

Abstract
Adenosine (ADO) is an essential biomolecule for life that provides critical regulation of energy utilization and homeostasis. Adenosine kinase (ADK) is an evolutionary ancient ribokinase derived from bacterial sugar kinases that is widely expressed in all forms of life, tissues and organ systems that tightly regulates intracellular and extracellular ADO concentrations. The facile ability of ADK to alter ADO availability provides a “site and event” specificity to the endogenous protective effects of ADO in situations of cellular stress. In addition to modulating the ability of ADO to activate its cognate receptors (P1 receptors), nuclear ADK isoform activity has been linked to epigenetic mechanisms based on transmethylation pathways. Previous drug discovery research has targeted ADK inhibition as a therapeutic approach to manage epilepsy, pain, and inflammation. These efforts generated multiple classes of highly potent and selective inhibitors. However, clinical development of early ADK inhibitors was stopped due to apparent mechanistic toxicity and the lack of suitable translational markers. New insights regarding the potential role of the nuclear ADK isoform (ADK-Long) in the epigenetic modulation of maladaptive DNA methylation offers the possibility of identifying novel ADK-isoform selective inhibitors and new interventional strategies that are independent of ADO receptor activation.
Keywords: Adenosine, Adenosine Kinase, Analgesia, Seizures, Inflammation, Motor Activity
Graphical Abstract
1. Introduction – Evolutionary Background
This review is dedicated to the memory of Professor Geoffrey Burnstock who discovered non-adrenergic, non-cholinergic (NANC) neurotransmission, and through his creativity, persistence, and scientific acumen, pioneered the realization that purine and pyrimidine nucleotides could function as extracellular messengers [1]. Geoff’s impact on the study of purinergic pharmacology cannot be overstated. His consistent open-minded scientific curiosity and excitement for new ideas are hallmarks of his six decades of scientific innovation. Geoff inspired and mentored hundreds of purine investigators. Their collective research efforts have not only greatly advanced our knowledge of the P1 and P2 receptor superfamilies but also led to the development of important new drug candidates [2].
The purine ribonucleoside adenosine (ADO) is a ubiquitous molecule at the core of fundamental functions of life. It is not only a part of RNA, but also of the energy metabolites ATP, ADP, and AMP, second messenger systems such as cAMP, and a variety of biomolecules including S-adenosylhomocysteine (SAH), S-adenosylmethionine (SAM), and the adenine-containing co-enzymes NAD and FAD. Based on experiments aimed at re-constructing conditions on our early planet leading to the formation of the “primordial soup” as the basis for the origin of life, it is likely that adenine (H5C5N5), created by the fusion of 5 molecules of hydrogen cyanide (HCN), was one of the first biomolecules playing a key role for the creation of life [3, 4]. One of the most important requirements for the formation of the first forms of life was most likely the development of a simple system to adjust energy consumption to energy supplies. In that sense ADO has been termed a “retaliatory metabolite” [5]. A situation of energy crisis, which can be as simple as a lack of nutrients or oxygen, or more complex, such as excessive energy consumption during a seizure, leads to the degradation of ATP into ADO, which – from an evolutionary perspective – is a rational choice to be used as a ubiquitous mediator to globally suppress mechanisms that consume energy, or to enhance those that conserve energy. Hence, the tight regulation and control of ADO levels is of crucial importance for the energy homeostasis of all living systems. ADK from Leishmania was found to be inactivated and activated by an aggregation/deaggregation cycle, with the inactive aggregate stabilized by ADP and the active monomer stabilized by a cyclophilin [6]. This mechanism is in line with the need to reduce ADK activity in situations of higher energy demand. Interestingly, ADK is also inhibited by its own substrate adenosine, a mechanism linked to the availability of K+. Thereby, ADK is inhibited by depletion of intracellular K+ and by higher concentrations of intracellular adenosine [7]. This means, the enzyme is able to modulate its activity based on sensing the energy status of a cell. It is now widely established that the high affinity, low capacity enzyme adenosine kinase (ADK: EC 2.7.1.20) plays a key role in the regulation and metabolic clearance of ADO [8–10]. ADK is an evolutionary ancient ribokinase derived from bacterial sugar kinases and likely assumed an early role in energy homeostasis. [11, 12]. Therefore it is no surprise that ADK is widely expressed in all forms of life, tissues and organ systems, which have been investigated to date [13]. Biochemically, ADK recycles and removes ADO by phosphorylation to form AMP. Thus, high levels of ADK lead to a reduction in the ambient levels of ADO and reduced activation of adenosine receptors (Figure 1). Conversely, blocking ADK through pharmacological agents or reducing or eliminating its expression through genetic means leads to an increase in ADO. The Adk gene in humans has a respectable size of 546 kb and is one of the largest genes known [14, 15]. Through alternative promoter use and splicing it produces two main transcripts encoding a cytoplasmic isoform ADK-S and a nuclear isoform ADK-L with a size of 38.7 and 40.5 kDa, respectively [16].
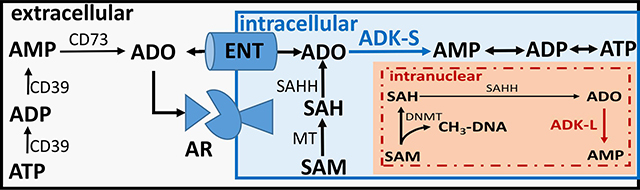
Figure 1: Biochemical pathways controlled by ADK.

Simplified scheme showing compartmentalization of the ADO system and major pathways controlled by ADK (for a comprehensive description of ADO metabolizing pathways, please see [7]). The main route for the extracellular production of ADO is degradation of ATP through a system of ectonucleotidases, with CD39 and CD73 shown here. Extra- and intracellular levels of ADO are equilibrated through equilibrative nucleoside transporters (ENT). Therefore, intracellular ADO metabolism through ADK can control adenosine receptor (AR) activation. In the intracellular compartment the ADO-ADK system is linked to S-adenosylmethionine (SAM) dependent methyltransferase (MT) reactions, which yield S-adenosylhomocysteine (SAH), which is a substrate for S-adenosylhomocysteine hydrolase (SAHH). By removing adenosine ADK-S in the cytoplasm and ADK-L in the nucleus drives the flux of methyl groups through the transmethylation pathway. In the intranuclear compartment ADK-L activity is needed to maintain DNA methyltransferase (DNMT) activity and DNA methylation.
