

Graphical abstract

Keywords: SARS-COV2, ADME studies, Crystal structure, DFT, Molecular docking
Abstract
Nowadays, over 200 countries face a wellbeing emergency because of epidemiological disease COVID-19 caused by the SARS-CoV-2 virus. It will cause a very high effect on the world’s economy and the worldwide health sector. The present work is an investigation of the newly synthesized 4-benzyl-1-(2,4,6-trimethyl-benzyl)-piperidine (M1BZP) molecule’s inhibitory potential against important protein targets of SARS-CoV-2 using computational approaches. M1BZP crystallizes in monoclinic type with P1211 space group. For the title compound M1BZP, spectroscopic characterization like 1H NMR, 13C NMR, FTIR, were carried out. The geometry of the compound had been optimized by the DFT method and its results were compared with the X-ray diffraction data. The calculated energies for the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO) showed the stability and reactivity of the title compound. Intermolecular interactions in the crystal network were determined using Hirshfeld surface analyses. The molecular electrostatic potential (MEP) picture was drawn using the same level of theory to visualize the chemical reactivity and charge distribution on the molecule. Molecular docking study performed for the synthesized compound revealed an efficient interaction with the COVID-19 protease and resulted in good activities. We hope the present study would help workers in the field to develop potential vaccines and therapeutics against the novel coronavirus. Virtual ADME studies were carried out as well and a relationship between biological, electronic, and physicochemical qualifications of the target compound was determined. Toxicity prediction by computational technique for the title compound was also carried out.
1. Introduction
The recently declared global emergency following the outbreak of COVID-19 has raised awareness of tackling the disease with more effective therapeutic means. Coronavirus Disease-2019 (COVID-19) is a respiratory disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) which is a novel single-stranded RNA virus of the Coronaviridae family [1]. Although the SARS-CoV-2 virus primarily affects the respiratory system, other organ systems are also involved. Lower respiratory tract infection related symptoms including fever, dry cough, and dyspnea were reported. It is now widely recognized that respiratory symptoms of COVID-19 are extremely heterogeneous, ranging from minimal symptoms to significant hypoxia with ARDS [2]. Being RNA viruses, CoVs readily evolve by mutation and homologous and non-homologous recombination, which expands their host range and facilitates the crossing of species barriers [3]. Coronaviruses are spherical in shape. Their most prominent feature is club-like projections on the virus surface which are referred to as “spikes”. Human coronaviruses are positive-sense RNA (30 kb) viruses. Two types of proteins characterize HCoVs, structural protein [spike (S), envelope (E), membrane (M), nucleocapsid (N)], and non-structural proteins (nsp1 up to nsp16) including the RNA dependent RNA polymerase [4], [5]. Among non-structural proteins, nsp12 (RNA-dependent RNA polymerase-RdRp) is important in the replication and transcription of the viral genome [6]. For SARS-CoV and SARS-CoV2, the S protein is the primary determinant for host tropism and pathogenicity. It is the main target for neutralizing antibodies and therefore of great interest in terms of immunological response and vaccine design [7]. When we set our sights on the broad-spectrum antiviral drugs, we found that a drug unlisted, Remdesivir, has demonstrated strength in trials related to MERS-CoV and Ebola virus infection. In the United States, the first patient with COVID-19 has shown significant improvement in clinical symptoms within 24 h of treatment with Remdesivir [8], [9], [10]. This case has convinced the public that Remdesivir could become a new “specific drug” for COVID-19. Because of the urgency and current need, scientists are involved in reusing already approved drug candidates to test in COVID-19 patients. This is carried out to reduce cost, time, and also the risks involved in the drug development process. No drug or vaccine has been approved yet. Pei Liu et al identified several N-substituted compounds as potent SARS-CoV-2 3C-like protease inhibitors [11]. Sathiskumar et al tested several coumarin analogues as an inhibitor of SARS-CoV-2 [12].
Computer-aided drug design technology minimizes wastage of time and financial burden in the drug discovery process. Research in the pharmaceutical industry has shown a great increase and progress in modern computational medicinal chemistry [13]. Also, pharmacodynamic (potency, affinity, efficacy, and selectivity), pharmacokinetic (ADME: absorption, distribution, metabolism, and excretion), and toxicity data of molecules can be evaluated and analysed using that technology [14]. As a result, all these methods contribute to an efficient and selective drug discovery process [15]. In this study, we report the synthesis of the title compound M1BZP and single crystal XRD structural investigation supported by various spectroscopic characterization techniques (1H NMR, 13C NMR, FT-IR) which is validated by DFT analysis. HOMO and LUMO, Mulliken charges, and MEP of the title compound were also accounted. Further M1BZP is evaluated for its potential against COVID-19 proteins through molecular docking. Based on the ligand docking technique, inhibition against the SARS-CoV-2 virus proteins is concluded. The drug-likeness and Toxicity screening of M1BZP were screened based on ADMET properties.
2. Experimental
2.1. Materials and methods
All the chemicals and solvents were acquired from commercial sources with high purity and analar grade. Melting points were obtained in open capillaries on a Gallen Kemp melting point apparatus (Sanyo). Pre-coated silica gel plates (silica gel 0.25 mm, 60 G F 254; Merck, Germany) were used for thin-layer chromatography and chloroform/acetone (9.5:0.5 mL) mixture for developing solvent system. The IR spectra were obtained in KBr discs using JASCO 4600 (Japan) spectrometer.1H and 13C NMR spectra (in DMSO‑d 6) were recorded on Bruker Avance III, 400 MHz, 9.4 Tesla superconducting magnet using DMSO as a solvent. Single crystals obtained were characterized using a BRUKER Quest X-ray (fixed-Chi geometry) diffractometer. The goniometer was controlled using the APEX3 software suite [16]. The absorption correction program SADABS [17] was employed to correct the data for absorption effects. A solution was obtained readily using XT/XS in APEX3. The absence of additional symmetry and voids was confirmed using PLATON (ADDSYM) [18], [19], [20]. The structure was refined (weighted least-squares refinement on F2) to convergence. Olex 2 was employed for the final data presentation and structure plots [21].
2.2. Synthesis of 2-bromomethylmesitylene (M1)
Mesitylene (12.0 g, 0.10 mol), paraformaldehyde (3.08 g, 0.10 mol), 50 mL of glacial acetic acid, and 20 mL of 31 wt% HBr/acetic acid solutions were stirred at 80 °C. After 2 h, the reaction mixture was poured into 100 mL of water. The product 2-bromomethyl mesitylene was filtered and vacuum-dried at room temperature [22].
2.3. Synthesis of 4-benzyl-1-(2,4,6-trimethyl-benzyl)-piperidine (M1BZP)
Dissolve 2-bromomethylmesitylene and 4-benzyl piperidine (1:1) was dissolved separately in methanol, and heated at 80 °C, and stirred continuously for 1 h. The solid was filtered and vacuum-dried (Scheme 1 ). The dried solid was recrystallized using chloroform. Single crystal of M1BZP formed in the solution has a single spot in TLC (Rf 0.60) in silica gel and chloroform: acetone (70:30%v/v) eluent. The qualitative analysis confirmed the absence of bromide and the presence of nitrogen. The yield was 85% with m.pt. 47–49 °C. FTIR (KBr, cm−1) (Fig.S1): 3722, 3026, 2905, 2745, 1941, 1868, 1442, 1388,1263, 696. 1H‐NMR (300 MHz, DMSO‐d6, δ) (Fig.S2): 1.47–1.94 (m, piperidine-H), 2.17–2.26 (s,9H), 3.31 (s,2H), 6.77–7.27 (m, Aromatic-H). 13C NMR (100 MHz, DMSO‐d6) (Fig.S3): 20.12, 20.98, 30.21, 42.93, 52.27, 59.11, 126.13–140.85 (Aromatic-C). The recorded single crystal XRD data were deposited in CCDC, no.:1951658.
Scheme 1.

