

Abstract
This expert opinion originally developed by a panel of the Italian Society of Thalassemias and Hemoglobinopathies (SITE), reviewed and adopted by the European Hematology Association (EHA) through the EHA Scientific Working Group on Red Cells and Iron, has been developed as priority decision-making algorithm on evidence and consensus with the aim to identify which patients with transfusion-dependent beta-thalassemia (TDT) could benefit from a gene therapy (GT) approach. Even if the wide utilized and high successful allogeneic hematopoietic stem-cell transplantation provides the possibility to cure several patients a new scenario has been opened by GT. Therefore, it is important to establish the patients setting for whom it is priority indicated, particularly in the early phase of the diffuse use outside experimental trials conducted in high selected centers. Moreover, actual price, limited availability, and resources disposal constitute a further indication to a rational and progressive approach to this innovative treatment. To elaborate this algorithm, the experience with allogeneic transplantation has been used has a predictive model. In this large worldwide experience, it has been clearly demonstrated that key for the optimal transplant outcome is optimal transfusion and chelation therapy in the years before the procedure and consequently optimal patient’s clinical condition. In the document, different clinical scenarios have been considered and analyzed for the possible impact on treatment outcome. According to the European Medicine Agency (EMA) for the GT product, this expert opinion must be considered as a dynamic, updatable, priority-based indications for physicians taking care of TDT patients.
Introduction
The Società Italiana Talassemie ed Emoglobinopatie (Italian Society of Thalassemias and Hemoglobinopathies [SITE]) has developed this expert opinion based on multidisciplinary discussions of a panel of experts to provide guidance on the identification and selection of patients with transfusion-dependent beta-thalassemia (β-TDT) who could benefit from gene therapy. The European Hematology Association (EHA), through the EHA Scientific Working Group (SWG) on Red Cells and Iron, has furthered the initiative through an international panel of reviewers and adopted these suggestions. This is the second version, March 2020; translated and edited, August 2020.
Currently, allogeneic transplantation of hematopoietic stem cells is the only curative and most widely used therapy treatment for β-TDT. However, recent trials of gene therapy have reported very promising results in terms of overall survival1–3 and thalassemia-free survival and are opening a new landscape of treatment. This algorithm for the selection of patients suitable for gene therapy and the supporting notes were formulated by consensus review after an evaluation of currently available scientific evidence using validated criteria. The evidence was interpreted with caution because clinical trial experience of gene therapy is currently limited, a conventional treatment is available for patients with β-TDT and the availability of gene therapy will, at least initially, be quite limited.
Clinical experience of allogeneic transplantation in β-TDT, which began in 1981, immediately showed the importance of patient risk stratification to achieve the best results4–6 (see the Pesaro experience and their classification of patients according to risk). Published data in the literature and the recent analysis of clinical evidence by the European Registry of Hemoglobinopathies of a large number of patients (2011 and 2018 analyses) confirm that young patient age (<14 yrs) and the availability of a human leukocyte antigen (HLA)-identical family donor are factors that offer the best outcome from allogeneic transplantation.6–10
Current knowledge of, and experience with, nonconventional treatments, such as allogeneic transplantation and gene therapy, are discussed to identify the best available treatment and indication for these patients according to their characteristics.
At this point in time, when we can see the emergence of “the age of gene therapy” and recent preliminary results on gene editing are promising,11 it is essential to establish the optimal patient setting in which gene therapy can be applied, or better, to define the setting that represents the most suitable indication for gene therapy, identify the patients who should have clinical priority for access to the procedure, and set out requirements and recommendations for the identification of qualified treatment centers for gene therapy. When considering changes to the treatment of patients with β-TDT, including gene therapy, it is essential that a detailed consultation is held with the patient and their caregiver/family to discuss all possible risks and potential benefits from the treatment. Discussion of this aspect of care is outside the scope of this expert opinion but remains an important element of patient care.
Methods
The current expert opinion describes the different clinical problems with practical conclusions on each aspect of the subject, and is summarized by a decision-making algorithm (Figure 1). A Supplemental Digital Appendix, http://links.lww.com/HS/A156, is also included in the supplementary information, providing details on the requirements and recommendations for the identification of qualified gene therapy treatment centers.
Figure 1.
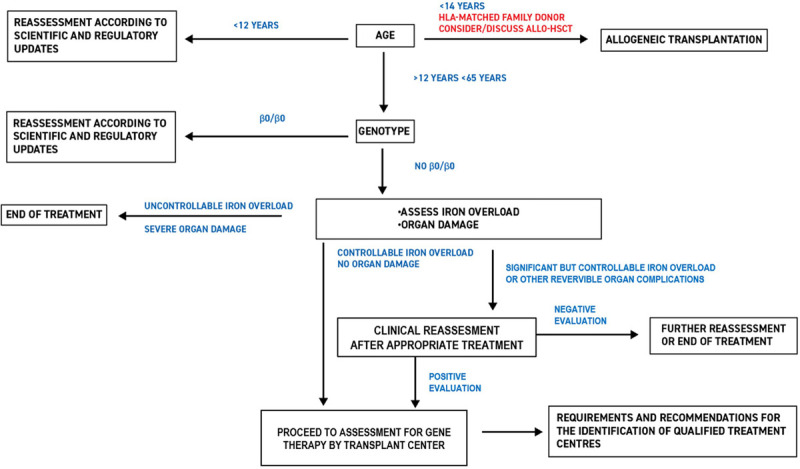
Algorithm for the selection of transfusion-dependent β-thalassemia patients for gene therapy.
The decision-making algorithm was developed by the SITE group, who defined the project and selected a multidisciplinary group of experts in hemoglobinopathies and/or transplantation to discuss the selection of β-TDT patients for gene therapy and draw up notes on the related clinical problems. The EHA, through the EHA SWG on Red Cells and Iron, has furthered the initiative through an international panel of reviewers and adopted these suggestions. The expert opinion has been prepared to be used by specialists in the centers of the Networks of Hemoglobinopathies.