2. Biology of Adenosine Kinase – Lessons from Knockouts and Human Mutations
Adenosine kinase is an essential enzyme and loss of function has severe consequences. The genetic knockout of the Adk gene in mice resulted in perinatal lethality, microvesicular hepatic steatosis, and stunted growth [17]. Further investigation of Adk-null mice led to the discovery that ADK plays an important role for the metabolic clearance of ADO, which is produced through SAH-dependent transmethylation reactions; importantly, metabolic clearance of ADO is needed to maintain transmethylation (e.g. to produce methylated lipids in the liver) (Figure 1). Consequently, the lack of ADK leads to a block in transmethylation pathways and the accumulation of non-methylated lipids in the liver [17]. Remarkably, an independent study published in the same year, revealed an almost identical biochemical phenotype in Arabidopsis engineered to lack ADK. In line with ADK knockout mice, those ADK deficient plants were characterized by stunted growth and similar transmethylation defects [18]. More recently, a few pedigrees of human patients with inborn Adk-deficiency were identified [19–21]. Again, a characteristic biochemical hallmark of those human Adk mutations was defects in transmethylation reactions associated with stunted growth, hepatic encephalopathy, developmental delay and psychomotor perturbation as well as seizures in some subjects [19, 20]. Together, those genetic data support a novel role of ADK as regulator of transmethylation reactions. The suggestion was made for clinicians to consider ADK deficiency in any neonate presenting with global developmental delay, hypotonia, dysmorphic features, and high methionine levels [21].
On the other hand, it has been shown that an excess of ADK is likewise detrimental. Maladaptive increases of ADK expression in conjunction with astrocyte activation have been described in epilepsy, Alzheimer’s and Parkinson’s disease, as well as in amyotrophic lateral sclerosis [22–26]. Overexpression of ADK in the brain has been associated with increased brain injury following a stroke and was shown to be sufficient to trigger spontaneous electrographic seizures [26–28]. Those studies have demonstrated that pathological increases of ADK in the brain reduce the thresholds for neuronal injury and seizures. More importantly, maladaptive changes in ADK expression following a precipitating challenge to the brain play a causal role in the development of epilepsy [26, 29]. Specifically, inflammatory processes in the brain lead to astrocyte activation and pathological overexpression of the astrocyte based enzyme ADK, resulting in progressive ADO deficiency and the development of epilepsy [26, 29]. Mechanistically, increased ADK expression drives increased DNA methylation as a critical component driving the epileptogenic cascade [29]. In line with the early evolutionary role of ADO to conserve energy, ADO is also a key regulator of sleep homeostasis [30]. In line with this notion, changes in ADK expression were shown to affect sleep. Mice with transgenic overexpression of ADK spent less time asleep compared to wild-type mice [31], whereas mice with an astrocyte-selective knockout of ADK exhibited enhanced expression of the ADO-mediated homeostatic sleep drive [32]. In addition, to brain pathologies, the equilibrium of ADK expression has become of growing interest for conditions as varied as diabetes [33], vascular diseases [34–36], and cancer [9, 37]. Because of the physiological relevance of ADK in a wide range of pathologies, it has become an important target for drug development.
3. Adenosine Kinase as a Drug Target
The ability of ADO to serve as a protective modulator of energy requirements in cells and tissues undergoing stress or trauma has been well documented [38–40]. Interventional strategies based on the nearly ubiquitous protective actions of ADO have driven the development of potent and selective P1 receptor agonists and enzymatic inhibitors along the purinergic cascade [10, 39, 41]. Given that the availability of intracellular and extracellular ADO concentrations is tightly regulated [42, 43], the concept of “site and event specificity” described the situational localization of ADO actions [44]. This concept was originally based on the ability of AICA riboside (5-amino-1-β-D-ribofuranosyl-imidazole-4-carboxamide, acadesine) to selectively enhance extracellular ADO levels in ischemic but not normal heart tissue [44]. Since adenosine kinase inhibition more effectively decreases cellular reuptake of ADO and thereby increases the local concentration of extracellular ADO in damaged as compared to normal tissues [43, 45], adenosine kinase inhibition was also hypothesized to function as a site and event specific ADO modulator as well [39, 46].
From a drug discovery perspective, the concept of adenosine kinase inhibition as a site and event specific therapeutic intervention was especially attractive given the reciprocal mechanism-based side effects associated with ADO receptor subtype agonists targeting either central nervous system or peripheral diseases [39]. Data supporting the selective beneficial actions of adenosine kinase inhibition were obtained in hippocampal and spinal cord slice preparations [47, 48] and in experimental models of inflammation [49]. In vivo microdialysis studies also showed that adenosine kinase inhibition could selectively enhance excitotoxic-evoked ADO release without altering basal ADO concentrations in rat striatum [50].
4. Evolution of Selective Adenosine Kinase Inhibitors
The first described ADK inhibitors were ribonucleosides including the prototypic inhibitors, 5’-deoxy-5’amino ADO (NH2d-ADO), 5-iodotubercidin (5-IT) and 5’d-5IT. (Figure 2). Rational drug design and optimization of high-throughput screening hits have resulted in the generation of numerous classes of potent nucleoside and non-nucleoside ADK inhibitors (Figure 2) [51–55]. The majority of these newer chemotypes (e.g. GP 3269 and ABT-702) are highly potent, selective and have improved cellular penetration compared to the early prototypic ADK inhibitors like NH2d-ADO [54, 55]. Both nucleoside and non-nucleoside ADK inhibitors are reversible and competitive blockers of the ADO recognition site and noncompetitively interact with the MgATP2− site [56, 57]. Multiple classes of potent ADK inhibitors block the long and short forms of ADK (ADK-S and ADK_L) with similar affinity and have equivalent potency across multiple mammalian species [56, 57]. Crystal structure determinations of ADK complexed with 5-IT and a structurally novel alkynylpyrimidine ADK inhibitor [58] revealed distinct binding modes for these inhibitors and significant rearrangement of the protein active site in the presence of these structurally different ADK inhibitors [59]. While much of research on the optimization of new ADK inhibitor scaffolds occurred in the late 1990s [10], there has been a renewed medicinal chemistry research to discover novel ADK inhibitors in recent years [60, 61].
Figure 2: Chemical structures of adenosine and potent nucleoside and non-nucleoside adenosine kinase inhibitors.

Early ADK inhibitors include NH2-dADO, 5-IT and 5’d-5IT [48]. Detailed structure-activity studies based on these molecules and optimization of novel chemotypes identified from high-throughput screening campaigns have led to the generation of the diverse array of ADK inhibitors shown here [45–55].
