

Abstract
Cancers must alter their metabolism to satisfy the increased demand for energy and building blocks that are required to create a rapidly growing tumor. Further, for cancer cells to thrive, they must also adapt to cope with an often changing tumor microenvironment that can present new metabolic challenges (ex. hypoxia) that are unfavorable for most other cells. As such, altered metabolism is now considered an emerging hallmark of cancer. Like many other malignancies, the metabolism of prostate cancer is considerably different compared to matched benign tissue. However, prostate cancers exhibit distinct metabolic characteristics that set them apart from many other tumor types. In this chapter, we will describe the known alterations in prostate cancer metabolism that occur during initial tumorigenesis and throughout the disease’s progression. In addition, we will highlight upstream regulators that control these metabolic changes. Finally, we will discuss how this new knowledge is being leveraged to improve patient care through the development of both novel biomarkers and new, metabolically targeted therapies.
Keywords: Prostate cancer, Metabolism, Androgen receptor, Imaging
Introduction
In the 1920s, Otto Warburg first described the ability of cancer cells to exhibit glycolysis even in the presence of oxygen (aerobic glycolysis), a phenomenon now commonly referred to as the Warburg effect [1,2]. Since then, the altered metabolism of cancers relative to benign tissue has been more broadly recognized and as such, is now considered one of the hallmarks of cancer [3]. Interestingly, while prostate cancers indeed have distinct metabolic phenotypes from normal prostate, they also exhibit atypical metabolism compared to many other cancer types. Hence, many of the generalities established for cancer metabolism are not pertinent for prostate cancer. But like many diseases, the metabolism of prostatic tumors is context and stage dependent.
The prostate is a reproductive gland, generating and releasing fluid that nourishes sperm. Sperm are nourished in large part by citrate that is produced and secreted from the luminal epithelial cells of the prostate. Luminal epithelial cells are able to produce and secrete large amounts of citrate as the result of a truncated tricarboxylic acid (TCA) cycle (also referred to as the citric acid or Krebs cycle) that is caused by the extraordinarily high levels of zinc that accumulate in these cells. To that end, the luminal epithelial cells of the prostate contain amongst the highest levels of zinc (~.8–1.5 mM) of any cell in the human body [4–9]. The high intracellular levels of zinc disrupt the TCA cycle by inhibiting the enzyme m-aconitase which converts citrate to isocitrate [7]. This truncated TCA cycle converts prostatic luminal epithelial cells from citrate-consuming to citrate-producing cells [7,10]. One of the first events to occur during the malignant transformation of prostate epithelial cells is a decrease in the expression of zinc transporters [11,12]. This results in decreased intracellular zinc accumulation and a derepression of aconitase and therefore the TCA cycle. As such, in contrast to what has been described for most cancers, the majority of prostate cancers exhibit high levels of glucose oxidation through increased TCA cycle flux (Figure 1).
Figure 1. Evolution of prostate cancer metabolism.
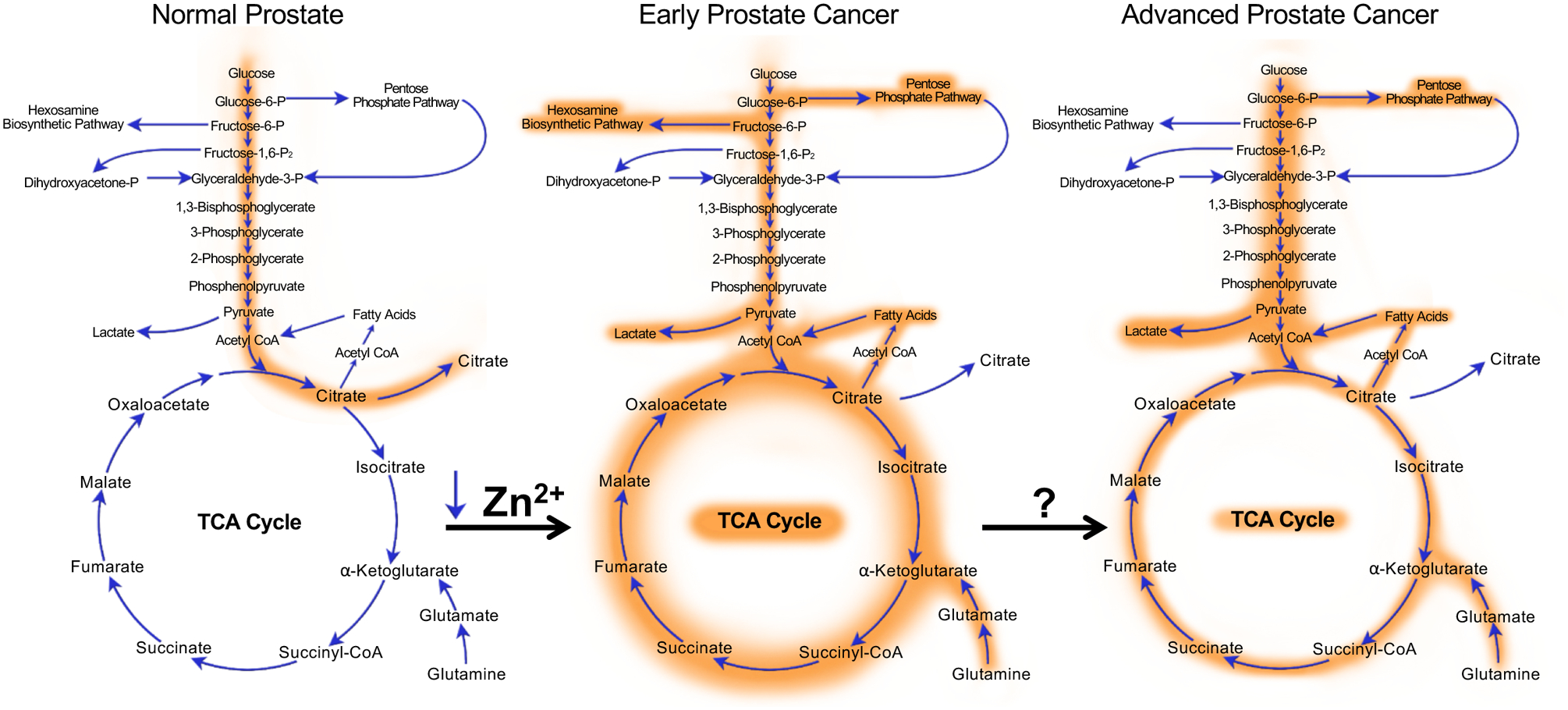
Normal prostate epithelial cells exhibit a truncated TCA cycle that results in the increased production and secretion of citrate. During the initial transformation towards malignancy, intracellular concentrations of zinc drop causing a derepression of aconitase (the enzyme that converts citrate to isocitrate) and subsequent increased flux through the TCA cycle. Concurrently, cancer cells start to exhibit aerobic glycolysis, elevated glutaminolysis and increased flux through the hexosamine biosynthetic and pentose phosphate pathways. Interestingly, another hallmark of prostate cancers is the concurrent increases in both de novo lipogenesis and fatty acid oxidation. When prostate cancers progress into the late stages of the disease, the classic Warburg effect becomes more pronounced while some pathways, such as the hexosamine biosynthetic pathway, may reverse. While the initial metabolic transformation of prostatic cells has been well described to result from alterations such as the decreases in intracellular zinc concentrations, many of the drivers of the metabolic changes that occur in advanced prostate cancer remain poorly understood. Shown here is only a brief snapshot of central carbon metabolism.
While the shift towards increased glucose oxidation during transformation has been known for over 20 years, what has become clear is that prostate cancers co-opt a number of other important metabolic processes, described below, to help satisfy the increased energetic and biosynthetic demands of a rapidly growing tumor (Figure 1). Further, these metabolic changes continue to change throughout disease progression. For example, many advanced, lethal prostate cancers will eventually demonstrate increased glycolytic flux, similar to the classic Warburg effect (Figure 1). Importantly, cancer cells must also adapt to survive the harsh tumor microenvironment that evolves in part due to the increased metabolic waste produced from the cancers themselves. Beyond their contribution to the production of energy, building blocks and redox homeostasis, new research is emerging that indicates the increased uptake of many nutrients contributes directly to the synthesis of new signaling molecules that can function as oncogenic signals to reprogram the cells and promote disease progression.
Our understanding of which nutrients are used by tumors, how and when they are metabolized and the regulation of these metabolic processes is required to translate these observations towards clinical utility. Importantly, the chemical nature of metabolism makes it possible to develop biomarkers (ex. imaging) that can assess when certain pathways have been altered in patients and therefore identify men who could benefit from emerging, metabolically targeted therapies.
Here, we describe the metabolic alterations that occur during the initiation and progression of prostate cancer. Further, we will highlight how key signaling pathways (ex. AR, PI3K, MYC) as well as other factors such as changes in the tumor microenvironment regulate these processes. Finally, we will discuss the clinical significance of this field. Accordingly, we will summarize the new metabolic-targeted therapies that are being tested for the treatment of prostate cancer. Importantly, we will also outline the emerging approaches being used to monitor metabolism in patients and how these could guide future clinical trials.
Metabolic Reprogramming in Prostate Cancer
Glucose Metabolism
The specific metabolic phenotype of normal prostate epithelial cells includes the accumulation of high zinc concentrations (~3–10 fold higher than in other tissues) that subsequently lead to a truncated TCA cycle and increased citrate production (~30–50 fold higher than other tissues), decreased oxidative phosphorylation and low energy metabolism [13]. Such inefficient metabolism cannot meet the energy requirements for rapidly growing prostate cancer cells. To adjust, prostate cancer cells are reprogrammed to have an efficient, energy-generating metabolism during their initial transformation. A notable metabolic shift during this transformation is an increase the levels of citrate oxidation as the malignant glands contain significantly lower concentrations of zinc compared to normal cells [14]. This shift allows cells to oxidize citrate and produce energy via a functional TCA cycle. This metabolic alteration can also protect prostate cancer cells from cell death [15]. In normal prostate epithelial cells, zinc accumulation facilitates Bax-associated mitochondrial pore formation which promotes cytochrome c release from mitochondria and subsequent caspase cascades as well as an inhibition of the anti-apoptotic protein NFkB [16,17]. Conversely, prostate cancer cells as less susceptible to mitochondrial induced apoptosis in the presence of low zinc concentrations. As noted above, zinc transporters are an important contributor to intracellular zinc regulation. The expression of ZIPs is significantly decreased or often absent altogether in prostate cancer [12,18]. Interestingly, differences in ZIP expression may also explain in part some of the racial disparities observed for prostate cancer. A study comparing African American and Caucasians suggested that ZIPs are expressed less in African Americans, preventing them from maintaining normal intracellular zinc concentrations [19]. Although it is not entirely clear how ZIPs are downregulated during prostate cancer progression, one potential explanation is epigenetic repression. In prostate cancer cells, higher methylation levels have been observed in the promoter region for the gene encoding activating enhancer binding protein 2 alpha (AP-2 alpha), leading to the decreased expression of AP-2 alpha, an important transcription factor of ZIPs [20]. Given the known role of zinc in prostate metabolism, interest has risen in the use of zinc dietary supplements and the development of ZIP inhibitors to treat prostate cancer [18]. Due to the metabolic shift towards citrate oxidation, some metabolic intermediates and genes in TCA cycle are also increased/hyperactivated during early transformation. A recent integrative proteomics study revealed that the TCA pathway proteins citrate synthase, aconitate hydratase, 2-ketoglutarate dehydrogenase complex, succinate-CoA ligase, fumarate hydratase and malate dehydrogenase, were all upregulated in primary prostate cancer compared to benign prostatic hyperplasia [21]. Furthermore, the corresponding intermediate metabolites in TCA cycle like malate, fumarate, succinate and 2-hydroxyglutaric acid were significantly elevated in tumors, suggesting a dependence of primary prostate cancers on oxidative phosphorylation (OXPHOS). Interestingly, because of the high OXPHOS in primary prostate cancers, only modest levels of glucose uptake and the Warburg effect are observed [22,23]. Consequently, [18F]fluorodeoxyglucose-positron emission tomography (FDG-PET) is a poor detector of primary prostate cancer.
Unlike most primary prostate cancers, late-stage prostate cancers can often be detected in FDG-PET scans. Accordingly, increased glycolytic metabolism has been correlated with disease progression and poor prognosis [24]. Though the exact mechanism of glucose metabolism regulation in prostate cancer has not been fully elucidated, emerging evidence suggests the regulation of multiple glycolytic enzymes in the advanced cancer stages.
Facilitative glucose transporter (GLUTs) control the first rate-limiting step of glucose metabolism by mediating glucose diffusion. To date, 14 members have been identified in the human GLUT family. Each are associated with different substrate affinities and tissue distributions. GLUT1 is overexpressed in several tumors including prostate cancer. High GLUT1 expression has been found elevated in prostate tissues compared to tumor-adjacent normal tissues and its expression is correlated with a shorted time to recurrence after radical prostatectomy [25,26]. Accordingly, higher expression of GLUT1 has been reported in androgen-independent prostate cancers [27]. Moreover, GLUT1 can be induced in a tissue-specific manner by androgens and glucose deprivation, which can help cancer cells survive in a low glucose environment [28]. Besides GLUT1, GLUT12 has also recently been shown to play a functional role in prostate cancer [29]. GLUT12 was required for androgen-induced glucose uptake and cell growth in LNCaP and VCaP cells. Further, it played a similar role in AR-negative PC-3 cells. Interestingly, GLUT12 subcellular trafficking was observed to be increased in multiple prostate cancer cell models, suggesting a functional regulation beyond mRNA and protein expression. Of note, other GLUT members such as GLUT3, 7 and 11 are reported to be overexpressed as well in prostate cancer [30]. However, the functional role of these transporters is less clear.
Hexokinases (HKs) catalyze the first irreversible step in glycolysis by converting glucose to glucose-6-phosphate (G6P). Out of the four isoforms identified, HK2 is a major contributor to the Warburg effect and is required for tumor growth. Recent work indicates that HK2 is significantly elevated in prostate tumors relative to normal tissue and it is significantly correlated to Gleason score [31,24,32,33]. However, dramatic variability in expression levels has been observed in individual CRPC patient samples, further evidence of the heterogeneity of prostate cancer [33]. Though our understanding of what increases HK2 expression during the disease evolution is incomplete, new findings have begun to reveal potential mechanisms of action. First, PTEN and p53 co-deletion/mutation have been correlated with high levels of HK2 in prostate cancer cell lines, xenograft and genetic mouse models and prostate cancer patient samples. On one hand, PTEN loss leads to mTOR signaling pathway activation and 4EBP1 phosphorylation, triggering cap-dependent translation of HK2 through facilitating the dissociation of 4EBP1 and 4IF4E [33]. On the other hand, HK2 expression can be further increased by decreased miR-143 expression, a miRNA whose biogenesis is promoted by p53 [34]. In support of these findings, a recent study revealed that increased HK2 expression and activity, caused by androgen deprivation, was associated with increased p-AKT in PTEN/p53 deficient mice [35]. AKT not only increased HK2 expression by mTORC1, but it also promoted the localization of HK2 to the mitochondria, increasing HK2 activity. Besides PTEN/p53, HK2 expression has been described to be regulated by EZH2 and PKA. EZH2 can upregulate HK2 and glycolysis in PC-3 cells through the inhibition of miR181b [36]. Whereas, PKA-CREB signaling can promote HK2 expression and glucose utilization following androgen treatment in LNCaP cells [37]. Importantly, systemic deletion of Hk2 in a genetic mouse model demonstrated a marked repression of tumor growth without any glucose homeostasis dysfunction in normal cells [38], suggesting a selective HK2 inhibitor could be exploited clinically with tolerant toxicities.
Pyruvate dehydrogenase complex (PDC) catalyzes the conversion of pyruvate to acetyl coenzyme A (acetyl CoA), thereby regulating the carbon flux from glycolysis into the TCA cycle. PDC is a multi-enzyme complex composed of three enzymes: pyruvate dehydrogenase (E1), dihydrolipoamide acetyltransferase (E2), and lipoamide dehydrogenase (E3). PDHA1 (E1 alpha subunit) is a major component of PDC. As such, PDHA1 plays a critical role in controlling the appropriate mitochondrial activity for the requirement of cell metabolism and growth. It is established that PDHA1 is regulated by phosphorylation [39,40]. PDHA1 can be phosphorylated at serine 293 by pyruvate dehydrogenase kinases (PDKs). PDKs inhibit PDC activity and therefore the TCA cycle. This shifts the cell’s metabolism away from OXPHOS and towards a Warburg effect. Conversely, dehydrogenase phosphatases (PDPs) can reverse such inhibition. Typically, PDKs are thought to be overexpressed in the glycolytic cancers [41]. In those cancers, the upregulated expression of PDKs partly inactivates PDC, and in turn reroutes pyruvate towards glycolysis and away from the mitochondrion for respiration. This metabolic alteration accelerates the Warburg effect and has frequently been associated with increased tumorigenesis. In agreement with this, Zhong et al. reported that reduced expression of PDHA1 in prostate cancer was correlated with poor prognosis [42]. Accordingly, knockout of PDHA1 significantly decreased mitochondrial OXPHOS but increased glycolysis, which contributed to fast tumor growth. Moreover, PDHA1 KO cells are able to expand a stem-like cell population, suggesting a potential role in chemotherapy resistance and migration. However, Chen et al. offered a different view by demonstrating the amplification and overexpression of PDHA1 and its phosphatase PDP1 in prostate cancer [43]. Here, the authors demonstrated that inhibition of PDC activity precluded the development of prostate cancer in mouse and human xenograft tumor models. They also provided evidence that compartmentalized PDCs have different functions, but that distinct pools could all still contribute to lipid biosynthesis for prostate cancer progression. For example, nuclear PDC promoted lipogenesis by regulating histone acetylation-mediated lipogenic gene expression [44]. Alternatively, mitochondrial PDC converted pyruvate to citrate for lipid anabolism. Taken together, it is still unclear whether PDHA1 and its regulators are tumor promotors or suppressors. Given the changing citrate-related metabolism during prostate cancer progression, it is very possible that PDC has context-dependent functions in prostate cancer.
Pyruvate kinase catalyzes the committed step that transfers the phosphate group from phosphoenolpyruvate (PEP) to ADP and generates pyruvate and ATP. PK muscle isozymes M1 and M2 are different splice variants encoded by the same gene, PKM. PKM2 is closely correlated to tumorigenesis. While PKM1 is expressed in differentiated tissues, PKM2 can be highly expressed in proliferating cells including embryonic and cancer cells [45]. Consistently, PKM2 is correlated with Gleason score and aggressive tumor types in prostate cancer [46,47]. There are some potential explanations for such isoform differences. First, PKM2 is an allosterically regulated isoform. Tetrameric PKM2 actively promotes the conversion of PEP into ATP and pyruvate. Conversely, dimeric PKM2 has low catalytic activity and instead promotes the entry of glycolytic intermediates into the glycolytic branch pathways such as the pentose phosphate pathway, by which cells can generate several key building blocks for their growth and proliferation [48]. This activity change is most evident in rapidly proliferating cells that appear to adhere to the Warburg effect. Second, PKM2 has non-metabolic functions that can modulate signaling and transcriptional activity to promote prostate cancer progression. For example, in CRPC, PKM2 may partner with KDM8 and co-translocate to the nucleus where they can function as coactivators of HIF-1α to upregulate glycolytic genes (GLUT1, HK2, PKM2, LDHA, etc.) and downregulate TCA cycle-related PDHA1 and B1 genes [49]. Moreover, the reciprocal regulation of PKM2 and HIF-1α could facilitate CRPC cell survival under hypoxic conditions and promote drug resistance [50,46,51]. Interestingly, PKM2 may also respond to extracellular signaling during prostate cancer metastasis. To that end, cancer associated fibroblasts can induce PKM2 post-translational modifications and nuclear translocation. In the nucleus, PKM2 worked with HIF-1α and DEC1 to deregulate miR-205 expression and in turn promote the epithelial mesenchymal transition [52]. Several PKM2 inhibitors and PKM2 tetramerization activators have been developed and exhibited efficacy, including overcoming drug resistance, in preclinical models [48,45]. Although little work has been done in this regards in prostate cancer, DASA-58, a PKM2 activator, did inhibit the lung metastasis of PC-3 cells in SCID mice [52], suggesting further studies are warranted in prostate cancer.
Lactate dehydrogenase (LDH) catalyzes the reversible conversion of pyruvate and lactate. LDH (mostly LDHA) has been identified as a potential target for prostate cancer therapy [53,54]. LDH is a tetramer composed of two subunits, LDHA and LDHB. LDHA has a high affinity to pyruvate and thereby favors the reaction from pyruvate to lactate. Studies have suggested that in most tumors, increased LDHA is a hallmark for overactivated glycolysis and advanced progression. LDHA knockdown in prostate cancer cell not only inhibited cell growth but sensitized cells to radiotherapy [53,55]. Conversely, LDHB exhibits context-dependent roles [56]. A recent study indicated that LDHA and LDHB have opposite roles in prostate cancer [57]. Abnormal LDH, hyperphosphorylation and high expression of LDHA and low expression of LDHB correlated with short overall survival and time to biochemical recurrence patients with prostate cancer. They also found this aberrant LDH status was regulated by fibroblast growth factor receptor 1 (FGFR1) signaling. Mechanistically, FGFR1 can phosphorylate four tyrosine residues on LDHA to stabilize the protein. This enhanced glycolysis and reduced oxygen consumption. However, FGFR can also repress LDHB transcription by inhibiting TET1 (demethylase) expression and subsequently increasing DNA methylation at its promoter. Such coordinated regulation allows cancer cells to develop highly glycolytic metabolism.
As noted above, late-stage prostate cancer cells often increase glycolysis. Thus, lactate builds up as a byproduct of excessive anaerobic metabolism. To avoid lactate-mediated toxicities and apoptosis, cancer cells express high levels monocarboxylate transporters (MCTs) to ensure the rapid efflux of intracellular lactate. MCTs are well-studied membrane transport proteins that are responsible for the transmembrane shuttling of small carboxylates like lactate, pyruvate and short-chain fatty acids [58]. While the expression of MCTs vary in prostate cancer patients, more MCT has generally been correlated with more aggressive disease and poor prognosis [14].
Of the 14 isoforms identified, MCT1, 2 and 4 have been implicated in prostate cancer progression. MCT4 was first identified as a lactate exporter specially in highly glycolytic cells [59]. The elevated expression of MCT4 has been confirmed by several studies and its high expression is strongly associated with high glycolytic rates in prostate cancer including CRPC and neuroendocrine prostate cancer [31,60,55]. However, a clinical pathology study argued against the relationship between increasing MCT4 with lactate efflux (also MCT2) in prostate cancer [61]. They observed a significant increase of MCT4 (and MCT2) expression in the cytoplasm of prostate cancer cells rather than at the plasma membrane, indicating the roles of MCTs in prostate cancer may be involved in organelle function instead of lactate shuttling. Although further investigation is required to demonstrate why prostate cancer cells are reprogramed to express more MCT4, MCT4 does appear to be a useful prognostic factor and potential target for prostate cancer. Regarding the latter, knockdown of MCT4 significantly reduced PC-3 cell proliferation [55]. Moreover, targeting MCT4 by antisense oligonucleotides inhibited glycolysis, lactate production and cell proliferation in advanced prostate cancer [60]. Furthermore, high MCT4 expression may be involved in cancer–stroma interactions thought to facilitate prostate cancer progression (discussed further below the Section “Influence of the tumor microenvironment”) [62].
In addition to MCT4, MCT2 promotes prostate cancer progression [63,64]. Elevated MCT2 levels in prostate cancer have been linked to two differentially methylated regions at the SLC16A7 locus (gene encoding MCT2). One locus, upstream of the promoter, was hypermethylated in patient samples and responsible for full length MCT2 expression. The other locus, within an internal promoter, was recurrently demethylated in patient samples and subsequently induced the expression of an alternative isoform of MCT2. This isoform contains a different set of 5’-UTR translation signals that are most likely related to the high MCT2 expression. In addition to expression, MCT2 has been demonstrated to translocate to peroxisomes during disease progression, where it bound to Pex19 and enhanced beta-oxidation for malignant transformation. Together, these findings suggest that prostate cancer cells are able to adaptively increase MCT2 by epigenetic and compartmentalized regulation to meet their metabolic demands.
MCT1 is a controversial target for prostate cancer therapy. In some glycolytic cancers, an MCT1 inhibitor is being investigated in clinical trials due to its potent ability to reduce tumor growth in preclinical studies [58]. In prostate cancer, unfortunately, the benefit is still undefined. Howard et al. showed that pharmacological inhibition of MCT1 by α-cyano-4-hydroxycinnamate (CHC, has a 10-fold selectivity for MCT1 compared to other MCTs) was associated with increased necrosis but not a significant difference in xenograft tumor size [65]. However, Pertega-Gomes et al. indicated that another MCT1 inhibitor, AR-C155858, decreased cell proliferation and increased cell apoptosis in Pten-deficient mouse tumor tissues without substantial side effects on benign tissue [31]. Regardless, the relationship between MCT1 and prostate cancer is complicated. Though a continued decrease of MCT1 has been found from benign prostatic tissue to metastatic prostate cancer, the expression of MCT1, MCT4 and CD147 were suggested to be markers of poor prognosis [61]. Interestingly, MCT1 expression is upregulated by hypoxia and glucose starvation [62,31], two common features of tumor microenvironments. However, it is unclear whether prostate cancer benefits from such MCT1 regulation.
The pentose phosphate pathway (PPP) is a glucose catabolic pathway that runs parallel to glycolysis. Evidence indicates that the PPP is important in cancer cell growth. This is largely due to PPP’s contribution to the production of nicotinamide adenine dinucleotide phosphate (NADPH) and ribose 5-phosphate for scavenging of reactive oxygen species, reductive biosynthesis and supplying of nucleotide precursors [66,67]. A recent study showed that transketolase-like protein 1, an enzyme involved in the non-oxidative phase of the PPP, was altered throughout prostate cancer progression [68]. Of note, metastatic prostate cancer tissue had the highest TKTL1 expression, suggesting the potential role of TKTL1 in prostate cancer metastasis. Moreover, glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme of the PPP, is also found to be a critical regulator for prostate cancer. Efrosini et al. revealed that G6PD is upregulated in prostate cancer and functionally was required for AR-mediated ROS modulation and cell growth [69]. At this time, the functional role of the PPP in metastasis is unknown.
Lipid Metabolism
Lipids are essential for many cell functions. They are used to form cell membranes and create membrane anchors, post-translationally modify proteins, promote various signaling pathways and can be used for energy storage. Lipid metabolic pathways produce compounds like fatty acids, steroids (including hormones and sterols like cholesterol), phospholipids and others. An important characteristic of prostate cancer cells is the dysregulation of lipid metabolic pathways. In normal cells, lipids are primarily acquired extracellularly, and can also be synthesized in specific tissues such as liver and adipose. Fatty acids produced in these tissues can be stored or transported to other areas. In cancer cells, there is an increase in de novo lipogenesis, which is necessary to accommodate the excessive cell growth and proliferation that occurs in cancer [70]. Prostate cancer cells undergo this characteristic shift from a reliance on extracellular fatty acid uptake to de novo lipogenesis. This shift is accompanied by the upregulation of multiple enzymes in the fatty acid synthesis pathway and is regulated by AR signaling. Upregulation of AR signaling in advancing cancer is accompanied by an increase in lipogenic enzymes [71]. This is largely dependent on the major lipogenesis transcription factor sterol regulatory element-binding protein (SREBP) [72]. Interestingly, a feedback mechanism also exists where AR transcription is regulated by SREBP [73]. Beyond AR, additional genomic alterations observed in prostate cancer such as PTEN and PML co-deletion can also hyperactivate SREBP-mediated lipogenesis and subsequent prostate cancer progression [74]. Thus, SREBP may represent a downstream conduit of several oncogenic signaling networks in prostate cancer.
SREBPs are important transcription factors regulating fatty acid metabolism. In mammals, SREBP exists as SREBP-1a and SREPB-1c (two variants of a single gene), and SREBP-2 [75,76]. SREBP associates with SREBP cleavage-activating protein (SCAP) in the ER membrane. In response to a decrease in intracellular sterols, the SREBP-SCAP complex translocates to the Golgi apparatus, where SREBP is cleaved and activated, allowing it to move to the nucleus. SREBP controls the transcription of several genes involved in fatty acid and cholesterol synthesis, including ACLY, ACACA, SCD1, and FASN [77,78]. SREBP is upregulated in various cancers and can be activated by androgens in prostate cancer, due to the effect of AR on SCAP transcription [79]. SREBP can, in turn, further activate AR expression by binding to a sterol regulatory element (SRE) in the AR gene [80].
Lipids have an important role in forming the structure of outer and inner cell membranes. They are used to form the phospholipid bilayer as well as cholesterol-rich membrane rafts within the plasma membrane, which are important for intracellular signaling and trafficking. Since cancer cells experience rapid proliferation as well as increased synthesis and uptake of materials, membrane expansion is necessary. In addition, the metabolic shift to de novo lipogenesis in cancer increases lipid saturation, which may prevent cell death from oxidative damage [81]. The lipid rafts are microdomains within the plasma membrane that contain cholesterol, sphingolipids and transmembrane proteins [82]. Receptors like tyrosine kinase receptors in these lipid rafts can be activated by ligands to promote phosphorylation cascades, while the microdomain of the raft allows for the accumulation of intracellular scaffolding and adaptors and can protect against phosphatases [83,84]. Cholesterol in lipid rafts regulates AKT signaling, a major driver of prostate cancer progression [85]. There is also evidence suggesting that AR can interact with AKT at lipid rafts to promote oncogenic signaling [86]. However, AR’s extragenomic roles in prostate cancer are still debated.
Intracellular fat can be stored in lipid droplets. Lipid droplets are composed of a phospholipid monolayer containing polar sterols and various transmembrane proteins, and internally, non-polar sterol esters and triacylglycerols [87]. In cancer, lipid droplets are increased due to the increased lipid accumulation. Lipid droplet biogenesis also occurs in response to cell stress, such as when cancer cells undergo metabolic or oxidative imbalance due to conditions like hypoxia or nutrient starvation [88–91]. In addition to their roles in resistance to stress and as a source for excess lipid storage, lipid droplets can interact with mitochondria to regulate β-oxidation, and have a role in maintenance of endoplasmic reticulum (ER) homeostasis. Androgens increase lipid droplets, which correlate with prostate cancer aggressiveness [92]. The increase in lipid droplets in prostate cancer is additionally related to the upregulation of the PI3K/AKT pathway, often following PTEN loss, which causes the accumulation of intracellular cholesterol esterases [93].
