

Abstract
Risk of chronic obstructive pulmonary disease (COPD) is determined by both cigarette smoking and genetic susceptibility, but little is known about gene-by-smoking interactions. We performed a genome-wide association analysis of 179,689 controls and 21,077 COPD cases from UK Biobank subjects of European ancestry recruited from 2006 to 2010, considering genetic main effects and gene-by-smoking interaction effects simultaneously (2-degrees-of-freedom (df) test) as well as interaction effects alone (1-df interaction test). We sought to replicate significant results in COPDGene (United States, 2008–2010) and SpiroMeta Consortium (multiple countries, 1947–2015) data. We considered 2 smoking variables: 1) ever/never and 2) current/noncurrent. In the 1-df test, we identified 1 genome-wide significant locus on 15q25.1 (cholinergic receptor nicotinic β4 subunit, or CHRNB4) for ever- and current smoking and identified PI*Z allele (rs28929474) of serpin family A member 1 (SERPINA1) for ever-smoking and 3q26.2 (MDS1 and EVI1 complex locus, or MECOM) for current smoking in an analysis of previously reported COPD loci. In the 2-df test, most of the significant signals were also significant for genetic marginal effects, aside from 16q22.1 (sphingomyelin phosphodiesterase 3, or SMPD3) and 19q13.2 (Egl-9 family hypoxia inducible factor 2, or EGLN2). The significant effects at 15q25.1 and 19q13.2 loci, both previously described in prior genome-wide association studies of COPD or smoking, were replicated in COPDGene and SpiroMeta. We identified interaction effects at previously reported COPD loci; however, we failed to identify novel susceptibility loci.
Keywords: chronic obstructive pulmonary disease, gene-environment interaction, gene-by-smoking interaction, genome-wide association study, smoking
Abbreviations
- df
degrees of freedom
- CHRNB4
cholinergic receptor nicotinic β4 subunit
- CI
confidence interval
- COPD
chronic obstructive pulmonary disease
- EGLN2
Egl-9 family hypoxia inducible factor 2
- FEV1
forced expiratory volume in 1 second
- FVC
forced vital capacity
- GWAS
genome-wide association study
- MECOM
myelodysplastic syndrome 1 and ecotropic viral integration site 1 complex locus
- OR
odds ratio
- SNP
single-nucleotide polymorphism
- SERPINA1
serpin family A member 1
SMPD3, sphingomyelin phosphodiesterase 3
- UKB
UK Biobank
Risk of chronic obstructive pulmonary disease (COPD) is determined by both cigarette smoking and genetic susceptibility. Adverse effects of smoking on risk of COPD might differ by an individual’s genetic susceptibility, which raises the potential for important gene-by-smoking interactions. However, little is known about gene-by-smoking interactions and COPD risk.
A significant interaction between PI*Z allele (rs28929474) of serpin family A member 1 (SERPINA1) and cigarette smoking has been reported for spirometric measures of lung function in European-ancestry subjects (1, 2). For COPD-related traits, genome-wide gene-by-smoking interaction studies have focused on quantitative measures of lung function from spirometry (3, 4). While spirometric measurements of lung function are used to diagnose COPD, no genome-wide studies have investigated gene-by-smoking interaction and the risk of COPD itself.
A recent large-scale genome-wide association study (GWAS) identified 82 distinct loci associated with COPD risk (5). However, these identified variants explained less than 10% of the phenotypic variability on a liability scale. To fill this gap in the risk of COPD explained by common variants, we included gene-by-smoking interactions in a GWAS model.
Studying gene-by-environment interactions requires much larger sample sizes, compared with conventional GWAS, to detect marginal effects of genes (6). The 2-degrees-of-freedom (df) joint test leverages genetic main effects and gene-by-environment interaction effects simultaneously and can provide better power than a standard interaction test, which is a 1-df test (7). Using a 2-df test, recent large-scale genome-wide gene-by-environment interaction studies of complex traits have identified new genetic factors as well as possible gene-by-environment interactions (3, 8–12). The availability of the large-scale UK Biobank (UKB) study, which collected a wide range of phenotypes as well as genetic data, provides a promising opportunity to detect possible gene-by-environment interactions. Here, we performed genome-wide gene-by-smoking interaction analyses of COPD in the UKB to identify novel genetic variants for risk of COPD while accounting for potential smoking interactions, and we assessed the impact of potential gene-by-smoking interactions on risk of COPD at known COPD and lung function GWAS loci.
METHODS
Study populations
The UKB is a population-based cohort for which 500,000 volunteers were originally recruited (13). We used UKB subjects as our discovery set and used 2 additional data sets (COPDGene Study and SpiroMeta Consortium) to further investigate significant results from the UKB. COPDGene recruited former and current smokers whose smoking history was at least 10 pack-years (14). SpiroMeta is comprised of a total of 79,055 individuals from 22 studies (15). All participants in these studies provided written informed consent and all studies were approved by local research ethics committees and/or institutional review boards.
Spirometric measures and genetic data
Details of quality controls of spirometric measures, genetic markers, and subjects in the UKB study have been previously described (5, 13, 15). Briefly, to determine lung function, measures of forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC) were derived from spirometry volume time-series data, subject to additional quality control based on American Thoracic Society/European Respiratory Society criteria (15, 16). Genotyping was performed using Axiom UK BiLEVE array and Axiom Biobank array (Affymetrix, Santa Clara, California) and imputed to the Haplotype Reference Consortium (http://www.haplotype-reference-consortium.org/) panel (version 1.1). We included independent subjects of European ancestry based on a combination of self-reported ethnicity and principal components data provided by UKB.
Measures of smoking exposure
We assigned smoking status to individuals in UKB based on their questionnaire responses. Never-smokers included noncurrent smokers or those who smoked less than 100 cigarettes in their lifetimes. Ever-smokers were defined as either current, most days (current or all days in the past), or smoked occasionally.
To test for possible gene-by-smoking interactions, we considered 2 smoking variables: ever/never and current/noncurrent smoker. For ever-/never-smokers, former and current smokers were in the exposed group. For current/noncurrent smokers, former and never smokers were in the unexposed group. Smoking variables were coded as 0 and 1 for unexposed and exposed groups, respectively. Here, we refer ever/never-smoker analysis as “G × Ever-smoking analysis” and current/noncurrent smoker analysis as “G × Current-smoking analysis.”