5. Neuroprotective Effects of ADK Inhibition
Consistent with the known actions of ADO [2, 8, 9, 40], a simple PubMed search shows that over the last three decades, ADK inhibitors have been investigated across a diverse array of model systems targeting central nervous system, cardiovascular, renal and immuno-oncological disorders (Table 1). Much of this research was enabled by the development of the potent and selective ADK inhibitors shown in Figure 2 as important tool compounds in these experimental model systems. Among disease indications, the protective effects of ADK inhibition have been most extensively studied in the context of epilepsy, pain and inflammation [55, 57, 62, 63]. ADK inhibitors dose-dependently attenuate seizures elicited by a variety of chemical and electrical stimuli and nociception induced by a variety of acute noxious events (thermal, pressure, chemical irritant) as well as exposure to chronic inflammation or nerve injury [55, 57, 62, 63]. The consistent ability of ADO receptor antagonists to fully block the protective effects of ADK inhibitors in in vivo disease models supports the hypothesis that ADK inhibition leads to increased local ADO concentrations as an underlying mechanism mediating the effects of ADK inhibitors in vivo [55]. This mechanistic interpretation of ADK inhibitor action on local ADO concentrations was directly demonstrated in microdialysis studies [50].
Table 1.
Protective effects of adenosine kinase inhibition in experimental model systems
| Organ Class | Experimental Model/Endpoint |
|---|---|
| Central Nervous System | Epilepsy – focal and generalized seizures |
| Pain – acute, chronic inflammatory and neuropathic | |
| Traumatic Brain Injury | |
| Neurotoxicity/neurodegeneration | |
| Ischemia | |
| Diabetic retinopathy | |
| Substance Abuse/drug withdrawal and dependence | |
| Psychiatric/schizophrenia | |
| Audition | |
| Sleep | |
| Cardiovascular | Atherosclerosis |
| Hypertension | |
| Myocardial Infarction | |
| Ischemia/reperfusion | |
| Cardiac remodeling | |
| Renal | Acute kidney injury |
| Chemotherapy induced nephrotoxicity | |
| Diabetic renal injury | |
| Immunological/Oncological | Inflammation/ acute and chronic |
| colitis | |
| Host defense | |
| B-cell proliferation | |
| T-cell production | |
| Melanoma |
While the protective effects of ADO receptor agonism have been well documented over the years [39, 41, 64], drug discovery efforts to optimize the benefit/risk profile of P1 agonists as therapeutic interventions have been largely unsuccessful given the ubiquitous nature of ADO signaling in mammalian physiology [64]. As noted above, the demonstrated “site and event” specificity of ADK inhibition offered a viable approach to expand the therapeutic index of modulating ADO for clinical benefit [10]. The potential of ADK inhibitors to reduce chronic inflammatory and neuropathic pain in experimental models at doses devoid of cardiovascular or psychomotor effects was demonstrated for both nucleoside and non-nucleoside ADK inhibitors [56, 65]. Further, the central nervous system sedative effects produced by high doses of ADK inhibitors was shown to be superior to direct-acting P1 receptor agonists and differentiated from classical sedatives like ethanol, benzodiazepines, and barbiturates [56]. However, ADK inhibition produces robust hypothermia in knockout mice lacking all ADO receptor subtypes indicating a complex role of individual P1 receptors in thermal homeostasis [66].
6. ADK Inhibitors as Clinical Candidates for Epilepsy and Pain
During the late 1990s, a nucleoside ADK inhibitor, GP-3269 (Metabasis/Gensia), and a non-nucleoside ADK inhibitor, ABT-702 (Abbott Laboratories) (Figure 2), were advanced into toxicology studies as candidates for the management of epileptic seizures and chronic pain, respectively [10]. Both clinical candidates are orally bioavailable and have appropriate drug-like pharmaceutical properties. However, the clinical development of both ADK inhibitors was independently halted by their respective sponsors due to compound-based and mechanism-based toxicological issues [10, 53, 67]. ABT-702 showed idiosyncratic clastogenic activity that is not representative of other pyridopyrimidine ADK inhibitors [67, 68]. Additionally, GP-3269 and other structurally diverse ADK inhibitors including at least one pyridopyrimidine ADK inhibitor were found to produce brain microhemorrhage foci in rats and dogs in sub-chronic toxicology studies [10, 53]. Further investigation of these toxicological findings revealed that this finding could be reproduced by multiple ADK inhibitor chemotypes, could be blocked by the ADO receptor antagonist, theophylline, and was not detectable using an inactive enantiomer of a potent ADK inhibitor [10]. While the toxicological mechanism(s) associated with systemically administered ADK inhibitors has not been precisely determined, the lack of validated clinical biomarkers or translational predictability of these potential harms led to the discontinuation of these ADK inhibitor development programs [10]. Chronic administration of other ribose-containing ADK inhibitors at analgesic doses have been reported to cause lethality in rats [69]. It is also noteworthy that genetic deletion of the ADK gene resulted in perinatal lethality attributed to deficits in thermoregulation and respiration, as well as liver failure [17].
7. Therapeutic Opportunities - ADK-L as a drug target?
Despite the earlier demonstrations of the reproducible toxicity of different chemical classes of potent ADK inhibitors discussed above, there is renewed interest in ADK inhibition as a therapeutic strategy with a focus on peripheral diseases [8, 9, 70–72]. Data from these newer studies is consistent with the beneficial effects of systemically administered ADK inhibitors in seizure and pain models. However, it is unknown whether targeting a peripheral disease will improve the benefit/risk profile of a small molecule ADK inhibitor or that the toxicological profile of ADK inhibitors is fully understood [10]. The available data from isolated enzymes in in vitro preparations indicate a similar pharmacology (i.e. competitive binding to the ADO recognition site of ADK) for all known ADK inhibitors [6–8].
However, it is important to note that the ADO system and its metabolic clearance through ADK is highly compartmentalized with extracellular, intracellular, and intra-nuclear compartments (Figure 1) [8, 9]. Therefore, specific roles and functions of the two major isoforms of ADK, namely ADK-S, which is located in the cytoplasm, and ADK-L, which is located in the cell nucleus [16], need to be considered. While there is no obvious difference in the kinetic properties between ADK-S and ADK-L [8], the relative contributions of selective inhibition of individual ADK isoforms in increasing extracellular ADO concentrations requires further investigation. Genetically it has been shown that the selective overexpression of ADK-S in the brain either in transgenic mice or through a viral construct, was sufficient to trigger spontaneous seizures and an increase in the vulnerability to brain injury [26, 27, 73, 74], whereas the transgenic overexpression of ADK-L in neurons of the forebrain did not impact injury severity but affected baseline- and injury-induced neurogenesis [75]. Conversely, in cultured baby hamster fibroblasts overexpression of ADK-L increased DNA methylation about 4-fold, whereas the overexpression of ADK-S increased DNA methylation only 0.5 fold [29]. Together these findings suggest that ADK-S and ADK-L have distinct roles in regulating extracellular adenosine and epigenetic outcomes, respectively. There is additional evidence that ADK can associate with protein interaction partners, such as S-adenosyl homocysteine hydrolase [76]. Compartment specific differences with protein interaction partners might influence binding affinities and kinetic properties of the enzyme. Earlier drug discovery initiatives using cell-based or in vivo assays were geared at maximizing extracellular adenosine availability and subsequent receptor activation, whereas the impact on epigenetic outcome was not assayed. Currently available ADK inhibitor chemotypes do not appear to functionally discriminate between the long and short forms of ADK in in vitro preparations [10], however it is conceivable that compartment or tissue specific protein interaction partners might provide a possible physiological basis for different binding properties of inhibitors in a living cell-based system.