Synthesis of M1BZP.
2.4. DFT studies
To procure an in-depth knowledge about the physical and chemical properties of the above investigated crystal structure, density functional theory (DFT) calculations were performed using a 6–311 + G(d,p) basis set by GAMESS (US) software [23]. The molecular orbital structures and molecular electrostatic potential diagram are visualized using Jmol 14.30.2 [24].
2.5. Hirshfeld surface analysis
The molecular Hirshfeld surfaces analysis was carried out to validate the various types of noncovalent interaction present in the crystal structure and their 2D fingerprint plots (FPs) were generated using CrystalExplorer 17.5 [25] software.
2.6. Molecular docking studies
To verify the binding effect of the title compound (ligand) with COVID-19 proteins, molecular docking technology was performed. MGL tools 1.5.6 with AutoDock Vina were used for the molecular docking analysis to detect the preferred binding sites [26], [27]. From the Protein Data Bank, three-dimensional structure of viral proteins Spike receptor protein (2AJF), Papin-like protease (PLpro) (PDB ID: 4OVZ), Spike glycoprotein with ACE2 receptor (PDB ID: 6ACD), Main protease (PDB ID:6LU7), RNA dependent RNA polymerase (PDB ID:6M71), spike binding domain with ACE2 receptor (PDB ID:6MOJ), Spike glycol protein (PDB ID:6VSB) and SARS-CoV Main protease (PDB ID:6Y84) were downloaded in PDB format. The docking simulations were performed with AutoDock Vina by the developer command script. The interactions in the ligand–protein complex were visualized and analyzed using Biovia Discovery Studio 2020 client [28].
2.7. ADME and Toxicity prediction
Physiochemical properties of M1BZP were calculated using Osiris data warrior software. The ADME properties (adsorption, distribution, metabolism, and excretion) of M1BZP were predicted using Swiss ADME online software [29]. The chemical structure of M1BZP is drawn on Marvin to generate SMILES and inserted directly on the webpage to initiate the prediction process. The SMILES of the compound were submitted to the website and toxicity mode was selected [30].
3. Results and discussion
3.1. Spectral characterization
The IR spectrum of M1BZP is given in Fig. S1. A peak at 3722 cm−1 is assigned for the polymorphism of M1BZP due to the sp3C-N and sp3C-sp2C rotation. Absorption around 3300–2900 cm−1 is assigned to sp3C-H, sp2CH stretch, and overtones. A weak abortion around 1735 cm−1 may be assigned for partial C N for resonance. Peaks around 1600–1400 cm−1 are for aromatic C C stretching, N—H band, and sp2CH overtones. A peak at 2745 cm−1 may be assigned for the hyperconjugation in mesitylene-sp3CH-N piperidine by donating the lone pair of electrons in the nitrogen to the sp3 bridging carbon between mesitylene and piperidine as the former is an electron withdrawing group, N is the good electron donor and sp3 is a medium of electron through induction. Absorption at 1263 cm−1 is for C—N stretching. Thus M1BZP is a molecular crystal with polymorphism through single bond rotation and resonance through hyperconjugation and induction.
The H1 NMR spectrum of M1BZP (fig. S2) has eleven signals. Singlet signals at 2.17 and 2.26 ppm are assigned to the hydrogen atoms in one and two methyl groups in the mesitylene ring. The methylene (–CH2-) group attached to the nitrogen atom is deshielded and the signal appears at 3.31 ppm as a singlet. All the hydrogen atoms in the piperidine ring occur between 1.47 and 1.94 ppm. The signals for the aromatic hydrogen atoms present in M1BZP appear between 6.77 and 7.27 ppm in the downfield. The 13C NMR spectrum of M1BZP (fig. S3) has nearly fifteen signals. The three aliphatic methyl carbon atoms in the mesitylene ring show signals at 20.12 and 20.98 ppm. Eight signals for the aromatic carbon atoms appear between 126.13 and 140.85 ppm in the downfield.
3.2. Crystal growth and XRD
Single crystal X-ray structural analysis confirmed that the M1BZP is crystallized in a monoclinic P 1 21 1 space group with two independent molecules in its unit cell (Z = 2). Fig. 1 emphasizes the thermal ellipsoid of the title compound. Structural and bond parameter values are given in Table 1 and supplementary table S1. Fig. 2 displays the cell content in M1BZP. It has a ‘U’ shape structure with two limbs which are benzyl and mesitylene rings with a difference of 72° from the piperidine ring in the base. The crystal structure of M1BZP revealed that the nitrogen atom of the piperidine ring distorts the planarity of the structure by twisting C10 about 0.148 Å from the plane of the mesityl ring. The distortion in planarity due to nitrogen atom is reflected from the dihedral angles −2.1(4) ° and 176.2(2) ° for C10-C1-C2-C7 and C10-N1-C11-C12 respectively. Bond lengths of N1-C10 and C1-C10 are 1.472(4) and 1.512(4) Å respectively. In M1BZP, the torsion angles of C14-C13-C16-C17 and C15-N1-C10-C1 are 61.6(3) ° and −60.2(3) ° respectively. It shows that the benzyl and mesityl rings are in an acute position from the central piperidine ring.
Fig. 1.

Thermal Ellipsoid View of M1BZP.
Table 1.
Crystal data and structure refinement for M1BZP.
| Identification code | m1bzpa | |
|---|---|---|
| Empirical formula | C22 H29 N | |
| Formula weight | 307.46 | |
| Temperature | 110.0 K | |
| Wavelength | 0.71073 Å | |
| Crystal system | Monoclinic | |
| Space group | P 1 21 1 | |
| Unit cell dimensions | a = 10.1081(4) Å | α = 90°. |
| b = 5.5022(2) Å | β = 96.6350(10)°. | |
| c = 16.2450(7) Å | γ = 90°. | |
| Volume | 897.44(6) Å3 | |
| Z | 2 | |
| Density (calculated) | 1.138 Mg/m3 | |
| Absorption coefficient | 0.065 mm−1 | |
| F(0 0 0) | 336 | |
| Crystal size | 0.483 × 0.416 × 0.059 mm3 | |
| Theta range for data collection | 2.262 to 24.997°. | |
| Index ranges | −12≤h≤12, −6≤k≤6, −19≤l≤19 | |
| Reflections collected | 21,462 | |
| Independent reflections | 3160 [R(int) = 0.0661] | |
| Completeness to theta = 24.997° | 99.8% | |
| Absorption correction | Semi-empirical from equivalents | |
| Max. and min. transmission | 0.7456 and 0.3105 | |
| Refinement method | Full-matrix least-squares on F2 | |
| Data / restraints / parameters | 3160 / 1 / 212 | |
| Goodness-of-fit on F2 | 1.041 | |
| Final R indices [I greater than 2sigma(I)] | R1 = 0.0599, wR2 = 0.1593 | |
| R indices (all data) | R1 = 0.0617, wR2 = 0.1616 | |
| Absolute structure parameter | −8.0(10) | |
| Extinction coefficient | 0.07(2) | |
| Largest diff. peak and hole | 0.179 and −0.185 e.Å−3 |
Fig. 2.