Outcome
We defined COPD cases based on prebronchodilator spirometry following the modified Global Initiative for Chronic Obstructive Lung Disease criteria for moderate airflow limitation: FEV1 less than 80% of predicted value (using reference equations from (17)), and the ratio of FEV1/FVC less than 0.7.
Genetic analysis
We included markers with minor allele frequency of ≥0.01 and imputation quality score (r2) ≥ 0.5. We performed a logistic regression analysis considering genetic main effects and gene-by-smoking interaction effects simultaneously (2-df joint test) as well as interaction effects alone (1-df interaction test), adjusting for age, sex, genotyping array, and the first 10 principal components. We used the 2-df joint test to search for new genetic variants of COPD and the 1-df interaction test to assess interaction effects alone. If a marker shows a significance in the 2-df joint test, it is associated with the outcome across exposure groups. If a marker shows a significance only in the 1-df interaction test, its genetic effect should differ by exposure group. Additionally, marginal GWAS were conducted, stratified by each smoking variable. All genome-wide analyses were performed using the PLINK software, version 2.0 (18). We created regional association plots via LocusZoom (http://locuszoom.org/), using 1,000 Genomes European reference data (November 2014 release) (19).
Conditional analysis
We defined distinct “loci” using a 1-Mb window (+/− 500kb) around the lead variant (i.e., most significant single-nucleotide polymorphism (SNP)). Because our joint analysis was likely to include substantial overlap with previously described association studies of marginal effects for risk of COPD, we performed conditional analysis of each lead variant to determine whether our signals were independent of known risk loci for COPD (5) or lung function (15). Given that the current genome-wide complex trait analysis (GCTA) tool does not account for gene-by-environment interactions in their conditional analysis, we took a stratified approach for this analysis (20). We stratified by smoking-exposed and -unexposed groups and conditioned on recognized SNPs from previous GWAS of COPD (5) or lung function (15) within 2-Mb of the lead variant. The conditioned 2-df test for genetic main effects and interaction effects was then calculated on the conditioned stratified results using the following equation (9, 21):
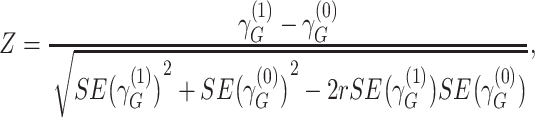 |
where 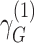 and
and 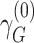 represent stratum-specific genetic effects for the 1-df test,
represent stratum-specific genetic effects for the 1-df test, 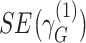 and
and 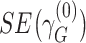 are their respective standard errors (SE), and r is the Spearman rank correlation coefficient between
are their respective standard errors (SE), and r is the Spearman rank correlation coefficient between 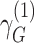 and
and 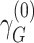 , calculated from genome-wide results. This Z statistic approximately follows a standard normal distribution under
, calculated from genome-wide results. This Z statistic approximately follows a standard normal distribution under 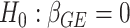 . For the 2-df test,
. For the 2-df test,
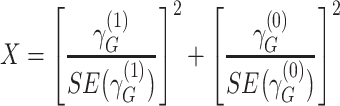 |
approximately follows a 2-df χ2 distribution under 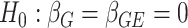 when the 2 strata are independent.
when the 2 strata are independent.
Dose-response analysis
To further characterize our significant results, we conducted a dose-response analysis in all subjects and in ever-smokers as a secondary analysis. We tested gene-by-smoking dose interaction using the standard 1-df test. We considered 3 quantitative measures of smoking dose: smoking duration, pack-years, and cigarettes per day. We considered exposures as both a quantitative and categorical variable grouped based on quartiles.
Replication
Because COPDGene cohort is enriched for heavy smokers, we hypothesized that SNPs presenting a stronger association among the exposed group in the UKB should also show some marginal effects on COPD risk in COPDGene subjects. For selected SNPs, we tested for a marginal association between each SNP and COPD risk, adjusting for age, sex, smoking status, pack-years, and genetic ancestry principal components in 5,342 non-Hispanic White COPDGene subjects. We further tested gene-by-smoking dose interaction based on the 1-df test. The Fagerström test for nicotine dependence (FTND) measure was collected from current smokers in COPDGene, so we also tested gene-by-FTND interaction. We considered both a quantitative FTND score and a categorical variable grouped into mild (0–3), moderate (4–6) and severe (7–10) (22).
We also attempted to replicate our results by lookup in a genome-wide association analysis of spirometric measures of lung function (FEV1, FVC, and FEV1/FVC) stratified by ever- and never-smoker groups in SpiroMeta (15). Using summary statistics from these stratified results, we calculated test statistics for a 1-df interaction test and a 2-df joint test based on the same approach used in our stratified conditional analysis (9, 21). Briefly, each study performed linear regression adjusting for age, age2, sex, and height by using rank-based inverse normal transformation, adjusting for population substructure, and performing separate analyses for ever- and never-smokers. Results were combined under a fixed-effects meta-analysis.
RESULTS
Subject characteristics
We analyzed 200,766 subjects of European ancestry, including 179,689 controls and 21,077 COPD cases from UKB (Table 1). These UKB subjects included 71,591 ever-smokers (former and current smokers combined) and 129,175 never-smokers, and 14,590 current smokers and 186,176 noncurrent smokers (never- and former smokers combined). Non-Hispanic White COPDGene subjects included 3,361 former smokers and 1,981 current smokers. While UKB subjects had a higher proportion of COPD cases among current smokers (31.5%) compared with former (13.8%) and never-smokers (6.7%), COPDGene subjects showed a higher proportion of COPD cases among former smokers (54.5%) than current smokers (49.4%).
Table 1.
