. Front. Microbiol. 2020; 11:1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P.. Integrative genomics viewer. Nat. Biotechnol. 2011; 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Putzer H., Condon C., Brechemier-Baey D., Brito R., Grunberg-Manago M.. Transfer RNA-mediated antitermination in vitro. Nucleic Acids Res. 2002; 30:3026–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Condon C., Putzer H., Grunberg-Manago M.. Processing of the leader mRNA plays a major role in the induction of thrS expression following threonine starvation in Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:6992–6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Condon C., Putzer H., Luo D., Grunberg-Manago M.. Processing of the Bacillus subtilis thrS leader mRNA is RNase E- dependent in Escherichia coli. J. Mol. Biol. 1997; 268:235–242. [DOI] [PubMed] [Google Scholar]
- 43. Even S., Pellegrini O., Zig L., Labas V., Vinh J., Brechemmier-Baey D., Putzer H.. Ribonucleases J1 and J2: two novel endoribonucleases in B. subtilis with functional homology to E. coli RNase E. Nucleic Acids Res. 2005; 33:2141–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shin J.H., Helmann J.D.. Molecular logic of the Zur-regulated zinc deprivation response in Bacillus subtilis. Nat. Commun. 2016; 7:12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ait-Bara S., Carpousis A.J.. RNA degradosomes in bacteria and chloroplasts: classification, distribution and evolution of RNase E homologs. Mol. Microbiol. 2015; 97:1021–1135. [DOI] [PubMed] [Google Scholar]
- 46. Ait-Bara S., Carpousis A.J., Quentin Y.. RNase E in the gamma-Proteobacteria: conservation of intrinsically disordered noncatalytic region and molecular evolution of microdomains. Mol. Genet. Genomics: MGG. 2015; 290:847–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Khemici V., Prados J., Linder P., Redder P.. Decay-initiating endoribonucleolytic cleavage by RNase Y is kept under tight control via sequence preference and sub-cellular localisation. PLos Genet. 2015; 11:e1005577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marincola G., Schafer T., Behler J., Bernhardt J., Ohlsen K., Goerke C., Wolz C.. RNase Y of Staphylococcus aureus and its role in the activation of virulence genes. Mol. Microbiol. 2012; 85:817–832. [DOI] [PubMed] [Google Scholar]
- 49. Linder P., Lemeille S., Redder P.. Transcriptome-wide analyses of 5'-ends in RNase J mutants of a gram-positive pathogen reveal a role in RNA maturation, regulation and degradation. PLoS Genet. 2014; 10:e1004207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Callaghan A.J., Marcaida M.J., Stead J.A., McDowall K.J., Scott W.G., Luisi B.F.. Structure of Escherichia coli RNase E catalytic domain and implications for RNA turnover. Nature. 2005; 437:1187–1191. [DOI] [PubMed] [Google Scholar]
- 51. Bandyra K.J., Luisi B.F.. RNase E and the high-fidelity orchestration of RNA metabolism. Microbiol Spectr. 2018; 6:doi:10.1128/microbiolspec.RWR-0008-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The NCBI Sequence Read Archive (SRA) submission number is SUB8491357