The published literature (Medline, PubMed, Embase, and Cochrane Library) was searched for high-quality evidence to define the best candidates for allogeneic transplantation who should not undergo gene therapy. The keywords used were beta-thalassemia; bone marrow transplantation; gene therapy; hematopoietic stem cell (HSC) transplantation; hemoglobinopathies; hepatitis; iron overload; liver complications; endocrine complications.
The literature evaluation/scientific evidence was reported and discussed by the SITE expert panel. The final version of the document was then assessed by a pool of external reviewers with modifications made as appropriate and the final version of the expert opinion will be uploaded onto the SITE website (www.site-italia.org). It is intended that the results of the process will be collected in the clinical electronic database used by the centers (eg, Webthal, International Health Repository) to carry out stratification.
The SITE group would like to emphasize the approach used for evaluating β-TDT patients’ access to gene therapy. Until a few decades ago, β-TDT was considered an unfavorable pathology. Today, however, β-TDT has an open prognosis thanks to advances in conventional treatment that have transformed it into a chronic disease. It was therefore decided, in this first phase of access to gene therapy, to give priority to patients in the best clinical condition who, as shown in the allogeneic transplantation setting, are those who are likely to obtain the most clinical benefit with the least risk. As mentioned previously this cautious approach is due to the limited clinical experience obtained during clinical trials and due to the probable limited availability of gene therapy, at least initially. The patient priority criteria for access to gene therapy defined in this algorithm and supporting notes could vary as new scientific evidence emerges, and therefore, the expert opinion will be reviewed regularly and updated when new clinical data are published and/or any changes are made to the European Medicines Agency (EMA) license for gene therapy.
Gene therapy: suitability requirements for patients with β-TDT
Suitable patients for gene therapy are included as follows:
Patients with β-thalassemia who are transfusion dependent.
Given the seriousness of their disease, many patients affected by β-thalassemia depend on a chronic regimen of blood transfusions both for their survival and to prevent complications associated with the disease. Transfusion dependence is defined as no transfusion-free period of >6 weeks in the last 2 years or the transfusion of a minimum 100 mL/kg of concentrated red blood cells/year in the last 2 years.2
Patients with β-TDT who are followed at “Centers experienced in the treatment of beta thalassaemia” according to the EMA prescription or at “Hemoglobinopathies Reference Centers” for the countries, like Italy, where a specialized network, recognized by institutions, exists.12,13 In consideration that not all countries have such specialist centers, we recommend that a summary of the below described information must be available.
At hemoglobinopathies specialized centers, it is possible to track clinical data, particularly data related to transfusion. At these centers, the patient also has a preliminary interview during which the available therapeutic options and the proposed procedures are discussed. The transplant center will carry out the final interview and final assessment and collect the patient’s informed consent form.
It is essential that patients are followed at a hemoglobinopathies specialized center for at least 2 years and that correct registration of medical data is guaranteed, with attention to data concerning comorbidities, laboratory and instrumental test results, and blood transfusions.
Traceability of blood transfusion data is particularly important to calculate annual transfusion and iron intake requirements, to register any alloantibodies and transfusion reactions, and to evaluate the need for any further therapy after gene therapy.
Patient age
Background and discussion
It is now clear that age is a risk factor for clinical outcomes in patients with hemoglobinopathies both during conventional therapy and in a transplant setting. In the 1980s, the Pesaro Group stratified thalassemic patients aged <16 years into 3 different risk groups that had a significant impact on the clinical outcome after transplantation. The statistically significant factors were hepatomegaly (<2 versus >2 cm from the costal margin), hepatic fibrosis (absent versus present), and chelation (regular versus irregular).4,5,14 This last parameter was linked to the use of deferoxamine, the only chelating drug then available and importantly it reflected the concept of duration and exposure to iron overload and toxicity. However, the Pesaro Classification was not predictive of clinical outcomes in adult patients with β-TDT, probably because they had already been exposed to iron chelation for too long. The correlation between the Pesaro Classification and our current understanding of the physiopathology of iron has now been recognized and has received fresh interpretation15,16; in itself, it underlines the pertinence today of the observation reported almost 30 years ago. However, the limited experience of gene therapy in clinical trials to date requires the adoption of a cautious approach when using the Pesaro classification of risk for patients undergoing gene therapy until novel evidence becomes available.
Age is also considered and established by the current regulations in force. In fact, gene therapy using LentiGlobin (Blubird Bio) has received the EMA approval for patients aged >12 years with β-TDT with non-β0/β0 genotype.17
It is important to note that the safety and efficacy of gene therapy in children aged <12 years has not been established.
Concerning children under the age of 12 they cannot be considered in this expert opinion because current EMA regulation excludes them from gene therapy because safety and efficacy of gene therapy in children aged <12 years has not been established. Children indication will require dedicated trials including a sufficient number of patients. It should be noted that the vector “GLOBE” that showed promising results in young children18 is actually not registered by any regulatory authority worldwide.
The upper age limit for gene therapy has not been fixed as it depends on the clinical condition (comorbidities/organ damage) of each individual. It is therefore up to the physician to examine the characteristics of their patient and evaluate the clinical priorities to be addressed.
Up to now, limited data are available regarding the impact of the aging process on medullary microenvironment in healthy subjects. Studies carried out in elderly subjects (aged >65 yrs) have shown how mobilization of CD34+ cells with granulocyte-colony stimulating factor can be reduced, but always on the basis of transplant requirements.19–21 In nonanemic older patients aged between 66 and 73 years, 2 independent studies have described a reduction in circulating CD34+ cells compared with younger subjects associated with age-dependent increased serum stem cell factor, indicating an age-dependent change in hematopoietic cytokine(s).22–24 Inadequate erythropoietin (EPO) production has been suggested to contribute to anemia in the elderly, but again limited and not conclusive studies are available. Furthermore, the presence of a comorbidity such as diabetes, arterial hypertension, or kidney failure might further alter the cellular response to EPO.21,25 Finally, bone marrow microenvironment is dynamically changing during aging and it might represent an additional factor to be taken into consideration in identification of candidate patients for gene therapy.26,27
Recommendations
Gene therapy products are today registered over the age of 11 years.