Subject Characteristics Stratified by Smoking Status in the UK Biobank (United Kingdom, 2006–2010) and COPDGene (United States, 2008–2010)
| UK Biobank | COPDGene | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Characteristic | Never (n = 129,175) | Former (n = 57,001) | Current (n = 14,590) | Former (n = 3,361) | Current (n = 1,981) | ||||||||||
| No. | % | Mean (SD) | No. | % | Mean (SD) | No. | % | Mean (SD) | No. | % | Mean (SD) | No. | % | Mean (SD) | |
| Moderate COPD | 8,631 | 6.70 | 7,857 | 13.80 | 4,589 | 31.50 | 1,831 | 54.50 | 978 | 49.40 | |||||
| Age, years | 55.55 (8.05) | 57.91 (7.64) | 54.12 (8.06) | 64.95 (8.23) | 57.48 (7.80) | ||||||||||
| Female sex | 50,194 | 38.90 | 29,175 | 51.20 | 6,941 | 47.60 | 1,620 | 48.20 | 907 | 45.80 | |||||
| No. of pack-years | 0.00 (0.00) | 19.21 (17.50) | 28.40 (18.25) | 47.15 (27.30) | 48.13 (24.49) | ||||||||||
| BMIa | 26.87 (4.56) | 27.95 (4.58) | 26.73 (4.68) | 28.97 (5.89) | 27.58 (5.79) | ||||||||||
| FEV1 % predicted | 96.44 (13.95) | 93.52 (16.30) | 85.10 (18.51) | 70.03 (29.35) | 75.29 (25.05) | ||||||||||
| FEV1/FVC | 0.77 (0.06) | 0.76 (0.07) | 0.72 (0.09) | 0.61 (0.19) | 0.65 (0.16) | ||||||||||
Abbreviations: BMI, body mass index; COPD, chronic obstructive pulmonary disease; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; SD, standard deviation.
a Weight (kg)/height (m2).
Genome-wide results
The analysis workflow is depicted in Figure 1.
Figure 1.
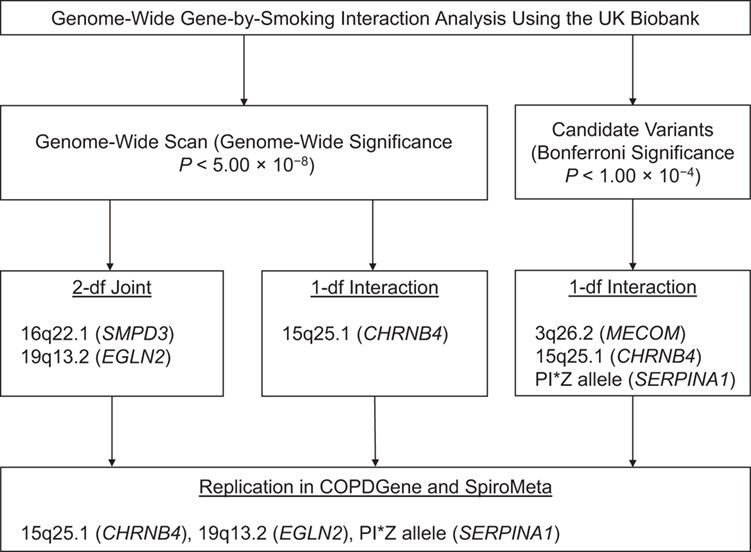
Analysis workflow for a genome-wide gene-by-smoking interaction analysis conducted in UK Biobank subjects, United Kingdom, 2006–2010. In the genome-wide scan, the genome-wide significance was set at P < 5.00 × 10−8. In the candidate variants, the significance threshold was considered as Bonferroni significance (P < 1.00 × 10−4). Candidate variants were selected from genome-wide association study of COPD (5) and lung function (15) and previously reported gene-by-smoking interactions of chronic obstructive pulmonary disease (1, 2, 4). Significant findings in the genome-wide scan and in candidate variants were selected to replicate in COPDGene (United States, 2008–2010) (14) and SpiroMeta consortium (multiple countries, 1947–2015) (15). CHRNB4, cholinergic receptor nicotinic β4 subunit; df, degrees of freedom; EGLN2, Egl-9 family hypoxia inducible factor 2; MECOM, myelodysplastic syndrome 1 and ecotropic viral integration site 1 complex locus; SERPINA1, serpin family A member 1; SMPD3, sphingomyelin phosphodiesterase 3.
2-df joint test.
We identified 48 loci for the G × Ever-smoking and 55 loci for the G × Current-smoking analysis (defined using 1-Mb windows) achieving genome-wide significance (P < 5.00 × 10−8) (Web Table 1 and Web Figure 1, available at https://doi.org/10.1093/aje/kwaa227). Thirty-five loci overlapped between G × Ever-smoking and G × Current-smoking analyses. Lead variants at 15 of these loci for G × Ever-smoking and 19 loci for G × Current-smoking analysis were previously identified in GWAS of COPD or lung function (5, 15). For the remaining loci, we conducted a conditional analysis to search for new signals (see Methods and Web Table 2). After adjusting for previously reported variants, 2 loci, 16q22.1, sphingomyelin phosphodiesterase 3 (SMPD3) (lead variant: rs141322661, P2-df = 3.92 × 10−9 from G × Ever-smoking and P2-df = 1.45 × 10−8 from the G × Current-smoking analysis), and 19q13.2, Egl-9 family hypoxia inducible factor 2 (EGLN2) (lead variant: rs2604894, P2-df = 5.87 × 10−9 from G × Current-smoking analysis), maintained genome-wide significance (Table 2 and Web Figure 2).
Table 2.