Unit cell packing arrangement viewed through crystallographic axis a*
Crystal structure analysis of the compound revealed the presence of a large number of non-covalent interactions such as inter and intramolecular hydrogen bond interactions, C—H…π, π… π stacking, and inter short interactions [31] (Fig. 3 ). Weak intermolecular hydrogen bonding interaction was observed in M1BZP between the hydrogen atom (H10) of the mesityl ring (donor) and the nitrogen atom (N1) of the neighbouring piperidine ring (acceptor). H-bonds are formed between C—H····π with an average length of 2.7 Å. The C—H····π interactions are observed between the π-system of the benzene ring with the C16-H16 atoms of the neighbouring piperidine ring. The van der Waals repulsion shows that the nitrogen atom was sandwiched between mesitylene and piperidine rings. M1BZP exhibited π-π stacking supramolecular interactions in the crystal due to their aromatic moieties with a short contact distance of < 3.2 Ǻ. There are three π-π stacking interactions [First π-π: 3.1 Ǻ, the centroid of mesityl ring….centroid of piperidine ring, second π-π: 2.95 Ǻ, centroid of piperidine…centroid of benzyl ring and third π-π: 2.840 Ǻ, centroid of benzyl ring…centroid of mesityl ring]. C—H…N and C—H…C intermolecular hydrogen bond interactions along with C..H short interactions played a significant role in stabilizing the molecular structure. These π-π stacking interactions created a perpendicular arrangement of piperidine moiety and benzyl rings. One M1BZP molecule is connected to five other molecules through weak C—H…N, C—H…π and π-π interactions. The weak intermolecular interactions and slip stacking arrangement [32] lead to the formation of different types of packing structures like sheets, tubes, and layered structures (Fig. 4 ). Nitrogen plays a major role in the formation of different packing structures by acting as a bridge between the layers.
Fig. 3.

Molecular interactions viewed through crystallographic axis a*
Fig. 4.

Different Packing arrangement of M1BZP.
3.3. Hirshfeld surface analysis
The Hirshfeld surface denotes the 3D image of intermolecular interactions that occur in the molecules. This study plays a valuable graphical tool to understand the intermolecular interactions in depth. Hirshfeld surface and fingerprint plots were generated for the title compound. A three-dimensional dnorm surface is a useful tool for analyzing and visualizing the intermolecular interactions. White color indicates medium proximity of outside atoms and red and blue colors indicate close or little proximity. Red spots on the Hirshfeld surface are related to the hydrogen bonding interactions. As shown in Fig. 5 (c), the red region on the Hirshfeld surface is prominent above the hydrogen atom in the C—H--N contacts and the H—C..H contacts, respectively. The red spots on the di (Fig. 5a) and de (Fig. 5b) surface indicate the existence of close contacts available inside and outside the surface. The shape-index (Fig. 5d) of the HS can visualize π-π stacking interactions by the presence of adjacent red and blue triangles. The curvedness (Fig. 5e) surface indicates the electron density surface around the molecular interactions. Moreover, the flat green region separated by blue edges in the curvedness surface is another characteristic of π-π stacking interactions.
Fig. 5.

View of the three-dimensional Hirshfeld surface of M1BZP.
The percentage contributions of intermolecular hydrogen bond in the molecule are calculated using a 2D fingerprint plot from Hirshfeld surface analysis. The intermolecular interaction pattern outlined as H….H / C…H is shown in Fig. 6 . The H….H contribution contact in the Hirshfeld surface of the studied molecule is 80.7%. The presence of C….H / H….C contribution in Hirshfeld surface is 19.3%. The most dominant interaction in the title molecule is indicated by the symmetrical narrow point spike with di + de ~ 2.2Ǻ corresponding to H….H interaction as shown in Fig. 6, similarly, the shark characteristic wing of di + de ~ 3.6 Ǻ corresponding to C…..H / H…..C are shown in Fig. 6.
Fig. 6.

The full two-dimensional fingerprint plots for the compound M1BZP.
3.4. DFT Studies:
The optimized geometry and electronic parameters (bond lengths, bond angles, EHOMO-ELUMO) of the compound were done using the B3LYP/6–311++G(d,p) level of theory. The optimized geometry of compound M1BZP is given in Fig. 7 . The optimized structure results are consistent with X-ray diffraction data. The geometrical parameters (bond lengths and bond angles) of the structure are compared and exhibited in the supplementary Table S2. FMO theory explicates the structure, reactivity, electrical and optical properties of compound [33], [34]. The stabilized ground state energy of the system with the minima point is found to be −910.520 Hartree. The HOMO and LUMO [35] structures are displayed in Fig. 8a . The HOMO-LUMO energy gap (Eg) of the title molecule is −5.7390 eV, which has a significant effect on the stability and reactivity of the molecule [36]. The large bandgap of the compound indicates high stability, less conjugation, and poor conductivity. The interaction taking place within the molecule is responsible for imparting bioactivity [37]. EHOMO and ELUMO values give details of the certain charge exchange collaboration inside the studied molecule. The HOMO energy determines the electron-donating ability. However, the LUMO energy provides the ability of a molecule to accept an electron [38]. The corresponding EHOMO and ELUMO values were found to be −5.6190 eV and 0.1200 eV respectively. The global reactive parameters such as ionization energy (I), electron affinity (A), electronegativity(χ), hardness (η), softness (σ), chemical potential (μ) and electrophilicity index (ω) of the molecule are calculated and recorded in Table 2 .
Fig. 7.