Patients >50 years old must be considered with caution.
We do not recommend gene therapy in patients aged over 55.
Allogeneic transplantation
Background and discussion
The 2 key factors that determine eligibility for allogeneic transplantation and affect clinical outcomes after transplantation are age and the availability of an HLA-identical family donor.
Currently, the European Hemoglobinopathies Registry states that allogeneic transplantation should take place in patients who are <14 years old. The correlation between age and clinical outcome is directly related to the iron-related organ damage that results from long-term repeated blood transfusions, despite the incredible improvements seen in treatment, iron chelation, and other supportive measures.
HLA compatibility is also extremely important in the transplant setting, both because it reduces the immunological complications related to antigenic disparity that results in graft-versus-host disease (GvHD) and because it allows less ablative and immunosuppressive conditioning regimens to be used, therefore reducing the transplant procedure-related toxicity and likelihood of infection complications.
Data from large cohort studies have confirmed that an HLA-identical family donor is the donor profile that guarantees the best outcome in terms of overall survival and thalassemia-free survival. Data from the European Hemoglobinopathies Registry show that patients aged <14 years with an HLA-identical family donor have an overall survival rate of 91.9% and thalassemia-free survival of 86%.9,28 Recently, experiences evaluating transplantation with matched unrelated donors and haploidentical donors in patients with β-TDT have been published with relevant results.8,29,30
Recommendations
Patient age <14 years and HLA-identical family donor are the factors associated with the best transplant outcomes in patients with β-TDT.
Genotype
A patient’s β-globin genotype must be registered on presentation of a report from a National Health accredited laboratory.
The HbVar database (http://globin.cse.psu.edu) can be used to define hematologic expression (β0 o β+ forms).
At present, regulatory authorities do not request studies of alfa, delta, and gamma globin genes. However, the complete molecular globin profile of a patient could be useful in assessing their expected response to therapy.
Recommendations
Identification of a patient’s complete molecular globin profile is strongly advised before starting gene therapy.
Iron overload
Background and discussion
The experience of the Pesaro group showed that constant monitoring of the level of iron accumulation is the determining factor for transplant outcome.10 The Pesaro classification14 (validated for patients < 18 yrs only), which stratifies patients on the basis of 3 independent factors (hepatomegaly, hepatic fibrosis, and length of exposure time to chelating therapy), remains pertinent today because it highlights the importance of monitoring iron accumulation throughout a patient’s lifetime.16,31 Moreover, an elevated LIC (as any elevated iron burden biomarker) is a sign of years of exposure to toxic forms of iron16 and parallel chelation compliance and tissue damage.
According to the formula recently proposed by Coates,15 iron-associated tissue toxicity is due to more than a single factor: the quantity of reactive species (non-transferrin bound iron, labile plasma iron, etc.); genetic antioxidant factors (superoxide dismutase, catalase, and glutathione peroxidase); environmental factors (food antioxidants, other metals such as copper and selenium); and length of time exposed to the toxic effects of iron.
Recent in vitro studies32,33 have shown that accumulation of iron can also influence the medullary bone marrow microenvironment and have a negative impact on both the quality and the quantity of the hematopoietic stem progenitor cells.
In relation to iron accumulation, when considering gene therapy, attention should be drawn to the paragraph in the Zynteglo EMA product approval documentation entitled “Risks associated with TDT and iron overload,” section 4.4 Special warnings and precautions for use in Supplemental Digital Appendix I, http://links.lww.com/HS/A156—Summary Of Product Characteristics used for gene therapy and approved by the EMA34: “Patients with TDT experience iron overload due to chronic red blood cell (RBC) transfusions that can lead to end organ damage. HSC transplantation with myeloablative conditioning is not appropriate for patients with TDT who have evidence of severely elevated iron in the heart i.e., patients with cardiac T2* <10 ms by magnetic resonance imaging (MRI). MRI of the liver should be performed on all patients prior to myeloablative conditioning. It is recommended that patients with MRI results demonstrating liver iron concentration (LIC) ≥15 mg/g undergo liver biopsy for further evaluation. If the liver biopsy demonstrates bridging fibrosis, cirrhosis, or active hepatitis, HSC transplantation with myeloablative conditioning is not appropriate.” Adopting a cautious approach, the SITE feels that more stringent iron accumulation criteria should be used than those indicated by the EMA (≥15 mgFe/g Liver, see above).
Recommendations
Patients with significant iron accumulation (LIC > 7 mgFe/g Liver) should have a “suspended indication” for gene therapy until values return to acceptable limits (LIC < 7 mgFe/g Liver).
Caution should be exercised and iron levels should be strictly monitored after adequate chelation therapy has been set up.
Cardiomyopathy
Background and discussion
In patients with β-TDT, cardiac complications and premature death due to cardiomyopathy represent a serious problem. Iron-related cardiac damage manifests as cardiac insufficiency, arrhythmias, sudden death, or progressive congestive heart failure. Overall, iron-related cardiac complications are the leading cause of death and the biggest cause of morbidity in these patients.35,36
In patients with β-TDT, the outcome of allogeneic transplantation is directly correlated with good performance status, adequate iron chelation without iron-related organ damage, and the absence of comorbidities.37 These same parameters also influence clinical outcomes of gene therapy in this patient population.37
The prevalence and predictive factors for cardiac complications in patients undergoing allogeneic transplantation are not known. In clinical trials of gene therapy in patients with β-TDT, a heart T2* MRI <10 ms and clinically significant pulmonary hypertension are conditions that should exclude access to gene therapy.2 In the light of observations on restrictive myocardiopathy in adult patients with preserved ejection fraction and without myocardial iron overload, the SITE panel consider that these factors should be included in the exclusion criteria.