ADK-L is an exciting new therapeutic target because its manipulation offers a unique opportunity to manipulate DNA methylation therapeutically. This epigenetic role of ADK-L was demonstrated by using baby hamster kidney (BHK) fibroblasts engineered to either lack both forms of ADK [77], or to selectively overexpress either ADK-S or ADK-L [29]. Compared to ADK knockout cells, ADK-L cells had global DNA methylation levels, as quantified by 5-methylcytosine levels, that were 400% higher, whereas global DNA methylation levels in the ADK-S cells were increased by only 50% [29]. This implies that preferential ADK-L inhibitor activity would be desirable for epigenetic therapies aimed at blocking maladaptive methylation changes in a wide range of pathologies. Additional strategies to capitalize on the epigenetic functions of intracellular adenosine might be the combination of an ADK inhibitor with a transport blocker. This strategy would be expected to trap adenosine inside the cell, maximizing cell autonomous epigenetic effects, while minimizing effects on extracellular adenosine and associated receptor mediate side effects.
Using ADK inhibitors for epigenetic therapeutic interventions has several possible applications. Because increased DNA methylation drives the process of the development of epilepsy and its progression (i.e. epileptogenesis) [8, 29, 78–81], and because overexpression of ADK is a pathological hallmark of epilepsy [22, 23, 25] and a target for the prediction and prevention of epileptogenesis [26, 82] there is a new opportunity to use intracellular adenosine augmentation therapies for epilepsy prevention. Thus, the short term delivery of ADO via silk-based adenosine releasing brain implants, designed to release 500 ng ADO per day for only ten days, prevented progression of epilepsy development in a rat model of progressive temporal lobe epilepsy for at least 3 months. Importantly, the therapy was effective after disease onset, implicating that the epileptogeneic process can be stopped after the diagnosis of epilepsy [29]. Prevention of seizure activity in this model was linked to profound inhibition of DNA methyltransferase activity during active adenosine release and restoration of normal global DNA methylation levels, an effect that was maintained even after cessation of ADO release from the polymers [29]. This study provided the first proof that ADO can prevent epileptogenesis via an epigenetic mechanism. Subsequently, it was shown that the ADK inhibitor 5-IT likewise had robust antiepileptogenic effects in the intrahippocampal kainic acid model of temporal lobe epilepsy. It was demonstrated that the transient systemic administration of 5-IT for only 5 days given between day 3 and 8 after triggering epilepsy prevented epilepsy almost completely in >50% of all mice [83]. The role of epigenetic alterations in epilepsy is a new research area [84]. In addition to interference with DNA methylation, inhibitors of histone deacetylases (HDAC) have shown some efficacy in preclinical model systems of epilepsy and its development [85, 86], however all FDA approved HDAC inhibitors carry class-effect warnings of thrombocytopenia, neutropenia, and fetal risk [87]. Since those drugs are indicated for cancer, the benefit/risk profile supports their use in some cancer patients, whereas it needs to be explored, whether the benefits would outweigh the risks for the treatment of epilepsy.
ADK-L might be a promising therapeutic target for a variety of other conditions. Beta cells in the pancreas exclusively express ADK-L, but not ADK-S, and it has been shown that ADK inhibitors promote beta cell replication as a possible therapeutic strategy for diabetes [33]. Likewise it was shown that the ADK inhibitor 5-IT promoted neurogenesis after a traumatic brain injury (TBI), an activity linked to ADK-L, because the selective deletion of ADK-L from dentate granular neurons enhanced neurogenesis after TBI. Because ADK-L contributes to the regulation of cell proliferation in the pancreas and in the brain, it is also a potential therapeutic target for cancer [9]. DNA hypermethylation is a pathological hallmark of many cancers [88, 89], and in particular of isocitrate dehydrogenase mutant gliomas. Thus blocking ADK-L might be a therapeutic strategy to reverse the DNA hypermethylation phenotype in cancer.
8. Outlook and conclusions
Although initial drug discovery efforts to identify ADK inhibitors 20 years ago yielded promising efficacy data, they were encumbered by toxicity issues, which prevented the further development of several promising compounds [10]. However, a wealth of new insights gleaned from genetic studies shows that the adenosine system is highly compartmentalized and that the two isoforms of ADK (ADK-L and ADK-S) have distinct physiological functions. The discovery of ADK-L as an epigenetic regulator offers the intriguing but challenging possibility to target ADK-L in the nucleus selectively. Consequently, the discovery of novel ADK inhibitors has received renewed interest and several new drug discovery programs have been launched [60, 90]. Future research is needed to determine the feasibility of selective pharmacological modulation of cytoplasmic ADK-S and nuclear ADK-L using either novel small molecule ADK inhibitors or biologic agents. The identification of protein interaction partners of ADK might offer additional opportunities for more selective targeting of specific isoforms of the enzyme.
Acknowledgements:
DB acknowledges generous support from Citizens United for Research in Epilepsy (CURE) and funding through the NIH via grants R01NS065957 and R01NS103740.