Optimized Molecular structure of M1BZP.
Fig. 8a.
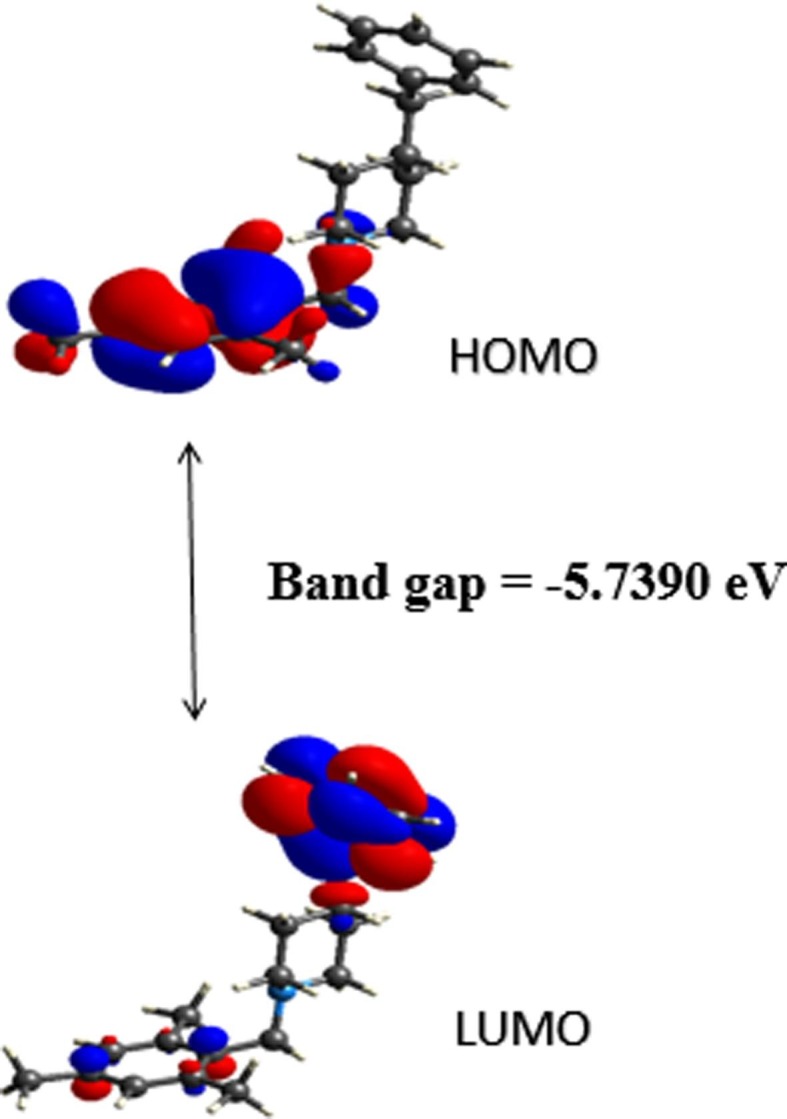
Calculated frontier molecular orbital profile.
Table 2.
The energy and related quantum chemical parameters values of M1BZP.
| Parameters | M1BZP |
|---|---|
| HOMO | −5.6190 |
| LUMO | 0.1200 |
| Band gap | −5.7390 |
| Ionisation potential | 5.6190 |
| Electron affinity | −0.1200 |
| Chemical potential | −2.7495 |
| Chemical hardness | 2.8695 |
| Global softness | 0.3485 |
| Electrophilic index | 10.8464 |
| Absolute electronegativity | 2.7495 |
| Q-max | 0.9582 |
The molecular electrostatic potential (MEP) was defined in terms of the total charge distribution of a molecule and it is a convenient tool used in drug design [39], [40], [41]. The optimized geometry at the B3LYP/6–311++G(d,p) level of theory was used to map the molecular electrostatic potential surface (MEPS) of the compound as shown in Fig. 8b . It helps to investigate the interaction between molecules, predict reaction sites, and molecular properties. Electron density is essential to understand the electrophilic or nucleophilic reactivity as well as non-bonding interactions [42] at the MEP surface. Different colours represent different electrostatic potential values. Red and blue colours indicate electron-rich and electron-poor regions respectively, and green indicates zero electrostatic potential.
Fig. 8b.

Graphical view of the molecular electrostatic potential (MEP) map.
Mulliken charges were calculated at the B3LYP/6–311++G(d,p) basis set are listed in Table 3 . The nitrogen atom of the piperidine ring is found to possess a high negative charge with a value of −0.4177e when compared with other atoms. This is mainly due to its participation in C—H…N hydrogen bonding interactions. The carbon atoms C3, C5, C7, C8, C12, C25, C26, C27, C28, C29, C32, C38, C39, C40, C41, C42 acquire a negative charge while other carbon atoms of the molecule acquire a positive charge. Among the carbon atoms, C4 possesses the highest positive value and C8 has the highest negative value. The highest positive charge on C4 is due to the adjacent electronegative carbon atoms. All the hydrogen atoms of the molecule are positively charged out of which H21 (0.1397 e) has the highest charge.
Table 3.
Mulliken Charge analysis of M1BZP.
| Atom | Charge | Atom | Charge |
|---|---|---|---|
| C1 | 0.0162 | C27 | −0.0566 |
| C2 | 0.1025 | C28 | −0.1805 |
| C3 | −0.1711 | C29 | −0.0222 |
| C4 | 0.1426 | H30 | 0.1093 |
| C5 | −0.1710 | H31 | 0.0998 |
| C6 | 0.1105 | C32 | −0.2411 |
| C7 | −0.0878 | H33 | 0.0977 |
| C8 | −0.3851 | H34 | 0.0915 |
| H9 | 0.0631 | H35 | 0.0921 |
| C10 | −0.3804 | H36 | 0.0971 |
| H11 | 0.0640 | C37 | 0.1228 |
| C12 | −0.3836 | C38 | −0.1256 |
| H13 | 0.1201 | C39 | −0.0839 |
| H14 | 0.1048 | C40 | −0.0875 |
| H15 | 0.1195 | C41 | −0.0849 |
| H16 | 0.1113 | C42 | −0.1171 |
| H17 | 0.1235 | H43 | 0.0795 |
| H18 | 0.1111 | H44 | 0.0827 |
| H19 | 0.0987 | H45 | 0.0821 |
| H20 | 0.1098 | H46 | 0.0825 |
| H21 | 0.1397 | H47 | 0.0765 |
| H22 | 0.0726 | H48 | 0.0643 |
| H23 | 0.0949 | H49 | 0.0848 |
| N24 | −0.4177 | H50 | 0.0818 |
| C25 | −0.0316 | H51 | 0.0944 |
| C26 | −0.1792 | H52 | 0.0642 |
3.5. Molecular docking studies
According to molecular docking studies, a ligand molecule may reduce or eliminate the SARS-CoV-2 by targeting different stages of the virus life cycle in the host cell. When the compound was analyzed by in silico computational docking, it was found to be successfully docked against the inhibitor region of the main protease, RNA dependent RNA polymerase, spike protein, and spike binding domain with ACE2 receptor of SARS-CoV-2 virus.
3.5.1. Interaction of spike receptor protein (2AJF) with M1BZP
The molecular docking analysis of M1BZP and Remdesivir with SARS-CoV-2 spike receptor protein [43] (Fig. 9 ) showed −8.2 and −8.0 KJ/mol free energy respectively, suggesting a high affinity for the binding pocket. It is observed that binding conformations of M1BZP are found in the active binding sites. The title compound binds with Trp349, Asp350, and His401 through hydrophobic interactions such as π-π stacking interactions. It has hydrogen bonding interactions with amino acid residues such as Asp382, Ala348, and Asn394 with an average distance of 2.15Ǻ. In the case of Remdesivir, hydrophobic interaction is observed between His401 and Hydrogen bonding interaction with Trp349. Thus, it is observed that M1BZP best binds with SARS Spike protein than Remdesivir.
Fig. 9.