Recommendations
The following heart conditions should be considered exclusion criteria:
myocardial iron overload T2* MRI <10 ms in the previous 6 months38,39;
pulmonary hypertension (determined by cardiac cathete- rization)40;
serious congestive heart failure (New York Heart Association class III or above);
significant arrhythmia requiring therapy, as defined by the European Heart Rhythm Association (EHRA) guidelines41;
myocardial ischemia in the previous 12 months, as defined by the European Society of Cardiology (ESC) guidelines41;
restrictive myocardiopathy, as defined by ESC guidelines.41
Liver disease
Background and discussion
Follow-up data on outcomes following gene therapy are lacking. It is therefore useful to refer to clinical experience from hematopoietic stem cell transplants in this setting. In 1990, Lucarelli et al5 identified fibrosis and hepatomegaly as factors predictive of a negative transplant outcome and, despite the subsequent controversy concerning the definition of hepatomegaly, these original criteria are still considered valid (ie, hepatomegaly is an expression of hepatic iron accumulation and of the duration of toxic iron exposure).16
Hepatic fibrosis
Hepatic fibrosis is a marker of exposure to iron and to viruses. Patients with liver disease, and particularly those with severe hepatic fibrosis or cirrhosis, are at higher risk of veno-occlusive disease (VOD) after myeloablative conditioning regimens and are also at increased risk of fatal liver failure even with reduced intensity conditioning.42 Assessment of the level of liver fibrosis should ideally be determined for all potential gene therapy candidates before treatment is started.
Hepatitis C virus infection
Data correlating hepatitis C virus (HCV) infection with gene therapy is lacking because patients who test positive for HCV-RNA are not usually enrolled into clinical trials. It is useful, therefore, to refer to the experience from hematopoietic stem cell transplants in patients with β-TDT to guide the selection of gene therapy candidates. HCV infection (ie, HCV-RNA positivity) appears to be clinically relevant in this setting, as hepatitis can worsen after immune reconstitution.43 The risk of death after virus reactivation is 8% and involves the allogeneic transplant setting only. HCV-positivity is therefore generally considered to be a significant risk factor in patients with β-TDT undergoing transplantation; patients with HCV have an increased risk of death after allogeneic stem cell transplantation even if they have normal or almost normal liver function.44 The association between HCV status and the risk of posttransplant VOD remains a subject of debate and is not supported by all experts. However, some experts consider HCV to be an independent risk factor for posttransplant VOD.45,46 An increase in the risk for fatal VOD in HCV-positive patients who had received cyclophosphamide and >12 Gy total body irradiation has been observed, but this seems to be correlated to liver inflammation and fibrosis and to the components of the conditioning regimen rather than the HCV itself.46 Considering the availability of effective and safe antiviral therapies that have demonstrated excellent results also in thalassemia patients, it does not seem justified to consider HCV-RNA-positive subjects as candidates for gene therapy.47–50
Hepatitis B virus infection
As stated previously, due to the lack of data for gene therapy in this setting, it is useful to refer to the experience of hematopoietic stem cell transplant to address this issue. In patients with β-TDT who have undergone hematopoietic stem cell transplant, hepatitis B virus (HBV) virus reactivation can be followed by normalization with seroconversion until HbsAg negativity is achieved in 25% of cases, even without antiviral therapy, while fulminant hepatitis is observed in a small percentage of patients (3%). In most cases, any new acute phase of hepatitis is only light and asymptomatic with a moderate and long-lasting increase in transaminase levels.51 The risk of reactivation of hepatitis B in HbsAg negative and anti-HBc positive subjects is around 6.5%. There are no observed differences between patients who are HBV-DNA positive and those who are HBV-DNA negative; among those with hematologic diseases, the risk of reactivation is higher in those treated with rituximab.52
Clinical presentation of HBV reactivation ranges from asymptomatic cases to acute liver failure and death. Mortality rates are higher in those patients who do not receive antiviral agents compared with those who do (~30% versus 12%, respectively); moreover, among those patients treated with antiviral agents, mortality rates are lower in those treated with entecavir compared to those treated with lamivudine.53 The use of anti-HBV prophylaxis is therefore recommended in patients who are HbsAg-negative, anti-HBc-positive with hematologic diseases, irrespective of the patient’s basal anti-HBs and HBV-DNA status. In addition, treatment with the second-generation nucleoside analogs (eg, entecavir/tenofovir) is strongly recommended for patients with active chronic HBV infection who are candidates for immunosuppressive therapy.54
Recommendations
The ideal candidates for gene therapy are patients with β-TDT who have no or only slight hepatic fibrosis, which in this context the SITE defines as level F1 based on Fibroscan or Ishak stage 0-1-(2) based on liver biopsy.
There are no reasons to consider anti-HCV-positive patients to be contraindicated for gene therapy if they have eliminated the virus either spontaneously or after anti-viral therapy, provided that they have no other hepatic or extra-hepatic contraindications.
Gene therapy is contraindicated in patients with chronic HBV infection, as defined by the European Association for the Study of Liver (EASL) guidelines.
Gene therapy can be considered for patients who have occult HBV infection, as defined by the EASL guidelines, provided that they accept appropriate prophylaxis and have no other contraindications to treatment.
Endocrinopathy
Background and discussion
Appropriate long-term medical treatment is the essential factor for good clinical outcomes after hematopoietic stem cell transplants,37 and this is probably also the case for patients undergoing gene therapy. Endocrinopathies (eg, diabetes, hypogonadism, short stature, and hypothyroidism) have been shown to be associated with higher ferritin levels,55 as in cases when patients started iron chelation therapy late,56 and cardiac overload,57 indicating that the presence of one or more endocrinological complications usually reflects an inadequate iron chelation therapy and therefore a high iron burden for the patient concerned.56,58
The term “diabetic stem cell mobilopathy” is used to indicate scarce mobilization of the bone marrow hematopoietic stem cells to the peripheral blood in patients with diabetes. This occurs because diabetes radically changes the bone marrow microenvironment resulting in a net reduction in the release of hematopoietic stem cells.59 In clinical trials of gene therapy, the presence of an endocrinopathy has never been an exclusion criterion. However, exclusion criteria have been reported to include the statement: “Any other evidence of severe iron overload that, in the Investigator’s opinion, warrants exclusion.”2
Endocrinological complications have not been shown to be relevant to the posttransplant outcome. These should, however, always be assessed before hematopoietic stem cell transplant to plan adequate monitoring of these conditions posttransplant.60
Recommendations
A cautious approach should be adopted at this stage given the limited clinical experience with gene therapy.