Abbreviations:
- ADK
Adenosine Kinase (ATP:adenosine 5’-phosphotransferase)
- ADO
Adenosine
- A-286501
N7-((1’R,2’S,3’R,4’S)-2’,3’-dihydroxy-4’-amino-cyclopentyl)-4-amino-5-bromo-pyrrolo[2,3-a]pyrimidine
- A-134974
N7-((1’R,2’S,3’R,4’S)-2’,3’-dihydroxy-4’-amino-cyclopentyl)-4-amino-5-iodopyrrolo[2,3-a]pyrimidine
- ABT-702
4-amino-5-(3-bromophenyl)-7-(6-morpholino-pyridin-3-yl)pyrido[2,3,-d]pyrimidine
- HDAC
histone deacetylase
- NH2dADO
5’amino,5’-deoxyadenosine
- 5-IT
5-iodotubercidin
- 5’d-5IT
5’-deoxy,5-iodotubercidin
- TBI
traumatic brain injury
Footnotes
Conflicts of interest: MFJ is an employee of Abbvie, Inc. and may hold stock in Abbott Laboratories and Abbvie, Inc. DB is a co-founder of PrevEp LLC.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Burnstock G, Purine and purinergic receptors, Brain Neurosci Adv 2 (2018) 2398212818817494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Burnstock G, The therapeutic potential of purinergic signalling, Biochem Pharmacol 151 (2018) 157–165. [DOI] [PubMed] [Google Scholar]
- [3].Miller SL, Urey HC, Origin of Life, Science 130(3389) (1959) 1622–1624. [DOI] [PubMed] [Google Scholar]
- [4].Miller SL, Urey HC, Organic compound synthesis on the primitive earth, Science 130(3370) (1959) 245–51. [DOI] [PubMed] [Google Scholar]
- [5].Newby AC, Adenosine and the concept of ‘retaliatory metabolites’. Trends Biochem Sci 9(2) (1984) 42–44. [Google Scholar]
- [6].Sen B, Chakraborty A, Datta R, Bhattacharyya D, Datta AK, Reversal of ADP-mediated aggregation of adenosine kinase by cyclophilin leads to its reactivation, Biochemistry 45(1) (2006) 263–271. [DOI] [PubMed] [Google Scholar]
- [7].de Oliveira RR, Morales-Neto R, Rocco SA, Sforca ML, Polo CC, Tonoli CCC, Mercaldi GF, Cordeiro AT, Murakami MT, Franchini KG, Adenosine Kinase couples sensing of cellular potassium depletion to purine metabolism, Sci Rep 8(1) (2018) 11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Boison D, Adenosine kinase: exploitation for therapeutic gain, Pharmacological Reviews 65 (2013) 906–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Boison D, Yegutkin GG, Adenosine metabolism: emerging concepts for cancer therapy, Cancer Cell 36 (2019) 582–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jarvis MF, Therapeutic potential of adenosine kinase inhibition-Revisited, Pharmacol Res Perspect 7(4) (2019) e00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Park J, van Koeverden P, Singh B, Gupta RS, Identification and characterization of human ribokinase and comparison of its properties with E. coli ribokinase and human adenosine kinase, FEBS Lett 581(17) (2007) 3211–6. [DOI] [PubMed] [Google Scholar]
- [12].Park J, Gupta RS, Adenosine kinase and ribokinase - the RK family of proteins, Cellular and Molecular Life Sciences 65(18) (2008) 2875–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Park J, Gupta RS, Adenosine metabolism, adenosine kinase, and evolution, in: Boison D, Masino MA (Eds.), Adenosine: a key link between metabolism and central nervous system activity, Springer, New York, 2013, pp. 23–54. [Google Scholar]
- [14].Singh B, Hao W, Wu Z.-c., Eigl B, Gupta RS, Cloning and characterization of cDNA for adenosine kinase from mammalian (Chinese hamster, mouse, human and rat) species. High frequency mutants of Chinese hamster ovary cells involve structural alterations in the gene., Eur. J. Biochem. 241 (1996) 564–571. [DOI] [PubMed] [Google Scholar]
- [15].Singh B, Gupta RS, Genomic organization and linkage via a bidirectional promoter of the AP-3 (adaptor protein-3) mu3A and AK (adenosine kinase) genes: deletion mutants of AK in Chinese hamster cells extend into the AP-3 mu3A gene, Biochem J 378(Pt 2) (2004) 519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cui XA, Singh B, Park J, Gupta RS, Subcellular localization of adenosine kinase in mammalian cells: The long isoform of AdK is localized in the nucleus, Biochem Biophys Res Commun 388(1) (2009) 46–50. [DOI] [PubMed] [Google Scholar]
- [17].Boison D, Scheurer L, Zumsteg V, Rülicke T, Litynski P, Fowler B, Brandner S, Mohler H, Neonatal hepatic steatosis by disruption of the adenosine kinase gene, Proc Natl Acad Sci USA 99(10) (2002) 6985–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Moffatt BA, Stevens YY, Allen MS, Snider JD, Pereira LA, Todorova MI, Summers PS, Weretilnyk EA, Martin-McCaffrey L, Wagner C, Adenosine kinase deficiency is associated with developmental abnormalities and reduced transmethylation, Plant Physiol 128(3) (2002) 812–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bjursell MK, Blom HJ, Cayuela JA, Engvall ML, Lesko N, Balasubramaniam S, Brandberg G, Halldin M, Falkenberg M, Jakobs C, Smith D, Struys E, von Dobeln U, Gustafsson CM, Lundeberg J, Wedell A, Adenosine kinase deficiency disrupts the methionine cycle and causes hypermethioninemia, encephalopathy, and abnormal liver function, Am J Hum Genet 89(4) (2011) 507–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Staufner C, Lindner M, Dionisi-Vici C, Freisinger P, Dobbelaere D, Douillard C, Makhseed N, Straub BK, Kahrizi K, Ballhausen D, la Marca G, Kolker S, Haas D, Hoffmann GF, Grunert SC, Blom HJ, Adenosine kinase deficiency: expanding the clinical spectrum and evaluating therapeutic options, J Inherit Metab Dis 39(2) (2016) 273–83. [DOI] [PubMed] [Google Scholar]
- [21].Alhusani A, Obaid A, Blom HJ, Wedell A, Alfadhel M, Adenosine Kinase Deficiency: Report and Review, Neuropediatrics 50(1) (2019) 46–50. [DOI] [PubMed] [Google Scholar]