Interaction of Spike receptor protein (2AJF) with M1BZP and Remdesivir.
3.5.2. Interaction of Papin-like protease (PLpro) (4OVZ) with M1BZP
The docking studies of M1BZP and Remdesivir with the papin-like protease (4OVZ) [44] were carried out (Fig. 10 ). M1BZP has π-π stacking interactions with the Tyr269, Tyr265, Glu204, Glu162, and Glu165 amino acids with an average bond length of 4.30Ǻ. It also has hydrogen bonding interactions with Tyr269, Arg167, Asn157, and Lys158 and hydrophobic interactions with Pro249 and Try208. The binding energy of papin-like protease with M1BZP is found to be −9.7 kcal/mole. In the case of standard drug, it has hydrophobic interactions with Tyr265, Tyr269, Leu163, Pro224, Met209, and hydrogen bonding interactions with the Asp165 amino acids. The binding energy of Remdesivir is found to be −9.4 kcal/mole. The above results revealed that M1BZP favours Papin-like protease over Remdesivir.
Fig. 10.

Interaction of Papin-like protease (PLpro) (4OVZ) with M1BZP and Remdesivir.
3.5.3. Interaction of spike glycoprotein with ACE2 receptor (6ACD) with M1BZP
For M1BZP, three types of interactions are observed with the spike glycoprotein [45] (Fig. 11 ). It has hydrogen bonding interactions with the amino acids Arg996, Gln939, Gln992, Arg100, and Gln936. It also has hydrophobic interaction with Ala940, Leu944, Ala940 involving the carbon atoms of the piperidine rings. Electrostatic interaction is observed between the nitrogen atom of the piperidine ring and Arg996 with a binding energy of −7.4 kcal/mole. On the other hand, Remdesivir gives a binding affinity of −6.0 kcal/mole on binding with spike glycoprotein. It has hydrogen bonding interactions with Leu944, Ala748, Arg747, Ala940, and one hydrophobic interaction with Thr988 amino acid. This result also has revealed that M1BZP showed good binding with the spike glycoprotein than Remdesivir.
Fig. 11.

Interaction of Spike glycoprotein with ACE2 receptor (6ACD) with M1BZP and Remdesivir.
3.5.4. Interaction of the Main protease (6LU7) with M1BZP
Molecular docking of M1BZP and Remdesivir with SARS-CoV-2 spike binding domain with Main protease [46] were analyzed and compared. Binding free energy of −6.2 and −5.4KJ/mol for M1BZP and Remdesivir (Fig. 12 ) in the grid box were explored. These results suggest the high affinity of M1BZP towards different amino acids Arg4, Lys137 with hydrophobic interactions. It has electrostatic interactions with Glu288, Arg4 amino acids. And hydrogen bonding interactions are observed between M1BZP with Lys5. In Remdesivir, there is a hydrogen bonding interaction with Phe291 and a hydrophobic interaction with Cys128. It is observed that the SARS-CoV-2 spike binding domain with the Main protease complex more favourably binds with M1BZP than Remdesivir.
Fig. 12.

Interaction of Main protease (6LU7) with M1BZP and Remdesivir.
3.5.5. Interaction of RNA dependent RNA polymerase (6 M71) with M1BZP
The docking analysis of M1BZP and Remdesivir with SARS-CoV-2 RNA-dependent RNA polymerase [47] were carried out and the results revealed that free energy of M1BZP (-7.4KJ/mol) dominates Remdesivir (-6.0KJ/mol) in the grid box, suggesting a high affinity towards the binding pocket (Fig. 13 ). M1BZP binds with the binding pockets of protein through hydrogen bonding interactions with Arg553, Arg555, Thr556, Lys621, Cys622, Asn691, Thr556 amino acids, and hydrophobic interactions with Cys622, Tyr445, Arg624. In Remdesivir hydrophobic interactions with Arg553, Lys621, Tyr455, Asp760, and hydrogen bonding interaction with Asp623 amino acid were found. In the case of RNA-dependent RNA polymerase, M1BZP binding is more favourable.
Fig. 13.

Interaction of RNA dependent RNA polymerase (6M71) with M1BZP and Remdesivir.
3.5.6. Interaction of spike binding domain with ACE2 receptor (6MOJ) with M1BZP
The docking analysis of the M1BZP and Remdesivir with SARS-CoV-2 spike binding domain with ACE2 receptor [48] (Fig. 14 ) results in −4.5 and −4.4 KJ/mol free energy respectively in the grid box, suggesting a high affinity for the binding pocket. All the binding modes of the compound are in the active binding pocket. The M1BZP binds with the amino acid residues Val151, Ala154, Leu121, Met158, His159, Ala154, Leu188 through hydrophobic interactions and Remdesivir binds with Val130, His159, Glu125 amino acids through hydrogen bonding interactions. Thus, it is observed that M1BZP favours SARS-CoV-2 spike binding domain with the ACE2 receptor complex over Remdesivir.
Fig. 14.

Interaction of spike binding domain with ACE2 receptor (6MOJ) with M1BZP and Remdesivir.
3.5.7. Interaction of spike glycol protein (6VSB) with M1BZP
The molecular docking analysis M1BZP and Remdesivir with SARS-CoV-2 spike glycol protein [49] revealed −3.9 and −3.6KJ/mol free energy respectively (Fig. 15 ), suggesting a high affinity for the binding pocket. It is observed that binding conformations of the M1BZP are in the active binding sites. M1BZP compound binds with Val100, Ala766, Leu763, and Leu101 through hydrophobic interactions. It also has hydrogen bonding interaction with Thr100, Gln100, Phe759, Gln762 with an average distance of 2.5Ǻ. Remdesivir has Hydrogen bonding interactions with Gln100, Thr100, Gln101 and hydrophobic interactions with Ala766, Leu763, Val100 amino acids. Thus, it is observed that M1BZP binds best with SARS Spike protein than Remdesivir.
Fig. 15.

Interaction of Spike glycol protein (6VSB) with M1BZP and Remdesivir.
3.5.8. Interaction of SARS-CoV Main protease (6Y84) with M1BZP
The molecular docking studies of SARS-CoV Main protease [50] with M1BZP and Remdesivir show binding affinity of −6.5 kcal/mol and −5.0 kcal/mol respectively. In M1BZP the binding pockets are Gln110 and Ser158 through hydrogen bonding interactions and Val104, Ile106, Cys160 through hydrophobic interactions. In Remdesivir, there is one hydrophobic interaction with Val104 and one hydrophobic interaction with Gln110. Thus, it is observed that the binding of M1BZP with SARS-CoV Main protease is more favourable than Remdesivir (Fig. 16 ).
Fig. 16.