Insulin-dependent patients should not be considered high-priority patients for gene therapy.
Nephropathy
Background and discussion
In patients with β-TDT, kidney damage can be related either to iron overload or to chronic hypoxia caused by anemia.60 In this setting, experience from allogeneic hematopoietic stem cell transplantation cannot be applied to the use of nephrotoxic immunosuppressors.
Different thresholds used to define chronic disease have been reported in the literature. Examples include: “kidney disease with a calculated creatinine clearance <30% normal value”60; “kidney disease with a baseline estimated glomerular filtration rate <70 mL/min/1.73 m2”2; or “adequate renal function as evidenced by serum creatinine <1.5 upper limit of normal.”18
Published clinical trials on the use of gene therapy in patients with β-TDT patients report that changes in kidney function are an exclusion criterion for clinical trial enrollment.
Recommendations
Kidney function must be assessed before starting gene therapy.
Patients with abnormal renal function should be considered for gene therapy with extreme caution and are not given priority at this stage.
Thrombophilic status
Background and discussion
Patients with β-TDT present with hypercoagulability (which is particularly evident in splenectomized patients) due to a high platelet count and peripheral erythroblastosis.60–64
Due to peripheral stem cell mobilization and the use of granulocyte-colony stimulating factor during gene therapy, patients with a history of thrombotic events must be identified and their thrombophilic status assessed before being considered eligible for gene therapy.18,65 Clinical studies of the use of gene therapy in patients with β-TDT have confirmed that a history of thrombotic events should be an exclusion criterion for enrollment; hypersplenism should also be a contraindication as it can have a negative impact on grafting.
Assessment of/screening for prothrombotic status before treatment is essential to confirm patient suitability and to promote good outcomes from gene therapy.
Recommendations
Patients with a “low-risk thrombophilic screen” (defined here as low levels of protein S, protein C, and/or antithrombin III) are not excluded from gene therapy as these may be related to thalassemic/sickle cell liver damage.18
Patients with a remote history of a thromboembolic event and no documented pulmonary hypertension can be evaluated for gene therapy after the suspension of anticoagulant therapy.
Patients with a negative history of significant previous thrombotic events are considered eligible for enrollment.18
Hypersplenism is a contraindication for gene therapy.2
Patients with white blood cell count <3 × 109/L, and/or platelet count <100 × 109/L are exclusion criteria (ie, pancytopenia postinfection).2
Lupus anticoagulant represents a contraindication to gene therapy; an uncorrected bleeding disorder (eg, low levels of Factor VII or VIII) is also an exclusion to treatment.
Summary of recommendations
Patient selection must be carried out through a consensus decision between the Center of the Network of Hemoglobinopathies and the treatment center qualified to carry out HSC transplantation.
Gene therapy should be restricted to centers experienced with myeloablative conditioning and with the treatment of transfusion-dependent β-thalassemia. Patients selected for gene therapy must fulfill the inclusion criteria established for LentiGlobin European Medicines Authority (EMA) license and be accredited by the Foundation for the Accreditation of Cellular Therapy and Joint Accreditation Committee (FACT-JACIE) for allotransplant.
It is the opinion of the expert panel that patients with β-TDT who could represent possible candidates for gene therapy with the only product currently available that is approved by the EMA (EMA/CHMP/166977/209; https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-zynteglo_en.pdf) (Zynteglo; https://www.zynteglo.eu/) must satisfy the criteria listed in the following summary Table 1.
Table 1.
Summary
| High-priority Patients | Ineligible Patients | Assessable Patients Undergoing Ongoing Changes to Therapy (ie, Potential Future Candidates for Gene Therapy) |
|---|---|---|
| • Patients followed by specialized hemoglobinopathies centers | • Patients not followed by specialized hemoglobinopathies centers | • Patients followed by specialized hemoglobinopathies centers |
| • β-TDT patients aged >12 and <55 yrs | ||
| • Patients eligible for allogeneic transplant with no HLA-identical family donor | • Patients aged <12 and >55 yrs | • β-TDT patients aged >12 and <55 yrs |
| • Patients with no significant iron overload | • Patients with uncontrolled iron overload | • Patients with a non-β0/β0 genotype |
| • Patients with no evidence of organ damage | • Patients with chronic organ damage, hepatopathy, insulin-dependent diabetes, nephropathy, and positive thrombophilic status | • Patients eligible for allogeneic transplant with no family HLA-identical donor |
| • Patients who are registered in a qualified transplant center that has experience in hematopoietic stem cell transplant and is connected to a center specialized in the treatment of patients with β-TDT | • Patients with iron overload: LIC > 7mgFe/g Liver dw—cardiac MRI T2* < 10 ms in the previous 6 mo | |
| • Patients with noninsulin-dependent diabetes | ||
| • Good compliance with treatment | ||
| • Patients with slight and/or reversible cardiopathy | ||
| • Patients who are HCV-RNA and/or HBV-DNA positive | ||
| • Good compliance with treatment |
β-TDT = transfusion-dependent β thalassemia; HCV = hepatitis C virus; HLA = human leukocyte antigen; LIC = liver iron concentration; MRI = magnetic resonance imaging.
Patients must be considered unsuitable for gene therapy if they:
meet exclusion criteria indicated by the regulatory authorities (see EMA/CHMP/166977/2019; https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-zynteglo_en.pdf);
have uncontrollable iron overload and/or chronic organ damage (eg, pulmonary hypertension).
Controllable iron overload requires reassessment. Caution must be exercised when assessing patients with complications and comorbidities.
Access to gene therapy must be reassessed according to scientific and regulatory updates.
In recognition of the continuous evolution of medical scientific knowledge, of new data being presented in the literature and, in specific cases, of changes to recommendations from the regulatory authorities, the SITE and EHA expert panels will update this algorithm and the supporting notes when new evidence is published that could modify the strength of the current recommendations. As these technologies will likely improve over time, the inclusion or change in some parameter may happen; for example, more effective vectors (β0 patients), less myeloablation, or nontoxic regimens, novel medications that improve some of the conditions related to iron overload.