- [22].Aronica E, Sandau US, Iyer A, Boison D, Glial adenosine kinase - A neuropathological marker of the epileptic brain, Neurochem Int 63 (2013) 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Aronica E, Zurolo E, Iyer A, de Groot M, Anink J, Carbonell C, van Vliet EA, Baayen JC, Boison D, Gorter JA, Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy, Epilepsia 52(9) (2011) 1645–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Boison D, Aronica E, Comorbidities in Neurology: Is adenosine the common link?, Neuropharmacology 97 (2015) 18–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gouder N, Scheurer L, Fritschy J-M, Boison D, Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis, J Neurosci 24(3) (2004) 692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li T, Ren G, Lusardi T, Wilz A, Lan JQ, Iwasato T, Itohara S, Simon RP, Boison D, Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice, J Clin Inv 118(2) (2008) 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li T, Quan Lan J, Fredholm BB, Simon RP, Boison D, Adenosine dysfunction in astrogliosis: cause for seizure generation?, Neuron Glia Biol 3(4) (2007) 353–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pignataro G, Simon RP, Boison D, Transgenic overexpression of adenosine kinase aggravates cell death in ischemia, J Cereb Blood Flow Metab 27(1) (2007) 1–5. [DOI] [PubMed] [Google Scholar]
- [29].Williams-Karnesky RL, Sandau US, Lusardi TA, Lytle NK, Farrell JM, Pritchard EM, Kaplan DL, Boison D, Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis, J Clin Inv 123(8) (2013) 3552–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bjorness TE, Greene RW, Adenosine and sleep, Curr Neuropharmacol 7(3) (2009) 238–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Palchykova S, Winsky-Sommerer R, Shen HY, Boison D, Gerling A, Tobler I, Manipulation of adenosine kinase affects sleep regulation in mice, J Neurosci 30(39) (2010) 13157–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bjorness TE, Dale N, Mettlach G, Sonneborn A, Sahin B, Fienberg AA, Yanagisawa M, Bibb JA, Greene RW, An Adenosine-Mediated Glial-Neuronal Circuit for Homeostatic Sleep, J Neurosci 36(13) (2016) 3709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Annes JP, Ryu JH, Lam K, Carolan PJ, Utz K, Hollister-Lock J, Arvanites AC, Rubin LL, Weir G, Melton DA, Adenosine kinase inhibition selectively promotes rodent and porcine islet beta-cell replication, Proc Natl Acad Sci U S A 109(10) (2012) 3915–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang Y, Xu Y, Yan S, Cao K, Zeng X, Zhou Y, Liu Z, Yang Q, Pan Y, Wang X, Boison D, Su Y, Jiang X, Patel VS, Fulton D, Weintraub NL, Huo Y, Adenosine kinase is critical for neointima formation after vascular injury by inducing aberrant DNA hypermethylation, Cardiovasc Res (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Xu Y, Wang Y, Yan S, Zhou Y, Yang Q, Pan Y, Zeng X, An X, Liu Z, Wang L, Xu J, Cao Y, Fulton DJ, Weintraub NL, Bagi Z, Hoda MN, Wang X, Li Q, Hong M, Jiang X, Boison D, Weber C, Wu C, Huo Y, Intracellular adenosine regulates epigenetic programming in endothelial cells to promote angiogenesis, EMBO Mol Med 9(9) (2017) 1263–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Xu Y, Wang Y, Yan S, Yang Q, Zhou Y, Zeng X, Liu Z, An X, Toque HA, Dong Z, Jiang X, Fulton DJ, Weintraub NL, Li Q, Bagi Z, Hong M, Boison D, Wu C, Huo Y, Regulation of endothelial intracellular adenosine via adenosine kinase epigenetically modulates vascular inflammation, Nat Commun 8(1) (2017) 943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].El-Kharrag R, Owen R, Boison D, Adenosine Kinase Deficiency Increases Susceptibility to a Carcinogen, J Caffeine Adenosine Res 9(1) (2019) 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Burnstock G, Purinergic nerves, Pharmacol Rev 24(3) (1972) 509–81. [PubMed] [Google Scholar]
- [39].Williams M, Jarvis MF, Purinergic and pyrimidinergic receptors as potential drug targets, Biochem Pharmacol 59(10) (2000) 1173–85. [DOI] [PubMed] [Google Scholar]
- [40].Jacobson KA, Gao ZG, Adenosine receptors as therapeutic targets, Nat Rev Drug Discov 5(3) (2006) 247–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jacobson KA, Jarvis MF, Williams M, Purine and pyrimidine (P2) receptors as drug targets, J Med Chem 45(19) (2002) 4057–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Engler R, Consequences of activation and adenosine-mediated inhibition of granulocytes during myocardial ischemia, Fed Proc 46(7) (1987) 2407–12. [PubMed] [Google Scholar]
- [43].Newby AC, Holmquist CA, Illingworth J, Pearson JD, The control of adenosine concentration in polymorphonuclear leucocytes, cultured heart cells and isolated perfused heart from the rat, Biochem J 214(2) (1983) 317–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mullane K, Acadesine: the prototype adenosine regulating agent for reducing myocardial ischaemic injury, Cardiovasc Res 27(1) (1993) 43–7. [DOI] [PubMed] [Google Scholar]
- [45].Davies LP, Jamieson DD, Baird-Lambert JA, Kazlauskas R, Halogenated pyrrolopyrimidine analogues of adenosine from marine organisms: pharmacological activities and potent inhibition of adenosine kinase, Biochem Pharmacol 33(3) (1984) 347–55. [DOI] [PubMed] [Google Scholar]
- [46].Kowaluk EA, Bhagwat SS, Jarvis MF, Adenosine kinase inhibitors, Current Pharmaceutical Design 4(5) (1998) 403–416. [PubMed] [Google Scholar]
- [47].Pak MA, Haas HL, Decking UKM, Schrader J, Inhibition of adenosine kinase increases endogenous adenosine and depresses neuronal activity in hippocampal slices, Neuropharmacol 33 (1994) 1049–1053. [DOI] [PubMed] [Google Scholar]
- [48].Golembiowska K, White TD, Sawynok J, Adenosine kinase inhibitors augment release of adenosine from spinal cord slices, Eur J Pharmacol 307(2) (1996) 157–62. [DOI] [PubMed] [Google Scholar]
- [49].Liu XJ, White TD, Sawynok J, Potentiation of formalin-evoked adenosine release by an adenosine kinase inhibitor and an adenosine deaminase inhibitor in the rat hind paw: a microdialysis study, Eur J Pharmacol 408(2) (2000) 143–52. [DOI] [PubMed] [Google Scholar]