PI3K/AKT signaling also leads to the upregulation of fatty acid synthase (FASN), which is overexpressed in prostate cancer [94,95] FASN is also regulated by AR signaling and correlates with Gleason score and PSA levels [96,97]. Inhibition of FASN with a small molecule inhibitor has been shown to suppress de novo fatty acid synthesis and tumor growth in part by targeting AR signaling [98]. FASN expression can also be increased in response to oxidative stress such as hypoxia. This effect is via activation of the AKT pathway and SREBP-1, which regulates FASN transcription [99]. FASN expression is also regulated by the p300 acetyltransferase, which increases FASN transcription and contributes to lipid accumulation and prostate cancer growth [100]. It remains to be seen whether p300’s effects are mediated through any of the above-mentioned transcription factors.
Lipids can have important roles in the post-translational modification of proteins. Acylation by the covalent addition of palmitate or myristate to proteins can facilitate oncogenic signaling [101]. In prostate cancer, myristoylation of Src kinase has been shown to increase oncogenic signaling and promote tumor progression [101]. In addition, palmitoylation of Src contributes to prostate cancer initiation [102]. Prenylation, the addition of isoprenoids such as farnesyl or geranylgeranyl to proteins, often causes membrane association [103]. Inhibition of geranylgeranylation has been shown to induce autophagy in prostate cancer cells and reduce prostate cancer metastasis in murine models [104,105].
While products from the TCA cycle are used for OXPHOS in normal cells, cancer cells upregulate alternative metabolic pathways to increase NADPH in part for fatty acid production. In this context, the citrate that is produced in the TCA cycle can be shuttled to the cytoplasm to be used for fatty acid synthesis. Cytosolic citrate production can also result from increased glutamine uptake, another characteristic of prostate cancer cells [106,29]. Glutamine can be converted glutamate and then to α-ketoglutarate, which can be used as a carbon source for fatty acid synthesis by conversion to cytosolic citrate via reductive metabolism by the enzymes IDH1 and ACO1 [107]. Alternately, α-ketoglutarate can replenish the carbon intermediates of the TCA cycle. This utilization of cytosolic glutamine for fatty acid synthesis and the TCA cycle is observed in cancer cells [108]. In addition, the oxidation of isocitrate to α-ketoglutarate can produce excess NADPH for fatty acid synthesis [109].
The process of fatty acid synthesis is mediated by a variety of enzymes that are involved in prostate cancer progression. Citrate is converted to acetyl-CoA in the cytoplasm by ATP citrate lyase (ACLY) [110]. ACLY activation can result from PI3K/AKT upregulation, which is common in prostate cancer [111]. Acetyl-CoA carboxylase (ACC1) converts acetyl-CoA to malonyl-CoA [112], which then is processed by fatty acid synthase (FASN), creating the 16-carbon fatty acid palmitate [113]. Enzymes such as stearoyl-CoA desaturase 1 (SCD1) or elongation enzymes can function to either insert a double bond in the fatty acid chain to generate a mono-unsaturated fatty acid, or to lengthen the carbon chain, respectively. Desaturation of fatty acids is required for prostate cancer cell growth, an effect that can be blocked by inhibition of SCD1 [114]. These fatty acids are then used in various cell functions including in catabolic pathways as a source of energy, membrane biogenesis, protein post-translational modifications, or regulation of oncogenic pathways. Cytosolic acetyl-CoA can alternately be used for the mevalonate pathway, in which it is converted to mevalonate by HMG-CoA reductase (HMGCR).
Mevalonate is a precursor for synthesis of isoprenoids and cholesterol, which can be used to produce steroids such as androgen [115]. Statins act to reduce cholesterol levels by inhibiting HMGCR, thereby preventing the formation of mevalonate from acetyl-CoA. Statins have been shown to prevent prostate cancer progression, biochemical recurrence, and mortality [116,117]. As such, they are under clinical evaluation for the treatment of prostate cancer. Statins cause an increase in the expression of low-density lipoprotein receptors (LDL-R) [118]. LDL-R is a plasma membrane protein that facilitates the intake of LDL into the cell via endocytosis, providing another source of cholesterol to the cell, and has been shown to regulate cholesterol homeostasis in prostate cancer cells [119]. The LDL-R pathway is regulated by various transcriptional and post-transcriptional factors. For example, expression of LDL-R is activated by SREBP proteins, mainly SREBP-2, through interaction with the SRE, while SREBP-2 activation is inhibited by high levels of intracellular cholesterol [120]. SREBP-2 also regulates transcription of HMGCR; together, this suggests that SREBP-2 contributes to cholesterol accumulation. SREBP-2 also has a role in regulating androgen production, which can be synthesized from cholesterol through this pathway. Interestingly, SREBP-2 can also be regulated by androgens, suggesting a feedback loop for androgen synthesis.
SREBP-2 has been shown to contribute to cholesterol accumulation, which opposes the effect of the nuclear receptor liver X receptors (LXR), another important factor in cholesterol homeostasis [121]. LXRs respond to excess cholesterol by modulating the transcription of various intermediates in the cholesterol and fatty acid synthesis pathways. Two isoforms exist (LXRα and LXRβ), which differ in their tissue localization. Oxysterols, an oxidized derivative of cholesterol, can activate LXRs, which eliminate cholesterol by reducing LDL uptake [122], regulating ATP-binding cassette (ABC) transporter expression [123], and converting cholesterol to bile acid [123,124]. In cells with increased levels of cholesterol, ABC transporters can mediate the release of cholesterol by the process of reverse cholesterol transport (RCT) which is mediated by LXRs [125]. LXR has been shown to promote a tumor suppressive role in prostate cancer [126,125]. In addition, the synthesis of steroids from cholesterol is regulated by the cytochrome P450 family of enzymes. CYP27A1 encodes a cytochrome P450 oxidase that converts cholesterol into 27HC and other oxysterols. This creates a system in which 27HC inhibits prostate cancer cell growth by depleting cellular stores of cholesterol [127] and possibly through the activation of LXR.
Interestingly, while prostate cancer has classically been characterized by increased fatty acid synthesis and lipid uptake, it is now clear that these tumors ironically also exhibit and require high levels of beta oxidation [128]. While fatty acid synthesis provides lipids for various purposes and pathways in the cell, beta oxidation can provide necessary energy from stored lipids, which can be used for cancer cell growth by providing ATP, as well as acetyl-CoA which can be recycled into the TCA cycle, used as second messenger or possibly rerouted for use in epigenetic regulation. The process of beta oxidation occurs mainly in the inner mitochondrial membrane, where fatty acids can be transported via carnitine transport. Carnitine palmitoyltransferase 1 (CPT1) has been demonstrated to be required for prostate cancer cell growth [129–131]. Blockade of beta oxidation at additional steps was also able to impair prostate cancer growth and metastasis in vivo by inhibiting CaMKII activation [132]. Beta oxidation also occurs in peroxisomes, after which oxidized lipids can be transported to the mitochondria or can enter different cell pathways [133]. An increase in beta oxidation in prostate cancer is supported by the upregulation of α-methylacyl-CoA racemase (AMACR), an enzyme involved in catalyzing the peroxisomal and mitochondrial beta oxidation of branched-chain fatty acids, in human prostate cancer [134,135]. D-bifunctional protein (DBP), which is involved in peroxisomal beta oxidation, is also upregulated in prostate cancer [136]. Taken together, prostate cancers exhibit dynamic lipid metabolism, indicating that both the synthesis and breakdown of fats may represent therapeutic vulnerabilities for the treatment of the disease.
Amino Acid Metabolism
While glutamine is not an essential amino acid, it plays an important role during cancer cell starvation as a source of energy, carbon and nitrogen [137–139]. SLC1A5 (ASCT2) is the primary glutamine transporter in cancer [140]. Other transporters such as SLC1A4 (ASCT1) may also play a role in glutamine transport under conditions of stress in the tumor microenvironment [141]. However, it is unclear at this time if this is due to direct or indirect transport. Both SLC1A4 and SLC1A5 are observed to be upregulated in prostate cancer by androgens [142,141]. However, SCL1A4 and SCL1A5 are not direct targets of the androgen receptor [141]. While MYC is a major regulator of glutamine metabolism in many cancer types [139,143,144], MYC appears to regulate glutamine metabolism in prostate cancer in a context-dependent manner that is potentially influenced by the PTEN status of the cell [141]. Interestingly, glutamine uptake in prostate cancer cells is driven by several oncogenic networks such as AR, MYC and mTOR signaling.
Due to the dependence of cancer cells on glutamine, several attempts have been made to exploit this vulnerability. The drug CB-839 (Calithera Biosciences), which targets mitochondrial glutaminase, is currently being tested in early phase clinical trials for the treatment of solid tumors in combination with chemotherapeutic drugs [145]. A recent study demonstrated that the aggressive prostate cancer cell line PC-3 and the metastatic derivative PC-3M are sensitive to CB-839 due to their dependence on glutamine metabolism [106]. While appealing to target the conversion of glutamine to glutamate, this approach has limitations as it does not consider the contributions of glutamine metabolism independent of glutaminolysis. First, glutamine can activate MAPK signaling independently of RAS [146]. Second, it was reported that amino acids, including glutamine, can activate CaMKK2 which in turn activates AMPK [147]. While AMPK was initially described as a tumor suppressor due to its upstream kinase LKB1 [148–150], a context-dependent oncogenic role for AMPK has emerged in recent years [131,151–153]. To account for these additional aspects of glutamine metabolism, the first selective inhibitor of SLC1A5/ASCT2, V-9302, was developed [154]. This inhibitor was tested across a panel of 29 cancer cell models and in vivo, resulting in decreased growth, increased cell death and oxidative stress [154]. However, it should be mentioned that glutamine is not the only amino acid transported by ASCT2 [154]. Hence, the effect of V-9302 might not be due to glutamine uptake alone. Further, even though ASCT2 is the major glutamine transporter [155], additional transporters such as LAT1 and 2, as well as SNAT1–5 can also transport glutamine [154]. LAT1 and 3 are upregulated in prostate cancer and while they are the main transporters for leucine, the transport of glutamine is also possible [142,154]. Thus, inhibition of other amino acid transporters besides ASCT2, such as LAT1, may also decrease tumor growth [142]. An advantage of the transporters is their cell surface localization, making them theoretically accessible for other types of potentially more selective targeting (ex. antibody-mediated delivery).
Another non-essential amino acid that has been targeted for cancer treatment is arginine. Many advanced cancers, including prostate cancer, demonstrate a loss of components of the urea cycle such as argininosuccinate synthease (ASS) [156–159]. ASS is needed for the eventual conversion of arginine from citrulline [160], which means that loss of ASS leads to a cellular dependence on the uptake of extracellular arginine. This vulnerability is targeted by introducing the enzyme arginine deiminase (ADI) conjugated to polyethylene glycol (the latter for decreased immune response and increased serum half-life), resulting in the depletion of circulating arginine from the serum [161]. In ASS-deficient prostate cancer cells such as CWR22Rv1, treatment with ADI-PEG20 alone or in combination with docetaxel led to reduced tumor growth in mouse xenograft models [157]. As expected, cells expressing low levels of ASS (PC-3) are responsive to ADI-PEG20, while those expressing high levels of ASS (LNCaP) are resistant to ADI-PEG20 [157]. While this approach shows promising results [157], it was reported that the deprivation of arginine can lead to resistance due to the compensatory induction of ASS expression [156]. Another approach to starve cancer of arginine is the use of recombinant arginase, which converts arginine into ornithine [162]. While this is dependent on the levels of ornithine carbamoyl transferase (OCT) expression, the study concluded that several prostate cancer cell lines with low levels of OCT are responsive to arginase treatment [162]. However, this approach has to be carefully considered due to the importance of the polyamine synthesis pathway which uses ornithine as a substrate. Several cancers have showed increased proliferation when the polyamine synthesis pathway was upregulated [163]. In prostate cancer, the enzyme ornithine decarboxylase (ODC), which converts ornithine to putrescine, is a direct target of AR [164]. Further, when ODC is overexpressed, regular prostate epithelial cells can be transformed into prostate cancer cell [165]. Inhibition of the uptake of polyamines by N1-spermine-L-lysine amide or ODC itself by α-difluoromethylornithine is sufficient to inhibit the growth of prostate cancer in culture and in vivo [166]. However, as noted below in the Section “Biofluids and Tissue Metabolism Biomarkers”, the role of the polyamines in prostate cancer may not mirror that observed for other cancer types. For example, the levels of polyamines, and especially spermine, are often decreased in primary prostate cancer relative to benign prostate [167–169]. Hence, whether the polyamines, or perhaps specific polyamines, have unique roles in prostate cancer and whether these roles vary further in different disease stages, remains poorly understood.
One-carbon metabolism connects two important pathways: the folate and methionine cycles which can then feed into the transsulfuration and polyamine synthesis pathways. As a result, one carbon metabolism regulates several cellular processes such as epigenetics via methylation (methionine cycle), DNA synthesis and repair (folate cycle) and protection against reactive oxygen species via glutathione (transsulfuration pathway) [170]. This intricate network is fueled by the amino acids serine and glycine [171]. Several key enzymes in these pathways are regulated by the androgen receptor [171]. For example, the conversion of glycine to N-methylglycine (sarcosine) is facilitated by the glycine-N-methyl transferase (GNMT) and has been shown to be important for cell invasion [172]. Knockdown of GNMT resulted in lower amounts of sarcosine and decrease in invasion using DU-145 cells [172]. In contrast, the knockdown of the enzyme sarcosine dehydrogenase (SARDH), responsible for the conversion of sarcosine to glycine, resulted in high levels of sarcosine and elevated invasion [173]. GNMT and SARDH are both regulated by AR and the TMPRSS2-ERG fusion [172,174].
Another important aspect of the one-carbon cycle is its role as a source for methyl groups to perform epigenetic modifications. Methylation of histones depends on S-adenosyl-methionine (SAM), a product of the methionine cycle [170]. It was shown that DNA is often hypermethylated in prostate cancer, regulating genes involved in cell cycle, DNA repair and apoptosis [175–184]. Thus, the main DNA methyltransferase 1 (DNMT1) is upregulated and activated in prostate cancer [185,186]. Consequently, tumor formation can be altered, using 5-azacitidine, an inhibitor of DNMT1 [187]. One-carbon metabolism is influenced by AR and fuels prostate epigenetic reprogramming by providing the substrate (SAM) to methyltransferases that are also upregulated by AR [170]. This is highly significant since the epigenetic reprogramming has been shown to lead to drug resistance in prostate cancer. Patients in phase I and II clinical trials demonstrated increased chemosensitivity to docetaxel in combination with 5-azacitidine [188]. In addition to SAM’s important role in epigenetics as a substrate for methyltransferases, it can also be diverted into the polyamine synthesis pathway [170]. Given the importance of the polyamines in the prostate and its association with cancer progression [163,189], the inhibition of methyltransferases may counterproductively lead to an increase in polyamine synthesis. To that end, it was reported that elevated levels of polyamines could lead to prostate cancer progression [190,191] as the reduction of polyamines in CRPC patients led to prolonged survival [192]. Interestingly, when SAM is utilized by the methyltransferases it creates S-adenosyl-homocysteine (SAH), which moves along the methionine cycle to intersect with the folate cycle to create methionine and regenerate SAM [170]. Along the methionine cycle SAM is converted to SAH and further to homocysteine by the enzyme S-adenosylhomocysteine hydrolase [171]. Homocysteine can then feed into the transsulfuration pathway, resulting in the production of glutathione and therefore altering the cellular response to ROS [170]. Taken together the one-carbon cycle represents a complex network of several pathways, of which we are only beginning to understand if, how and when to target.
Hexosamine Biosynthetic Pathway
The Hexosamine Biosynthetic Pathway (HBP) branches from the traditional glycolytic pathway to synthesize UDP-GlcNAc, the essential substrate for N- and O-linked protein glycosylation and glycosaminoglycan, proteoglycan, and glycolipid production. While the HBP shunts only a small percentage of fructose 6-phosphate away from glycolysis, it is viewed as a readout of total cellular energy levels because it incorporates metabolites from several key metabolic pathways, including nucleotide (uracil), amino acid (glutamine), fatty acid (acetyl-CoA), and glucose (F6P) metabolism [193]. Additionally, the UDP in UDP-GlcNAc is an energetic compound that can serve as a non-ATP readout of cellular energy availability [193].
In prostate cancer, the HBP is upregulated in hormone-sensitive, localized tumor samples, as the mRNA and protein levels of the first and rate-limiting enzyme in the HBP, glutamine fructose-6-phosphate amidotransferase (GFAT/GFPT1), and the final enzyme in the HBP, UDP-N-Acetylglucosamine Pyrophosphorylase 1 (UAP1) are elevated in cancerous prostate tissues compared to matched benign samples. GFAT and UAP1 levels are also increased in response to androgens in LNCaP and VCaP cells [194,195]. High-performance liquid chromatography (HPLC)-based evaluation of the levels of sugar nucleotides in cells revealed UDP-GlcNAc levels are high in early-stage, AR-positive cell lines (LNCaP and VCaP), but low in nontransformed human prostate cell lines (RWPE-1 and PNT2) as well as AR-negative PC-3 cells. In addition, AR-positive cell lines express ~50% more enzymes involved in the HBP compared to AR-negative cell lines. This indicates that AR mediates the upregulation of HBP enzymes, and, therefore, the flux of metabolites through the HBP in early, localized prostate cancer.
N-linked glycosylation utilizes UDP-GlcNAc to add complex sugar conjugates to proteins in the ER and Golgi bound for the outer membrane which can, amongst other effects, influence the localization and stability of those proteins [196]. The decrease in the expression of AR target genes KLK3 and CAMKK2 (cytosolic proteins that are not N-glycosylated) in the presence of the N-linked glycosylation inhibitor tunicamycin suggests that AR activity may be dependent on positive crosstalk with a membrane bound protein that relies on N-linked glycosylation for activity and/or stability. The main candidates for this are receptor tyrosine kinases (RTKs), specifically insulin-like growth factor 1 receptor (IGF-1R). N-linked glycosylation increases RTK membrane retention time resulting in longer RTK signaling. In LNCaPs and VCaPs, glycosylation of IGF-1R increased AR-mediated transcription of IGF-1R indicating there is a positive feedback between the two that requires glycosylation-mediated IGF-1R membrane retention and subsequent IGF-1R-mediated AR activation.
O-linked β-N-acetylglucosamine transferase (OGT), the enzyme that utilizes UDP-GlcNAc to mediate O-GlycNAcylation (O-GlcNAc) of protein substrates, is also upregulated in clinical samples of localized prostate cancer and is associated with poor prognosis [195,197,198]. O-GlcNAc modification of Ser/Thr residues on proteins can act similarly to phosphorylation to change the activity of a given target protein. Inhibition of OGT either pharmacologically or molecularly led to a decrease in the viability of LNCaP, VCaP and PC-3 cells. It also led to a decrease in tumor size in mouse xenografts of prostate cancer and other cancer types. This could be due in large part to the destabilization of c-MYC caused when OGT is lost. When glycosylated by OGT, c-MYC remains stable, but when c-MYC remains unglycosylated, it becomes vulnerable to ubiquitin mediated proteasomal degradation.
Paradoxically, in CRPC, expression of the HBP enzyme glucosamine-phosphate N-acetyltransferase 1 (GNPNAT1) is significantly decreased compared to localized prostate cancer [199]. Loss of GNPNAT1 expression increased the aggressiveness and proliferation of CRPC. This was mediated by the expression of several oncogenic cell cycle genes through activation of the PI3K-AKT pathway in cells expressing full-length androgen receptor (AR) or by specific protein 1 (SP1)-regulated expression of carbohydrate response element-binding protein (ChREBP) in cells containing the AR-V7 variant. Accordingly, addition of UDP-GlcNAc to CRPC cells decreased proliferation in cell culture and tumor growth in vivo. UDP-GlcNAc treatments also sensitized CRPC cells to enzalutamide. Taken together, activation of the HBP or addition of its end product UDP-GlcNAc, particularly in conjunction with a standard of care drug like enzalutamide, could have clinical efficacy in the treatment of CRPC.
Metabolic Scavenging
Autophagy is a cellular recycling process that can provide metabolites under conditions of cellular stress such as starvation. It can also help mitigate ROS by clearing out dysfunctional organelles (ex. mitochondria), and clear the cell of protein aggregates and damaged proteins that interfere with normal cell operations. Initially, autophagy was categorized as a tumor suppressive process due to its ability to shut down proliferation and induce cell death when hyper-activated. However, recent studies indicate autophagy is contextual, exhibiting both pro-cancer and anti-cancer functions [200,201]. In prostate cancer, autophagy has been shown to provide advanced prostate cancers protection against starvation and hypoxia while promoting resistance to cancer therapies [202]. Moving forward, the intricate relationship between autophagy core components, its upstream regulators, altered AR signaling, and conditions in the tumor microenvironment must be defined to determine if, how, and when modulators of autophagy could be used to treat prostate cancer.
A study of melanoma showed that knockout of the core autophagy gene Atg7 led to a decrease in tumor growth due to DNA damage and activation of senescence [203]. Another study of melanoma showed monoallelic loss of Atg5 led to tumor metastasis while, in contrast, total loss of Atg5 led to increased sensitivity to BRAF inhibitors and decreased tumor burden [204]. These findings suggest that there are dose-dependent effects of autophagy in cancer. This seems to be the case in prostate cancer as well. Using genetic mouse models, deletion of Atg7 specifically in prostate epithelia was sufficient to impair cancer progression in Pb-Cre, Ptenf/f in both intact and castrate conditions [205]. Interestingly, co-targeting of HK2 and ULK1-dependent autophagy suppressed the growth of PTEN- and TP53- deficient CRPC [185], indicating that autophagy inhibitors may have improved efficacy under induced conditions of metabolic stress.
It is becoming increasingly clear that autophagy plays a critical role in resistance to treatments of advanced prostate cancers [206,207]. To that end, enzalutamide resistance in CRPC cells can be overcome by inhibition of autophagy [208–210]. Docetaxel induces autophagy through Beclin1-Vps34-Atg14 complex formation in CRPC cells without effecting mTOR or p-mTOR expression [211]. Inhibition of autophagy boosted sensitivity to the chemotherapy in CRPC cells, and interestingly, it has been found that activation of STAT3 by IL-6 could inhibit autophagy and improve chemotherapeutic efficacy [211–213]. Additionally, autophagy provided protection against the anti-cancer drug diindolylmethane (DIM) in LNCaP and C4–2B cell lines and PC-3 xenograft mouse models [214,215]. Treatment of DIM in combination with the ULK1 inhibitor MRT 67307, chloroquine (CQ), or siRNAs against the oncogene AEG-1 or AMPK significantly reduced cell proliferation in culture and tumor growth in vivo. Further, when PC-3 cells were treated with the cyclooxygenase-2 (COX-2) inhibitor celecoxib, c-Jun N-terminal kinase (JNK) mediated the activation of autophagy to protect cells from celecoxib-induced apoptosis [216]. Finally, curcumin can activate autophagy and apoptosis in CRPC. Curcumin treatment in conjunction with inhibitors of autophagy increased apoptotic cell death and mitigated the protective effect of autophagy [217]. To date, however, significant anti-cancer efficacy of autophagy-targeted therapies has not been observed in prostate cancer patients. However, this could be due to the lack of potent and selective inhibitors of autophagy, a major limitation of the field.
Recently, loss of the transcription factor repressor element-1 silencing transcription factor (REST) combined with induction of autophagy was found to promote the neuroendocrine differentiation (NED) of prostate cancer. Monoamine oxidase A (MAOA), a mitochondrial enzyme, was identified to be downregulated by REST [218]. Downregulation of MAOA resulted in decreased autophagic flux (specifically mitophagy). Cells with downregulated MAOA were also identified to have fewer NED characteristics. Clinically, MAOA expression tracked with prostate cancer relapse in patient samples. Thus, MAOA expression could induce neuroendocrine transdifferentiation in part through induction of autophagy/mitophagy. Like MAOA, tumor necrosis factor α-inducible protein 8 (TNFAIP8) has been shown to be expressed highly in AR-negative PC-3 cells and expression is associated with prostate cancer survival [219]. Overexpression of TNFAIP8 in PC-3 cells tracked with autophagic flux and biomarkers of NED. Another new possible regulator of autophagy is dCTP pyrophosphatase 1 (DCTPP1) [220]. DCTPP1 typically is involved in hydrolyzing dCTP to dCMP. High DCTPP1 levels track with prostate cancer progression and Gleason score as well as the progression of other cancer types. A bioinformatics study has linked DCTPP1 tumor promoting actions to autophagy, but further research is still needed to determine how DCTPP1 is mechanistically linked to autophagy.
While still controversial, most data support a tumor suppressive role of autophagy in the early stages of the disease and a tumor-supportive role of autophagy in the late stages [202]. Current evidence suggests that inhibiting autophagy, or its upstream regulators, in combination with antiandrogens, taxanes, or other targeted therapies may have clinical efficacy in the advanced stages of prostate cancer. Recent interesting findings point toward a link between autophagy and NED. How autophagy can induce this differentiation or if autophagy is just a bystander during NED is still unclear. As new ways to accurately measure autophagic flux in vivo become available, understanding what type of autophagy, when autophagy is activated, and where autophagy is activated will provide the needed insights into how autophagy is contributing to disease progression and therapy resistance. Further, discovery of new regulators of autophagy and efficacious in vivo inhibitors will undoubtedly be needed to push this therapeutic approach in the clinic.
During a process known as macropinocytosis, cells engulf nearby extracellular substances and transport them to lysosomes for degradation to yield smaller metabolites that provide cells with additional nutrients. In the context of cancer, macropinocytosis can provide starved and stressed cells with additional nutrients much like autophagy, even sharing some of the same components and regulatory pathways [221]. However, unlike autophagy, macropinocytosis can scavenge extracellular nutrients, easing the burden on cells to recycle their own components which will eventually become depleted. Given that cancer cells can utilize macropinocytosis to survive in nutrient starved environments, targeting its upstream regulators and core machinery could have therapeutic value.
Macropinocytosis requires PI3K-mediated production of PIP3 for membrane enclosure and RAC1 activation to prompt cytoskeletal remodeling and membrane ruffling necessary for engulfment of extracellular materials [222]. The PI3K inhibitor PTEN is the most commonly deleted tumor suppressor in prostate cancer. Concurrently, the RAC1 activator AMPK is often highly activated in prostate cancer due to the increased expression of the AR target gene and AMPK activator CaMKK2 [223]. Taken together, macropinocytosis can be highly activated in prostate cancer. Recently, it was found that macropinocytosis in PTEN-deficient prostate cancer differs from RAS-driven cancers as it did not account for all of the albumin uptake into the cell [222]. However, it was shown that necrotic debris was exclusively taken into the cell via macropinocytotic engulfment. This discovery was elegantly accomplished using fluorescently labeled murine hematopoietic cell “corpses” introduced into the media of prostate cancer cells. Necrotic debris is often available in the tumor microenvironment due to starvation-induced death initiated by the tumor cell itself or by cancer therapies that kill some but not all tumor cells. Containing organelles, proteins and other nutrients, engulfed necrotic tissue is broken down into metabolites to sustain growth, proliferation, and survival. Proteins from the necrotic debris provide amino acids for biomass. Besides increased biomass, the replenished amino acid pool activates mTOR signaling. Further, lipids are also derived from the necrotic tissue and supplement lipid biosynthesis and subsequent lipid metabolism that is critical to many prostate cancers. Hence, micropinocytosis is an emerging biological process in cancer that may warrant drug development efforts. To that end, the requirement for lysosomal activity in both autophagy and micropinocytosis may account for some of the efficacy of the reported autophagy inhibitors that function via disrupting lysosomal functions.
Regulation of Metabolic Reprogramming
Signal Transduction
The regulation of cancer metabolism by signal transduction pathways has garnered considerable attention over the past two decades. It is clear that aberrant signaling, from both intracellular and extracellular stimuli, converge to alter a cancer cell’s central metabolism to support the high demands for energy production and building blocks. In the context of prostate cancer, AR activation has been tightly coupled with global metabolic alterations. Also, MYC amplification, PTEN loss and aberrant activation PI3K/AKT/mTOR signaling, all common events in advanced prostate cancer, have profound effects on metabolic adaptation. Here, we summarized the association of several key signaling pathway regulators with metabolic reprogramming in prostate cancer.
As the major driver of prostate cancer, AR’s influence on diverse metabolic pathways has significant implications for prostate cancer progression. Transcriptional upregulation of enzymatic genes is one of the important ways that AR works in metabolic rewiring. A common mechanism is AR directly binding to the promoters of these genes and increasing their transcription. The expression of these critical enzymes promotes a metabolic shift that facilitates cell growth, survival and migration [224,153,225,226]. A detailed description of known, direct AR metabolic target genes has been previously described [225]. Additionally, some important metabolic regulators are downstream targets of AR. For example, AR actives a CaMKK2-AMPK-mediated cascade. CAMKK2 is the direct target of AR and is overexpressed and over-activated in prostate cancer [151–153,227]. CaMKK2, the predominant upstream kinase of AMPK in the prostate, helps cells to adapt to various energetic stresses. AMPK-mediated metabolic changes have been correlated with increasing intracellular ATP levels, glycolysis, glucose uptake and PGC-1α-mediated mitochondrial biogenesis [131,227,153,29]. HIF1α also coordinates with AR to mediate metabolic adaptation to hypoxia and help cells maintain redox balance and cell survival under hypoxia. In a low androgen environment, HIF1 α directly upregulated AR expression in the presence of hypoxia [228]. Meanwhile, AR can stabilize and activate HIF1α through an autocrine loop of PI3K/AKT in a hypoxia-independent manner [229]. This crosstalk provides rationale for the joint inhibition of AR and HIF-1α to treat prostate cancer by blocking metabolic adaption to varied androgen or oxygen levels. Of note, AR splice variants can also regulate prostate cancer cell metabolism. For example, AR-V7 can promote cell growth, migration, and glycolysis [173]. Like AR, in CRPC cells, AR-V7 can drive de novo lipogenesis [71]. However, AR-V7 did exhibit some unique metabolic regulatory behavior. A metabolic profile showed that in AR-V7-stimulated cells, there were some striking differences in the levels of TCA cycle intermediates [173]. Notably, AR-V7 promoted higher levels of citrate oxidation, similar to what was observed in CRPC patient samples [173]. Further, AR-V7 increased glutaminolysis and reductive carboxylation.
MYC is another common oncogene that drives prostate cancer tumorigenesis. Amplification and mutations of MYC are frequently seen in advanced prostate cancer and associated with poor prognosis in a subset of cases [230]. Similar to AR, MYC contributes to metabolic reprogramming partially through the activation and expression of metabolic enzymes. Mitochondrial glutaminase, GLS1, has been identified as a MYC downstream effector for glytaminolysis in PC-3 cells via miR-23a/b [143]. Additionally, glutamine uptake was regulated by MYC in a PTEN-dependent manner [141]. Many MYC-mediated effects are exerted through complex interactions. MYC-E2F1 had a greater regulation of nucleotide metabolism while MYC-HIF-1α was more involved in glucose metabolism [231]. Moreover, MYC may also play a role in lipid metabolism. Oncogene-mediated metabolic signatures in prostate cancer revealed that dysregulated lipid metabolism was induced by MYC overexpression [232].