Significant Results of 2-Degrees-of-Freedom Joint Test and 1-Degree-of-Freedom Interaction Test in UK Biobank, United Kingdom, 2006–2010
| Genetic Main | Interaction a | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs ID | Chromosome Position | NearestGene | Effect/Reference Allele | EAF | Smoking Exposure | OR | 95% CI | P Value | OR | 95% CI | P Value | 2-df Joint b P Value |
| Significant From 2-df Joint Test c | ||||||||||||
| rs141322661 | 16:68398875 | SMPD3 | G/A | 0.01 | Ever smoking | 0.76 | 0.65, 0.88 | 2.09 × 10−4 | 0.94 | 0.78, 1.15 | 5.72 × 10−1 | 3.92 × 10−9 |
| rs141322661 | 16:68398875 | SMPD3 | G/A | 0.01 | Current smoking | 0.77 | 0.69, 0.86 | 1.47 × 10−6 | 0.81 | 0.62, 1.07 | 1.45 × 10−1 | 1.45 × 10−8 |
| rs2604894 | 19:41292404 | EGLN2 | A/G | 0.45 | Current smoking | 0.96 | 0.94, 0.98 | 1.28 × 10−3 | 0.9 | 0.84, 0.95 | 4.11 × 10−4 | 5.87 × 10−9 |
| Significant From 1-df Interaction Test c | ||||||||||||
| rs7170068 | 15:78912943 | CHRNA3 | A/G | 0.22 | Current smoking | 0.96 | 0.93, 0.98 | 2.38 × 10−3 | 0.79 | 0.74, 0.86 | 1.82 × 10−9 | 6.08 × 10−16 |
| rs12440014 | 15:78926726 | CHRNB4 | G/C | 0.24 | Ever smoking | 1.02 | 0.98, 1.06 | 3.14 × 10−1 | 0.83 | 0.79, 0.88 | 8.96 × 10−12 | 6.96 × 10−18 |
| Significant From 1-df Interaction Test in Candidate Variants d | ||||||||||||
| rs7642001 | 3:168746145 | MECOM | A/G | 0.37 | Current smoking | 1.07 | 1.04, 1.09 | 3.20 × 10−7 | 1.12 | 1.05, 1.19 | 3.65 × 10−4 | 1.73 × 10 −14 |
| rs28929474 | 14:94844947 | SERPINA1 | T/C | 0.02 | Ever smoking | 0.95 | 0.85, 1.07 | 4.02 × 10−1 | 1.3 | 1.12, 1.52 | 6.70 × 10−4 | 1.10 × 10−4 |
Abbreviations: CHRNA3, cholinergic receptor nicotinic α3 subunit; CHRNB4, cholinergic receptor nicotinic β4 subunit; CI, confidence interval; df, degrees of freedom; EAF, effect allele frequency; EGLN2, Egl-9 family hypoxia inducible factor 2; ID, identification; MECOM, myelodysplastic syndrome 1 and ecotropic viral integration site 1 complex locus; OR, odds ratio; SERPINA1, serpin family A member 1; SMPD3, sphingomyelin phosphodiesterase 3.
a 1-df interaction test.
b 2-df test of genetic main effects and gene-by-smoking interaction effects.
c Genome-wide statistical significance (P < 5.00 × 10−8) applied.
d Bonferroni-corrected statistical significance (P < 1.00 × 10−4) applied.
In a previous UKB GWAS of COPD examining only marginal genetic effects (5), rs141322661 at 16q22.1 reached genome-wide significance (P = 1.88 × 10−9), but not in a meta-analysis of UKB and the International COPD Genetics Consortium (P = 1.90 × 10−8), and rs2604894 at 19q13.2 did not reach genome-wide significance (P = 1.17 × 10−4) (5). Other signals from our analyses were attenuated and did not reach genome-wide significance.
1-df interaction test.
We identified 1 locus, 15q25.1 (defined using 1-Mb windows), as achieving genome-wide significance (P < 5.00 × 10−8) for both G × Ever- and G × Current-smoking analyses (Table 2 and Web Figure 3 and 4). In the G × Ever-smoking analysis, the lead variant (rs12440014 in the cholinergic receptor nicotinic β4 subunit gene (CHRNB4)) showed P1-df interaction = 8.96 × 10−12, presenting as a significant association among ever-smokers (odds ratio (OR) = 0.85, 95% confidence interval (CI): 0.82, 0.88; P = 3.39 × 10−19; data not shown) but not among never-smokers.
Interaction of previously reported variants
We examined possible gene-by-smoking interactions for risk of COPD at 82 known COPD-associated loci, 279 known lung function–associated loci, and 2 loci previously reporting smoking interactions for either lung function or COPD (Web Table 3). Because results from the 2-df test for these known loci predominantly showed genetic main effects, we evaluated results from the 1-df interaction test under Bonferroni-corrected significance thresholds.
At known loci for risk of COPD, rs55676755 in the cholinergic receptor nicotinic α3 subunit gene (CHRNA3) and rs28534575 in CHRNB4 significantly interacted with smoking, presenting as significant associations in ever-smokers (rs55676755: OR = 1.19, 95% CI: 1.15, 1.22, P = 8.74 × 10−28; and rs28534575: OR = 0.85, 95% CI: 0.82, 0.88, P = 2.53 × 10−18) but not in never-smokers (Web Table 4). SNP rs7642001 at 3q26.2, myelodysplastic syndrome 1 and ecotropic viral integration site 1 complex locus (MECOM), significantly interacted with current smoking (P1-df interaction = 3.65 × 10−4) but not with ever-smoking. At known loci for lung function, there was no evidence of significant interactions with smoking. At loci previously reporting smoking interactions, PI*Z allele (rs28929474) in SERPINA1 significantly interacted with ever-smoking (P1-df interaction = 6.70 × 10−4). (See Web Tables 3 and 4.)
Selected SNPs
To further investigate significant results, we selected SNPs at 5 loci (Table 3 and Figure 2). In the 2-df joint test, rs141322661 at 16q22.1 (SMPD3) and rs2604894 at 19q13.2 (EGLN2) reached genome-wide significance, independent of previously described loci for either COPD or lung function (Figure 2A). In the 1-df interaction test, we included rs12440014 at 15q25.1 (CHRNB4). Among previously reported variants, rs7642001 at 3q26.2 (MECOM) and rs28929474 in SERPINA1, showed evidence of interaction (Figure 2B).
Table 3.