Should no such evidence emerge, the SITE and EHA expert panel will review and update the algorithm and supporting notes every 2 years.
Reviewer panel
| Emanuele Angelucci | U.O. Ematologia e Centro trapianti—IRCCS Ospedale Policlinico San Martino, Genova, Italy |
| Maria Domenica Cappellini | Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico Malattie Rare, Milano, Italy |
| Holger Cario | Center for Rare Diseases Ulm (ZSE Ulm), Ulm University Medical Center, Germany |
| Marina Cavazzana | Necker Hospital, Paris Descartes University, France |
| Achille Iolascon | CEINGE Università Federico II di Napoli, Italy |
| Antonis Kattamis | National and Kapodistrian University of Athens, “Aghia Sophia” Children’s Hospital, Athens, Greece |
| Andreas Kulozik | University of Heidelberg, Germany |
| Aurelio Maggio | U.O.C. Ematologia II con Talassemia, Ospedali Riuniti P.O. Cervello Palermo, Palermo, Italy |
| Martina Muckenthaler | University of Heidelberg, Germany |
| Antonio Giulio Piga | Ospedale San Luigi Gonzaga, Università di Torino Orbassano, Torino, Italy |
| David Rees | Department of Haematology, King’s College Hospital NHS Foundation Trust, London, UK |
| Ali Taher | American University of Beirut Medical Center, Beirut, Lebanon |
| Hannah Tamary | Schneider Children’s Medical Center of Israel, Felsenstain Medical Research Center, Sackler School of Medicine, Tel-Aviv University, Israel |
| Isabelle Thuret | CHU de Marseille—Hôpital de la Timone, Marseille, France |
| Sule Unal | Hacettepe University Center for Fanconi Anemia and Other Inherited BMF Syndromes, Ankara, Turkey |
Disclosures
GLF reports advisory board consultation with Novartis, Celgene, and BlueBirdBio. DB reports nothing to disclose. MC reports payments for lectures and advisory boards from Novartis Pharma. LDF reports payments for an advisory board with Novartis. GG reports nothing to disclose. RO reports speaker honoraria from Novartis; fees for advisory boards from Celgene and BlueBird Bio. PR reports nothing to disclose. VP reports employment/consultation with Novartis, BlueBird Bio. MM reports consultation with Gilead srl; payments for lectures from Amgen, Novartis. IA reports advisory board consultation with Celgene and BlueBird Bio. All the other authors have no conflicts of interest to disclose.
Supplementary Material
Footnotes
Supplemental digital content is available for this article.
References
- 1.Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010; 467:318–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson AA, Walters MC, Kwiatkowski J, et al. Gene Therapy in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2018; 378:1479–1493 [DOI] [PubMed] [Google Scholar]
- 3.Kunz JB, Kulozik AE. Gene therapy of the hemoglobinopathies. HemaSphere. 2020; 4:e479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lucarelli G, Galimberti M, Polchi P, et al. Marrow transplantation in patients with advanced thalassemia. N Engl J Med. 1987; 316:1050–1055 [DOI] [PubMed] [Google Scholar]
- 5.Lucarelli G, Galimberti M, Polchi P, et al. Bone marrow transplantation in patients with thalassemia. N Engl J Med. 1990; 322:417–421 [DOI] [PubMed] [Google Scholar]
- 6.Fleischhauer K, Locatelli F, Zecca M, et al. Graft rejection after unrelated donor hematopoietic stem cell transplantation for thalassemia is associated with nonpermissive HLA-DPB1 disparity in host-versus-graft direction. Blood. 2006; 107:2984–2992 [DOI] [PubMed] [Google Scholar]
- 7.Anurathapan U, Hongeng S, Pakakasama S, et al. Hematopoietic stem cell transplantation for homozygous β-thalassemia and β-thalassemia/hemoglobin E patients from haploidentical donors. Bone Marrow Transplant. 2016; 51:813–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun Q, Wu B, Lan H, et al. Haploidentical haematopoietic stem cell transplantation for thalassaemia major based on an FBCA conditioning regimen. Br J Haematol. 2018; 182:554–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baronciani D, Angelucci E, Potschger U, et al. Hemopoietic stem cell transplantation in thalassemia: a report from the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Registry, 2000–2010. Bone Marrow Transplant. 2016; 51:536–541 [DOI] [PubMed] [Google Scholar]
- 10.Angelucci E. Hematopoietic stem cell transplantation in thalassemia. Hematology Am Soc Hematol Educ Program. 2010; 2010:456–462 [DOI] [PubMed] [Google Scholar]
- 11.Frangoul H, Altshuler D, Cappellini MD, et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N Engl J Med. 2021; 384:252–260 [DOI] [PubMed] [Google Scholar]
- 12.Forni GL, Puntoni M, Boeri E, et al. The influence of treatment in specialized centers on survival of patients with thalassemia major. Am J Hematol. 2009; 84:317–318 [DOI] [PubMed] [Google Scholar]
- 13.Forni GL, Barella S, Cappellini M, Maggio A, Piga AG. Architettura Della Rete Italiana Talassemie Ed Emoglobinopatie. 2018. Available at: http://www.site-italia.org/forza_download.php?file=Architettura_Rete_Italiana.pdf. Accessed August 2, 2020
- 14.Lucarelli G, Galimberti M, Polchi P, et al. Marrow transplantation in patients with thalassemia responsive to iron chelation therapy. N Engl J Med. 1993; 329:840–844 [DOI] [PubMed] [Google Scholar]
- 15.Coates TD. Physiology and pathophysiology of iron in hemoglobin-associated diseases. Free Radic Biol Med. 2014; 72:23–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Angelucci E, Pilo F, Coates TD. Transplantation in thalassemia: revisiting the Pesaro risk factors 25 years later. Am J Hematol. 2017; 92:411–413 [DOI] [PubMed] [Google Scholar]