PTEN loss and subsequent hyperactivation of PI3K/AKT/mTOR signaling are also common events in advanced prostate cancer. As a master regulator of metabolism, the PI3K/AKT/mTOR pathway controls nutrient uptake and utilization as well as metabolic scavenging. PI3K/AKT activation has been strongly linked to aerobic glycolytic metabolism [232]. Further, mTORC1 promotes glycolysis by increasing HK2 translation and upregulating the expression of HIF-1α [233,234]. The mTORC2 complex further augmented glycolysis through AKT-dependent HK2 activation [233]. In addition, activation of AKT via PTEN-deficiency has been indicated to increase glucose metabolism by increasing HK2 phosphorylation and expression which in turn increased intracellular ROS-mediated cell growth [235]. Moreover, AKT/mTORC1 has been suggested to influence fatty acid synthesis through the activation of SREBP and upregulation of FASN [95,236]. Inhibition of AKT in PTEN-deficient cells modulated the activation of ACLY. This repression limited the conversion of citrate to acetyl-CoA which ultimately reduced histone acetylation and epigenetic regulation [237,234]. PTEN loss also led to cholesterol ester accumulation which has been linked with more aggressive diseases [93,238]. Therapeutically, Naguib et al. suggested that inhibition of mitochondrial complex I is an effective strategy to decrease PTEN loss-induced cell growth [239]. This was attributed to the fact that PTEN-null cells are more dependent on consuming ATP through mitochondrial complex V. Additionally, mTOR signaling modulates amino acid metabolism in prostate cancer through its regulation of glutamine uptake, glutamine utilization, and polyamine biosynthesis [141,240,241]. Importantly, PI3K/AKT/mTOR signaling can function in part as a nutrient sensor by responding to changes in cellular energy status. For instance, leucine deprivation inhibited proliferation and induced apoptosis in CRPC via blocking mTORC1 signaling [242].
Of note, signaling pathways rarely work in isolation. Instead, they are greatly influenced by one another. AR has been demonstrated to regulate MYC expression in a context-dependent manner [141]. Blocking either AR or the PI3K/AKT/mTOR signaling pathway can mutually stimulate the other pathway to support cancer cell proliferation, particularly in the context of CRPC [243–245]. Meanwhile, AR activation induced mTOR nuclear localization and reprogramed its genomic binding. In this scenario, mTOR acted as a transcriptional integrator to facilitate androgen-dependent metabolic rewiring [246]. In addition, AMPK activation is essential for PTEN loss-increased macropinocytosis [222]. Also, mTOR signaling promoted prostate cancer stem cell survival, an effect that was modulated by HIF-1α [247]. Considering the abundance of multiple feedback mechanisms, future therapeutic regimens may benefit from combinatorial treatment strategies, especially for overcoming drug resistance [244,245,248].
Non-coding RNAs
MicroRNAs (miRNAs) are small, endogenous non-coding RNAs of 18~25 nucleotides in length, which act as gene regulators. Different from transcription factors, miRNAs regulate gene expression by directly binding to the 3’-untranslated region (3’UTR) of mRNAs and inducing mRNA degradation and/or inhibiting translation. Therefore, miRNAs have been associated with a number of biological processes including proliferation, apoptosis and metabolism. Emerging evidence has revealed that the altered metabolism in cancers including prostate cancer is strongly mediated by miRNAs. They can either directly target the transporters, kinases and enzymes in established metabolic pathways or indirectly manipulate important signaling pathways that regulate cancer metabolic shifts. Here, we focus on the direct regulation and will briefly summarize miRNAs and their related metabolic targets in prostate cancer (Table 1).
Table 1:
miRNAs Regulating Prostate Cancer Metabolism.
| miRNAs | Regulation | Target Genes | Direct | miRNA Function in Metabolism | Tissues/Cell Lines | Reference (PMID) |
|---|---|---|---|---|---|---|
| miR-132 | down | GLUT1 | Yes | inhibit glucose uptake, lactate secretion and glycolysis | PC-3, DU-145, LNCaP, prostate cancer tissue | 27398313 |
|
PKM2 HK2 |
unknown | |||||
| miR-181b | down | HK2 | Yes | inhibit glycolysis | PC-3 | 28184935 |
| miR-143 | down | HK2 | Yes | glucose metabolism | GSE21032 dataset, PC-3 | 26269764 |
| miR-421 | down | PFKFB2 | Yes | inhibit glycolysis | LNCaP, 22Rv1, PC-3, LNCaP, prostate cancer tissue | 26269764 |
| miR-205 | down |
HK2 GLUT1 |
unknown | promote metabolic shift from glycolysis to OXPHOS, Docetaxel resistance | docetaxel-resistant PC-3 and DU-145 | 27542265 |
| MDH2 | Yes | inhibit MDH2 expression | different stages of prostate cancer tissue | 29563510 | ||
|
miR185 miR-342 |
down |
SREBP1 SREBP2 |
Yes | inhibit the expression of FASN and HMGCT, inhibit lipogenesis and cholesterogenesis | LNCaP, C4–2B | 23951060 |
| miR23a/b | down | GLS1 | yes | increase glutamine catabolism | PC-3 | 19219026 |
| miR22b-3p | up | PRODH | inhibit proline catabolism | PC-3 | 22615405 | |
| miR-22 | down | ACLY | yes | inhibit de novo lipid synthesis | PC-3 | 27317765 |
| down | MDH2 | Yes | inhibit MDH2 expression | primary prostate cancer, CRPC, PC-3 | 29563510 | |
| miR-17/92 cluster | up | PPARA | unknown | increase lipogenesis | LNCaP | 23059473 |
|
miR-1 miR-206 |
down |
G6PD TKT PGD GPD2 |
unknown | inhibit glycolysis | DU-145 | 23921124 |
| miR-29c | down | SLC2A3 | Yes | inhibit glucose metabolism | prostate cancer tissue | 29715514 |
In contrast to the largely global increase of miRNAs in prostate cancer [249], miRNAs directly regulating prostate cancer glucose metabolism are mostly downregulated. Such inhibition facilitates the stability and expression of metabolism-related mRNAs. Decreased expression of miR-132, observed in prostate cancer, can promote a metabolic shift towards glycolysis by increasing the expressions of GLUT1, HK2 and PKM2 [250]. Targeting miR-132 using an inhibitor was sufficient to stimulate glucose uptake, increase lactate secretion and boost cell proliferation. Similarly, miR-181b, 142, 421, 205 and 143 are also associated with the regulation of glycolysis (Table 1).
In addition, miRNAs also regulate the TCA cycle and OXPHOS. Malate dehydrogenase 2 (MDH2), a TCA cycle enzyme, has been associated with miR-22 and miR-205 [21]. Through comparing RNA-seq and proteomics data from benign, untreated primary prostate cancer and CRPC samples, a recent study showed that the protein expression of MDH2, which persistently increased during prostate cancer progression, was not correlated with its mRNA level. Strikingly, miR-22 and miR-205 were found to directly bind to the mRNA of MDH2 and suppress its protein translation. miR-205 also contributed to docetaxel resistance in prostate cancer by promoting a metabolic shift from glycolysis to OXPHOS. OXPHOS engagement may be a hallmark of docetaxel resistance in PC-3 cells. Compared to parental PC-3 cells, in docetaxel-resistant derivative PC-3 cells, OXPHOS-related genes are upregulated while glycolytic genes are downregulated. Accordingly, restoration of miR-205 led to the increase of HK2 and GLUT1 mRNA and made cells more sensitive to the docetaxel. However, it remains unknown how miR-205 upregulates HK2 and GLUT1.
Further, miRNAs can target the PPP to provide building blocks for nucleotide biosynthesis as well as NADPH for anabolic metabolism and ROS homeostasis. Singh et al. demonstrated that miR-1 and its identical paralog miR-206 are associated with prostate cancer metabolic alterations through targeting three key PPP genes (G6PD, PGD, and TKT) and one carbohydrate/lipid metabolism regulation gene (glycerol-3-phosphate dehydrogenase, GPD2) [251]. Nuclear factor erythroid-2-related factor 2 (NRF2) was reported to promote tumor growth by attenuating miR-1 and miR-206 expression, activating the PPP pathway, which in turn accelerated cell proliferation.
SREBP-1, −2, ACLY and PPARA have been identified as the direct targets of miRNAs [252]. For instance, ACLY was identified as a direct target of miR-22 [253]. MiR-22 bound to the 3’UTR (seed sequence GGCAGCU) of ACLY to decrease it subsequent de novo lipid synthesis. As a result, miR-22 treatment was able to inhibit PC-3 cell growth and metastasis in cell and xenograft models. Moreover, SREBP-1 and −2, master transcription factors for lipogenesis and cholesterogenesis, are controlled by miR-185 and miR-342 in prostate cancer [254]. Through repressing SREBP-1 and −2, these two miRNAs block the expression of FASN and HMGCR, two genes important for fatty acid and cholesterol synthesis. Compared to the non-cancerous prostate epithelial cell line RWPE-1, LNCaP and C4–2B cells had lower expression of both miR-185 and 342. Restoration of miR-185 and 342 not only decreased the amounts of fatty acid and cholesterol, but inhibited tumorigenesis, cell growth, migration and invasion in the prostate cancer cells.
MiR-23a and miR-23b are two miRNAs identified to regulate glutamine catabolism in prostate cancer. Gao et al. reported that miR-23a and miR-23b directly targeted mitochondrial glutaminase to influence cell survival [143]. Mechanistically, MYC transcriptionally impeded the expression of miR-23a and miR-23b, which increased the expression of glutaminase. This promoted the conversion of glutamine to glutamate. Glutamate could then serve as a substrate for ATP production or glutathione synthesis, both of which could impact the cell proliferation. However, MYC was also reported to upregulate miR-23b-3p and consequently proline dehydrogenase expression [255]. Since proline dehydrogenase can induce apoptosis, the decreased level of proline dehydrogenase could protect cells against oxidative stress and increase cell survival. Clearly, additional work needs to be done to fully understand MYC’s regulation of this set of miRNAs.
Influence of the Tumor Microenvironment
While cancer cell intrinsic mutations and signaling aberrations undoubtedly drive metabolic reprogramming in prostate cancer, it is now appreciated that the cancer microenvironment, including fibroblasts, adipocytes, immune cells as well as endothelial cells, can greatly influence metabolism and disease progression [256]. Cancer initiation, progression and metastasis all require adaptation to the harsh host microenvironments that can include a lack of nutrients, high oxidative pressure and hypoxia. Meanwhile, by interaction or signal secretion, cancer cells are able to remodel the extracellular matrix (ECM), repurpose the surrounding non-malignant cells and eventually leverage their neighbors to support their rapid proliferation. Therefore, the crosstalk between cancer cells and surrounding cells helps determines the fate of cancer and thus may provide an attractive target for cancer therapy. Here, we will focus on the metabolic interaction between prostate cancer and its microenvironment.
Hypoxia is an important factor that influences cancer progression, metastasis and drug resistance. Evidence supports the existence of a hypoxic area in prostate cancer and linked it to higher clinical disease stages [257]. Hypoxia promotes epigenetic and genetic adaptation and therefore induces corresponding biological changes including metabolic reprograming that support rapid cancer cell growth. The key regulator of this process is the HIF-1 complex, an oxygen-dependent transcriptional factor. Overexpression of HIF-1 has been detected in both primary and metastatic prostate cancer. HIF-1 can trigger a number of metabolic alterations including the induction of glycolysis to maintain ATP levels and provide biosynthetic building blocks. We have discussed the regulatory role of HIF-1 on glycolytic genes including HK2, PDK1, PKM2, LDHA and MCT4 above. The events ensure the glucose supply for anaerobic respiration, promote lactate excretion to prevent the inhibition of glycolysis and maintain the redox balance for proliferation and invasion. HIF-1 mediated-glycolysis enhancement has been associated with ADT resistance [258]. ADT decreased the PPP in prostate cancer, while hypoxia and HIF-1 maintained the glucose uptake and lactate production. Further, ADT increased the expression of glucose-6-phosphate isomerase (GPI) specifically under hypoxic conditions. These data suggest that targeting GPI or glycolysis is a potential strategy to overcome ADT resistance in hypoxic prostate tumors. In hypoxic conditions, glutamine also becomes a more significant carbon source for lipid synthesis. To that end, HIF-1-mediated PDK1 activation repressed the production of citrate, which in turn strongly increased the α-ketoglutarate to citrate ratio and altered glutamine conversion from oxidative to reductive [107]. It is still not fully understood how hypoxia affects fatty acid metabolism in prostate cancer, but hypoxia induced-CPT1C expression and fatty acid oxidation may be one mechanism that helps explain how prostate cancer cells could combat metabolic stress [259]. However, there are many other regulators of the hypoxic response that need to be considered including, but not limited to, p53, Myc and mTOR. To date, their relative contributions to the hypoxic response in prostate cancer are only beginning to be defined.
Oxidative stress can also induce metabolic reprogramming and contribute to the progression of prostate cancer. Whilst reactive oxygen species (ROS) commonly cause damage to normal cells, they facilitate tumor growth and malignant progression by inducing DNA damage and genetic alteration, and eventually, by reprogramming cancer cell metabolism to adapt to the stressful tumor environment. Compared to normal prostate cells, prostate cancer cells harbor high levels of ROS. Evidence suggests that several signaling pathways contribute to the observed increased ROS. First, androgen can promote ROS production by enhancing NADPH oxidase (NOX) expression, a major generator for extramitochondrial ROS, as well as the six transmembrane protein of prostate 2 (STAMP2) [260,261]. Such ROS generation was essential for prostate cancer cell growth and invasion. Second, the loss of antioxidant proteins resulting from Nrf2 deregulation and inactivation of the glutathione S-transferase family members promoted increased ROS [262,263]. The upregulation of thioredoxin-1 (TRX1) is one mechanism that protected CRPCs from oxidative stress following ADT [264]. Consequently, Khandrika et al. proposed that the elevated ROS may lead to the accumulation of mtDNA mutations, which in turn changed the cellular metabolism [263]. However, more direct evidence is still needed to confirm this intriguing hypothesis. Meanwhile, ROS is a dual sword for prostate cancer cells given that high ROS makes them to be more vulnerable to cell death. As such, further increasing ROS in prostate cancer has been tested as a treatment strategy [265,264]. Interestingly, a recent study indicated that the inactivation of T cells in prostate cancer is partially due to the increased ROS that accumulates in T cells. In the tumor microenvironment, T cell activation was not only inhibited by nutrient deprivation and microenvironment acidification, but 1-pyrroline-5-carboxylate released by prostate cancer cells also repressed T cell proliferation and function by increasing ROS production and decreasing ATP production in T cells [266,256].
Cancer associated fibroblasts (CAFs) are the most abundant non-cancer cells in tumors. They are derived from the resident fibroblasts or other precursor cells but maintain a perpetually activated phenotype [267]. CAFs have been indicated to promote proliferation, invasion, and metastasis of prostate cancer, along with development of chemotherapy resistance. The reciprocal activation between cancer cells and CAFs has been reported in prostate cancer, resulting in cancer “stemness” and EMT [268]. PKM2 was one of the key regulators for this crosstalk. The close contact of prostate cancer cells to CAFs triggered PKM2 translocation to the nucleus where it formed a trimeric complex with HIF-1α and DEC1 to stimulate the EMT and an OXPHOS phenotype. In addition, cancer cells exert reciprocal effects on CAFs to influence their metabolism. The exposure of CAFs to cancer cells induced a metabolic shift in the CAFs mimicking the “Warburg effect” with higher glucose consumption and lactate production and export [269–271]. The expression of GLUT1 and MCT4 contributed to this metabolic reprogramming [62,270]. Interestingly, lactate from the CAFs became an energy source to fuel cancer cell OXPHOS. Using co-culture experiments, prostate cancer cells were reported to decrease glucose uptake via changes in GLUT1 expression but increased lactate influx via MCT1 [62,270]. Accordingly, MCT expression differences between prostate cancer cells and CAFs has been associated with poor clinical prognosis [272]. However, an IHC staining study in 96 node-negative prostate cancer specimens, while supporting the role of CAFs in promoting prostate cancers, proposed an opposite metabolic symbiotic relationship between prostate cancer and CAFs [273]. They suggested the cancer cells preferred to produce lactate and undergo anaerobic metabolism with high LDH5 (LDHA) expression while CAFs favored aerobic mentalism by LDH1 (LDHB) expression. Whether there is reversible metabolic switching that occurs between prostate cancer cells and CAFs is not known. The expression of carbonic anhydrase IX (CAIX) in CAFs is another other important mediator for cancer progression. The cancer cells can stimulate the expression of CAIX in CAFs [272,274,275]. Since CAIX catalyzes the reversible reaction from carbon dioxide to bicarbonate and protons, it is involved in regulating the pH of the tumor microenvironment. An acidic microenvironment has been correlated with cancer invasion, dissemination and drug resistance [271]. Bicarbonate can serve as a monocarbonic intermediator for growth needs [275]. Therefore, cancer induced–CAIX expression in CAFs could facilitate the establishment of an environment supportive of proliferation and metastasis. Finally, CAFs can protect prostate cancer cells against chemotherapy-induced cell death by increasing intracellular glutathione (GSH) levels [276]. CAF co-culture or conditioned medium can increase GSH levels ~30% in LNCaP cells. This prevented cells from doxorubicin-induced oxidative stress and ultimately apoptosis.
Similar to CAFs, adipocytes can influence cancer cell metabolism. While adipocytes are sparse in the prostate gland, the peri-prostatatic adipose tissue (PPAT) and adipocytes in bone marrow are thought to provide a reciprocal interaction between adipocytes and prostate cancer cells that promotes disease progression [277,18,278]. In this regard, the prognostic value of PPAT quantity has been evaluated in several clinical studies and associated with prostate cancer aggressiveness [277]. Adipocytes may help fuel prostate cancer cells with glycerol and fatty acids via lipolysis. Using Fourier-transform infrared spectroscopy, marrow adipocytes were shown to provide lipids to the mCRPCs [279]. This work is supported by a recent publication that indicated extracellular lipids provide a much greater fraction of carbons to intracellular lipid pools than previously appreciated [280]. This cell interaction may directly contribute to the growth, morphology and cytokine expression of prostate cancer mCRPCs [281]. Interestingly, HIF-1α may play an important role in metabolic shifts after such lipid translocation. Paracrine lipids were reported to induce HIF-1α-mediated glycolysis in an oxygen-independent manner [282]. Co-culture or conditioned media induced a significant upregulation of glycolytic-associated genes including PDK1, ENO2, HK2, GLUT1, and LDHA in prostate cancer, which, in turn, promoted lactate and ATP production. The p62 scaffolding protein is also an important regulator for adipocyte/prostate cancer cell crosstalk [278]. Loss of p62 in adipocytes not only inhibited its own energy expenditure, but increased osteopontin secretion, which resulted in a lipid-rich environment that could be utilized by tumors. This metabolic reprogramming promoted the tumorigenesis and invasiveness in the TRAMP mouse model. Interestingly, prostate cancer cells were capable of elevating the adipose triglyceride lipase in adipocytes, resulting in the activation of lipolysis and the subsequent supply of substrates to support cancer cell metabolism.
Exploiting Metabolic Alterations for Prostate Cancer Therapy
Diagnostic Imaging of Prostate Cancer
Prostate-specific antigen (PSA) level, Gleason score, and clinical stage are routinely used metrics to optimize prostate cancer patient treatments. Advances in biomedical research over the past decade, including gene expression profiling applied to prostate cancer classification (ProlarisScore, OncotypeDx), have provided valuable static molecular signatures, but they do not fully account for cancer progression’s complexity. Since the biological behavior of prostate cancer varies widely, and markers for early disease progression are not clearly established, no consensus currently exists for the active surveillance of early onset disease. Selection criteria that control eligibility for early monitoring and the biometric changes that constitute cancer progression are lacking. Nonetheless, active surveillance has emerged as a viable management option for selected cases. However, given the physical and psychological burdens associated with active surveillance including the need for repeat biopsies [283], the development of non-invasive, in vivo molecular imaging for risk stratification and surveillance remains an unmet clinical need.
Molecular imaging is broadly defined as techniques that visualize, characterize and measure the biological processes at the molecular and cellular levels in living systems. In recent years, there has been a growing interest among clinicians and basic researchers in imaging cancer metabolism as a viable diagnostic tool in oncology. This resurgent interest includes the need to understand the molecular and cellular changes that occur in metabolism, how the tumor microenvironment directly affects metabolism and how metabolic vulnerabilities can affect treatment response. Metabolic changes are emerging as hallmarks of cancer that can be exploited as imaging biomarkers and employed as a reporter of cancer grades and severity [284]. Oncogenic mutations play a pivotal role in altering the metabolism in cancer. Real-time metabolic imaging provides the opportunity to visualize the perturbation of metabolism as the cancer initiates, progresses and responds to therapy.
Hyperpolarized Imaging in Prostate Cancer:
Hyperpolarized 13C Magnetic Resonance (HP-MR) is emerging as a non-toxic, non-radioactive method for interrogating tissue metabolism [285]. Hyperpolarization allows for over 10,000-fold signal enhancement relative to conventional magnetic resonance imaging (MRI) or spectroscopy (MRS). After hyperpolarization, the signal enhancement can be retained on the metabolites of the hyperpolarized molecules for several minutes. Many laboratories are working on techniques to extend this relaxation time so that more detailed metabolic studies can be considered. Dynamic HP-MR has been utilized for non-invasive assessment of the downstream metabolic product of glycolysis.
The Warburg effect is a hallmark of tumor growth, and detecting it provides extremely useful information in both the detection and characterization of cancer. Clinically, FDG-PET uptake is often used as a surrogate marker of the Warburg effect. In this regard, FDG-PET is useful for the diagnosis of a number of cancers in the clinic. However, slow-growing prostate tumors do not show an appreciable difference in FDG uptake compared to normal or abnormal prostate tissue; therefore, FDG-PET has poor diagnostic value in local staging of prostate cancer. Furthermore, location of the malignancy in a high background region (bladder) can hinder tumor detection. As such, an alternative metabolic imaging technique is needed that can be utilized where FDG-PET fails [286]. In addition, FDG reveals only glucose uptake and phosphorylation; downstream metabolic processing of glucose cannot be detected. Changes in downstream glucose metabolism have been seen between indolent versus aggressive tumors. Using hyperpolarization, not only can small molecules important in key metabolic pathways be imaged, their metabolites can be imaged as well. One example is pyruvate and its breakdown product lactate, which are central to ATP production. Methods of interrogating in vivo metabolic changes in real time would enable a more detailed understanding of tumor metabolism. Hyperpolarized metabolic imaging has the potential of being a complementary diagnostic tool to the FDG-PET.
The mostly widely used method for hyperpolarized metabolic imaging is dynamic nuclear polarization (DNP). DNP is a solid-state polarization method where the imaging compound is mixed with a matrix and irradiated with microwaves at low temperature (near absolute zero Kelvin) and in a high magnetic field. While performing imaging or spectroscopy at increasingly higher field strength raises the relative fraction of polarized nuclei to several parts per million (ppm), the hyperpolarization process can raise the polarized fraction into the high percentages (80–90%), many orders of magnitude higher (10,000–100,000 fold). After the compound is hyperpolarized, it is dissolved in media and the imaging compound is released and readied for injection. GE Healthcare’s SPINLab polarizer has been approved for clinical use and has been used in a Phase I trial for the diagnosis of prostate cancer.
To date, the most studied hyperpolarized 13C compound is pyruvate. The utility of DNP polarized pyruvate in metabolic imaging has been explored extensively [287–289,106,290–295]. Hyperpolarized pyruvate can be used to follow the metabolism of pyruvate to alanine, lactate and bicarbonate. The rate of hyperpolarized lactate production has been revealed as a marker for cancer in multiple animal studies. In addition to pyruvate, the utility of several metabolic imaging compounds in cancer diagnosis and other modalities of hyperpolarization are under investigation.
Although it is anticipated that hyperpolarized metabolic imaging could improve early diagnosis and determining the efficacies of therapies in many areas of oncology, to date most clinical effort in this area has been concentrated on prostate cancer in part due to the above-described unique metabolism of the disease. There are two widely used methods for diagnosing prostate cancer: serum PSA value and biopsy with the determined Gleason score. Both methods have limited sensitivity in determining the level of aggressiveness of the cancer. The PSA score can range significantly due to age or volume changes in relation to benign prostatic hyperplasia, it can be undetectable or low even in the malignant form of prostate cancer, and it cannot localize the disease if present. The determination of the Gleason score from a biopsy is the preferred method of determining the aggressiveness of prostate cancer. A tumor’s Gleason score can be used to predict how that particular cancer will behave; however, due to the heterogeneous nature of prostate tumors, the diagnosis of the biopsied tissue might not reflect the overall aggressiveness of the disease. Sampling error can lead to high-grade cancer lesions being missed. Hyperpolarized pyruvate in mice has been shown to be able to accurately detect prostate cancer in mouse models. Moreover, the ratio of pyruvate to its metabolite lactate potentially distinguishes high-grade from low-grade disease and provides spatial information on the aggressive lesions.
Preclinical Hyperpolarized Metabolic Imaging in Prostate Cancer:
The conversion of hyperpolarized [1-13C] pyruvate to lactate has been used as a valuable tool to detect prostate cancer from surrounding healthy tissue as well as to differentiate between prostate cancer types and progression of disease. Interestingly, a comparison of hyperpolarized pyruvate imaging between mice implanted with the CRPC cell line PC-3 and mice implanted with its more highly metastatic counterpart PC-3M revealed significantly higher lactate production in the PC-3 mice suggesting a greater role for OXPHOS in highly aggressive cancer cells [106]. Nuclear Magnetic Resonance (NMR) analysis of ex vivo tumor samples revealed significantly higher steady-state concentrations of lactate and taurine in the PC-3 tumors while aspartate, glutamate, glutamine, and succinate were significantly lower in PC-3 compared to PC-3M. Further analysis of cell culture media revealed significantly higher lactate production, but lower glutamine consumption in PC-3 cells compared to PC-3M. Thus, PC-3 cells appear more glycolytic while PC-3M heavily utilize glutamine as a source for the TCA cycle. Further, AR+ PDX tumors produced significantly more lactate following hyperpolarized [1-13C] pyruvate injections compared to AR- PDXs [290]. Follow-up NMR analysis of ex vivo tumor samples confirmed the significantly higher concentrations of lactate and succinate in AR+ tumors compared to AR−. In a separate study, the uptake of [1-13C]pyruvate and its conversion into alanine and lactate were correlated with tumors of varying histological grade in the TRAMP mouse model of spontaneous prostate cancer [293]. Importantly, lactate production was able to stratify regions of interest into normal prostate, low-grade tumor, and high-grade tumor with high specificity and was verified via histopathology of excised tissue. While not as extensive, changes in alanine production followed the same trend as lactate across tumor grades. A follow-up study demonstrated similar findings using hyperpolarized lactate production to discriminate tumors into early and advanced disease groups [296]. In addition, a hyperpolarized dual agent of [1-13C]pyruvate and 13C urea was used to measure metabolic flux and blood perfusion in low- and high-grade prostate tumors of TRAMP mice [297]. Pyruvate and lactate signals were modeled to generate the rate constant between pyruvate and lactate (kPL). Urea signal was modeled to generate the area under dynamic curve (AUC), which evaluates the distribution of the tracer in tissue, and the volume transfer constant (Ktrans), which represents tissue permeability and perfusion. High-grade tumors were characterized by an AUC-kPL mismatch possessing significantly lower urea AUC but significantly higher kPL compared to low-grade tumors. High-grade tumors also possessed significantly higher Ktrans than low-grade tumors. The contrast between high Ktrans and low AUC suggests that high-grade tumors are very permeable with high intake and clearance rates. These metabolic and perfusion findings were supplemented with histopathologic analysis which revealed significantly higher expression of HIF-1α, LDHA/LDHB ratio, VEGF, MCT1, and MCT4 in the high-grade tumors.
The ability to test the efficacy of a multitude of different therapies is a major strength of hyperpolarized pyruvate imaging. Tumor properties of TRAMP mice were imaged at several time-points following high-intensity focused ultrasound (HIFU) ablation [298]. Metabolism was measured with hyperpolarized [1-13C] pyruvate, perfusion with hyperpolarized 13C urea and gadolinium dynamic contrast-enhanced (DCE) MRI, and cellularity with diffusion-weighted magnetic resonance imaging (DWI). In the fully ablated zone, the ratio of lactate/pyruvate, the mean 13C urea signal, and gadolinium DCE parameters Ktrans and AUC were all significantly reduced from baseline values by 3–4 hours and remained so when measured at 1 day and 5 days post-treatment. In the partially ablated zones in the margin of the focused ultrasound beam, these values were initially significantly reduced from baseline by 3–4 hours post-treatment but had recovered by the 1-day and 5-day time-points. Hyperpolarized lactate production was significantly decreased by 3-fold in LNCaP prostate cancer cells treated with the AKT inhibitor MK2206 [299]. Platelet derived growth factor receptor (PDGFR) was inhibited with imatinib in mice implanted with the human PCa cell line PC-3MM2 [300]. Hyperpolarized lactate production in the tumor significantly dropped after two days following treatment along with a significant drop of LDH activity and c-Myc protein levels. Notably, tumor volume did not significantly change in this time although there was a significant reduction long-term. NAMPT, an enzyme needed to produce NAD, was targeted with the drug GNE-617 in PC-3 prostate cancer cells and interrogated with hyperpolarized [1-13C]pyruvate and [18F]FDG-PET [301]. Lactate production significantly decreased by 6 hours while FDG standardized uptake value (SUV) was reduced later by 24 hours. Two human prostate cancer cell lines, DU-145 and PC-3, were implanted in mice, and metabolic assays were performed before and after the administration of the LDH inhibitor FX-11 [302]. Following the injection of hyperpolarized [1-13C]pyruvate, lactate production was significantly higher in the DU-145 mice compared to PC-3 mice. Interestingly, both in vivo and ex vivo analysis of steady-state metabolism revealed no significant differences in pyruvate and lactate concentrations between the cell lines. FX-11 was administered to both tumor types, and following hyperpolarized [1-13C]pyruvate injections, lactate production was significantly reduced in the DU-145 mice, but not in the PC-3 mice. This was accompanied with significantly reduced tumor growth rate in the DU-145 mice but not in the PC-3 mice. Thus, imaging with hyperpolarized [1-13C]pyruvate was able to both predict and evaluate treatment response to an LDH inhibitor. In another study, the MEK inhibitor U0126 was administered to prostate cancer PC-3 and breast cancer MCF-7 cell lines and followed with hyperpolarized [1-13C]pyruvate perfusion [303]. While pyruvate-to-lactate flux significantly decreased in the MCF-7 cells, it significantly increased in PC-3 cells following treatment. The contrast in these findings were attributed to the combination of LDH, the enzyme that catalyzes the conversion between pyruvate and lactate, and MCT1, the membrane protein which transports monocarboxylates such as pyruvate across the cell membrane. Intracellular lactate concentration and LDH activity were significantly increased following treatment in both cell lines, but MCT1 levels decreased in MCF-7 cells while remaining unchanged in PC-3. Thus, it is important to understand the mechanism behind these molecular therapies in order to accurately interpret readouts of hyperpolarized MRI assays.