Replications of Selected Variants in Data From COPDGene (United States, 2008–2010) and SpiroMeta Consortium (Multiple Countries, 1947–2015)
| COPDGene NHW | SpiroMeta FEV 1 c | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Marginal Association b | Never-Smoker | Ever-Smoker | ||||||||||||||
| rs ID | Chromosome Position | Nearest Gene | Effect/Reference Allele | EAF a | Smoking Exposure | OR | 95% CI | P Value | β | 95% CI | P Value | β | 95% CI | P Value | Interaction d P Value | 2-df Joint e P Value |
| Main Effects | ||||||||||||||||
| rs141322661 | 16:68398875 | SMPD3 | G/A | 0.01 | Ever smoking | 0.99 | 0.69, 1.41 | 9.52 × 10−1 | 0.01 | −0.05, 0.08 | 6.45 × 10−1 | 0.06 | 0.00, 0.13 | 6.03 × 10−2 | 1.07 × 10−1 | 1.54 × 10−1 |
| rs2604894 | 19:41292404 | EGLN2 | A/G | 0.45 | Current smoking | 0.90 | 0.82, 0.98 | 1.62 × 10−2 | 0.001 | −0.02, 0.02 | 9.12 × 10−1 | 0.003 | −0.01, 0.02 | 6.91 × 10−1 | 7.66 × 10−1 | 9.18 × 10−1 |
| Evidence With Interaction Effects | ||||||||||||||||
| rs7642001 | 3:168746145 | MECOM | A/G | 0.37 | Current smoking | 1.10 | 1.01, 1.20 | 3.63 × 10−2 | −0.04 | −0.05, −0.02 | 3.31 × 10−5 | −0.01 | −0.03, 0.01 | 1.93 × 10−1 | 1.07 × 10−3 | 7.79 × 10−5 |
| rs28929474 | 14:94844947 | SERPINA1 | T/C | 0.02 | Ever smoking | 1.34 | 1.00, 1.81 | 5.08 × 10−2 | 0.04 | −0.02, 0.09 | 2.04 × 10−1 | 0.02 | −0.04, 0.08 | 4.65 × 10−1 | 5.53 × 10−1 | 3.41 × 10−1 |
| rs12440014 | 15:78926726 | CHRNB4 | G/C | 0.24 | Ever smoking | 0.76 | 0.69, 0.85 | 3.41 × 10−7 | 0.01 | −0.01, 0.03 | 4.95 × 10−1 | 0.03 | 0.01, 0.05 | 1.46 × 10−3 | 7.33 × 10−3 | 5.00 × 10−3 |
Abbreviations: CHRNB4, cholinergic receptor nicotinic β4 subunit; CI, confidence interval; df, degrees of freedom; EAF, effect allele frequency; EGLN2, Egl-9 family hypoxia inducible factor 2; FEV1, forced expiratory volume in 1 second; ID, identification; MECOM, myelodysplastic syndrome 1 and ecotropic viral integration site 1 complex locus; NHW, non-Hispanic White; OR, odds ratio; SERPINA1, Serpin family A member 1; SMPD3, sphingomyelin phosphodiesterase 3.
a EAF is from UK Biobank study. EAFs from COPDGene and SpiroMeta were similar.
b Marginal association between each selected variant and chronic obstructive pulmonary disease was tested in COPDGene.
c We performed lookups of selected variants in genome-wide association study of FEV1 stratified by never- and ever-smoker groups in SpiroMeta.
d 1-degree-of-freedom interaction test.
e 2-degree-of-freedom test of genetic main effects and gene-by-smoking interaction effects.
Figure 2.

Statistical significances of selected variants in multiple data sets. A) Variants presenting main effects based on the 2-degrees-of-freedom (df) joint test. Smoking exposures of rs141322661 and rs2604894 are ever-smoking and current smoking, respectively. B) Variants presenting the evidence of interaction effects based on the 1-df interaction test. Smoking exposures of rs7642001, rs28929474, and rs12440014 are current smoking, ever-smoking, and ever-smoking, respectively. For selected variants presenting the genome-wide significance from a 2-df joint test and a 1-df interaction test in the genome-wide gene-by-smoking interaction analysis, we compared the P values for a marginal genome-wide association study of chronic obstructive pulmonary disease (5) (marginal genome-wide association study (GWAS)), stratified analysis of marginal GWAS by smoking exposure (exposed and unexposed), 1-df interaction test and 2-df joint test. Summary statistics of “Marginal GWAS” are from the meta-analysis of UK Biobank (2006–2010) and the International COPD Genetics Consortium (multiple countries, 1987–2013) (5). Summary statistics of analysis testing of “Exposed,” “Unexposed,” “1-df Interaction,” and “2-df Joint” are from the analysis of UK Biobank (2006–2010).
To examine whether selected SNPs were associated with smoking behavior, we checked regions of selected SNPs in the most recent and largest GWAS of smoking itself (23) (Web Table 5). Markers at 15q25.1 and 19q13.2 were associated with cigarettes per day and current smoking.
To further characterize these potential gene-by-smoking interactions, we conducted a dose-response analysis (Web Table 6). SNPs rs7642001 at 3q26.2 (MECOM), rs28929474 in SERPINA1, rs12440014 at 15q25.1 (CHRNB4) and rs2604894 at 19q13.2 (EGLN2) showed nominally significant interactions with smoking duration on COPD risk (P < 0.05). In a dose-response analysis among ever-smokers, the significance of these SNPs was attenuated, but rs7642001 and rs12440014 were still nominally significant (P < 0.05).
Replication
To replicate our findings, we used COPDGene and summary statistics from SpiroMeta (Table 3).
COPDGene.
Given that COPDGene is enriched for heavy smokers, any SNPs showing a stronger association among the exposed group in the UKB should also show some marginal associations with COPD risk among non-Hispanic White COPDGene subjects. SNPs rs7642001 at 3q26.2 (MECOM), rs28929474 in SERPINA1, rs12440014 at 15q25.1 (CHRNB4), and rs2604894 at 19q13.2 (EGLN2) were nominally significantly associated with COPD risk (P< 0.05). In a dose-response analysis, a stronger association between rs7642001 at 3q26.2 (MECOM) and COPD was observed with longer duration of smoking (P1-df interaction = 6.20 × 10−4) (Web Table 6). For 1,937 current smokers in COPDGene with scores on the Fagerström test for nicotine dependence, rs12440014 at 15q25.1 (CHRNB4) showed evidence of interaction with higher nicotine dependence (P1-df interaction = 4.37 × 10−2).
SpiroMeta.