- [50].Britton DR, Mikusa J, Lee CH, Jarvis MF, Williams M, Kowaluk EA, Site and event specific increase of striatal adenosine release by adenosine kinase inhibition in rats, Neurosci Lett 266(2) (1999) 93–6. [DOI] [PubMed] [Google Scholar]
- [51].Lee CH, Jiang M, Cowart M, Gfesser G, Perner R, Kim KH, Gu YG, Williams M, Jarvis MF, Kowaluk EA, Stewart AO, Bhagwat SS, Discovery of 4-amino-5-(3-bromophenyl)-7-(6-morpholino-pyridin-3-yl)pyrido[2,3-d]pyrimidine, an orally active, non-nucleoside adenosine kinase inhibitor, J Med Chem 44(13) (2001) 2133–8. [DOI] [PubMed] [Google Scholar]
- [52].Erion MD, Ugarkar BG, Dare J, Castellino AJ, Fujitaki JM, Dixon R, Appleman JR, Wiesner JB, Design, Synthesis and Anticonvulsant Activity Of the Potent Adenosine Kinase Inhibitor Gp3269, Nucleosides & Nucleotides 16(7–9) (1997) 1013–1021. [Google Scholar]
- [53].Erion MD, Wiesner JB, Rosengren S, Ugarkar BG, Boyer SH, Tsuchiya M, Therapeutic potential of adenosine kinase inhibitors as analgesic agents., Drug Dev Res 50 (2000) S14–06. [Google Scholar]
- [54].McGaraughty S, Cowart M, Jarvis MF, Recent developments in the discovery of novel adenosine kinase inhibitors: mechanism of action and therapeutic potential, CNS Drug Rev 7(4) (2001) 415–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].McGaraughty S, Cowart M, Jarvis MF, Berman RF, Anticonvulsant and antinociceptive actions of novel adenosine kinase inhibitors, Curr Top Med Chem 5(1) (2005) 43–58. [DOI] [PubMed] [Google Scholar]
- [56].Jarvis MF, Mikusa J, Chu KL, Wismer CT, Honore P, Kowaluk EA, McGaraughty S, Comparison of the ability of adenosine kinase inhibitors and adenosine receptor agonists to attenuate thermal hyperalgesia and reduce motor performance in rats, Pharmacol Biochem Behav 73(3) (2002) 573–81. [DOI] [PubMed] [Google Scholar]
- [57].Jarvis MF, Yu H, Kohlhaas K, Alexander K, Lee CH, Jiang M, Bhagwat SS, Williams M, Kowaluk EA, ABT-702 (4-amino-5-(3-bromophenyl)-7-(6-morpholinopyridin-3-yl)pyrido[2, 3-d]pyrimidine), a novel orally effective adenosine kinase inhibitor with analgesic and anti-inflammatory properties: I. In vitro characterization and acute antinociceptive effects in the mouse, J Pharmacol Exp Ther 295(3) (2000) 1156–64. [PubMed] [Google Scholar]
- [58].Gomtsyan A, Didomenico S, Lee CH, Matulenko MA, Kim K, Kowaluk EA, Wismer CT, Mikusa J, Yu H, Kohlhaas K, Jarvis MF, Bhagwat SS, Design, synthesis, and structure-activity relationship of 6-alkynylpyrimidines as potent adenosine kinase inhibitors, J Med Chem 45(17) (2002) 3639–48. [DOI] [PubMed] [Google Scholar]
- [59].Muchmore SW, Smith RA, Stewart AO, Cowart MD, Gomtsyan A, Matulenko MA, Yu H, Severin JM, Bhagwat SS, Lee CH, Kowaluk EA, Jarvis MF, Jakob CL, Crystal structures of human adenosine kinase inhibitor complexes reveal two distinct binding modes, J Med Chem 49(23) (2006) 6726–31. [DOI] [PubMed] [Google Scholar]
- [60].Toti KS, Osborne D, Ciancetta A, Boison D, Jacobson KA, South (S)- and North (N)-Methanocarba-7-Deazaadenosine Analogues as Inhibitors of Human Adenosine Kinase, Journal of Medicinal Chemistry 59(14) (2016) 6860–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kose M, Schiedel AC, Bauer AA, Poschenrieder H, Burbiel JC, Akkinepally RR, Stachel HD, Muller CE, Focused screening to identify new adenosine kinase inhibitors, Bioorg Med Chem 24(21) (2016) 5127–5133. [DOI] [PubMed] [Google Scholar]
- [62].Kowaluk EA, Mikusa J, Wismer CT, Zhu CZ, Schweitzer E, Lynch JJ, Lee CH, Jiang M, Bhagwat SS, Gomtsyan A, McKie J, Cox BF, Polakowski J, Reinhart G, Williams M, Jarvis MF, ABT-702 (4-amino-5-(3-bromophenyl)-7-(6-morpholino-pyridin- 3-yl)pyrido[2,3-d]pyrimidine), a novel orally effective adenosine kinase inhibitor with analgesic and anti-inflammatory properties. II. In vivo characterization in the rat, J Pharmacol Exp Ther 295(3) (2000) 1165–74. [PubMed] [Google Scholar]
- [63].Wiesner JB, Ugarkar BG, Castellino AJ, Barankiewicz J, Dumas DP, Gruber HE, Foster AC, Erion MD, Adenosine kinase inhibitors as a novel approach to anticonvulsant therapy, J Pharmacol Exp Ther 289(3) (1999) 1669–77. [PubMed] [Google Scholar]
- [64].Chen JF, Eltzschig HK, Fredholm BB, Adenosine receptors as drug targets--what are the challenges?, Nat Rev Drug Discov 12(4) (2013) 265–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kowaluk EA, Jarvis MF, Therapeutic potential of adenosine kinase inhibitors, Expert Opin Investig Drugs 9(3) (2000) 551–64. [DOI] [PubMed] [Google Scholar]
- [66].Xiao C, Liu N, Jacobson KA, Gavrilova O, Reitman ML, Physiology and effects of nucleosides in mice lacking all four adenosine receptors, PLoS Biol 17(3) (2019) e3000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Matulenko MA, Lee CH, Jiang M, Frey RR, Cowart MD, Bayburt EK, Didomenico S, Gfesser GA, Gomtsyan A, Zheng GZ, McKie JA, Stewart AO, Yu H, Kohlhaas KL, Alexander KM, McGaraughty S, Wismer CT, Mikusa J, Marsh KC, Snyder RD, Diehl MS, Kowaluk EA, Jarvis MF, Bhagwat SS, 5-(3-Bromophenyl)-7-(6-morpholin-4-ylpyridin-3-yl)pyrido[2,3-d]pyrimidin-4-ylamin e: structure-activity relationships of 7-substituted heteroaryl analogs as non-nucleoside adenosine kinase inhibitors, Bioorg Med Chem 13(11) (2005) 3705–20. [DOI] [PubMed] [Google Scholar]
- [68].Matulenko MA, Paight ES, Frey RR, Gomtsyan A, DiDomenico S Jr., Jiang M, Lee CH, Stewart AO, Yu H, Kohlhaas KL, Alexander KM, McGaraughty S, Mikusa J, Marsh KC, Muchmore SW, Jakob CL, Kowaluk EA, Jarvis MF, Bhagwat SS, 4-amino-5-aryl-6-arylethynylpyrimidines: structure-activity relationships of non-nucleoside adenosine kinase inhibitors, Bioorg Med Chem 15(4) (2007) 1586–605. [DOI] [PubMed] [Google Scholar]