In addition to pyruvate, prostate cancer models have been studied with many alternative hyperpolarized 13C agents. The role of glutaminolysis in prostate cancer was interrogated by monitoring the production of glutamate following the perfusion of hyperpolarized [5-13C]glutamine in PC-3 and DU-145 cell lines [304]. DU-145 cells appeared to upregulate glutaminolysis, as four times as much glutamate production was observed in these cells compared to the PC-3 cell line. The natural anticancer drugs resveratrol and sulforaphane were administered which can act on glutamine-dependent cellular regulators such as PI3K. Both treatments reduced the cell count of each cell line by approximately 50% which correlated with a significant reduction of hyperpolarized glutamate production. Alternatively, the redox status of prostate tumors in TRAMP mice was probed with the injection of hyperpolarized [1–13C]dehydroascorbate (DHA) and monitoring its reduction to vitamin C [305]. This reaction was significantly increased by 2.5-fold in the tumor compared to surrounding healthy prostate tissue which correlated to a significant increase in glutathione concentration. Additionally, there was a 3-fold increase of [18F]-FDG SUV in the tumor compared to surrounding healthy prostate tissue imaged with PET. In a proof-of-concept study, [2-13C]fructose was polarized and injected into TRAMP mice [306]. Its metabolic product β-fructofuranose- 6-phosphate was observed in the tumor with sufficient signal-to-noise ratio to be accurately quantified. D-[1,2,3,4,5,6,6-13C6]glucose-d7 solution was polarized and perfused into MCF7 breast cancer and PC-3 prostate cancer cells to measure downstream metabolic products in the glycolytic and pentose phosphate pathways [307]. The pentose phosphate products 6-phophogluconate and 6-phosphogluconolactone were observed as well as the glycolytic products dihydroxyacetone phosphate, pyruvate, lactate, and bicarbonate. These resonances were fit to a kinetic model and free cytosolic [NAD+]/[NADH] ratio was calculated which was found to be approximately 3-fold higher in PC-3 cells compared to MCF-7. Further, simultaneous hyperpolarization and imaging of multiple 13C-enriched compounds was demonstrated in TRAMP mice [308]. A solution containing co-polarized [1-13C]pyruvate and 13C-sodium bicarbonate was injected into mice, and pyruvate, lactate, bicarbonate, and carbon dioxide peaks could all be detected. Through incorporation into the Henderson–Hasselbalch equation, the bicarbonate and carbon dioxide signals could be used to form voxel-based pH maps in the tumor. Moreover, it was shown that it is possible to simultaneously polarize [1-13C]pyruvate, 13C sodium bicarbonate, [1,1-13C]fumaric acid, and [1-13C]urea. Injection of a solution containing these hyperpolarized compounds into TRAMP mice resulted in spectra where pyruvate, lactate, bicarbonate, carbon dioxide, fumarate, and urea could all be resolved. Thus, through advanced data processing and modeling, simultaneous assays of pyruvate-to-lactate flux, pH, necrosis, and perfusion could be performed, all of which are relevant to prostate cancer detection, staging, and therapeutic response.
Clinical Hyperpolarized Metabolic Imaging in Prostate Cancer:
The first clinical study using HP-MR was reported by the University of California San Francisco (UCSF) in 2013 [294]. This imaging study evaluated the safety and feasibility of hyperpolarized [1-13C]pyruvate as an agent for noninvasively characterizing alterations in tumor metabolism for patients with prostate cancer. The study population comprised of patients with biopsy-proven prostate cancer, with 31 subjects injected with hyperpolarized [1-13C]pyruvate. No dose-limiting toxicities were observed, and the highest dose (0.43 ml/kg of 230 mM agent) gave the best signal-to-noise ratio for hyperpolarized [1-13C]pyruvate. The results were promising in not only confirming the safety of the agent but also showing elevated [1-13C]lactate/[1-13C]pyruvate in regions of biopsy-proven cancer. The follow-up human study on prostate cancer by the same group was reported recently [295]. Hyperpolarized [1-13C]-pyruvate MRI was employed to detect an early metabolic response to androgen deprivation therapy (ADT) in prostate cancer [295]. After six weeks of ADT, the patient’s tumor showed a significant reduction of lactate production following the injection of hyperpolarized pyruvate. Although, there was negligible change in size of tumor on T2-weighted MRI and only a modest change on ADC (apparent diffusion coefficient) imaging, this study demonstrated the ability of HP 13C MRI to detect early metabolic responses. Translation of this technology into humans encouraged additional sites to initiate clinical trials in prostate cancer. To date, seven sites have performed clinical hyperpolarization trials using the SPINlab polarizer, and more than 20 such polarizers have been installed around the world.
Challenges of Clinical Translation of Hyperpolarized Metabolic Imaging:
Dynamic HP-MRI data acquisition.
The high signal-to-noise ratio provided by this imaging technique makes high-resolution acquisitions feasible. However, rapid metabolism and short longitudinal relaxation times (T1) of the HP imaging agents can limit the matrix size and thus the spatial resolution and coverage possible with conventional MRI. The MR acquisition techniques used for initial animal studies and the first human trial were limited in spatial coverage (typically < 8 cm) and dynamic temporal information. New acquisition and analysis techniques are needed for volumetric, dynamic HP MR data with improved spatial coverage and temporal resolution.
Post-image data processing and quantification:
Dynamic MRS data can be resolved in three spatial dimensions, the spectral dimension, and time with adequate coverage and speed to enable reliable quantification of metabolic parameters. Spatial data reduction strategies, including parallel and constrained imaging methods, are crucial for reducing the number of excitations that are necessary to reconstruct dynamically changing data. It will be important moving forward to develop acquisition strategies that minimize uncertainty while maximizing the efficiency of spatial, temporal, and spectral encoding.
Reproducibility and kinetic modeling:
Clinical studies for HP imaging, like FDG-PET studies, should include tests of repeatability and precision to measure the qualitative and quantitative variability of HP imaging results using test/retest methods. There are a few kinetic modeling techniques (both unidirectional and bi-directional) that provide insights about underlying biology. But more fine-tuning is needed with these kinetic modeling approaches before the data can be presented to radiologists in a meaningful, reliable manner.
Future directions:
Clinical oncology practice relies increasingly on anatomic imaging at different stages of patient care. HP-MR has the potential to provide a new dimension and understanding of the underlying tumor biology, thus allowing a more personalized, patient-centric approach. Despite its proven feasibility in humans and its significant potential in clinical oncology, HP metabolic imaging will still have to prove itself against established and emerging clinical techniques such as PET and demonstrate its added value in clinical practice.
PET Imaging in Prostate Cancer:
PET imaging with the glucose analogue 2-fluoro-deoxy-glucose (FDG) verified Warburg’s hypothesis of altered glucose metabolism in cancer cells. As described above, it has been observed over the years that [18F]FDG-PET is not particularly effective in imaging patients with prostate cancer [309]. Alternatively, other PET-based molecular agents including 11C-choline, 11acetate, 18F-fluciclovine, [18F]-PSMA have been successfully employed for prostate cancer imaging (Table 2). Prostate cancer patients exhibit elevated total choline, primarily due to an increase in phosphocholine and glycerophosphocholine. As a PET tracer, 11C-choline is readily incorporated into cells through phosphorylcholine synthesis and integration largely in membrane phospholipids [310,311]. 11C-acetate is primarily viewed as an indirect biomarker of fatty acid synthesis, which is also upregulated in prostate cancer. Imaging of prostate cancer with 11C-acetate thus provides information about biosynthesis [312]. [18F]-fluciclovine and [18F]-prostate specific membrane antigen (PSMA) are also currently being used in PET imaging [313,314]. Fluciclovine is a synthetic amino acid (an analog of L-leucine) that is preferentially taken up by prostate cancer cells and can predict disease relapse following ADT. PSMA is a transmembrane protein that is overexpressed in many prostate cancers. PSMA-PET appears to be a promising new agent, especially for the detection of metastatic prostate cancer. Table 2 highlights some of PET based molecular imaging agents currently employed for diagnostic imaging of prostate cancer.
Table 2:
PET-imaging Agents Currently Employed in the Diagnostic Imaging of Prostate Cancer.
| PET-imaging agents for prostate cancer | Reference (PMID) |
|---|---|
| 18F-FDG | 12209157 |
| 11C-choline | 9627331 11007527 |
| 11C-acetate | 15235071 |
| 18F-fluciclovine | 28267449 |
| 18F-PSMA | 27789722 |
Biofluid and Tissue Metabolite Biomarkers
LDH Test:
Though LDH is an intracellular enzyme involved in metabolism, it can also be released into the bloodstream when tissues are injured or during diseases like cancer. Therefore, serum LDH, often detected with an enzyme-linked immunosorbent assay (ELISA), is an established prognostic indicator for progression and overall survival probability in many malignancies including prostate cancer [315]. Although LDH level cannot monitor the early stages of prostate cancer [22], several clinical studies demonstrate elevated LDH levels strongly associate with poor outcome in mCRPC patients. Halabi et al. collected 1101 mCRPC patients’ data from 1991 to 2001 and established a prognostic model that consisted of LDH, PSA, alkaline phosphatase, Gleason score, Eastern Cooperative Oncology Group performance status, hemoglobin, and the presence of visceral disease to predict patient survival. They found this model can be very closely used to predict patient survival and stratify mCRPC patients [316]. In addition, Katsuya et al. investigated 165 patients from 1998 to 2003 and noticed abnormally high serum LDH levels in patients with bone metastasis. Notably, a combination of biomarkers that includes LDH is now being tested as a surrogate for survival in clinical trials. A successful example is a phase III trial of abiraterone acetate plus prednisone versus prednisone alone in patients with metastatic CRPC previously treated with docetaxel. Circulating tumor cell counts combined with LDH levels were found to satisfy the statistical surrogacy requirements [317]. However, in a clinical study for radium-223 safety and efficacy with mCRPC patients, total alkaline phosphatase and LDH were indicated to be correlated with overall survival but did not meet the surrogacy requirements [318]. Hence, while undoubtedly useful as a clinical biomarker, LDH’s value as a bona fide surrogate marker is still unresolved.
Nuclear Magnetic Resonance (NMR) Spectroscopy-based Metabolomics in Prostate Cancer:
NMR spectroscopy is a routinely employed analytical technique in metabolomics. NMR has been used extensively in biomarker discovery to detect, grade, and intervene in the therapy of prostate cancer. Researchers generally use blood serum and tissue biopsies for NMR-based metabolomics to understand the prostate cancer metabolism. In NMR-based metabolomics studies detection of proton (1H) nuclei is abundantly used with the next detected nuclei in descending order being 13C, 31P, and 23Na. Studies involve either extracting metabolites from liquid biopsies such as blood serum, fluids and urine or intact tissues in rotors for high resolution-magic angle spinning (HR-MAS) NMR spectroscopy. The metabolite extraction, data acquisition and analysis are explained in detail in many NMR-based metabolomics reviews which we suggest for further reading [319–322].
In vivo magnetic resonance spectroscopy (MRS) and ex vivo HR-MAS have shown altered total choline, creatine, polyamines, myo-inositol, and citrate as a biomarkers to detect prostate cancer [323,167,324]. In addition, lactate and alanine are shown to increase in concentration in prostate cancer tumors compared to normal tissues [325]. Conversely, citrate and polyamines are reported to decrease in concentration within primary prostate cancer tumors compared to healthy prostate. To that end, it has also been shown that citrate and spermine can be used to distinguish indolent from malignant prostate cancer [167–169]. This is consistent with the proposal that spermine could be an endogenous inhibitor of primary prostate cancer. In another NMR-based metabolomics study, analysis from 102 serum samples consisting of 40 low-grade prostate cancers, 30 high-grade prostate cancers and 32 healthy control samples demonstrated that the combination of biomarkers sarcosine, pyruvate, alanine, and glycine was able to detect and differentiate low-grade prostate cancer, high-grade prostate cancer and healthy controls [326]. Using HR-MAS spectroscopy, ethanolamine was also reported to be decreased in prostate cancer compared to benign tissues [327]. An in vitro 31P NMR study has shown that phosphocholine, glycerophosphocholine, phosphoethanolamine and glycerophosphoethanolamine were significantly altered in prostate cancer tissues compared to benign prostatic hyperplasia [328].
Mass Spectrometry-based Metabolomics in Prostate Cancer: Mass spectrometry is an effective tool to analyze the molecular composition of clinical prostate cancer biofluids and biospecimens [329–331]. Liquid chromatography/mass spectrometry (LC/MS) is frequently used for targeted metabolomics studies because of high sample throughput, assay sensitivity and ability to measure more molecules in a complex biologic matrix. A commonly used approach for LC/MS analysis is multiple reaction monitoring (MRM) using a triple quadrupole mass spectrometer [332,333]. An LC/MS MRM platform can unambiguously identify and quantify prostate cancer metabolites in metabolic pathways such as glycolysis, respiration, the TCA cycle, steroid and lipid metabolism, amino acid and nucleotide metabolism [334,159]. To determine individual metabolites, there are various open-source and commercial software packages, such as Mass Profiler (Agilent), available for raw LC/MS data analysis. Alternatively, an internal reference library including m/z ratios, retention time and parent/product spectra is used from authentic chemical standards and isotopically labeled standards to characterize specific metabolites. Sample normalization is achieved by adding internal standards and sample variation due to batch effects can be limited using pooled quality control samples [335]. As described above, there are a number of unique metabolic alterations in prostate cancer that can be readily detected using MS-based approaches. Thus, small molecule biomarkers determined by LC/MS show promise in differentiating aggressive prostate cancer phenotypes and may help guide future treatment strategies.
Drug Development
Prostate tumors are regarded as metabolically distinct among solid tumors owing to their enhanced reliance on mitochondrial oxidative phosphorylation [9] and distinct lipogenic character [336]. While current approaches for the management of metastatic disease are primarily focused on the inhibition of AR, new insights into prostate tumor metabolism are identifying metabolic dependencies that may be leveraged for therapeutic benefit. Moreover, AR signaling itself has dramatic effects on cellular metabolism [153], suggesting strategies to inhibit AR-driven metabolic processes may persist as durable targets in AR-positive, CRPC. While clinical translation of metabolic inhibitors is an active area of interest, clinical trials are ongoing and no drugs specifically targeting metabolic endpoints have received FDA approval for the treatment of prostate cancer at this time. This section will briefly review emerging strategies to pharmacologically perturb prostate tumor metabolism with a focus on oxidative phosphorylation, lipogenesis, and glutaminolysis (Table 3).
Table 3:
Emerging Treatments for Prostate Cancer that Target Metabolism.
| Compound | Target | Preclinical Model | Effect | Reference (PMID) |
|---|---|---|---|---|
| Oxidative Phosphorylation | ||||
| Metformin | ETC Complex I, Pleiotropic | Multiple preclinical studies, retrospective analyses, and multiple ongoing clinical trials | Inhibits tumor proliferation in multiple tumor types, potential survival benefit in large retrospective prostate tumor cohort, ongoing clinical trials. | 29940252 30150001 28444639 29075616 27746051 |
| IACS-010759 | ETC Complex I | Brain cancer and acute myeloid leukemia | Inhibits proliferation, depletes macromolecule pools, and induces apoptosis. In vitro and in vivo data, ongoing clinical trial. | 29892070 |
| MSDC-0160 | Mitochondrial Pyruvate Carrier | Hormoneresponsive and castrate-resistant AR-positive prostate cancer | Decreases mitochondrial oxygen consumption, depletes TCA intermediates, inhibits lipogenesis, activates integrated stress response, suppresses cell proliferation and tumor growth. In vitro and in vivo data. | https://doi.org/10.1038/s42255-018-0002-y |
| Lipogenesis | ||||
| IPI-9119 | FASN | Hormone responsive and castrate resistant AR positive prostate cancer | Antagonizes growth through metabolic reprogramming and results in reduced protein expression and transcriptional activity of full-length AR and splice variant AR-V7. In vitro and in vivo data. | 30578319 |
| ND-646 | Acetyl-CoA Carboxylase | Non-small-cell lung cancer | Suppresses fatty acid synthesis, inhibits tumor growth in KRAS p53 and KRAS Lkb1 autochthonous mouse models. | 27643638 |
| MT 63–78 | AMPK | Hormone responsive and castrate resistant prostate cancer. AR positive and AR negative. | Inhibits cell proliferation, induces mitotic arrest and apoptosis. Constitutively activates AMPK and suppresses lipogenesis. In vitro and in vivo data. | 24497570 |
| Fatostatin | SREBP-SCAP | Metastatic and non-metastatic autochthonous models of mouse prostate cancer | Inhibits lipogenesis, blocks tumor growth and metastatic spread. | 29335545 |
| Statins | HMG-CR | Multiple preclinical studies, retrospective analyses, and multiple ongoing clinical trials | Inhibits tumor proliferation in multiple tumor types, potential survival benefit in large retrospective prostate tumor cohort, ongoing clinical trials. | 28806117 20377474 17179483 |
| Glutaminolysis | ||||
| CB839 | Glutaminase | triple-negative breast cancer, lung cancer | Antiproliferative activity and decreased glutamine consumption in triple negative breast cancer models. In vitro and in vivo data. Radiosensitization in lung tumor models. In vitro and in vivo data | 24523301 30557074 |
| V-9302 | SLC1A5/ASCT2 | Multiple cell line models, colon cancer xenografts | Attenuates proliferation, increases apoptosis and oxidative stress. In vitro and in vivo data. | 29334372 |
| Autophagy | ||||
| Chloroquine | Lysosomal function | Multiple preclinical studies and multiple ongoing clinical trials | Inhibits tumor proliferation and survival in multiple tumor types, ongoing clinical trials. | 25134829 22241682 23670050 27322458 |
The biguanide metformin has antidiabetic characteristics that have made it a frontline agent for the management of type 2 diabetes for decades. A variety of epidemiological studies indicated metformin use was associated with decreased cancer incidence in diabetic patients, sparking interest in metformin as an anticancer agent [337,338]. However, subsequent retrospective studies did not find any link between metformin use and decreased cancer risk [339–350] and, in fact, some studies even suggested that increased metformin use correlated with more aggressive prostate cancer [339,344]. As of January 2019, there are 9 actively recruiting clinical trials for the use of metformin in prostate cancer on ClinicalTrials.gov. Importantly, the first prospective clinical trials directly testing the effect of metformin on prostate cancer have recently been completed and showed limited efficacy [351,352]. Although disappointing, these results may be due to our incomplete understanding of how best to choose a patient population that would benefit the most from such an approach. Metformin acts pleiotropically, though the major molecular target is thought to be complex I of the electron transport chain [353]. At this time, it is unclear whether the potential anti-cancer effects of metformin would be due primarily to direct tumor effects or tumor-indirect/host effects. Recent data suggests metformin can exert direct anti-cancer effects in ovarian cancer through the inhibition of cell-intrinsic mitochondrial metabolism [354]. Whether this will also be the case in prostate cancer is currently not known. But, given the dependency of prostate cancer on mitochondrial OXPHOS, a similar mechanism of anti-cancer effects could be possible [355]. Certainly, the ongoing prospective, randomized controlled clinical trials will be necessary to evaluate these associations and define the clinical utility of metformin in prostate cancer.
OXPHOS is an emerging target in cancer therapy [356] and the metabolic properties of prostate cancer suggest agents inhibiting oxidative phosphorylation may be a particularly effective strategy for the treatment of this disease. Though OXPHOS is known to be required for cellular proliferation, the molecular mechanisms underpinning this requirement remained unclear until recently. One major role of respiration in proliferating cells is to provide electron acceptors for aspartate biosynthesis, which in turn enables nucleotide biosynthesis [357,358]. Likewise, respiration is intimately coupled with TCA function, and TCA cycling supplies a variety of anabolic pathways via cataplerosis [359]. OXPHOS may be inhibited by directly targeting components of the electron transport chain, or indirectly by preventing generation of the reducing equivalents required to power the electron transport chain. For example, IACS-010759 is a recently reported complex I inhibitor that dramatically inhibits OXPHOS and, by extension, tumor growth in a variety of pre-clinical models of brain cancer and acute myeloid leukemia [360]. Ongoing efforts to examine the efficacy of IACS-010759 in prostate cancer are underway. Indirect inhibition of OXPHOS and the electron transport chain may also be achieved by interrupting the production of reducing equivalents in the TCA. For example, administration of MSDC-0160, which inhibits the mitochondrial pyruvate carrier, restricts oxidative phosphorylation by depleting metabolic intermediates in the TCA and dramatically decreasing reducing equivalents. The net effect of MSDC-0160 administration is growth restriction in pre-clinical models of hormone-responsive and castrate-resistant prostate adenocarcinoma [361]. Moreover, the mitochondrial pyruvate carrier is directly regulated by AR, suggesting mitochondrial pyruvate import may represent a required metabolic process for AR-mediated proliferation in the hormone-responsive and castrate-resistant settings. Regardless of the way in which it is achieved, inhibition of mitochondrial oxidative phosphorylation in prostate cancer is expected to hold therapeutic promise and clinical trials to gauge efficacy are warranted.
Another prominent metabolic feature of prostate cancer is the de novo synthesis and accumulation of lipids in prostate tumors. Therefore, inhibition of lipogenesis in prostate cancer has been a therapeutic goal and multiple points along the lipogenic pathway are targets for drug design. For example, direct inhibition of fatty acid synthase in prostate cancer using a novel small molecule, IPI-9119, repressed xenograft growth and elicited endoplasmic reticulum stress, which resulted in the downregulation of AR itself. Similarly, direct inhibition of acetyl-CoA carboxylase, a rate-limiting lipogenic enzyme, using the allosteric inhibitor ND-646 resulted in the repression of tumor growth in preclinical models of non-small-cell lung cancer. The lipogenic pathway can also be suppressed by preventing the master transcriptional regulator of lipogenesis, SREBP, from interacting with its activating protein, SCAP, using Fatostatin. Fatostatin blocked both prostate tumor growth and metastasis in Pten-deficient autochthonous mouse models of prostate cancer [74]. Lipogenesis may also be repressed by constitutively activating AMPK (which in turn applies an inhibitory phosphorylation to acetyl-CoA carboxylase) or by depleting the cellular citrate pool (which serves as the primary lipogenic substrate). AMPK activation has been achieved with the direct AMPK activator MT 63–78 while citrate depletion has been demonstrated using MSDC-0160. In both cases, cellular lipid pools were depleted and prostate tumor growth suppressed. Inhibition of lipogenesis may be achieved at multiple points along the pathway and demonstrates significant anti-tumor efficacy in the preclinical setting with the expectation of clinical trials on the horizon.
In addition to OXPHOS and lipogenesis, some models of prostate cancer demonstrate glutamine reliance and this property may be exploited for therapeutic benefit. Though previous work demonstrated therapeutic efficacy of inhibiting glutamine uptake in prostate cancer models [362,141], future studies using current generation glutaminase inhibitors (e.g. CB-839) or glutamine uptake inhibitors (e.g. V-9302) are warranted to gauge the translational potential of glutamine restriction in prostate cancer. Curiously, some AR-positive cell line models do not require glutamine for growth in culture while AR-negative cells such as PC-3 and DU-145 are heavily reliant on this amino acid. These data suggest fundamental metabolic differences may exist between AR-positive and AR-negative prostate cancer. However, glutamine can be used to maintain the TCA cycle during impaired mitochondrial pyruvate transport [363]. In accordance, recent work demonstrated inhibition of the mitochondrial pyruvate carrier dramatically sensitized AR-positive prostate tumor models to glutamine restriction [361]. Though glutamine reliance in prostate tumor models is heterogeneous in culture, additional work in this area is expected to clarify the requirement for glutamine in prostate tumors in vivo.
Greater understanding of the unique metabolic properties of prostate cancer continues to uncover new opportunities for therapeutic intervention. With its low cost and excellent safety profile, metformin is currently in several clinical trials as an adjuvant to current therapies. In addition, multiple drugs that inhibit OXPHOS have been described and demonstrate impressive efficacy in the preclinical setting. IACS-010759 has begun early phase clinical trials for the treatment of leukemia and solid tumors including prostate cancer. Lipogenesis remains a promising therapeutic target and multiple new agents targeting various points along this pathway demonstrate encouraging preclinical results. Last, glutamine reliance in prostate cancer is heterogeneous but current observations indicate it may be most prominent in aggressive models of the disease, suggesting agents that restrict glutamine metabolism may be useful for the management of treatment emergent, aggressive variant prostate cancer. Prostate tumor metabolism is complex, dynamic, and not yet fully understood. Nevertheless, multiple new drugs targeting critical pathways including OXPHOS, lipogenesis, and glutaminolysis have been described and are nearing or entering clinical trials.
Conclusions and Future Directions
The field of cancer metabolism has seen a resurgence in recent years. This is in part due to realization that many of the alterations observed in oncogenic and tumor suppressive pathways impact tumorigenesis through shifting the cell’s metabolism. These changes allow cancers to use a greater array of nutrients to feed anabolic tumor processes and withstand the harsh tumor microenvironment. Importantly, the unique metabolism of cancers relative to benign tissue offers new opportunities for therapeutic intervention. The most direct benefit to patient care will likely come from the establishment of new clinical biomarkers and development of novel treatment modalities. Tumors that become overly dependent on select metabolic pathways should be more susceptible to disruption of these pathways. Further, the differential use of nutrients and production of new metabolites should also yield new biomarkers that can distinguish patient populations based on their underlying biology. At a minimum, these biomarkers would provide correlative data points that are prognostic. However, biomarkers that accurately provide a readout of known pathways could also be used as pharmacodynamic (PD) markers of new treatments that target these same pathways. For example, monitoring hypoxia using 18F-FAZA-PET imaging may hold promise as a non-invasive approach to assess the response to novel OXPHOS inhibitors that are currently under clinical investigation [360]. Importantly, if biomarkers can be developed and validated to detect the causal metabolic changes that drive the disease, this would create a powerful new precision medicine approach that could guide the selection of patients that would benefit the most from new metabolic-targeted therapies.
Several major challenges lie ahead for the field of prostate cancer metabolism. First, linking correlative changes in metabolism to causal driver events remains a major obstacle. Metabolite levels have been assessed in large patient cohorts and uncovered correlative changes that track with prostate cancer [364–367]. However, finding causal events within these studies has been elusive. It should be noted that many of these larger clinical studies rely heavily on the measurement of metabolites from serum which will be heavily influenced by other systemic differences between patients. Alternatively, the direct analysis of metabolites from patient tumor samples is more technically challenging and thus rarer.
A second major challenge is that methods currently do not exist that can separate out cell type-specific metabolism from a heterogeneous tumor cell population. This inability to account for the metabolic crosstalk between different cell types within a tumor (ex. a metabolite secreted from a tumor-associated macrophage is taken up and used by the cancer cell) limits our understanding of the important relationship a cancer cell can have with its metabolic milieu. To overcome this, researchers could focus on transcriptomics to assess alterations in the expression of metabolic genes. This would allow the application of laser capture dissection and single cell sequencing to overcome issues regarding heterogeneity. A major drawback of this approach is that it would miss metabolic changes that occurred as the result of posttranscriptional alterations. Clearly, the development of new approaches that could define the different metabolic phenotypes in a heterogeneous sample would be a major breakthrough.
A third challenge in the field is the difficultly in delineating the etiology of an observed change in metabolism. One of the first questions that often arises following the determination of a significant change in metabolite levels is whether the change occurred from alterations in the production of the metabolite or whether this was the result of changes in the breakdown/further metabolism of the metabolite. To date, much of the metabolomics data reported for prostate cancer is limited to steady state metabolomics. This is because this type of metabolomics can be done on flash frozen tissues without the prior need for additional labelling. While amenable to the study of archived samples, a limitation of this approach is that it only provides a snapshot in time of the metabolite levels. To overcome this limitation, the use of labeled isotopes, either radioactive or stable (the latter becoming increasingly common in metabolomics), can provide additional information on the flux of a particular metabolite, and more specifically the labeled atoms on the metabolite, through a specific metabolic pathway. As a result, techniques such as stable isotope tracing measured using NMR or MS can provide additional context to any changes observed using steady state metabolomics. Therefore, these two approaches can complement one another to generate a deeper understanding of cancer metabolism.
Related to the previous point, a fourth challenge is that isotope tracing studies require, by definition, a prior labeling step. Although this is relatively straightforward for cell culture experiments, it is significantly more difficult to do in vivo. This is due to the constant exchange of nutrients and waste products throughout the body. Despite the technical challenges that in vivo isotope tracing pose, the potential differences in tumor metabolism in vivo compared to in cell culture [368,369] indicate these types of analyses will still be critical to complete our understanding of prostate cancer metabolism in the important context of the tumor microenvironment. To overcome these issues, new methods have been developed to perform stable isotope tracing in preclinical models of cancer [370]. These methods use constant infusions of a labeled nutrient of interest (ex. 13C-labeled glucose) often infused over time until steady state concentrations are achieved in both the blood and tumor. Then tumors/tissues are rapidly harvested/frozen for subsequent NMR or MS analysis. Importantly, because these tracers are not radioactive, they are amenable for use in patients where they have begun to yield important insights and importantly, debunk previously accepted dogma [371,369,372].
Finally, while the general metabolic changes that occur during the initiation of prostate cancer have been well described, our understanding of the shifts that occur in the later, lethal stages of the disease is limited. Importantly, new subtypes of prostate cancer have emerged (ex. neuroendocrine-like prostate cancer) as the unwanted by-product/mechanism of resistance of new and improved AR-targeted drugs such as enzalutamide [373]. Our understanding of the underlying biology of these emerging subtypes, including their metabolic dependencies, is still in its infancy (Figure 2). Given that these cancers are amongst the deadliest forms of the disease and that they are becoming more common, large efforts are ongoing to define their molecular drivers. The significant genomic, transcriptomic and even morphological differences between many of these aggressive cancers and their potential adenocarcinoma origins suggest that they may have profoundly unique metabolic requirements. Thus, these new forms of prostate cancer may be excellent targets for the development of novel metabolic-based biomarkers and/or therapies to identify and treat this advanced stage of the disease.
Figure 2. Continued evolution of prostate cancer metabolism.