We replicated a significant interaction for rs12440014 at 15q25.1 (CHRNB4) with ever-smoking on FEV1 (P1-df interaction = 7.33 × 10−3), presenting as a stronger association among ever-smokers compared with never-smokers (Table 3). We observed a significant interaction for rs7642001 at 3q26.2 (MECOM) on FEV1 (P1-df interaction = 1.07 × 10−3). However, the direction of this apparent interaction effect was opposite between UKB and SpiroMeta. Allele “A” at rs7642001 was more significantly associated with decreased FEV1 among never-smokers (β = −0.04, 95% CI: −0.05, −0.02, P = 3.31 × 10−5) compared with ever-smokers (β = −0.01, 95% CI: −0.03, 0.01, P = 1.93 × 10−1). In the UKB, this SNP was more significantly associated with increased risk of COPD among current smokers (OR = 1.20, 95% CI: 1.13, 1.27, P = 4.54 × 10−10) compared with noncurrent smokers (OR = 1.07, 95% CI: 1.04, 1.09, P = 3.09 × 10−7) (Web Table 3). The stratified analyses for other measures of lung function are listed in Web Table 7.
DISCUSSION
We conducted a genome-wide association analysis of COPD accounting for a gene-by-smoking interaction to identify novel susceptibility loci and to assess the potential gene-by-smoking interactions in UKB subjects of European ancestry. Most of the significant signals in the 2-df joint test were also significant for genetic marginal effects, aside from 16q22.1 (SMPD3) and 19q13.2 (EGLN2). In the 1-df interaction test, we identified 1 genome-wide significant locus, 15q25.1 (CHRNB4) and identified PI*Z allele at SERPINA1 and 3q26.2 (MECOM) in an analysis of previously reported COPD risk loci. The estimated effects at 15q25.1 and 19q13.2, both previously described in prior GWAS of COPD or smoking, were replicated in COPDGene and SpiroMeta.
SNP rs141322661 at 16q22.1 reached genome-wide significance in previous GWAS of COPD in UKB but not in the meta-analysis of UKB and International COPD Genetics Consortium (5). SNP rs141322661, an intronic variant of the gene for SMPD3, has a low frequency in European populations (the G allele has a frequency 0.01–0.02), which makes replication difficult. Further investigation of this 16q22.1 region is required.
SNP rs2604894 at 19q13.2 could only be observed through genome-wide association analysis accounting for current-smoking interaction in UKB subjects. In the previous UKB GWAS of COPD (which did not incorporate interaction into the model), rs2604894 did not reach genome-wide significance (5). In a conventional GWAS of COPD using cohorts enriched for smokers, rs2604894 was reported to be significantly associated with COPD risk (OR = 0.74, 95% CI: 0.65, 0.84; P = 3.41 × 10−8) (24). This study included cohorts such as COPDGene (14) and ECLIPSE (25) designed to identify genetic factors for COPD by recruiting exclusively former and current smokers. The different study design between the UKB study, a population-based cohort, and cohorts at high risk of COPD because of their smoking history might have attenuated the statistical significance for rs2604894 association and hindered the replication in previous marginal GWAS in the UKB. Our finding highlights the importance of accounting for heterogeneity in genetic effects across exposure groups in association discovery studies.
SNP rs2604894 at 19q13.2 is an intronic variant of EGLN2, a gene known to be involved in regulating hypoxia tolerance and apoptosis in cardiac and skeletal muscle. Markers at 19q13.2 were reported to be associated with CPD and current smoking (23). Significant lung expression quantitative traits loci (but not including rs2604894) have been detected at 19q13.2 (26). Further functional studies of the 19q13.2 region is clearly warranted.
We identified genome-wide significant gene-by-smoking interaction effects at 15q25.1 in UKB and replicated these findings in SpiroMeta, revealing associations primarily in ever-smokers. The cholinergic receptor nicotinic α5 subunit (CHRNA5)/A3/B4 gene cluster on 15q25.1 encodes the nicotinic acetylcholine receptor subunits α5, α3, and β4. Variants in this gene cluster have been robustly associated with several lung-related traits, such as lung cancer (27) and COPD (5), as well as smoking-related phenotypes, such as smoking quantity (23, 28–30) and nicotine dependence (28). Because smoking is the most important environmental risk factor for COPD, it is quite likely that the association of variants in 15q25.1 region with COPD mediates through smoking behavior (31). Although our data does not support a direct effect of the 15q25.1 region on risk of COPD, not mediated by smoking, we note that other studies have described such a direct effect (31, 32). The 15q25.1 region also contains the gene for iron-responsive element binding protein 2 (IREB2), which has been shown to have smoking-independent effects on COPD risk (33, 34).
We noted a significant dose response for rs12440014 at 15q25.1 in UKB but not in COPDGene. This might simply be due to smaller sample sizes of COPDGene. However, given that COPDGene was enriched for severe COPD cases compared with UKB (a population-based cohort), our results could reflect other possibilities: 1) the genetic susceptibility of the 15q25.1 region on COPD could be substantial at relatively low levels of smoking exposure, and/or 2) COPD patients might be more likely to quit smoking as symptoms worsen, diluting any association between markers in 15q25.1 and COPD.
We confirmed a known gene-by-smoking interaction for COPD, a SERPINA1 (PI*Z allele)-by-smoking interaction in our study population (1, 2). In a previous study, a PI*Z-by-smoking interaction was identified for FEV1 (P = 0.03) and COPD status (P = 0.01) in subjects of European ancestry (2). SERPINA1, which encodes the alpha-1 antitrypsin protein, influences the risk of COPD (35). Homozygosity for PI*Z allele is the most common cause of alpha-1 antitrypsin deficiency. Although it is a Mendelian syndrome, there is marked variability in the development and severity of COPD among PI*ZZ individuals. Our replication in the UKB study helps to understand variable manifestations of COPD risk among individuals with alpha-1 antitrypsin deficiency.