Interaction of SARS-CoV Main protease (6Y84) with M1BZP and Remdesivir.
Based on the docking studies, M1BZP shows good orientations with all the proteins of SARS-Cov and also shows good binding affinities. This may be due to the flexible nature of M1BZP having U shaped structure.
3.6. ADME and Toxicity Prediction:
Many drugs fail in the drug development process due to their poor pharmacokinetics and toxicity problems. Drawbacks arising during drug development could be addressed at the early stage in the pipeline of this process [51]. To analyse this issue of new chemical compounds, in silico ADMET methods are the first step in this pipeline process. These methods avoid wasting time on a lead compound that would be toxic or metabolized by the body into an inactive form and unable to cross membranes. These ADMET parameters disclose the behaviour of the chemical compounds in a living organism. Understanding physicochemical properties are necessary for designing new pharmacological compounds. A drug-likeness profile can be evaluated through the parameters of the molecule such as molecular weight, TPSA, number of heavy atoms, HBA, HBD, rotatable bonds, and molar refractivity. The physicochemical parameters of M1BZP were calculated using Osiris data warrior and depicted in Table 4 . Bioavailability radar shows that all parameters such as XLOGP3 = 4.25 (-0.7 to + 5.0), Molecular weight = 307.47 g/mol (150 to 500 g/mol), Solubility (Log S) = -5.36 (log S not higher than 6), Flexibility (FLEX) = 4 (not more than 9 rotatable bonds) lie within the acceptable range except Saturation (INSATU) = 0.45 (not<0.25) [29], [52]. The M1BZP shows drug-likeness from all methods such as Lipinski, Ghose, Veber, Egan, and Muegge. The results of these predictions showed that the compound obeyed all the five rules along with the standard without any violation of the bioactivity score of 0.55. Further, the synthetic accessibility of the compound was assessed to quantify the complexity of the molecular structure. The results showed that the compound does not have a complex synthetic route based on their score in the range of 2.52.
Table 4.
Physicochemical parameters generated using Osiris datawarrior.
| Property Name | Property Value |
|---|---|
| Molecular Weight | 307.47 g/mol |
| Clog P | 5.1584 |
| Clog S | −4.413 |
| Hydrogen Bond Donor Count | 0 |
| Hydrogen Bond Acceptor Count | 1 |
| Rotatable Bond Count | 4 |
| Total Surface Area | 261.66 |
| Relative PSA | 0.01356 |
| PSA | 3.24 |
| Shape Index | 0.6521 |
| Molecular Flexibility | 0.4815 |
| Molecular Complexity | 0.6920 |
| Stereo Centres | 0 |
| Druglikness | 4.2521 |
The mean predicted lipophilicity values were assessed to decide the non-aqueous solubility of the compound and were calculated by considering the consensus log Po/w. According to this, if a molecule is more soluble, then its consensus log Po/w values will be more negative. Results showed that the compound is not soluble in a non-aqueous medium and to estimate the aqueous solubility log S scale was used: According to the log S scale the compound is moderately soluble (-5.3). (if log S < − 10 - poorly soluble, < −6 - moderately soluble, < −4 - soluble, < −2 - very soluble, and < 0 highly soluble) [53], [54], [55], [56].
The pharmacokinetic parameters like absorption, skin permeation, distribution, and metabolism/biotransformation/excretion were predicted. For gastrointestinal absorption and passive brain permeation Brain Or IntestinaL EstimateD permeation method (BOILED-Egg) is known to predict an accurate result. According to this predictive model (Fig. 17 ) molecule falls in the yellow region which indicates passive brain permeation. It is evident from this predictive result that the compound had high GI absorption and high BBB permeant as tabulated in Table 5 . The compound being blood–brain permeant there may be a possibility of causing harmful toxicants in the brain and bloodstream when metabolized. The skin permeability model can predict and identify potential drugs for oral and transdermal administration. As per the model, if a molecule has more negative log Kp value it is said to be less skin permeant. Based on this the compound was found to be the least skin permeant.
Fig. 17.