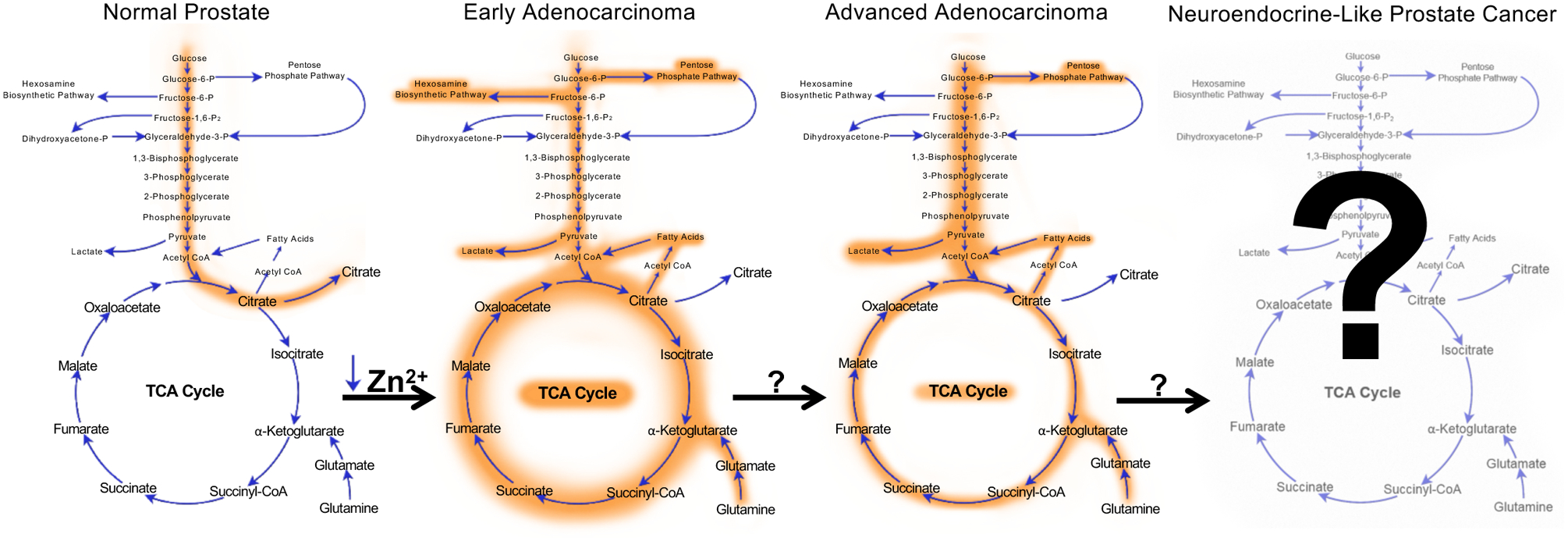
The emergence of a new subtype of advanced prostate cancer termed neuroendocrine-like prostate cancer (NEPC; also commonly referred to as small cell-like prostate cancer (SCPC) and/or aggressive variant prostate cancer (AVPC) depending on its features) raises new questions regarding the metabolic phenotype of this form of the disease. Is the metabolism of NEPCs similar to that of advanced adenocarcinomas? Are there unique features that could be exploited for detection or treatment purposes?
Acknowledgements
We apologize to all the authors whose work we could not cite due to space limitations. We also thank Kelly Kage (UT MD Anderson Cancer Center) for assistance with the figures. This work was supported by grants from the National Institutes of Health (R01CA184208 to D.E.F.; P50CA094056, U54CA151668 and R21CA185536 to P.B.), American Cancer Society (RSG-16-084-01-TBE to D.E.F.), an Institutional Research Grant (to P.B.), startup grants from the University of Texas MD Anderson Cancer Center (to P.B. and D.E.F.), a grant from the Gulf Coast Consortium (to P.B.) and generous philanthropic contributions to The University of Texas MD Anderson Moon Shots Program (to D.E.F.) and Koch Foundation Genitourinary Medical Oncology Funds (to P.B.). This work was also supported by an Antje Wuelfrath Gee and Harry Gee, Jr. Family Legacy Scholarship (to C.L.), an American Legion Auxiliary Fellowship (to D.A.), a CPRIT Research Training Grant Award (RP170067 to T.C.S.) and a GCC/Keck Center CCBTP postdoctoral fellowship (CPRIT RP170593 to S.P.). M.T. also acknowledges support from the Neubauer Family Foundation.
References
- 1.Warburg O (1956) On the origin of cancer cells. Science 123 (3191):309–314. doi: 10.1126/science.123.3191.309 [DOI] [PubMed] [Google Scholar]
- 2.Warburg O, Wind F, Negelein E (1927) The Metabolism of Tumors in the Body. The Journal of general physiology 8 (6):519–530. doi: 10.1085/jgp.8.6.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144 (5):646–674. doi: 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 4.Costello LC, Franklin RB (2016) A comprehensive review of the role of zinc in normal prostate function and metabolism; and its implications in prostate cancer. Arch Biochem Biophys 611:100–112. doi: 10.1016/j.abb.2016.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costello LC, Franklin RB, Feng P (2005) Mitochondrial function, zinc, and intermediary metabolism relationships in normal prostate and prostate cancer. Mitochondrion 5 (3):143–153. doi: 10.1016/j.mito.2005.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costello LC, Franklin RB (2011) Zinc is decreased in prostate cancer: an established relationship of prostate cancer. J Biol Inorg Chem 16 (1):3–8. doi: 10.1007/s00775-010-0736-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costello LC, Franklin RB (1981) Aconitase activity, citrate oxidation, and zinc inhibition in rat ventral prostate. Enzyme 26 (6):281–287. doi: 10.1159/000459195 [DOI] [PubMed] [Google Scholar]
- 8.Costello LC, Franklin RB (1998) Novel role of zinc in the regulation of prostate citrate metabolism and its implications in prostate cancer. Prostate 35 (4):285–296. doi: [DOI] [PubMed] [Google Scholar]
- 9.Costello LC, Franklin RB (2006) The clinical relevance of the metabolism of prostate cancer; zinc and tumor suppression: connecting the dots. Mol Cancer 5:17. doi: 10.1186/1476-4598-5-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costello LC, Liu Y, Franklin RB, Kennedy MC (1997) Zinc inhibition of mitochondrial aconitase and its importance in citrate metabolism of prostate epithelial cells. J Biol Chem 272 (46):28875–28881. doi: 10.1074/jbc.272.46.28875 [DOI] [PubMed] [Google Scholar]
- 11.Desouki MM, Geradts J, Milon B, Franklin RB, Costello LC (2007) hZip2 and hZip3 zinc transporters are down regulated in human prostate adenocarcinomatous glands. Mol Cancer 6:37. doi: 10.1186/1476-4598-6-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franklin RB, Feng P, Milon B, Desouki MM, Singh KK, Kajdacsy-Balla A, Bagasra O, Costello LC (2005) hZIP1 zinc uptake transporter down regulation and zinc depletion in prostate cancer. Mol Cancer 4:32. doi: 10.1186/1476-4598-4-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franz MC, Anderle P, Burzle M, Suzuki Y, Freeman MR, Hediger MA, Kovacs G (2013) Zinc transporters in prostate cancer. Mol Aspects Med 34 (2–3):735–741. doi: 10.1016/j.mam.2012.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eidelman E, Twum-Ampofo J, Ansari J, Siddiqui MM (2017) The Metabolic Phenotype of Prostate Cancer. Front Oncol 7:131. doi: 10.3389/fonc.2017.00131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng P, Liang JY, Li TL, Guan ZX, Zou J, Franklin R, Costello LC (2000) Zinc induces mitochondria apoptogenesis in prostate cells. Mol Urol 4 (1):31–36 [PubMed] [Google Scholar]
- 16.Feng P, Li TL, Guan ZX, Franklin RB, Costello LC (2002) Direct effect of zinc on mitochondrial apoptogenesis in prostate cells. Prostate 52 (4):311–318. doi: 10.1002/pros.10128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franklin RB, Costello LC (2007) Zinc as an anti-tumor agent in prostate cancer and in other cancers. Arch Biochem Biophys 463 (2):211–217. doi: 10.1016/j.abb.2007.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cutruzzola F, Giardina G, Marani M, Macone A, Paiardini A, Rinaldo S, Paone A (2017) Glucose Metabolism in the Progression of Prostate Cancer. Front Physiol 8:97. doi: 10.3389/fphys.2017.00097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rishi I, Baidouri H, Abbasi JA, Bullard-Dillard R, Kajdacsy-Balla A, Pestaner JP, Skacel M, Tubbs R, Bagasra O (2003) Prostate cancer in African American men is associated with downregulation of zinc transporters. Appl Immunohistochem Mol Morphol 11 (3):253–260 [DOI] [PubMed] [Google Scholar]
- 20.Makhov PB, Golovine KV, Kutikov A, Canter DJ, Rybko VA, Roshchin DA, Matveev VB, Uzzo RG, Kolenko VM (2011) Reversal of epigenetic silencing of AP-2alpha results in increased zinc uptake in DU-145 and LNCaP prostate cancer cells. Carcinogenesis 32 (12):1773–1781. doi: 10.1093/carcin/bgr212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Latonen L, Afyounian E, Jylha A, Nattinen J, Aapola U, Annala M, Kivinummi KK, Tammela TTL, Beuerman RW, Uusitalo H, Nykter M, Visakorpi T (2018) Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression. Nat Commun 9 (1):1176. doi: 10.1038/s41467-018-03573-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naruse K, Yamada Y, Aoki S, Taki T, Nakamura K, Tobiume M, Zennami K, Katsuda R, Sai S, Nishio Y, Inoue Y, Noguchi H, Hondai N (2007) Lactate dehydrogenase is a prognostic indicator for prostate cancer patients with bone metastasis. Hinyokika Kiyo 53 (5):287–292 [PubMed] [Google Scholar]
- 23.Elia I, Schmieder R, Christen S, Fendt SM (2016) Organ-Specific Cancer Metabolism and Its Potential for Therapy. Handb Exp Pharmacol 233:321–353. doi: 10.1007/164_2015_10 [DOI] [PubMed] [Google Scholar]
- 24.Wang Jun L J, Li Xiaojuan, Peng Shubin, Li Jun, Yan Weixin, Cui Yubin, Xiao Hengjun, Wen Xingqiao (2017) Increased expression of glycolytic enzymes in prostate cancer tissues and association with Gleason score. Int J Clin Exp Pathol 10 (11):11080–11089 [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao H, Wang J, Yan W, Cui Y, Chen Z, Gao X, Wen X, Chen J (2018) GLUT1 regulates cell glycolysis and proliferation in prostate cancer. Prostate 78 (2):86–94. doi: 10.1002/pros.23448 [DOI] [PubMed] [Google Scholar]
- 26.Jans J, van Dijk JH, van Schelven S, van der Groep P, Willems SH, Jonges TN, van Diest PJ, Bosch JL (2010) Expression and localization of hypoxia proteins in prostate cancer: prognostic implications after radical prostatectomy. Urology 75 (4):786–792. doi: 10.1016/j.urology.2009.08.024 [DOI] [PubMed] [Google Scholar]
- 27.Vaz CV, Alves MG, Marques R, Moreira PI, Oliveira PF, Maia CJ, Socorro S (2012) Androgen-responsive and nonresponsive prostate cancer cells present a distinct glycolytic metabolism profile. Int J Biochem Cell Biol 44 (11):2077–2084. doi: 10.1016/j.biocel.2012.08.013 [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez-Menendez P, Hevia D, Alonso-Arias R, Alvarez-Artime A, Rodriguez-Garcia A, Kinet S, Gonzalez-Pola I, Taylor N, Mayo JC, Sainz RM (2018) GLUT1 protects prostate cancer cells from glucose deprivation-induced oxidative stress. Redox Biol 17:112–127. doi: 10.1016/j.redox.2018.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.White MA, Tsouko E, Lin C, Rajapakshe K, Spencer JM, Wilkenfeld SR, Vakili SS, Pulliam TL, Awad D, Nikolos F, Katreddy RR, Kaipparettu BA, Sreekumar A, Zhang X, Cheung E, Coarfa C, Frigo DE (2018) GLUT12 promotes prostate cancer cell growth and is regulated by androgens and CaMKK2 signaling. Endocr Relat Cancer 25 (4):453–469. doi: 10.1530/ERC-17-0051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez-Menendez P, Hevia D, Mayo JC, Sainz RM (2018) The dark side of glucose transporters in prostate cancer: Are they a new feature to characterize carcinomas? Int J Cancer 142 (12):2414–2424. doi: 10.1002/ijc.31165 [DOI] [PubMed] [Google Scholar]
- 31.Pertega-Gomes N, Felisbino S, Massie CE, Vizcaino JR, Coelho R, Sandi C, Simoes-Sousa S, Jurmeister S, Ramos-Montoya A, Asim M, Tran M, Oliveira E, Lobo da Cunha A, Maximo V, Baltazar F, Neal DE, Fryer LG (2015) A glycolytic phenotype is associated with prostate cancer progression and aggressiveness: a role for monocarboxylate transporters as metabolic targets for therapy. J Pathol 236 (4):517–530. doi: 10.1002/path.4547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng Y, Lu J (2015) Targeting hexokinase 2 in castration-resistant prostate cancer. Mol Cell Oncol 2 (3):e974465. doi: 10.4161/23723556.2014.974465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, Guo X, Chang LJ, Zhang Y, You MJ, Koochekpour S, Saleem M, Huang H, Lu J, Deng Y (2014) Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep 8 (5):1461–1474. doi: 10.1016/j.celrep.2014.07.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou P, Chen WG, Li XW (2015) MicroRNA-143 acts as a tumor suppressor by targeting hexokinase 2 in human prostate cancer. Am J Cancer Res 5 (6):2056–2063 [PMC free article] [PubMed] [Google Scholar]
- 35.Martin PL, Yin JJ, Seng V, Casey O, Corey E, Morrissey C, Simpson RM, Kelly K (2017) Androgen deprivation leads to increased carbohydrate metabolism and hexokinase 2-mediated survival in Pten/Tp53-deficient prostate cancer. Oncogene 36 (4):525–533. doi: 10.1038/onc.2016.223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tao T, Chen M, Jiang R, Guan H, Huang Y, Su H, Hu Q, Han X, Xiao J (2017) Involvement of EZH2 in aerobic glycolysis of prostate cancer through miR-181b/HK2 axis. Oncol Rep 37 (3):1430–1436. doi: 10.3892/or.2017.5430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moon JS, Jin WJ, Kwak JH, Kim HJ, Yun MJ, Kim JW, Park SW, Kim KS (2011) Androgen stimulates glycolysis for de novo lipid synthesis by increasing the activities of hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 in prostate cancer cells. Biochem J 433 (1):225–233. doi: 10.1042/BJ20101104 [DOI] [PubMed] [Google Scholar]
- 38.Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, Chandel N, Laakso M, Muller WJ, Allen EL, Jha AK, Smolen GA, Clasquin MF, Robey B, Hay N (2013) Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 24 (2):213–228. doi: 10.1016/j.ccr.2013.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stacpoole PW (2017) Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J Natl Cancer Inst 109 (11). doi: 10.1093/jnci/djx071 [DOI] [PubMed] [Google Scholar]
- 40.Kolobova E, Tuganova A, Boulatnikov I, Popov KM (2001) Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J 358 (1):69–77. doi: 10.1042/bj3580069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sutendra G, Michelakis ED (2013) Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front Oncol 3:38. doi: 10.3389/fonc.2013.00038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhong Y, Li X, Ji Y, Li X, Li Y, Yu D, Yuan Y, Liu J, Li H, Zhang M, Ji Z, Fan D, Wen J, Goscinski MA, Yuan L, Hao B, Nesland JM, Suo Z (2017) Pyruvate dehydrogenase expression is negatively associated with cell stemness and worse clinical outcome in prostate cancers. Oncotarget 8 (8):13344–13356. doi: 10.18632/oncotarget.14527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen J, Guccini I, Di Mitri D, Brina D, Revandkar A, Sarti M, Pasquini E, Alajati A, Pinton S, Losa M, Civenni G, Catapano CV, Sgrignani J, Cavalli A, D’Antuono R, Asara JM, Morandi A, Chiarugi P, Crotti S, Agostini M, Montopoli M, Masgras I, Rasola A, Garcia-Escudero R, Delaleu N, Rinaldi A, Bertoni F, Bono J, Carracedo A, Alimonti A (2018) Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat Genet 50 (2):219–228. doi: 10.1038/s41588-017-0026-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E, Michelakis ED (2014) A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 158 (1):84–97. doi: 10.1016/j.cell.2014.04.046 [DOI] [PubMed] [Google Scholar]
- 45.Hsu MC, Hung WC (2018) Pyruvate kinase M2 fuels multiple aspects of cancer cells: from cellular metabolism, transcriptional regulation to extracellular signaling. Mol Cancer 17 (1):35. doi: 10.1186/s12943-018-0791-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siddiqui FA, Prakasam G, Chattopadhyay S, Rehman AU, Padder RA, Ansari MA, Irshad R, Mangalhara K, Bamezai RNK, Husain M, Ali SM, Iqbal MA (2018) Curcumin decreases Warburg effect in cancer cells by down-regulating pyruvate kinase M2 via mTOR-HIF1alpha inhibition. Sci Rep 8 (1):8323. doi: 10.1038/s41598-018-25524-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wong N, Yan J, Ojo D, De Melo J, Cutz JC, Tang D (2014) Changes in PKM2 associate with prostate cancer progression. Cancer Invest 32 (7):330–338. doi: 10.3109/07357907.2014.919306 [DOI] [PubMed] [Google Scholar]
- 48.Dong G, Mao Q, Xia W, Xu Y, Wang J, Xu L, Jiang F (2016) PKM2 and cancer: The function of PKM2 beyond glycolysis. Oncol Lett 11 (3):1980–1986. doi: 10.3892/ol.2016.4168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang HJ, Pochampalli M, Wang LY, Zou JX, Li PS, Hsu SC, Wang BJ, Huang SH, Yang P, Yang JC, Chu CY, Hsieh CL, Sung SY, Li CF, Tepper CG, Ann DK, Gao AC, Evans CP, Izumiya Y, Chuu CP, Wang WC, Chen HW, Kung HJ (2018) KDM8/JMJD5 as a dual coactivator of AR and PKM2 integrates AR/EZH2 network and tumor metabolism in CRPC. Oncogene. doi: 10.1038/s41388-018-0414-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yasumizu Y, Hongo H, Kosaka T, Mikami S, Nishimoto K, Kikuchi E, Oya M (2018) PKM2 under hypoxic environment causes resistance to mTOR inhibitor in human castration resistant prostate cancer. Oncotarget 9 (45):27698–27707. doi: 10.18632/oncotarget.25498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hasan D, Gamen E, Abu Tarboush N, Ismail Y, Pak O, Azab B (2018) PKM2 and HIF-1alpha regulation in prostate cancer cell lines. PLoS One 13 (9):e0203745. doi: 10.1371/journal.pone.0203745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Giannoni E, Taddei ML, Morandi A, Comito G, Calvani M, Bianchini F, Richichi B, Raugei G, Wong N, Tang D, Chiarugi P (2015) Targeting stromal-induced pyruvate kinase M2 nuclear translocation impairs oxphos and prostate cancer metastatic spread. Oncotarget 6 (27):24061–24074. doi: 10.18632/oncotarget.4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xian ZY, Liu JM, Chen QK, Chen HZ, Ye CJ, Xue J, Yang HQ, Li JL, Liu XF, Kuang SJ (2015) Inhibition of LDHA suppresses tumor progression in prostate cancer. Tumour Biol 36 (10):8093–8100. doi: 10.1007/s13277-015-3540-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koukourakis MI, Giatromanolaki A, Panteliadou M, Pouliliou SE, Chondrou PS, Mavropoulou S, Sivridis E (2014) Lactate dehydrogenase 5 isoenzyme overexpression defines resistance of prostate cancer to radiotherapy. Br J Cancer 110 (9):2217–2223. doi: 10.1038/bjc.2014.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi SY, Xue H, Wu R, Fazli L, Lin D, Collins CC, Gleave ME, Gout PW, Wang Y (2016) The MCT4 Gene: A Novel, Potential Target for Therapy of Advanced Prostate Cancer. Clin Cancer Res 22 (11):2721–2733. doi: 10.1158/1078-0432.CCR-15-1624 [DOI] [PubMed] [Google Scholar]
- 56.Doherty JR, Cleveland JL (2013) Targeting lactate metabolism for cancer therapeutics. J Clin Invest 123 (9):3685–3692. doi: 10.1172/JCI69741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu J, Chen G, Liu Z, Liu S, Cai Z, You P, Ke Y, Lai L, Huang Y, Gao H, Zhao L, Pelicano H, Huang P, McKeehan WL, Wu CL, Wang C, Zhong W, Wang F (2018) Aberrant FGFR Tyrosine Kinase Signaling Enhances the Warburg Effect by Reprogramming LDH Isoform Expression and Activity in Prostate Cancer. Cancer Res 78 (16):4459–4470. doi: 10.1158/0008-5472.CAN-17-3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones RS, Morris ME (2016) Monocarboxylate Transporters: Therapeutic Targets and Prognostic Factors in Disease. Clin Pharmacol Ther 100 (5):454–463. doi: 10.1002/cpt.418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wilson MC, Jackson VN, Heddle C, Price NT, Pilegaard H, Juel C, Bonen A, Montgomery I, Hutter OF, Halestrap AP (1998) Lactic acid efflux from white skeletal muscle is catalyzed by the monocarboxylate transporter isoform MCT3. J Biol Chem 273 (26):15920–15926. doi: 10.1074/jbc.273.26.15920 [DOI] [PubMed] [Google Scholar]
- 60.Choi SYC, Ettinger SL, Lin D, Xue H, Ci X, Nabavi N, Bell RH, Mo F, Gout PW, Fleshner NE, Gleave ME, Collins CC, Wang Y (2018) Targeting MCT4 to reduce lactic acid secretion and glycolysis for treatment of neuroendocrine prostate cancer. Cancer Med. doi: 10.1002/cam4.1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pertega-Gomes N, Vizcaino JR, Miranda-Goncalves V, Pinheiro C, Silva J, Pereira H, Monteiro P, Henrique RM, Reis RM, Lopes C, Baltazar F (2011) Monocarboxylate transporter 4 (MCT4) and CD147 overexpression is associated with poor prognosis in prostate cancer. BMC Cancer 11:312. doi: 10.1186/1471-2407-11-312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanita P, Capulli M, Teti A, Galatioto GP, Vicentini C, Chiarugi P, Bologna M, Angelucci A (2014) Tumor-stroma metabolic relationship based on lactate shuttle can sustain prostate cancer progression. BMC Cancer 14:154. doi: 10.1186/1471-2407-14-154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pertega-Gomes N, Vizcaino JR, Felisbino S, Warren AY, Shaw G, Kay J, Whitaker H, Lynch AG, Fryer L, Neal DE, Massie CE (2015) Epigenetic and oncogenic regulation of SLC16A7 (MCT2) results in protein over-expression, impacting on signalling and cellular phenotypes in prostate cancer. Oncotarget 6 (25):21675–21684. doi: 10.18632/oncotarget.4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Valenca I, Pertega-Gomes N, Vizcaino JR, Henrique RM, Lopes C, Baltazar F, Ribeiro D (2015) Localization of MCT2 at peroxisomes is associated with malignant transformation in prostate cancer. J Cell Mol Med 19 (4):723–733. doi: 10.1111/jcmm.12481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim HS, Masko EM, Poulton SL, Kennedy KM, Pizzo SV, Dewhirst MW, Freedland SJ (2012) Carbohydrate restriction and lactate transporter inhibition in a mouse xenograft model of human prostate cancer. BJU Int 110 (7):1062–1069. doi: 10.1111/j.1464-410X.2012.10971.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang P, Du W, Wu M (2014) Regulation of the pentose phosphate pathway in cancer. Protein Cell 5 (8):592–602. doi: 10.1007/s13238-014-0082-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cho ES, Cha YH, Kim HS, Kim NH, Yook JI (2018) The Pentose Phosphate Pathway as a Potential Target for Cancer Therapy. Biomol Ther (Seoul) 26 (1):29–38. doi: 10.4062/biomolther.2017.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.da Costa IA, Hennenlotter J, Stuhler V, Kuhs U, Scharpf M, Todenhofer T, Stenzl A, Bedke J (2018) Transketolase like 1 (TKTL1) expression alterations in prostate cancer tumorigenesis. Urol Oncol 36 (10):472.e421–472.e427. doi: 10.1016/j.urolonc.2018.06.010 [DOI] [PubMed] [Google Scholar]
- 69.Tsouko E, Khan AS, White MA, Han JJ, Shi Y, Merchant FA, Sharpe MA, Xin L, Frigo DE (2014) Regulation of the pentose phosphate pathway by an androgen receptor-mTOR-mediated mechanism and its role in prostate cancer cell growth. Oncogenesis 3:e103. doi: 10.1038/oncsis.2014.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ookhtens M, Kannan R, Lyon I, Baker N (1984) Liver and adipose tissue contributions to newly formed fatty acids in an ascites tumor. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 247 (1):R146–R153. doi: 10.1152/ajpregu.1984.247.1.R146 [DOI] [PubMed] [Google Scholar]
- 71.Han W, Gao S, Barrett D, Ahmed M, Han D, Macoska JA, He HH, Cai C (2018) Reactivation of androgen receptor-regulated lipid biosynthesis drives the progression of castration-resistant prostate cancer. Oncogene 37 (6):710–721. doi: 10.1038/onc.2017.385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Swinnen JV, Heemers H, de Sande TV, Schrijver ED, Brusselmans K, Heyns W, Verhoeven G (2004) Androgens, lipogenesis and prostate cancer. The Journal of Steroid Biochemistry and Molecular Biology 92 (4):273–279. doi: 10.1016/j.jsbmb.2004.10.013 [DOI] [PubMed] [Google Scholar]
- 73.Huang W-C, Li X, Liu J, Lin J, Chung LWK (2012) Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol Cancer Res 10 (1):133–142. doi: 10.1158/1541-7786.MCR-11-0206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen M, Zhang J, Sampieri K, Clohessy JG, Mendez L, Gonzalez-Billalabeitia E, Liu XS, Lee YR, Fung J, Katon JM, Menon AV, Webster KA, Ng C, Palumbieri MD, Diolombi MS, Breitkopf SB, Teruya-Feldstein J, Signoretti S, Bronson RT, Asara JM, Castillo-Martin M, Cordon-Cardo C, Pandolfi PP (2018) An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat Genet 50 (2):206–218. doi: 10.1038/s41588-017-0027-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yokoyama C, Wang X, Briggs MR, Admon A, Wu J, Hua X, Goldstein JL, Brown MS (1993) SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 75 (1):187–197. doi: 10.1016/S0092-8674(05)80095-9 [DOI] [PubMed] [Google Scholar]
- 76.Shao W, Espenshade PJ (2012) Expanding roles for SREBP in metabolism. Cell metabolism 16 (4):414–419. doi: 10.1016/j.cmet.2012.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, Nelson CC (2004) Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res 64 (6):2212–2221 [DOI] [PubMed] [Google Scholar]
- 78.Heemers H, Verrijdt G, Organe S, Claessens F, Heyns W, Verhoeven G, Swinnen JV (2004) Identification of an androgen response element in intron 8 of the sterol regulatory element-binding protein cleavage-activating protein gene allowing direct regulation by the androgen receptor. J Biol Chem 279 (29):30880–30887. doi: 10.1074/jbc.M401615200 [DOI] [PubMed] [Google Scholar]
- 79.Swinnen JV, Ulrix W, Heyns W, Verhoeven G (1997) Coordinate regulation of lipogenic gene expression by androgens: evidence for a cascade mechanism involving sterol regulatory element binding proteins. Proc Natl Acad Sci 94 (24):12975–12980. doi: 10.1073/pnas.94.24.12975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang W-C, Zhau HE, Chung LWK (2010) Androgen Receptor Survival Signaling Is Blocked by Anti-β2-microglobulin Monoclonal Antibody via a MAPK/Lipogenic Pathway in Human Prostate Cancer Cells. Journal of Biological Chemistry 285 (11):7947–7956. doi: 10.1074/jbc.M109.092759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, Van Veldhoven PP, Waltregny D, Daniëls VW, Machiels J, Vanderhoydonc F, Smans K, Waelkens E, Verhoeven G, Swinnen JV (2010) De novo Lipogenesis Protects Cancer Cells from Free Radicals and Chemotherapeutics by Promoting Membrane Lipid Saturation. Cancer Res 70 (20):8117. doi: 10.1158/0008-5472.CAN-09-3871 [DOI] [PubMed] [Google Scholar]
- 82.Simons K, Toomre D (2000) Lipid rafts and signal transduction. Nature Reviews Molecular Cell Biology 1:31. doi: 10.1038/35036052 [DOI] [PubMed] [Google Scholar]
- 83.Li L, Ren CH, Tahir SA, Ren C, Thompson TC (2003) Caveolin-1 maintains activated Akt in prostate cancer cells through scaffolding domain binding site interactions with and inhibition of serine/threonine protein phosphatases PP1 and PP2A. Molecular and cellular biology 23 (24):9389–9404. doi: 10.1128/MCB.23.24.9389-9404.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Young RM, Holowka D, Baird B (2003) A Lipid Raft Environment Enhances Lyn Kinase Activity by Protecting the Active Site Tyrosine from Dephosphorylation. Journal of Biological Chemistry 278 (23):20746–20752. doi: 10.1074/jbc.M211402200 [DOI] [PubMed] [Google Scholar]
- 85.Rice KR, Koch MO, Cheng L, Masterson TA (2012) Dyslipidemia, statins and prostate cancer. Expert Review of Anticancer Therapy 12 (7):981–990. doi: 10.1586/era.12.75 [DOI] [PubMed] [Google Scholar]
- 86.Cinar B, Mukhopadhyay NK, Meng G, Freeman MR (2007) Phosphoinositide 3-Kinase-independent Non-genomic Signals Transit from the Androgen Receptor to Akt1 in Membrane Raft Microdomains. Journal of Biological Chemistry 282 (40):29584–29593. doi: 10.1074/jbc.M703310200 [DOI] [PubMed] [Google Scholar]
- 87.Farese RV, Walther TC (2009) Lipid Droplets Finally Get a Little R-E-S-P-E-C-T. Cell 139 (5):855–860. doi: 10.1016/j.cell.2009.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Petan T, Jarc E, Jusović M (2018) Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 23 (8). doi: 10.3390/molecules23081941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bensaad K, Favaro E, Lewis Caroline A, Peck B, Lord S, Collins Jennifer M, Pinnick Katherine E, Wigfield S, Buffa Francesca M, Li J-L, Zhang Q, Wakelam Michael JO, Karpe F, Schulze A, Harris Adrian L (2014) Fatty Acid Uptake and Lipid Storage Induced by HIF-1α Contribute to Cell Growth and Survival after Hypoxia-Reoxygenation. Cell Reports 9 (1):349–365. doi: 10.1016/j.celrep.2014.08.056 [DOI] [PubMed] [Google Scholar]
- 90.Cabodevilla AG, Sánchez-Caballero L, Nintou E, Boiadjieva VG, Picatoste F, Gubern A, Claro E (2013) Cell Survival during Complete Nutrient Deprivation Depends on Lipid Droplet-fueled β-Oxidation of Fatty Acids. Journal of Biological Chemistry 288 (39):27777–27788. doi: 10.1074/jbc.M113.466656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Koizume S, Miyagi Y (2016) Lipid Droplets: A Key Cellular Organelle Associated with Cancer Cell Survival under Normoxia and Hypoxia. International journal of molecular sciences 17 (9):1430. doi: 10.3390/ijms17091430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Swinnen JV, Verhoeven G (1998) Androgens and the control of lipid metabolism in human prostate cancer cells11Proceedings of the 13th International Symposium of the Journal of Steroid Biochemistry & Molecular Biology “Recent Advances in Steroid Biochemistry & Molecular Biology” Monaco, 25–28 May 1997. The Journal of Steroid Biochemistry and Molecular Biology 65 (1):191–198. doi: 10.1016/S0960-0760(97)00187-8 [DOI] [PubMed] [Google Scholar]