- [69].Bookser BC, Ugarkar BG, Matelich MC, Lemus RH, Allan M, Tsuchiya M, Nakane M, Nagahisa A, Wiesner JB, Erion MD, Adenosine kinase inhibitors. 6. Synthesis, water solubility, and antinociceptive activity of 5-phenyl-7-(5-deoxy-beta-D-ribofuranosyl)pyrrolo[2,3-d]pyrimidines substituted at C4 with glycinamides and related compounds, J Med Chem 48(24) (2005) 7808–20. [DOI] [PubMed] [Google Scholar]
- [70].Kohler D, Streienberger A, Morote-Garcia JC, Granja TF, Schneider M, Straub A, Boison D, Rosenberger P, Inhibition of Adenosine Kinase Attenuates Acute Lung Injury, Crit Care Med 44(4) (2016) e181–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wahlman C, Doyle TM, Little JW, Luongo L, Janes K, Chen Z, Esposito E, Tosh DK, Cuzzocrea S, Jacobson KA, Salvemini D, Chemotherapy-induced pain is promoted by enhanced spinal adenosine kinase levels through astrocyte-dependent mechanisms, Pain 159(6) (2018) 1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zhu CZ, Gopalakrishnan S, Doyle K, Nikkel AL, Olson L, Abraham VC, Leys L, Widomski D, Salte K, Putman B, Pratt S, Ma J, Su Z, Gopalakrishnan M, Lee CH, McGaraughty SP, A-306989, an inhibitor of adenosine kinase, is renoprotective in rodent models of podocyte, basement membrane, and obstructive injury, Eur J Pharmacol 788 (2016) 1–11. [DOI] [PubMed] [Google Scholar]
- [73].Shen HY, Lusardi TA, Williams-Karnesky RL, Lan JQ, Poulsen DJ, Boison D, Adenosine kinase determines the degree of brain injury after ischemic stroke in mice, J Cereb Blood Flow Metab 31(7) (2011) 1648–1659; PMCID: PMC3137468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Shen HY, Sun H, Hanthorn MM, Zhi Z, Lan JQ, Poulsen DJ, Wang RK, Boison D, Overexpression of adenosine kinase in cortical astrocytes and focal neocortical epilepsy in mice, J Neurosurg 120(3) (2014) 628–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Gebril HM, Rose RM, Gesese R, Emond MP, Huo Y, Aronica E, Boison D, Adenosine kinase inhibition promotes proliferation of neural stem cells after traumatic brain injury, Brain Commun 2(1) (2020) fcaa017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Lee S, Doxey AC, McConkey BJ, Moffatt BA, Nuclear targeting of methyl-recycling enzymes in Arabidopsis thaliana is mediated by specific protein interactions, Mol Plant 5(1) (2012) 231–48. [DOI] [PubMed] [Google Scholar]
- [77].Huber A, Padrun V, Deglon N, Aebischer P, Mohler H, Boison D, Grafts of adenosine-releasing cells suppress seizures in kindling epilepsy, Proc. Natl. Acad. Sci. USA 98(13) (2001) 7611–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kobow K, Blumcke I, The methylation hypothesis: do epigenetic chromatin modifications play a role in epileptogenesis?, Epilepsia 52 Suppl 4 (2011) 15–9. [DOI] [PubMed] [Google Scholar]
- [79].Kobow K, Kaspi A, Harikrishnan KN, Kiese K, Ziemann M, Khurana I, Fritzsche I, Hauke J, Hahnen E, Coras R, Muhlebner A, El-Osta A, Blumcke I, Deep sequencing reveals increased DNA methylation in chronic rat epilepsy, Acta Neuropathol 126(5) (2013) 741–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kobow K, Blumcke I, Epigenetic mechanisms in epilepsy, Prog Brain Res 213 (2014) 279–316. [DOI] [PubMed] [Google Scholar]
- [81].Klein P, Dingledine R, Aronica E, Bernard C, Blumcke I, Boison D, Brodie MJ, Brooks-Kayal AR, Engel J Jr., Forcelli PA, Hirsch LJ, Kaminski RM, Klitgaard H, Kobow K, Lowenstein DH, Pearl PL, Pitkanen A, Puhakka N, Rogawski MA, Schmidt D, Sillanpaa M, Sloviter RS, Steinhauser C, Vezzani A, Walker MC, Loscher W, Commonalities in epileptogenic processes from different acute brain insults: Do they translate?, Epilepsia 59(1) (2018) 37–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Diamond ML, Ritter AC, Jackson EK, Conley YP, Kochanek PM, Boison D, Wagner AK, Genetic variation in the adenosine regulatory cycle is associated with posttraumatic epilepsy development, Epilepsia 56(8) (2015) 1198–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Sandau US, Yahya M, Bigej R, Friedman JL, Saleumvong B, Boison D, Transient use of a systemic adenosine kinase inhibitor attenuates epilepsy development in mice, Epilepsia 60(4) (2019) 615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Boison D, Rho JM, Epigenetics and epilepsy prevention: The therapeutic potential of adenosine and metabolic therapies, Neuropharmacology 167 (2020) 107741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Citraro R, Leo A, Santoro M, D’Agostino G, Constanti A, Russo E, Role of Histone Deacetylases (HDACs) in Epilepsy and Epileptogenesis, Curr Pharm Des 23(37) (2017) 5546–5562. [DOI] [PubMed] [Google Scholar]
- [86].Basu T, O’Riordan KJ, Schoenike BA, Khan NN, Wallace EP, Rodriguez G, Maganti RK, Roopra A, Histone deacetylase inhibitors restore normal hippocampal synaptic plasticity and seizure threshold in a mouse model of Tuberous Sclerosis Complex, Sci Rep 9(1) (2019) 5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Matsuoka H, Unami A, Fujimura T, Noto T, Takata Y, Yoshizawa K, Mori H, Aramori I, Mutoh S, Mechanisms of HDAC inhibitor-induced thrombocytopenia, Eur J Pharmacol 571(2–3) (2007) 88–96. [DOI] [PubMed] [Google Scholar]
- [88].Huang WY, Hsu SD, Huang HY, Sun YM, Chou CH, Weng SL, Huang HD, MethHC: a database of DNA methylation and gene expression in human cancer, Nucleic Acids Res 43(Database issue) (2015) D856–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Gokul G, Khosla S, DNA methylation and cancer, Subcell Biochem 61 (2013) 597–625. [DOI] [PubMed] [Google Scholar]
- [90].Iqbal J, Burbiel JC, Muller CE, Development of off-line and on-line capillary electrophoresis methods for the screening and characterization of adenosine kinase inhibitors and substrates, Electrophoresis 27(12) (2006) 2505–17. [DOI] [PubMed] [Google Scholar]