- 17.Zynteglo. European Medicines Agency. 2019. Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/zynteglo. Accessed August 2020
- 18.Marktel S, Scaramuzza S, Cicalese MP, et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ß-thalassemia. Nat Med. 2019; 25:234–241 [DOI] [PubMed] [Google Scholar]
- 19.Price EA. Aging and erythropoiesis: current state of knowledge. Blood Cells Mol Dis. 2008; 41:158–165 [DOI] [PubMed] [Google Scholar]
- 20.Ballester G, Tirona MT, Ballester O. Hematopoietic stem cell transplantation in the elderly. Oncology (Williston Park). 2007; 21:1576–1583 [PubMed] [Google Scholar]
- 21.Chatta GS, Price TH, Allen RC, et al. Effects of in vivo recombinant methionyl human granulocyte colony-stimulating factor on the neutrophil response and peripheral blood colony-forming cells in healthy young and elderly adult volunteers. Blood. 1994; 84:2923–2929 [PubMed] [Google Scholar]
- 22.Egusa Y, Fujiwara Y, Syahruddin E, et al. Effect of age on human peripheral blood stem cells. Oncol Rep. 1998; 5:397–400 [DOI] [PubMed] [Google Scholar]
- 23.Bagnara GP, Bonsi L, Strippoli P, et al. Hemopoiesis in healthy old people and centenarians: well-maintainedresponsiveness of CD34+ cells to hemopoietic growth factors and remodelingof cytokine network. J Gerontol Ser A Biol Sci Med Sci. 2000; 55:B61–B66 [DOI] [PubMed] [Google Scholar]
- 24.Baraldi-Junkins CA, Beck AC, Rothstein G. Hematopoiesis and cytokines. Relevance to cancer and aging. Hematol Oncol Clin North Am. 2000; 14:45–61 [DOI] [PubMed] [Google Scholar]
- 25.Ershler WB, Sheng S, McKelvey J, et al. Serum erythropoietin and aging: a longitudinal analysis. J Am Geriatr Soc. 2005; 53:1360–1365 [DOI] [PubMed] [Google Scholar]
- 26.Lee GY, Jeong SY, Lee HR, et al. Age-related differences in the bone marrow stem cell niche generate specialized microenvironments for the distinct regulation of normal hematopoietic and leukemia stem cells. Sci Rep. 2019; 9:1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aprile A, Gulino A, Storto M, et al. Hematopoietic stem cell function in β-thalassemia is impaired and is rescued by targeting the bone marrow niche. Blood. 2020; 136:610–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baronciani D, Boumendil A, Dalissier A, et al. Hematopoietic cell transplantation in thalassemia and sickle cell disease: report from the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Registry: 2000-2017. Blood. 2018; 132suppl 1168–168 [Google Scholar]
- 29.Anurathapan U, Hongeng S, Pakakasama S, et al. Hematopoietic stem cell transplantation for severe thalassemia patients from haploidentical donors using a novel conditioning regimen. Biol Blood Marrow Transplant. 2020; 26:1106–1112 [DOI] [PubMed] [Google Scholar]
- 30.He Y, Jiang H, Li C, et al. Long-term results of the <scp>NF-08-TM</scp> protocol in stem cell transplant for patient with thalassemia major: a multi-center large-sample study. Am J Hematol. 2020; 95:E297, E299 [DOI] [PubMed] [Google Scholar]
- 31.Angelucci E, Brittenham GM, McLaren CE, et al. Correction: hepatic iron concentration and total body iron stores in thalassemia major. N Engl J Med. 2000; 343:1740. [DOI] [PubMed] [Google Scholar]
- 32.Chai X, Li D, Cao X, et al. ROS-mediated iron overload injures the hematopoiesis of bone marrow by damaging hematopoietic stem/progenitor cells in mice. Sci Rep. 2015; 5:10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chai X, Li D, Cao X, et al. Erratum: corrigendum: ROS-mediated iron overload injures the hematopoiesis of bone marrow by damaging hematopoietic stem/progenitor cells in mice. Sci Rep. 2017; 7:10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Summary of product characteristics of Zynteglo. 2019. Available at: https://www.ema.europa.eu/en/documents/product-information/zynteglo-epar-product-information_en.pdf. Accessed August 2020
- 35.Borgna-Pignatti C, Rugolotto S, De Stefano P, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica. 2004; 89:1187–1193 [PubMed] [Google Scholar]
- 36.Piga A, Longo F, Musallam KM, et al. Assessment and management of iron overload in β-thalassaemia major patients during the 21st century: a real-life experience from the Italian WEBTHAL project. Br J Haematol. 2013; 161:872–883 [DOI] [PubMed] [Google Scholar]
- 37.Angelucci E, Pilo F. Management of iron overload before, during, and after hematopoietic stem cell transplantation for thalassemia major. Ann N Y Acad Sci. 2016; 1368:115–121 [DOI] [PubMed] [Google Scholar]
- 38.Kirk P, Roughton M, Porter JB, et al. Cardiac T2* magnetic resonance for prediction of cardiac complications in thalassemia major. Circulation. 2009; 120:1961–1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carpenter JP, He T, Kirk P, et al. On T2* magnetic resonance and cardiac iron. Circulation. 2011; 123:1519–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Derchi G, Galanello R, Bina P, et al. Prevalence and risk factors for pulmonary arterial hypertension in a large group of β-thalassemia patients using right heart catheterization: a Webthal study. Circulation. 2014; 129:338–345 [DOI] [PubMed] [Google Scholar]
- 41.European Society of Cardiology. Clinical practice guidelines. 2020. Available at: https://www.escardio.org/Guidelines/Clinical-Practice-Guidelines. Accessed August 2020
- 42.Hogan WJ, Maris M, Storer B, et al. Hepatic injury after nonmyeloablative conditioning followed by allogeneic hematopoietic cell transplantation: a study of 193 patients. Blood. 2004; 103:78–84 [DOI] [PubMed] [Google Scholar]