- 93.Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, Cheng L, Masterson TA, Liu X, Ratliff TL, Cheng JX (2014) Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab 19 (3):393–406. doi: 10.1016/j.cmet.2014.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Swinnen JV, Roskams T, Joniau S, Van Poppel H, Oyen R, Baert L, Heyns W, Verhoeven G (2002) Overexpression of fatty acid synthase is an early and common event in the development of prostate cancer. Int J Cancer 98 (1):19–22. doi: 10.1002/ijc.10127 [DOI] [PubMed] [Google Scholar]
- 95.Van de Sande T, Roskams T, Lerut E, Joniau S, Van Poppel H, Verhoeven G, Swinnen JV (2005) High-level expression of fatty acid synthase in human prostate cancer tissues is linked to activation and nuclear localization of Akt/PKB. The Journal of Pathology 206 (2):214–219. doi: 10.1002/path.1760 [DOI] [PubMed] [Google Scholar]
- 96.Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, Inazuka F, Grisanzio C, Palescandolo E, Shin E, Fiore C, Xie W, Kung AL, Febbo PG, Subramanian A, Mucci L, Ma J, Signoretti S, Stampfer M, Hahn WC, Finn S, Loda M (2009) Fatty Acid Synthase: A Metabolic Enzyme and Candidate Oncogene in Prostate Cancer. Journal of the National Cancer Institute 101 (7):519–532. doi: 10.1093/jnci/djp030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hamada S, Horiguchi A, Kuroda K, Ito K, Asano T, Miyai K, Iwaya K (2014) Increased fatty acid synthase expression in prostate biopsy cores predicts higher Gleason score in radical prostatectomy specimen. BMC Clinical Pathology 14 (1):3. doi: 10.1186/1472-6890-14-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zadra G, Ribeiro CF, Chetta P, Ho Y, Cacciatore S, Gao X, Syamala S, Bango C, Photopoulos C, Huang Y, Tyekucheva S, Bastos DC, Tchaicha J, Lawney B, Uo T, D’Anello L, Csibi A, Kalekar R, Larimer B, Ellis L, Butler LM, Morrissey C, McGovern K, Palombella VJ, Kutok JL, Mahmood U, Bosari S, Adams J, Peluso S, Dehm SM, Plymate SR, Loda M (2019. ) Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc Natl Acad Sci 116 (2):631–640. doi: 10.1073/pnas.1808834116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Furuta E, Pai SK, Zhan R, Bandyopadhyay S, Watabe M, Mo Y-Y, Hirota S, Hosobe S, Tsukada T, Miura K, Kamada S, Saito K, Iiizumi M, Liu W, Ericsson J, Watabe K (2008) Fatty Acid Synthase Gene Is Up-regulated by Hypoxia via Activation of Akt and Sterol Regulatory Element Binding Protein-1. Cancer Res 68 (4):1003. doi: 10.1158/0008-5472.CAN-07-2489 [DOI] [PubMed] [Google Scholar]
- 100.Gang X, Yang Y, Zhong J, Jiang K, Pan Y, Karnes RJ, Zhang J, Xu W, Wang G, Huang H (2016) P300 acetyltransferase regulates fatty acid synthase expression, lipid metabolism and prostate cancer growth. Oncotarget 7 (12):15135–15149. doi: 10.18632/oncotarget.7715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim S, Yang X, Li Q, Wu M, Costyn L, Beharry Z, Bartlett MG, Cai H (2017) Myristoylation of Src kinase mediates Src-induced and high-fat diet–accelerated prostate tumor progression in mice. Journal of Biological Chemistry 292 (45):18422–18433. doi: 10.1074/jbc.M117.798827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cai H, Smith DA, Memarzadeh S, Lowell CA, Cooper JA, Witte ON (2011) Differential transformation capacity of Src family kinases during the initiation of prostate cancer. Proc Natl Acad Sci 108 (16):6579–6584. doi: 10.1073/pnas.1103904108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang FL, Casey PJ (1996) Protein Prenylation: Molecular Mechanisms and Functional Consequences. Annual Review of Biochemistry 65 (1):241–269. doi: 10.1146/annurev.bi.65.070196.001325 [DOI] [PubMed] [Google Scholar]
- 104.Wasko BM, Dudakovic A, Hohl RJ (2011) Bisphosphonates Induce Autophagy by Depleting Geranylgeranyl Diphosphate. Journal of Pharmacology and Experimental Therapeutics 337 (2):540. doi: 10.1124/jpet.110.175521 [DOI] [PubMed] [Google Scholar]
- 105.Reilly JE, Neighbors JD, Hohl RJ (2016) Targeting protein geranylgeranylation slows tumor development in a murine model of prostate cancer metastasis. Cancer biology & therapy 18 (11):872–882. doi: 10.1080/15384047.2016.1219817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zacharias NM, McCullough C, Shanmugavelandy S, Lee J, Lee Y, Dutta P, McHenry J, Nguyen L, Norton W, Jones LW, Bhattacharya PK (2017) Metabolic Differences in Glutamine Utilization Lead to Metabolic Vulnerabilities in Prostate Cancer. Scientific Reports 7 (1):16159. doi: 10.1038/s41598-017-16327-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fendt SM, Bell EL, Keibler MA, Olenchock BA, Mayers JR, Wasylenko TM, Vokes NI, Guarente L, Vander Heiden MG, Stephanopoulos G (2013) Reductive glutamine metabolism is a function of the alpha-ketoglutarate to citrate ratio in cells. Nat Commun 4:2236. doi: 10.1038/ncomms3236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ (2011) Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481 (7381):385–388. doi: 10.1038/nature10642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, Stephanopoulos G (2011) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481 (7381):380–384. doi: 10.1038/nature10602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zaidi N, Swinnen JV, Smans K (2012) ATP-citrate lyase: a key player in cancer metabolism. Cancer Res 72 (15):3709–3714. doi: 10.1158/0008-5472.CAN-11-4112 [DOI] [PubMed] [Google Scholar]
- 111.Shah S, Carriveau WJ, Li J, Campbell SL, Kopinski PK, Lim HW, Daurio N, Trefely S, Won KJ, Wallace DC, Koumenis C, Mancuso A, Wellen KE (2016) Targeting ACLY sensitizes castration-resistant prostate cancer cells to AR antagonism by impinging on an ACLY-AMPK-AR feedback mechanism. Oncotarget 7 (28):43713–43730. doi: 10.18632/oncotarget.9666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, Verhoeven G, Swinnen JV (2007) Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res 67 (17):8180–8187. doi: 10.1158/0008-5472.CAN-07-0389 [DOI] [PubMed] [Google Scholar]
- 113.Swinnen JV, Van Veldhoven PP, Timmermans L, De Schrijver E, Brusselmans K, Vanderhoydonc F, Van de Sande T, Heemers H, Heyns W, Verhoeven G (2003) Fatty acid synthase drives the synthesis of phospholipids partitioning into detergent-resistant membrane microdomains. Biochemical and Biophysical Research Communications 302 (4):898–903. doi: 10.1016/S0006-291X(03)00265-1 [DOI] [PubMed] [Google Scholar]
- 114.Peck B, Schug ZT, Zhang Q, Dankworth B, Jones DT, Smethurst E, Patel R, Mason S, Jiang M, Saunders R, Howell M, Mitter R, Spencer-Dene B, Stamp G, McGarry L, James D, Shanks E, Aboagye EO, Critchlow SE, Leung HY, Harris AL, Wakelam MJO, Gottlieb E, Schulze A (2016) Inhibition of fatty acid desaturation is detrimental to cancer cell survival in metabolically compromised environments. Cancer & Metabolism 4 (1):6. doi: 10.1186/s40170-016-0146-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS (2008) Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res 68 (11):4447–4454. doi: 10.1158/0008-5472.CAN-08-0249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mener DJ (2010) Prostate specific antigen reduction following statin therapy: Mechanism of action and review of the literature. IUBMB Life 62 (8):584–590. doi: 10.1002/iub.355 [DOI] [PubMed] [Google Scholar]
- 117.Raval AD, Thakker D, Negi H, Vyas A, Salkini MW (2016) Association between statins and clinical outcomes among men with prostate cancer: a systematic review and meta-analysis. Prostate Cancer And Prostatic Diseases 19:151. doi: 10.1038/pcan.2015.58 [DOI] [PubMed] [Google Scholar]
- 118.Sirtori CR (2014) The pharmacology of statins. Pharmacological Research 88:3–11. doi: 10.1016/j.phrs.2014.03.002 [DOI] [PubMed] [Google Scholar]
- 119.Chen Y, Hughes-Fulford M (2001) Human prostate cancer cells lack feedback regulation of low-density lipoprotein receptor and its regulator, SREBP2. International Journal of Cancer 91 (1):41–45. doi: [DOI] [PubMed] [Google Scholar]
- 120.Zhang Y, Ma KL, Ruan XZ, Liu BC (2016) Dysregulation of the Low-Density Lipoprotein Receptor Pathway Is Involved in Lipid Disorder-Mediated Organ Injury. International journal of biological sciences 12 (5):569–579. doi: 10.7150/ijbs.14027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Krycer JR, Brown AJ (2011) Cross-talk between the Androgen Receptor and the Liver X Receptor. Journal of Biological Chemistry 286 (23):20637–20647. doi: 10.1074/jbc.M111.227082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zelcer N, Hong C, Boyadjian R, Tontonoz P (2009) LXR Regulates Cholesterol Uptake Through Idol-Dependent Ubiquitination of the LDL Receptor. Science 325 (5936):100. doi: 10.1126/science.1168974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Repa JJ, Turley SD, Lobaccaro JMA, Medina J, Li L, Lustig K, Shan B, Heyman RA, Dietschy JM, Mangelsdorf DJ (2000) Regulation of Absorption and ABC1-Mediated Efflux of Cholesterol by RXR Heterodimers. Science 289 (5484):1524. doi: 10.1126/science.289.5484.1524 [DOI] [PubMed] [Google Scholar]
- 124.Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro J-MA, Hammer RE, Mangelsdorf DJ (1998) Cholesterol and Bile Acid Metabolism Are Impaired in Mice Lacking the Nuclear Oxysterol Receptor LXRα. Cell 93 (5):693–704. doi: 10.1016/S0092-8674(00)81432-4 [DOI] [PubMed] [Google Scholar]
- 125.Pommier AJC, Alves G, Viennois E, Bernard S, Communal Y, Sion B, Marceau G, Damon C, Mouzat K, Caira F, Baron S, Lobaccaro JMA (2010) Liver X Receptor activation downregulates AKT survival signaling in lipid rafts and induces apoptosis of prostate cancer cells. Oncogene 29:2712. doi: 10.1038/onc.2010.30 [DOI] [PubMed] [Google Scholar]
- 126.Lin C-Y, Gustafsson J-Å (2015) Targeting liver X receptors in cancer therapeutics. Nature Reviews Cancer 15:216. doi: 10.1038/nrc3912 [DOI] [PubMed] [Google Scholar]
- 127.Alfaqih MA, Nelson ER, Liu W, Safi R, Jasper JS, Macias E, Geradts J, Thompson JW, Dubois LG, Freeman MR, Chang CY, Chi JT, McDonnell DP, Freedland SJ (2017) CYP27A1 Loss Dysregulates Cholesterol Homeostasis in Prostate Cancer. Cancer Res 77 (7):1662–1673. doi: 10.1158/0008-5472.CAN-16-2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu Y (2006) Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate cancer and prostatic diseases 9 (3):230–234 [DOI] [PubMed] [Google Scholar]
- 129.Flaig TW, Salzmann-Sullivan M, Su LJ, Zhang Z, Joshi M, Gijon MA, Kim J, Arcaroli JJ, Van Bokhoven A, Lucia MS, La Rosa FG, Schlaepfer IR (2017) Lipid catabolism inhibition sensitizes prostate cancer cells to antiandrogen blockade. Oncotarget 8 (34):56051–56065. doi: 10.18632/oncotarget.17359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Schlaepfer IR, Rider L, Rodrigues LU, Gijon MA, Pac CT, Romero L, Cimic A, Sirintrapun SJ, Glode LM, Eckel RH, Cramer SD (2014) Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol Cancer Ther 13 (10):2361–2371. doi: 10.1158/1535-7163.MCT-14-0183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tennakoon JB, Shi Y, Han JJ, Tsouko E, White MA, Burns AR, Zhang A, Xia X, Ilkayeva OR, Xin L, Ittmann MM, Rick FG, Schally AV, Frigo DE (2014) Androgens regulate prostate cancer cell growth via an AMPK-PGC-1α-mediated metabolic switch. Oncogene 33:5251–5261. doi: 10.1038/onc.2013.463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yu G, Cheng CJ, Lin SC, Lee YC, Frigo DE, Yu-Lee LY, Gallick GE, Titus MA, Nutt LK, Lin SH (2018) Organelle-Derived Acetyl-CoA Promotes Prostate Cancer Cell Survival, Migration, and Metastasis via Activation of Calmodulin Kinase II. Cancer Res 78 (10):2490–2502. doi: 10.1158/0008-5472.CAN-17-2392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wanders RJA (2014) Metabolic functions of peroxisomes in health and disease. Biochimie 98:36–44. doi: 10.1016/j.biochi.2013.08.022 [DOI] [PubMed] [Google Scholar]
- 134.Luo J, Zha S, Gage WR, Dunn TA, Hicks JL, Bennett CJ, Ewing CM, Platz EA, Ferdinandusse S, Wanders RJ, Trent JM, Isaacs WB, De Marzo AM (2002) α-Methylacyl-CoA Racemase A New Molecular Marker for Prostate Cancer. Cancer Res 62 (8):2220–2226 [PubMed] [Google Scholar]
- 135.Lloyd MD, Yevglevskis M, Lee GL, Wood PJ, Threadgill MD, Woodman TJ (2013) α-Methylacyl-CoA racemase (AMACR): Metabolic enzyme, drug metabolizer and cancer marker P504S. Progress in Lipid Research 52 (2):220–230. doi: 10.1016/j.plipres.2013.01.001 [DOI] [PubMed] [Google Scholar]
- 136.Zha S, Ferdinandusse S, Hicks JL, Denis S, Dunn TA, Wanders RJ, Luo J, De Marzo AM, Isaacs WB (2005) Peroxisomal branched chain fatty acid β-oxidation pathway is upregulated in prostate cancer. The Prostate 63 (4):316–323. doi: 10.1002/pros.20177 [DOI] [PubMed] [Google Scholar]
- 137.Wise DR, Thompson CB (2010) Glutamine addiction: a new therapeutic target in cancer. Trends in Biochemical Sciences 35 (8):427–433. doi: 10.1016/j.tibs.2010.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Daye D, Wellen KE (2012) Metabolic reprogramming in cancer: Unraveling the role of glutamine in tumorigenesis. Seminars in Cell & Developmental Biology 23 (4):362–369. doi: 10.1016/j.semcdb.2012.02.002 [DOI] [PubMed] [Google Scholar]
- 139.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci 105 (48):18782–18787. doi: 10.1073/pnas.0810199105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Altman BJ, Stine ZE, Dang CV (2016) From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 16 (10):619–634. doi: 10.1038/nrc.2016.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.White MA, Lin C, Rajapakshe K, Dong J, Shi Y, Tsouko E, Mukhopadhyay R, Jasso D, Dawood W, Coarfa C, Frigo DE (2017) Glutamine Transporters Are Targets of Multiple Oncogenic Signaling Pathways in Prostate Cancer. Mol Cancer Res 15 (8):1017–1028. doi: 10.1158/1541-7786.MCR-16-0480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang Q, Tiffen J, Bailey CG, Lehman ML, Ritchie W, Fazli L, Metierre C, Feng YJ, Li E, Gleave M, Buchanan G, Nelson CC, Rasko JE, Holst J (2013) Targeting amino acid transport in metastatic castration-resistant prostate cancer: effects on cell cycle, cell growth, and tumor development. J Natl Cancer Inst 105 (19):1463–1473. doi: 10.1093/jnci/djt241 [DOI] [PubMed] [Google Scholar]
- 143.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458 (7239):762–765. doi: 10.1038/nature07823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, Thomas GV, Sawyers CL (2003) Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 4 (3):223–238. doi: 10.1016/s1535-6108(03)00197-1 [DOI] [PubMed] [Google Scholar]
- 145.Harding JJ, Telli ML, Munster PN, Le MH, Molineaux C, Bennett MK, Mittra E, Burris HA, Clark AS, Dunphy M, Meric-Bernstam F, Patel MR, DeMichele A, Infante JR (2015) Safety and tolerability of increasing doses of CB-839, a first-in-class, orally administered small molecule inhibitor of glutaminase, in solid tumors. Journal of Clinical Oncology 33 (15_suppl):2512–2512. doi: 10.1200/jco.2015.33.15_suppl.2512 [DOI] [Google Scholar]
- 146.Rhoads JM, Argenzio RA, Chen W, Graves LM, Licato LL, Blikslager AT, Smith J, Gatzy J, Brenner DA (2000) Glutamine metabolism stimulates intestinal cell MAPKs by a cAMP-inhibitable, RAF-independent mechanism. Gastroenterology 118 (1):90–100. doi: 10.1016/S0016-5085(00)70417-3 [DOI] [PubMed] [Google Scholar]
- 147.Dalle Pezze P, Ruf S, Sonntag AG, Langelaar-Makkinje M, Hall P, Heberle AM, Razquin Navas P, van Eunen K, Tölle RC, Schwarz JJ, Wiese H, Warscheid B, Deitersen J, Stork B, Fäßler E, Schäuble S, Hahn U, Horvatovich P, Shanley DP, Thedieck K (2016) A systems study reveals concurrent activation of AMPK and mTOR by amino acids. Nat Commun 7:13254. doi: 10.1038/ncomms13254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Mäkelä TP, Alessi DR, Hardie DG (2003) Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol 2 (4):28. doi: 10.1186/1475-4924-2-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D (2003) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 13 (22):2004–2008. doi: 10.1016/j.cub.2003.10.031 [DOI] [PubMed] [Google Scholar]
- 150.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC (2004) The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci 101 (10):3329–3335. doi: 10.1073/pnas.0308061100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Frigo DE, Howe MK, Wittmann BM, Brunner AM, Cushman I, Wang Q, Brown M, Means AR, McDonnell DP (2011) CaM kinase kinase beta-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res 71 (2):528–537. doi: 10.1158/0008-5472.CAN-10-2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Karacosta LG, Foster BA, Azabdaftari G, Feliciano DM, Edelman AM (2012) A Regulatory Feedback Loop Between Ca(2+)/Calmodulin-dependent Protein Kinase Kinase 2 (CaMKK2) and the Androgen Receptor in Prostate Cancer Progression. The Journal of Biological Chemistry 287 (29):24832–24843. doi: 10.1074/jbc.M112.370783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, Warren A, Scott H, Madhu B, Sharma N, Bon H, Zecchini V, Smith DM, Denicola GM, Mathews N, Osborne M, Hadfield J, Macarthur S, Adryan B, Lyons SK, Brindle KM, Griffiths J, Gleave ME, Rennie PS, Neal DE, Mills IG (2011) The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J 30 (13):2719–2733. doi: 10.1038/emboj.2011.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, Kondo J, Coffey RJ, Johnson MO, Rathmell JC, Sharick JT, Skala MC, Smith JA, Berlin J, Washington MK, Nickels ML, Manning HC (2018) Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med 24 (2):194–202. doi: 10.1038/nm.4464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Liu Y, Zhao T, Li Z, Wang L, Yuan S, Sun L (2018) The role of ASCT2 in cancer: A review. European Journal of Pharmacology 837:81–87. doi: 10.1016/j.ejphar.2018.07.007 [DOI] [PubMed] [Google Scholar]
- 156.Feun L, You M, Wu CJ, Kuo MT, Wangpaichitr M, Spector S, Savaraj N (2008) Arginine deprivation as a targeted therapy for cancer. Current pharmaceutical design 14 (11):1049–1057. doi: 10.2174/138161208784246199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kim RH, Coates JM, Bowles TL, McNerney GP, Sutcliffe J, Jung JU, Gandour-Edwards R, Chuang FYS, Bold RJ, Kung H-J (2009) Arginine Deiminase as a Novel Therapy for Prostate Cancer Induces Autophagy and Caspase-Independent Apoptosis. Cancer Res 69 (2):700. doi: 10.1158/0008-5472.can-08-3157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Keshet R, Szlosarek P, Carracedo A, Erez A (2018) Rewiring urea cycle metabolism in cancer to support anabolism. Nat Rev Cancer 18 (10):634–645. doi: 10.1038/s41568-018-0054-z [DOI] [PubMed] [Google Scholar]
- 159.Lee JS, Adler L, Karathia H, Carmel N, Rabinovich S, Auslander N, Keshet R, Stettner N, Silberman A, Agemy L, Helbling D, Eilam R, Sun Q, Brandis A, Malitsky S, Itkin M, Weiss H, Pinto S, Kalaora S, Levy R, Barnea E, Admon A, Dimmock D, Stern-Ginossar N, Scherz A, Nagamani SCS, Unda M, Wilson DM 3rd, Elhasid R, Carracedo A, Samuels Y, Hannenhalli S, Ruppin E, Erez A (2018) Urea Cycle Dysregulation Generates Clinically Relevant Genomic and Biochemical Signatures. Cell 174 (6):1559–1570 e1522. doi: 10.1016/j.cell.2018.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Husson A, Brasse-Lagnel C, Fairand A, Renouf S, Lavoinne A (2003) Argininosuccinate synthetase from the urea cycle to the citrulline–NO cycle. European Journal of Biochemistry 270 (9):1887–1899. doi: 10.1046/j.1432-1033.2003.03559.x [DOI] [PubMed] [Google Scholar]
- 161.Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA (2002) Pegylated arginine deiminase (ADI-SS PEG20,000 mw) inhibits human melanomas and hepatocellular carcinomas in vitro and in vivo. Cancer Res 62 (19):5443–5450 [PubMed] [Google Scholar]
- 162.Hsueh EC, Knebel SM, Lo W-H, Leung Y-C, Cheng PN-M, Hsueh C-T (2012) Deprivation of arginine by recombinant human arginase in prostate cancer cells. Journal of hematology & oncology 5:17. doi: 10.1186/1756-8722-5-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nowotarski SL, Woster PM, Casero RA (2013) Polyamines and cancer: Implications for chemoprevention and chemotherapy. Expert reviews in molecular medicine 15:e3. doi: 10.1017/erm.2013.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bai GE, Kasper S, Matusik RJ, Rennie PS, Moshier JA, Krongrad A (1998) Androgen Regulation of the Human Ornithine Decarboxylase Promoter in Prostate Cancer Cells. Journal of Andrology 19 (2):127–135. doi: 10.1002/j.1939-4640.1998.tb01981.x [DOI] [PubMed] [Google Scholar]
- 165.Shukla-Dave A, Castillo-Martin M, Chen M, Lobo J, Gladoun N, Collazo-Lorduy A, Khan FM, Ponomarev V, Yi Z, Zhang W, Pandolfi PP, Hricak H, Cordon-Cardo C (2016) Ornithine Decarboxylase Is Sufficient for Prostate Tumorigenesis via Androgen Receptor Signaling. American Journal of Pathology. doi: 10.1016/j.ajpath.2016.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Devens BH, Weeks RS, Burns MR, Carlson CL, Brawer MK (2000) Polyamine depletion therapy in prostate cancer. Prostate Cancer And Prostatic Diseases 3:275. doi: 10.1038/sj.pcan.4500420 [DOI] [PubMed] [Google Scholar]
- 167.Giskeødegård GF, Bertilsson H, Selnæs KM, Wright AJ, Bathen TF, Viset T, Halgunset J, Angelsen A, Gribbestad IS, Tessem M-B (2013) Spermine and Citrate as Metabolic Biomarkers for Assessing Prostate Cancer Aggressiveness. PLOS ONE 8 (4):e62375. doi: 10.1371/journal.pone.0062375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Swanson MG, Zektzer AS, Tabatabai ZL, Simko J, Jarso S, Keshari KR, Schmitt L, Carroll PR, Shinohara K, Vigneron DB, Kurhanewicz J (2006) Quantitative analysis of prostate metabolites using 1H HR-MAS spectroscopy. Magnetic Resonance in Medicine 55 (6):1257–1264. doi: 10.1002/mrm.20909 [DOI] [PubMed] [Google Scholar]
- 169.Cheng LL, Wu C-l, Smith MR, Gonzalez RG (2001) Non-destructive quantitation of spermine in human prostate tissue samples using HRMAS 1H NMR spectroscopy at 9.4 T. FEBS letters 494 (1–2):112–116. doi: 10.1016/S0014-5793(01)02329-8 [DOI] [PubMed] [Google Scholar]
- 170.Corbin MJ, Ruiz-Echevarría JM (2016) One-Carbon Metabolism in Prostate Cancer: The Role of Androgen Signaling. International Journal of Molecular Sciences 17 (8). doi: 10.3390/ijms17081208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Shuvalov O, Petukhov A, Daks A, Fedorova O, Vasileva E, Barlev NA (2017) One-carbon metabolism and nucleotide biosynthesis as attractive targets for anticancer therapy. Oncotarget 8 (14):23955–23977. doi: 10.18632/oncotarget.15053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, Laxman B, Mehra R, Lonigro RJ, Li Y, Nyati MK, Ahsan A, Kalyana-Sundaram S, Han B, Cao X, Byun J, Omenn GS, Ghosh D, Pennathur S, Alexander DC, Berger A, Shuster JR, Wei JT, Varambally S, Beecher C, Chinnaiyan AM (2009) Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 457:910. doi: 10.1038/nature07762 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 173.Shafi AA, Putluri V, Arnold JM, Tsouko E, Maity S, Roberts JM, Coarfa C, Frigo DE, Putluri N, Sreekumar A, Weigel NL (2015) Differential regulation of metabolic pathways by androgen receptor (AR) and its constitutively active splice variant, AR-V7, in prostate cancer cells. Oncotarget 6 (31):31997–32012. doi: 10.18632/oncotarget.5585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ottaviani S, Brooke GN, O’Hanlon-Brown C, Waxman J, Ali S, Buluwela L Characterisation of the androgen regulation of glycine N-methyltransferase in prostate cancer cells. Journal of molecular endocrinology 51 (3):301–312. doi: 10.1530/jme-13-0169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Nelson WG, De Marzo AM, Yegnasubramanian S (2009) Epigenetic alterations in human prostate cancers. Endocrinology 150 (9):3991–4002. doi: 10.1210/en.2009-0573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yegnasubramanian S, Kowalski J, Gonzalgo ML, Zahurak M, Piantadosi S, Walsh PC, Bova GS, De Marzo AM, Isaacs WB, Nelson WG (2004) Hypermethylation of CpG Islands in Primary and Metastatic Human Prostate Cancer. Cancer Res 64 (6):1975. doi: 10.1158/0008-5472.can-03-3972 [DOI] [PubMed] [Google Scholar]
- 177.Jerónimo C, Henrique R, Hoque MO, Mambo E, Ribeiro FR, Varzim G, Oliveira J, Teixeira MR, Lopes C, Sidransky D (2004) A Quantitative Promoter Methylation Profile of Prostate Cancer. Clinical Cancer Research 10 (24):8472. doi: 10.1158/1078-0432.ccr-04-0894 [DOI] [PubMed] [Google Scholar]
- 178.Maruyama R, Toyooka S, Toyooka KO, Virmani AK, Zöchbauer-Müller S, Farinas AJ, Minna JD, McConnell J, Frenkel EP, Gazdar AF (2002) Aberrant Promoter Methylation Profile of Prostate Cancers and Its Relationship to Clinicopathological Features. Clinical Cancer Research 8 (2):514. [PubMed] [Google Scholar]
- 179.Florl AR, Steinhoff C, Müller M, Seifert HH, Hader C, Engers R, Ackermann R, Schulz WA (2004) Coordinate hypermethylation at specific genes in prostate carcinoma precedes LINE-1 hypomethylation. British journal of cancer 91 (5):985–994. doi: 10.1038/sj.bjc.6602030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Padar A, Sathyanarayana UG, Suzuki M, Maruyama R, Hsieh JT, Frenkel EP, Minna JD, Gazdar AF (2003) Inactivation of cyclin D2 gene in prostate cancers by aberrant promoter methylation. Clin Cancer Res 9 (13):4730–4734 [PubMed] [Google Scholar]
- 181.Yamanaka M, Watanabe M, Yamada Y, Takagi A, Murata T, Takahashi H, Suzuki H, Ito H, Tsukino H, Katoh T, Sugimura Y, Shiraishi T (2003) Altered methylation of multiple genes in carcinogenesis of the prostate. Int J Cancer 106 (3):382–387. doi: 10.1002/ijc.11227 [DOI] [PubMed] [Google Scholar]
- 182.Ellinger J, Bastian PJ, Jurgan T, Biermann K, Kahl P, Heukamp LC, Wernert N, Müller SC, von Ruecker A (2008) CpG Island Hypermethylation at Multiple Gene Sites in Diagnosis and Prognosis of Prostate Cancer. Urology 71 (1):161–167. doi: 10.1016/j.urology.2007.09.056 [DOI] [PubMed] [Google Scholar]
- 183.Lodygin D, Epanchintsev A, Menssen A, Diebold J, Hermeking H (2005) Functional Epigenomics Identifies Genes Frequently Silenced in Prostate Cancer. Cancer Res 65 (10):4218. doi: 10.1158/0008-5472.can-04-4407 [DOI] [PubMed] [Google Scholar]
- 184.Antoinette SP, Ruth F, Karen W, Mark L (2006) The emerging roles of DNA methylation in the clinical management of prostate cancer. Endocrine-Related Cancer Endocr Relat Cancer 13 (2):357–377. doi: 10.1677/erc.1.01184 [DOI] [PubMed] [Google Scholar]
- 185.Lee E, Wang J, Yumoto K, Jung Y, Cackowski FC, Decker AM, Li Y, Franceschi RT, Pienta KJ, Taichman RS (2016) DNMT1 Regulates Epithelial-Mesenchymal Transition and Cancer Stem Cells, Which Promotes Prostate Cancer Metastasis. Neoplasia 18 (9):553–566. doi: 10.1016/j.neo.2016.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Morey Kinney SR, Moser MT, Pascual M, Greally JM, Foster BA, Karpf AR (2010) Opposing Roles of Dnmt1 in Early- and Late-Stage Murine Prostate Cancer. Molecular and Cellular Biology 30 (17):4159. doi: 10.1128/mcb.00235-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.McCabe MT, Low JA, Daignault S, Imperiale MJ, Wojno KJ, Day ML (2006) Inhibition of DNA Methyltransferase Activity Prevents Tumorigenesis in a Mouse Model of Prostate Cancer. Cancer Res 66 (1):385. doi: 10.1158/0008-5472.can-05-2020 [DOI] [PubMed] [Google Scholar]