(a) The BOILED-Egg model and (b) Bioavailability radar of M1BZP.
Table 5.
Pharmacokinetics of M1BZP evaluated using SwissADME tool.
| Parameters | Pharmacokinetic |
|---|---|
| GI Absorption | High |
| BBB Permeant | Yes |
| P-gp Substrate | No |
| CYP1A2 Inhibitor | No |
| CYP2C19 Inhibitor | No |
| CYP2C9 Inhibitor | No |
| CYP2D6 Inhibitor | Yes |
| CYP3A4 Inhibitor | No |
| Log KP (Skin permeation) | −4.24 cm/s |
The permeability glycoprotein (P-gp) is an important protein that plays a significant role in drug absorption and disposition. Hence, the compound was evaluated to determine whether the compound can act as a substrate or an inhibitor of P-gp and results revealed that the compound is found to be non-substrates of P-gp. Interaction of molecules with cytochrome P450 (CYP) enzymes is essential because these isoenzymes are involved in drug elimination through metabolic transformation. Inhibition of these isoenzymes may result in unwanted adverse side-effects by lowering the solubility and the accumulation of the drug or its metabolites. Prediction reveals that the compound act as a non-inhibitor for CYP2C9 whereas CYP1A2 acts as inhibitors of CYP2D6. Compound M1BZP is predicted to be CYP3A4 non-substrate and for the isoenzyme, CYP2C19 were non-inhibitors.
Toxicity prediction by computational technique is one of the new techniques in the drug development process, the modern era which consumes less time and low cost. Toxicity prediction of chemical entities is important to predict the amount of tolerability of the chemical entities before entering into in-vivo studies. The maximum tolerated dose of M1BZP is 0.383(log mg/kg_bw/day). The compound M1BZP shows chronic toxicity is 1.233(log mg/kg_bw/day). The Predicted LD50 values ranged from 2.77 to 2.783 mol/kg. The liver plays a critical role in energy exchanges and the biotransformation of xenobiotics and drugs. Liver suffering from damage always disrupts normal metabolism and could even lead to liver failure. The hepatotoxicity descriptor predicted that a molecule could present hepatotoxicity. The toxicity was predicted using the pkCSM online tool [57] and its results indicate that the compound M1BZP is less toxic (Table 6 ). Thus, these in silico properties are appreciable. Therefore, further studies of in vitro and clinical studies dealing with SARS-CoV-2 would be carried out.
Table 6.
Toxicology of M1BZP evaluated using SwissADME tool.
| Parameters | Toxicity |
|---|---|
| AMES Toxicity | Less toxic |
| Max. Tolerated Dose (human) | 0.383 |
| HERG 1 Inhibitor | No |
| HERG 2 Inhibitor | Yes |
| Oral Rate Acute Toxicity (LDSO) | 2.783 mol/kg |
| Oral Rate Chronic Toxicity (LOAEL) | 1.233(log mg/kg_bw/day) |
| Hepatotoxicity | Yes |
| Skin Sensitization | No |
| T. Pyriformis Toxicity | 1.727 log mg/L |
| Minnow Toxicity | −0.109 log mM |
3.7. Conclusion
The structures of potential targets of COVID-19 were chosen based upon the literature of the coronavirus life cycle. In the aim of discovering a new potent drug against COVID-19, M1BZP was applied against eight proteins and found to be higher activity than expected. Molecular docking was performed to predict the binding modes between the targets and M1BZP. The docking results produced by autodock vina models reveal the important sites where steric, electrostatic, and hydrophobic interactions might significantly influencing the activity of the molecules. X-ray structural analysis of M1BZP revealed several intermolecular interactions (hydrogen bond, short interactions, C—H…π and π… π) which greatly contributed to the ligand–protein binding process. These interactions are well quantified by Hirshfeld surface analysis. The compound was predicted to display low toxicity levels and showed drug-likeness according to the Lipinski and Veber rules. Thus, these in silico properties are appreciable. The compound shows good binding with the papin like protease. Based on the comparison with Remdesivir, the M1BZP molecule showed good interaction with target proteins, which may be a competitive drug alternative for Remdesivir.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioorg.2021.104967.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Song Z., Yanfeng X.u., Bao L., Zhang L., Pin Y.u., Yajin Q.u., Zhu H.a., Zhao W., Han Y., Qin C. From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses. 2019;11:59. doi: 10.3390/v11010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson B.T., Chambers R.C., Liu K.D. Acute respiratory distress syndrome. N. Engl. J. Med. 2017;377:562–572. doi: 10.1056/NEJMra1608077. [DOI] [PubMed] [Google Scholar]
- 3.Simon J Anthony, Christine K Johnson, Denise J Greig, Sarah Kramer, Xiaoyu Che, Heather Wells, Allison L Hicks, Damien O Joly, Nathan D Wolfe, Peter Daszak, William Karesh, W I Lipkin, Stephen S Morse, PREDICT Consortium; Jonna A K Mazet, Tracey Goldstein, Global patterns in coronavirus diversity, Virus Evol, 2017 3 (1) vex012. [DOI] [PMC free article] [PubMed]
- 4.Fehr A.R., Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol. Biol. 2015;1282:1–23. doi: 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elfiky A.A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sciences. 2020;248 doi: 10.1016/j.lfs.2020.117477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romano M., Ruggiero A., Squeglia F., Maga G., Berisio R. A Structural View of SARS-CoV-2 RNA Replication Machinery. RNA Synthesis, Proofreading, and Final Capping. Cells. 2020;9:1267. doi: 10.3390/cells9051267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hulswit R.J., de Haan C.A., Bosch B.J. Coronavirus Spike Protein and Tropism Changes. Adv. Virus Res. 2016;96:29–57. doi: 10.1016/bs.aivir.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J. Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. Journal of Chemical Information and Modeling. 2020;60:3277–3286. doi: 10.1021/acs.jcim.0c00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vijayan V., Pant P., Vikram N., Kaur P., Singh T.P., Sharma S., Sharma P. Identification of promising drug candidates against NSP16 of SARS-CoV-2 through computational drug repurposing study. Journal of Biomolecular Structure and Dynamics. 2020:1–15. doi: 10.1080/07391102.2020.1802349. [DOI] [PubMed] [Google Scholar]
- 10.Fischer A., Sellner M., Neranjan S., Smieško M., Lill M.A. Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds. International Journal of Molecular Sciences. 2020;21:3626. doi: 10.3390/ijms21103626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pei Liu, Hongbo Liu, Qi Sun, Hao Liang, Chunmei Li, Xiaobing Deng, Ying Liu, Luhua Lai, Potent inhibitors of SARS-CoV-2 3C-like protease derived from N-substituted isatin compounds, European Journal of Medicinal chemistry, 2020 (206) 112702. [DOI] [PMC free article] [PubMed]
- 12.Chidambaram S., El-Sheikh M.A., Alfarhan A.H., Radhakrishnan S., Akbar I. Synthesis of novel coumarin analogues: Investigation of molecular docking interaction of SARS-CoV-2 proteins with natural and synthetic coumarin analogues and their pharmacokinetics studies. Saudi Journal of Biological Sciences. 2021;28:1100–1108. doi: 10.1016/j.sjbs.2020.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ooms F. Molecular modeling and computer-aided drug design. Examples of their applications in medicinal chemistry. Curr. Med. Chem. 2000;7:141–158. doi: 10.2174/0929867003375317. [DOI] [PubMed] [Google Scholar]
- 14.S.V. Jain, M. Ghate, K.S. Bhadoriya, S.B. Bari, A. Chaudhari, J.S. Borse, 2D, 3D- QSAR and docking studies of 1, 2, 3-thiadiazole thioacetanilides analogues as potent HIV-1 non-nucleoside reverse transcriptase inhibitors, Organic Med. Chem. Lett. 2 2012 (1) 22. [DOI] [PMC free article] [PubMed]
- 15.Zhang J., Shan Y., Pan X., Wang C., Xu W., He L. Molecular docking, 3D-QSAR Studies, and in silico ADME prediction of p-aminosalicylic acid derivatives as neuraminidase inhibitors. Chem. Biol. Drug Des. 2011;78:709–717. doi: 10.1111/j.1747-0285.2011.01179.x. [DOI] [PubMed] [Google Scholar]
- 16.APEX3 “Program For Data Collection On Area Detectors” BRUKER AXS Inc., 5465 East Cheryl Parkway, Madison, USA WI 53711–5373.
- 17.SADABS, G.M. Sheldrick, “Program For Absorption Correction of Area Detector Frames”, BRUKER AXS Inc., 5465 East Cheryl Parkway, Madison, WI 53711–5373 USA.