Our objective was to identify genetic loci associated with COPD risk, which might have been missed when considering only genetic main effects in the conventional GWAS approach used in Sakornsakolpat et al. (5). However, our study incorporating potential smoking interactions did not reveal novel loci. There are several possible explanations for our lack of novel findings. First, power for detecting gene-by-smoking interactions and discovering novel genetic risk factors might be limited even in this large sample size (36, 37). It is possible that most gene-by-smoking interaction effects are relatively small, and even if presenting over a large number of genes, such interactions could be difficult to identify (11). To overcome this issue, use of a polygenic risk score, an aggregate measure of genetic variants with relatively small effect sizes, might be helpful in testing for interaction. Second, we included only independent subjects of European ancestry. Investigation of more ethnically diverse populations might allow more robust inferences of gene-by-environment interaction by increasing diversity of not only environmental exposure but also genetic determinants (38). Third, we used self-reported smoking history. Measurement errors of smoking exposure could lead to our lack of findings of gene-by-smoking interactions (6).
Despite our large sample size, there are limitations in our study. First, our use of 2 smoking measures (ever-smoking and current smoking) in genome-wide investigation might have limited interpretation of our results. Smokers with more severe COPD are more likely to reduce or quit smoking, and those without symptoms are more likely to continue smoking (often described as the “healthy smoker effect”). Such a phenomenon is highly possible in COPDGene and might also be relevant for UKB (39). Second, a “healthy volunteer” selection bias exists in UKB. The UKB cohort is not fully representative of the general population; its participants are less likely to smoke and have fewer self-reported health conditions compared with the general population of the United Kingdom (40). However, generalizability is not necessary to draw inferences about associations. The large sample size and heterogeneity of smoking exposures should still make our findings valid. Third, our use of the 2-df joint test might restrict our understanding of gene-by-smoking interaction for COPD risk or, more broadly, gene-by-environment interaction. Integration of genetic markers and other “-omics” data (transcriptomic, proteomic, or epigenomic data) could be helpful.
In summary, our genome-wide investigation incorporating smoking interaction did not identify novel susceptibility loci of COPD. However, we identified interaction effects at previously reported COPD loci, 15q25.1 (CHRNB4) and PI*Z allele in SERPINA1, on COPD risk. Cigarette smoking is the most important environmental risk factor for COPD, but individuals vary in their susceptibility to the damaging effects of cigarette smoke; it raises the possibility of detectable gene-by-smoking interactions, but we identified few significant interactions in our large-scale study. Considering diverse populations and other approaches might better help further elucidate gene-by-environment interactions on COPD risk.
Supplementary Material
ACKNOWLEDGMENTS
Author affiliations: Department of Epidemiology, Johns Hopkins School of Public Health, Baltimore, Maryland, United States (Woori Kim); Genetics and Aging Research Unit, Department of Neurology, Massachusetts General Hospital, Boston, Massachusetts, United States (Dmitry Prokopenko); Department of Medicine, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand (Phuwanat Sakornsakolpat); Channing Division of Network Medicine, Brigham and Women’s Hospital, Boston, Massachusetts, United States (Brian D. Hobbs, Edwin K. Silverman, Michael H. Cho); Division of Pulmonary and Critical Care Medicine, Brigham and Women’s Hospital, Boston, Massachusetts, United States (Brian D. Hobbs, Edwin K. Silverman, Michael H. Cho); PRecisiOn Medicine Translational Research (PROMoTeR) Center, Department of Population Medicine, Harvard Medical School and Harvard Pilgrim Health Care, Boston, Massachusetts, United States (Sharon M. Lutz); Colorado School of Public Health, University of Colorado Denver, Aurora, Colorado, United States (John E. Hokanson); Department of Health Sciences, University of Leicester, Leicester, United Kingdom (Louise V. Wain, Carl A. Melbourne, Nick Shrine, Martin D. Tobin); National Institute for Health Research, Leicester Respiratory Biomedical Research Centre, Glenfield Hospital, Leicester, United Kingdom (Louise V. Wain, Martin D. Tobin); and Department of Epidemiology, Johns Hopkins School of Public Health, Baltimore, Maryland, United States (Terri H. Beaty). W.K. is currently at the Systems Biology and Computer Science Program, Ann Romney Center for Neurological Diseases, Department of Neurology, Brigham and Women's Hospital, Boston, Massachusetts, United States.
This research was conducted by using the UK Biobank resource under application numbers 29050 (W.K.) and 20915 (M.H.C.). B.D.H. is supported by the National Institutes of Health (grant K08 HL136928) and the Parker B Francis Research Opportunity Award. S.M.L. is supported by the National Heart, Lung, and Blood Institute (grant K01HL125858). M.D.T. and L.V.W. have been supported by the Medical Research Council (award MR/N011317/1). The research was partially supported by the National Institute for Health Research, Leicester Biomedical Research Centre. M.D.T. is supported by a Wellcome Trust Investigator Award (WT202849/Z/16/Z). M.H.C was supported by the National Heart, Lung, and Blood Institute (grants R01HL113264, R01HL137927, R01HL135142, and P01HL132825). The COPDGene project was supported by the National Heart, Lung, and Blood Institute (awards U01 HL089897 and U01 HL089856). The COPDGene project is also supported by the COPD Foundation through contributions made to an Industry Advisory Board comprised of AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Novartis, Pfizer, Siemens, and Sunovion.
The views expressed are those of the author(s) and not necessarily those of the National Health Service, the National Institute for Health Research, or the Department of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
L.V.W. holds a GSK/British Lung Foundation Chair in Respiratory Research. M.H.C. has received grant funding from GSK and consulting fees from Genentech. E.K.S. has received grant support from GlaxoSmithKline and Bayer. The other authors report no conflicts.