- 43.Angelucci E, Muretto P, Nicolucci A, et al. Effects of iron overload and hepatitis C virus positivity in determining progression of liver fibrosis in thalassemia following bone marrow transplantation. Blood. 2002; 100:17–21 [DOI] [PubMed] [Google Scholar]
- 44.Ramos CA, Saliba RM, de Pádua L, et al. Impact of hepatitis C virus seropositivity on survival after allogeneic hematopoietic stem cell transplantation for hematologic malignancies. Haematologica. 2009; 94:249–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frickhofen N, Wiesneth M, Jainta C, et al. Hepatitis C virus infection is a risk factor for liver failure from veno-occlusive disease after bone marrow transplantation. Blood. 1994; 83:1998–2004 [PubMed] [Google Scholar]
- 46.Strasser SI, Myerson D, Spurgeon CL, et al. Hepatitis C virus infection and bone marrow transplantation: a cohort study with 10-year follow-up. Hepatology. 1999; 29:1893–1899 [DOI] [PubMed] [Google Scholar]
- 47.Origa R, Ponti ML, Filosa A, et al. Italy for THAlassemia and hepatitis C Advance - Società Italiana Talassemie ed Emoglobinopatie (ITHACA-SITE). Treatment of hepatitis C virus infection with direct-acting antiviral drugs is safe and effective in patients with hemoglobinopathies. Am J Hematol. 2017; 92:1349–1355 [DOI] [PubMed] [Google Scholar]
- 48.Ponti ML, Comitini F, Murgia D, et al. Impact of the direct-acting antiviral agents (DAAs) on chronic hepatitis C in Sardinian patients with transfusion-dependent Thalassemia major. Dig Liver Dis. 2019; 51:561–567 [DOI] [PubMed] [Google Scholar]
- 49.Sinakos E, Kountouras D, Koskinas J, et al. Treatment of chronic hepatitis C with direct-acting antivirals in patients with β-thalassaemia major and advanced liver disease. Br J Haematol. 2017; 178:130–136 [DOI] [PubMed] [Google Scholar]
- 50.Marzano A, Angelucci E, Astegiano M, et al. AISF position paper on HCV in immunocompromised patients. Dig Liver Dis. 2019; 51:10–23 [DOI] [PubMed] [Google Scholar]
- 51.Locasciulli A, Bruno B, Alessandrino EP, et al. Italian Cooperative Group for Blood and Marrow Transplantation. Hepatitis reactivation and liver failure in haemopoietic stem cell transplants for hepatitis B virus (HBV)/hepatitis C virus (HCV) positive recipients: a retrospective study by the Italian group for blood and marrow transplantation. Bone Marrow Transplant. 2003; 31:295–300 [DOI] [PubMed] [Google Scholar]
- 52.Cholongitas E, Haidich AB, Apostolidou-Kiouti F, et al. Hepatitis B virus reactivation in HBsAg-negative, anti-HBc-positive patients receiving immunosuppressive therapy: a systematic review. Ann Gastroenterol. 2018; 31:480–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roche B, Samuel D. The difficulties of managing severe hepatitis B virus reactivation. Liver Int. 2011; 3suppl 1104–110 [DOI] [PubMed] [Google Scholar]
- 54.Lampertico P, Agarwal K, Berg T, et al. EASL 2017 clinical practice guidelines on the management of hepatitis B virus infection. J Hepatol. 2017; 67:370–398 [DOI] [PubMed] [Google Scholar]
- 55.Shalitin S, Carmi D, Weintrob N, et al. Serum ferritin level as a predictor of impaired growth and puberty in thalassemia major patients. Eur J Haematol. 2005; 74:93–100 [DOI] [PubMed] [Google Scholar]
- 56.De Sanctis V, Elsedfy H. Clinical and biochemical data of adult thalassemia major patients (TM) with multiple endocrine complications (MEC) versus TM patients with normal endocrine functions: a long-term retrospective study (40 years) in a tertiary care center in Italy. Mediterr J Hematol Infect Dis. 2016; 8:2016022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Noetzli LJ, Panigrahy A, Mittelman SD, et al. Pituitary iron and volume predict hypogonadism in transfusional iron overload. Am J Hematol. 2012; 87:167–171 [DOI] [PubMed] [Google Scholar]
- 58.Casale M, Forni GL, Cassinerio E, et al. Risk factors for endocrine complications in transfusion-dependent thalassemia patients on chelation therapy with deferasirox: a risk assessment study from a multicentre nation-wide cohort. Haematologica. 2021. January 7. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fadini GP, DiPersio JF. Diabetes mellitus as a poor mobilizer condition. Blood Rev. 2018; 32:184–191 [DOI] [PubMed] [Google Scholar]
- 60.Cappellini MD, Cohen A, Porter J, Taher A, Viprakasit V. Cappellini MD, Cohen A, Porter J, Taher A, Viprakasit V. Chapter 8: Endocrine; Chapter 3: Liver Disease; Chapter 6: The Spleen. In:. Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT) [Internet]. 2014; 146–157, 3rd ed. Nicosia, Cyprus: Thalassaemia International Federation; 42–97,–126–133 [Google Scholar]
- 61.Rund D, Rachmilewitz E. Beta-thalassemia. N Engl J Med. 2005; 353:1135–1146 [DOI] [PubMed] [Google Scholar]
- 62.Taher A, Isma’eel H, Cappellini MD. Thalassemia intermedia: revisited. Blood Cells Mol Dis. 2006; 37:12–20 [DOI] [PubMed] [Google Scholar]
- 63.Taher AT, Musallam KM, Karimi M, et al. Splenectomy and thrombosis: the case of thalassemia intermedia. J Thromb Haemost. 2010; 8:2152–2158 [DOI] [PubMed] [Google Scholar]
- 64.Taher AT, Musallam KM, El-Beshlawy A, et al. Age-related complications in treatment-naïve patients with thalassaemia intermedia. Br J Haematol. 2010; 150:486–489 [DOI] [PubMed] [Google Scholar]
- 65.Lidonnici MR, Aprile A, Frittoli MC, et al. Plerixafor and G-CSF combination mobilizes hematopoietic stem and progenitors cells with a distinct transcriptional profile and a reduced in vivo homing capacity compared to plerixafor alone. Haematologica. 2017; 102:e120–e124 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.