- 18.Sheldrick G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2008;A64:112–122. doi: 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheldrick G.M. SHELXT –Integrated space-group and crystal-structure. Acta Cryst. 2015;A71:3–8. doi: 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheldrick G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015;C71:3–8. doi: 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dolomanov O.V., Bourhis L.J., Gildea R.J., Howard J.A.K., Puschmann H. OLEX2 < /i>; a complete structure solution, refinement, and analysis program. J. Appl. Cryst. 2009;42:339–341. [Google Scholar]
- 22.Van der Made A.W., Van der Made R.H. A convenient procedure for bromomethylation of aromatic compounds: selective mono-, bis-, or trisbromomethylation. J. Org. Chem. 1993;58:1262–1263. [Google Scholar]
- 23.Schmidt M.W., Baldridge K.K., Boatz J.A., Elbert S.T., Gordon M.S., Jensen J.H., Koseki S., Matsunaga N., Nguyen K.A., Su S., Windus T.L., Dupuis M., Montgomery J.A. General atomic and molecular electronic structure system. Journal of Computational Chemistry. 1993;14:1347–1363. [Google Scholar]
- 24.Hanson R.M. ‘Jmol SMILES and Jmol SMARTS: specifications and applications’, Journal of. Cheminformatics. 2016;8:50. doi: 10.1186/s13321-016-0160-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKinnon J.J., Jayatilaka D., Spackman M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007;37:3814–3816. doi: 10.1039/b704980c. [DOI] [PubMed] [Google Scholar]
- 26.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trott O., Olson A.J. Auto Dock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herowatia R., Widodo G.P. Molecular docking studies of chemical constituents of Tinosporacordifolia on glycogen phosphorylase. Procedia Chem. 2014;13:63–68. [Google Scholar]
- 29.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific reports. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pires D.E., Blundell T.L., Ascher D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. Journal of medicinal chemistry. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeffrey G.A., Saenger W. Springer; London: 1994. Hydrogen Bonding in Biological Structures. [Google Scholar]
- 32.Yao Z.F., Wang J.Y., Pei J. Control of p–p stacking via crystal engineering in organic conjugated small molecule crystals. Cryst. Growth Des. 2018;18:7–15. [Google Scholar]
- 33.Almeida M.O., Barros D.A., Araujo S.C., Faria S.H., Maltarollo V.G., Honorio K.M. Study on molecular structure, spectroscopic properties (FTIR and UV–Vis), NBO, QTAIM, HOMO-LUMO energies and docking studies of 5-fluorouracil, a substance used to treat cancer. Spectrochim. Acta A. Mol. Biomol. Spectrosc. 2017;5:169–176. doi: 10.1016/j.saa.2017.04.070. [DOI] [PubMed] [Google Scholar]
- 34.Rajaei I., Mirsattari S.N. Spectroscopic characteristic (FT-IR, 1H, 13C NMR and UV–Vis) and theoretical calculations (MEP, DOS, HOMO-LUMO, PES, NBO analysis and keto-enol tautomerism) of new tetradentate N, N′-bis (4-hydroxysalicylidene)-1, 4-phenylenediamine ligand as a chelating agent for the synthesis of dinuclear Co (II), Ni (II), Cu (II) and Zn (II) complexes. J. Mol. Struct. 2018;1163:236–251. [Google Scholar]
- 35.Soltani A., Ghari F., Khalaji A.D., Tazikeh Lemeski E., Fejfarova K., Dusek M., Shikhi M. Crystal structure, spectroscopic and theoretical studies on two Schiff base compounds of 2,6-dichlorobenzylidene-2,4-dichloroaniline and 2,4-dichlorobenzylidene-2,4-dichloroaniline. Spectrochim. Acta A. Mol. Biomol. Spectrosc. 2015;139:271–278. doi: 10.1016/j.saa.2014.10.099. [DOI] [PubMed] [Google Scholar]
- 36.Helal M.H., El-Awdan S.A., Salem M.A., Abd-Elaziz T.A., Moahamed Y.A., El-Sherif A.A., Mohamed G.A. Synthesis, biological evaluation and molecular modeling of novel series of pyridine derivatives as anticancer, anti-inflammatory and analgesic agents. Spectrochim. Acta A. Mol. Biomol. Spectrosc. 2015;25:764–773. doi: 10.1016/j.saa.2014.06.145. [DOI] [PubMed] [Google Scholar]
- 37.Singh H. ADFT approach for theoretical and experimental study of structure, electronic, Hirshfeld surface and spectroscopic properties of 12-(4bromophenyl)-2-(prop-2-ynyloxy)-9,10-dihydro-8H-benzo[a]xanthen11(12H)-on single crystal. Chemical Physics. 2019;524:1–13. [Google Scholar]
- 38.Sakthivel S., Alagesan T., Muthu S., Abraham C.S., Geetha E. Quantum mechanical, spectroscopic study (FT-IR and FT - Raman), NBO analysis, HOMO-LUMO, first order hyperpolarizability, and docking studies of a non-steroidal anti-inflammatory compound. Journal of Molecular Structure. 2018;1156:645–656. [Google Scholar]
- 39.Pepe G., Siri D., Reboul J.-P. The Molecular Electrostatic Potential, and Drug Design. Journal of Molecular Structure. 1992;256:175–185. [Google Scholar]
- 40.Matta C.F. Modeling biophysical and biological properties from the characteristics of the molecular electron density, electron localization and delocalization matrices, and the electrostatic potential. J. Comput. Chem. 2014;35:1165–1198. doi: 10.1002/jcc.23608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sebastian S.R., Attia M.I., Almutairi M.S., El-Emam A.A., Panicker C.Y., Van Alsenoy C. FT-IR, FT-Raman, molecular structure, first order hyperpolarizability, HOMO and LUMO analysis, MEP and NBO analysis of 3-(adamantan-1-yl)-4-(prop-2en-1-yl)-1H-1, 2, 4-triazole-5 (4H)-thione, a potential bioactive agent. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014;11:295–304. doi: 10.1016/j.saa.2014.04.177. [DOI] [PubMed] [Google Scholar]
- 42.Hartwar V.R., Sist M., Jorgensen M.R.V., Mamakhel A.H., Wang X., Hoffmann C.M., Sugimoto K., Overgaard J., Iversen B.B. Quantitative analysis of intermolecular interactions in orthorhombic rubrene. IUCrJ. 2015;2:563–574. doi: 10.1107/S2052252515012130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with the receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 44.Baez-Santos Y.M., Barraza S.J., Wilson M.W., Agius M.P., Mielech A.M., Davis N.M., Baker S.C., Larsen S.D., Mesecar A.D. X-ray Structural and Biological Evaluation of a Series of Potent and Highly Selective Inhibitors of Human Coronavirus Papain-like Proteases. J Med Chem. 2014;57:2393–2412. doi: 10.1021/jm401712t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song W., Gui M., Wang X., Xiang Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018;14 doi: 10.1371/journal.ppat.1007236. e1007236 e1007236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mprofrom SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 47.Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L., Wang T., Sun Q., Ming Z., Zhang L., Ge J., Zheng L., Zhang Y., Wang H., Zhu Y., Zhu C., Hu T., Hua T., Zhang B., Yang X., Li J., Yang H., Liu Z., Xu W., Guddat L.W., Wang Q., Lou Z., Rao Z. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368:779–782. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mohan K., Ueda G., Kim A.R., Jude K.M., Fallas J.A., Guo Y., Hafer M., Miao Y., Saxton R.A., Piehler J., Sankaran V.G., Baker D., Garcia K.C. Topological control of cytokine receptor signaling induces differential effects in hematopoiesis. Science. 2019;364:6442. doi: 10.1126/science.aav7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Owen C.D., Lukacik P., Strain-Damerell C.M., Douangamath A., Powell A.J., Fearon D., Brandao-Neto J., Crawshaw A.D., Aragao D., Williams M., Flaig R., Hall D.R., McAuley K.E., Mazzorana M., Stuart D.I., von Delft F., Walsh M.A. SARS-CoV-2 main protease with unliganded active site (2019-nCoV, coronavirus disease. 2019;COVID-19):2020. [Google Scholar]
- 51.Cook C., McDonald S., Karim A. Importance of pharmacokinetic and physicochemical data in the discovery and development of novel antiarrhythmic drugs. Xenobiotica. 1993;23:1299–1309. doi: 10.3109/00498259309059440. [DOI] [PubMed] [Google Scholar]
- 52.Daina A., Michielin O., Zoete V. iLOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014;54:3284–3301. doi: 10.1021/ci500467k. [DOI] [PubMed] [Google Scholar]
- 53.Debnath B., Ganguly S. Synthesis, biological evaluation, in silico docking, and virtual ADME studies of 2-[2-Oxo-3-(arylimino) indolin-1-yl]-N-arylacetamides as potent anti-breast cancer agents. Monatshefte für Chemie-Chemical Monthly. 2016;147:565–574. [Google Scholar]
- 54.Lipinski C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 55.C.A. Lipinski, Lead-and drug-like compounds: the rule-of-five revolution, Drug Discov. Today: Technol. 1 2004 (4) 337–341. [DOI] [PubMed]
- 56.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 57.Daina A., Michielin O., Zoete V. SwissTargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019;47:W357. doi: 10.1093/nar/gkz382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.