- 188.Singal R, Ramachandran K, Gordian E, Quintero C, Zhao W, Reis IM (2015) Phase I/II Study of Azacitidine, Docetaxel, and Prednisone in Patients With Metastatic Castration-Resistant Prostate Cancer Previously Treated With Docetaxel-Based Therapy. Clinical Genitourinary Cancer 13 (1):22–31. doi: 10.1016/j.clgc.2014.07.008 [DOI] [PubMed] [Google Scholar]
- 189.Casero RA Jr, Marton LJ (2007) Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nature Reviews Drug Discovery 6:373. doi: 10.1038/nrd2243 [DOI] [PubMed] [Google Scholar]
- 190.Gerner EW, Meyskens FL Jr (2004) Polyamines and cancer: old molecules, new understanding. Nature Reviews Cancer 4:781. doi: 10.1038/nrc1454 [DOI] [PubMed] [Google Scholar]
- 191.Soda K (2011) The mechanisms by which polyamines accelerate tumor spread. Journal of experimental & clinical cancer research : CR 30 (1):95–95. doi: 10.1186/1756-9966-30-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cipolla BG, Havouis R, Moulinoux J-P (2010) Polyamine reduced diet (PRD) nutrition therapy in hormone refractory prostate cancer patients. Biomedicine & Pharmacotherapy 64 (5):363–368. doi: 10.1016/j.biopha.2009.09.022 [DOI] [PubMed] [Google Scholar]
- 193.Fantus IG, Goldberg HJ, Whiteside CI, Topic D (2006) The Hexosamine Biosynthesis Pathway. In: Cortes P, Mogensen CE (eds) The Diabetic Kidney. Humana Press, Totowa, NJ, pp 117–133. doi: 10.1007/978-1-59745-153-6_7 [DOI] [Google Scholar]
- 194.Itkonen HM, Engedal N, Babaie E, Luhr M, Guldvik IJ, Minner S, Hohloch J, Tsourlakis MC, Schlomm T, Mills IG (2015) UAP1 is overexpressed in prostate cancer and is protective against inhibitors of N-linked glycosylation. Oncogene 34 (28):3744–3750. doi: 10.1038/onc.2014.307 [DOI] [PubMed] [Google Scholar]
- 195.Itkonen HM, Minner S, Guldvik IJ, Sandmann MJ, Tsourlakis MC, Berge V, Svindland A, Schlomm T, Mills IG (2013) O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res 73 (16):5277–5287. doi: 10.1158/0008-5472.CAN-13-0549 [DOI] [PubMed] [Google Scholar]
- 196.Itkonen HM, Mills IG (2013) N-linked glycosylation supports cross-talk between receptor tyrosine kinases and androgen receptor. PLoS One 8 (5):e65016. doi: 10.1371/journal.pone.0065016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kamigaito T, Okaneya T, Kawakubo M, Shimojo H, Nishizawa O, Nakayama J (2014) Overexpression of O-GlcNAc by prostate cancer cells is significantly associated with poor prognosis of patients. Prostate Cancer Prostatic Dis 17 (1):18–22. doi: 10.1038/pcan.2013.56 [DOI] [PubMed] [Google Scholar]
- 198.Lynch TP, Ferrer CM, Jackson SR, Shahriari KS, Vosseller K, Reginato MJ (2012) Critical role of O-Linked beta-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J Biol Chem 287 (14):11070–11081. doi: 10.1074/jbc.M111.302547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kaushik AK, Shojaie A, Panzitt K, Sonavane R, Venghatakrishnan H, Manikkam M, Zaslavsky A, Putluri V, Vasu VT, Zhang YQ, Khan AS, Lloyd S, Szafran AT, Dasgupta S, Bader DA, Stossi F, Li HW, Samanta S, Cao XH, Tsouko E, Huang SX, Frigo DE, Chan L, Edwards DP, Kaipparettu BA, Mitsiades N, Weigel NL, Mancini M, McGuire SE, Mehra R, Ittmann MM, Chinnaiyan AM, Putluri N, Palapattu GS, Michailidis G, Sreekumar A (2016) Inhibition of the hexosamine biosynthetic pathway promotes castration-resistant prostate cancer. Nat Commun 7. doi: 10.1038/ncomms11612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, Kimmelman A, Kumar S, Levine B, Maiuri MC, Martin SJ, Penninger J, Piacentini M, Rubinsztein DC, Simon HU, Simonsen A, Thorburn AM, Velasco G, Ryan KM, Kroemer G (2015) Autophagy in malignant transformation and cancer progression. Embo Journal 34 (7):856–880. doi: 10.15252/embj.201490784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Goldsmith J, Levine B, Debnath J (2014) Autophagy and cancer metabolism. Methods Enzymol 542:25–57. doi: 10.1016/B978-0-12-416618-9.00002-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ziparo E, Petrungaro S, Marini ES, Starace D, Conti S, Facchiano A, Filippini A, Giampietri C (2013) Autophagy in Prostate Cancer and Androgen Suppression Therapy. International Journal of Molecular Sciences 14 (6):12090–12106. doi: 10.3390/ijms140612090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Xie X, Koh JY, Price S, White E, Mehnert JM (2015) Atg7 Overcomes Senescence and Promotes Growth of BrafV600 E-Driven Melanoma. Cancer Discov 5 (4):410–423. doi: 10.1158/2159-8290.CD-14-1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Garcia-Fernandez M, Karras P, Checinska A, Canon E, Calvo GT, Gomez-Lopez G, Cifdaloz M, Colmenar A, Espinosa-Hevia L, Olmeda D, Soengas MS (2016) Metastatic risk and resistance to BRAF inhibitors in melanoma defined by selective allelic loss of ATG5. Autophagy 12 (10):1776–1790. doi: 10.1080/15548627.2016.1199301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Santanam U, Banach-Petrosky W, Abate-Shen C, Shen MM, White E, DiPaola RS (2016) Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev 30 (4):399–407. doi: 10.1101/gad.274134.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Farrow JM, Yang JC, Evans CP (2014) Autophagy as a modulator and target in prostate cancer. Nat Rev Urol 11 (9):508–516. doi: 10.1038/nrurol.2014.196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chen X, Lu J, Xia L, Li G (2018) Drug Resistance of Enzalutamide in CRPC. Curr Drug Targets 19 (6):613–620. doi: 10.2174/1389450118666170417144250 [DOI] [PubMed] [Google Scholar]
- 208.Nguyen HG, Yang JC, Kung HJ, Shi XB, Tilki D, Lara PN Jr., DeVere White RW, Gao AC, Evans CP (2014) Targeting autophagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene 33 (36):4521–4530. doi: 10.1038/onc.2014.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Parikh M, Hyunh JC, Lara P, Pan CX, Robles D, Evans CP (2018) Enzalutamide and metformin combination therapy to overcome autophagy resistance in castration resistant prostate cancer (CRPC): Current results from a phase I study. Journal of Clinical Oncology 36 (6). doi:DOI 10.1200/JCO.2018.36.6_suppl.281 [DOI] [Google Scholar]
- 210.Kranzbuhler B, Salemi S, Mortezavi A, Sulser T, Eberli D (2019) Combined N-terminal androgen receptor and autophagy inhibition increases the antitumor effect in enzalutamide sensitive and enzalutamide resistant prostate cancer cells. Prostate 79 (2):206–214. doi: 10.1002/pros.23725 [DOI] [PubMed] [Google Scholar]
- 211.Hu F, Zhao Y, Yu Y, Fang JM, Cui R, Liu ZQ, Guo XL, Xu Q (2018) Docetaxel-mediated autophagy promotes chemoresistance in castration-resistant prostate cancer cells by inhibiting STAT3. Cancer Lett 416:24–30. doi: 10.1016/j.canlet.2017.12.013 [DOI] [PubMed] [Google Scholar]
- 212.Cristofani R, Montagnani Marelli M, Cicardi ME, Fontana F, Marzagalli M, Limonta P, Poletti A, Moretti RM (2018) Dual role of autophagy on docetaxel-sensitivity in prostate cancer cells. Cell Death Dis 9 (9):889. doi: 10.1038/s41419-018-0866-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wang Q, He WY, Zeng YZ, Hossain A, Gou X (2018) Inhibiting autophagy overcomes docetaxel resistance in castration-resistant prostate cancer cells. Int Urol Nephrol 50 (4):675–686. doi: 10.1007/s11255-018-1801-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Draz H, Goldberg AA, Titorenko VI, Tomlinson Guns ES, Safe SH, Sanderson JT (2017) Diindolylmethane and its halogenated derivatives induce protective autophagy in human prostate cancer cells via induction of the oncogenic protein AEG-1 and activation of AMP-activated protein kinase (AMPK). Cell Signal 40:172–182. doi: 10.1016/j.cellsig.2017.09.006 [DOI] [PubMed] [Google Scholar]
- 215.Draz H, Goldberg AA, Tomlinson Guns ES, Fazli L, Safe S, Sanderson JT (2018) Autophagy inhibition improves the chemotherapeutic efficacy of cruciferous vegetable-derived diindolymethane in a murine prostate cancer xenograft model. Invest New Drugs 36 (4):718–725. doi: 10.1007/s10637-018-0595-8 [DOI] [PubMed] [Google Scholar]
- 216.Zhu X, Zhou M, Liu GY, Huang XL, He WY, Gou X, Jiang T (2017) Autophagy activated by the c-Jun N-terminal kinase-mediated pathway protects human prostate cancer PC3 cells from celecoxib-induced apoptosis. Experimental and Therapeutic Medicine 13 (5):2348–2354. doi: 10.3892/etm.2017.4287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Yang C, Ma X, Wang Z, Zeng X, Hu Z, Ye Z, Shen G (2017) Curcumin induces apoptosis and protective autophagy in castration-resistant prostate cancer cells through iron chelation. Drug Des Devel Ther 11:431–439. doi: 10.2147/DDDT.S126964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Lin YC, Chang YT, Campbell M, Lin TP, Pan CC, Lee HC, Shih JC, Chang PC (2017) MAOA-a novel decision maker of apoptosis and autophagy in hormone refractory neuroendocrine prostate cancer cells. Sci Rep 7:46338. doi: 10.1038/srep46338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Niture S, Ramalinga M, Kedir H, Patacsil D, Niture SS, Li J, Mani H, Suy S, Collins S, Kumar D (2018) TNFAIP8 promotes prostate cancer cell survival by inducing autophagy. Oncotarget 9 (42):26884–26899. doi: 10.18632/oncotarget.25529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Lu J, Dong W, He H, Han Z, Zhuo Y, Mo R, Liang Y, Zhu J, Li R, Qu H, Zhang L, Wang S, Ma R, Jia Z, Zhong W (2018) Autophagy induced by overexpression of DCTPP1 promotes tumor progression and predicts poor clinical outcome in prostate cancer. Int J Biol Macromol 118 (Pt A):599–609. doi: 10.1016/j.ijbiomac.2018.06.005 [DOI] [PubMed] [Google Scholar]
- 221.Commisso C, Debnath J (2018) Macropinocytosis Fuels Prostate Cancer. Cancer Discov 8 (7):800–802. doi: 10.1158/2159-8290.CD-18-0513 [DOI] [PubMed] [Google Scholar]
- 222.Kim SM, Nguyen TT, Ravi A, Kubiniok P, Finicle BT, Jayashankar V, Malacrida L, Hou J, Robertson J, Gao D, Chernoff J, Digman MA, Potma EO, Tromberg BJ, Thibault P, Edinger AL (2018) PTEN Deficiency and AMPK Activation Promote Nutrient Scavenging and Anabolism in Prostate Cancer Cells. Cancer Discov 8 (7):866–883. doi: 10.1158/2159-8290.CD-17-1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Khan AS, Frigo DE (2017) A spatiotemporal hypothesis for the regulation, role, and targeting of AMPK in prostate cancer. Nature Reviews Urology 14 (3):164–180. doi: 10.1038/nrurol.2016.272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Vaz CV, Marques R, Alves MG, Oliveira PF, Cavaco JE, Maia CJ, Socorro S (2016) Androgens enhance the glycolytic metabolism and lactate export in prostate cancer cells by modulating the expression of GLUT1, GLUT3, PFK, LDH and MCT4 genes. J Cancer Res Clin Oncol 142 (1):5–16. doi: 10.1007/s00432-015-1992-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Awad D, Pulliam TL, Lin C, Wilkenfeld SR, Frigo DE (2018) Delineation of the androgen-regulated signaling pathways in prostate cancer facilitates the development of novel therapeutic approaches. Curr Opin Pharmacol 41:1–11. doi: 10.1016/j.coph.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Butler LM, Centenera MM, Swinnen JV (2016) Androgen control of lipid metabolism in prostate cancer: novel insights and future applications. Endocr Relat Cancer 23 (5):R219–227. doi: 10.1530/ERC-15-0556 [DOI] [PubMed] [Google Scholar]
- 227.Dadwal UC, Chang ES, Sankar U (2018) Androgen Receptor-CaMKK2 Axis in Prostate Cancer and Bone Microenvironment. Front Endocrinol (Lausanne) 9:335. doi: 10.3389/fendo.2018.00335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Mitani T, Yamaji R, Higashimura Y, Harada N, Nakano Y, Inui H (2011) Hypoxia enhances transcriptional activity of androgen receptor through hypoxia-inducible factor-1alpha in a low androgen environment. J Steroid Biochem Mol Biol 123 (1–2):58–64. doi: 10.1016/j.jsbmb.2010.10.009 [DOI] [PubMed] [Google Scholar]
- 229.Mabjeesh NJ, Willard MT, Frederickson CE, Zhong H, Simons JW (2003) Androgens stimulate hypoxia-inducible factor 1 activation via autocrine loop of tyrosine kinase receptor/phosphatidylinositol 3’-kinase/protein kinase B in prostate cancer cells. Clin Cancer Res 9 (7):2416–2425 [PubMed] [Google Scholar]
- 230.Rebello RJ, Pearson RB, Hannan RD, Furic L (2017) Therapeutic Approaches Targeting MYC-Driven Prostate Cancer. Genes (Basel) 8 (2). doi: 10.3390/genes8020071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Dang CV, Le A, Gao P (2009) MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res 15 (21):6479–6483. doi: 10.1158/1078-0432.CCR-09-0889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Priolo C, Pyne S, Rose J, Regan ER, Zadra G, Photopoulos C, Cacciatore S, Schultz D, Scaglia N, McDunn J, De Marzo AM, Loda M (2014) AKT1 and MYC induce distinctive metabolic fingerprints in human prostate cancer. Cancer Res 74 (24):7198–7204. doi: 10.1158/0008-5472.CAN-14-1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Mossmann D, Park S, Hall MN (2018) mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer 18 (12):744–757. doi: 10.1038/s41568-018-0074-8 [DOI] [PubMed] [Google Scholar]
- 234.Yu JS, Cui W (2016) Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 143 (17):3050–3060. doi: 10.1242/dev.137075 [DOI] [PubMed] [Google Scholar]
- 235.Nogueira V, Patra KC, Hay N (2018) Selective eradication of cancer displaying hyperactive Akt by exploiting the metabolic consequences of Akt activation. Elife 7. doi: 10.7554/eLife.32213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Giunchi Francesca F M, Lodab Massimo (2018) The Metabolic Landscape of Prostate Cancer. European Urology Oncology In Press. doi: 10.1016/j.euo.2018.06.010 [DOI] [PubMed] [Google Scholar]
- 237.Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S, Jackson E, Aiello NM, Haas NB, Rebbeck TR, Judkins A, Won KJ, Chodosh LA, Garcia BA, Stanger BZ, Feldman MD, Blair IA, Wellen KE (2014) Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab 20 (2):306–319. doi: 10.1016/j.cmet.2014.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Toren P, Zoubeidi A (2014) Targeting the PI3K/Akt pathway in prostate cancer: challenges and opportunities (review). Int J Oncol 45 (5):1793–1801. doi: 10.3892/ijo.2014.2601 [DOI] [PubMed] [Google Scholar]
- 239.Naguib A, Mathew G, Reczek CR, Watrud K, Ambrico A, Herzka T, Salas IC, Lee MF, El-Amine N, Zheng W, Di Francesco ME, Marszalek JR, Pappin DJ, Chandel NS, Trotman LC (2018) Mitochondrial Complex I Inhibitors Expose a Vulnerability for Selective Killing of Pten-Null Cells. Cell Rep 23 (1):58–67. doi: 10.1016/j.celrep.2018.03.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, Jeong SM, Dempsey JM, Parkhitko A, Morrison T, Henske EP, Haigis MC, Cantley LC, Stephanopoulos G, Yu J, Blenis J (2013) The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 153 (4):840–854. doi: 10.1016/j.cell.2013.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Zabala-Letona A, Arruabarrena-Aristorena A, Martin-Martin N, Fernandez-Ruiz S, Sutherland JD, Clasquin M, Tomas-Cortazar J, Jimenez J, Torres I, Quang P, Ximenez-Embun P, Bago R, Ugalde-Olano A, Loizaga-Iriarte A, Lacasa-Viscasillas I, Unda M, Torrano V, Cabrera D, van Liempd SM, Cendon Y, Castro E, Murray S, Revandkar A, Alimonti A, Zhang Y, Barnett A, Lein G, Pirman D, Cortazar AR, Arreal L, Prudkin L, Astobiza I, Valcarcel-Jimenez L, Zuniga-Garcia P, Fernandez-Dominguez I, Piva M, Caro-Maldonado A, Sanchez-Mosquera P, Castillo-Martin M, Serra V, Beraza N, Gentilella A, Thomas G, Azkargorta M, Elortza F, Farras R, Olmos D, Efeyan A, Anguita J, Munoz J, Falcon-Perez JM, Barrio R, Macarulla T, Mato JM, Martinez-Chantar ML, Cordon-Cardo C, Aransay AM, Marks K, Baselga J, Tabernero J, Nuciforo P, Manning BD, Marjon K, Carracedo A (2017) mTORC1-dependent AMD1 regulation sustains polyamine metabolism in prostate cancer. Nature 547 (7661):109–113. doi: 10.1038/nature22964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Wang Q, Bailey CG, Ng C, Tiffen J, Thoeng A, Minhas V, Lehman ML, Hendy SC, Buchanan G, Nelson CC, Rasko JE, Holst J (2011) Androgen receptor and nutrient signaling pathways coordinate the demand for increased amino acid transport during prostate cancer progression. Cancer Res 71 (24):7525–7536. doi: 10.1158/0008-5472.CAN-11-1821 [DOI] [PubMed] [Google Scholar]
- 243.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, Scardino PT, Rosen N, Sawyers CL (2011) Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 19 (5):575–586. doi: 10.1016/j.ccr.2011.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Edlind MP, Hsieh AC (2014) PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J Androl 16 (3):378–386. doi: 10.4103/1008-682X.122876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Crumbaker M, Khoja L, Joshua AM (2017) AR Signaling and the PI3K Pathway in Prostate Cancer. Cancers (Basel) 9 (4). doi: 10.3390/cancers9040034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Audet-Walsh E, Dufour CR, Yee T, Zouanat FZ, Yan M, Kalloghlian G, Vernier M, Caron M, Bourque G, Scarlata E, Hamel L, Brimo F, Aprikian AG, Lapointe J, Chevalier S, Giguere V (2017) Nuclear mTOR acts as a transcriptional integrator of the androgen signaling pathway in prostate cancer. Genes Dev 31 (12):1228–1242. doi: 10.1101/gad.299958.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Marhold M, Tomasich E, El-Gazzar A, Heller G, Spittler A, Horvat R, Krainer M, Horak P (2015) HIF1alpha Regulates mTOR Signaling and Viability of Prostate Cancer Stem Cells. Mol Cancer Res 13 (3):556–564. doi: 10.1158/1541-7786.MCR-14-0153-T [DOI] [PubMed] [Google Scholar]
- 248.Barfeld SJ, Itkonen HM, Urbanucci A, Mills IG (2014) Androgen-regulated metabolism and biosynthesis in prostate cancer. Endocr Relat Cancer 21 (4):T57–66. doi: 10.1530/ERC-13-0515 [DOI] [PubMed] [Google Scholar]
- 249.Fabris L, Ceder Y, Chinnaiyan AM, Jenster GW, Sorensen KD, Tomlins S, Visakorpi T, Calin GA (2016) The Potential of MicroRNAs as Prostate Cancer Biomarkers. Eur Urol 70 (2):312–322. doi: 10.1016/j.eururo.2015.12.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Qu W, Ding SM, Cao G, Wang SJ, Zheng XH, Li GH (2016) miR-132 mediates a metabolic shift in prostate cancer cells by targeting Glut1. FEBS Open Bio 6 (7):735–741. doi: 10.1002/2211-5463.12086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Singh A, Happel C, Manna SK, Acquaah-Mensah G, Carrerero J, Kumar S, Nasipuri P, Krausz KW, Wakabayashi N, Dewi R, Boros LG, Gonzalez FJ, Gabrielson E, Wong KK, Girnun G, Biswal S (2013) Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J Clin Invest 123 (7):2921–2934. doi: 10.1172/JCI66353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Shih JW, Wang LY, Hung CL, Kung HJ, Hsieh CL (2015) Non-Coding RNAs in Castration-Resistant Prostate Cancer: Regulation of Androgen Receptor Signaling and Cancer Metabolism. Int J Mol Sci 16 (12):28943–28978. doi: 10.3390/ijms161226138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Xin M, Qiao Z, Li J, Liu J, Song S, Zhao X, Miao P, Tang T, Wang L, Liu W, Yang X, Dai K, Huang G (2016) miR-22 inhibits tumor growth and metastasis by targeting ATP citrate lyase: evidence in osteosarcoma, prostate cancer, cervical cancer and lung cancer. Oncotarget 7 (28):44252–44265. doi: 10.18632/oncotarget.10020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Li X, Chen YT, Josson S, Mukhopadhyay NK, Kim J, Freeman MR, Huang WC (2013) MicroRNA-185 and 342 inhibit tumorigenicity and induce apoptosis through blockade of the SREBP metabolic pathway in prostate cancer cells. PLoS One 8 (8):e70987. doi: 10.1371/journal.pone.0070987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Liu W, Le A, Hancock C, Lane AN, Dang CV, Fan TW, Phang JM (2012) Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc Natl Acad Sci 109 (23):8983–8988. doi: 10.1073/pnas.1203244109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Lyssiotis CA, Kimmelman AC (2017) Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol 27 (11):863–875. doi: 10.1016/j.tcb.2017.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Marignol L, Rivera-Figueroa K, Lynch T, Hollywood D (2013) Hypoxia, notch signalling, and prostate cancer. Nat Rev Urol 10 (7):405–413. doi: 10.1038/nrurol.2013.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Geng H, Xue C, Mendonca J, Sun XX, Liu Q, Reardon PN, Chen Y, Qian K, Hua V, Chen A, Pan F, Yuan J, Dang S, Beer TM, Dai MS, Kachhap SK, Qian DZ (2018) Interplay between hypoxia and androgen controls a metabolic switch conferring resistance to androgen/AR-targeted therapy. Nat Commun 9 (1):4972. doi: 10.1038/s41467-018-07411-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J, Huang P, Sawyer SK, Fuerth B, Faubert B, Kalliomaki T, Elia A, Luo X, Nadeem V, Bungard D, Yalavarthi S, Growney JD, Wakeham A, Moolani Y, Silvester J, Ten AY, Bakker W, Tsuchihara K, Berger SL, Hill RP, Jones RG, Tsao M, Robinson MO, Thompson CB, Pan G, Mak TW (2011) Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev 25 (10):1041–1051. doi: 10.1101/gad.1987211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Jin Y, Wang L, Qu S, Sheng X, Kristian A, Maelandsmo GM, Pallmann N, Yuca E, Tekedereli I, Gorgulu K, Alpay N, Sood A, Lopez-Berestein G, Fazli L, Rennie P, Risberg B, Waehre H, Danielsen HE, Ozpolat B, Saatcioglu F (2015) STAMP2 increases oxidative stress and is critical for prostate cancer. EMBO Mol Med 7 (3):315–331. doi: 10.15252/emmm.201404181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Tam NN, Gao Y, Leung YK, Ho SM (2003) Androgenic regulation of oxidative stress in the rat prostate: involvement of NAD(P)H oxidases and antioxidant defense machinery during prostatic involution and regrowth. Am J Pathol 163 (6):2513–2522. doi: 10.1016/S0002-9440(10)63606-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Frohlich DA, McCabe MT, Arnold RS, Day ML (2008) The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene 27 (31):4353–4362. doi: 10.1038/onc.2008.79 [DOI] [PubMed] [Google Scholar]
- 263.Khandrika L, Kumar B, Koul S, Maroni P, Koul HK (2009) Oxidative stress in prostate cancer. Cancer Lett 282 (2):125–136. doi: 10.1016/j.canlet.2008.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Samaranayake GJ, Troccoli CI, Huynh M, Lyles RDZ, Kage K, Win A, Lakshmanan V, Kwon D, Ban Y, Chen SX, Zarco ER, Jorda M, Burnstein KL, Rai P (2017) Thioredoxin-1 protects against androgen receptor-induced redox vulnerability in castration-resistant prostate cancer. Nat Commun 8 (1):1204. doi: 10.1038/s41467-017-01269-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Li K, Zheng Q, Chen X, Wang Y, Wang D, Wang J (2018) Isobavachalcone Induces ROS-Mediated Apoptosis via Targeting Thioredoxin Reductase 1 in Human Prostate Cancer PC-3 Cells. Oxid Med Cell Longev 2018:1915828. doi: 10.1155/2018/1915828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Yan Y, Chang L, Tian H, Wang L, Zhang Y, Yang T, Li G, Hu W, Shah K, Chen G, Guo Y (2018) 1-Pyrroline-5-carboxylate released by prostate Cancer cell inhibit T cell proliferation and function by targeting SHP1/cytochrome c oxidoreductase/ROS Axis. J Immunother Cancer 6 (1):148. doi: 10.1186/s40425-018-0466-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, Takeyama H (2015) Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers (Basel) 7 (4):2443–2458. doi: 10.3390/cancers7040902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, Chiarugi P (2010) Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res 70 (17):6945–6956. doi: 10.1158/0008-5472.CAN-10-0785 [DOI] [PubMed] [Google Scholar]
- 269.Lopes-Coelho F, Gouveia-Fernandes S, Serpa J (2018) Metabolic cooperation between cancer and non-cancerous stromal cells is pivotal in cancer progression. Tumour Biol 40 (2):1010428318756203. doi: 10.1177/1010428318756203 [DOI] [PubMed] [Google Scholar]
- 270.Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, Lanciotti M, Serni S, Cirri P, Chiarugi P (2012) Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res 72 (19):5130–5140. doi: 10.1158/0008-5472.CAN-12-1949 [DOI] [PubMed] [Google Scholar]
- 271.Chiarugi P, Paoli P, Cirri P (2014) Tumor microenvironment and metabolism in prostate cancer. Semin Oncol 41 (2):267–280. doi: 10.1053/j.seminoncol.2014.03.004 [DOI] [PubMed] [Google Scholar]
- 272.Pertega-Gomes N, Vizcaino JR, Attig J, Jurmeister S, Lopes C, Baltazar F (2014) A lactate shuttle system between tumour and stromal cells is associated with poor prognosis in prostate cancer. BMC Cancer 14:352. doi: 10.1186/1471-2407-14-352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Giatromanolaki A, Koukourakis MI, Koutsopoulos A, Mendrinos S, Sivridis E (2012) The metabolic interactions between tumor cells and tumor-associated stroma (TAS) in prostatic cancer. Cancer Biol Ther 13 (13):1284–1289. doi: 10.4161/cbt.21785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Fiaschi T, Giannoni E, Taddei ML, Cirri P, Marini A, Pintus G, Nativi C, Richichi B, Scozzafava A, Carta F, Torre E, Supuran CT, Chiarugi P (2013) Carbonic anhydrase IX from cancer-associated fibroblasts drives epithelial-mesenchymal transition in prostate carcinoma cells. Cell Cycle 12 (11):1791–1801. doi: 10.4161/cc.24902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Santi A, Caselli A, Paoli P, Corti D, Camici G, Pieraccini G, Taddei ML, Serni S, Chiarugi P, Cirri P (2013) The effects of CA IX catalysis products within tumor microenvironment. Cell Commun Signal 11:81. doi: 10.1186/1478-811X-11-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Cheteh EH, Augsten M, Rundqvist H, Bianchi J, Sarne V, Egevad L, Bykov VJ, Ostman A, Wiman KG (2017) Human cancer-associated fibroblasts enhance glutathione levels and antagonize drug-induced prostate cancer cell death. Cell Death Dis 8 (6):e2848. doi: 10.1038/cddis.2017.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Nassar ZD, Aref AT, Miladinovic D, Mah CY, Raj GV, Hoy AJ, Butler LM (2018) Peri-prostatic adipose tissue: the metabolic microenvironment of prostate cancer. BJU Int 121 Suppl 3:9–21. doi: 10.1111/bju.14173 [DOI] [PubMed] [Google Scholar]
- 278.Huang J, Duran A, Reina-Campos M, Valencia T, Castilla EA, Muller TD, Tschop MH, Moscat J, Diaz-Meco MT (2018) Adipocyte p62/SQSTM1 Suppresses Tumorigenesis through Opposite Regulations of Metabolism in Adipose Tissue and Tumor. Cancer Cell 33 (4):770–784 e776. doi: 10.1016/j.ccell.2018.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Gazi E, Dwyer J, Lockyer NP, Gardner P, Shanks JH, Roulson J, Hart CA, Clarke NW, Brown MD (2007) Biomolecular profiling of metastatic prostate cancer cells in bone marrow tissue using FTIR microspectroscopy: a pilot study. Anal Bioanal Chem 387 (5):1621–1631. doi: 10.1007/s00216-006-1093-y [DOI] [PubMed] [Google Scholar]
- 280.Balaban S, Nassar ZD, Zhang AY, Hosseini-Beheshti E, Centenera MM, Schreuder M, Lin HM, Aishah A, Varney B, Liu-Fu F, Lee LS, Nagarajan SR, Shearer RF, Hardie RA, Raftopulos NL, Kakani MS, Saunders DN, Holst J, Horvath LG, Butler LM, Hoy AJ (2019) Extracellular Fatty Acids are the Major Contributor to Lipid Synthesis in Prostate Cancer. Mol Cancer Res. doi: 10.1158/1541-7786.MCR-18-0347 [DOI] [PubMed] [Google Scholar]
- 281.Tokuda Y, Satoh Y, Fujiyama C, Toda S, Sugihara H, Masaki Z (2003) Prostate cancer cell growth is modulated by adipocyte-cancer cell interaction. BJU Int 91 (7):716–720. doi: 10.1046/j.1464-410X.2003.04218.x [DOI] [PubMed] [Google Scholar]