REFERENCES
- 1. Silverman EK, Province MA, Campbell EJ, et al. . Family study of α1-antitrypsin deficiency: effects of cigarette smoking, measured genotype, and their interaction on pulmonary function and biochemical traits. Genet Epidemiol. 1992;9(5):317–331. [DOI] [PubMed] [Google Scholar]
- 2. Castaldi PJ, Demeo DL, Hersh CP, et al. . Impact of non-linear smoking effects on the identification of gene-by-smoking interactions in COPD genetics studies. Thorax. 2011;66(10):903–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hancock DB, Soler Artigas MM, Gharib SA, et al. . Genome-wide joint meta-analysis of SNP and SNP-by-smoking interaction identifies novel loci for pulmonary function. PLoS Genet. 2012;8(12):e1003098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Park B, Koo S-M, An J, et al. . Genome-wide assessment of gene-by-smoking interactions in COPD. Sci Rep. 2018;8(1):9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sakornsakolpat P, Prokopenko D, Lamontagne M, et al. . Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nat Genet. 2019;51(3):494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aschard H, Lutz S, Maus B, et al. . Challenges and opportunities in genome-wide environmental interaction (GWEI) studies. Hum Genet. 2012;131(10):1591–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kraft P, Yen Y-CC, Stram DO, et al. . Exploiting gene-environment interaction to detect genetic associations. Hum Hered. 2007;63(2):111–119. [DOI] [PubMed] [Google Scholar]
- 8. Vries PS, Brown MR, Bentley AR, et al. . Multiancestry genome-wide association study of lipid levels incorporating gene-alcohol interactions. Am J Epidemiol. 2019;188(6):1033–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bentley AR, Sung YJ, Brown MMRM, et al. . Multi-ancestry genome-wide gene-smoking interaction study of 387,272 individuals identifies new loci associated with serum lipids. Nat Genet. 2019;51(4):636–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu J, Gaddis NC, Bartz TM, et al. . Omega-3 fatty acids and genome-wide interaction analyses reveal DPP10-pulmonary function association. Am J Respir Crit Care Med. 2019;199(5):631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sung YJ, Winkler TW, Las Fuentes L, et al. . A large-scale multi-ancestry genome-wide study accounting for smoking behavior identifies multiple significant loci for blood pressure. Am J Hum Genet. 2018;102(3):375–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Justice AE, Winkler TW, Feitosa MF, et al. . Genome-wide meta-analysis of 241,258 adults accounting for smoking behaviour identifies novel loci for obesity traits. Nat Commun. 2017;8:14977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bycroft C, Freeman C, Petkova D, et al. . The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Regan EA, Hokanson JE, Murphy JR, et al. . Genetic epidemiology of COPD (COPDGene) study design. COPD. 2010;7(1):32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shrine N, Guyatt AL, Erzurumluoglu AM, et al. . New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat Genet. 2019;51(3):481–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vogelmeier CF, Criner GJ, Martinez FJ, et al. . Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 Report. GOLD Executive Summary. Am J Respir Crit Care Med. 2017;195(5):557–582. [DOI] [PubMed] [Google Scholar]
- 17. Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. 1999;159(1):179–187. [DOI] [PubMed] [Google Scholar]
- 18. Chang CC, Chow CC, Tellier LCAM, et al. . Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pruim RJ, Welch RP, Sanna S, et al. . LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang J, Ferreira T, Morris AP, et al. . Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet. 2012;44(4):369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sung YJ, Winkler TW, Manning AK, et al. . An empirical comparison of joint and stratified frameworks for studying G × E interactions: systolic blood pressure and smoking in the CHARGE Gene-Lifestyle Interactions Working Group. Genet Epidemiol. 2016;40(5):404–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hancock DB, Reginsson GW, Gaddis NC, et al. . Genome-wide meta-analysis reveals common splice site acceptor variant in CHRNA4 associated with nicotine dependence. Transl Psychiatry. 2015;5(10):e651–e651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu M, Jiang Y, Wedow R, et al. . Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51(2):237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cho MH, Castaldi PJ, Wan ES, et al. . A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Hum Mol Genet. 2012;21(4):947–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vestbo J, Anderson W, Coxson HO, et al. . Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE). Eur Respir J. 2008;31(4):869–873. [DOI] [PubMed] [Google Scholar]
- 26. Lamontagne M, Couture C, Postma DS, et al. . Refining susceptibility loci of chronic obstructive pulmonary disease with lung eQTLs. PLoS One. 2013;8(7):e70220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hung RJ, McKay JD, Gaborieau V, et al. . A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452(7187):633–637. [DOI] [PubMed] [Google Scholar]
- 28. Thorgeirsson TE, Geller F, Sulem P, et al. . A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452(7187):638–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Furberg H, Kim Y, Dackor J, et al. . Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet. 2010;42(5):441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lutz SM, Frederiksen B, Begum F, et al. . Common and rare variants genetic association analysis of cigarettes per day among ever-smokers in chronic obstructive pulmonary disease cases and controls. Nicotine Tob Res. 2019;21(6):714–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lutz SM, Hokanson JE. Genetic influences on smoking and clinical disease: understanding behavioral and biological pathways with mediation analysis. Ann Am Thorac Soc. 2014;11(7):1082–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wilk JB, Shrine NRG, Loehr LR, et al. . Genome-wide association studies identify CHRNA5/3 and HTR4 in the development of airflow obstruction. Am J Respir Crit Care Med. 2012;186(7):622–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cloonan SM, Glass K, Laucho-Contreras ME, et al. . Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med. 2016;22(2):163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Siedlinski M, Tingley D, Lipman PJ, et al. . Dissecting direct and indirect genetic effects on chronic obstructive pulmonary disease (COPD) susceptibility. Hum Genet. 2013;132(4):431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. DeMeo DL, Silverman EK. Alpha1-antitrypsin deficiency. 2: genetic aspects of alpha(1)-antitrypsin deficiency: phenotypes and genetic modifiers of emphysema risk. Thorax. 2004;59(3):259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Burton PR, Hansell AL, Fortier I, et al. . Size matters: just how big is BIG?: quantifying realistic sample size requirements for human genome epidemiology. Int J Epidemiol. 2009;38(1):263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aschard H, Tobin MD, Hancock DB, et al. . Evidence for large-scale gene-by-smoking interaction effects on pulmonary function. Int J Epidemiol. 2017;46(3):894–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ritz BR, Chatterjee N, Garcia-Closas M, et al. . Lessons learned from past gene-environment interaction successes. Am J Epidemiol. 2017;186(7):778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Becklake MR, Lalloo U. The ‘healthy smoker’: a phenomenon of health selection? Respiration. 1990;57(3):137–144. [DOI] [PubMed] [Google Scholar]
- 40. Fry A, Littlejohns TJ, Sudlow C, et al. . Comparison of sociodemographic and health-related characteristics of UK Biobank participants with those of the general population. Am J Epidemiol. 2017;186(9):1026–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.