- 282.Diedrich JD, Rajagurubandara E, Herroon MK, Mahapatra G, Huttemann M, Podgorski I (2016) Bone marrow adipocytes promote the Warburg phenotype in metastatic prostate tumors via HIF-1alpha activation. Oncotarget 7 (40):64854–64877. doi: 10.18632/oncotarget.11712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Fujita K, Landis P, McNeil BK, Pavlovich CP (2009) Serial prostate biopsies are associated with an increased risk of erectile dysfunction in men with prostate cancer on active surveillance. The Journal of Urology 182 (6):2664–2669. doi: 10.1016/j.juro.2009.08.044 [DOI] [PubMed] [Google Scholar]
- 284.Vander Heiden MG (2011) Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov 10 (9):671–684. doi: 10.1038/nrd3504 [DOI] [PubMed] [Google Scholar]
- 285.Kurhanewicz J, Vigneron DB, Brindle K, Chekmenev EY, Comment A, Cunningham CH, DeBerardinis RJ, Green GG, Leach MO, Rajan SS (2011) Analysis of cancer metabolism by imaging hyperpolarized nuclei: prospects for translation to clinical research. Neoplasia 13 (2):81–97. doi: 10.1593/neo.101102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Wibmer AG, Burger IA, Sala E, Hricak H, Weber WA, Vargas HA (2015) Molecular Imaging of Prostate Cancer. Radiographics 36 (1):142–159. doi: 10.1148/rg.2016150059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Ardenkjaer-Larsen JH (2009) Hyperpolarized 13C magnetic resonance imaging-principles and applications. Molecular Imaging: Principles and Practice:377–388 [Google Scholar]
- 288.Bhattacharya P, Ross BD, Bünger R (2009) Cardiovascular applications of hyperpolarized contrast media and metabolic tracers. Experimental Biology and Medicine 234 (12):1395–1416. doi: 10.3181/0904-MR-135 [DOI] [PubMed] [Google Scholar]
- 289.Brindle K (2008) New approaches for imaging tumour responses to treatment. Nature Reviews Cancer 8 (2):94. [DOI] [PubMed] [Google Scholar]
- 290.Zacharias N, Lee J, Ramachandran S, Shanmugavelandy S, McHenry J, Dutta P, Millward S, Gammon S, Efstathiou E, Troncoso P, Frigo DE, Piwnica-Worms D, Logothetis CJ, Maity SN, Titus MA, Bhattacharya P (2018) Androgen Receptor Signaling in Castration-Resistant Prostate Cancer Alters Hyperpolarized Pyruvate to Lactate Conversion and Lactate Levels In Vivo. Mol Imaging Biol. doi: 10.1007/s11307-018-1199-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Dutta P, Martinez GV, Gillies RJ (2013) A new horizon of DNP technology: application to in-vivo 13 C magnetic resonance spectroscopy and imaging. Biophysical reviews 5 (3):271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Dutta P, Le A, Vander Jagt DL, Tsukamoto T, Martinez GV, Dang CV, Gillies RJ (2013) Evaluation of LDH-A and glutaminase inhibition in vivo by hyperpolarized 13C-pyruvate magnetic resonance spectroscopy of tumors. Cancer Res. doi: 10.1158/0008-5472.CAN-13-0465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Albers MJ, Bok R, Chen AP, Cunningham CH, Zierhut ML, Zhang VY, Kohler SJ, Tropp J, Hurd RE, Yen Y-F (2008) Hyperpolarized 13C lactate, pyruvate, and alanine: noninvasive biomarkers for prostate cancer detection and grading. Cancer Res 68 (20):8607–8615. doi: 10.1158/0008-5472.CAN-08-0749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Nelson SJ, Kurhanewicz J, Vigneron DB, Larson PE, Harzstark AL, Ferrone M, van Criekinge M, Chang JW, Bok R, Park I (2013) Metabolic imaging of patients with prostate cancer using hyperpolarized [1–13C] pyruvate. Science translational medicine 5 (198):198ra108. doi: 10.1126/scitranslmed.3006070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Aggarwal R, Vigneron DB, Kurhanewicz J (2017) Hyperpolarized 1-[13C]-Pyruvate Magnetic Resonance Imaging Detects an Early Metabolic Response to Androgen Ablation Therapy in Prostate Cancer. European Urology 72 (6):1028–1029. doi: 10.1016/j.eururo.2017.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Lupo JM, Chen AP, Zierhut ML, Bok RA, Cunningham CH, Kurhanewicz J, Vigneron DB, Nelson SJ (2010) Analysis of hyperpolarized dynamic 13C lactate imaging in a transgenic mouse model of prostate cancer. Magnetic Resonance Imaging 28 (2):153–162. doi: 10.1016/j.mri.2009.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Chen H-Y, Larson PE, Bok RA, von Morze C, Sriram R, Santos RD, Santos JD, Gordon JW, Bahrami N, Ferrone M (2017) Assessing prostate cancer aggressiveness with hyperpolarized dual-agent 3D dynamic imaging of metabolism and perfusion. Cancer Res:canres. 2083. doi: 10.1158/0008-5472.CAN-16-2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Lee JE, Diederich CJ, Bok R, Sriram R, Santos RD, Noworolski SM, Salgaonkar VA, Adams MS, Vigneron DB, Kurhanewicz J (2018) Assessing high-intensity focused ultrasound treatment of prostate cancer with hyperpolarized 13C dual-agent imaging of metabolism and perfusion. NMR in Biomedicine:e3962. doi: 10.1002/nbm.3962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Tee SS, Suster I, Truong S, Jeong S, Eskandari R, DiGialleonardo V, Alvarez JA, Aldeborgh HN, Keshari KR (2018) Targeted AKT Inhibition in Prostate Cancer Cells and Spheroids Reduces Aerobic Glycolysis and Generation of Hyperpolarized [1-(13)C] Lactate. Mol Cancer Res 16 (3):453–460. doi: 10.1158/1541-7786.MCR-17-0458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Dafni H, Larson PE, Hu S, Yoshihara HA, Ward CS, Venkatesh HS, Wang C, Zhang X, Vigneron DB, Ronen SM (2010) Hyperpolarized 13C spectroscopic imaging informs on hypoxia-inducible factor-1 and myc activity downstream of platelet-derived growth factor receptor. Cancer Res:0008–5472. CAN-0010–0883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Keshari KR, Wilson DM, Van Criekinge M, Sriram R, Koelsch BL, Wang ZJ, VanBrocklin HF, Peehl DM, O’brien T, Sampath D (2015) Metabolic response of prostate cancer to nicotinamide phophoribosyltransferase inhibition in a hyperpolarized MR/PET compatible bioreactor. The Prostate 75 (14):1601–1609. doi: 10.1002/pros.23036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Scroggins BT, Matsuo M, White AO, Saito K, Munasinghe JP, Sourbier C, Yamamoto K, Diaz V, Takakusagi Y, Ichikawa K (2018) Hyperpolarized [1–13C]-pyruvate magnetic resonance spectroscopic imaging of prostate cancer in vivo predicts efficacy of targeting the Warburg effect. Clinical Cancer Research. doi: 10.1158/1078-0432.CCR-17-1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Lodi A, Woods SM, Ronen SM (2013) Treatment with the MEK inhibitor U0126 induces decreased hyperpolarized pyruvate to lactate conversion in breast, but not prostate, cancer cells. NMR in Biomedicine 26 (3):299–306. doi: 10.1002/nbm.2848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Canapè C, Catanzaro G, Terreno E, Karlsson M, Lerche MH, Jensen PR (2015) Probing treatment response of glutaminolytic prostate cancer cells to natural drugs with hyperpolarized [5–13C] glutamine. Magnetic Resonance in Medicine 73 (6):2296–2305. doi: 10.1002/mrm.25360 [DOI] [PubMed] [Google Scholar]
- 305.Keshari KR, Sai V, Wang ZJ, VanBrocklin HF, Kurhanewicz J, Wilson DM (2013) Hyperpolarized [1–13C] dehydroascorbate MR spectroscopy in a murine model of prostate cancer: comparison with 18F-FDG PET. Journal of Nuclear Medicine 54 (6):922–928. doi: 10.2967/jnumed.112.115402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Keshari KR, Wilson DM, Chen AP, Bok R, Larson PE, Hu S, Criekinge MV, Macdonald JM, Vigneron DB, Kurhanewicz J (2009) Hyperpolarized [2–13C]-fructose: a hemiketal DNP substrate for in vivo metabolic imaging. Journal of the American Chemical Society 131 (48):17591–17596. doi: 10.1021/ja9049355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Christensen CE, Karlsson M, Winther JR, Jensen PR, Lerche MH (2014) Non-invasive in-cell determination of free cytosolic [NAD+]/[NADH] ratios using hyperpolarized glucose show large variations in metabolic phenotypes. Journal of Biological Chemistry 289 (4):2344–2352. doi: 10.1074/jbc.M113.498626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Wilson DM, Keshari KR, Larson PE, Chen AP, Hu S, Van Criekinge M, Bok R, Nelson SJ, Macdonald JM, Vigneron DB (2010) Multi-compound polarization by DNP allows simultaneous assessment of multiple enzymatic activities in vivo. Journal of Magnetic Resonance 205 (1):141–147. doi: 10.1016/j.jmr.2010.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Gambhir SS (2002) Molecular imaging of cancer with positron emission tomography. Nature Reviews Cancer 2 (9):683. [DOI] [PubMed] [Google Scholar]
- 310.Hara T, Kosaka N, Kishi H (1998) PET imaging of prostate cancer using carbon-11-choline. Journal of Nuclear Medicine 39 (6):990–995 [PubMed] [Google Scholar]
- 311.Kotzerke J, Prang J, Neumaier B, Volkmer B, Guhlmann A, Kleinschmidt K, Hautmann R, Reske SN (2000) Experience with carbon-11 choline positron emission tomography in prostate carcinoma. European Journal of Nuclear Medicine 27 (9):1415–1419. doi: 10.1007/s002590000309 [DOI] [PubMed] [Google Scholar]
- 312.Seltzer MA, Jahan SA, Sparks R, Stout DB, Satyamurthy N, Dahlbom M, Phelps ME, Barrio JR (2004) Radiation dose estimates in humans for 11C-acetate whole-body PET. Journal of Nuclear Medicine 45 (7):1233–1236 [PubMed] [Google Scholar]
- 313.Savir-Baruch B, Zanoni L, Schuster DM (2018) Imaging of prostate cancer using fluciclovine. Urologic Clinics of North America 45 (3):489–502. doi: 10.1016/j.ucl.2018.03.015 [DOI] [PubMed] [Google Scholar]
- 314.Cardinale J, Schäfer M, Benešová M, Bauder-Wüst U, Leotta K, Eder M, Neels OC, Haberkorn U, Giesel FL, Kopka K (2017) Preclinical evaluation of 18F-PSMA-1007, a new prostate-specific membrane antigen ligand for prostate cancer imaging. Journal of Nuclear Medicine 58 (3):425–431. doi: 10.2967/jnumed.116.181768 [DOI] [PubMed] [Google Scholar]
- 315.Liu R, Cao J, Gao X, Zhang J, Wang L, Wang B, Guo L, Hu X, Wang Z (2016) Overall survival of cancer patients with serum lactate dehydrogenase greater than 1000 IU/L. Tumour Biol 37 (10):14083–14088. doi: 10.1007/s13277-016-5228-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Halabi S, Small EJ, Kantoff PW, Kattan MW, Kaplan EB, Dawson NA, Levine EG, Blumenstein BA, Vogelzang NJ (2003) Prognostic model for predicting survival in men with hormone-refractory metastatic prostate cancer. J Clin Oncol 21 (7):1232–1237. doi: 10.1200/JCO.2003.06.100 [DOI] [PubMed] [Google Scholar]
- 317.Scher HI, Heller G, Molina A, Attard G, Danila DC, Jia X, Peng W, Sandhu SK, Olmos D, Riisnaes R, McCormack R, Burzykowski T, Kheoh T, Fleisher M, Buyse M, de Bono JS (2015) Circulating tumor cell biomarker panel as an individual-level surrogate for survival in metastatic castration-resistant prostate cancer. J Clin Oncol 33 (12):1348–1355. doi: 10.1200/JCO.2014.55.3487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Sartor O, Coleman RE, Nilsson S, Heinrich D, Helle SI, O’Sullivan JM, Vogelzang NJ, Bruland O, Kobina S, Wilhelm S, Xu L, Shan M, Kattan MW, Parker C (2017) An exploratory analysis of alkaline phosphatase, lactate dehydrogenase, and prostate-specific antigen dynamics in the phase 3 ALSYMPCA trial with radium-223. Ann Oncol 28 (5):1090–1097. doi: 10.1093/annonc/mdx044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Euceda LR, Andersen MK, Tessem M-B, Moestue SA, Grinde MT, Bathen TF (2018) NMR-Based Prostate Cancer Metabolomics. In: Prostate Cancer. Springer, pp 237–257 [DOI] [PubMed] [Google Scholar]
- 320.Reo NV (2002) NMR-based metabolomics. Drug and Chemical Toxicology 25 (4):375–382. doi: 10.1081/DCT-120014789 [DOI] [PubMed] [Google Scholar]
- 321.Markley JL, Brüschweiler R, Edison AS, Eghbalnia HR, Powers R, Raftery D, Wishart DS (2017) The future of NMR-based metabolomics. Current Opinion in Biotechnology 43:34–40. doi: 10.1016/j.copbio.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Kim HK, Choi YH, Verpoorte R (2010) NMR-based metabolomic analysis of plants. Nature Protocols 5 (3):536. [DOI] [PubMed] [Google Scholar]
- 323.García-Martín ML, Adrados M, Ortega M, Fernández González I, López-Larrubia P, Viano J, García-Segura J (2011) Quantitative 1H MR spectroscopic imaging of the prostate gland using LCModel and a dedicated basis-set: Correlation with histologic findings. Magnetic Resonance in Medicine 65 (2):329–339. doi: 10.1002/mrm.22631 [DOI] [PubMed] [Google Scholar]
- 324.Serkova NJ, Gamito EJ, Jones RH, O’donnell C, Brown JL, Green S, Sullivan H, Hedlund T, Crawford ED (2008) The metabolites citrate, myo-inositol, and spermine are potential age-independent markers of prostate cancer in human expressed prostatic secretions. The Prostate 68 (6):620–628. doi: 10.1002/pros.20727 [DOI] [PubMed] [Google Scholar]
- 325.Tessem MB, Swanson MG, Keshari KR, Albers MJ, Joun D, Tabatabai ZL, Simko JP, Shinohara K, Nelson SJ, Vigneron DB (2008) Evaluation of lactate and alanine as metabolic biomarkers of prostate cancer using 1H HR-MAS spectroscopy of biopsy tissues. Magnetic Resonance in Medicine 60 (3):510–516. doi: 10.1002/mrm.21694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Kumar D, Gupta A, Mandhani A, Sankhwar SN (2015) Metabolomics-Derived Prostate Cancer Biomarkers: Fact or Fiction? Journal of Proteome Research 14 (3):1455–1464. doi: 10.1021/pr5011108 [DOI] [PubMed] [Google Scholar]
- 327.Swanson MG, Keshari KR, Tabatabai ZL, Simko JP, Shinohara K, Carroll PR, Zektzer AS, Kurhanewicz J (2008) Quantification of choline- and ethanolamine-containing metabolites in human prostate tissues using 1H HR-MAS total correlation spectroscopy. Magnetic Resonance in Medicine 60 (1):33–40. doi: 10.1002/mrm.21647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Komoroski RA, Holder JC, Pappas AA, Finkbeiner AE (2011) 31P NMR of phospholipid metabolites in prostate cancer and benign prostatic hyperplasia. Magnetic Resonance in Medicine 65 (4):911–913. doi:doi: 10.1002/mrm.22677 [DOI] [PubMed] [Google Scholar]
- 329.Zadra G, Loda M (2018) Metabolic Vulnerabilities of Prostate Cancer: Diagnostic and Therapeutic Opportunities. Cold Spring Harbor Perspectives in Medicine 8 (10). doi: 10.1101/cshperspect.a030569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Cajka T, Fiehn O (2015) Toward merging untargeted and targeted methods in mass spectrometry-based metabolomics and lipidomics. Analytical Chemistry 88 (1):524–545. doi: 10.1021/acs.analchem.5b04491 [DOI] [PubMed] [Google Scholar]
- 331.Wishart DS (2016) Emerging applications of metabolomics in drug discovery and precision medicine. Nature Reviews Drug Discovery 15 (7):473. [DOI] [PubMed] [Google Scholar]
- 332.Xu Y-F, Lu W, Rabinowitz JD (2015) Avoiding misannotation of in-source fragmentation products as cellular metabolites in liquid chromatography–mass spectrometry-based metabolomics. Analytical Chemistry 87 (4):2273–2281. doi: 10.1021/ac504118y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Kitteringham NR, Jenkins RE, Lane CS, Elliott VL, Park BK (2009) Multiple reaction monitoring for quantitative biomarker analysis in proteomics and metabolomics. Journal of Chromatography B 877 (13):1229–1239 [DOI] [PubMed] [Google Scholar]
- 334.Thapar R, Titus MA (2014) Recent advances in metabolic profiling and imaging of prostate cancer. Current Metabolomics 2 (1):53–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Guo B, Chen B, Liu A, Zhu W, Yao S (2012) Liquid chromatography-mass spectrometric multiple reaction monitoring-based strategies for expanding targeted profiling towards quantitative metabolomics. Current drug metabolism 13 (9):1226–1243 [DOI] [PubMed] [Google Scholar]
- 336.Zadra G, Photopoulos C, Loda M (2013) The fat side of prostate cancer. Biochim Biophys Acta 1831 (10):1518–1532. doi: 10.1016/j.bbalip.2013.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Whitburn J, Edwards CM, Sooriakumaran P (2017) Metformin and Prostate Cancer: a New Role for an Old Drug. Current Urology Reports 18:1–7. doi: 10.1007/s11934-017-0693-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Richards KA, Liou Ji, Cryns VL, Downs TM, Abel EJ, Jarrard DF (2018) Metformin Use is Associated with Improved Survival for Patients with Advanced Prostate Cancer on Androgen Deprivation Therapy. Journal of Urology 200:1256–1263. doi: 10.1016/j.juro.2018.06.031 [DOI] [PubMed] [Google Scholar]
- 339.Allott EH, Abern MR, Gerber L, Keto CJ, Aronson WJ, Terris MK, Kane CJ, Amling CL, Cooperberg MR, Moorman PG, Freedland SJ (2013) Metformin does not affect risk of biochemical recurrence following radical prostatectomy: results from the SEARCH database. Prostate Cancer Prostatic Dis 16 (4):391–397. doi: 10.1038/pcan.2013.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Rieken M, Xylinas E, Kluth L, Crivelli JJ, Chrystal J, Faison T, Lotan Y, Karakiewicz PI, Fajkovic H, Babjuk M, Kautzky-Willer A, Bachmann A, Scherr DS, Shariat SF (2013) Association of diabetes mellitus and metformin use with oncological outcomes of patients with non-muscle-invasive bladder cancer. BJU Int 112 (8):1105–1112. doi: 10.1111/bju.12448 [DOI] [PubMed] [Google Scholar]
- 341.Wang SY, Chuang CS, Muo CH, Tu ST, Lin MC, Sung FC, Kao CH (2013) Metformin and the incidence of cancer in patients with diabetes: a nested case-control study. Diabetes Care 36 (9):e155–156. doi: 10.2337/dc13-0708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Soranna D, Scotti L, Zambon A, Bosetti C, Grassi G, Catapano A, La Vecchia C, Mancia G, Corrao G (2012) Cancer risk associated with use of metformin and sulfonylurea in type 2 diabetes: a meta-analysis. Oncologist 17 (6):813–822. doi: 10.1634/theoncologist.2011-0462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Azoulay L, Dell’Aniello S, Gagnon B, Pollak M, Suissa S (2011) Metformin and the incidence of prostate cancer in patients with type 2 diabetes. Cancer Epidemiol Biomarkers Prev 20 (2):337–344. doi: 10.1158/1055-9965.EPI-10-0940 [DOI] [PubMed] [Google Scholar]
- 344.Bensimon L, Yin H, Suissa S, Pollak MN, Azoulay L (2014) The use of metformin in patients with prostate cancer and the risk of death. Cancer Epidemiol Biomarkers Prev 23 (10):2111–2118. doi: 10.1158/1055-9965.EPI-14-0056 [DOI] [PubMed] [Google Scholar]
- 345.Currie CJ, Poole CD, Gale EA (2009) The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 52 (9):1766–1777. doi: 10.1007/s00125-009-1440-6 [DOI] [PubMed] [Google Scholar]
- 346.Kaushik D, Karnes RJ, Eisenberg MS, Rangel LJ, Carlson RE, Bergstralh EJ (2014) Effect of metformin on prostate cancer outcomes after radical prostatectomy. Urol Oncol 32 (1):43 e41–47. doi: 10.1016/j.urolonc.2013.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Margel D, Urbach D, Lipscombe LL, Bell CM, Kulkarni G, Austin PC, Fleshner N (2013) Association between metformin use and risk of prostate cancer and its grade. J Natl Cancer Inst 105 (15):1123–1131. doi: 10.1093/jnci/djt170 [DOI] [PubMed] [Google Scholar]
- 348.Patel T, Hruby G, Badani K, Abate-Shen C, McKiernan JM (2010) Clinical outcomes after radical prostatectomy in diabetic patients treated with metformin. Urology 76 (5):1240–1244. doi: 10.1016/j.urology.2010.03.059 [DOI] [PubMed] [Google Scholar]
- 349.Tsilidis KK, Capothanassi D, Allen NE, Rizos EC, Lopez DS, van Veldhoven K, Sacerdote C, Ashby D, Vineis P, Tzoulaki I, Ioannidis JP (2014) Metformin does not affect cancer risk: a cohort study in the U.K. Clinical Practice Research Datalink analyzed like an intention-to-treat trial. Diabetes Care 37 (9):2522–2532. doi: 10.2337/dc14-0584 [DOI] [PubMed] [Google Scholar]
- 350.Feng T, Sun X, Howard LE, Vidal AC, Gaines AR, Moreira DM, Castro-Santamaria R, Andriole GL, Freedland SJ (2015) Metformin use and risk of prostate cancer: results from the REDUCE study. Cancer Prev Res (Phila) 8 (11):1055–1060. doi: 10.1158/1940-6207.CAPR-15-0141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Joshua AM, Zannella VE, Downes MR, Bowes B, Hersey K, Koritzinsky M, Schwab M, Hofmann U, Evans A, van der Kwast T, Trachtenberg J, Finelli A, Fleshner N, Sweet J, Pollak M (2014) A pilot ‘window of opportunity’ neoadjuvant study of metformin in localised prostate cancer. Prostate Cancer Prostatic Dis 17 (3):252–258. doi: 10.1038/pcan.2014.20 [DOI] [PubMed] [Google Scholar]
- 352.Medicine UNLo (2015) ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01433913.
- 353.Vancura A, Bu P, Bhagwat M, Zeng J, Vancurova I (2018) Metformin as an Anticancer Agent. Trends in Pharmacological Sciences 39:867–878. doi: 10.1016/j.tips.2018.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Liu X, Romero IL, Litchfield LM, Lengyel E, Locasale JW (2016) Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell Metabolism 24:728–739. doi: 10.1016/j.cmet.2016.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Zingales V, Distefano A, Raffaele M, Zanghi A, Barbagallo I, Vanella L (2017) Metformin: A Bridge between Diabetes and Prostate Cancer. Frontiers in Oncology 7:1–7. doi: 10.3389/fonc.2017.00243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS (2018) Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clinical cancer research : an official journal of the American Association for Cancer Research 24:2482–2490. doi: 10.1158/1078-0432.CCR-17-3070 [DOI] [PubMed] [Google Scholar]
- 357.Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG (2015) Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162 (3):552–563. doi: 10.1016/j.cell.2015.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM (2015) An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162 (3):540–551. doi: 10.1016/j.cell.2015.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.DeBerardinis RJ, Chandel NS (2016) Fundamentals of cancer metabolism. Science Advances 2:e1600200. doi: 10.1126/sciadv.1600200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, McAfoos T, Morlacchi P, Ackroyd J, Agip A-nA, Al-Atrash G, Asara J, Bardenhagen J, Carrillo CC, Carroll C, Chang E, Ciurea S, Cross JB, Czako B, Deem A, Daver N, de Groot JF, Dong J-w, Feng N, Gao G, Gay J, Do MG, Greer J, Giuliani V, Han J, Han L, Henry VK, Hirst J, Huang S, Jiang Y, Kang Z, Khor T, Konoplev S, Lin Y-h, Liu G, Lodi A, Lofton T, Ma H, Mahendra M, Matre P, Mullinax R, Peoples M, Petrocchi A, Rodriguez-Canale J, Serreli R, Shi T, Smith M, Tabe Y, Theroff J, Tiziani S, Xu Q, Zhang Q, Muller F, DePinho RA, Toniatti C, Draetta GF, Heffernan TP, Konopleva M, Jones P, Di Francesco ME, Marszalek JR (2018) An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nature Medicine 24:1036–1046. doi: 10.1038/s41591-018-0052-4 [DOI] [PubMed] [Google Scholar]
- 361.Bader DA, Hartig SM, Putluri V, Foley C, Hamilton MP, Smith EA, Saha PK, Panigrahi A, Walker C, Zong L, Martini-Stoica H, Chen R, Rajapakshe K, Coarfa C, Sreekumar A, Mitsiades N, Bankson JA, Ittmann MM, O’Malley BW, Putluri N, McGuire SE (2018) Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nature Metabolism 1:70–85. doi: 10.1038/s42255-018-0002-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Wang Q, Hardie RA, Hoy AJ, Van Geldermalsen M, Gao D, Fazli L, Sadowski MC, Balaban S, Schreuder M, Nagarajah R, Wong JJL, Metierre C, Pinello N, Otte NJ, Lehman ML, Gleave M, Nelson CC, Bailey CG, Ritchie W, Rasko JE, Holst J (2015) Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. Journal of Pathology 236:278–289. doi: 10.1002/path.4518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Yang C, Ko B, Hensley CT, Jiang L, Wasti AT, Kim J, Sudderth J, Calvaruso MA, Lumata L, Mitsche M, Rutter J, Merritt ME, DeBerardinis RJ (2014) Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Molecular Cell 56:414–424. doi: 10.1016/j.molcel.2014.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Adams C, Richmond RC, Santos Ferreira DL, Spiller W, Tan VY, Zheng J, Wurtz P, Donovan JL, Hamdy FC, Neal DE, Lane JA, Davey Smith G, Relton CL, Eeles RA, Henderson BE, Haiman CA, Kote-Jarai Z, Schumacher FR, Amin Al Olama A, Benlloch S, Muir K, Berndt SI, Conti DV, Wiklund F, Chanock SJ, Gapstur SM, Stevens VL, Tangen CM, Batra J, Clements JA, Gronberg H, Pashayan N, Schleutker J, Albanes D, Wolk A, West CML, Mucci LA, Cancel-Tassin G, Koutros S, Sorensen KD, Maehle L, Travis RC, Hamilton R, Ingles SA, Rosenstein BS, Lu YJ, Giles GG, Kibel AS, Vega A, Kogevinas M, Penney KL, Park JY, Stanford JL, Cybulski C, Nordestgaard BG, Brenner H, Maier C, Kim J, John EM, Teixeira MR, Neuhausen SL, DeRuyck K, Razack A, Newcomb LF, Lessel D, Kaneva RP, Usmani N, Claessens F, Townsend P, Gago Dominguez M, Roobol MJ, Menegaux F, Khaw KT, Cannon-Albright LA, Pandha H, Thibodeau SN, Martin RM (2018) Circulating Metabolic Biomarkers of Screen-Detected Prostate Cancer in the ProtecT Study. Cancer Epidemiol Biomarkers Prev. doi: 10.1158/1055-9965.EPI-18-0079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Huang J, Mondul AM, Weinstein SJ, Koutros S, Derkach A, Karoly E, Sampson JN, Moore SC, Berndt SI, Albanes D (2016) Serum metabolomic profiling of prostate cancer risk in the prostate, lung, colorectal, and ovarian cancer screening trial. Br J Cancer 115 (9):1087–1095. doi: 10.1038/bjc.2016.305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Mondul AM, Moore SC, Weinstein SJ, Karoly ED, Sampson JN, Albanes D (2015) Metabolomic analysis of prostate cancer risk in a prospective cohort: The alpha-tocolpherol, beta-carotene cancer prevention (ATBC) study. Int J Cancer 137 (9):2124–2132. doi: 10.1002/ijc.29576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Mondul AM, Moore SC, Weinstein SJ, Mannisto S, Sampson JN, Albanes D (2014) 1-stearoylglycerol is associated with risk of prostate cancer: results from serum metabolomic profiling. Metabolomics 10 (5):1036–1041. doi: 10.1007/s11306-014-0643-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Mayers JR, Vander Heiden MG (2015) Famine versus feast: understanding the metabolism of tumors in vivo. Trends Biochem Sci 40 (3):130–140. doi: 10.1016/j.tibs.2015.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, Gui DY, Sullivan LB, Wasylenko TM, Subbaraj L, Chin CR, Stephanopolous G, Mott BT, Jacks T, Clish CB, Vander Heiden MG (2016) Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab 23 (3):517–528. doi: 10.1016/j.cmet.2016.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Reisz JA, D’Alessandro A (2017) Measurement of metabolic fluxes using stable isotope tracers in whole animals and human patients. Curr Opin Clin Nutr Metab Care 20 (5):366–374. doi: 10.1097/MCO.0000000000000393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, Li H, Huet G, Yuan Q, Wigal T, Butt Y, Ni M, Torrealba J, Oliver D, Lenkinski RE, Malloy CR, Wachsmann JW, Young JD, Kernstine K, DeBerardinis RJ (2017) Lactate Metabolism in Human Lung Tumors. Cell 171 (2):358–371 e359. doi: 10.1016/j.cell.2017.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, Wodzak M, Klimko C, McMillan E, Butt Y, Ni M, Oliver D, Torrealba J, Malloy CR, Kernstine K, Lenkinski RE, DeBerardinis RJ (2016) Metabolic Heterogeneity in Human Lung Tumors. Cell 164 (4):681–694. doi: 10.1016/j.cell.2015.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Davies AH, Beltran H, Zoubeidi A (2018) Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat Rev Urol 15 (5):271–286. doi: 10.1038/nrurol.2018.22 [DOI] [PubMed] [Google Scholar]